
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New Fe-doped two-dimensional BiVO4 nanosheets for direct methane conversion to methyl oxygenates†
Catherine
Afriyie
 *ab and
Xingwang
Zhang
a
*ab and
Xingwang
Zhang
a
aCollege of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: 22028182@zju.edu.cn
bDepartment of Chemical Engineering, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana
First published on 19th March 2024
Abstract
Photocatalytic methane conversion is a sustainable approach to transform methane under mild conditions into its value-added derivatives for the petrochemical industry. However, obtaining high productivity and selectivity of desired products still remains a great challenge. In this work, high-efficiency Fe-doped two-dimensional BiVO4 nanosheets have been synthesized and evaluated for photocatalytic methane conversion with H2O, O2 or H2O2 as the oxidizer. The Fe-doped and undoped samples were obtained by hydrothermal synthesis using 3-dodecylbenzenesulfonate (SDBS) as the morphology-directing agent, and examined by XRD, SEM, EDS, TEM, XPS, UV-DRS and BET characterization techniques. The replacement of some Bi3+ with Fe3+ was a successful approach to enhance the photoexcitation process and the adsorption properties of BiVO4, resulting in an improvement in the yield of methyl oxygenates. Complete selectivity of methyl oxygenates (CH3OH and CH3OOH) was achieved, and the yield reached 217.6 μmol gcat−1 h−1 in 2 h under visible light irradiation under mild conditions over 1.0 wt% Fe-doped BiVO4 nanosheets in the presence of H2O2, outperforming most reported studies on this reaction. This work provides new insight into the choice of a suitable oxidizer for methane conversion in the BiVO4 photocatalyst system by projecting H2O2 as the relevant oxidant.
1. Introduction
As the simplest hydrocarbon molecule, methane (CH4) has abundant natural reserves with an increased production from shale gas and tight oil, which has resulted in the reduction of the cost of methane-rich natural gas compared to crude oil. Meanwhile, CH4 is the second most relevant greenhouse gas, as its global warming potential is 25 times higher than CO2.1,2 Also, it is inconvenient to liquefy methane gas for transportation due to its low boiling-point (109 K) at atmospheric pressure.2–4 Hence, it is highly desired to develop efficient techniques for on-site upgrading of methane to easily transportable and storable value-added liquid oxygenated products, especially methanol. However, the direct partial oxidation of methane remains a major challenge because of the extremely high dissociation energy of methane's C–H bond (439.3 kJ mol−1), negligible electron affinity (−1.9 eV), and low polarizability (2.8 × 10−40 C2 m2 J−1).3,5–8 In classical thermal catalysis, activation of the C–H bond in the methane molecule requires high temperature (>500 °C) which makes it energy intensive, leads to poor selectivity of useful oxygenates, and puts high requirements on the stability of the catalyst whereby the catalyst can be easily coked and sintered.2–7 Photocatalysis is one of the green alternative techniques and a promising approach for direct methane conversion where the energy of photons provides an alternative energy source for methane activation, which may occur under irradiation even at room temperature.1–9 Hence, it is worth developing stable photocatalytic systems in a cost-effective, novel, and eco-friendly approach for highly selective conversion of methane to useful oxygenates.To drive methane photocatalytic conversion, oxide semiconductors which are abundant in nature are widely used.10 Most reported studies on these photocatalytic systems for this reaction employed binary oxides loaded with noble-metal cocatalysts.11–14 These systems can activate O2 conversion into highly reactive oxygen species which sometimes result in the overoxidation of desired products. In addition, the high cost of precious metals impedes their practical application. Bismuth vanadate (BiVO4) with its low cost, nontoxicity, and narrow band gap (2.4 eV) presents a promising alternative to binary oxide semiconductors such as TiO2, ZnO and WO3 for visible light photocatalytic applications.15,16 Theoretically, the conduction band edge of BiVO4 is positioned at a more negative potential (E = ∼−0.7 V vs. NHE at pH = 7) than the reduction potential of O2 to produce ˙O2− (O2/˙O2− = −0.33 V) or H2O2 to produce ˙OH (H2O2/˙OH = 0.06 V) through interaction with photogenerated electrons to activate CH4 to undergo oxidation.5,8 There are three crystal structures of BiVO4, the tetragonal zircon phase with a band gap (2.9 eV), the monoclinic scheelite phase with a band gap of 2.4 eV, and the tetragonal scheelite phase.15,17 Among these crystalline structures, monoclinic scheelite phase BiVO4 exhibits greater photocatalytic performance under visible-light illumination owing to the Bi–O polyhedron distortion by a 6s2 lone pair of Bi3+.15,18,19
Murcia-Lopez et al.20 were the first to report on Bi2WO6, BiVO4 and Bi2WO6/TiO2–P25 for the partial photo-oxidation of methane using pure water as the sole oxidant. Their experimental results suggested that BiVO4, in spite of its lower specific surface area as compared to Bi2WO6 and Bi2WO6/TiO2–P25, is the most promising photocatalyst for this reaction, displaying higher CH3OH selectivity with C2H6 and CO2 as the by-products, being more stable than the others. Their group also reported on evaluating the activity of BiVO4 for partial oxidation of methane in the presence of low concentration of nitrite.21 Furthermore, CH4 transformation with water over a V2O5/BiVO4/beta zeolite has been studied by their group.22 Quite recently, Zhu et al.23 fabricated BiVO4 microcrystals with bipyramidal and platelet morphologies for the transformation of methane with water. The most recent studies reported aerobic conversion of CH4 using quantum-sized BiVO4 (q-BiVO4) nanoparticles in the presence of water.24
Some previous studies on BiVO4 highlight that the photocatalyst can activate the C–H bond and implement CH4 transformation with only H2O as the oxidizer.20,22,23 However, theoretical deductions suggest that the top of the valence band of BiVO4 is positioned less positively in energy (E = ∼+2.4 V vs. NHE at pH = 7) than the oxidation potential of water (H2O/˙OH = 2.8 V) to activate CH4 through ˙OH produced by the interaction of photogenerated holes with H2O.5,8 Chen et al.25 in their preliminary studies on finding a suitable single component semiconductor photocatalyst fabricated a range of semiconductors including SrTiO3, KNbO3, CdS, Cu2O, BiVO4, g-C3N4, Ag3PO4, and TiO2–P25, for oxidative methane conversion through the production of ˙OH from water cleavage under light irradiation by photogenerated holes. Upon examining their performance on driving methane photo-oxidation, none of the aforementioned semiconductors including BiVO4 showed any activity for CH4 photo-oxidation with H2O, except TiO2–P25 which showed a moderate photoactivity. These conflicting findings inspired the need for further study on BiVO4 semiconductor photocatalysts for the partial oxidation of methane. Furthermore, a single component BiVO4 photocatalyst suffers from low charge separation efficiency and fast recombination of photoinduced electron–hole pairs, as well as weak surface adsorption due to low specific surface area.15–20,26,27
Doping with metal plays a role in introducing a new energy level into the band structure to extend the light absorption of photocatalysts and help trap electrons or holes, leading to reduced recombination of photogenerated charge carriers.8,28 It is speculated that there is a shift in the Fermi level of doped BiVO4 towards the conduction band, thus accelerating the charge separation by increasing the band bending.15,17,29 Zhang et al.30 proposed the synthesis of monoclinic BiVO4 nanosheets by means of a hydrothermal method assisted by a morphology-directing agent. This nanosheet shape material exhibited much higher photocatalytic activity than the bulk material. Motivated by these findings, this work aims to investigate for the first time the performance of Fe-doped two-dimensional BiVO4 nanosheets for photocatalytic partial methane selective oxidation by utilizing green oxidants (H2O, O2 or H2O2). Complete selectivity of methyl oxygenates was achieved with the nanosheet morphology, and Fe3+ doping of BiVO4 nanosheets led to enhancement in the yield of methyl oxygenates in the presence of H2O2 at 40 °C within 2 h. The increase in productivity of methyl oxygenates was attributed to improved charge separation, optical and surface adsorption properties of the Fe-doped BiVO4 nanosheets as compared to the undoped sample. This work represents the rational design of a high-efficiency, low-cost 2-dimensional semiconductor photocatalyst for visible light photocatalytic application in the selective oxidative conversion of methane into its primary oxygenated derivatives.
2. Experimental section
2.1. Materials
Bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O), ammonium metavanadate (NH4VO3), sodium 3-dodecylbenzenesulfonate (SDBS) (C18H29NaO3S), nitric acid (HNO3), sodium hydroxide (NaOH), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O), absolute ethanol (C2H5OH) and hydrogen peroxide (H2O2) 30% (w/w) were purchased from Sinopharm Chemical Reagent Co., Ltd, Shanghai Aladdin Biochem. Tech. Co., Ltd, and Shanghai Macklin Biochemical Co., Ltd. All other reagents and chemicals were commercially obtained and used without further purification.2.2. Preparation of Fe doped and undoped BiVO4 nanosheets
The catalysts were synthesized using a facile hydrothermal method with sodium 3-dodecylbenzenesulfonate (SDBS) as the structure-directing agent, and calcination. The undoped BiVO4 nanosheets were synthesized by following a procedure reported by Zhang et al.,30 with modification of hydrothermal conditions. Firstly, the Fe doped BiVO4 nanosheets were prepared by dissolving 3.0 mmol Bi(NO3)3·5H2O and 2.16 mmol C18H29NaO3S (SDBS) in 30.0 mL (4.0 mol L−1) HNO3 solution and the resultant solution was denoted as solution A. In another beaker, 3.0 mmol NH4VO3 was added to 30.0 mL (2.0 mol L−1) NaOH solution, and the resultant solution was denoted as solution B. The solutions were stirred for 30 min in order to ensure the complete dissolution of the chemicals. Then, solution B was added to solution A under vigorous stirring. 70 mg of Fe(NO3)3·9H2O was added to the mixture to give a solution with theoretical Fe doping of 1.0 wt%. The pH of the mixed solution was adjusted to about 8 with 2 M NaOH solution and stirred for 2 h. The mixture was poured into three different 50 mL Teflon-lined stainless-steel autoclaves until 80% of the volume of each autoclave was occupied. The Teflon-lined stainless-steel autoclaves were sealed and heated in an oven at 180 °C for 3 h. After the hydrothermal treatment, the autoclaves were cooled naturally to room temperature. After cooling, each sample was transferred to a centrifuge tube, centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min during each centrifugation cycle, and washed with deionized water three times and once with absolute ethanol. The brownish-yellow precipitate was dried in an oven at 80 °C overnight before collecting the powder. The dried sample was calcined at 200 °C for 2 h in a muffle furnace. 0.5, 1.5 and 2.0 wt% theoretical Fe doping amounts were also prepared by changing the amount of Fe(NO3)3·9H2O. The vivid-yellow undoped BiVO4 powder was prepared by the same procedure except for the addition of Fe(NO3)3·9H2O.
000 rpm for 5 min during each centrifugation cycle, and washed with deionized water three times and once with absolute ethanol. The brownish-yellow precipitate was dried in an oven at 80 °C overnight before collecting the powder. The dried sample was calcined at 200 °C for 2 h in a muffle furnace. 0.5, 1.5 and 2.0 wt% theoretical Fe doping amounts were also prepared by changing the amount of Fe(NO3)3·9H2O. The vivid-yellow undoped BiVO4 powder was prepared by the same procedure except for the addition of Fe(NO3)3·9H2O.
2.3. Characterization
The crystalline structure of the Fe doped and undoped BiVO4 was characterized using an X-ray diffraction technique on a Bruker D8 advance with Cu Kα radiation (λ = 1.5406 Å) over a 2θ range of 10°–43° and 10°–80° respectively. The morphology and microstructure of the samples were characterized using a scanning electron microscope (SEM, Hitachi SU8010) equipped with an energy dispersive X-ray spectrometer (EDS) to determine the elemental composition and distribution of the elements in the 1.0 wt% Fe-doped sample. A field emission transmission electron microscope (FE-TEM) model JEM-2100F (JEOL) with an accelerating voltage of 200 kV was used for better observation of morphology of the undoped BiVO4. A high-resolution transmission electron microscope (HR-TEM) was used to measure the lattice spacing of the sample. Before testing, the sample was dispersed in ethanol under sonication for 2 h. The resulting suspended powder was dropped on a thin copper grid covered with a carbon film. In order to better understand the chemical state of the Bi, V, and O present in undoped BiVO4 and Bi, V, O, and Fe elements present in 1.0 wt% Fe-doped BiVO4, the samples were analyzed by X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha) with an Al Kα X-ray source. All binding energies of the elements in the Fe-doped and undoped samples were calibrated with respect to C 1s spectral peak at 284.8 eV, corresponding to surface adventitious carbon which was used as a reference. The light harvesting ability of Fe–BiVO4 and BiVO4 was measured by UV-Vis diffuse reflectance spectroscopy (Agilent Cray 5000) in the wavelength range of 200–800 nm, with BaSO4 as the reference. The textural properties of Fe doped and undoped BiVO4 were analyzed by means of N2 sorption–desorption curves by using a Quantachrome Autosorb IQ3 instrument. The surface areas and pore-size distributions were determined via N2 adsorption at 77.35 K. Prior to measurements, the catalysts were degassed at 200 °C for 8 h. The specific surface area was calculated according to the Brunauer–Emmett–Teller method and the pore size distribution was derived from the desorption branch of the isotherm with the Barrett–Joyner–Halenda (BJH) model.2.4. Photocatalytic reaction
The photocatalytic partial methane oxidation tests were conducted in a 50 mL batch-type reactor (IKA RCT basic, CHEMN) equipped with a quartz window to allow light irradiation as shown in Fig. S1, ESI.† The photocatalytic activity tests with O2 proceeded by dispersing 20 mg of the photocatalyst powder into 20 mL of deionized water. Then the mixture was added to the reaction cell, and the reaction cell was placed in the batch-reactor. Prior to illumination, the reactor suspension was purged with oxygen gas at a partial pressure of 2 bar for 5 min to remove air. Then CH4 was introduced into the reactor at a total pressure of 20 bar (CH4:O2 = 9:1), and the photocatalytic reaction proceeded at temperatures of 40, 50, and 65 °C for 2 h. UV-vis light, Xe lamp (MC-XS500, Testmart) 300 W (λ ≥ 420 nm) was used to provide incident light at an intensity of 100 mW cm−2. A temperature probe was connected to the reactor to directly detect the temperature of the liquid solution. The solution was constantly stirred at a rotation of 800 rpm during the reaction to maintain homogeneity. For the reaction involving H2O2, the photocatalytic activity tests proceeded by dispersing 20 mg of the photocatalyst powder in 20 mL of deionized water and 100 μL of H2O2 30% (w/w) was added. Then the mixture was added to the reaction cell, and the reaction cell was placed in the batch-reactor. Prior to illumination, the reactor suspension was purged with CH4 gas at a pressure of 2 bar for 5 min to remove air, and the CH4 pressure was increased to 20 bar afterward. The photocatalytic reaction was conducted at a temperature of 40 °C for 1, 2, and 3 h with the same light source and intensity as the reaction with O2. Higher reaction temperatures of 60 and 80 °C were also tested. After each reaction, the reactor was subjected to cooling to room temperature, and centrifugation of the reaction mixture at 10000 rpm was carried out for 5 min. 400 μL of the reaction liquid, 150 μL of D2O and 1 μL of DMSO were collected in an NMR tube for quantitative 1H-NMR analysis (Agilent DD2 600). During the NMR measurements, a solvent suppression program was run in order to minimize the signal originating from H2O. The gaseous components were injected and examined with a gas chromatography system (GC-2014, Shimadzu) equipped with a BID detector. Furthermore, control tests were conducted without light irradiation, and without the catalyst while maintaining every other reaction condition. It should be noted that before testing the photocatalytic activity of the catalysts in the presence of O2 or H2O2, preliminary anaerobic experiments were conducted with water as the sole oxidant.
The productivity of products was calculated according to the following equation:
 | (1) |
3. Results and discussion
The XRD patterns of undoped BiVO4 and Fe–BiVO4 are shown in Fig. 1. With reference to the XRD patterns of the standard BiVO4 (JCPDS PDF card no. 14-0688), it can be deduced that undoped BiVO4 has a single-phase monoclinic scheelite structure. As shown in Fig. 1a, the main diffraction peaks at 2θ values for BiVO4 of 15.1°, 18.9°, 28.8°, 30.5°, 34.4°, 35.2°, 39.7°, 42.4°, 46.0°, 47.3°, 50.3°, 53.2°, 54.5, 58.5°, and 59.2° could be well indexed to (020), (011), (−121), (040), (200), (002), (211), and (051), (132), (042), (202), (−161), (013), (321), and (123) which match the planes of pure monoclinic BiVO4 (JCPDS card no. 14-0688). From Fig. 1b, the addition of the Fe3+ dopant resulted in the appearance of characteristic peaks for the tetragonal zircon phase of BiVO4 at 2θ = 24.3° and 32.6° that matched the standard JCPDS card no. 14-0133. This result suggests that the presence of Fe3+ ions might induce the formation of a monoclinic–tetragonal heterostructure. As the Fe doping content was increased from 0.5 wt%, it was observed that the highest peak of the monoclinic phase at 2θ = 28.8° assigned to the lattice (−121) decreased gradually in intensity. Meanwhile, the intensity of the tetragonal phase peak at 2θ = 24.3° increased with increasing Fe doping content. This demonstrates that, as the Fe doping amount increases, the monoclinic phase decreases with a rising tetragonal phase. No characteristic peaks for iron or iron oxides were observed. This result may be attributed to the lower concentration of Fe3+ ions in the catalyst. The peaks at 2θ = 18.6° and 18.9° are assigned to the (110) and (011) lattices of the monoclinic scheelite phase, these peaks distorted in the presence of the Fe3+ dopant. Also, the peaks at 34.4° and 35.2°, which are indexed to the lattices (200) and (002) of the monoclinic scheelite phase shifted closer to each other in the presence of 0.5 and 1.0 wt% Fe3+ dopant, and they completely merged into a single peak when the concentration of the Fe3+ dopant reaching 1.5 and 2.0 wt%. These deformations might be caused by a substitutional defect, where Fe3+ ions (r = 0.064 nm) replace Bi3+ ions (r = 0.103 nm), consistent with the literature.31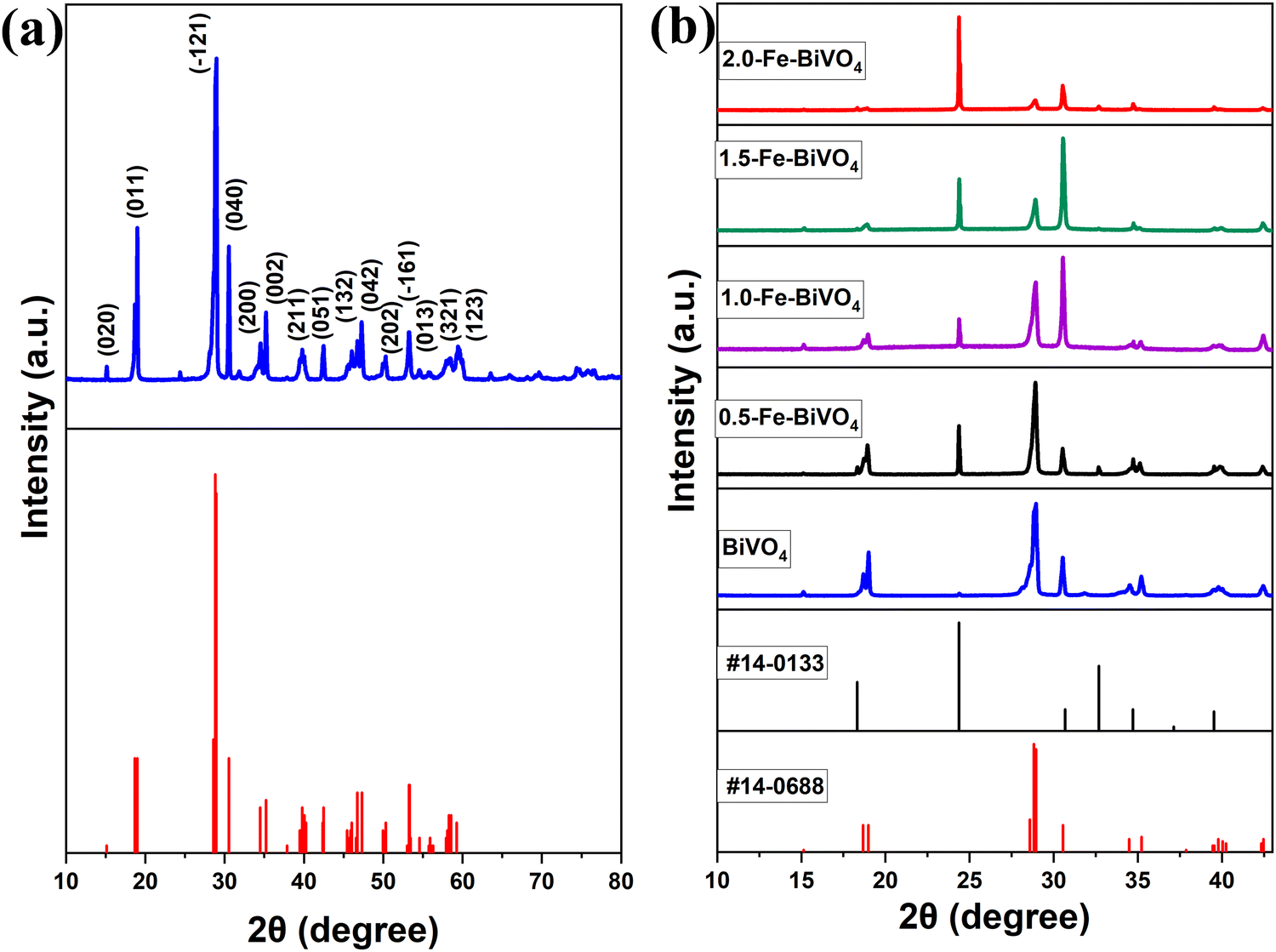 | ||
| Fig. 1 XRD patterns of (a) undoped BiVO4 nanosheets compared with the JCPDS PDF standard card of monoclinic scheelite BiVO4, and (b) undoped BiVO4 and Fe doped BiVO4 nanosheets compared with JCPDS PDF standard cards of monoclinic scheelite and tetragonal zircon BiVO4. | ||
The surface morphology and microstructure of the undoped and 1.0 wt% Fe doped BiVO4 samples were studied using SEM, and the representative micrographs are displayed in Fig. 2a and b. The Fe doped BiVO4 exhibited a smoother surface nanosheet morphology as compared to the rough surface nanosheet morphology of the undoped BiVO4. The particle size of the Fe doped BiVO4 ranges from 180–350 nm, which is smaller than the undoped (250–400 nm). These results indicate that the iron doping did not significantly affect the morphology of BiVO4, because both doped and undoped samples maintained the nanosheet structure. However, the nanosheets are more well defined in the Fe doped sample with a decrease in particle size. Studies have demonstrated that the larger the particle size, the greater the surface recombination of photogenerated charge carriers.15,32 Hence, the smaller particle size distribution of the Fe doped sample as compared to the undoped signifies less surface charge recombination. For better observation of morphology of the undoped BiVO4, TEM analysis was performed and the monographs are displayed in Fig. 2c and d, which confirmed the nanosheet morphology observed in the SEM. The diameter of the single BiVO4 nanosheet in the TEM micrographs is 280 nm, which falls within the range and is consistent with SEM observation. The selective area electron diffraction (SAED) pattern of a single nanosheet in Fig. 2e shows well-defined bright dotted ED rings indicating the single-crystalline nature of the nanosheet. The ED pattern can be indexed to the diffraction patterns of (121) which represents the highest and sharpest peak in the XRD of monoclinic scheelite BiVO4, and (321) which is another characteristic peak of monoclinic scheelite BiVO4. The HR-TEM image of the nanosheet shown in Fig. 2f also confirms the single-crystalline nature. The lattice distances of 0.1714 nm (161), and 0.1683 nm (013) are characteristic planes identified in the XRD of monoclinic BiVO4. Fig. 3 shows the EDS mapping of the 1.0 wt% Fe–BiVO4 and the elemental distribution of Bi, V, O, and Fe in the sample. The EDS analysis of the percent content of the elements revealed that Bi, V, O and Fe are 44.090, 13.680, 41.092 and 1.138 wt%, respectively. Furthermore, the SEM-EDS mapping confirms the presence of Bi, V, O, and Fe in the 1.0-Fe–BiVO4 sample, and indicates that these elements are distributed uniformly in the sample which implies a successful synthesis of Fe-doped BiVO4 nanosheets.
 | ||
| Fig. 2 (a) Low-magnification SEM images of BiVO4 nanosheets, and (b) 1.0-Fe–BiVO4 nanosheets; (c) low-magnification and (d) high-magnification TEM images (e) SAED pattern of a single nanosheet; and (f) HR-TEM image, inset is FFT image of BiVO4 nanosheet. | ||
 | ||
| Fig. 3 Elemental mapping EDS composition of 1.0-Fe–BiVO4 nanosheets (a) 1.0-Fe–BiVO4 (b) Bi, (c) V, (d) O, (e) Fe, and (f) elemental composition. | ||
The chemical states and binding energies of elements present in the undoped and 1.0 wt% Fe doped BiVO4 were analyzed by X-ray photoelectron spectroscopy (XPS). The survey spectra shown in Fig. 4a demonstrate the coexistence of Bi, O, and V elements in BiVO4, and Bi, O, V and Fe elements in 1.0-Fe–BiVO4. It can clearly be seen with the appearance of twelve peaks corresponding to Bi 5d, Bi 4f, Bi 4f5, C 1s, Bi 4d5, Bi 4d3, V 2p, O 1s, V 2s, Bi 4p3, Bi 4p1, and Bi 4s of the undoped BiVO4. Thirteen peaks including Fe 2p were observed with the survey spectra of the Fe doped BiVO4, though the Fe 2p peak is not very intense due to the low doping amount of Fe. In terms of both peak position and shape, the XPS peaks of O 1s and V 2p remained unchanged between undoped BiVO4 and the 1.0 wt% Fe doped sample. However, the peak for Bi 4f is more intense and sharper at the edge in the XPS survey spectra of the Fe doped BiVO4 sample than the undoped one. This may also be attributed to substitutional defect of Fe3+ ions (r = 0.064 nm) replacing Bi3+ ions (r = 0.103 nm). For 1.0-Fe–BiVO4, it is observed from Fig. 4b that the Bi 4f spectrum exhibited two symmetric peaks at BE = 158.38 and 163.68 eV, assignable to the spin–orbit splitting of Bi 4f7/2 and 4f5/2 of Bi3+, respectively, which indicate that all bismuth ions in the sample existed in tri-valency.17,33 The asymmetrical V 2p spectrum shown in Fig. 4c could be decomposed into two components at BE = 515.98 and 523.58 eV, assignable to the spin–orbit splitting of V 2p3/2 and V 2p1/2 of V5+ and V4+, respectively. The asymmetrical O 1s spectrum shown in Fig. 4d could be fitted to two peaks centered at 530.42 and 529.08 eV. The component with the lower value of binding energy was assigned to the lattice oxygen species, while the one with the higher binding energy value was ascribed to the surface-adsorbed oxygen species. Similarly, as shown in Fig. 4e, the XPS spectrum of Fe 2p revealed two dominant peaks: Fe 2p3/2 at 710.18 eV and Fe 2p1/2 at 724.18 eV. Moreover, the fitted Fe 2p3/2 peak showed an additional peak at 713.28 eV, which was assigned to the Fe3+ state.31 The binding energies of Bi 4f, V 2p and O 1s of the undoped BiVO4 showed a negligible difference between the Fe doped sample because of the same valency of Fe3+ and Bi3+. The quantity of chemisorbed oxygen in 1.0-Fe–BiVO4 and undoped BiVO4 was found to be nearly identical, which implies that doping with Fe has no effect on the formation of surface defects. Hence, the formation of oxygen vacancies could not be confirmed from XPS data.
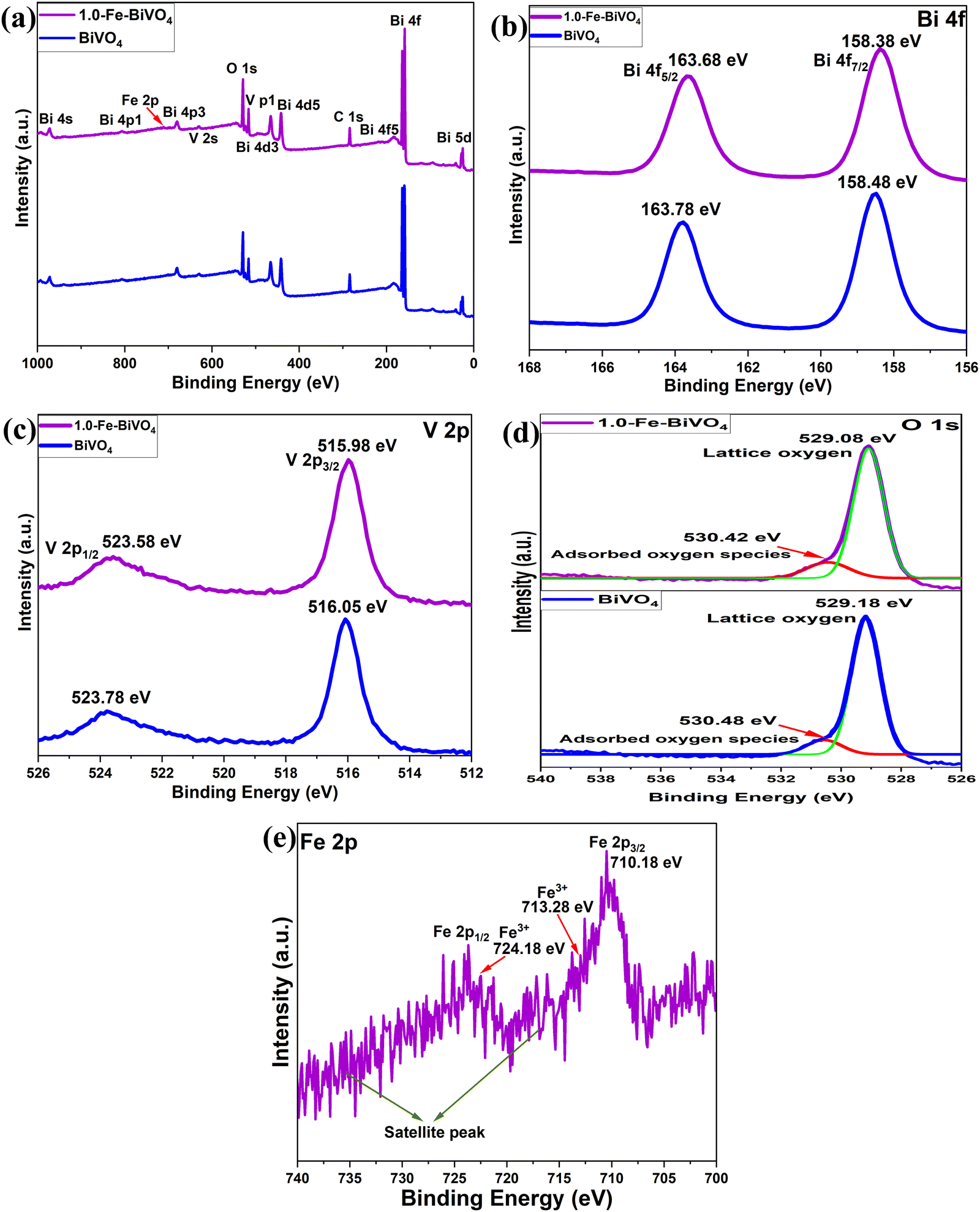 | ||
| Fig. 4 (a) XPS survey spectra of undoped BiVO4 and 1.0-Fe–BiVO4; XPS spectra of (b) Bi 4f, (c) V 2p, and (d) O 1s of undoped BiVO4 compared with 1.0-Fe–BiVO4; (e) Fe 2p XPS spectrum of 1.0-Fe–BiVO4. | ||
Fig. 5 shows the UV-vis diffuse reflectance spectra of Fe doped and undoped BiVO4. All samples showed an absorption band from the UV range up to the visible range. The absorption edge shifted from 520 to 600 nm with the addition of 1.0 wt% of the Fe dopant. The 1.0 wt% Fe-doped BiVO4 showed the highest absorption in the visible region, while the 2.0 wt% Fe doped sample followed by the 1.5 wt% showed the least absorption in the visible range, which may be due to the presence of a significant amount of the tetragonal phase in the higher Fe-doped samples as confirmed by XRD. The band gap energy (Eg) value was calculated from UV-vis DRS using the equation Eg = 1240/λ, where λ is the wavelength of the absorption onset. The band gap energy was determined from Fig. 5b by the extrapolation of the linear part of (αhv)2versus (hv). The Eg values of Fe–BiVO4 samples were narrower; 2.34, 2.28, 2.30 and 2.36 eV for 0.5, 1.0, 1.5 and 2.0 wt% Fe-doped samples, respectively, than the undoped BiVO4, 2.4 eV.
 | ||
| Fig. 5 (a) UV-Vis DRS of undoped BiVO4 and Fe–BiVO4 and (b) plot with the corresponding band gap energy. | ||
The textural properties of Fe doped and undoped BiVO4 were analyzed by means of N2 sorption–desorption. The N2 adsorption isotherms plot and the plot of pore size distribution of Fe doped BiVO4 samples are illustrated and compared with those of undoped BiVO4 in Fig. 6. From Fig. 6a, all the Fe-doped and undoped BiVO4 samples exhibited a typical type III isotherm sorption curve according to the division method of the International Union of Pure and Applied Chemistry (IUPAC). The BET specific surface areas of the Fe-doped samples is 10.366, 10.420, 21.228, and 15.621 m2 g−1 for 0.5, 1.0, 1.5 and 2.0 wt% Fe doping, respectively, which are significantly higher than that of undoped BiVO4 (3.556 m2 g−1). This signifies that the presence of the Fe dopant in the Bi and V precursor mixture may have influenced the formation of crystalline particles through a characteristic nucleation–dissolution–recrystallization effect during the hydrothermal treatment process.31 It was observed that the specific surface areas of 0.5 and 1.0 wt% Fe doped samples are almost the same. Interestingly, the specific surface area increased greatly with an Fe doing content of 1.5 wt%. However, an Fe doping content exceeding 1.5 wt% resulted in a significant decrease in the specific surface area, but still much higher than that of the 0.5 and 1.0 wt% Fe doped samples. These results indicate that the right Fe doping content is an important factor for optimized control of the specific surface area of Fe–BiVO4. Generally, the larger specific surface area means better dispersion of catalyst particles and exposure of active sites. The pore size distribution curves of Fe doped and undoped BiVO4 in Fig. 6b–f, were derived by the Barrett–Joyner–Halenda (BJH) method with average pore volumes of 0.038, 0.021, 0.096, and 0.069 cm3 g−1 for 0.5, 1.0, 1.5 and 2.0 wt% Fe doping, respectively, and pore diameters of 14.0777, 13.819, 17.776, and 19.397 nm for 0.5, 1.0, 1.5 and 2.0 wt% Fe doping, respectively, indicative of the characteristics of mesopores. The larger pore volume and diameter of the Fe-doped samples as compared to the undoped (0.009 cm3 g−1 and 4.294 nm) signifies better adsorption capacity of reactants.
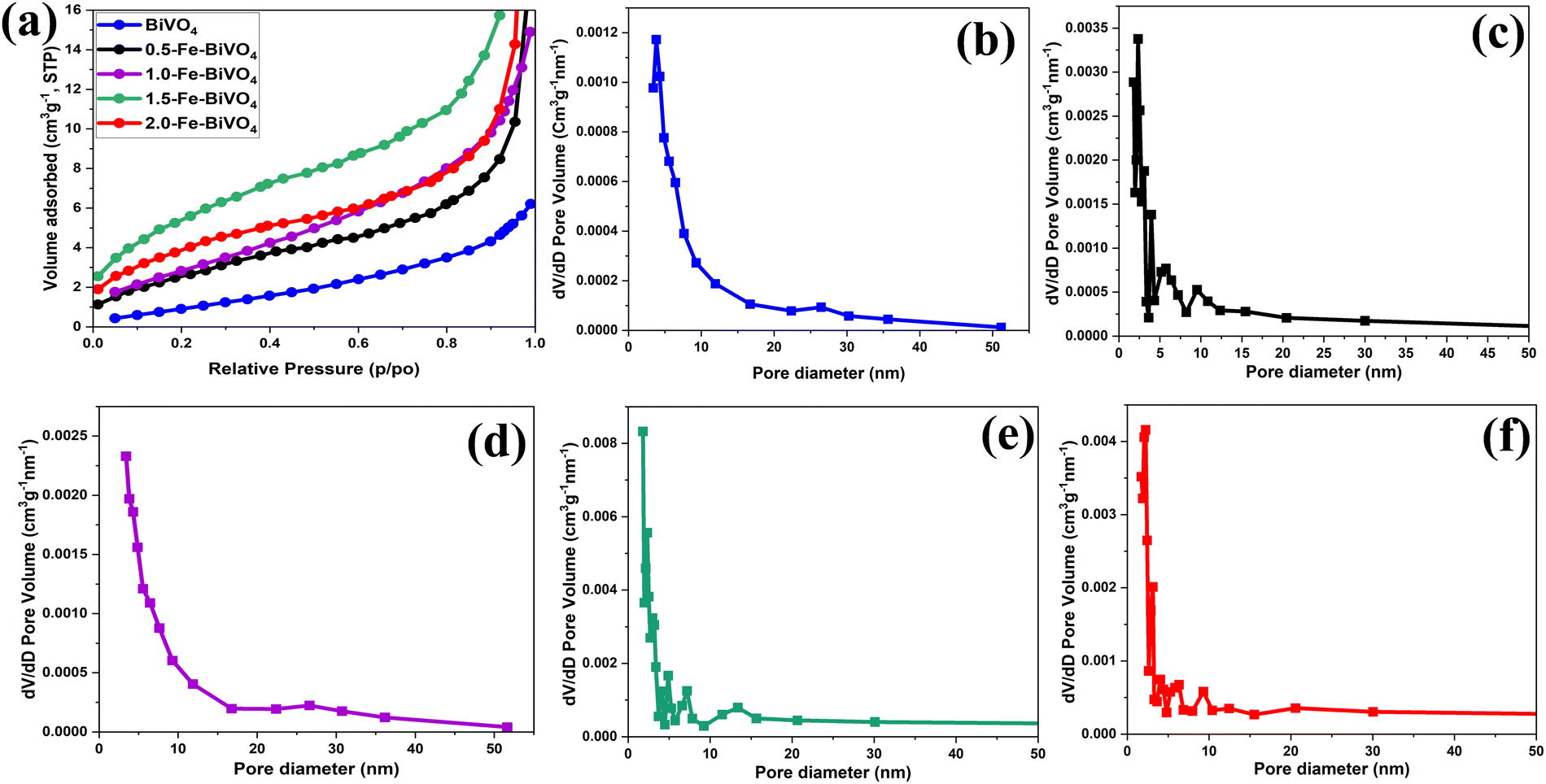 | ||
| Fig. 6 (a) BET N2 adsorption isotherms of Fe-doped and undoped BiVO4 nanosheets; BJH pore size distribution curve of (b) undoped BiVO4; (c) 0.5; (d) 1.0; (e) 1.5; and (f) 2.0 wt% Fe–BiVO4 nanosheets. | ||
The performance of Fe-doped and undoped BiVO4 nanosheets for the partial oxidation of methane with O2 in the presence of water was tested. The quantitative 1H-NMR spectra collected for the reaction liquid after 2 h of light irradiation at 40, 50 and 65 °C suggested the absence of oxygenated products. The anaerobic reaction involving H2O as the sole oxidant did not produce any oxygenated product as well. The result of the BiVO4 photocatalyst being inactive for methane conversion with H2O is expected because of its theoretical valence band maximum position, and in agreement with the result of Chen et al.25 In contrast, our experimental observations suggested that the photogenerated electrons of BiVO4 are not capable of reducing O2 to produce ˙O2− which could attack the strong C–H bond to activate methane to selectively produce useful oxygenates. In addition, the mechanism of photocatalytic methane conversion to methanol and formaldehyde as reported by Fan et al.24 over q-BiVO4 nanoparticles in an aqueous medium by using O2 followed the path of ˙OH radical generation. Their report suggested that the photo holes of q-BiVO4 were capable of oxidizing H2O to produce ˙OH. The photogenerated electrons then reacted with O2 and H+ to generate hydroxyl radicals which facilitated methane conversion. Hence, the mechanism for oxygenate formation over q-BiVO4 in the presence of O2 did not follow the path of CH4 activation by ˙O2−.
As a comparative study, we synthesized monoclinic phase BiVO4 microcrystals according to the procedure reported by Zhu et al.,23 (see the ESI†). Microcrystals of BiVO4 have been previously used to photo-catalytically oxidize CH4 when dispersed in water, which produced methanol and CO2.23 Photocatalytic tests of methane with H2O and O2 over BiVO4 microcrystals after 2 h of irradiation at 40, 50 and 65 °C did not yield any oxygenated product. The photocatalytic activation of CH4 to oxygenates is a complex process, which is affected by many factors, including reaction conditions, oxidants, and light source.5,28 The reaction temperatures employed in this study were 40, 50 and 65 °C, and the reported reaction temperature of the BiVO4 photocatalyst producing oxygenates from methane conversion via ˙OH generated from H2O oxidation by photogenerated holes ranges from 25 to 70 °C. Therefore, the reaction temperature can be ruled out from being the cause of the current result. 20 mL of water containing 20 mg of the catalyst was used in this study, which corresponds to 1 g L−1 used in other studies on BiVO4 for this reaction.20–22 Indicating that the current result may not be due to insufficient amount of water or catalyst. Furthermore, a 450 W mercury lamp with UVC-vis radiation, a 350 W xenon lamp (λ = 350–800 nm), and visible light (λ = 400–780 nm) were employed as the source of light irradiation in all the reported studies on BiVO4 for methane conversion.20–24 The UV-vis 300 W xenon lamp light source (λ ≥ 420 nm at an intensity of 100 mW cm−2) employed in this study is relatively comparable to that of the other studies. Hence, the current results may not be attributed to the light source as well. Also, the molar ratio used for the partial pressures of CH4 to O2 (9:1) is rational because O2 possess relatively higher solubility in water than CH4, and the current result may not have been affected by insufficient proportional amount of the reactants. Since one of the major factors that affect methane conversion to oxygenates is the oxidant, the reactivity of the Fe-doped and undoped BiVO4 nanosheets toward partial oxidation of methane was further investigated with H2O2 as the oxidizer.
The photocatalytic reaction was conducted as illustrated in Fig. S3, ESI.† Typical 1H-NMR spectra (Fig. S4a and b, ESI†) at 40 °C for 2 h of irradiation in the presence of H2O2 indicated methanol (δ = 3.25 ppm) and methyl hydroperoxide (δ = 3.75 ppm) as the identified oxygenated products. No COx products were observed for all the Fe-doped and undoped BiVO4 nanosheets, and therefore 100% selectivity of primary liquid oxygenates (CH3OH and CH3OOH) was obtained. The 1.0 wt% Fe-doped sample showed the highest productivity, whereby the total yield of the primary products reached 217.6 μmol gcat−1 h−1, followed by 188.8, 176.4, 169.2 and 149.6 μmol gcat−1 h−1 for 1.5-Fe–BiVO4, 0.5-Fe–BiVO4, 2.0-Fe–BiVO4, and undoped BiVO4, respectively. Therefore, an enhancement in the photocatalytic activity of the Fe-doped BiVO4 was confirmed relative to that determined for the undoped BiVO4 as presented in Fig. 7. The lower total productivity of the primary products below 1.0 wt% Fe doping amount may be attributed to the lack of maximum separation of charge carriers at lower Fe doping concentrations. The total productivity of primary products gradually reduced with an Fe content higher than 1.0 wt% because excessive Fe doping resulted in the appearance of a significant amount of the tetragonal phase of BiVO4 according to the XRD data, which has less photocatalytic activity compared to the monoclinic phase, and this is also reflected in their lower visible-light absorption capacity according to the UV-DRS data.
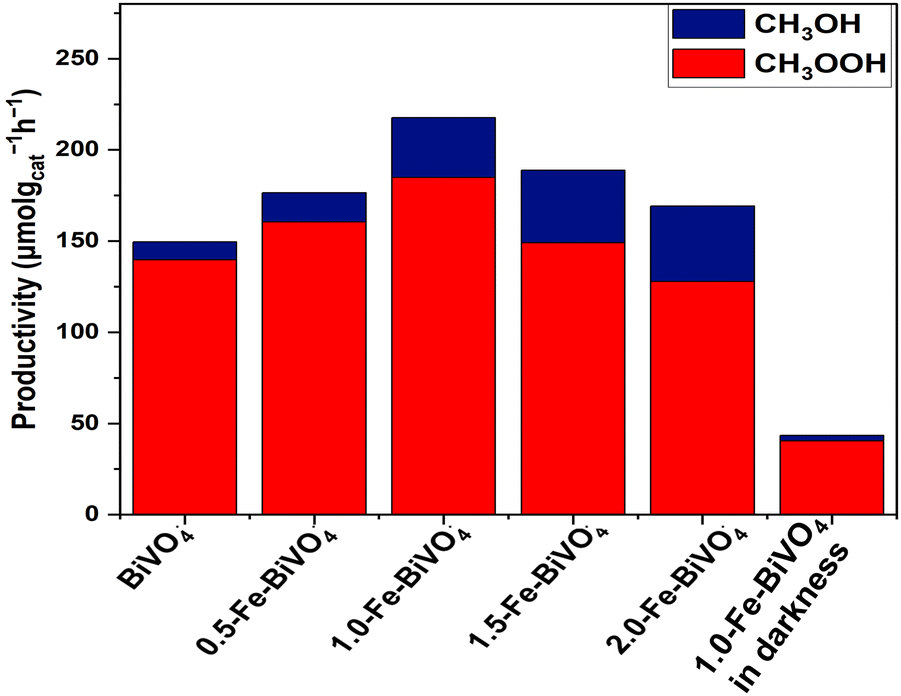 | ||
| Fig. 7 Productivity of methyl oxygenates over 20 mg of Fe–BiVO4 or BiVO4 nanosheets, and 20 mL of H2O in the presence of 100 μL 30% (w/w) H2O2 at 2 h of irradiation at 40 °C. | ||
To confirm that methane conversion is from the photocatalytic process by 1.0-Fe–BiVO4, control experiments were conducted in the absence of light. The IH-NMR spectra (Fig. S4c, ESI†) revealed that CH3OOH was mainly produced with negligible traces of CH3OH, and a productivity of about 43.5 μmol gcat−1 h−1 in 2 h. Implying that more than five times the amount of methyl oxygenates were produced over 1.0-Fe–BiVO4 in the presence of light as compared to the results in darkness. This shows that, CH4 conversion was mainly photo-driven, and some amount of CH4 was converted by the catalytic decomposition of H2O2 at a reaction temperature of 40 °C in darkness. Furthermore, control experiments without the catalyst while maintaining all other reaction conditions constant did not yield any oxygenated product.
Our study of the effect of the reaction time with 1.0-Fe–BiVO4 nanosheets showed that the productivity of methyl oxygenates increased steadily with increasing reaction time up to 2 h, while their total productivity dropped slightly at a reaction duration of 3 h with increased CH3OH content but a drop in the CH3OOH content as shown in Fig. 8a. This is an indication that a longer reaction period may affect the product distribution. Evaluation of 1.0-Fe–BiVO4 nanosheets at higher reaction temperatures of 60 and 80 °C for 2 h resulted to an increase in the total yield of methyl oxygenates, while the selectivity dropped with CO2 production accompanying the rising temperature as shown in Fig. 8b. Hence, methanol experienced further oxidation which resulted in the formation of CO2 but at low yields. It should be noted that, reactions at temperatures such as 20 or 30 °C were difficult to control in this study since there was no means to circulate cooling water with the type of batch reactor design employed. To evaluate the stability of 1.0-Fe–BiVO4 nanosheets, recycling tests were conducted for 1 and 2 h in a run, after each run the catalyst was dried at 80 °C overnight to evaporate all absorbed reactants and products. The productivity of methyl oxygenates remained almost unchanged after three runs as presented in Fig. 8c, demonstrating the good durability of 1.0-Fe–BiVO4 nanosheets for methane conversion with H2O2. Furthermore, there was no change in coloration of the spent catalyst after the recycling tests.
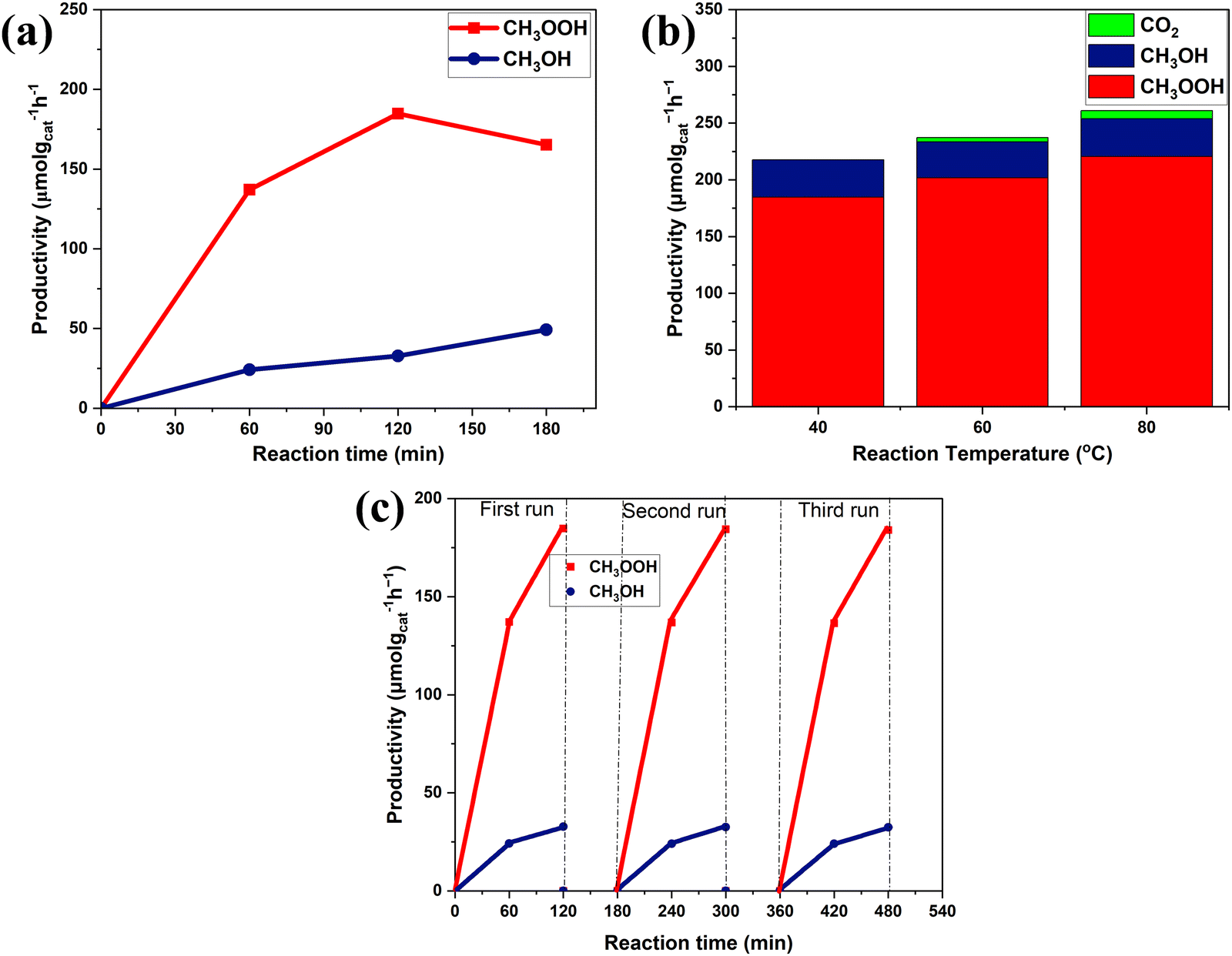 | ||
| Fig. 8 (a) Effect of the reaction time at 40 °C with 1.0-Fe–BiVO4; (b) effect of the reaction temperature on methane oxidation products for 2 h with 1.0-Fe–BiVO4; and (c) stability evaluation with 1.0-Fe–BiVO4 nanosheets at 40 °C. | ||
It has been demonstrated in the previous experiments of this study that the photo holes generated by BiVO4 upon photon excitation do not have the ability to directly attack the C–H bond of methane. Therefore, it can be deduced that the possible mechanism of methane activation in the presence of H2O2 was mainly by the production of ˙OH from H2O2 interaction with photo-electrons upon light irradiation, and partly from H2O2 catalytic decomposition at the reaction temperature. The reaction pathway of methane to form oxygenated products via semiconductor photocatalysis in the presence of H2O2 has been previously reported.4,34,35 The generation of methyl radicals (˙CH3) is realized when highly active ˙OH species oxidize CH4 by the abstraction of a hydrogen atom. Further reaction of undissociated H2O2 in solution with ˙OH results in the formation of hydroperoxyl radicals (˙OOH) which couple with ˙CH3 to produce CH3OOH. The methyl radical can react with water or another hydroxyl radical, resulting in CH3OH formation. Photogenerated electrons can reduce CH3OOH to produce CH3OH. Meanwhile, at room temperature, the facile reduction of CH3OOH with NaBH4 can proceed to form CH3OH.36 CH3OH can undergo further oxidation with ˙OH to form formaldehyde, formic acid, CO, and CO2. However, the mild oxidative environment of the Fe-doped and undoped BiVO4 nanosheet photocatalyst system prevented overoxidation of CH3OH at a lower reaction temperature of 40 °C.
The enhancement in the photocatalytic activity of Fe-doped BiVO4 as compared to undoped may be attributed to the formation of a monoclinic-tetragonal heterostructure which promoted the interfacial charge transfer between phases and slowed down the internal recombination of the photoinduced charge carriers.15,31 Also, the increased specific surface area, larger pore volume and diameter of Fe–BiVO4 signifies better dispersion of catalyst particles and exposure of active sites, as well as better adsorption capacity of reactants. The smaller particle size distribution of the 1.0-Fe doped sample as compared to the undoped sample signifies less surface charge recombination.15,32 Furthermore, the enhanced light absorption in the visible range beyond the optical response of undoped BiVO4 and other Fe doping amounts may have contributed to the higher photocatalytic performance of 1.0-Fe–BiVO4 nanosheets. A comprehensive summary from the literature of the photocatalytic conversion of methane to primary oxygenated products in BiVO4 and other photocatalyst systems in comparison to this work is presented (Table S1, ESI†).
4. Conclusions
In summary, this work explored the synthesis of the BiVO4 nanosheet morphology in a cost-effective manner by doping with Fe, and testing the photocatalytic performance in selective partial oxidation of methane alternatingly with H2O, O2, or H2O2 as the oxidizer. Although some reported studies suggest that BiVO4 is active for methane conversion with H2O, theoretical deductions of the valence band maximum position of BiVO4, and the experimental observations of this study suggest otherwise. The conduction band minimum position of BiVO4 projects O2 and H2O2 as the possible oxidizers in the BiVO4 photocatalyst system for methane conversion. However, the experimental findings of this study indicate that the synthesized catalysts showed no conversion of methane in the presence of O2. It was concluded that this result may not have been affected by the reaction conditions and light source. The possible reason may be due to the position of the conduction band edge of BiVO4 being very close to the reduction potential of O2 to produce ˙O2− through interaction with photo-electrons. Methane conversion was achieved when H2O2 was used as the oxidant under mild conditions, with complete selectivity of methyl oxygenates, and Fe doping of BiVO4 nanosheets was a successful approach to enhance the photocatalytic and adsorption properties of BiVO4, which resulted in improvement in the yield of methyl oxygenates. The Fe–BiVO4 nanosheets also demonstrated good stability for this reaction after three runs of recycling tests. Hence, the experimental observations of this study project H2O2 as the relevant oxidant for the generation of oxidative species for methane conversion in the BiVO4 photocatalyst system. For economic reasons regarding the large scale applicability of this system, modification of BiVO4 for in situ generation of H2O2 from H2 and O2 should be considered in future work.Author contributions
Catherine Afriyie: conceptualization; investigation; and writing – original draft. Xingwang Zhang: conceptualization; funding acquisition; supervision; resources; and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Fundamental Research Funds for the Central Universities (2020QNA4035). The authors are grateful to Prof. Pengfei Xie (Zhejiang University) for guidance and support on this research topic.References
- M. A. R. da Silva, J. C. Gil and N. V. Tarakina, et al. , Chem. Commun., 2022, 58, 7419–7422 RSC.
- Z. Wang, Z. Zhang and Z. Wang, et al. , Appl. Catal., A, 2023, 654, 119082 CrossRef CAS.
- H. Song, X. Meng and Z. Wang, et al. , Joule, 2019, 3, 1606–1636 CrossRef CAS.
- J. Yang, J. Hao and J. Wei, et al. , Fuel, 2020, 266, 117104 CrossRef CAS.
- Y. Tian, L. Piao and X. Chen, Green Chem., 2021, 23, 3526–3541 RSC.
- X. Li, C. Wang and J. Tang, Nat. Rev. Mater., 2022, 7, 617–632 CrossRef CAS.
- X. Meng, X. Cui and N. P. Rajan, et al. , Chem, 2019, 5, 2296–2325 CAS.
- G. Yuniar, W. H. Saputera and D. Sasongko, et al. , Molecules, 2022, 27, 5496 CrossRef CAS PubMed.
- D. Hu, V. V. Ordomsky and A. Y. Khodakov, Appl. Catal., B, 2021, 286, 119913 CrossRef CAS.
- M. M. Khan, S. F. Adil and A. Al-Mayouf, J. Saudi Chem. Soc., 2015, 19, 462–464 CrossRef.
- H. Song, X. Meng and S. Wang, ACS Catal., 2020, 10, 14318–14326 CrossRef CAS.
- H. Song, X. Meng and S. Wang, et al. , J. Am. Chem. Soc., 2019, 141(51), 20507–20515 CrossRef CAS PubMed.
- X. Cai, S. Fang and Y. H. Hu, J. Mater. Chem. A, 2021, 9, 10796–10802 RSC.
- L. Luo, Z. Gong and Y. Xu, et al. , J. Am. Chem. Soc., 2022, 144, 740–750 CrossRef CAS PubMed.
- A. Malathi, J. Madhavan and M. Ashokkumar, et al. , Appl. Catal., A, 2018, 555, 47–74 CrossRef CAS.
- X. Liu, S. Gu and Y. Zhao, et al. , J. Mater. Sci. Technol., 2020, 56, 45–68 CrossRef CAS.
- I. Abdellaoui, 2021, https://www.researchgate.net/publication/354825063.
- Y. Luo, G. Tann and G. Dong, et al. , Ceram. Int., 2015, 41, 3259–3268 CrossRef CAS.
- S. Saxena, A. Verma and N. K. Biswas, et al. , Mater. Chem. Phys., 2021, 267, 124675 CrossRef CAS.
- S. Murcia-López, K. Villa and T. Andreu, et al. , ACS Catal., 2014, 4, 3013–3019 CrossRef.
- S. Murcia-López, K. Villa and T. Andreu, et al. , Chem. Commun., 2015, 51, 7249–7252 RSC.
- S. Murcia-López, M. C. Bacariza and K. Villa, et al. , ACS Catal., 2017, 7, 2878–2885 CrossRef.
- W. Zhu, M. Shen and G. Fan, et al. , ACS Appl. Nano Mater., 2018, 1(12), 6683–6691 CrossRef CAS.
- Y. Fan, W. Zhou and X. Qiu, et al. , Nat. Sustainability, 2021, 4, 509–515 CrossRef.
- X. Chen, Y. Li and X. Pan, et al. , Nat. Commun., 2016, 7, 12273 CrossRef CAS PubMed.
- X. Wei, J. Zhang and L. Wang, et al. , Chem. Eng. J., 2024, 482, 149114 CrossRef CAS.
- J. Zhang, X. Wei and J. Zhao, et al. , Chem. Eng. J., 2023, 454, 140081 CrossRef CAS.
- Q. Li, Y. Ouyang and H. Li, et al. , Angew. Chem., Int. Ed., 2022, 61, e202108069 CrossRef CAS PubMed.
- V. I. Merupo, S. Velumani and M. Bizarro, et al., Electrical Engineering, Computing Science and Automatic Control (CCE), 12th International Conference, IEEE, Mexico City, Mexico, 2015.
- L. Zhang, D. Chen and X. Jiao, J. Phys. Chem. B, 2006, 110, 2668–2673 CrossRef CAS PubMed.
- C. Regmi, Y. K. Kshetri and T.-H. Kim, et al. , Mol. Catal., 2017, 432, 220–231 CrossRef CAS.
- D. Zhao, W. Zong and Z. Fan, et al. , J. Nanopart. Res., 2017, 19, 124 CrossRef.
- J. Zhang, Y. Lu and L. Ge, et al. , Appl. Catal., B, 2017, 204, 385–393 CrossRef CAS.
- J. Xie, R. Jin and A. Li, et al. , Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
- D. Premachandra and M. D. Heagy, Methane, 2023, 2, 103–112 CrossRef.
- N. Agarwal, Simon J. Freakley and R. U. McVicker, et al. , Science, 2017, 358, 223–227 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00069b |
| This journal is © The Royal Society of Chemistry 2024 |