
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pyrrolo[3,2-b]pyrrole core containing a covalent triazine-based framework (CTF) for photocatalytic H2 production†
Osman
Ali
a,
Anupam
Jana
a,
Aruntima
Das
a,
Sandeep Kumar
Dey
b and
Asamanjoy
Bhunia
 *a
*a
aDepartment of Chemistry, Inorganic Chemistry Section, Jadavpur University, Kolkata 700032, India. E-mail: abhunia.chemistry@jadavpuruniversity.in
bMaterial Science and Technology Division, CSIR-North East Institute of Science and Technology, Jorhat, Assam 785006, India. E-mail: sandeep@neist.res.in
First published on 13th April 2024
Abstract
The pursuit of developing photocatalysts for visible-light-driven water splitting has garnered considerable attention within the research community. In this context, covalent triazine-based frameworks (CTFs) have emerged as the most important materials in the realm of photocatalytic water splitting owing to their predictive structural design, well-defined long-range ordering, tunable porosity, and remarkable stability. In this study, we present a pyrrolo[3,2-b]pyrrole core-containing CTF, named CTF-TPP, which was fabricated from 1,2,4,5-tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (TPP) by a solvothermal method. This synthesized CTF-TPP served as a dye-sensitized photocatalyst for visible light-induced water splitting that exhibits H2 production of 612 μmol g−1 in 4 h. In addition, the hydrogen evolution rate (HER) was enhanced by introducing in situ generated Pt and Pd nanoparticles (NPs) into the CTF-TPP solid matrix in different proportions. Among them, CTF-TPP@Pd0.5 exhibited superior H2 evolution compared to its Pt counterpart, achieving a hydrogen production rate of 7980 μmol g−1 in 4 h with an apparent quantum efficiency (AQY) of 13.97% at 420 nm. Additionally, it demonstrated a good turnover number (TON) exceeding 173 for 4 h of photocatalysis. This enhanced performance was attributed to the strong interaction between the Pd NPs and CTF-TPP, facilitating efficient photoelectron transfer. Furthermore, we have also given a plausible mechanism based on the photophysical and electrochemical study to efficiently elucidate the enhanced photocatalytic H2 production. This study offers an idea for developing a highly efficient photocatalyst based on CTF in the context of H2 production.
Introduction
Photocatalytic hydrogen (H2) evolution holds great promise as a sustainable method for transforming solar energy into environmentally friendly chemicals.1 The motivation behind developing such technologies stems from the rapid depletion of fossil fuels and the environmental issues associated with their usage. In the past few decades, considerable attention has been directed toward the strategic design and synthesis of proficient photocatalysts. Since the initial discovery of TiO2's photocatalytic properties,2 various inorganic semiconductors have been examined for their suitability in the field of water splitting.3–5 In the early stages of exploration, inorganic metal-based semiconductors with precisely defined crystal structures, such as metal sulfides,6 metal oxides,7 and oxometallates,8 were extensively studied and continued to be fascinating in recent years. However, these inorganic photocatalysts often face limitations due to their narrow intrinsic band structures, primarily absorbing ultraviolet light.9 To solve this issue, recently non-metallic semiconductors have evolved as favourable alternatives for photocatalytic hydrogen evolution, including graphitic carbon nitrides,10 small molecular organic dyes,11 linear conjugated polymers,12 and several other organic scaffolds. These organic photocatalysts can be customized with precisely arranged band structures, enabling them to efficiently capture a significant amount of visible light. However, these organic photocatalysts face a challenge in separating photo-generated charges.13 One of the most promising approaches to enhance charge separation is by constructing extended frameworks. Consequently, a range of porous organic polymers (POPs) like conjugated microporous polymers (CMPs),14 covalent organic frameworks (COFs),15 and covalent triazine-based frameworks (CTFs),16 have been established in the discipline of photocatalysis. Framework-based photocatalysts offer numerous adjustable photoactive sites and the flexibility to tailor the pore structures. These two advantages make porous organic frameworks highly promising for applications in photocatalytic water splitting, which has influenced considerable attention in recent years.Among these POPs, CTFs stand out as an interesting class of organic semiconductor materials and have recently gained significant interest due to their distinctive structural features including a nitrogen-rich composition, remarkable chemical stability, fully conjugated skeleton, and tunable surface area. These exceptional properties enhance the appeal of CTFs for diverse applications, particularly in the realm of photocatalysis.17–20 The photocatalytic properties of CTFs are highly influenced by the synthetic methods. To date, CTFs have been synthesized by various methods such as (1) the ionothermal method (ZnCl2),21 (2) the P2O5 catalyzed method,22 (3) the polycondensation method,23 (4) the Friedel–Crafts method,24 and (5) the superacid-catalyzed method.25 Despite their high BET surface area and porous structure, CTFs synthesized by the ionothermal and P2O5 catalyzed methods are not favorable candidates for photocatalytic application due to their high-temperature synthesis that leads to partial carbonization of the materials, and hence, results in structural defects. Friedel–craft methods for CTF synthesis suffer from side reactions (i.e. the Scholl reaction).26 On the other hand, polycondensation methods are limited by the inaccessible and often expensive monomers rather than utilizing readily available nitrile-containing monomers. Hence, the aspiration to achieve CTFs via an efficient method under mild conditions remains a challenging task. So, there is a need for an alternative milder synthetic strategy to prevent the mismatch band structures in the dark colored and carbonized CTFs that restrict their photocatalytic activity. In this context, the superacid-catalyzed strategy has proven to be more effective, yielding colored CTFs that preserve the semiconductive nature due to the absence of carbonization. This favors the absorption of visible light and charge separation due to the π-stacked structure.27,28 Hence, selecting an appropriate synthetic method is crucial to ensure the effective preparation of CTFs for improved photocatalytic performance. Cooper et al. synthesized CTFs (CTF-1 to CTF-4) using trifluoromethanesulfonic acid (CF3SO3H) as a Brønsted acid catalyst, demonstrating varying H2 evolution rates of 35, 296, 45, and 0 μmol g−1 h−1 under visible light irradiation.29 Li et al. synthesized CTF-BT/Th-x molecular heterostructures using CF3SO3H-catalyzed polymerization, integrating Th and BT into CTF skeletons.30 CTF-BT/Th-1 showed efficient charge separation, achieving a notable H2 evolution rate of 6600 μmol g−1 h−1. Taking into account all considerations, we chose the Brønsted acid-catalyzed method among various synthetic approaches to produce efficient photocatalysts, aiming for enhanced H2 production.
In this work, we report the visible light-induced hydrogen evolution reaction (HER) by using a newly synthesized 1,2,4,5-tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (TPP)-based CTF, named CTF-TPP, that was synthesized by the solvothermal method using a Brønsted acid catalyst. The HER activity of CTF-TPP exhibited a substantial 15-fold improvement in the presence of an eosin Y (EY) photosensitizer. To the best of our knowledge, this is the first report on an EY-sensitized CTF photocatalyst for H2 production (Table S4, ESI†). The HER activity was further enhanced by incorporating in situ synthesized Pd or Pt NPs into the CTF-TPP polymeric scaffold. It was observed that all CTF-TPP@Pd0.5 exhibited superior photocatalytic activity when compared to CTF-TPP@Pt0.5. This enhanced performance could be attributed to the smaller size of the Pd NPs. These smaller nanoparticles offer enhanced electron transfer capabilities and improved separation of photogenerated electron–hole pairs, contributing to their superior photocatalytic performance compared to Pt-supported CTFs.
Results and discussion
Physicochemical characterization of CTFs
1,2,4,5-Tetrakis(4-cyanophenyl)-1,4-dihydropyrrolo[3,2-b]pyrrole (TPP) was synthesized by mixing 4-cyanobenzaldehyde and 4-cyanoaniline in the presence of iron(III) perchlorate hydrate as a catalyst.31 The synthesized ligand (TPP) was employed for the cyclotrimerization reaction using trifluoromethanesulfonic acid (CF3SO3H) as the Brønsted acid catalyst via the solvothermal method to obtain the desired CTF, named CTF-TPP (Scheme 1 and ESI†).32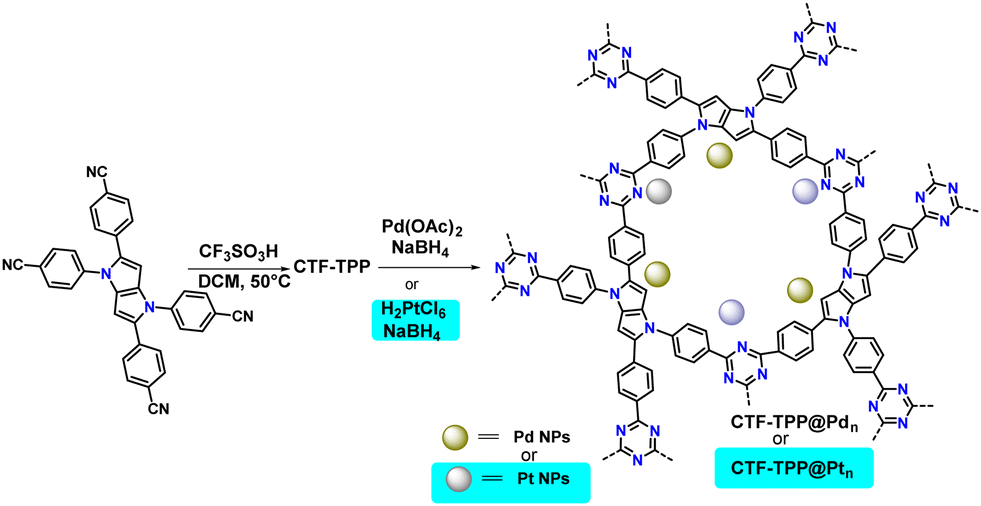 | ||
| Scheme 1 Schematic illustration of CTF-TPP and its Pd NP or Pt NP incorporation. | ||
The exclusive formation of a triazine framework was confirmed by Fourier transform infrared spectroscopy (FT-IR) and solid-state 13C NMR spectroscopy (Fig. 1a and b).
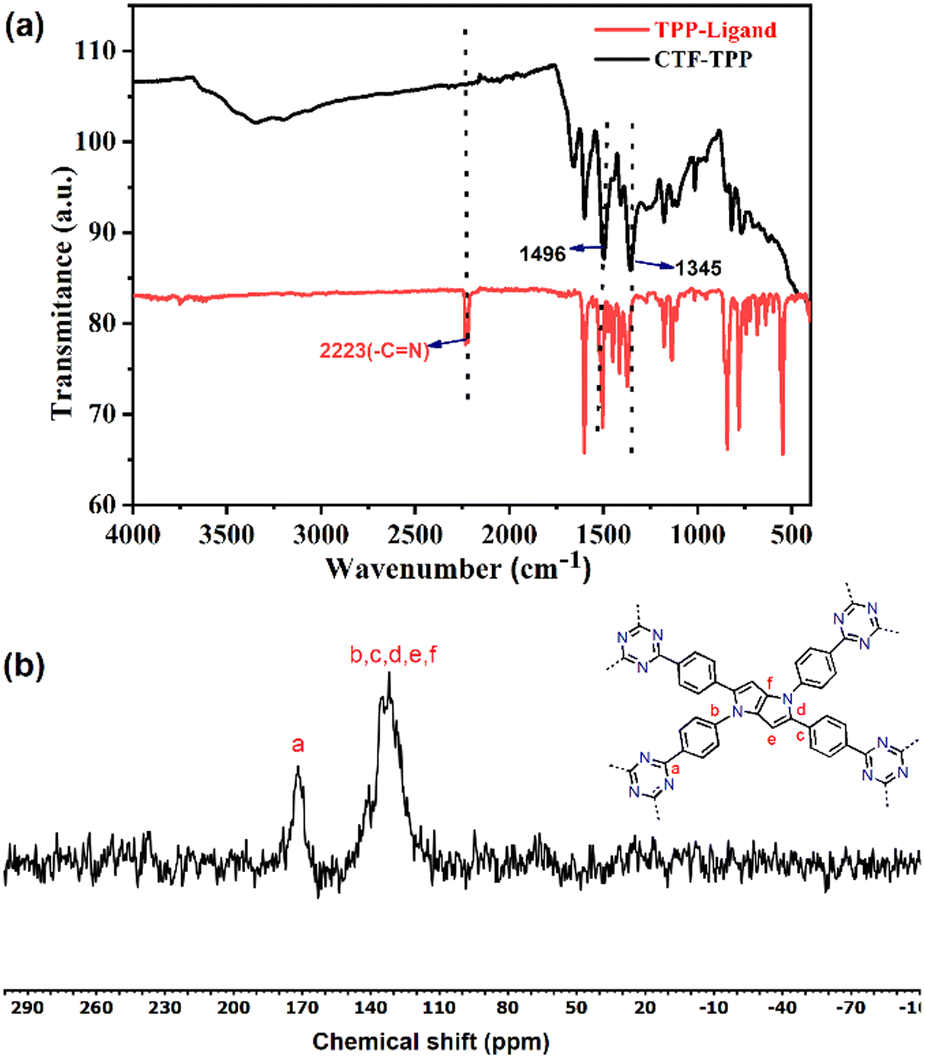 | ||
| Fig. 1 (a) FT-IR spectra of the TPP monomer and CTF-TPP, and (b) solid-state CP/MAS 13C NMR spectrum of CTF-TPP. | ||
FT-IR studies established the formation of triazine units in the obtained material and the absence of free nitrile groups in the TPP-based CTF.
The FT-IR spectrum of CTF-TPP displayed a stretching and breathing band at around 1496 cm−1 and 1345 cm−1 for a molecular triazine ring. Simultaneously, the intense nitrile (–CN) stretching band around 2226 cm−1 almost evanesced.33,34 These results proved a successful trimerization of the nitrile groups (–CN). The confirmation of triazine unit formation was further supported by solid-state 13C/MAS NMR, where the signal at 169 ppm indicated the distinct carbon signals of the triazine ring.35 The carbon atoms within the pyrrole moieties and benzene rings exhibited signals in the range of 110 to 145 ppm.36,37
The powder X-ray diffraction (PXRD) pattern of the CTF-TPP material was found to have broad peaks at 5°, 21°, and 44° suggesting the amorphous nature of the synthesized material (Fig. S1a, ESI†).34 Thermogravimetric analysis (TGA) revealed that the thermal stability of the synthesized CTF-TPP is up to a temperature of 320 °C. The initial weight loss of the material might be attributed to the removal of water molecules or guest solvent molecules present in the pores (Fig. S1b, ESI†). Furthermore, a weight loss beyond 320 °C could be ascribed to the framework degradation. X-ray photoelectron spectroscopy (XPS) was employed to analyse the elemental composition of the CTF-TPP, which confirmed the presence of carbon and nitrogen atoms (Fig. 2a, b and Fig. S1c, ESI†). In the C1s spectrum of CTF-TPP, two peaks were detected, and the one with a binding energy (BE) of 284.6 eV represents the benzene ring and the other with BE of 286.1 eV represents carbon in the triazine ring, respectively (Fig. 2a). Furthermore, the distribution of nitrogen atoms was determined by deconvoluting the high-resolution spectral regions for the N1s signal (Fig. 2b). The N1s spectra were successfully deconvoluted into two distinct peaks, representing different types of nitrogen atoms. The binding energies of 398.6 and 400.3 eV corresponded to triazine N and pyrrolic N in the CTF-TPP, respectively.38,39 On the other hand, a peak at 288.7 was observed, which corresponds to the C–F bond of the residual CF3SO3H (Fig. 2a).40 Furthermore, the morphology of CTF-TPP was characterized through scanning electron microscopy (SEM), which exhibited the disordered shape (Fig. 2c). The porosity analysis of the CTF-TPP was conducted through a N2 adsorption–desorption experiment at 77 K (Fig. S2a and b in ESI†). The adsorption isotherm was observed to be type II, which indicated the nonporous nature of the frameworks.34,41 The Brunauer–Emmett–Teller (BET) surface area of CTF-TPP was determined to be 37 m2 g−1. Additionally, an electron paramagnetic resonance (EPR) experiment was conducted in the solid state at room temperature. The EPR spectrum of CTF-TPP showed a solitary Lorentzian line with a g-value of 1.999 (Fig. S10, ESI†). This signal is attributed to the unpaired electrons within the π-conjugated framework.42
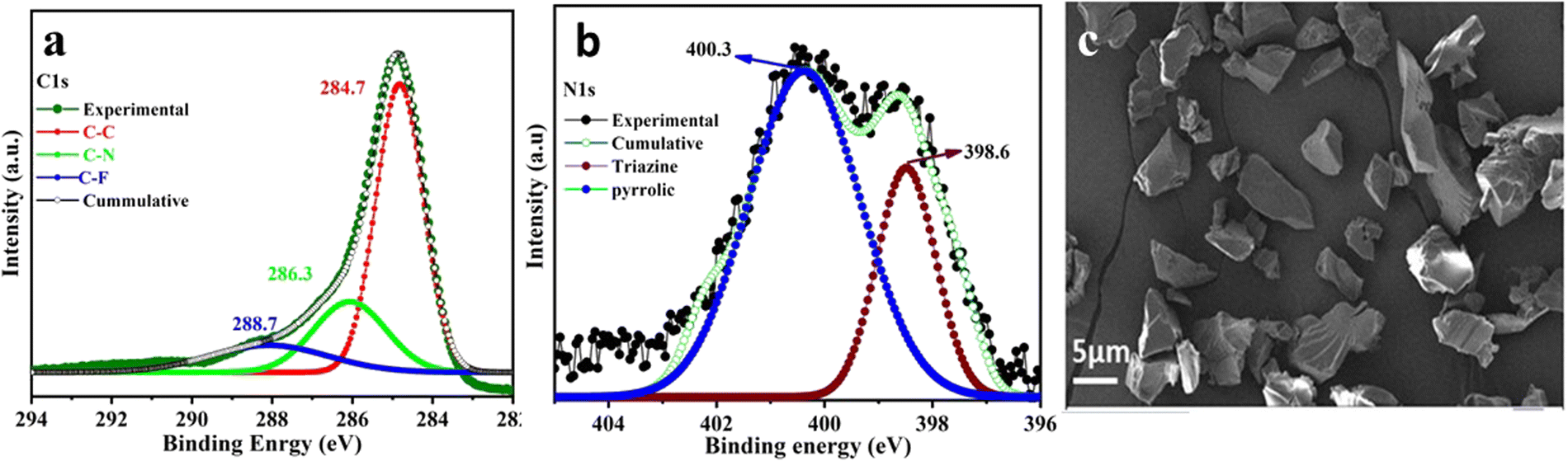 | ||
| Fig. 2 (a) XPS spectra depicting C1s. (b) XPS spectra depicting N1s, and (c) FE-SEM image of CTF-TPP. | ||
After the successful synthesis of CTF-TPP, Pd, and Pt NPs were incorporated by in situ reduction of Pd(II)/Pt(II) salts into the CTF-TPP matrix, named as CTF-TPP@Pdn or CTF-TPP@Ptn (where, n = wt% of Pd and Pt NPs), respectively (Scheme 1 and ESI† for synthesis details). Among all the prepared photocatalysts, CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5 were considered for detailed structural analysis due to their high photocatalytic activity under optimum conditions. The metal NP content in CTF-TPP was examined using inductively coupled plasma optical emission spectroscopy (ICP-OES). The loading values of Pd and Pt were determined to be 0.25, 0.5, and 1.7 wt% (Table S1, ESI†). The immobilization of Pd NPs and Pt NPs into CTF-TPP was confirmed through a combination of SEM and energy-dispersive X-ray spectroscopic (EDX) elemental mapping analysis. The SEM images revealed the morphology and structure of the CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5 samples, which displayed irregular shapes (Fig. S3(a–d), ESI†). Furthermore, EDX elemental mapping unveiled the uniform distribution of carbon, nitrogen, Pd, and Pt across the selected area, indicating the successful creation of metal-semiconductor-CTF nanomaterials (Fig. S4 and S5 in ESI†).
The successful immobilization of Pd and Pt NPs onto the surface of CTF-TPP was identified by TEM analysis (Fig. S6a and S7a in ESI†). The particle size distribution curve demonstrates that the average size of the Pt nanoparticles (NPs) is approximately 32 nm, while for Pd nanoparticles, it is around 2.5 nm (Fig. S6b and S7b, in ESI†).
Photoelectrochemical properties
To gain a deeper understanding of the photocatalytic activity of CTF-TPP, CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5 a comprehensive investigation of their optical and electronic properties has been carried out. The optical characteristics of the as-prepared materials, including CTF-TPP, CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5 heterojunctions, as measured by UV-vis spectroscopy, are depicted in Fig. 3a. The absorption edges of these materials have been observed at 357 nm, 532 nm, and 410 nm (approx.) for CTF-TPP, CTF-TPP@Pd0.5, and CTF-TPP@Pd0.5, respectively. It is worth noting that the absorption edges of the CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5 heterojunctions have been red-shifted with a notable enhancement in absorption intensity in the visible region. These results suggested that the incorporation of Pd and Pt NPs into CTF-TPP broadens the absorption spectrum of the heterojunctions into the visible range. However, it is important to mention that the absorption intensity of these metal-anchored CTF-TPP materials was relatively low. Consequently, these materials exhibited reduced visible light absorption. | ||
| Fig. 3 (a) UV-Vis spectra, (b) Tauc plot, and (c) potential energy diagram of CTF-TPP, CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5. (d) Nyquist diagrams of electrochemical impedance spectra, (e) transient photocurrent response, and (f) time-resolved photoluminescence spectra of CTF-TPP, CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5. | ||
To overcome this limitation and enhance the visible light absorption capacity of the materials, a dye was utilized that led to a significant increase in their photocatalytic H2 evolution activity. The band gap energies (Eg) of CTF-TPP, CTF-TPP@Pt0.5, and CTF-TPP@Pd0.5 have been estimated from Tauc plots43,44 to be 2.90 eV, 2.70 eV, and 2.50 eV, respectively (Fig. 3b and Fig. S8a in ESI†). On the other hand, density functional theory (DFT) measurement was carried out using Gaussian 09 software. The ground state geometry was fully optimized using the B3LYP/6-31G basis set to gain insight into the electronic structures and visualize the corresponding frontier molecular orbitals. The electronic distribution of the energy levels from quantum chemical calculation reveals that the electron density of the HOMO is mainly localized on the pyrrole-pyrrole unit, whereas the electron density of the LUMO is present on the phenyl-triazine units (Fig. S9 in ESI†). The phenyl-triazine segment linked to the pyrrole-pyrrole units acts as an acceptor while the pyrrole moieties act as electron donors. The theoretically computed optical band gap for the CTF-TPP was determined to be 2.85 eV, demonstrating good agreement with the experimentally obtained optical band gap from the Tauc plot (2.90 eV). The band structures of the samples were determined via Mott–Schottky plots. The flat band potentials of CTF-TPP@Pd0.5 CTF-TPP@Pt0.5, and CTF-TPP have been estimated to be approximately −0.83 eV, −0.91 eV, and −1.02 eV versus an Ag/Ag+ electrode (Fig. S8 in ESI†), respectively, corresponding to −0.63 eV, −0.71 eV, and −0.82 eV versus the normal hydrogen electrode (NHE). Furthermore, the valence band (VB) potentials of CTF-TPP@Pd0.5, CTF-TPP@Pt0.5 and CTF-TPP were 1.87 eV, 1.99 eV and 2.08 eV calculated by the band gap energy (Eg = EVB − ECB) (Fig. 3c). Electrochemical impedance spectroscopy (EIS) was utilized to analyze both the electrical conductivity and internal resistance involved in the charge transfer process within the photocatalysts. In the context of the equivalent circuit, a smaller arc radius on the Nyquist plot signified a reduced resistance.45 The EIS plots showed that the charge transfer resistance of CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5 was lower than that of CTF-TPP, reflecting the fastest interfacial charge transfer between CTF-TPP and Pd or Pt NPs (Fig. 3d). These results revealed that the CTF-TPP@Pd0.5 catalyst had excellent electron transfer efficiency and charge separation ability, which might lead to enhanced photocatalytic activity. Furthermore, a photocurrent test was conducted with periodic visible-light irradiation to monitor the generation and movement of photoinduced charges (Fig. 3e). The prepared photocatalysts including CTF-TPP, CTF-TPP@Pd0.5 and CTF-TPP@Pt0.5 exhibited an increase in current when exposed to light. The photocurrent response revealed a greater current density in the Pd and Pt-incorporated CTF-TPP compared to the pristine CTF when exposed to visible light. Among the tested photocatalysts, CTF-TPP@Pd0.5 displayed the highest photocurrent response among them. This outcome was in line with the order of photocatalytic activity, which was arranged as CTF-TPP@Pd0.5 > CTF-TPP@Pt0.5 > CTF-TPP. Time-resolved photoluminescence spectroscopy was utilized to understand the charge transfer efficiency (Fig. 3f). The decay curves of CTF-TPP exhibited a good fit with a bi-exponential function, and the photoluminescence average lifetimes (τavg) for CTF-TPP@Pd0.5, CTF-TPP@Pt0.5 and CTF-TPP were determined to be 2.51, 1.82 and 1.71 ns, respectively. (Table S3 in ESI†). A prolonged time-resolved photoluminescence (TRPL) lifetime suggested a slower recombination of electron–hole pairs. Additionally, an extended lifetime of the excited state was advantageous for increasing the probability of electron transfer from the conduction band (CB) to the co-catalyst Pd and Pt NPs.
Photocatalytic hydrogen evolution activity
The photocatalytic HER activity of the synthesized materials was evaluated in an aqueous medium using triethanolamine (TEOA) as an electron donor and EY as a photosensitizer under visible light irradiation with a wavelength of 420 nm. The gaseous products for the above mentioned experiments were analysed after irradiation with visible light (>400 nm) for 2 h using GC (Fig. S14–S16, ESI†). However, several metal-free organic dyes including EY, rhodamine B and rose Bengal were employed for HER activity (Fig. S11, ESI†). Among them, EY dye exhibited the highest HER activity. Hence, EY was selected for the water splitting reaction. We conducted several control experiments to confirm the essential contribution of each component (TEOA, light and photocatalyst) in the process of photocatalytic hydrogen production.We observed that the absence of any of these components prevented the HER by water splitting (Fig. 4b and Fig. S14a–c in ESI†). Under the optimum conditions (e.g., 2 mg catalyst, 4 mg EY, 1 mL TEOA and 5 mL H2O), CTF-TPP, CTF-TPP@Pt0.5, and CTF-TPP@Pd0.5 displayed H2 production of 220, 1216, and 2520 μmol g−1 in 2 h (Fig. 4a and Table S1 in ESI†). However, the photocatalytic H2O splitting performance has also been observed without EY, which generated H2 of 14, 180 and 416 μmol g−1 in 2 h for CTF-TPP, CTF-TPP@Pt0.5 and CTF-TPP@Pd0.5, respectively (Table S2, ESI†). The reduced effectiveness of CTF-TPP in H2 evolution is due to the higher rate of electron–hole pair recombination during the photocatalytic process.
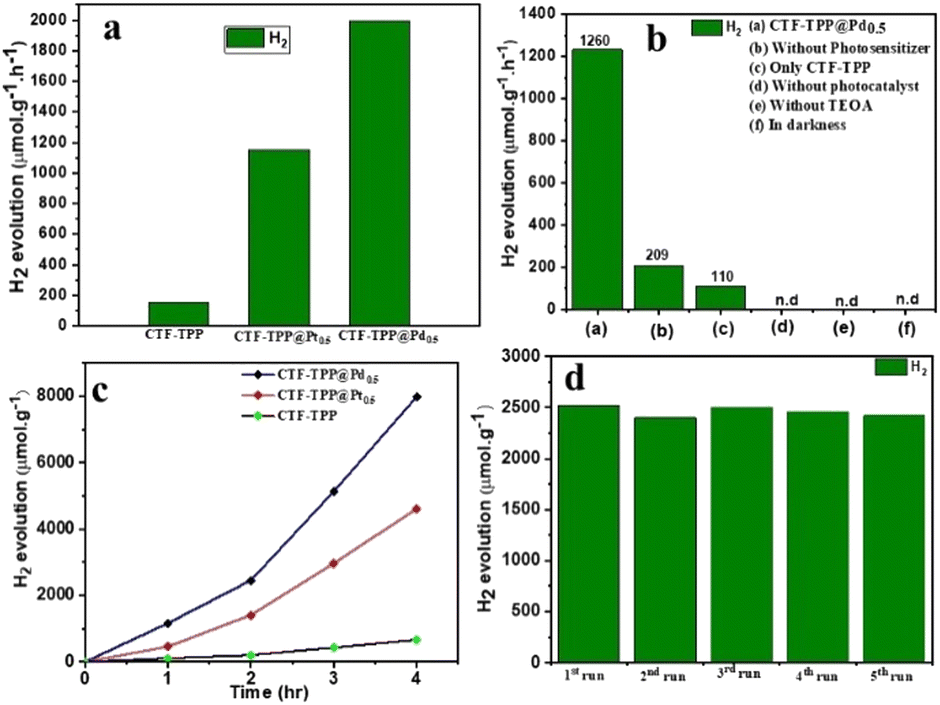 | ||
| Fig. 4 Catalytic performance of photocatalytic H2O splitting. (a) The evolution rate of H2 production over the as-synthesized CTF-TPP, CTF-TPP@Pt0.5 and CTF-TPP@Pd0.5. (b) Control experiments over the CTF-TPP@Pd0.5 photocatalyst. (c) Long run H2 production of CTF-TPP, CTF-TPP@Pt0.5 and CTF-TPP@Pd0.5. (d) Cycling production of H2 with the CTF-TPP@Pd0.5 photocatalyst in 2 h. | ||
The photocatalytic activity of CTF-TPP was further enhanced by depositing different amounts of in situ generated Pd or Pt NPs (see ESI† for synthesis details). However, at the same wt% of NP loading, CTF-TPP@Pd0.5 exhibited a higher catalytic performance than CTF-TPP@Pt0.5 (Table S1 ESI†). The higher photocatalytic activity of CTF-TPP@Pd0.5 might be credited to the growth of smaller size Pd NPs on the CTF-TPP (Fig. S6a and S7a in ESI†). These results emphasized the pivotal role of Pt and Pd sites as the primary catalytic centers, with CTF-TPP playing a crucial part in facilitating electron transfer from the dye to the active sites and stabilizing metal sites in the catalytic process. Moreover, it was found that CTF-TPP@Pd0.5 exhibited superior photocatalytic efficiency, generating an impressive H2 production rate of 1995 μmol g−1 h−1 (Fig. 4a). On the other hand, prolonged photocatalytic experiments with CTF-TPP@Pd0.5 revealed a significant H2 evolution of 7980 μmol g−1 with TON ≥ 173 over a 4 h run (Fig. 4c) which is comparable to many previously reported CTF-based photocatalysts (Table S4, ESI†). To the best of our knowledge, this is the first report on an EY-sensitized CTF photocatalyst for H2 production (Table S4, ESI†).
In further support of the light-driven nature of the reaction, the apparent quantum yield (AQY) for H2 generation over CTF-TPP@Pd0.5 was analyzed, revealing a value of 13.97% at 420 nm. However, it had been observed that at higher Pd NP concentrations (1.7 wt% Pd loading), the catalytic efficiency of CTF-TPP@Pd1.7 diminishes, resulting in an H2 production of only 560 μmol g−1. This decline could be attributed to the possibility that an increased concentration of Pd might lead to the formation of even larger Pd NPs. This highlighted the critical role of Pd concentration in driving H2 generation and emphasized the importance of optimizing the conditions for optimal catalytic efficiency. The H2 production rate of CTF-TPP@Pd0.5 exhibited minimal variance even after completing five cycles (Fig. 4d). This observation served as confirmation of the sustained, long-term stability and cycling durability for H2 evolution. Even after extended irradiation, the PXRD patterns and FT-IR spectra remained unchanged, indicating the stable nature of the photocatalyst (Fig. S12 and S13 in ESI†). The Pd content after five recycling tests of CTF-TPP@Pd0.5 was performed using an ICP-OES experiment. The analysis revealed that the Pd content of the recycled materials was determined to be 0.38 wt%. This result indicated that the Pd NPs were released from the CTF-TPP support during the H2 production process.
Plausible mechanism
The direction of electron transfer in photovoltaic devices and photocatalysis often relies on the Fermi level difference between the photosensitizer and acceptor materials.46 When EY molecules directly bind to CTF-TPP and are excited by visible light, a significant energy band offset forms between EY* and CTF-TPP. This setup suggested that excited electrons were more likely to transfer to the CTF. Supporting this assumption, the photoluminescence spectra of EY and EY-CTF-TPP were examined (Fig. 5a). EY solution emits a strong peak at 551 nm due to its conjugated xanthene structure, resulting in intense fluorescence upon visible light excitation. However, upon introducing CTF-TPP into the EY solution, a notable decrease in fluorescence intensity was observed, indicating a significant quenching effect and a little blue shift (Fig. 5a). The subtle change and quenching might arise from the transfer of electrons at the interface from EY* molecules attached to the CTF. It has also been proven from the electrochemical study that the estimated potential of the EY* (−3.45 eV)47 is more negative than the CB edge of the CTF-TPP (−0.72 eV) as determined from Mott–Schottky experiment. Such a difference in the energy levels should be sufficient to drive the electron transfer from the photoexcited EY* to the CTF-TPP.48 Moreover, photoelectrochemical experiments also validated the photoinduced electron transfer processes. The observed transient photocurrent of the EY-CTF-TPP electrode was noticeable and exhibited clear responsiveness to visible light during on–off irradiation cycles (Fig. 5b). Conversely, the CTF-TPP electrode, operating under similar experimental conditions, generated a negligible photocurrent. This strongly indicated the effective transfer of electrons from the photoexcited EY (EY−˙) to the CTF-TPP. Moreover, when the CTFs were anchored with metal NPs, the trapped electrons in the CTF network migrated to the Pd or Pt NPs, assuming that they act as electron acceptors and facilitators, leading to a notable reduction in the recombination rate of photoexcited pairs. Consequently, this enhanced the efficacy of photoelectric conversion and photocatalysis.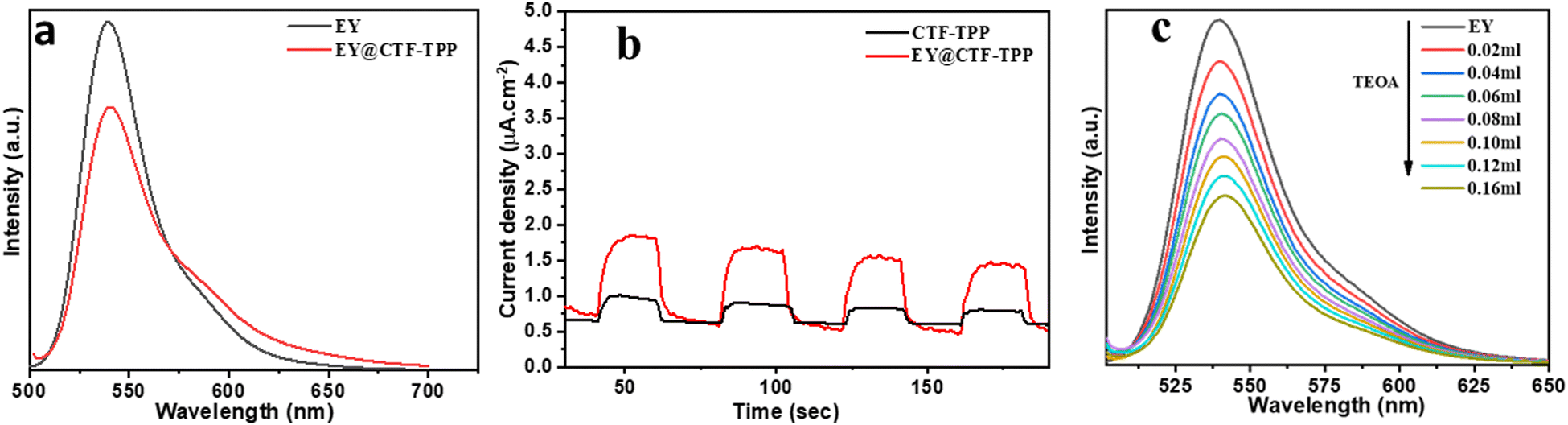 | ||
| Fig. 5 (a) Fluorescence quenching of EY (1 × 10−5 M) by CTF-TPP in aqueous TEOA solution. (b) Photocurrent–time diagram of CTF-TPP and EY@CTF-TPP in a solution of 0.1 M NaSO4 (≥420 nm). (c) Fluorescence quenching of EY by aqueous TEOA solution. | ||
Based on the above results, the most plausible mechanism for photocatalytic H2 evolution within the EY-CTF-TPP@Pd/Pt system is illustrated in Scheme 2.
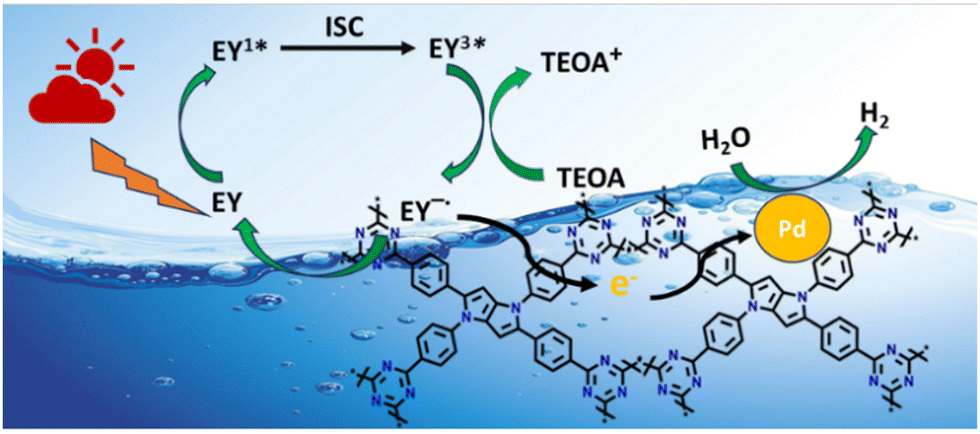 | ||
| Scheme 2 Plausible photocatalytic mechanism for H2 evolution over CTF-TPP@Pdn (Pt NPs not shown in the figure for better clarity). | ||
In the CTF-TPP@Pd/Pt nanohybrid, a particular portion of EY molecules might be adsorbed on the CTF surface due to the noncovalent π–π interaction between EY and CTF-TPP.49
Under the visible light spectrum, the adsorbed EY molecules might gain a photon and be converted into a singlet excited state EY1*. Afterwards, the excited state could further go to a lower-energy triplet excited state EY3* through an intersystem crossing (ISC). The gradual quenching of the fluorescence intensity of EY upon incremental addition of TEOA indicated that the EY3* state might undergo reductive quenching upon interaction with the sacrificial donor TEOA (Fig. 5c), resulting in the formation of EY−˙ species.50–52 These EY−˙ species might favorably donate their electrons to the CTF-TPP, promoting the effective spatial segregation of photogenerated charges.47,49,53 These electrons on the CTF-TPP surface could shift to the Pd or Pt cocatalyst. This cocatalyst facilitated an efficient reduction of protons, consequently driving facile H2 generation. However, the highly reductive EY−˙ species might rapidly recombine with TEOA+ or dye species in the absence of CTF-TPP resulting in reduced catalytic activity during photocatalytic H2 evolution. Thus, the presence of CTF-TPP might significantly enhance the catalytic H2 evolution of the CTF-TPP@Pd0.5/Pt0.5 by extending the lifespan of photogenerated charges and thereby, upgrading the charge separation efficiency.
Conclusions
In summary, we prepared a new pyrrolo[3,2-b]pyrrole core-containing CTF through the solvothermal method and fabricated a series of photocatalysts by immobilizing Pt and Pd NPs onto the synthesized CTF for photocatalytic water splitting. Among all the photocatalysts, the CTF-TPP@Pd composite with 0.5 wt% Pd loading displayed an optimum photocatalytic H2 production rate of 1995 μmol g−1 h−1. This rate was significantly elevated, demonstrating a 13-fold increase compared to pure CTF-TPP and a 2-fold increase when compared to the Pt-counterpart. In contrast, the performance of CTF-TPP@Pd was least favorable at 0.25 wt% and 1.7 wt% Pd loadings. The presence of smaller Pd NPs played a crucial role in effectively separating electron–hole pairs. The robust interaction between Pd metal and pyridinic nitrogen created a closely bonded interface between the Pd nanoparticles and CTF support, facilitating rapid photoelectron transfer and making CTF-TPP@Pd0.5 a promising catalyst. This study presented the potential of CTFs as a photocatalyst in the realm of renewable fuel production by water splitting using solar energy.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was financially supported by DST-INSPIRE (DST/INSPIRE/04/2017/001072), SERB (EEQ/2020/000357), and a UGC (No. F. 30-552/2021(BSR)) start-up grant. O. A. and A. J. acknowledge UGC and CSIR for the fellowship. We thank Mr Nabin Parui (IACS, Kolkata) for help with TEM.References
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Sivula and R. Van De Krol, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS PubMed.
- L. Tong, L. Ren, A. Fu, D. Wang, L. Liu and J. Ye, Chem. Commun., 2019, 55, 12900–12903 RSC.
- X. Chen, S. Shen, L. Guo and S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- Y. Liu, Y. Li, G. Chen, X. F. Wang, R. Fujii, Y. Yamano, O. Kitao, T. Nakamura and S. I. Sasaki, Adv. Mater. Interfaces, 2021, 8, 2101303 CrossRef CAS.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J. L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, M. Sachs, R. Clowes, J. R. Durrant, D. J. Adams and A. I. Cooper, Chem. Commun., 2016, 52, 10008–10011 RSC.
- T. Banerjee, F. Haase, G. Savasci, K. Gottschling, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2017, 139, 16228–16234 CrossRef CAS PubMed.
- Z. A. Lan, M. Wu, Z. Fang, Y. Zhang, X. Chen, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202201482 CrossRef CAS PubMed.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 RSC.
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. A. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC.
- X. Jiang, P. Wang and J. Zhao, J. Mater. Chem. A, 2015, 3, 7750–7758 RSC.
- Y. Shen, C. Zhu, S. Song, T. Zeng, L. Li and Z. Cai, Environ. Sci. Technol., 2019, 53, 9091–9101 CrossRef CAS PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- T. Chen, W. Q. Li, W. B. Hu, W. J. Hu, Y. A. Liu, H. Yang and K. Wen, RSC Adv., 2019, 9, 18008–18012 RSC.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS PubMed.
- S. Dey, A. Bhunia, D. Esquivel and C. Janiak, J. Mater. Chem. A, 2016, 4, 6259–6263 RSC.
- T. Yasuda, T. Shimizu, F. Liu, G. Ungar and T. Kato, J. Am. Chem. Soc., 2011, 133, 13437–13444 CrossRef CAS PubMed.
- S. Gu, J. Guo, Q. Huang, J. He, Y. Fu, G. Kuang, C. Pan and G. Yu, Macromolecules, 2017, 50, 8512–8520 CrossRef CAS.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- D. Wu, F. Xu, B. Sun, R. Fu and H. He, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J. S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef CAS.
- W. Huang, Q. He, Y. Hu and Y. Li, Angew. Chem., Int. Ed., 2019, 58, 8676–8680 CrossRef CAS PubMed.
- M. Tasior, O. Vakuliuk, D. Koga, B. Koszarna, K. Gorski, M. Grzybowski, L. Kielesinski, M. Krzeszewski and D. T. Gryko, J. Org. Chem., 2020, 85, 13529–13543 CrossRef CAS PubMed.
- L. Huang, Z. Luo, Y. N. Zhou, Q. Zhang, H. Zhu and S. Zhu, Mater. Today Chem., 2021, 20, 100475 CrossRef CAS.
- A. Bhunia, D. Esquivel, S. Dey, R. Fernández-Terán, Y. Goto, S. Inagaki, P. Van Der Voort and C. Janiak, J. Mater. Chem. A, 2016, 4, 13450–13457 RSC.
- I. D. Wessely, A. M. Schade, S. Dey, A. Bhunia, A. Nuhnen, C. Janiak and S. Bräse, Materials, 2021, 14, 3214 CrossRef CAS PubMed.
- L. Guo, Y. Niu, H. Xu, Q. Li, S. Razzaque, Q. Huang, S. Jin and B. Tan, J. Mater. Chem. A, 2018, 6, 19775–19781 RSC.
- A. Hryniewicka, J. Breczko, G. Siemaszko, K. Brzezinski, A. Ilnicka, A. P. Terzyk and M. E. Plonska-Brzezinska, Mater. Adv., 2024, 5, 1065–1077 RSC.
- A. Bhunia, I. Boldog, A. Möller and C. Janiak, J. Mater. Chem. A, 2013, 1, 14990–14999 RSC.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345 RSC.
- S. Abednatanzi, S. Gohari, P. Derakhshandeh, K. Leus, H. Vrielinck, F. Callens, J. Schmidt, A. Savateev and P. Van Der Voort, Sci. Adv., 2020, 6, eaaz2310 CrossRef CAS PubMed.
- Z. Yang, H. Chen, S. Wang, W. Guo, T. Wang, X. Suo, D. E. Jiang, X. Zhu, I. Popovs and S. Dai, J. Am. Chem. Soc., 2020, 142, 6856–6860 CrossRef CAS PubMed.
- A. Jana, A. Maity, A. Sarkar, B. Show, P. A. Bhobe and A. Bhunia, J. Mater. Chem. A, 2024, 12, 5244–5253 RSC.
- W. Huang, N. Huber, S. Jiang, K. Landfester and K. A. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18368–18373 CrossRef CAS PubMed.
- T. Sick, A. G. Hufnagel, J. Kampmann, I. Kondofersky, M. Calik, J. M. Rotter, A. Evans, M. Döblinger, S. Herbert, K. Peters and D. Bohm, J. Am. Chem. Soc., 2017, 140, 2085–2092 CrossRef PubMed.
- F. A. Rahimi, S. Dey, P. Verma and T. K. Maji, ACS Catal., 2023, 13, 5969–5978 CrossRef CAS.
- T. Huang, X. Lin, Y. Liu, J. Zhao, H. Lin, Z. Xu, S. Zhong, C. Zhang, X. Wang, X. Fu and J. Long, ChemSusChem, 2020, 13, 672–676 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- S. Min and G. Lu, J. Phys. Chem. C, 2011, 115, 13938–13945 CrossRef CAS.
- A. Wojcik and P. V. Kamat, ACS Nano, 2010, 4, 6697–6706 CrossRef CAS PubMed.
- S. Min and G. Lu, J. Phys. Chem. C, 2012, 116, 25415–25424 CrossRef CAS.
- T. Shimidzu, T. Iyoda and Y. Koide, J. Am. Chem. Soc., 1985, 107, 35–41 CrossRef CAS.
- T. Lazarides, T. McCormick, P. W. Du, G. G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- S. De, S. Das and A. Girigoswami, Spectrochim. Acta, Part A, 2005, 61, 1821–1833 CrossRef PubMed.
- S. Min and G. Lu, Int. J. Hydrogen Energy, 2012, 37, 10564–10574 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR, TGA, PXRD, XPS, UV-vis, N2 uptake, TEM, SEM, EPR, and Mott–Schottky plot. See DOI: https://doi.org/10.1039/d4ma00205a |
| This journal is © The Royal Society of Chemistry 2024 |