
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing direct hydroxylation of benzene to phenol on Fe1/PMA single-atom catalyst: a comparative study of H2O2vs. O2-assisted reactions†
Beenish
Bashir
 ab,
Shamraiz Hussain
Talib
*cd,
Muhammad Ajmal
Khan
b,
Sharmarke
Mohamed
cd,
Ahsanulhaq
Qurashi
cd,
Hai
Xiao
a and
Jun
Li
*ae
ab,
Shamraiz Hussain
Talib
*cd,
Muhammad Ajmal
Khan
b,
Sharmarke
Mohamed
cd,
Ahsanulhaq
Qurashi
cd,
Hai
Xiao
a and
Jun
Li
*ae
aDepartment of Chemistry and Engineering Research Center of Advanced Rare-Earth Materials of Ministry of Education, Tsinghua University, Beijing 100084, China. E-mail: junli@tsinghua.edu.cn; Fax: +86(10)6279747; Tel: +86(10)62795381
bDepartment of Chemistry and Biochemistry, George Mason University, 4400 University Drive, Fairfax, VA 22030, USA
cCenter for Catalysis and Separations, Khalifa University of Science and Technology, P.O. Box 127788, Abu Dhabi, United Arab Emirates. E-mail: dal16@tsinghua.org.cn
dDepartment of Chemistry, Khalifa University of Science and Technology, Abu Dhabi 127788, United Arab Emirates
eDepartment of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
First published on 23rd May 2024
Abstract
In this study, systematic first-principles calculations were carried out to ascertain the optimal single-atom catalyst (SAC) among the 3d-transition metals (TM1 = Fe1, Co1, Ni1, Cu1, and Zn1) anchored on a phosphomolybdic acid (PMA) cluster for efficient benzene oxidation to phenol, which is otherwise challenging at ambient temperature. Strong binding due to substantial charge transfer between Fe1 and PMA, and the adsorption energies of H2O2 and O2 oxidants enabled significant bonding within the Fe1/PMA cluster, facilitating enhanced catalytic performance compared to that of 3d-TM1 (Co1, Ni1, Cu1, and Zn1). The Fe1/PMA cluster demonstrated enhanced reactivity towards H2O2 supported by lower activation barriers and rate-determining steps for H2O2 (0.84 and 0.67 eV) compared to O2 (1.02 and 0.66 eV). The spontaneous dissociation of H2O2 on Fe1/PMA, in contrast to O2, is a crucial step to initiate iron–oxo (Fe1–O) active site formation, easing benzene to phenol oxidation at ambient temperature. Thus, the proficient coordination environment of Fe1 atoms as SACs adsorbed on the PMA cluster is found to influence catalytic performance, especially in the case of H2O2. The proposed mechanism is reminiscent of hydrocarbon hydroxylation in enzymatic processes, establishing Fe1/PMA as an environmentally friendly, heterogeneous and non-noble metal green catalyst for electrocatalytic phenol production.
1. Introduction
The selective transformation of hydrocarbons, such as xylene, toluene, benzene, etc., into valuable compounds like phenol by the oxidation of benzene under normal working conditions is one of the most widely researched fundamental topics in applied catalysis.1–3 However, high-energy benzene activation significantly reduces the production yield of phenol, increasing the conversion cost and making it challenging from the research viewpoint.4,5 Currently, phenol is produced by the direct H2O2-based oxidation of benzene as described by the three-step cumene process developed in the 1950s.6 Despite the considerable success of the cumene process, being energy deficient, producing a large number of by-products and pollutants, and especially low selectivity for phenol are obvious concerns regarding the efficiency of the process.7 To overcome the drawbacks of benzene to phenol indirect conversion, direct hydroxylation is considered a potential, economical, and environment-friendly route.7,8 The direct hydroxylation of benzene is carried out using different catalysts, such as mesoporous molecular sieves attached with titanium, Fe-zeolites, vanadium-substituted phosphomolybdate, palladium membranes, etc. by one-step phenol production.1,8–10 The direct hydroxylation of benzene has also been extensively studied using transition metals such as Fe, Cu, and V,11,12 while the Fenton reagent (Fe2+–H2O2)13 has been most widely used as a catalyst in processes involving benzene conversion to phenol. Besides, several studies have reported Fe–g-C3N4 and TiO2 as being active catalysts for the benzene oxidation reaction occurring under visible or ultraviolet light.14,15 During the last two decades, the direct hydroxylation of benzene has been of key research interest and extensively studied for phenol production,16,17 which is attributed to the independence of the market price of acetone for phenol production via a one-step process.6,18,19Numerous researchers have adopted direct hydroxylation of benzene to phenol using different green oxidants such as molecular oxygen (O2),20–22 hydrogen peroxide (H2O2),8,23,24 a mixture of oxygen (O) and hydrogen (H),1,25,26 and nitrous oxide (N2O),27–30via three main pathways. High-temperature requirements led to the over-oxidation of benzene, decreasing the phenol selectivity and the process yield.30,31 Among different green oxidants, N2O has been adopted as a commercial catalytic oxidant, and it can be retrieved as a cheap by-product at the end of the process.32 In contrast to the other catalysts, the generation of water as a by-product is a clear advantage of H2O2 over the rest, therefore utilizing a H2O2 oxidant under a titanium-containing zeolite-catalyst for the direct hydroxylation of benzene to phenol offers outstanding catalytic efficiency even without provisional reaction conditions.9 Meanwhile, developing low-cost and green heterogeneous catalysts to produce phenol via one-step hydroxylation of benzene under normal working conditions is highly imperative from the industrial viewpoint.
Single-atom catalysis (SAC) is a new branch of heterogeneous catalysis in which transition metal active centers are dispersed over the surface of selective metal oxide or conducting surfaces to better utilize atoms to improve selectivity and enhance the reaction kinetics.33,34 Despite the atomic scale dispersion, post-use separation of catalysts is not required during industrial chemical reactions. One of the greatest advantages of the SAC is an extensive increase in catalytic efficiency and selectivity, where a catalyst present as a single atom significantly reduces the demand for costly metals and requires minimal quantity.35 Significant downsizing of a catalyst from nanoparticle catalysts, nanoclusters, and dots to SAC substantially changes the bonds and coordination environments of electronic shell configurations that exclusively changes the chemical phenomena and result in an efficient catalytic response. However, the SAC structure strongly depends on the processing pressure and temperature, while the catalytic activity quenches with the formation of nanoparticles on the support. The accumulation of metal atoms occurs due to the high surface energy of the metal atoms, which results in the formation of giant clusters of metal oxide. Therefore, developing an appropriate, microscopically smooth, and efficiently active support surface is a prerequisite to understanding the true nature of SACs, especially while working under ambient conditions.35,36 In this regard, several computational and experimental research studies have reported stable metal oxide supports that effectively supported SACs.34,37,38 Whereas the broad spectrum of SAC applications such as electro-catalysis, photo-electrocatalysis, photo-catalysis, and thermo-catalysis is evidence of the exceptional catalytic potential based on the specific electronic structure, especially for the case of C–H bond activation.39,40 Similarly, non-noble metal SACs are also good at benzene hydroxylation with H2O2, but their efficiency is very low and needs improvement.11–13,41 Besides, the complex composition of catalysts results in a poor understanding of their catalytic active sites and structure–activity relationship that ultimately leads to relatively low efficiencies of the catalytic reactions.
Recently, heteropolyacid oxo-anions known as polyoxometalate (POM) clusters based on their excellent catalytic characteristics and well-defined structures are being considered the most promising and highly stable metal oxide supports for SACs, and they provide coordination sites for single atom anchoring and protect leaching of active metal centers.42,43 Recently, researchers have highlighted the promising potential of POM-based materials and their derivatives for their application in electrocatalysis and energy storage.44 The study is based on systematic experiments, conducted at elevated temperatures (150–400 °C), in which surface oxygen species derived from the polyoxometalate (POM) component act as the oxidizing agent for the conversion of CO to CO2.45 Another study explored a novel strategy for SAC creation. By employing POM cluster traps, researchers were able to disperse cobalt atoms and create highly active and stable SACs for the oxygen reduction reaction (ORR).46 One of the distinctive features of the POM family members like H3PMo12O40 and PMA is their keggin-type structure which is considered very effective in stabilizing single Pt atoms.47 That is the reason why the POMs studied as heterogeneous catalysts offer a perfect platform to investigate the structure–activity relationship in SACs.48,49
Iron-based (Fe) surfaces in catalysis have remarkably changed the world. The Haber–Bosch process is an important example in which Fe-based catalysts usually convert molecular nitrogen into ammonia which is the building block for food production with an ever-growing global population.50 The formal oxidation states of Fe range from −II to +VI and present a wide window of redox reactions. Lewis acidity varies from moderate to high and its cations can bind with N- and O- species N-based heterocycles. Iron-based catalysts played a critical role in direct benzene oxidation to phenol on Fe-based surfaces.51 Earlier copper-based complexes were used for direct oxidation but yield was quite low.52 Moreover, supports are also critical in heterogeneous systems. ZSM-5 and other porous supports were used for Fe-based catalysts to perform direct oxidation of benzene to phenol.53 The present study proposed a new path of using a PMA cluster as a support to avoid leaching and access abundant sites for SAC.
Based on the proposed features of the heterogeneous SAC in preceding studies, current work focuses on the extensive density functional theory (DFT) calculations of benzene oxidation to phenol using Fe1 atoms anchored on the PMA cluster support under ambient working conditions. In addition to the electronic structure, geometry stability, and Fe1/PMA catalytic activity calculations, the H2O2 and O2 oxidant absorption is also studied. Eventually, the present study aims to investigate the SAC mechanism dominating the benzene oxidation to phenol by H2O2 and O2 as active oxidants under ambient conditions.
2. Computational details
In this study, the energy calculations and geometrical optimizations are conducted using the Vienna ab initio simulation package (VASP)54,55 through spin-polarized DFT with the GGA-PBE exchange–correlation functional.56,57 To explain the interactions between the ionic core and valence electrons, the projector augmented wave (PAW) method was utilized.58 The valence electrons designated to the corresponding atoms are 3d-TMs-Fe1, Co1, Ni1, Cu1, Zn1 (3dx 4sx), Mo (4d5 5s1), O (2s2 2p4), P (3s2 3p3), and H (1s1). The kinetic energy cutoff was set to be 400 eV for a plane-wave basis set expansion of the electron eigenfunction. An appropriate 20 Å vacuum space was preset to avoid the interactivity among different periodic systems under study. A widely used empirical dispersion correction method, the Grimme DFT-D3 method, was used to investigate the long-range van der Waals interactions. The accuracy of the Brillouin zone integration for the PMA was enhanced by using a 1 × 1 × 1 k sampling point during the geometry optimization of the PMA itself, the binding of a specific TM1, and the adsorption of H2O2 and O2 oxidants on the PMA cluster for benzene oxidation to phenol, as shown in Fig. 1 (for more details, see Table S1 in the ESI†).33,42 Moreover, convergence for the total energy was achieved at 10−5 eV, and the maximum allowed relaxation force was set to less than 0.03 eV Å−1. The matrix-based Bader analysis was used for the computation of electron charge exchange.59 The binding energy of the TM1 atom on the PMA is given as follows:| Ebin(TM1) = Etot (TM1−PMA) − E(PMA) − E(TM1) | (1) |
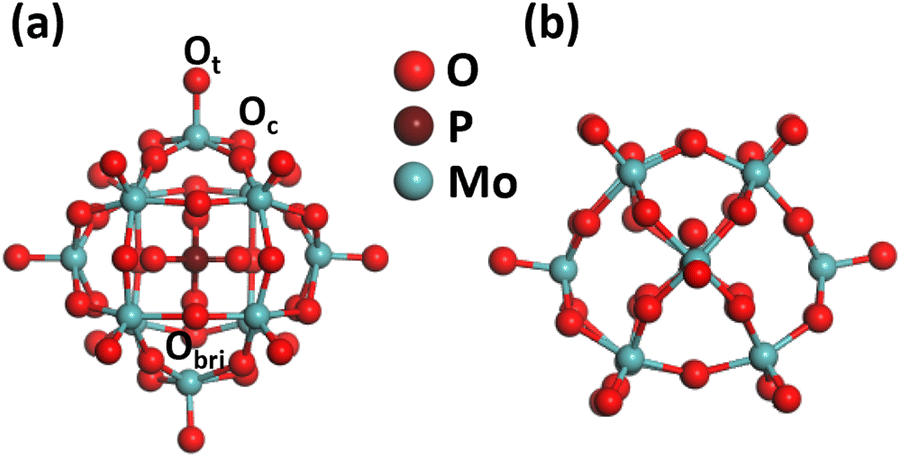 | ||
| Fig. 1 Optimized structure of PMA: (a) top view; (b) side view. Color code: Ot/Ob, red; Mo, dark cyan; and P, pink. | ||
The adsorption energy is defined as follows:
| ΔEads = E(adsorbate+catalyst) − E(adsorbate) − E(catalyst) | (2) |
3. Results and discussion
3.1. Exploring coordination environments in PMA for SACs
Phosphomolybdic acid (PMA) establishes a kegging-type structure where the corners of 12 MO6 octahedrons are connected to the PO4 tetrahedron pinned at the center. The cubic unit cell at the epicenter of the classical keggin comprises 12 Mo atoms, 40 O atoms, and 1 P atom. The oxygen atoms present on the surface of the PMA cluster offer different coordination sites, such as the terminal (Ot), bridging (Obri), and at the sides of the Keggin structure (Oc). Therefore, the PMA cluster can provide single to multifold binding sites for metal atoms such as the single corner, Oc–Obr-bridge, three-fold hollow (3-H_Oc and 3-H_Obr), and four-fold hollow (4-H) sites. The optimized structure of PMA is shown in Fig. 1, and the possible coordination sites are presented in Fig. S1 of the ESI.† The optimized key geometry parameters are presented in Table S2 (ESI†).3.2. Screening binding energies of 3d-TMs on PMA
A series of 3d-TMs in the periodic table (3d-TMs = Fe1, Co1, Ni1, Cu1, and Zn1) were investigated for an ideal catalytic system for benzene oxidation to phenol. As shown in Fig. S2 (ESI†), all the considered 3d-TM atoms are expected to anchor at the 4H sites on the PMA cluster, which is consistent with previous theoretical calculations.33,63 The effectiveness of heterogeneous catalysis relies heavily on the interaction between the adsorbed metal and its supporting material, determining the catalytic activity and stability of the catalytic system. Our study aimed to evaluate the catalytic potential of 3d-TM1, including Fe1, Co1, Ni1, Cu1, and Zn1, by analyzing their interactions with a PMA cluster. The computational results revealed negative binding and formation energies for the interactions between 3d-TM1 adatoms and the PMA cluster. These negative values generally decrease as the atomic size of the adatoms increases, as presented in Table S3 (ESI†). Notably, Fe1 exhibited the highest binding energy at −10.47 eV, while Zn1 displayed the lowest binding energy at −5.46 eV. The substantial binding and formation energies of Fe1 develop a robust bond with the PMA cluster, making it more stable compared to the other 3d-TMs (Co1, Ni1, Cu1, and Zn1).Besides, Bader charge analysis was conducted for gaining deeper insights into the strong binding between the 3d-TMs and the PMA cluster. The results, outlined in Table S3 (ESI†), consistently showed that as the atomic size of the 3d-TMs increased, there was a decrease in the electron transfer from these metals to the PMA cluster. A strong correlation between binding energies and charge transfer was observed for transition metals adsorbed on the PMA cluster. This correlation suggests that a higher degree of charge transfer from the transition metal atoms corresponds to stronger binding energies of these atoms. In this context, the substantial charge transfer between Fe1 and the PMA cluster played a pivotal role in establishing a strong binding energy observed, especially when compared to other 3d-TM1 (Co1, Ni1, Cu1, and Zn1). This strong metal-cluster binding interaction, driven by significant charge transfer, suggests that the Fe1/PMA catalyst may exhibit exceptional catalytic activity and stability in this specific reaction.
3.3. Screening 3d-TM1/PMA clusters via adsorption of H2O2 and O2 oxidants for unveiling active phenol catalysis
In the context of heterogeneous catalysis, the adsorption of reactant molecules plays a pivotal role. Therefore, before delving into the detailed mechanism of benzene oxidation to phenol, we initiated an individual investigation into the adsorption of H2O2 and O2 molecules, which serve as oxidants, on Fe1, Co1, Ni1, Cu1, and Zn1/PMA clusters under ambient reaction conditions. The optimized structures of H2O2 on the 3d-TM1/PMA clusters are visually represented in Fig. S3 (ESI†), and the corresponding adsorption energies are provided in Table S4 (ESI†). From the outcomes of these calculations, it is evident that H2O2 exhibits relatively weak adsorption on Zn1/PMA, with an adsorption energy of merely −0.33 eV. In contrast, the most substantial interaction is observed with Fe1/PMA, where the adsorption energy amounts to −0.53 eV.Regarding O2 adsorption on the 3d-TM1/PMA surfaces, different binding configurations are observed as shown in Fig. S4 (ESI†). O2 molecules tend to align in a parallel fashion over the Fe1 and Co1 atoms, with both oxygen atoms of O2 forming connections with the metal atom (referred to as the side-on coordination). Conversely, for Ni1, Cu1, and Zn1, only one oxygen atom establishes a connection with the metal atom (termed the end-on coordination). The most robust interaction is likewise witnessed with Fe1/PMA, where the adsorption energy is measured at −0.41 eV, while Cu1/PMA exhibits the least favorable adsorption energy at −0.04 eV. The results indicate that among the 3d-TM1/PMA clusters, Fe1/PMA demonstrates the highest adsorption energies for both H2O2 and O2. Based on the computational results, Fe1/PMA is a highly active and stable catalyst for benzene oxidation to phenol due to its strong binding interactions with both the PMA cluster and the oxidant molecules, as detailed in Table S4 (ESI†).
3.4. Electronic properties of Fe1/PMA
According to the optimized geometry structure of the Fe1/PMA cluster shown in Fig. 2(a) and (b), the Fe1 atoms are allocated at the most stable 4-Hollow (4-H) sites of the PMA cluster, which is more favorable (Table S5 of the ESI†), where Fe1 develops Fe1–O bonding (1.84 and 1.81 Å) with the four adjacent oxygen atoms. The calculated binding energy (−10.47 eV) testifies the strong interaction between the Fe1 atom and PMA which makes it a compelling candidate for achieving a chemically stable SAC. Based on the Bader charge analysis, there exists a charge difference between neighboring oxygen atoms (−1.16|e|) and Fe1 atoms (1.66|e|) due to which the electron density tends to transfer from Fe1 to PMA and the surface acts as an electron-withdrawing support, consequently, the Fe1 single atom stabilizes and the binding energy between the Fe1 atom and the support surface increases. Because of having a more positive charge, the Fe1 atom not only stabilizes the adsorption of gases, which is important for a catalytic reduction but also provokes the catalytic reaction of benzene oxidation due to being in a higher oxidation state. Moreover, the charge density differences of the Fe1/PMA cluster were also calculated to gain insights into the nature of chemical bonding between the Fe1 atom and PMA cluster, and the results are shown in Fig. 2(c) and (d). As seen from Fig. 2(c) and (d), a significant charge density accumulates around Fe1 and neighboring O atoms because of the Fe1/PMA cluster assimilation, significantly reorganizing electrons. The magnetic moment of Fe1 atoms (2.90μB) and oxygen atoms (−0.07μB) also suggests strong spin density on the Fe1 atoms compared to O atoms. That is why the H2O and O2 molecules can easily activate and coordinate with free electrons on the Fe1 atoms. Spin-polarized partial density of states (PDOS) results shown in Fig. 2(e) further confirm the preceding arguments, where the overlapping among the peaks of Fe1 and O atoms is quite significant. The strong coupling of Fe1 atom orbitals (3d and 4s) with O atom orbitals (2p) near the Fermi level (EF) is clear evidence of the covalent metal–support interactions (CMSI) at the 4-H sites of PMA. Similar results for SAC have been reported by Qiao et al.64 Despite the asymmetric spin-up and spin-down PDOS of the Fe 3d orbital, its presence near the EF is evidence of the high reactivity which might be responsible for the activation of adsorbates during catalytic reactions.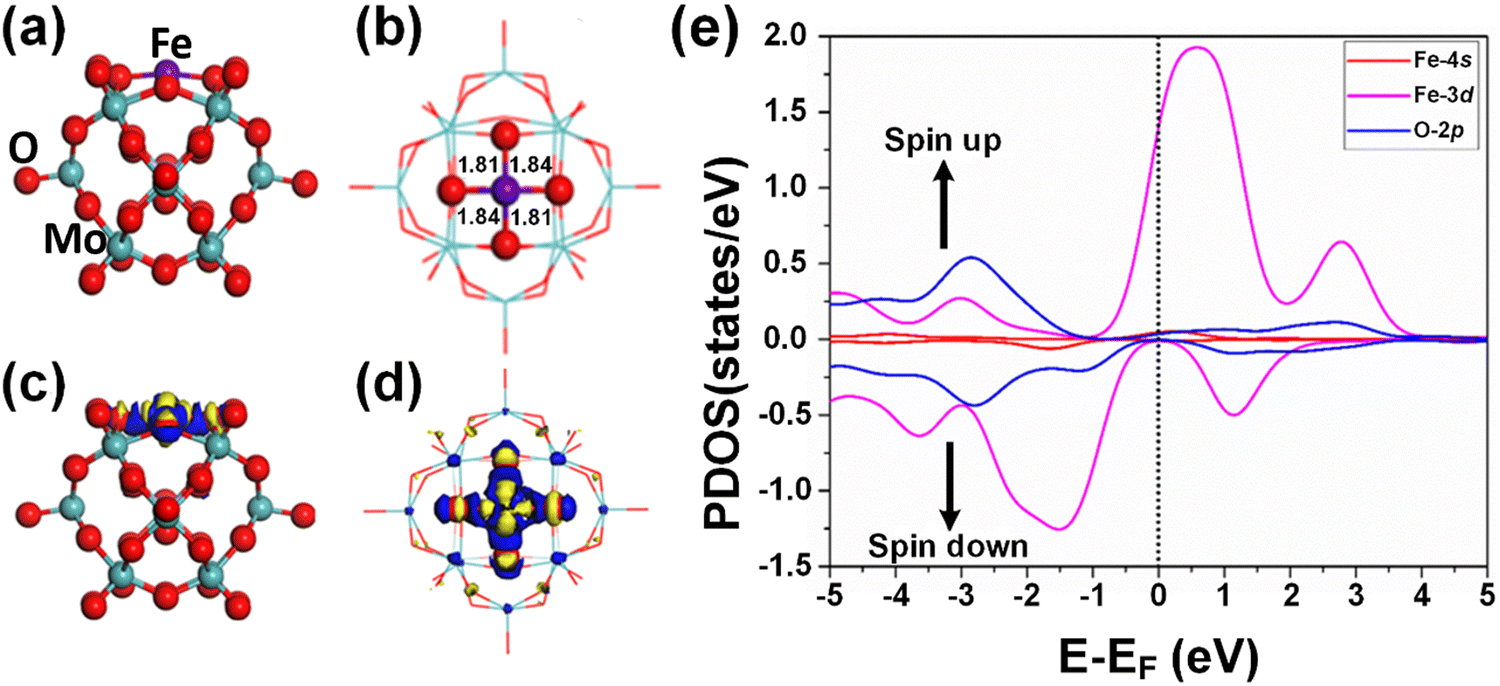 | ||
| Fig. 2 Optimized geometry of Fe1 embedded on the PMA cluster, side view (a) top view (b), charge density difference (CDD); (c) and (d) the counter value of the CDD is ±0.05 a.u., where charge accumulation is rendered in blue and charge depletion is shown yellow in the contour plots; and (e) the spin-polarized density of states on Fe1-3d (purple), Fe1-4s (red), and O-2p (blue) orbitals. The Fermi level (EF) is set to be zero. | ||
3.5. Adsorption of H2O2 and O2 on Fe1/PMA
The adsorption of the reactants during heterogeneous catalysis is of key importance to proceed with the mechanism, therefore before going deep into the mechanism of benzene oxidation to phenol, at first, we individually investigate the adsorption of H2O2 and O2 molecules as oxidants on the Fe1/PMA support surface under ambient reaction conditions. The calculated value of adsorption energy Eads = −0.53 eV of the H2O2 molecule on the Fe1/PMA surface shows that the reaction is exothermic, which is important for the adsorption of the H2O2 molecule on the surface (Fig. 3a). The bond distance between Fe1–O is 2.12 Å, whereas the O–O bond distance for the case of adsorbed H2O2 (1.47 Å) is larger than that for the free form of H2O2 (1.45 Å) by an amount of 0.02 Å. According to the Bader charge analysis, although the H2O2 molecule gained 0.79|e| charge from the Fe1/PMA cluster the Fe1 atoms remain positively charged (1.89|e|). The charge density difference represented in Fig. 3b shows that on the one side the charge density accumulates between the Fe1 and O atoms, while on the other side, it decreases between the two oxygen atoms, as seen by the blue and yellow areas, respectively. A slight decrease in the magnetic moment of Fe1/PMA from 2.90 to 2.78μB is observed after the H2O2 adsorption, which might be attributed to a large bond distance of 2.12 Å between the Fe1 atom and the O atom from H2O2. In Fig. 3c, the PDOS calculations for the Fe1/PMA surface adsorbed with H2O2 molecules, and Fe1-4s/3d and O-2p orbitals further confirm the Fe1-3d and O-2p orbital mixing. However, the orbital mixing is not very significant, which is again because of the large bond distance of 2.12 Å of Fe1–O. Besides, the wide range overlap between the major peaks of Fe1-3d and O-2p orbitals below the Fermi level shows significant interactions between Fe1 and H2O2 molecules.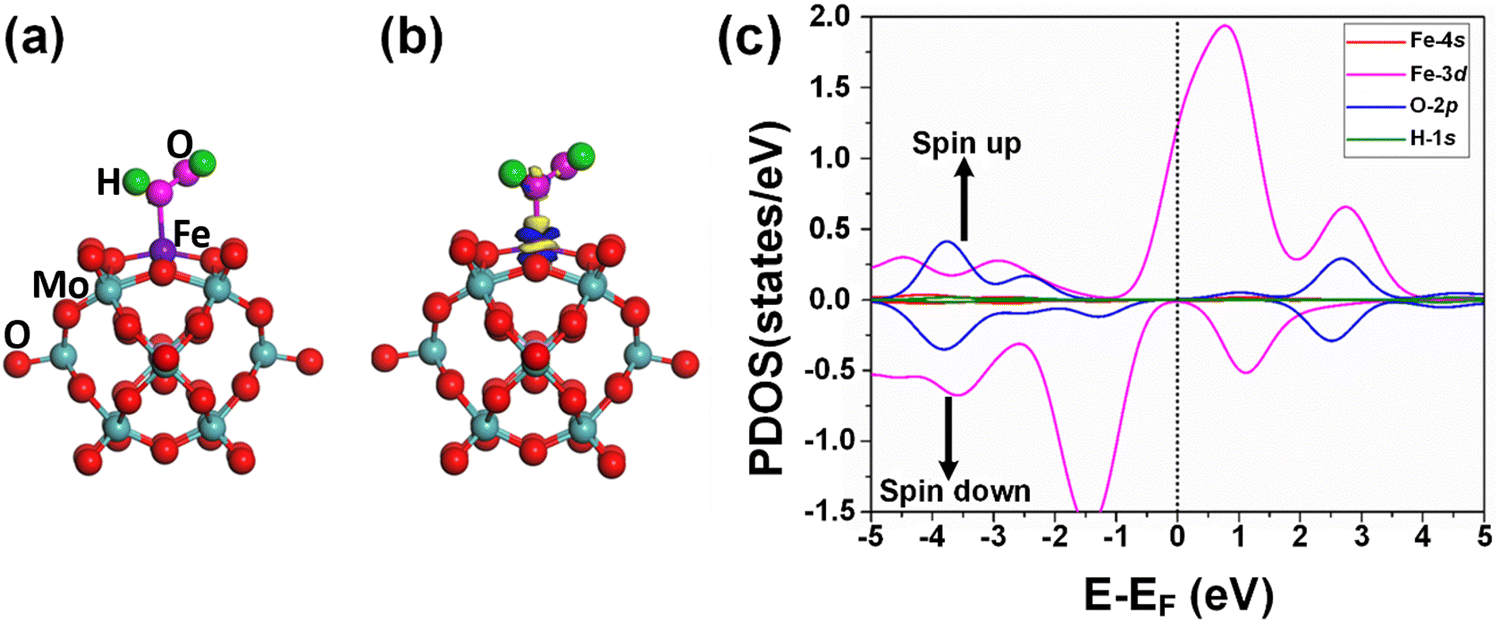 | ||
| Fig. 3 (a) Top view of the optimized geometry of H2O2 embedded in the Fe1/PMA cluster, CDD, for the contour plots, the electron accumulation regions are represented in blue while the electron depletion regions are shown in yellow; (b) the counter value of the CDD is ±0.05 a.u.; (c) the spin-polarized PDOS projected on Fe1-3d (purple), Fe1-4s (red), H-1s (green) and O-2p (blue) states. The EF was set to be zero. | ||
Besides, the O2 molecules adsorbed with Eads = −0.41 eV on the Fe1/PMA cluster were found to be situated parallel to the Fe1 atoms developing side-on coordination, as shown in Fig. 4a. The higher value of the O–O bond length (1.33 Å) is evidence of O2 acting as superoxide (O2−) on the Fe1/MA cluster when compared with the free O2 molecule bond length (1.23 Å). The O–O distance varies according to the reduction state, that is why the O–O distance for free di-oxygen is 1.21 Å, that for superoxide is 1.28 Å, and that for peroxide is 1.50 Å,65 hence the O2 molecule was accordingly adsorbed and activated as superoxide on the Fe1/PMA cluster. The calculated interatomic distances for Fe1–O are 1.84 and 1.81 Å, while 1.33 Å for O–O making it 0.12 Å longer than 1.21 Å for free O2 molecules. The Bader charge analysis of O2 absorbed on the Fe1/PMA cluster shows that Fe1 atoms gained a positive charge (+1.82|e|), while absorbed O2 gained a negative charge (−0.2|e|) from the cluster and became activated for the proceeding reactions. Therefore, the Fe1 atoms act as charge donors while O2 molecules as acceptors due to having positive and negative charges, respectively. The charge density difference results of O2 on Fe1/PMA, as shown in Fig. 4b, also validate the argument of charge transfer from the surface to the adsorbed O2. The excessive charge present on the O2 molecule fills the 2pπ* antibonding orbital and accumulates between the O–O bond, thus the O–O bond length increases which also demonstrates the activation of the O2 molecule by the Fe1/PMA cluster.
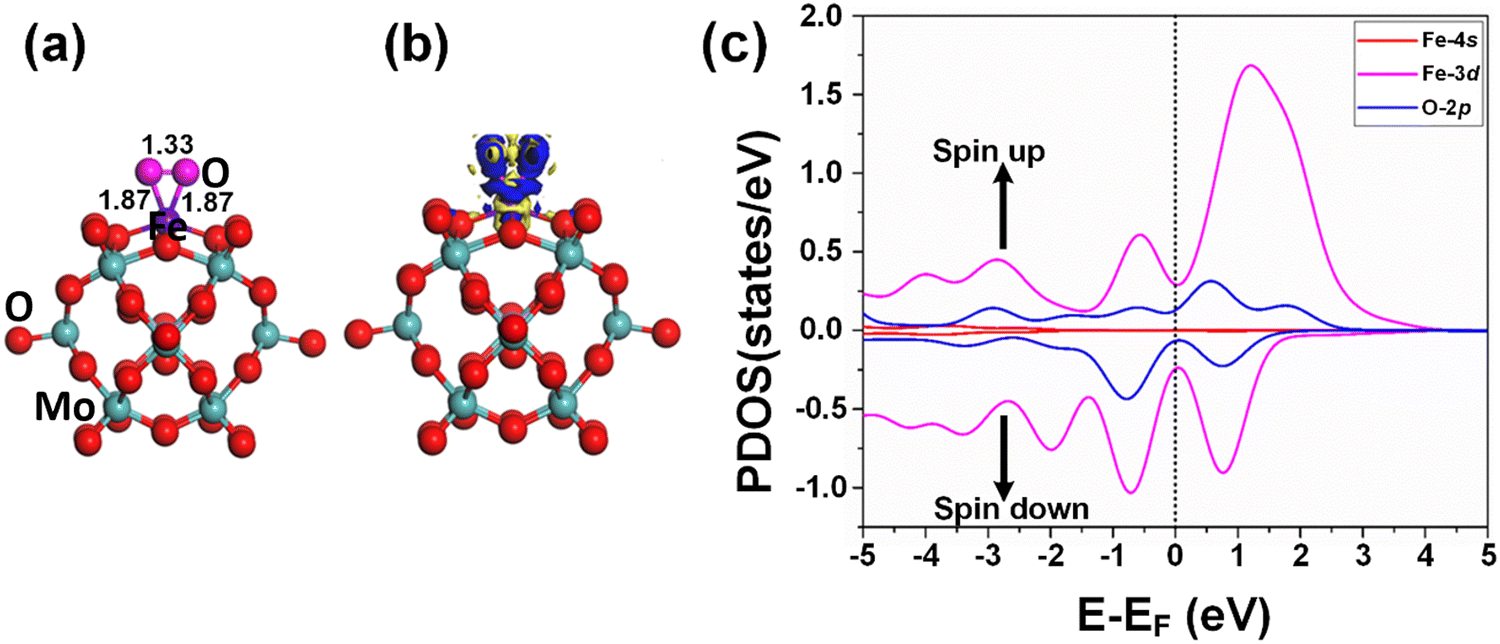 | ||
| Fig. 4 (a) Top view of the optimized geometry of O2 embedded in the Fe1/PMA cluster; (b) CDD, for the contour plots, the electron accumulation regions are represented in blue while the electron depletion regions are shown in yellow, while the counter value of the CDD is ±0.05 a.u.; and (c) the spin-polarized PDOS projected on Fe1/3d (red) Fe1/4s (red) and adsorbed O2 2p (blue) states. The EF was set to be zero. | ||
After the adsorption of the O2 molecule on Fe1/PMA, although the magnetic moment of Fe1/PMA decreased from 2.9 to 2.06μB, the majority of the magnetic moments were still located around the Fe1 atom. The apparent decrease in the unpaired electrons might be attributed to the orbital mixing from Fe1 (3d) and O (2p), as seen from the spin-polarized PDOS of O2/Fe1/PMA in Fig. 4c. Near the Fermi level, the extreme closeness of the Fe1 (3d) orbital with the O (2p) of the adsorbed O2 molecule on the single Fe1 atom is because of their relatively strong interaction that consequently influences the electron transfer from the Fe1/PMA catalyst to the O2 (2p) orbital. Due to the partial occupation of the O2 (2p) antibonding orbital, on the one side the O–O bond gets undermined while on the other side, the bonding between O2 and Fe1/PMA strengthens. That is the reason why the O–O bond stretches because of the accumulation of charge and filling of the O2 (2p) antibonding orbital.
Hence, despite the small difference in the adsorption energies of H2O2 (−0.53 eV) and O2 (−0.41 eV), the adsorption of H2O2 is preferred when compared with the adsorption of the O2 molecule on the Fe1/PMA cluster. Therefore, it can be inferred that relatively strong adsorption between H2O2 and the Fe1/PMA cluster is key to the efficient commencement of the benzene oxidation to phenol. Aiming for a better understanding, a further detailed reaction mechanism of benzene oxidation to phenol is investigated computationally via DFT to explore the efficiency of Fe1/PMA SAC under ambient working conditions.
3.6. Oxidation of benzene to phenol on Fe1/PMA using H2O2
The adsorption of the H2O2 oxidant on the active Fe1 sites of the Fe1/PMA cluster is shown in the potential energy profile in Fig. 5(a), where the activated H2O2 oxidant efficiently decomposes into a water molecule (H2O) and active surface oxygen (O*) by the Fe1/PMA cluster under ambient conditions, as shown in TS1; the initial state (IS), intermediate state (IM), transition state (TS), and final state (FS) are shown in Fig. 5(b). Initially, the Fe1–O bond is formed due to the adsorption of the H2O2 oxidant on an active Fe1 site with a bond distance of 2.12 Å. In the meantime, the O–O bond is activated by expanding the bond length to 1.47 Å from 1.45 Å in the gas phase. Furthermore, an increase in the O–O bond length from 1.47 to 1.74 Å and a decrease in the O–H bond length from 1.92 to 1.61 Å show that the current variations in the bond lengths are associated with the dissociation of the H2O2 oxidant into H2O and active O* species on the Fe1/PMA cluster. The potential energy profile also shows that the first elementary reaction i.e., the H2O2 decomposition into H2O and O* viaIS1 → FS1 (Fig. 5) having an energy barrier of 0.84 eV (TS1, imaginary frequency 470i cm−1) is highly exothermic (−1.25 eV). Besides, the low adsorption energy (0.13 eV) of H2O on OFe1/PMA is evidence of the greater probability of forming an OFe1/PMA catalyst. Accordingly, the H2O molecule departs, leaving iron–oxo (Fe1–O) species intact on the surface to form OFe1/PMA, which further reacts as a catalyst for the oxidation of benzene. The potential energy profile including IS, IM, TS, and FS of the benzene oxidation through the OFe1/PMA catalyst is shown in Fig. 6(a) and (b).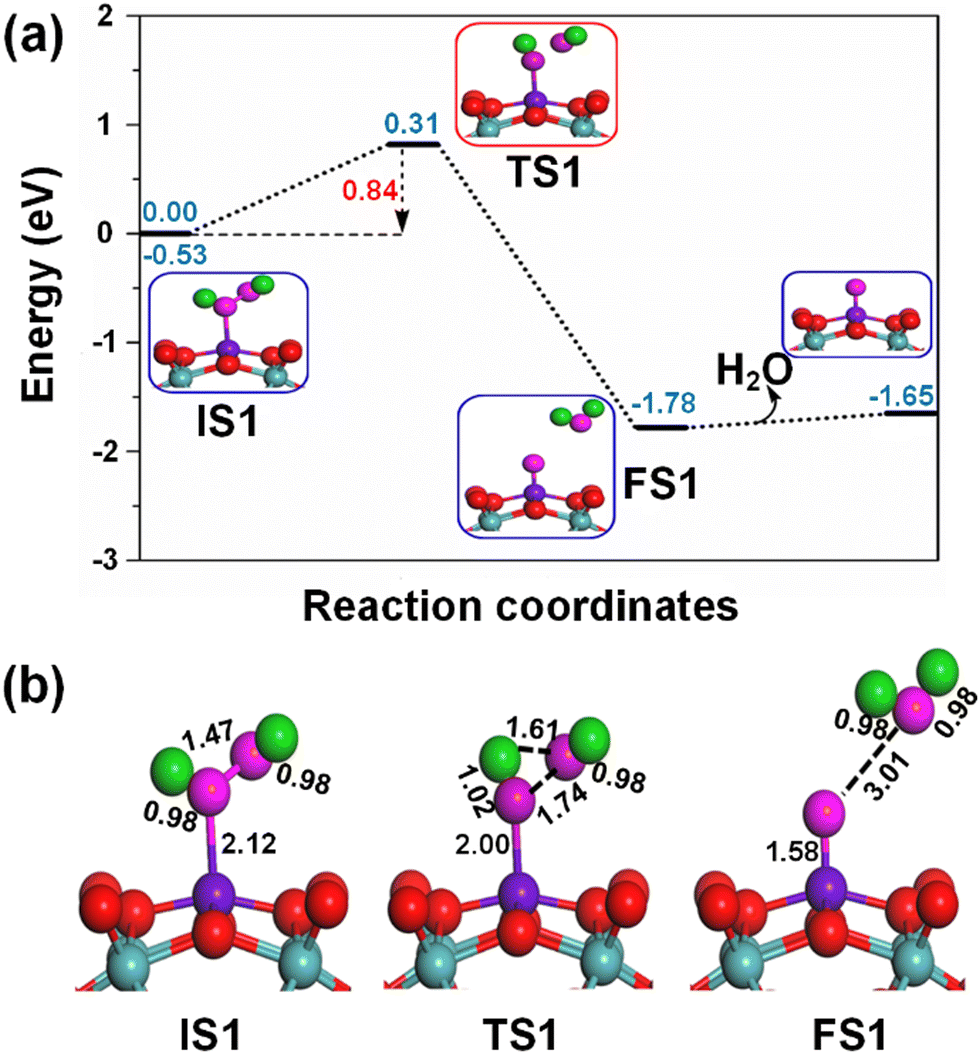 | ||
| Fig. 5 (a) Potential energy profile of the efficient decomposition of H2O2 on Fe1/PMA, all DFT energies are in eV; (b) the optimized geometries of IS, TS, and FS of the H2O2 decomposition on Fe1/PMA, all the calculated bond lengths are given in Å. | ||
 | ||
| Fig. 6 (a) Potential energy profile of the efficient decomposition of direct oxidation of benzene to phenol on Fe1/PMA (in the case of a H2O2 oxidant), all DFT energies are in eV; (b) optimized geometries of IS, TS, and FS of the benzene oxidation on Fe1/PMA, all the calculated bond lengths are given in Å. | ||
In our study, we also examined the performance of other investigated 3d-TM1/PMA clusters (3d-TM1 = Co1, Ni1, Cu1, and Zn1). Our findings revealed that the energy barriers for the initial step of the H2O2 decomposition reaction into H2O and O* were notably higher for these cluster systems, specifically, 1.11 eV for Co1, 1.21 eV for Ni1, 2.25 eV for Cu1, and 1.68 eV for Zn1, when compared to the energy barriers for Fe1/PMA (as detailed in Fig. S5, ESI†). These results strongly suggest that Fe1/PMA is a more effective catalyst for facilitating the H2O2 decomposition reaction, primarily due to its significantly lower energy barrier, which can be advantageous in the direct conversion of benzene oxidation to phenol.
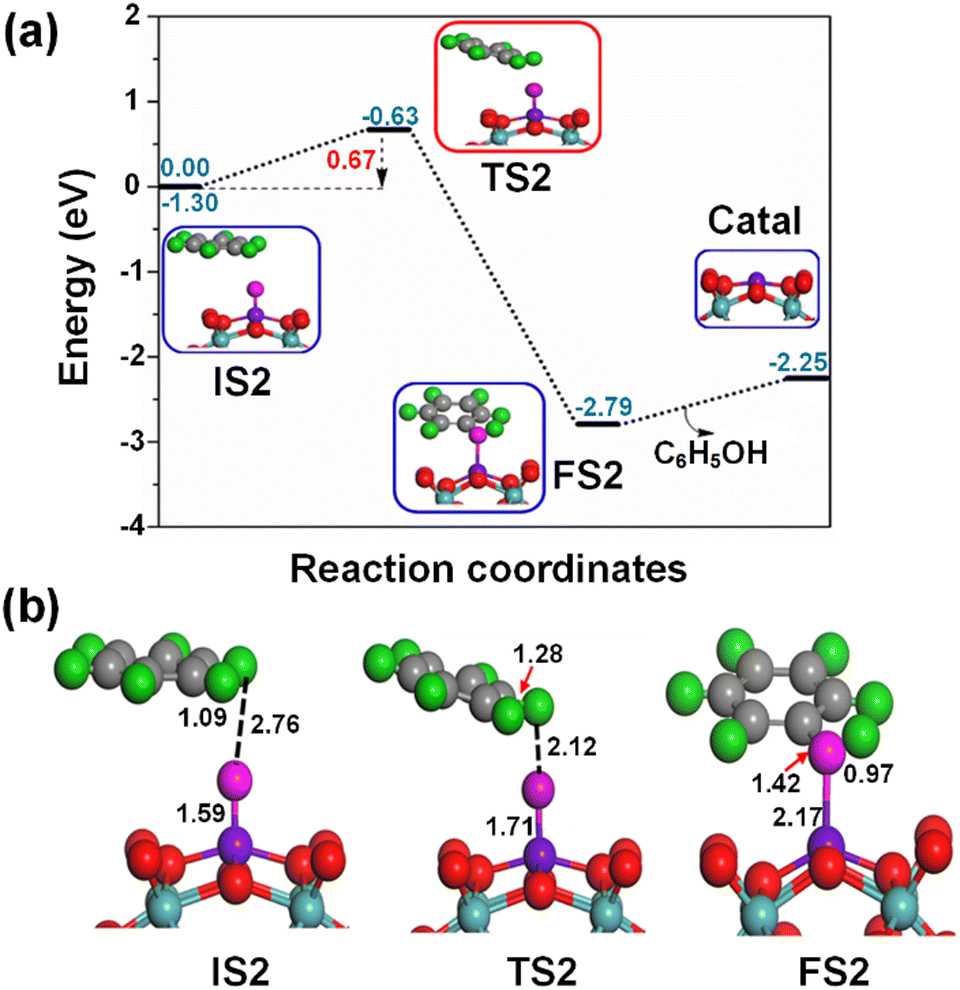
Fig. 6 shows the formation of a π type iron–oxo–benzene complex due to the adsorption of benzene by van der Waals interaction of C–H⋯O on the OFe1/PMA cluster during the second elementary step (IS2). The adsorption energy of benzene is 0.35 eV which shows that the adsorption is of the physical type, while the C–H and O⋯H bond lengths in IS2 are 1.09 and 2.76 Å.
Notably, understanding chemisorption is critical for obtaining a comprehensive picture of the interaction between benzene and the metal atoms in our single-atom catalysts. To check the direct interaction (chemisorption) between benzene and the metal atoms, we also considered the adsorption energies of these systems. According to the results, the adsorption energies for the chemisorption of benzene on the studied single-atom catalysts varied significantly (See Fig. S6 and Table S6 in the ESI†). Further details regarding the chemisorption of benzene on all studied TM1 are provided in the ESI.† And the results indicated that the adsorption energy of benzene on Fe1/PMA is 0.35 eV for physisorption and −1.62 eV for chemisorption (Table S6 in the ESI†). Higher chemisorption energy makes it difficult for the next oxidation step to occur. The results suggest that physisorption facilitates benzene conversion to phenol on Fe1/PMA. Subsequently, the detachment of the H atom from the benzene molecule occurs via a TS2 transition state reaction, and a hydroxo intermediate is formed simultaneously by a C⋯H–O interaction, with the activation of the C–H bond of C6H6 on the surface of OFe1/PMA. Besides, due to the C⋯H–O type interaction with the FeO complex, benzene appears a little distorted. The TS2 transition state having 0.67 eV activation energy and 164i cm−1 imaginary frequency is responsible for the dissociation of the C–H bond from benzene and the formation of an O–H bond. Furthermore, the iron–oxo-benzene complex connects the hydroxo complex during the TS2 transition state. An increase in the C–H bond distance from 1.09 to 1.28 Å in C6H6 and a decrease in the O–H bond distance from 2.76 to 2.12 Å are also evidence for the feasible transition state structure which is responsible for the cleavage of C–H and the bifurcation of the O–H bond. Eventually, the phenol complex [Fe1(C6H5OH)] is formed which results in the dissociation of the Fe1–O bond and the association of the O–H bond. Therefore, the phenol complex and the hydroxo intermediate are connected to the OFe1/PMA cluster during the FS2 reaction. Similarly, the C–H bond length in C6H6 increased to 1.42 Å while the O–H bond length decreased to 0.97 Å. Finally, the phenol desorption from the surface occurred with the restoration of the Fe1/PMA cluster at the last step of the reaction. The progression from IS1 to FS2 is highly exothermic as evident from ΔE = −2.26 eV. The low adsorption energy (Eads = 0.54 eV) between phenol and the Fe1–PMA surface during FS2 is suitable for the desorption of phenol from Fe1(C6H5OH)/PMA leaving behind the Fe1/PMA catalyst. It should be noted that the phenol moiety in the complex structure is quite similar to that of free phenol. Besides, a comparison of the energy barriers in the first and second steps shown, respectively, in Fig. 5a and 6a define the decomposition of H2O2 in the first elementary reaction as the rate-limiting step.
The spin magnetic moment calculated for the elementary steps in the benzene oxidation to phenol provides valuable insights about the IS, TS, and species description. The spin magnetic moment calculated at the initial stage of the mechanism shows that Feδ+ is an active center of the Fe1/PMA cluster. When H2O2 is absorbed on the Fe1/PMA cluster, the magnetic moment of Feδ+ changed slightly from 2.90 to 2.78μB (see IS1 in Fig. 5a). A small change in the magnetic moment of Feδ+ might be related to the large separation of 2.12 Å between Fe1 and adsorbed H2O2, which results in a very weak interaction between them. With the physisorption of H2O2 on the Fe1/PMA cluster (see TS1 in Fig. 5) Feδ+ is reduced having a spin magnetic moment of 1.44μB. However, decreasing the spin magnetic moment of FS1 to 0.53μB indicates a reduction in Feδ+, as shown in Fig. 5. The reduction in the magnetic moment is related to the interaction of the surface oxygen with the Fe1 atom of the Fe1/PMA cluster.
During IS2 and TS2, with the introduction of benzene on OFe1/PMA, a hydroxo complex forms, and the magnetic moments for IS2 and TS2 are 0.66μB and 0.77μB, respectively, as shown in Fig. 6. Therefore, due to the C⋯H–O type interaction with the Fe1O complex, benzene gets a little distorted, as discussed earlier. With the formation of the phenol complex on the Fe1/PMA cluster, Feδ+ is oxidized, and the magnetic moment is increased to 1.63μB. Similarly, the magnetic moment increases to 3.56μB with the desorption of phenol from the Fe1/PMA cluster, indicating re-oxidation of Feδ+.
When examining the PDOS of Fe 3d-orbitals for each elementary step in the reaction a strong correlation emerges between the changes in the magnetic moment of Fe and the behavior of the Fe 3d orbital near the Fermi level, as shown in Fig. S8 of the ESI.† Specifically, after the initial adsorption of benzene (IS2), there seems to be a decrease in the availability of states for Fe 3d-orbitals near the Fermi level compared to the initial state. As the reaction progresses up to the formation of phenol, the PDOS near the Fermi level indicate an increase in the available states. Fig. S8 of the ESI† reveals a significant discrepancy between the spin-up and spin-down PDOS of Fe 3d-orbitals. This asymmetry suggests a non-zero magnetic moment for Fe, with the magnitude potentially proportional to the degree of separation between the spin-up and spin-down peaks. The large asymmetry in the Fe1/PMA PDOS, with a dominant contribution from spin-down states, indicates a strong magnetic moment for the reference material. The minimal change in the asymmetry for the initial state (IS1) compared to Fe1/PMA suggests a relatively small variation in the magnetic moment during the initial stage of the reaction. The higher spin-up contribution might be linked to a decrease in the net spin magnetic moment of the final state (IF1). A less pronounced asymmetry in IS2 compared to IS1 could imply a less magnetic moment on Fe1 at this stage. The formation of the phenol complex on the Fe1/PMA cluster in FS2 might lead to the observed discrete molecular levels in the Fe1 PDOS. The increased contribution from spin-down states in this final state could indicate a change in the magnetic moment compared to the initial state. Overall, the analysis of the PDOS plots provides valuable qualitative information about the magnetic properties of Fe1 during the reaction. Furthermore, the shifting and broadening of the high valence bands around the Fermi level across the elementary steps suggest a change in the overall electronic structure of the catalyst surface (OFe1/PMA). This shift in the d-band center, as reflected in the PDOS plots, aligns with the reactivity of the catalyst for benzene oxidation.66
The foregoing discussion reveals that the H2O2 molecule can be dissociated with an activation energy of 0.84 eV on the confined Fe sites, subsequently forming a Fe1–O active center by releasing the H2O molecule from the surface. Fe1–O having active surface oxygen (O*) successfully absorbs the benzene molecule and forms C–O and O–H bonds at low temperatures with 0.67 eV activation energy. Therefore, it can be concluded that benzene oxidation to phenol is a direct process rather than being controlled by species or ionic intermediates.
3.7. Oxidation of benzene to phenol on Fe1/PMA using O2
When oxygen is an oxidant, at first place the Fe1/PMA cluster activates the O2 molecule as superoxide, and then it is activated as an oxidizing catalyst, as shown in Fig. 4. As described previously, the O2 while staying on the Fe1 atom in PMA is activated as superoxide and prefers to develop side-on coordination. The calculated potential energy landscape of benzene oxidation mediated by the O2⋯Fe1/PMA catalyst ((IS), intermediate (IM), (TS), and (FS)) is shown in Fig. 7. The calculated adsorption energy of benzene attacking the adsorbed O2 molecule on Fe1/PMA is −0.05 eV, which is higher than that of the initial state of O2⋯Fe1/PMA (IS3), and the reaction is thermodynamically feasible as described by the highly exothermic (−0.78 eV) adsorption of O2 on Fe1/PMA. The iron–oxo–benzene complex (IS3) is formed between O2Fe1/PMA and benzene because of the van der Waals interactions between C–H⋯O–O, where the bond lengths of C–H and O⋯H of IS3 are 1.09 and 2.80 Å. Afterward, one of the oxygen atoms from adsorbed O2 molecules on the Fe1/PMA cluster transfers to benzene and forms a new hydroxo-intermediate complex by a C⋯H–O–O interaction along with the activation of the C–H bond of C6H6 on the surface of O2Fe1/PMA. It can be inferred that during the transition state (TS3) when benzene is connected with one of the O atoms, the side-on configuration of O atoms transmuted to an end-on configuration.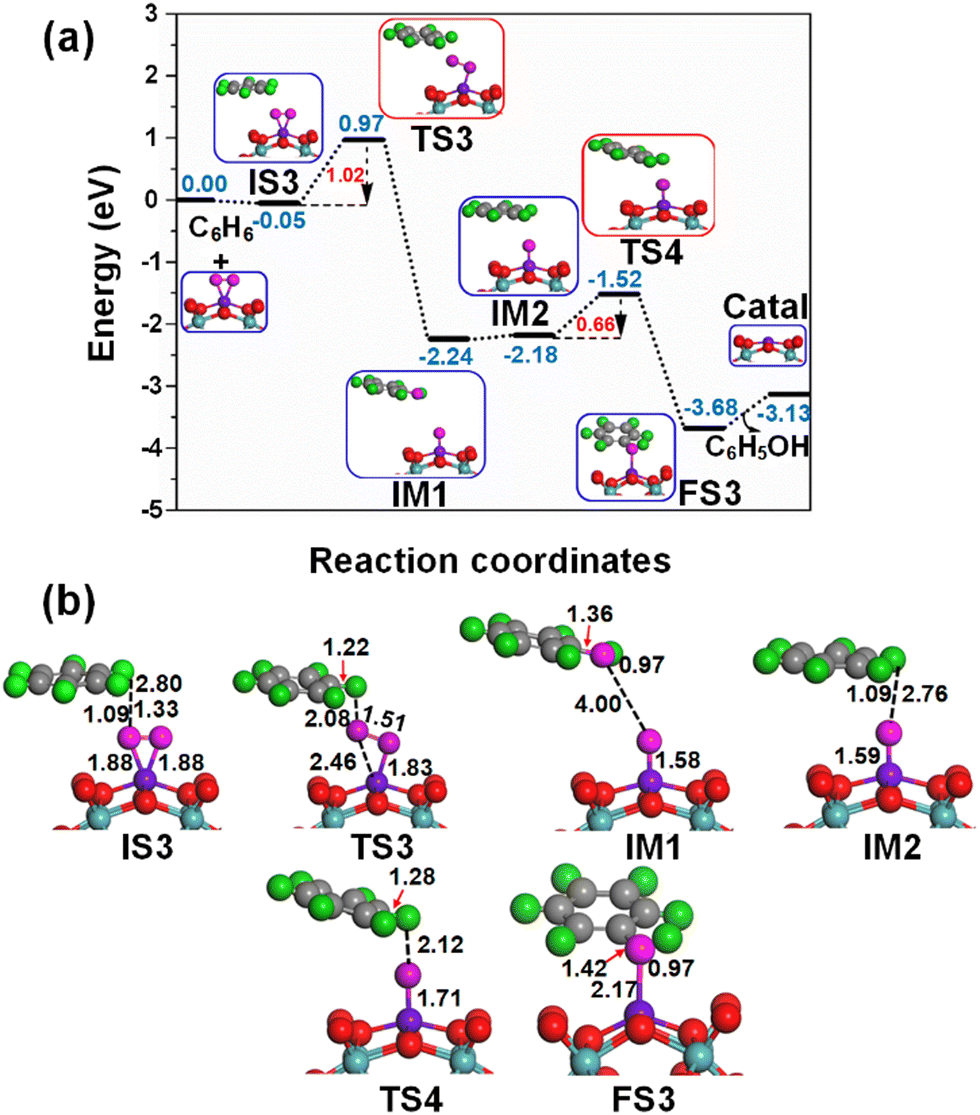 | ||
| Fig. 7 (a) Potential energy profile of the efficient decomposition of direct oxidation of benzene to phenol on Fe1/PMA in the case of an O2 oxidant, all DFT energies are in eV; (b) optimized geometries of IS, IM, TS, and FS of the benzene oxidation on Fe1/PMA, all the calculated bond lengths are given in Å. | ||
Ranging from the dissociation of the C–H bond from benzene and the formation of the O–H bond, the TS3 transition state having 1.02 eV activation energy and one imaginary frequency of 209i cm−1 is responsible for many electronic processes. Furthermore, the iron–oxo–benzene complex connects the hydroxo complex during the TS3 transition state. Besides, an increase in the C–H bond distance from 1.09 to 1.22 Å in C6H6 and a decrease in the O–H bond distance from 2.8 to 2.08 Å are quite appropriate for the TS3 structure to cleavage the C–H bond and to bifurcate the O–H bond. Whereas the formation of a hydroxo intermediate is of key importance in the process of benzene oxidation to phenol, which is associated with the breakage of the O–O bond and the formation of the O–H bond occurring between TS3 and IM1. Eventually, the phenol complex [Fe1O⋯(C6H5OH)] is formed which results in the dissociation of the Fe1–O⋯O bond and the association of the O–H bond. Therefore, the phenol complex and the hydroxo intermediate are connected to the OFe1/PMA cluster during the IM1 reaction. Similarly, the C–H bond length in C6H6 increased to 1.42 Å while the O–H bond length decreased to 0.97 Å. Finally, the phenol desorption from the surface occurred with the restoration of the PMA cluster at the last step of the reaction. The progression from IS3 to IM1 is highly exothermic as evident from ΔE = −2.19 eV. Due to the weak adsorption energy (Eads = 0.06 eV), the phenol leaves the surface making Fe1/PMA covered with the O atom and being available for another cycle of oxidation of benzene.
Successively, the oxygen atom pre-adsorbed on the Fe1/PMA cluster reacts with the benzene during IM2, and the benzene molecule physiosorbed at 2.76 Å away from the surface O atom is taken as an underlying structure in IM3. The reaction between the surface O atom and benzene at the Fe1/PMA cluster produces a second phenol during FS3 through TS4, while the catalyst is recovered as the cluster. The TS4 having 0.66 eV of activation energy and 164i cm−1 imaginary frequency is responsible for the dissociation of the C–H bond and the formation of the O–H bond. However, the formation of new C–O and O–H bonds with 1.42 and 0.97 Å bond lengths, respectively, is responsible for the arising of the final state (FS). The progression from IM2 to FS3 is highly exothermic as evident from ΔE = −1.50 eV. Due to the weak adsorption energy (Eads = 0.55 eV), the phenol leaves the surface thus regenerating the Fe1/PMA catalyst, whereas the phenol moiety in the complex structure is quite similar to that of free phenol. Moreover, the formation of the first phenol during IS3 to IM1 (see Fig. 7) on the Fe1/PMA cluster and the activation of C–H bond are the rate-limiting steps having an activation energy of 1.02 eV in phenol production from benzene oxidation.
According to the spin magnetic moment calculations, the Fe1/PMA cluster has a Feδ+ active center at the start of the mechanism, which is the same as that of the Fe1/PMA catalyst. However, with the physisorption of O2 on Fe1/PMA during IS1 (see Fig. 4 for benzene oxidation), the oxidation state of Feδ+ was reduced having a spin magnetic moment of 2.06μB. The introduction of benzene on O2Fe1–PMA during IS3 and TS3 (see Fig. 7) forms a hydroxo complex while Fe1 has spin magnetic moments of 2.05μB and 1.88μB during IS3 and TS3, respectively. However, Feδ+ further reduced after the formation of the phenol complex during IM1, while having a magnetic moment of 0.65μB. In IM2 and TS4 (see Fig. 7), when the second benzene is introduced on OFe1–PMA, the magnetic moments of Feδ+ changed to 0.66μB and 0.77μB, respectively. In conclusion, the benzene and hydroxo complex was distorted because of their interaction with the O atom present on the surface of OFe1/PMA.
During FS3, with the formation of the phenol complex on the surface of the Fe1/PMA cluster, Feδ+ oxidized with the increase of the magnetic moment to 1.63μB. Besides, Feδ+ is re-oxidized as the magnetic moment increased to 3.56μB after the desorption of phenol from the surface. According to the PDOS calculation shown in Fig. S9 of the ESI,† the Fe1 3d orbital near the Fermi level is a key factor behind changes in the magnetic moment of the Fe1 atom during all the fundamental steps. In Fig. S9 of the ESI,† the Fe 3d-orbitals exhibit significant asymmetry between the spin-up and spin-down PDOS, indicating a non-zero magnetic moment for Fe1. In IS3, PDOS displays a strong magnetic moment of Fe1, with spin-down states dominating, while IM1 and IM2 exhibit less asymmetry, suggesting a low magnetic moment. In FS3, the phenol complex formation results in discrete molecular levels in Fe1 PDOS, with increased spin-down contribution indicating a change in the magnetic moment. In summary, PDOS analysis offers valuable qualitative insights into the magnetic properties of Fe1 during the reaction. Furthermore, a gradual change in the d-band center with the shifting of high and broad valence bands around Fermi energy indicates the reactivity of the O2Fe1/PMA surface for benzene oxidation (Fig. S9 of the ESI†). We also investigated the spin configuration of the Co1/PMA cluster, detailed in Fig. S10 of the ESI.†
The preceding discussion reveals that it is easy to activate the O2 molecule on the confined Fe1 sites by forming iron–superoxide species (Fe1O2) as an active center. Still, it offers comparatively weak adsorption of benzene molecules for the formation of C–O and O–H bonds with a 1.02 eV activation energy barrier of the rate-limiting step. The above results indicate that the Fe1O active center plays an important role in improving benzene oxidation activity as compared to the Fe1O2 active center.
3.8. Microkinetic study for oxidation of benzene to phenol on Fe1/PMA
To better understand the catalytic benzene oxidation reaction on the Fe1/PMA cluster, the rate constants k (s−1) for each fundamental step in the decomposition of H2O2, oxidation of benzene to phenol via the H2O2 oxidant, and the O2 oxidant can be calculated using the transition state theory (TS).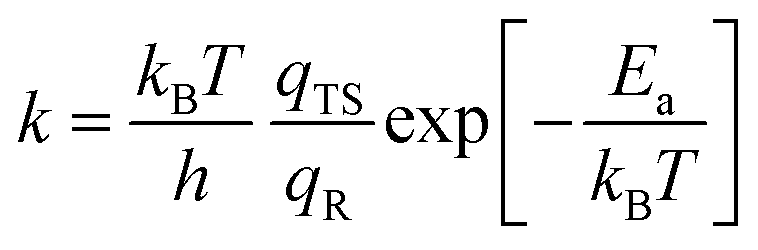 | (3) |
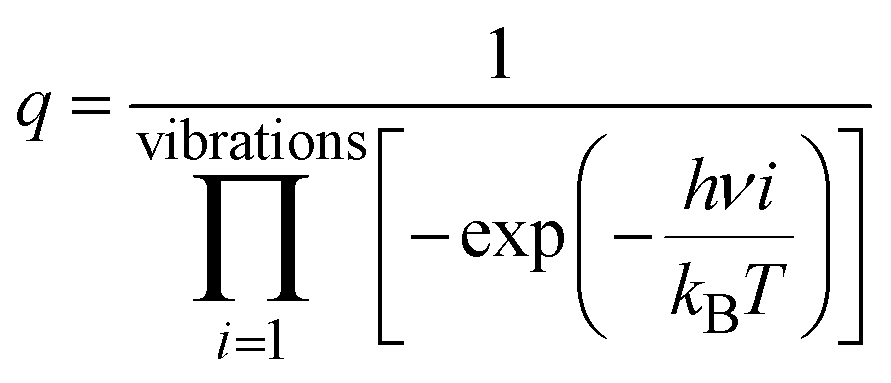 | (4) |
In this context, vi signifies the vibrational frequency. It is important to remember that imaginary frequencies are omitted for TS. Table 1 shows the rate constants for all the rate-determining steps elaborated in the aforementioned mechanisms at various temperatures. The rate constants (k) of the rate-determining step in the oxidation of benzene to phenol via the H2O2 oxidant are considerably higher than those for the decomposition of H2O2 and oxidation of benzene to phenol via the O2 oxidant. The results are consistent with the DFT calculations that predicted benzene to phenol oxidation via the H2O2 oxidant would progress more efficiently than other mechanisms. In contrast, rate constants for many reactions increase as temperature increases, suggesting that Fe1/PMA can accelerate benzene oxidation. The cryogenic atmosphere inhibits the oxidation of benzene to phenol with H2O2, while using the O2 oxidant the reaction rate becomes even slower at 100 K.
| T (K) | 100 | 200 | 298.15 | 400 | 500 | 600 | |
|---|---|---|---|---|---|---|---|
| H2O2 Decomposition | H2O2* → H2O + O* | 6.06 × 10−30 | 1.78 × 10−8 | 0.24 × 100 | 1.36 × 103 | 2.23 × 105 | 6.91 × 106 |
| H2O2 Oxidant | O* + C6H6 → C6H5OH + * | 2.24 × 10−21 | 0.00 | 1.84 × 102 | 1.89 × 105 | 1.15 × 107 | 1.85 × 108 |
| O2 Oxidant | C6H6 + O2* → C6H5OH + O* | 5.14 × 10−39 | 5.19 × 10−13 | 0.000 | 7.31 × 100 | 3.42 × 103 | 2.12 × 105 |
| C6H6 + O* →C6H5OH + * | 7.21 × 10−21 | 0.000 | 2.72 × 102 | 2.53 × 105 | 1.45 × 107 | 2.24 × 108 |
3.9. Intrinsic electronic features of Fe1O and Fe1O2 active centers
The electronic features of Fe1O and Fe1O2 are further investigated regarding their reactivity to influence the benzene oxidation because different coordination environments of SAC result in distinct catalytic performance as suggested by Gao et al.67 Fe1O is a coordinatively oxygen-unsaturated iron center having a coordination number of 5 around the Fe1 metal, which is less than that of Fe1O2 having a coordinatively oxygen-saturated iron center due to a coordination number of about 6 around the Fe1 metal. Moreover, the interaction of benzene with coordinatively oxygen-saturated iron center Fe1O2 (0.05 eV) is weaker than that with coordinatively oxygen-unsaturated iron center Fe1O (0.35 eV), which may result in a higher C–H bond activation barrier of benzene for coordinatively oxygen-saturated Fe1O2 (1.01 eV) than its oxygen-unsaturated counterpart (0.84 eV). Therefore, the probability of Fe1O2 coordinatively oxygen-saturated iron center to activate the C–H bond is relatively lower than the coordinatively oxygen-unsaturated iron center Fe1O, which is important for the benzene oxidation to phenol at ambient temperature.Besides, the electron localization function (ELF) is an important parameter to differentiate the charge transfer reactivity, electron delocalization in molecules and solids, and bond classification.62,68 According to the ELF of Fe1O shown in Fig. 8a, the Fe1–O bond exhibits covalent characteristics. At the same time, the lone pair electron property of oxygen becomes more significant due to the surrounding non-spherical charge distribution, whereas a significant charge transfer occurs from Fe1 to O thus reducing Fe1 (II) to Fe1 (I) as described by the IS1 to FS1 in Fig. 5. Consequently, the reactivity of surface O atoms is enhanced due to the development of radical-like characteristics. After the formation of the iron–oxo–benzene complex (see Fig. 6, IS2), benzene appears a little distorted due to the C–H⋯O interaction on the OFe1/PMA cluster (see Fig. 8b). Therefore, the adsorption of benzene on the O site of Fe1–O is more promising (0.35 eV). These results are also consistent with the charge density difference calculation results shown in Figure S11 of the ESI.†
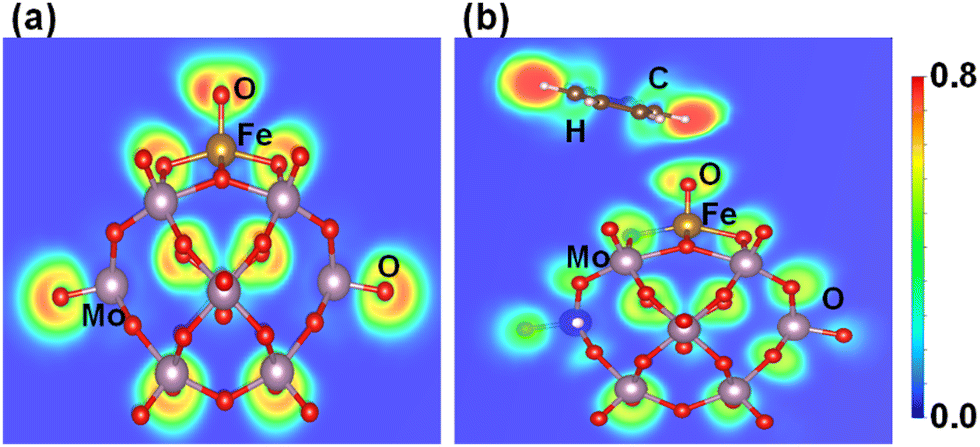 | ||
| Fig. 8 Electron localization functions after geometry optimization for (a) OFe1/PMA and (b) C6H6–OFe1/PMA. | ||
Similarly, the ELF of Fe1O2 shows that the charge density is localized between the O2 molecules, while Fe1–O has covalent bond characteristics where the strong localization of orbitals increases its polarizability, as shown in Fig. 9a. Consequently, the interaction between the benzene and Fe1O2 becomes very weak (0.05 eV) due to the very poor charge transfer from the O site to the benzene (see Fig. 9b). These results also agree with the CDD calculation results shown in Fig. S12 of the ESI.† Besides, the higher Bader charge population of O atoms in Fe1–O (−0.51|e|) as compared to that of Fe1O2 (−0.20|e|) also indicates better catalytic activity for benzene oxidation in the case of the Fe1O active center.
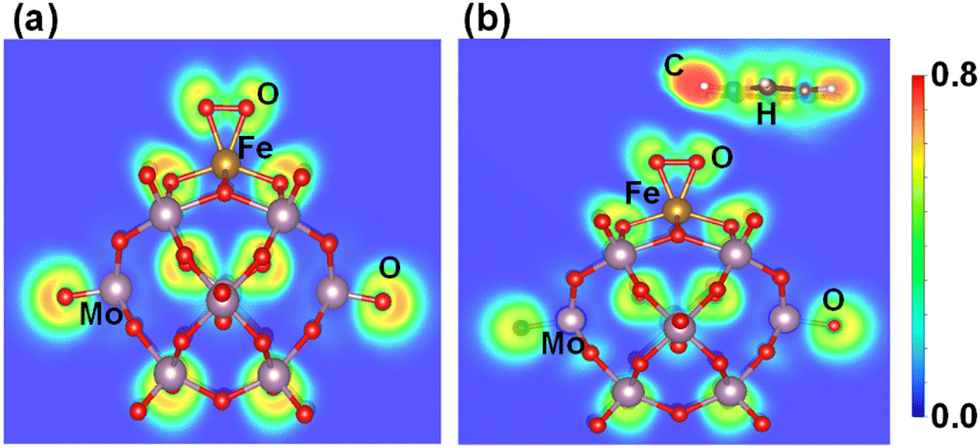 | ||
| Fig. 9 (a) Electron localization functions after geometry optimization for (a) O2Fe1/PMA and (b) C6H6–OFeO1/PMA. | ||
4. Conclusions
In this study, density functional theory was used to investigate the mechanism of direct benzene oxidation to phenol under ambient conditions, focusing on a non-noble metal Fe1 SAC dispersed on a PMA support (Fe1/PMA) compared to other 3d-TM1 (Co1, Ni1, Cu1, and Zn1). The results demonstrate that Fe1/PMA exhibits strong binding interactions with both the PMA cluster and the oxidant molecules (H2O2 and O2), making it a highly active and stable catalyst for benzene oxidation to phenol. Based on the theoretical calculations, the key conclusions obtained are as follows:(1) The comparable adsorption energies −0.53 and −0.41 eV on the Fe1/PMA cluster and low energy barriers 0.84 and 1.02 eV for rate-limiting steps of H2O2 and O2 molecules, respectively, make them ideal oxidants for the oxidation of benzene to produce phenol under ambient conditions.
(2) H2O2, having a relatively low energy barrier, adheres to spontaneous dissociation on the Fe1/PMA surface to initiate the active site Fe1–O, which is responsible for the catalytic activity. Meanwhile, the adsorption of benzene on the surface O site of the Fe1–O active center is more promising because of the coordinative oxygen unsaturated Fe1 center. While for the O2 oxidant, the O atoms from the Fe1-superoxide (Fe1–O2) are comparatively less active to support benzene adsorption because of the coordinative oxygen-saturated Fe1 center. It was observed that the interaction of benzene with the oxygen-saturated iron center Fe1–O2 (0.05 eV) is weaker than that with the oxygen-unsaturated iron center Fe1–O (0.35), which may result in a higher C–H bond activation barrier of benzene for oxygen-saturated O–Fe1–O (1.01 eV) than its oxygen-unsaturated counterpart (0.67 eV).
(3) Besides, the reactivity of Fe1O and Fe1O2 (oxo and super oxo active centers) was also explored through coordination states of the Fe1 metal atom on the PMA surface. It was found that the probability of the coordinative unsaturated (CUS) Fe1 atom to activate H2O2 for the formation of Fe1–O is relatively higher than that to activate the O2 molecule, which is important for the benzene oxidation to phenol. In summary, the results demonstrate that coordination patterns influence not only the structure and electronic features but also the catalytic reaction pathway and the formation of key oxidative species.
(4) Finally, the energy barrier of 0.84 eV for the rate-limiting step of Fe1O is lower than that (1.01 eV) for the rate-limiting step of Fe1O2, suggesting that Fe1O2 species is relatively less active for the oxidation of benzene to phenol.
In the future perspective, the current study might flag the new structure–reactivity descriptors contributing to the next generation of functional condensed phase catalysis at a molecular level.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 22033005 to J. L. and grant no. 21903047 to H. X.), NSFC Center for Single-Atom Catalysis (22388102), and Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002). The calculations were performed using the supercomputers at Tsinghua National Laboratory for Information Science and Technology.References
- Q. Wei, H. Fan, F. Qin, Q. Ma and W. Shen, Metal-free honeycomb-like porous carbon as catalyst for direct oxidation of benzene to phenol, Carbon, 2018, 133, 6–13 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Carbocatalysis by graphene-based materials, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed.
- G. Wen, S. Wu, B. Li, C. Dai and D. S. Su, Active sites and mechanisms for direct oxidation of benzene to phenol over carbon catalysts, Angew. Chem., Int. Ed., 2015, 54, 4105–4109 CrossRef CAS PubMed.
- G. Ding, W. Wang, T. Jiang, B. Han, H. Fan and G. Yang, Highly selective synthesis of phenol from benzene over a vanadium-doped graphitic carbon nitride catalyst, ChemCatChem, 2013, 5, 192–200 CrossRef CAS.
- H. Zhang, X. Pan, X. Han, X. Liu, X. Wang, W. Shen and X. Bao, Enhancing chemical reactions in a confined hydrophobic environment: An NMR study of benzene hydroxylation in carbon nanotubes, Chem. Sci., 2013, 4, 1075–1078 RSC.
- G. Centi and S. Perathoner, One-step H2O2 and phenol syntheses: examples of challenges for new sustainable selective oxidation processes, Catal. Today, 2009, 143, 145–150 CrossRef CAS.
- G. Panov, Advances in oxidation catalysis; oxidation of benzene to phenol by nutrous oxide, Cattech, 2000, 4, 18–31 CrossRef CAS.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Titanium-containing mesoporous molecular sieves for catalytic oxidation of aromatic compounds, Nature, 1994, 368, 321–323 CrossRef CAS PubMed.
- L. Balducci, D. Bianchi, R. Bortolo, R. D'Aloisio, M. Ricci, R. Tassinari and R. Ungarelli, Direct oxidation of benzene to phenol with hydrogen peroxide over a modified titanium silicalite, Angew. Chem., 2003, 115, 5087–5090 CrossRef.
- J. Chen, S. Gao and J. Xu, Direct hydroxylation of benzene to phenol over a new vanadium-substituted phosphomolybdate as a solid catalyst, Catal. Commun., 2008, 9, 728–733 CrossRef CAS.
- G. Tanarungsun, W. Kiatkittipong, P. Praserthdam, H. Yamada, T. Tagawa and S. Assabumrungrat, Ternary metal oxide catalysts for selective oxidation of benzene to phenol, J. Ind. Eng. Chem., 2008, 14, 596–601 CrossRef CAS.
- J.-S. Choi, T.-H. Kim, K.-Y. Choo, J.-S. Sung, M. Saidutta, S.-D. Song and Y.-W. Rhee, Transition metals supported on activated carbon as benzene hydroxylation catalysts, J. Porous Mater., 2005, 12, 301–310 CrossRef CAS.
- X. Fu, X. Gu, S. Lu, V. K. Sharma, M. L. Brusseau, Y. Xue, M. Danish, G. Y. Fu, Z. Qiu and Q. Sui, Benzene oxidation by Fe (III)-activated percarbonate: matrix-constituent effects and degradation pathways, Chem. Eng., 2017, 309, 22–29 CrossRef CAS PubMed.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, Fe-g-C3N4-catalyzed oxidation of benzene to phenol using hydrogen peroxide and visible light, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- C. Basheer, Nanofiber-membrane-supported TiO2 as a catalyst for oxidation of benzene to phenol, J. Chem., 2013, 2013, 562305 Search PubMed.
- H. Yang, J. Li, L. Wang, W. Dai, Y. Lv and S. Gao, Exceptional activity for direct synthesis of phenol from benzene over PMoV@ MOF with O2, Catal. Commun., 2013, 35, 101–104 CrossRef CAS.
- M. Tani, T. Sakamoto, S. Mita, S. Sakaguchi and Y. Ishii, Hydroxylation of benzene to phenol under air and carbon monoxide catalyzed by molybdovanadophosphoric acid, Angew. Chem., 2005, 117, 2642–2644 CrossRef.
- H. Yamanaka, R. Hamada, H. Nibuta, S. Nishiyama and S. Tsuruya, Gas-phase catalytic oxidation of benzene over Cu-supported ZSM-5 catalysts: an attempt of one-step production of phenol, J. Mol. Catal. A: Chem., 2002, 178, 89–95 CrossRef CAS.
- Y. Dong, X. Niu, W. Song, D. Wang, L. Chen, F. Yuan and Y. Zhu, Facile synthesis of vanadium oxide/reduced graphene oxide composite catalysts for enhanced hydroxylation of benzene to phenol, Catalysts, 2016, 6, 74 CrossRef.
- Y.-Y. Gu, X.-H. Zhao, G.-R. Zhang, H.-M. Ding and Y.-K. Shan, Selective hydroxylation of benzene using dioxygen activated by vanadium–copper oxide catalysts supported on SBA-15, Appl. Catal., A, 2007, 328, 150–155 CrossRef CAS.
- Y. Ichihashi, Y.-h Kamizaki, N. Terai, K. Taniya, S. Tsuruya and S. Nishiyama, One-Step oxidation of benzene to phenol over Cu/Ti/HZSM-5 Catalysts, Catal. Lett., 2010, 134, 324–329 CrossRef CAS.
- A. B. Ene, T. Archipov and E. Roduner, Competitive adsorption and interaction of benzene and oxygen on Cu/HZSM5 zeolites, J. Phys. Chem. C, 2011, 115, 3688–3694 CrossRef CAS.
- B. Guo, L. Zhu, X. Hu, Q. Zhang, D. Tong, G. Li and C. Hu, Nature of vanadium species on vanadium silicalite-1 zeolite and their stability in hydroxylation reaction of benzene to phenol, Catal. Sci. Technol., 2011, 1, 1060–1067 RSC.
- P. Chammingkwan, W. Hoelderich, T. Mongkhonsi and P. Kanchanawanichakul, Hydroxylation of benzene over TS-PQ™ catalyst, Appl. Catal.,A, 2009, 352, 1–9 CrossRef CAS.
- T. Miyake, M. Hamada, Y. Sasaki and M. Oguri, Direct synthesis of phenol by hydroxylation of benzene with oxygen and hydrogen, Appl. Catal., A, 1995, 131, 33–42 CrossRef CAS.
- W. Laufer and W. F. Hoelderich, New direct hydroxylation of benzene with oxygen in the presence of hydrogen over bifunctional ion-exchange resins, Chem. Commun., 2002, 1684–1685 RSC.
- I. Yuranov, D. A. Bulushev, A. Renken and L. Kiwi-Minsker, Benzene to phenol hydroxylation with N2O over Fe-Beta and Fe-ZSM-5: Comparison of activity per Fe-site, Appl. Catal., A, 2007, 319, 128–136 CrossRef CAS.
- S. M. Hosseini, M. Ghiaci, S. A. Kulinich, W. Wunderlich, H. Farrokhpour, M. Saraji and A. Shahvar, Au-Pd@ g-C3N4 as an efficient photocatalyst for visible-light oxidation of benzene to phenol: experimental and mechanistic study, J. Phys. Chem. C, 2018, 122, 27477–27485 CrossRef CAS.
- G. Panov, G. Sheveleva, A. E. A. Kharitonov, V. Romannikov and L. A. Vostrikova, Oxidation of benzene to phenol by nitrous oxide over Fe-ZSM-5 zeolites, Appl. Catal., A, 1992, 82, 31–36 CrossRef CAS.
- Y. Luo, J. Xiong, C. Pang, G. Li and C. Hu, Direct hydroxylation of benzene to phenol over TS-1 catalysts, Catalysts, 2018, 8, 49 CrossRef.
- S. Feng, S. Pei, B. Yue, L. Ye, L. Qian and H. He, Synthesis and characterization of V-HMS employed for catalytic hydroxylation of benzene, Catal. Lett., 2009, 131, 458–462 CrossRef CAS.
- A. Koekkoek, W. Kim, V. Degirmenci, H. Xin, R. Ryoo and E. Hensen, Catalytic performance of sheet-like Fe/ZSM-5 zeolites for the selective oxidation of benzene with nitrous oxide, J. Catal., 2013, 299, 81–89 CrossRef CAS.
- S. H. Talib, Z. Lu, X. Yu, K. Ahmad, B. Bashir, Z. Yang and J. Li, Theoretical inspection of M1/PMA single-atom electrocatalyst: ultra-high performance for water splitting (HER/OER) and oxygen reduction reactions (OER), ACS Catal., 2021, 11, 8929–8941 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- J.-C. Liu, Y. Tang, Y.-G. Wang, T. Zhang and J. Li, Theoretical understanding of the stability of single-atom catalysts, Natl. Sci. Rev., 2018, 5, 638–641 CrossRef CAS.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Single-atom electrocatalysts, Angew. Chem. Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong and Y. Wang, Design of single-atom Co–N5 catalytic site: a robust electrocatalyst for CO2 reduction with nearly 100% CO selectivity and remarkable stability, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- M. D. Marcinkowski, M. T. Darby, J. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Pt/Cu single-atom alloys as coke-resistant catalysts for efficient C–H activation, Nat. Chem., 2018, 10, 325–332 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, C–H bond activation enables the rapid construction and late-stage diversification of functional molecules, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- M. Zhang, Y.-G. Wang, W. Chen, J. Dong, L. Zheng, J. Luo, J. Wan, S. Tian, W.-C. Cheong and D. Wang, Metal (hydr) oxides@ polymer core–shell strategy to metal single-atom materials, J. Am. Chem. Soc., 2017, 139, 10976–10979 CrossRef CAS PubMed.
- S. H. Talib, X. Yu, Z. Lu, K. Ahmad, T. Yang, H. Xiao and J. Li, A polyoxometalate cluster-based single-atom catalyst for NH3 synthesis via an enzymatic mechanism, J. Mater. Chem. A, 2022, 10, 6165–6177 RSC.
- B. Zhang, H. Asakura and N. Yan, Atomically dispersed rhodium on self-assembled phosphotungstic acid: structural features and catalytic CO oxidation properties, Ind. Eng. Chem. Res., 2017, 56, 3578–3587 CrossRef CAS.
- Y. Zhang, J. Liu, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Polyoxometalate-based materials for sustainable and clean energy conversion and storage, EnergyChem, 2019, 1, 100021 CrossRef.
- M. J. Hülsey, B. Zhang, Z. Ma, H. Asakura, D. A. Do, W. Chen, T. Tanaka, P. Zhang, Z. Wu and N. Yan, In situ spectroscopy-guided engineering of rhodium single-atom catalysts for CO oxidation, Nat. Commun., 2019, 10, 1330 CrossRef PubMed.
- Y. Wu, S. Niu, Z. Wei, L. Meng and W. Wei, Steric construction and modulation of CoNx single-atom electrocatalysts via polyoxometalate clusters integration, Appl. Catal., B, 2024, 352, 124014 CrossRef CAS.
- Q. Liu, P. He, H. Yu, L. Gu, B. Ni, D. Wang and X. Wang, Single molecule–mediated assembly of polyoxometalate single-cluster rings and their three-dimensional superstructures, Sci. Adv., 2019, 5, eaax1081 CrossRef CAS PubMed.
- B. Zhang, G. Sun, S. Ding, H. Asakura, J. Zhang, P. Sautet and N. Yan, Atomically dispersed Pt1–polyoxometalate catalysts: how does metal–support interaction affect stability and hydrogenation activity?, J. Am. Chem. Soc., 2019, 141, 8185–8197 CrossRef CAS PubMed.
- X. López, J. J. Carbó, C. Bo and J. M. Poblet, Structure, properties and reactivity of polyoxometalates: a theoretical perspective, Chem. Soc. Rev., 2012, 41, 7537–7571 RSC.
- T. D. Rapson and C. C. Wood, Analysis of the Ammonia Production Rates by Nitrogenase, Catalyst, 2022, 12, 844 CrossRef CAS.
- G. Altinay and R. B. Metz, Vibrational spectroscopy of intermediates in benzene-to-phenol conversion by FeO+, J. Am. Soc. Mass Spectrom., 2010, 21, 750–757 CrossRef CAS PubMed.
- Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, Direct hydroxylation of benzene to phenol using hydrogen peroxide catalyzed by nickel complexes supported by pyridylalkylamine ligands, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS PubMed.
- M. Tada, R. Bal, T. Sasaki, Y. Uemura, Y. Inada, S. Tanaka, M. Nomura and Y. Iwasawa, Novel Re-cluster/HZSM-5 catalyst for highly selective phenol synthesis from benzene and O2: performance and reaction mechanism, J. Phys. Chem. C, 2007, 111, 10095–10104 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- B. Bashir, B. Zhang, M.-H. Lee, S. Pan and Z. Yang, DFT-Based Comparative Study about the Influence of Fluorine and Hydroxyl Anions on Opto-Electric Properties of Borate Crystals: Choice for Better Anion, Inorg. Chem., 2017, 56, 5636–5645 CrossRef CAS PubMed.
- B. Bashir, B. Zhang, S. Pan and Z. Yang, Combination of d10-cations and fluorine anion as active participants to design novel borate/carbonate nonlinear optical materials, J. Alloys Compd., 2018, 758, 85–90 CrossRef CAS.
- P. E. Blöchl, J. Kästner and C. J. Först, Electronic structure methods: Augmented waves, pseudopotentials and the projector augmented wave method, Handbook of Materials Modeling: Methods, 2005, pp. 93–119 Search PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved grid-based algorithm for Bader charge allocation, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, A simple measure of electron localization in atomic and molecular systems, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- S.-S. Wang and G.-Y. Yang, Recent advances in polyoxometalate-catalyzed reactions, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Ultrastable single-atom gold catalysts with strong covalent metal-support interaction (CMSI), Nano Res., 2015, 8, 2913–2924 CrossRef CAS.
- J. Su, S. Hu, W. Huang, M. Zhou and J. Li, On the oxidation states of metal elements in MO3-(M = V, Nb, Ta, Db, Pr, Gd, Pa) anions, Sci. China: Chem., 2016, 59, 442–451 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Electronic factors determining the reactivity of metal surfaces, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, Single atom (Pd/Pt) supported on graphitic carbon nitride as an efficient photocatalyst for visible-light reduction of carbon dioxide, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- J. K. Burdett and T. A. McCormick, Electron localization in molecules and solids: the meaning of ELF, Phys. Chem. A, 1998, 102, 6366–6372 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the optimized geometry of PMA with various possible coordination sites, the optimized Fe1/PMA structure, spin-polarized partial density of states (PDOS) of all the elementary steps, d-band center, and charge density plots. See DOI: https://doi.org/10.1039/d4ma00238e |
| This journal is © The Royal Society of Chemistry 2024 |