
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmeliorating La0.5Sr1.5MnO4 with Ni-doping to enhance cathode electrocatalysis for proton-conducting solid oxide fuel cells†
Jiapeng
Xu
a,
Junyi
Gong
a,
Kunpeng
Du
a,
Wei
Liu
b and
Jie
Hou
 *a
*a
aSchool of Resource Environment and Safety Engineering, University of South China, Hengyang 421001, China. E-mail: jiehou@usc.edu.cn
bCAS Key Laboratory of Materials for Energy Conversion and Collaborative Innovation Center of Suzhou Nano Science and Technology, University of Science and Technology of China, Hefei 230026, China
First published on 15th April 2024
Abstract
Cathodes with high electrocatalytic activity are critical for commercializing proton-conducting solid oxide fuel cells (H-SOFCs). Herein, B site acceptor Ni-doping is first attempted to ameliorate manganite-based Ruddlesden–Popper phase oxide La0.5Sr1.5MnO4+δ (LSMO), introducing more oxygen vacancies and new effective Ni2+–O–Ni3+ electron-hopping transition paths with an irregular structural deformation, ultimately enhancing the electrocatalysis with a 2.23 to 2.60 times improvement of electrical conductivity at 500–700 °C. The significantly enhanced oxygen/proton diffusion in the La0.5Sr1.5Mn0.7Ni0.3O4+δ (LSMN) sample, verified via electrical conductivity relaxation results, can enhance electrocatalytic activity, enabling more efficient oxygen reduction reaction kinetics. Hence, the Ni-ameliorated LSMN cathode exhibits a prominent power output of 1342 and 668 mW cm−2 at 700 and 600 °C on a H-SOFC, surpassing the cells with the Ni-free LSMO cathode and other LSMO-based and Ln2NiO4-based cathodes in the literature. On balance, the excellent power density and polarization performance, together with the superior operation stability demonstrates that the LSMN candidate is a preferable alternative to H-SOFC cathodes. This work is an attempt to design highly active electrodes by regulating the crystal structure derived from B-site acceptor-doping, which is also beneficial for new material designs in H-SOFC and related electrocatalytic fields.
1. Introduction
Developing sustainable low-carbon energy generation technologies to reduce CO2 emissions and the reliance on fossil fuels is crucial for the green transition of our society. As proton migration is much quicker than for oxygen ions due to their smaller size and mass, proton-conducting solid oxide fuel cells (H-SOFCs) could hold great potential. They can operate at low temperatures (LTs, ≤600 °C) with adequate electrolyte conductivity (≥0.01 S cm−1) and low activation energy (Ea < 0.5 eV), making them commercially competitive.1–4 However, their limited cathode electrocatalytic activity, derived from notoriously sluggish oxygen reduction reaction (ORR) kinetics, dominates the cell polarization loss and performance degradation when the operation temperature decreases.5–7 Then, developing new highly catalytic active cathode materials with good durability is significant, which is also a main topic in this field.Though cathode materials based on Ln2NiO4+δ (Ln = La, Pr, and Nd) nickelate have been widely used and intensively investigated for H-SOFCs in the literature,8 attention has rarely been paid to Ln2MnO4+δ manganite, which possesses the same K2NiF4-type layered structure. The open lattice permits a great number of hyper-stoichiometric oxygens in the interstices of the Ln2O2 layer, leading to rapid oxygen ionic diffusion;8,9 however, the electrical conductivity in the Ln2MnO4+δ manganite is extremely low. Yet, Mn-based La2−xSrxMnO4+δ electrode materials adopted for oxygen-ion conducting SOFCs10–15 show inferior performance; thus, La0.5Sr1.5MnO4+δ (LSMO) could be interesting due to its relatively satisfactory electrochemical behavior and good redox durability.10,11,15,16 Seeing that the rate-limiting step for ORR in the LSMO cathode is a charge transfer process,15 coupling with high electronic conductor La0.5Sr0.5MnO3−δ (LSM) was attempted for H-SOFC,17 revealing a preferable electrochemical performance as a result of the fine charge transfer ability in LSM and facile oxygen ionic diffusion in LSMO. This is the first application of La2−xSrxMnO4+δ in H-SOFC, indicating the acceleration of charge transfer is projected to enhance the catalytic activity of typical LSMO. In addition, partially B-site substitution of transition elements, e.g. Fe, Co, Ni, and Cu, could alter electronic states, affecting the charge transfer behavior.8,18–20 Furthermore, the variant oxidation and spin states of transition metal ions and their ionic radius play a crucial role in the physical and chemical properties in LSMO.21–23 In the interest of exploring the suitability of single Mn-based layered Ln2MnO4+δ as a cathode for H-SOFCs, equivalent state Fe3+/Fe4+ cations and low-valence state Cu+/Cu2+ cations were used to modify LSMO, both achieving an acceptable performance.18,24 Thus, using a B-site transition metal to tailor LSMO could be a potential way.
As Ni-doping could perform a valence change in the B-site transition metals of Mn-based perovskites, by altering the numbers of Mn3+/Mn4+ couples and introducing Ni2+/Ni3+ couples, which act as hopping sites for electrons/holes,25,26 the electrocatalysis could be significantly enhanced. Herein, in this assignment, layered LSMO is first modified with Ni cations, leading to replacement of MnO6 octahedrons with NiO4 planes in the crystal, causing the irregular deformation in structure and introducing a highly efficient electron-hopping transition path Ni2+–O–Ni3+. This could increase the average Mn valence state and motivate the formation of oxygen vacancies and electron holes, thus advancing the proton (H+)/oxygen ion (O2−) transfer and electrode electrocatalysis dramatically. Hence, the modified La0.5Sr1.5Mn0.7Ni0.3O4+δ (LSMN) cathode exhibits exceptional electrochemical performance accompanied by excellent durability on a single cell. All these demonstrate that LSMN is a preferable H-SOFC cathode alternative, and the strategy using lower-valence Ni metal ions to regulate the structure and material properties is feasible, providing a path for designing highly active electrode materials in related electrocatalytic fields.
2. Experimental section
K2NiF4-type LSMN/LSMO and perovskite structured BaZr0.1Ce0.7Y0.2O3−δ (BZCY) powders were synthesized via a citric acid-nitrate gel combustion process described elsewhere in detail.5,18 The raw materials are as follows, La2O3, Sr(NO3)3, MnCO3, Ni(NO3)3·6H2O, BaCO3, Zr(NO2)4·5H2O, Ce(NO3)3·6H2O, and Y(NO4)3·6H2O. The three pure phase materials were obtained by calcining corresponding as-prepared ash-like powders at 1100, 1100 and 1000 °C for 3 h in air, and their phase structure parameters were analyzed by an X-ray diffractometer (XRD, DX-2700BH). The crystallinity and morphology of the as-prepared powders were identified via transmission electron microscopy (TEM, Tecnai, G2, F20). X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha) was utilized to analyze surface chemical states for LSMO and LSMN. The electrical conductivity relaxation (ECR) tests using a four-probe approach were performed to investigate the conductivity and time of LSMO and LSMN bars to reach equilibrium when abruptly switching the gas from air to oxygen (50% N2) and dry air to moist air. The surface exchange and bulk diffusion coefficient for oxygen ions and protons could be extracted by fitting ECR data according to diffusion equations mentioned by Huang et al.27 and Ren et al.28 Both bars were fabricated by dry-pressing followed by calcination at 1300 °C for 5 h, and conductivity values were measured at 400–800 °C each at 50 °C.To study the effect of Ni amelioration for LSMO on the H-SOFC performance, BZCY-based half-cells were prepared, in which NiO-BZCY (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35 weight ratio) and BZCY were used as the anode and electrolyte, respectively. Green BZCY-based half-cells were calcined at 1350 °C for 6 h after co-pressing. LSMN and LSMO cathode ink attained by mixing thoroughly with a 10 wt% ethylcellulose-terpineol binder were painted onto a dense BZCY surface and later fired at 1000 °C for 3 h in air to obtain porous cathode layers. Eventually, the single cell NiO-BZCY|BZCY|LSMN (LSMO) is obtained. The assembled single cells were evaluated using an electrochemical workstation based on a four-probe configuration (Squidstat Plus, Admiral Instruments) at 500–700 °C with wet hydrogen (∼3% H2O, 30 ml min−1) and static ambient air as the fuel and oxidant, respectively. Electrochemical impedance spectroscopy (EIS) was measured under open circuit conditions using the electrochemical workstation with a frequency of 10 MHz–0.1 Hz and an AC amplitude of 5 mV. The cell microstructures were investigated by scanning electron microscopy (SEM, Phenom XL G2).
:35 weight ratio) and BZCY were used as the anode and electrolyte, respectively. Green BZCY-based half-cells were calcined at 1350 °C for 6 h after co-pressing. LSMN and LSMO cathode ink attained by mixing thoroughly with a 10 wt% ethylcellulose-terpineol binder were painted onto a dense BZCY surface and later fired at 1000 °C for 3 h in air to obtain porous cathode layers. Eventually, the single cell NiO-BZCY|BZCY|LSMN (LSMO) is obtained. The assembled single cells were evaluated using an electrochemical workstation based on a four-probe configuration (Squidstat Plus, Admiral Instruments) at 500–700 °C with wet hydrogen (∼3% H2O, 30 ml min−1) and static ambient air as the fuel and oxidant, respectively. Electrochemical impedance spectroscopy (EIS) was measured under open circuit conditions using the electrochemical workstation with a frequency of 10 MHz–0.1 Hz and an AC amplitude of 5 mV. The cell microstructures were investigated by scanning electron microscopy (SEM, Phenom XL G2).
3. Results and discussion
XRD Rietveld refinement profiles [Fig. 1(a) and (b)] reveal that LSMN and LSMO possess a tetragonal K2NiF4 structure with I4/mmm space group symmetry. Two separate phases in the LSMN-BZCY pattern [Fig. 1(c)] indicate a good chemical compatibility between LSMN and BZCY at the cathode calcination temperature. Surprisingly, the XRD peak in LSMN reveals lower and higher angle shifts after Ni-incorporation [Fig. 1(d)], illustrating an irregular structure deformation. Good crystallized LSMN particles [150–500 nm, Fig. 1(e)] are helpful for achieving a fine porous cathode, favoring fast gaseous diffusion and efficient electrochemical reactions. Additionally, SAED results [Fig. 1(f)] give further evidence of good crystallinity, possessing lattice spacings of 3.66, 2.70, 2.48, 1.83, 1.69, and 1.58 Å for (101), (110), (112), (202), (211), and (213) lattice planes in LSMN. The d-spacing values for these six lattice planes in LSMO (PDF 01-083-1896) are 3.68, 2.72, 2.49, 1.84, 1.71 and 1.59 Å, respectively, which are almost identical to that obtained via the Rietveld method [Fig. 1(b) and Table S1 in (ESI†)]. | ||
| Fig. 1 Rietveld refinement plot of (a) LSMN and (b) LSMO using X-ray powder diffraction data. (c) XRD patterns of LSMO, LSMN, and BZCY and the chemical compatibility between LSMN and BZCY at 1000 °C. (d) Peak enlarged view of LSMN and LSMO from 28 to 34 degrees. (e) Bright-field TEM of LSMN powders and (f) Corresponding selected area electron diffraction (SAED) patterns. (g) HRTEM image at the (103) lattice plane. A schematic illustration of (h) (004) and (110) lattice planes in the LSMO structure with (i) Corresponding MnO6 and (j) NiO2 plane after Ni2+ entering. (k) TEM HADDF-EDS for LSMN. | ||
Furthermore, the HRTEM image [Fig. 1(g)] confirms the lattice spacing of 2.81 Å for the (103) lattice plane, which is a little lower than the value of 2.82 Å in LSMO (PDF 01-083-1896). Though SAED and HRTEM results only give compressed lattice planes with smaller lattice spacings, the irregular deformation in LSMN compared with LSMO could be affirmed by Rietveld refinement results (Table S1 in ESI†), which are closely related to the Ni2+ substitution of Mn. As illustrated in Fig. 1(h)–(j), when the acceptor dopant Ni replaces Mn in LSMO, the MnO6 octahedrons [Fig. 1(i)] will be replaced by NiO4 or NiO6, facilitated by the presence of Ni2+ (bonding with four oxygens) and Ni3+ (bonding with six oxygens), as verified by the Ni 3p XPS result, detailed later. As shown in Fig. 1(j), to maintain the crystal structure, there tends to be NiO4 plane, which should be compressed under the crystal stress. Thus, the crystal structure is compressed in the a × b dimensional plane and stretched in the vertical direction. This could lead the (004) and (110) lattice plane to be stretched with higher d-spacing and compressed with lower lattice spacing, respectively, which is also corroborated by the XRD result shown in Fig. 1(d). Moreover, the entering of the NiO4 plane in LSMO could partially eliminate surrounding oxygens and introduce oxygen deficiencies, which is beneficial for the ORR occurrence. Notably, the element homogeneity is important, closely interrelating with crystal components. Consequently, TEM energy dispersive X-ray spectroscopy (EDS) elemental mapping results shown in Fig. 1(k) provide confirmation that all metal elements containing La, Sr, Mn, and Ni are uniformly distributed with no visible element segregations, suggesting that the incorporation of Ni into LSMO, and only one homogeneous phase existed.
The surface chemical states were analyzed by XPS. Due to the partial overlapping of La 3d and Ni 2p peaks, it is hard to accurately determine the Ni2+/Ni3+ ratio. Alternatively, Ni 3p spectra are not interfered by other elements and the accuracy could be guaranteed.29 As depicted in Fig. 2(a), the Ni 3p core-level spectra of LSMN powders exhibit typically two doublets: a pair of peaks for Ni2+ at the binding energy (BE) of 67.7 eV (3p3/2) and 69.2 eV (3p1/2), with an extra doublet peak of Ni3+ located at the BE of 71.1 eV (3p3/2) and 73.2 eV.30,31 The calculated Ni2+/Ni3+ ratio is 1.37. This could give confirmation that the MnO6 octahedrons in LSMO are only replaced by planar NiO4 or octahedral NiO6, and new highly efficient transition paths Ni2+–O–Ni3+ for electron-hopping are successfully introduced. Actually, the Ni entry into the Mn-site affects the numbers of Mn3+/Mn4+ couples for it could change the valence, thus dominating the electron-hopping behavior via Mn3+–O–Mn4+ transition paths, which is proved by Mn 2p spectra shown in Fig. 2(b). Both spectra could be fitted into four split peaks at 641.9 eV (Mn3+ 2p3/2), 643 eV (Mn4+ 2p3/2), 653.5 eV (Mn3+ 2p1/2), and 654.7 eV (Mn4+ 2p1/2),18,32,33 and the ratio of Mn4+ cations increased from 28% in LSMO (Mn3+: Mn4+ = 7.2:2.8) to 84% in LSMN (Mn3+: Mn4+ = 1.6:8.4). Apparently, inadequate Mn4+ in LSMO is the cause of the low electron-hopping efficiency. Interestingly, shown in Fig. 2(c) is the XPS spectra of O 1s for LSMN and LSMO, indicating different oxygen species at surface:lattice oxygen, highly oxidative oxygen species (O−/O22−), surface hydroxyl group or adsorbed oxygen (−OH or O2), and adsorbed water (H2O). The slightly higher BEs for all oxygen species in LSMN are correlative with the intricate B-site environment after Ni-incorporation, which connect with oxygens directly. Anyhow, the highly oxidative oxygen species (O−/O22−) are closely related to the surface oxygen vacancies34–36 and the calculated relative concentration of O−/O22− species increased from 8.2% for LSMO to 16.5% for LSMN, which indicates that B-site Ni substitution could increase the surface oxygen vacancy concentration, thus promoting the adsorption and activation of oxygen at the active site and facilitating the ORR process.34,35
 | ||
| Fig. 2 XPS spectra of (a) Ni 3p, (b) Mn 2p, and (c) O 1s; (d) EC comparation of LSMN and LSMO at 400–800 °C; ECR results for LSMN and LSMO measured at 600 °C under the conditions of abruptly changing (e) from air to oxygen (50% N2) and (f) from dry air to wet air (3% H2O). | ||
As shown in Fig. 2(d), the electrical conductivity (EC) of LSMN is 10.69–17.83 S cm−1 at 500–700 °C, exhibiting a 2.23–2.6 times improvement compared with that of LSMO with values of 4.11–7.98 S cm−1. LSMN and LSMO present a semiconducting-like conduction behavior, in which EC increases with temperature from 400 to 800 °C. Although the relatively high Mn4+ concentration (84%) in LSMN may not have a positive effect for a little less Mn3+/Mn4+ couples, newly introduced Ni2+–O–Ni3+ transition paths could primarily govern electron-hopping, thus improving the electrical conductivity and further enhancing the electrocatalysis.37 Distinctly, analysis via ECR tests shows the surface exchange coefficient (KO) is enhanced by one order of magnitude (1.4 × 10−4 cm s−1 for LSMO and 1.6 × 10−3 cm s−1 for LSMN), while the oxygen bulk diffusion coefficient (DO) only shows a mild improvement from 6.0 × 10−5 cm2 s−1 for LSMO to 7.9 × 10−5 cm2 s−1 for LSMN [Fig. 2(e)]. This could be explained by the improved concentration of oxygen vacancies in LSMN compared to LSMO as Ni2+ ions can only bond with four oxygen ions, as indicated by the O 1s XPS analysis. The improved EC also donates to this for advancing the charge transfer and oxygen transmission. Furthermore, the response for proton defect diffusion in LSMN is more rapid, only requiring about 340 s to reach conductivity equilibrium, whereas the equilibrium time for LSMO is 1660 s [Fig. 2(f)]. Then, fitting results indicate that the proton surface exchange coefficient (KH) exhibits an increase of approximately two fold, while the proton diffusion coefficient (DH) reveals an enhancement of two orders of magnitude, exhibiting the KH of 6.5 × 10−4 cm s−1 and DH of 4.2 × 10−3 cm2 s−1 for LSMN with the corresponding KH of 3.4 × 10−4 cm s−1 and DH of 2.2 × 10−5 cm2 s−1 for LSMO. The proton absorption can occur through a hydrogenation reaction ( ), which actually involves a mixture of oxidation (
), which actually involves a mixture of oxidation ( ) and hydration reaction (
) and hydration reaction ( ), leading to proton absorption at the expense of holes. The high hole conductivity in LSMN, derived from the existence of Ni3+–O–Ni2+ and Mn4+–O–Mn3+ electron-hopping transition paths, along with the higher oxygen vacancy concentration, as evidenced by the O 1s XPS peak, accelerates the proton diffusion process drastically.38–40
), leading to proton absorption at the expense of holes. The high hole conductivity in LSMN, derived from the existence of Ni3+–O–Ni2+ and Mn4+–O–Mn3+ electron-hopping transition paths, along with the higher oxygen vacancy concentration, as evidenced by the O 1s XPS peak, accelerates the proton diffusion process drastically.38–40
To assess the effect of Ni-amelioration on LSMO, LSMO and LSMN cathodes based on H-SOFC applications were estimated comparably. The LSMN cell outputs the peak power densities (PPDs) of 1342, 971, 668, 434 and 260 mW cm−2 with the corresponding PPD values of 982, 679, 430, 364 and 146 mW cm−2 for the LSMO cell at 700, 650, 600, 550 and 500 °C, respectively [Fig. 3(a) and (b)]. Explicitly, the cell power performance reveals a significantly improvement when switching the cathode from LSMO to LSMN [Fig. 3(c)]. As both the cells are identical except for the cathode, which are obtained by the same manner, the cell performance discrepancy could be ascribed to the cathode variation. Moreover, as shown in Fig. 3(d), the PPD performance of the LSMN cell was compared with the reported LSMO-based cathodes and Ln2NiO4-based single cathodes, e.g. La0.5Sr1.5MnO4+δ–La0.5Sr0.5MnO3−δ,17 La0.5Sr1.5Mn0.8Cu0.2O4+δ,18 La0.5Sr1.5Mn0.8Fe0.2O4+δ,24 La2Ni0.75Zn0.25O4+δ,41 La1.6Sr0.4Cu0.6Ni0.4O4+δ,42 La1.5Ca0.5NiO4+δ,43 and La1.2Sr0.8Ni0.4Fe0.6O4+δ.44 Here, only cells with PPD exceeding 700 mW cm−2 for the single Ln2NiO4-based cathodes were selected, and the LSMN cell possesses the highest cell power output. However, the recently reported La1.2Sr10.8Ni0.5Cu0.5O4+δ presents a higher power performance for much smaller cathode particles derived from lower powder calcination and cathode assembly temperature19 but was not listed here due to the relatively big difference in preparation conditions. Even so, the highest PPD output among the LSMO-based cathode cells and single Ln2NiO4-based cathode cells under similar conditions could indicate the predominance of Ni-modified LSMO. Except for the enhanced power performance, the LSMN cell also possesses an excellent durability with almost no degradation to work for 100 h [Fig. 3(e)], demonstrating the good compatibility of operation stability and high performance.
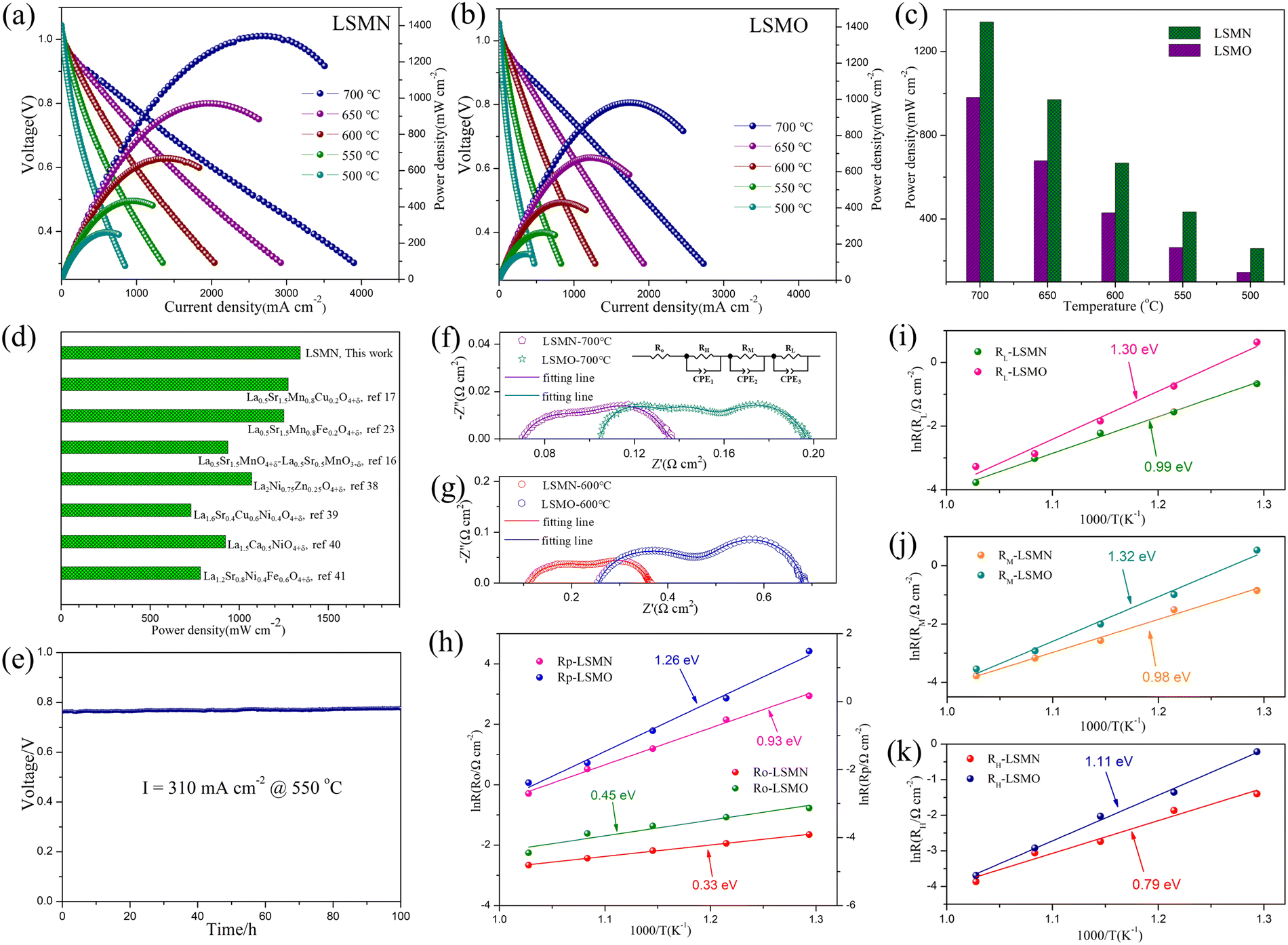 | ||
| Fig. 3 I–V and power density curves for H-SOFCs using (a) LSMN and (b) LSMO single cathode; (c) PPD performance comparison between LSMN and LSMO cells; (d) power output comparison for BaCeO3-based cells employing LSMO-based cathodes and Ln2NiO4-based single cathodes at 700 °C in the literature; (e) durability assessment for LSMN cell at 550 °C; EIS plots for LSMN and LSMO cells tested at (f) 700 °C and (g) 600 °C with the equivalent circuit for fitting given in the inset image; and the comparison of Arrhenius plots for (h) RO and RP, (i) RL, (j) RM, and (k) RH of two single cells at 550–700 °C. | ||
Furthermore, the enhanced cathode electrocatalysis of LSMN could be further evidenced by the typical EIS. As depicted in Fig. 3(f) and (g), the high-frequency intercept represents the cell ohmic resistance (RO), consisting of the resistance originating from the electrolyte and electrodes, and contact resistance concerning the interfaces of the components, while the difference between two intercepts corresponds to the interfacial polarization resistance (RP). The LSMN cell reveals the RP of 0.067, 0.138, 0.251, 0.589, and 1.187 Ω cm2 at 700, 650, 600, 550 and 500 °C, respectively. However, the LSMO cell possesses the RP of 0.092, 0.165, 0.426, 1.107, and 4.413 Ω cm2 at corresponding temperatures. The significantly smaller polarization loss demonstrates that the LSMN cathode permits a faster cathode reaction, producing lower RP with improved fuel cell performance. Similarly, RO for the LSMN cell shows a similar trend with values of 0.07, 0.088, 0.113, 0.144, and 0.192 at 700–500 °C, while corresponding RO values for the LSMO cell are 0.105, 0.2, 0.256, 0.342, and 0.462 Ω cm2. The noticeable decrease in ohmic loss could be attributed to the 2.23–2.6 times increased EC in LSMN [refer to Fig. 2(d)], and inferior EC in LSMO [refer to Fig. 2(d)] makes the cathode largely contribute to the RO values. As the restricted charge transfer derived from low EC dominates the ORR process, limited oxygen ions at the electrolyte-LSMO cathode interface would retard the proton transfer, thus generating higher ohmic loss, which is the main cause for higher Ro. Ro and RP decrease with increasing temperatures, showing clearly that the electrochemical reaction processes are thermally activated. As shown from Arrhenius relations shown in Fig. 3(h), the activation energy (Ea) for RP decreases from 1.26 eV to 0.93 eV when using the Ni-modified LSMN cathode, while Ea for RO shows an identical trend with the Ea value decreasing from 0.45 eV to 0.33 eV. This phenomenon could be mainly ascribed to the cathode ORR process. In addition, EIS plots fitted with an LR(RHQH)(RMQM)(RLQL) equivalent circuit were used to further analyze cathode processes. Three electrode processes corresponding to resistances at low frequency (RL), middle frequency (RM) and high-frequency (RH) exist in both the cells [Fig. 3(f)–(g)]. All resistances containing RL, RM and RH possess a decrease in the LSMN cell, revealing faster electrode processes (refer to Table 1). RL corresponds to the transportation and adsorption of O2, while RM and RH are interrelated with the oxygen diffusion and proton transfer process, respectively.17,45–47 As depicted in Fig. 3(i)–(k), all the three electrode processes corresponding to three different arcs in the LSMN cell exhibit slower performance deterioration, as indicated by significantly reduced Ea. The apparently lower RL with greatly depressed Ea from 1.30 eV to 0.99 eV [Fig. 3(i)] is correlative with the improved surface oxygen vacancies [Fig. 2(c)] and enhanced oxygen surface exchange abilities [Fig. 2(e)]. Meanwhile, similar Ea variation for RM from 1.32 eV to 0.98 eV [Fig. 3(j)] could be attributed to increased EC [Fig. 2(d)], which advances the charge transfer process for generating oxygen ions (Oad + e− → Oad−, Oad− + e− → Oad2−), and thus promotes the oxygen diffusion. Moreover, as shown in Fig. 3(k), RH has a profound reduction in the LSMN cell, and the Ea decrease reveals a bigger reaction potential between protons and oxygen ions. This difference could be attributed to the relatively insufficient oxygen ions and weak proton transmission in LSMO [Fig. 2(f)]. Thus, the improved proton migration can increase the charge carrier (proton) transportation at the reaction active area, leading to the decline of RH for LSMN in comparison with that for LSMO.
| R H, RM and RL (Ω cm2) (°C) | R L | R M | R H | |||
|---|---|---|---|---|---|---|
| LSMN | LSMO | LSMN | LSMO | LSMN | LSMO | |
| 700 | 0.023 | 0.038 | 0.023 | 0.029 | 0.021 | 0.025 |
| 650 | 0.049 | 0.057 | 0.042 | 0.054 | 0.047 | 0.054 |
| 600 | 0.109 | 0.159 | 0.077 | 0.135 | 0.065 | 0.132 |
| 550 | 0.212 | 0.476 | 0.222 | 0.373 | 0.155 | 0.258 |
| 500 | 0.512 | 1.896 | 0.428 | 1.709 | 0.247 | 0.808 |
Fig. 4(a) and (b) present the cell microstructures for both cells. The similar morphology with good adherence in LSMN and LSMO cells suggests that the cell performance discrepancy could be attributed to the enhanced electrocatalysis of LSMN not to the concerns of the microstructure. Interestingly, grains in the LSMN cathode layer are almost identical to the starting powder [Fig. 1(e)], indicating that the lower cathode assembly temperature (1000 °C) has a weak sintering effect. In LSMN and LSMO cathode layers, the particles interconnect well with each other, facilitating gas diffusion and electrochemical reactions. Besides, the particle size in the LSMN cathode (150–550 nm, average value 332 nm) is much larger than that in the LSMO cathode (100–300 nm, average value 187 nm), which may result in generation of less electrochemical reaction sites that are adverse factors. Even so, the much better cell power and polarization performance in the LSMN cell indicate the predomination of Ni amelioration in LSMO. Based on all the above, the reaction mechanism at the cathode interface is proposed. For low EC and proton/oxygen transfer ability in LSMO, the ORR should be mainly restricted at the LSMO/BZCY interface [Fig. 4(c)]. However, the one order of magnitude enhancement of KO, the two-fold increased KH accompanied by the two orders of magnitude improved DH in LSMN enable an exceedingly larger cathode reaction area extending to the overall cathode due to the high protonation of this material. As shown in Fig. 4(d), the improved ORR at the LSMN surface shows a much more rapid oxygen ionic generation and transfer, and the reformative protonation capability permits a longer proton migration distance, increasing the effective cathode reaction area and accelerating the overall fuel cell reaction 2H2 + O2 → 2H2O. Ultimately, the distinctive structure transformation and favourable proton/oxygen diffusion properties in LSMN give rise to the integrated preeminent cell electrochemical performance with good durability.
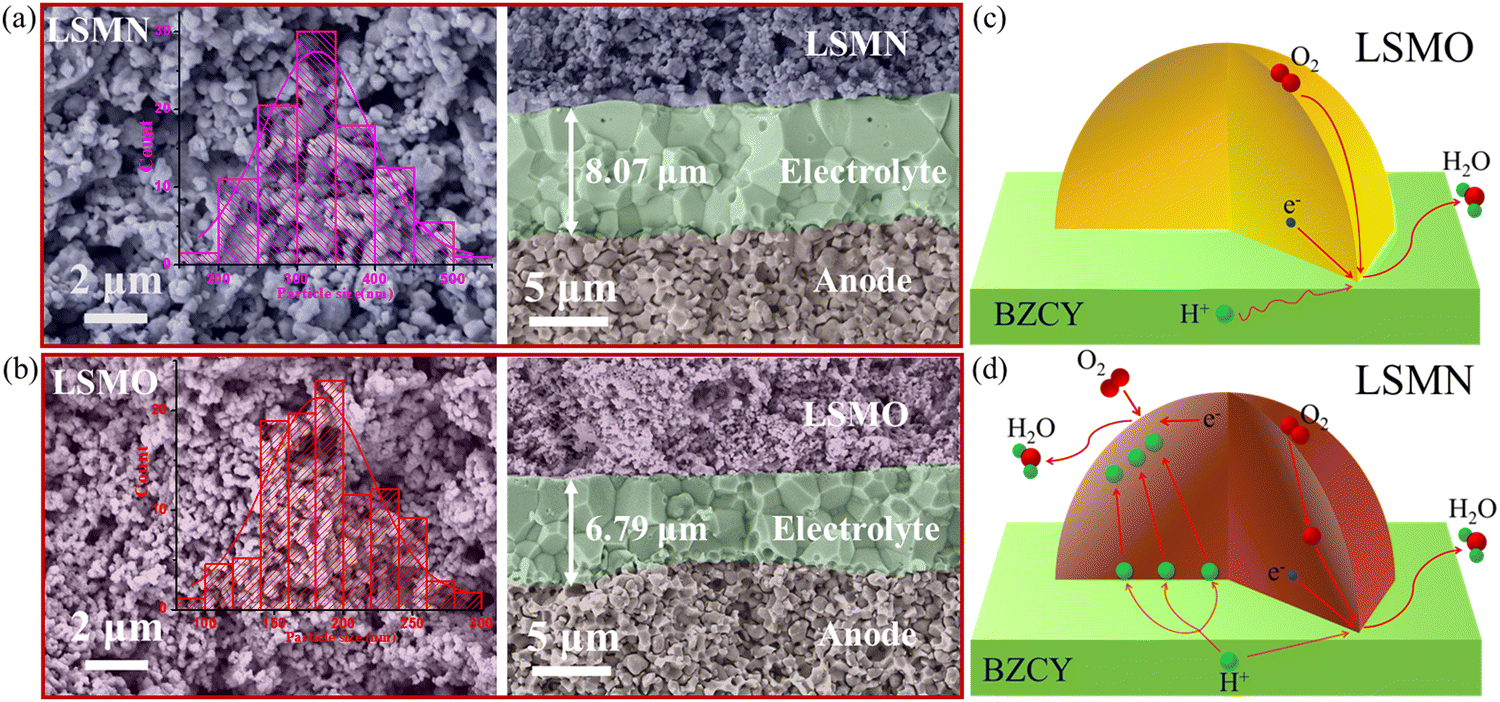 | ||
| Fig. 4 Cross-sectional SEM views for (a) LSMN and (b) LSMO cells after testing. Schemes for cathode reactions at (c) BZCY-LSMO and (d) BZCY-LSMN interfaces. | ||
4. Conclusions
The development of highly active cathodes with high proton and oxygen diffusivity is crucial for enhancing H-SOFC performance, compelling researchers to explore new materials, strategies and designs. One potential candidate is the Ln2MnO4+δ manganite, which possesses a typical K2NiF4-type layered structure that is rarely utilized in H-SOFCs. In this assignment, the amelioration of layered LSMO is attempted by introducing a small amount of Ni dopant, which results in an irregular structure deformation. The incorporation of Ni into B-sites creates effective Ni2+–O–Ni3+ electron-hopping transition paths, greatly increasing the electronic conductivity, and thus enhancing the cathode electrocatalysis. As a result, the successfully ameliorated LSMN demonstrates a favorable cell performance in H-SOFC, possessing a power output of 1342 and 668 mW cm−2, along with the RP of 0.067 and 0.251 Ω cm2 at 700 and 600 °C, respectively. This superior cell performance could be attributed to the higher proton/oxygen diffusion rate, as well as the more efficient ORR kinetics. Comparatively, the LSMN cell outperforms the LSMO cell significantly. Though LSMO is first ameliorated using the acceptor Ni-doping, its performance surpasses that of H-SOFCs employing LSMO-based cathodes and single Ln2NiO4-based cathodes in the literature. Importantly, the favorable power and polarization performance do not compromise its durability, indicating the high potential for utilization of LSMN in H-SOFCs. This use of the lower-valence B-site acceptor Ni-dopant to regulate the LSMO structure, which produces oxygen vacancies and new Ni3+/Ni2+ couples for high-efficient electron-hopping, could provide a path towards new material designs in H-SOFC and related electrocatalytic fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos: 51802200) and Startup Funding for Talents at University of South China.References
- Y. Gao, M. Zhang, M. Fu, W. Hu, H. Tong and Z. Tao, Energy Rev., 2023, 2, 100038 CrossRef
.
- W. Bian, W. Wu, B. Wang, W. Tang, M. Zhou, C. Jin, H. Ding, W. Fan, Y. Dong, J. Li and D. Ding, Nature, 2022, 604, 479–485 CrossRef CAS PubMed
- Y. Chen, B. deGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. H. Kim, Y. Ding, P. Hu, F. F. Tao and M. Liu, Nat. Energy, 2018, 3, 1042–1050 CrossRef CAS
- Y. Yin, H. Dai, S. Yu, L. Bi and E. Traversa, SusMat, 2022, 2, 607–616 CrossRef CAS
- J. Hou, J. Gong and J.-L. Luo, Chem. Eng. J., 2023, 459, 141459 CrossRef CAS
- S. Choi, C. J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H.-I. Ji and S. M. Haile, Nat. Energy, 2018, 3, 202–210 CrossRef CAS
- Y. Yin, Y. Zhou, Y. Gu and L. Bi, J. Adv. Ceram., 2023, 12, 587–597 CrossRef CAS
- A. P. Tarutin, J. G. Lyagaeva, D. A. Medvedev, L. Bi and A. A. Yaremchenko, J. Mater. Chem. A, 2021, 9, 154–195 RSC
- A. Tarancón, M. Burriel, J. Santiso, S. J. Skinner and J. A. Kilner, J. Mater. Chem., 2010, 20, 3799–3813 RSC
- M. V. Sandoval, C. Cárdenas, E. Capoen, C. Pirovano, P. Roussel and G. H. Gauthier, Electrochim. Acta, 2019, 304, 415–427 CrossRef CAS
- M. V. Sandoval, C. Pirovano, E. Capoen, R. Jooris, F. Porcher, P. Roussel and G. H. Gauthier, Int. J. Hydrogen Energy, 2017, 42, 21930–21943 CrossRef CAS
- J. Shen, G. Yang, Z. Zhang, W. Zhou, W. Wang and Z. Shao, J. Mater. Chem. A, 2016, 4, 10641–10649 RSC
- J. Zhou, G. Chen, K. Wu and Y. Cheng, J. Power Sources, 2014, 270, 418–425 CrossRef CAS
- C. Jin, Z. Yang, H. Zheng, C. Yang and F. Chen, Electrochem. Commun., 2012, 14, 75–77 CrossRef CAS
- S. Liping, H. Lihua, Z. Hui, L. Qiang and C. Pijolat, J. Power Sources, 2008, 179, 96–100 CrossRef
- S. Li-Ping, L. Qiang, H. Li-Hua, Z. Hui, Z. Guo-Ying, L. Nan, J.-P. Viricelle and C. Pijolat, J. Power Sources, 2011, 196, 5835–5839 CrossRef
- J. Hou, Q. Wang, J. Li, Y. Lu, L. Wang, X.-Z. Fu and J.-L. Luo, J. Power Sources, 2020, 466, 228240 CrossRef CAS
- D. Yang, J. Gong and J. Hou, Electrochim. Acta, 2023, 461, 142668 CrossRef CAS
- C. Jin, L. Ma, J. Gong, W. Liu and J. Hou, Ceram. Int., 2023, 49, 35830–35836 CrossRef CAS
- Y. Yin, D. Xiao, S. Wu, E. H. Da'as, Y. Gu and L. Bi, SusMat, 2023, 3, 697–708 CrossRef CAS
- D. Singh, S. Sharma, A. Mahajan, S. Singh and R. Singh, Ionics, 2013, 19, 1297–1302 CrossRef CAS
- D. Singh and A. Mahajan, J. Solid State Chem., 2013, 207, 126–131 CrossRef CAS
- L. Malavasi, M. C. Mozzati, C. Ritter, V. Pomjakushin, C. Tealdi, C. B. Azzoni and G. Flor, J. Phys. Chem. B, 2006, 110, 17430–17436 CrossRef CAS PubMed
- L. Ma, J. Gong, C. Jin, D. Yang and J. Hou, J. Alloys Compd., 2023, 945, 169359 CrossRef CAS
- S. Y. Lee, J. Yun and W.-P. Tai, Adv. Powder Technol., 2018, 29, 2423–2428 CrossRef CAS
- P. Thamilmaran, M. Arunachalam, S. Sankarrajan and K. Sakthipandi, J. Magn. Magn. Mater., 2015, 396, 181–189 CrossRef CAS
- Y. L. Huang, C. Pellegrinelli, K. T. Lee, A. Perel and E. D. Wachsman, J. Electrochem. Soc., 2015, 162, F965 CrossRef CAS
- R. Ren, X. Yu, Z. Wang, C. Xu, T. Song, W. Sun, J. Qiao and K. Sun, Appl. Catal., B, 2022, 317, 121759 CrossRef CAS
- Y. Sun, R. Li, X. Chen, J. Wu, Y. Xie, X. Wang, K. Ma, L. Wang, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, Adv. Energy Mater., 2021, 11, 2003755 CrossRef CAS
- Y. Wang, C. Huang, K. Chen, Y. Zhao, J. He, S. Xi, P. Chen, X. Ding, X. Wu, Q. Kong, X. An, F. Raziq, X. Zu, Y. Du, H. Xiao, K. H. L. Zhang and L. Qiao, ACS Appl. Mater. Interfaces, 2021, 13, 58566–58575 CrossRef CAS PubMed
- L. Qiao and X. Bi, Europhys. Lett., 2011, 93, 57002 CrossRef
- C. Zhang, C. Wang, W. Zhan, Y. Guo, Y. Guo, G. Lu, A. Baylet and A. Giroir-Fendler, Appl. Catal., B, 2013, 129, 509–516 CrossRef CAS
- Q. Wang, J. Hou, Y. Fan, X.-A. Xi, J. Li, Y. Lu, G. Huo, L. Shao, X.-Z. Fu and J.-L. Luo, J. Mater. Chem. A, 2020, 8, 7704–7712 RSC
- J. Gong and J. Hou, Appl. Surf. Sci., 2023, 158385 CrossRef CAS
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS
- R. Zhou, Y. Yin, H. Dai, X. Yang, Y. Gu and L. Bi, J. Adv. Ceram., 2023, 12, 1189–1200 CrossRef CAS
- S. Yi, Y. Shen, H. Zhao, Z. Du, N. Chen and B. Huang, Electrochim. Acta, 2016, 219, 394–400 CrossRef CAS
- P. Wang, D. Xu, J. Cheng and T. Hong, Ionics, 2021, 27, 1185–1192 CrossRef CAS
- Y. Chen, T. Hong, P. Wang, K. Brinkman, J. Tong and J. Cheng, J. Power Sources, 2019, 440, 227122 CrossRef CAS
- D. Poetzsch, R. Merkle and J. Maier, Faraday Discuss., 2015, 182, 129–143 RSC
- X. Yang, X. Xu, S. Wu, S. Yu and L. Bi, Ceram. Int., 2022, 48, 19626–19632 CrossRef CAS
- J. Ma, Y. Pan, Y. Wang and Y. Chen, J. Power Sources, 2021, 509, 230369 CrossRef CAS
- C.-Y. Gu, X.-S. Wu, J.-F. Cao, J. Hou, L.-N. Miao, Y.-P. Xia, F. Chao and W. Liu, Int. J. Hydrogen Energy, 2020, 45, 23422–23432 CrossRef CAS
- L. Miao, J. Hou, Z. Gong, Z. Jin and W. Liu, Int. J. Hydrogen Energy, 2019, 44, 7531–7537 CrossRef CAS
- F. He, T. Wu, R. Peng and C. Xia, J. Power Sources, 2009, 194, 263–268 CrossRef CAS
- E. Fabbri, L. Bi, D. Pergolesi and E. Traversa, Energy Environ. Sci., 2011, 4, 4984–4993 RSC
- S. Wu, X. Xu, X. Li and L. Bi, Sci. China Mater., 2022, 65, 675–682 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01133j |
| This journal is © The Royal Society of Chemistry 2024 |