
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and development of a new flowable and photocurable lactide and caprolactone-based polymer for bone repair and augmentation†
A. S.
Hamidi
a,
M. A.
Hadis
a,
R. L.
Williams
b,
L. M.
Grover
 b and
W. M.
Palin
*a
b and
W. M.
Palin
*a
aDental and Biomaterials Science, School of Dentistry, College of Medical and Dental Science, Institute of Clinical Sciences, University of Birmingham, 5 Mill Pool Way, Edgbaston, Birmingham, B5 7EG, UK. E-mail: w.m.palin@bham.ac.uk; Tel: +44 0121 466 55 47
bHealthcare Technologies Institute (HTI), School of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 29th February 2024
Abstract
With a global aging population, there is a high demand for new biomaterials that provide regenerative or fixation modalities following a bone injury. Here, the design and development of newly synthesised poly(L-lactic acid)-dimethacrylate (PLLA-DM) and poly(caprolactone-co-fumarate)-dimethacrylate (PCF-DM) monomer systems serves to address some of the main medical challenges and requirements of surgeons during application and better postoperative outcomes of new bone-healing biomaterials. Synthesis of PLLA-DM and PCF-DM via ring opening polymerisation (ROP) and polycondensation routes led to low MW ‘flowable’ and resorbable monomers that polymerise in-situ at up to 6 mm curing depth. Tensile testing of photocured PLLA-DM/PCF-DM formulations at strain rate 0.05 s−1, revealed elastic moduli of 4.4 ± 0.5 to 11.7 ± 2.5 (SD) GPa, with ultimate tensile strength ranging between 29.7 ± 4.9 to 76.1 ± 13.5 (SD) MPa. Resazurin-based metabolic activity studies via an indirect contact method involving Saos-2 osteoblast-like cell lines revealed enhanced cytocompatibility with metabolic activity of treated Saos-2 cells increasing by up to 20% compared with respective untreated control groups. Attachment of Saos-2 cells on PLLA-DM/PCF-DM specimen surfaces revealed cellular structures such as filopodia extending beyond lamellipodia, indicative of remarkable cell adhesion and favouring colonization. The initial development of the polymer chemistry presented here provides the potential for the design and further development towards a new resorbable biomaterial with enhanced mechanical properties for bone repair and augmentation involving both orthopaedic (bone cement) and restorative dentistry applications.
1. Introduction
Bone is the second most transplanted tissue following blood transfusion.1 With a growing geriatric population worldwide and the rise of traffic and sports-related injuries, there exists significant demand for advanced and innovative osteo-biologic biomaterials and/or fixation modalities for bone injury. In the USA alone, over 6 million bone fractures are reported annually, with a further 2.2 million bone grafting surgeries for treatment of bone-related diseases such as osteoarthritis and osteoporosis.2,3Polymers such as polyacrylic acid (PLA) and cyanoacrylates account for the majority of resin-based bone augmentation materials currently available commercially.4,5 COPAL®, OSTEOPAL®, PALACOS®, Simplex P®, Spineplex® and IlluminOss are a collection of polymethyl-methacrylate (PMMA)-based chemically curing bone cements that are currently being marketed for bone augmentation procedures ranging from femural neck fracture (arthroplasty) to vertebroplasty.6–11 Other bone cements include CortOss® and CompO6™ composites containing silica, calcium phosphates and a mixture of dimethacrylate-based polymer matrices.6,12–14 However, there exists several concerns involving suitability and biological safety of these materials. This includes poor biomechanical suitability and cytotoxicity caused by unreacted monomers and other additives leaching out whilst in service. In addition to potential damage to the local tissue caused by the high thermal exposure that occur during mixing and high polymerisation exotherms.11,15–20
Ideally, bone augmentation biomaterials are required to aid repair and promote bone regeneration in conjunction with providing adequate mechanical support and stability at the defected/damaged site, following implantation. Equally, the implanted (fixation) device ought to degrade during the healing process into non-cytotoxic products that can be either metabolized (resorbed) or excreted directly from the body without the further need for surgical intervention. For clinical application, the material should exhibit appropriate mouldability, flow characteristics and fast setting properties to quickly conform to the defective site to provide adequate bone-implant adhesion, and avoid implant loosening or premature failure.21 Over the last ∼20 years, numerous efforts have been made to develop photocurable systems using biocompatible synthetic polymers for biomedical and bone tissue engineering applications.22,23 However, much of the research have been focused on the design and prefabrication of (porous) scaffold structures and composites using thermal or chemical polymerisation initiators in conjunction with crosslinking agents such as fumarate and hydroxyethyl methacrylate (HEMA). However, relatively slow polymerisation reactions and the presence of non-degradable diluents have led to these materials being unsuitable in the clinical context.24–33 The development of photopolymerisable systems allows single paste formulation, and spatial and temporal control of the placement and setting reaction.
PLA is an aliphatic biocompatible and biodegradable polyester with high stiffness Tg of 40–70 °C34–36 with an elastic modulus and tensile strength ranging from 1.4–9.2 GPa and 27.6–82.7 MPa, respectively.34,37 PLA has been extensively studied for applications such as drug delivery and biodegradable implants.23,38,39 Polycaprolactone (PCL) is an aliphatic semi-crystalline biodegradable polyester with a Tg of −60 °C34 and flexible with rubber-like properties at physiological temperatures (37 °C), thus providing high fracture toughness and elastic modulus that is several orders of magnitude smaller when compared with PLA.40 Depending on MW, crystallinity and network structure of the starting polymer, degradation may take up to 2–3 years in vivo due to its hydrophobicity.41,42 The use of PCL in biomedical applications is increasing due to its versatility within copolymer formulations, tuneability in terms of crystallinity, rates of degradation and clinical approval for human use by regulatory authorities.23,38
Ho & Young, developed photopolymerisable and biodegradable tri-block monomers involving polypropylene glycol (PPG), lactide and glycolide units at varying compositions with degree of conversion (DC (%)), reportedly more than 90% depending on the length and ratio of lactide to PPG in monomer chain.43 Subsequent studies involving various bioceramics were conducted to assess cytotoxicity, degradation, in-vivo suitability, and mechanical properties of both the polymer and derived composites. It was concluded that the new polymer-based material could potentially be used to provide immediate early fixation for relatively minor low-load bearing bone fractures, where limited flexural movement would be anticipated.44–50 However, commercial and clinical aspect of the polymer system are yet to be elucidated.
Similarly, Jabari et al., developed caprolactone fumarate-based polymer systems for bone and tissue engineering applications. Subsequent studies including, the modification of monomers structures, were carried out to investigate cytotoxicity, degradation, polymerisation and mechanical properties. Eventually, polymer system was put forward for nerve conduit development, due to its high flexibility for stretching.26,27,51 In another study, Lipik et al., developed an array of PCL-co-PLA thermoplastic elastomers to investigate mechanical, degradation and elastomeric characteristics. The authors concluded the biodegradable elastomers to be better suited for stent applications.52 The aim of this study was to design and formulate a new block copolymer biomaterial using PLA and PCL that would ultimately facilitate tissue regeneration and allow natural bone development following a traumatic injury or irreparable bone defect. In addition, the new ‘tuneable’ polymer system is proposed to act as a standalone bone fixation device with sufficient mechanical stability and structural integrity during the early stages of the healing process. Furthermore, the new polymer design is aimed to rapidly photopolymerise in situ, using conventional photochemistry and light curing technology, to meet key material handling requirements such as extended working time and significantly faster polymerisation.
2. Materials and methods
Unless specified otherwise, consumables were purchased form Sigma Aldrich, Gillingham, UK.2.1 Synthesis of monomers
Poly(L-lactic acid)-dimethacrylate (PLLA-DM) and poly(caprolactone-co-fumarate)-dimethacrylate (PCF-DM) monomers were provided by Specific Polymers (Montpelier, France), and used as received.For methacrylation, PLLA-OH was evaporated twice with tetrahydrofuran (THF) to remove all traces of water, then dissolving triethylamine (Et3N) in dry THF. The mixture was cooled to 0 °C, before adding distilled methacryloyl chloride (CH3–C2HOCl) dropwise and leaving it stirring at room temperature for several hours. 1H NMR was used to evaluate the extent of functionalisation reaction. Et3N salt was removed by filtration and THF was evaporated leaving behind the crude, which was dissolved in DCM and washed once with HCl solution and deionised water. The organic layer was then dried with Na2SO4 and filtered to obtain PLLA-DM as a clear viscous gel.
Methacrylation was achieved by co-evaporating PCF-OH with THF to remove any traces of water. PCF-OH was then dissolved in dry DCM before adding Et3N. The mixture was cooled to 0 °C before the dropwise addition of freshly distilled CH3–C2HOCl. The mixture was left stirring at room temperature for 3 to 5 hours. The extent of reaction was evaluated by 1H NMR. The organic layer was washed with HCl solution and deionized water, before drying with Na2SO4. After evaporating solvents under reduced pressure relative to the atmosphere, PCF-DM was obtained as a viscous oily fluid.
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n) and weight-average (w) molecular weights of both PLLA-DM and PCF-DM were determined using Varian PL-GPC-50 size exclusion chromatographer equipped with a polypore column and a refractive index (RI) detector. Polystyrene and THF were used as a standard and the mobile phase, respectively.
n) and weight-average (w) molecular weights of both PLLA-DM and PCF-DM were determined using Varian PL-GPC-50 size exclusion chromatographer equipped with a polypore column and a refractive index (RI) detector. Polystyrene and THF were used as a standard and the mobile phase, respectively.
2.2 Preparation of photocurable formulations
Monomer formulations were prepared by mixing PCF-DM into PLLA-DM under gentle heating (55 to 60 °C) until an even blend was achieved involving different compositions of PLLA-DM/PCF-DM by weight (Table 1). A co-monomer mixture of bisphenol-A-glycidyldimethacrylate (bisGMA), and triethylene glycol dimethacrylate (TEGDMA) at 80/20 wt/wt%, was used as a comparison example in the present study.| Formulations | PLLA-DM/PCF-DM (wt/wt%) | bisGMA/TEDGDMA(wt/wt%) |
|---|---|---|
| F1 | 50/50 | |
| F2 | 60/40 | |
| F3 | 70/30 | |
| F4 | 80/20 | |
| F5 | 90/10 | |
| BT2 | 80/20 |
To allow light-induced polymerisation, monomer formulations were incorporated with 0.4 wt% camphorquinone (CQ) λmax: 469 nm, Molar Absorptivity: 42 ± 2 cm−1/(mol L−1) (Fig. 1(a); ESI,† S1B), and 0.80 wt% dimethylaminoethyl methacrylate (DMAEMA) as the photosensitiser (PI) and co-initiator (Co-I), respectively.
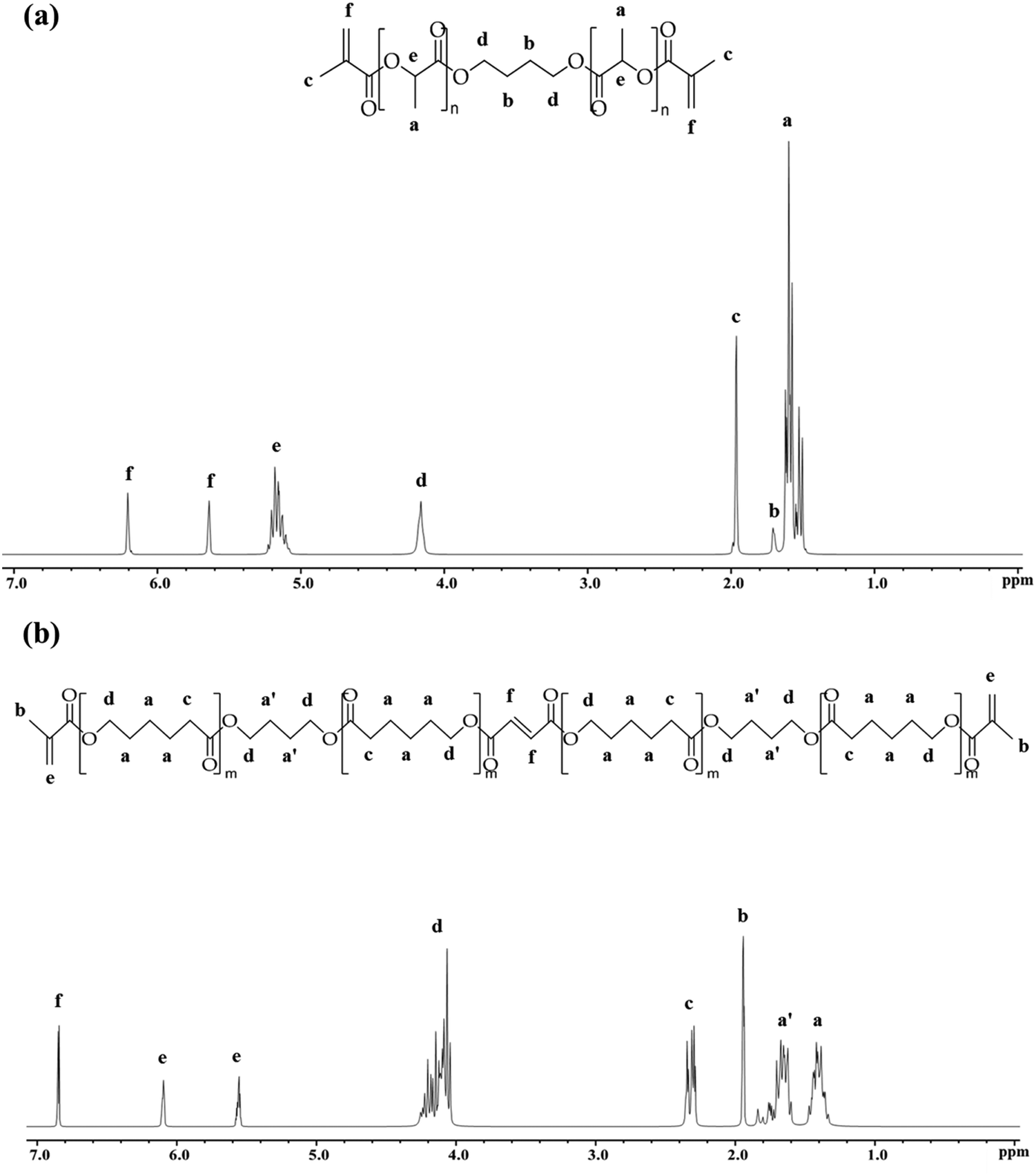 | ||
| Fig. 1 Characterisation of (a) PLLA-DM and, (b) PCF-DM using 1H NMR (300 MHz, chloroform-d (CDCl3)). | ||
2.3 Light sources and photopolymerisation
Samples were photopolymerised using an AURA light engine® (Lumencor®, Beaverton, USA), equipped with a liquid light guide (active tip diameter, 6 mm) which produced a homogeneous beam profile (λmax = 469 ± 2 nm (SD)). The light engine (source) was calibrated to deliver an absolute irradiance of 1012 ± 0.20 mW cm−2 from 7 mm distance. The absolute irradiance was determined, by integrating area under the curve, and was used throughout the study for sample preparations and measuring real-time polymerisation. For tensile testing, specimens were prepared using ESPE Visio Beta Vario (3M, Bracknell, UK) light curing oven, equipped with four fluorescent tubes, to deliver a total absolute irradiance of 20.23 ± 0.90 mW cm−2 (350 to 615 nm). Due to relatively low irradiance of the curing oven, specimens were irradiated for an equivalent radiant exposure used to photocure with AURA to ensure equivalent radiant exposure delivery. Absolute spectral irradiances of AURA and ESPE Vario, are given in suplementary file (Fig. 1 and ESI,† S1B).Real-time photopolymerisation reaction of monomer formulations were measured using Fourier transformed Infra-red spectroscopy (FTIRS) (Nicolet 6700 FT-IR; Thermo Scientific, Hemel Hampstead, UK), equipped with an attenuated total reflectance (ATR) detector consisted of a horizontal multiple-reflection diamond crystal and an internal built-in 45° mirror angle. Temperature of the ATR platform was controlled and adjusted accordingly to the required operating temperature (25/37 ± 0.5 °C), using a Peltier plate temperature regulator (Pike Technologies, Fitchburg, USA).
Dynamic measurements of degree of conversion (DC %) (n = 3) of monomer formulations were assessed at various curing depths using Teflon moulds (inner diameter: 10 mm; thickness: 0.5, 3 and 6 mm). The upper surfaces of samples were covered with an acetate film to minimise oxygen interference on free-radicals and irradiated at 7 mm distance (AURA light engine; 1012 ± 0.2 mW cm−2).
Real-time DC (%) was calculated by monitoring the height of the aliphatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration (1637 cm−1) peak and bending mode of methylene group(s) (CH2)58 (1453 cm−1), as the isosbestic reference for synthesised resins. For BT2, the aromatic CC stretching (1608 cm−1) was used as the isosbestic ref. 59 (eqn (1)).
C stretching vibration (1637 cm−1) peak and bending mode of methylene group(s) (CH2)58 (1453 cm−1), as the isosbestic reference for synthesised resins. For BT2, the aromatic CC stretching (1608 cm−1) was used as the isosbestic ref. 59 (eqn (1)).
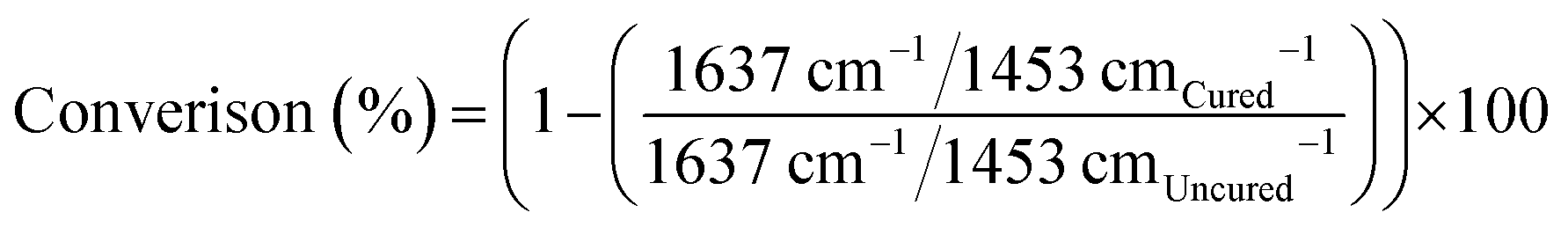 | (1) |
2.4 Tensile testing
Tensile testing was performed using Zwick/Roell Z030 mechanical tester (Hertfordshire, UK), equipped with a 50 kN load Cell. Tests were conducted until failure using strain rate of 0.05 s−1, which corresponds to the maximum strain rate during sprinting and/or downhill running.60 Specimens were prepared using a split mould (ASTM (D638), Type V) with length 31.75 mm, gauge length: 3.81 mm, width: 1.65 mm. Unpolymerised formulations were pipetted into the mould and covered with an acetate film during photopolymerisation. To circumvent overlapping caused during spot curing, specimens (n = 5) were prepared by photocuring formulations using the ESPE Visio Beta Vario light curing oven. Elastic moduli of photocured specimens were determined via a line of regression between two distinct points in the proportional elastic region. Tensile yield strengths were measured at a tolerance point of 0.2% strain from elastic moduli.2.5 Cellular attachment and cytocompatibility studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and incubated at 37 °C under 5% CO2 humidified atmosphere for 24 h. Subsequently, old media was aspirated and wells were washed three times with DPBS before treating cells with 50 μL of aged (24 h/7 d) media extracts per well. An additional 50 μL of freshly supplemented DMEM was added to give a total volume of 100 μL (media extract to fresh DMEM ratio = 1:1), per well. Cells were incubated for further 24 h under standard conditions before performing the assay, which involved removing the old media and washing the wells with DPBS prior to supplementing with DMEM containing 10% working solution (0.15 mg mL−1 in DPBS) of AB dye. Cells were then incubated for 4 h before measuring absorbance at 570 nm (Sparks Plate Reader, Tecan Group Ltd, Switzerland). Absorbance measurements at 600 nm were used as reference and reductions of AB into resorufin were calculated as percentage differences between treated and untreated cells (control groups).65 Cytocompatibility studies were performed in triplicates with two biological repeats (2 different cell passages).
000 cells per well and incubated at 37 °C under 5% CO2 humidified atmosphere for 24 h. Subsequently, old media was aspirated and wells were washed three times with DPBS before treating cells with 50 μL of aged (24 h/7 d) media extracts per well. An additional 50 μL of freshly supplemented DMEM was added to give a total volume of 100 μL (media extract to fresh DMEM ratio = 1:1), per well. Cells were incubated for further 24 h under standard conditions before performing the assay, which involved removing the old media and washing the wells with DPBS prior to supplementing with DMEM containing 10% working solution (0.15 mg mL−1 in DPBS) of AB dye. Cells were then incubated for 4 h before measuring absorbance at 570 nm (Sparks Plate Reader, Tecan Group Ltd, Switzerland). Absorbance measurements at 600 nm were used as reference and reductions of AB into resorufin were calculated as percentage differences between treated and untreated cells (control groups).65 Cytocompatibility studies were performed in triplicates with two biological repeats (2 different cell passages).
Once fixed and dried, specimens were carefully removed and mounted on aluminium stubs covered with adhesive carbon tabs (diameter: 12 mm) (Agar Scientific, UK) and copper taped at one side. Specimens were sputter coated (Emitech K550X, Quorum Technologies, UK) with two 15 nm gold layers, before mounting them onto the EVO MA10 SEM chamber (Carl Zeiss AG, Germany). Cell attachment images were captured using secondary electrons with electron beam operating at 20 kV under high vacuum.
Finally, separate one-way ANOVAs and post-hoc Bonferroni multiple comparison tests were carried out to establish statistical significances (P < 0.05) in AB reduction (%) by Saos-2 cells between groups, following treatment with aged media extracts (24 h/7 d).
3. Results
3.1 Synthesis and characterisation of PLLA-DM and PCF-DM
Fig. 1(a) depicts the 1H NMR spectrum, with Table 2 summarising the MW of PLLA-DM and PCF-DM ascertained by NMR and GPC (Chromatograms are given in ESI,† S1B). Chemical shifts (δ) at 1.69 and 4.14 ppm correspond to the inner and outer protons of methylene segment, 1,4-di(λ-oxidaneyl) (O–C4H8–O), respectively.67 Equally, chemical shifts at 1.57 and 5.15 ppm correspond to the methyl and methine protons of lactyl repeating units, respectively.54 Resonances at 5.62 and 6.18 ppm, can be assigned to alkene (CC) protons of the two terminus (methacrylate) groups. Finally, the chemical shift at 1.94 ppm corresponds to methyl (CH3) protons present in methacrylate moieties.68Mn,NMR corresponds to monomer number-averaged MW determined via1H NMR end-group analysis method,57 and given in Table 2. Based on Mn,NMR, the average ratio of lactyl with respect to BDO unit is 2n:1, where n ranges between 4–5.
| Monomer | M n,NMR |
n
|
w
|
PDI |
|---|---|---|---|---|
| PLLA-DM | 933 ± 16.52 | 1099 ± 75.34 | 1392 ± 91.94 | 1.3 ± 0.04 |
| PCF-DM | 1017 ± 24.43 | 1581 ± 179.57 | 2646 ± 492.24 | 1.7 ± 0.12 |
Fig. 1(b) shows the chemical structure of co-polymer PCF-DM, characterised by 1H NMR. Chemical shifts (δ) at 1.38 and 1.65 (1.86–1.53) ppm correspond to the inner protons of methylene segments of PCL and O–C4H8–O, respectively.26,67 Chemical shift at 1.91 ppm represents 6 protons of the two methyl groups in methacrylate moieties. Similarly, chemical shift at 2.29 ppm corresponds to magnetically equivalent protons adjacent to CO group in PCL units.26 Resonance at 4.11 (4.31–3.96) ppm can be assigned to equivalent outer protons of O–C4H8–O and PCL units.26,51 Equally, chemical shifts at 6.06 and 5.52 ppm correspond to the alkene protons of methacrylate-end groups. Finally, chemical shift at 6.81 ppm represents the two protons of carbon double bond of the fumarate unit. The ratio of butanediol (BDO) with respect to PCL units in a typical PCF-DM chain was 1:2. Equally, 1H NMR revealed the presence of ∼1 fumarate and 4 caprolactone units (1:4 ratio), per monomer chain.
3.2 Photopolymerisation of formulations
Fig. 2 depicts photopolymerisation conversion profiles at 25/37 °C and various curing depths including ‘dark’ curing, where polymerisation continued even after light source (AURA) was turned ‘-off-’ at 120 seconds. Two-way ANOVA revealed significant differences (P < 0.05) in DC120s (%) as a function of curing depth and operating temperature. Subsequent post-hoc Bonferroni comparison tests also revealed significant differences (P < 0.05) in DC120s values following an increase in temperature and curing depth. For instance, DC120s of F4 increased by 8% when curing depth was increased from 0.5 to 3 at 25 °C. This was further augmented by almost 10%, when the operating temperature was raised to 37 °C (Fig. 2).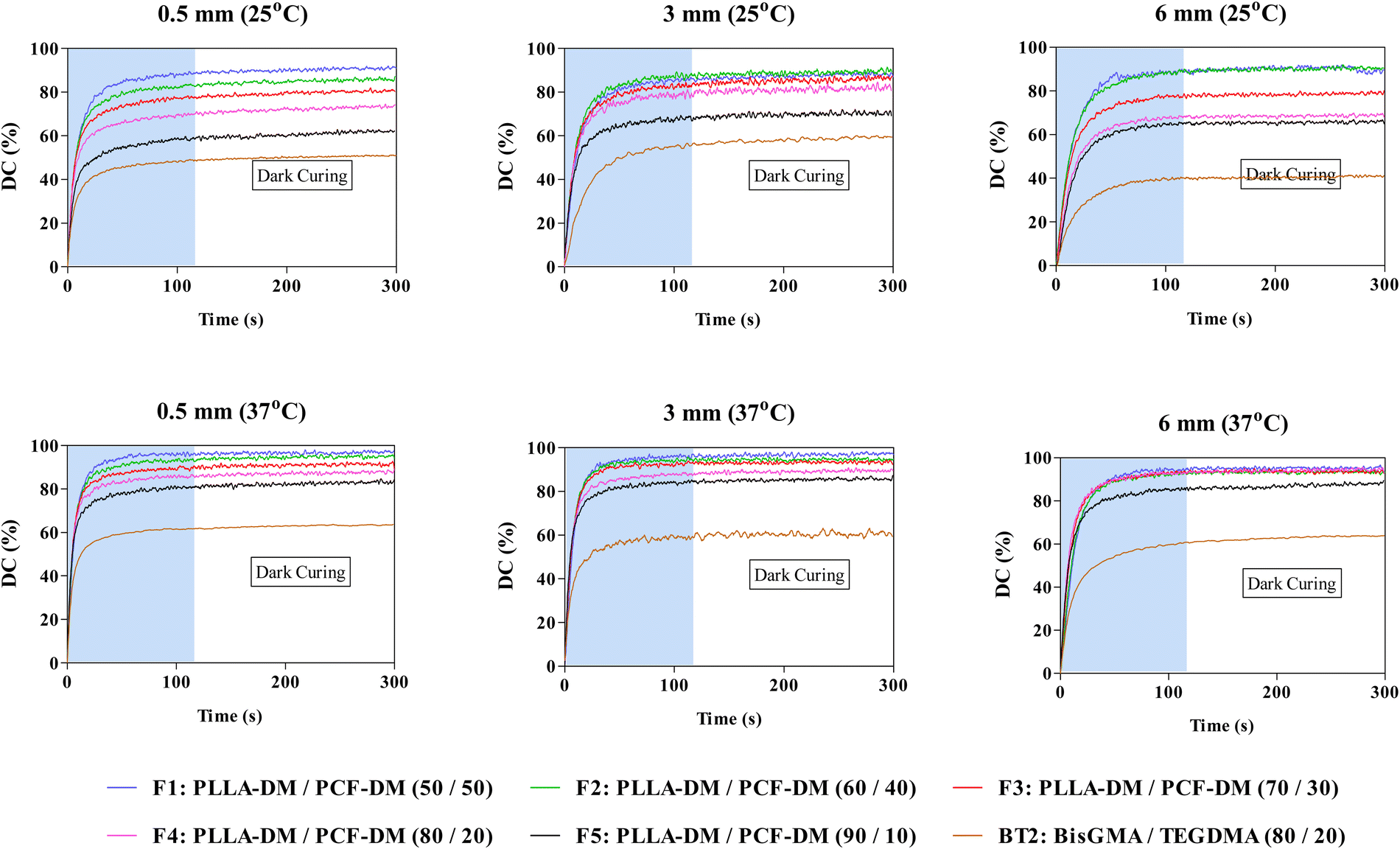 | ||
| Fig. 2 Photopolymerisation of monomer formulations (F1–F5 (wt/wt%)) at different curing depths and operating (environmental) temperatures (25 and 37 °C). Blue shaded areas represent photocuring period (120 s), followed by post-irradiation period for 180 s (dark curing). | ||
Generally, the extent of polymerisation conversion, during the 120 s photocuring period (DC120s (%)), began to decrease with subsequent increase in PLLA-DM weight fraction from 50% in F1 to 90% in F5. However, when compared at different curing depths DC120s (%) generally decreased at 0.5 mm curing depth before increasing at 3 and 6 mm curing depths with F1 being the exception whereby, DC120s (%) decreased from ∼90% at 0.5 mm to 85% at 3 mm curing depth (P < 0.05), under 25 °C operating temperature (Fig. 2). Noteworthy, increase in DC120s (%) at 6 mm curing depth was observed for formulations with initial low viscosities only (F1 to F3, Fig. 4 and ESI,† S1B), as shown in Fig. 2. This trend continued into the dark curing stage (180 s post-irradiation) with a further conversion of up to 5% observed for some formulations. BT2 exhibited similar behaviour except for polymerisation extent involving DC120s (%) and dark curing at various curing depths where, increases were relatively marginal when compared with PLLA-DM/PCF-DM formulations (post-hoc Bonferroni, P < 0.05).
3.3 Tensile testing of photocured formulations
Fig. 3 depicts the three parameters for all formulations under tensile testing. The amount of applied stress increased progressively with E-modulus increasing significantly from 4.4 ± 0.5 (SD) for F1, to 11.7 ± 2.5 (SD) GPa for F4 (P < 0.05; Fig. 3(a)). F4 and F5 formulations exhibited the highest ultimate tensile strengths of 73.1 ± 4.4 (SD) and 76.1 ± 13.5 (SD) MPa, both of which were significantly higher (P < 0.05), when compared with F1 (Fig. 3(c)).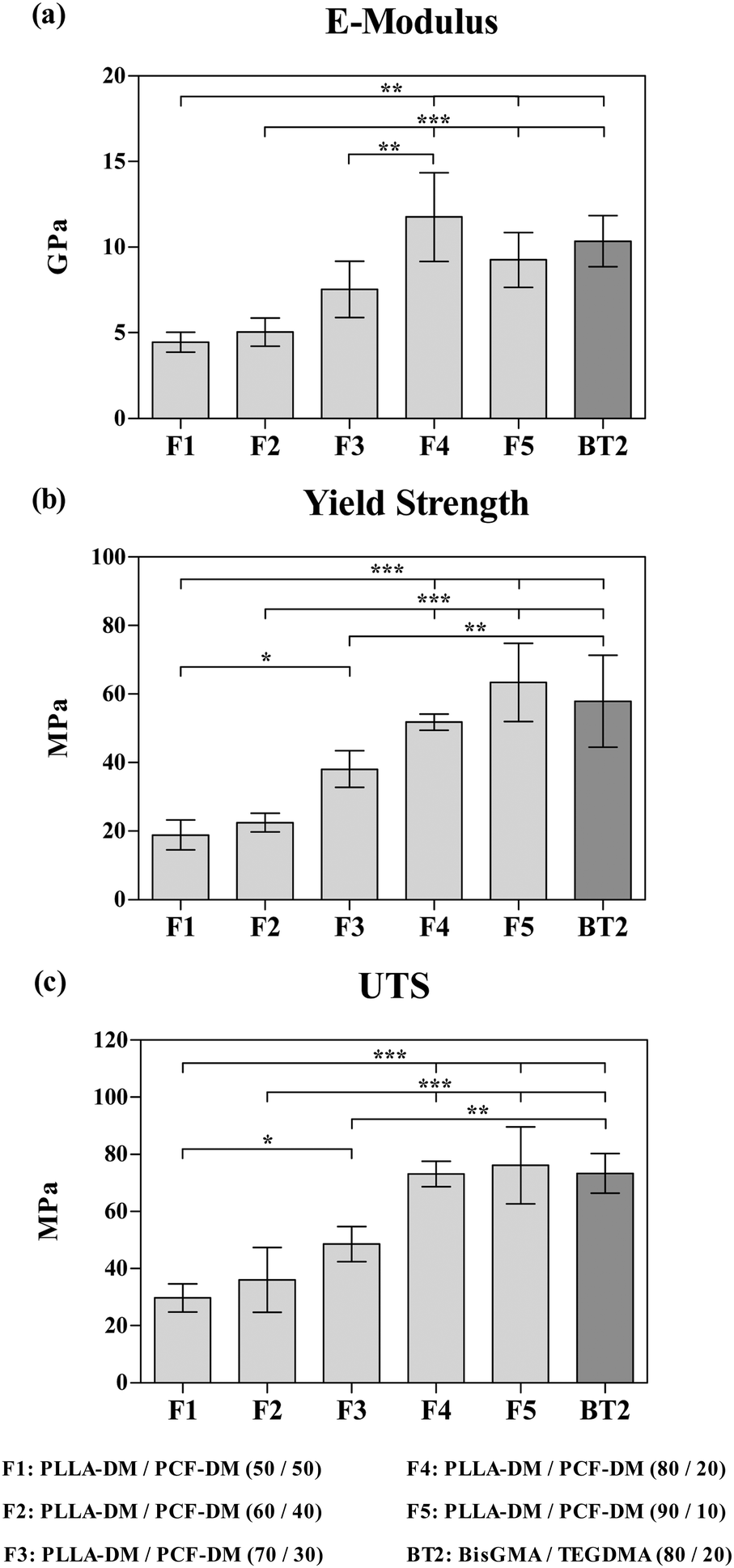 | ||
| Fig. 3 Mechanical properties of photocured formulations (F1–F5 (wt/wt%)), including BT2 under tensile testing (ASTM (D638) Type V) at 0.05 s−1 strain rate. UTS represents ultimate tensile strength (MPa). Columns and error bars represent mean and standard deviation, respectively. Asterisks (*p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001) indicate statistically significant differences between groups, based on post-hoc Bonferroni multiple comparison tests following one-way ANOVA. | ||
3.4 In vitro cytocompatibility of Saos-2 cells
Fig. 4 depicts reduction in AB (%) as a function of metabolic activity of Saos-2 cells, following treatment with media extracts of neat monomers and their corresponding formulations. One-way ANOVA revealed statistically significant (P < 0.05) variation in post-treatment metabolic activities of Saos2 cells. Generally, treatment with neat PLLA-DM, PCF-DM, F4 media extracts exhibited improved cytocompatibility with relative metabolic activity appeared to have increased following treatment with both media extract formats (24 h and 7 d). Exposure to 24 h media extracts (Fig. 4(a)) at 1:1 ratio (v/v) to supplemented DMEM, led to an increase of 8.11, 14.93 and 10.14% in resorufin production when compared with Control group, respectively. Equally, treatment with 7 d media extracts (Fig. 4(b)) led to an increase of 10.86, 19.86 and 9.94% in resorufin production, when compared with corresponding control group, respectively. Post-hoc Bonferroni multiple comparison test following one-way ANOVA revealed that increase in metabolic activity of Saos-2 cells following treatment with the 7 d PCF-DM media extract was significant (P < 0.05), when compared with the respective control group. The highest relative reduction in metabolic activity was registered for Saos-2 cells treated with TEGDMA extracts. Treatment with 24 h and 7 d media extracts led to resorufin production reducing to 66.92 and 33.59% (P < 0.05), when compared with corresponding control groups, respectively (Dashed lines; Fig. 4). Cytotoxic effects of TEGDMA can be further realised on Saos-2 cells, treated with media extracts of uncured BT2 following 7 d media extract (P < 0.05; Fig. 4(b)). In contrast, Saos-2 cells exhibited relatively less cytotoxicity when treated with bisGMA extracts. Relative resorufin production reduced to 93.87 and 94.01%, when treated with respective 24 h and 7 d old media extracts of bisGMA. Both treatment outcomes were not statistically significant (P > 0.05), when compared with respective control groups (Fig. 4).
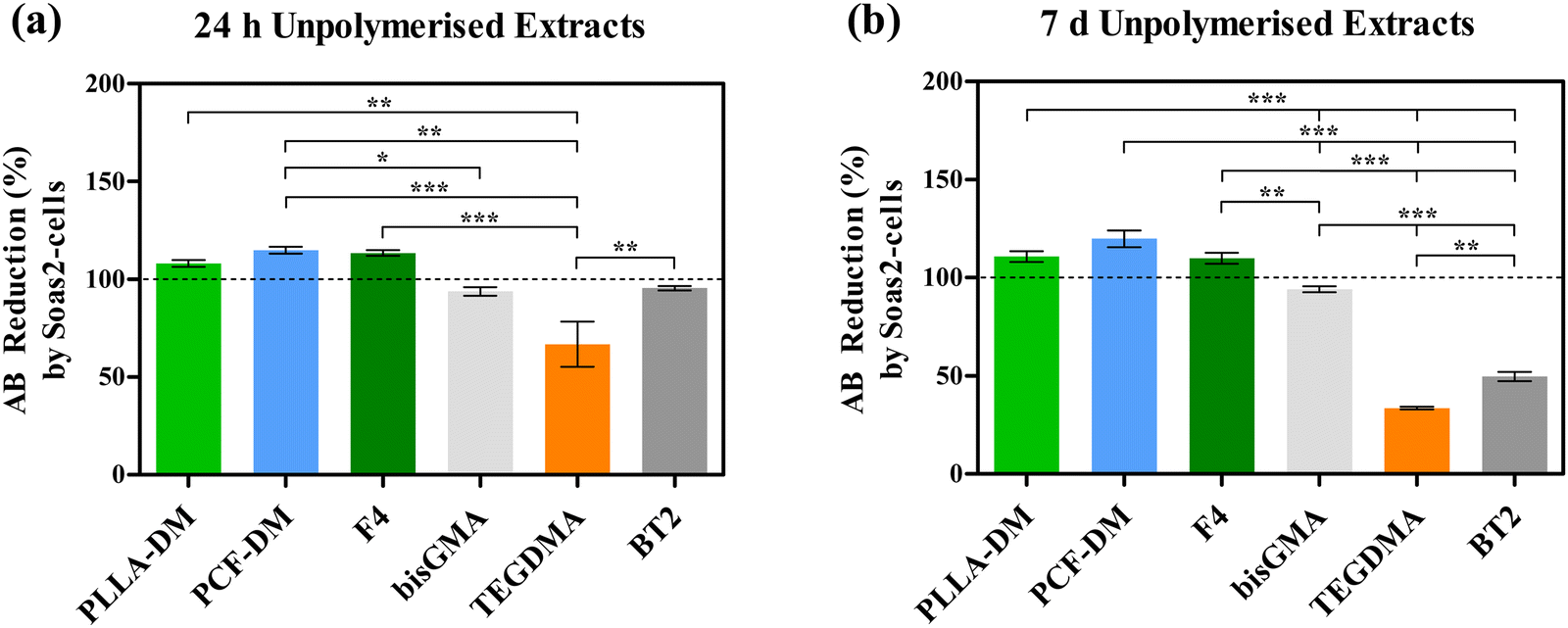 | ||
| Fig. 4 Reduction of AB (Resazurin) into resorufin (%) by Saos-2 cells treated with 24 h (a) and 7 d extracts (b) of unpolymerised (neat) monomers and their corresponding formulations, in supplemented DMEM. Dashed lines represent untreated groups (control). Columns and error bars represent mean and standard deviation, respectively. Asterisks (*p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001) indicate statistically significant differences between groups, based on post-hoc Bonferroni multiple comparison tests following one-way ANOVA. | ||
Fig. 5 depicts AB reduction of resazurin (%) as a function of Saos-2 cells metabolic activity, following exposure to photocured F4 media extracts (24 h and 7 d), under standard incubating conditions. One-way ANOVA did not reveal statistically significant (P > 0.05) variation in post-treatment metabolic activities of Saos-2 cells. Nonetheless, treatment with both photocured F4 media extracts (24 h/7 d) appeared to have improved cytocompatibility, due to cellular activities mentioned above, when compared with respective control groups. On average, resorufin production (%) increased by 15.65% and 19.10%, following treatment with 24 h (Fig. 5(a)) and 7 d (Fig. 5(b)) media extracts when compared with corresponding control groups, respectively.
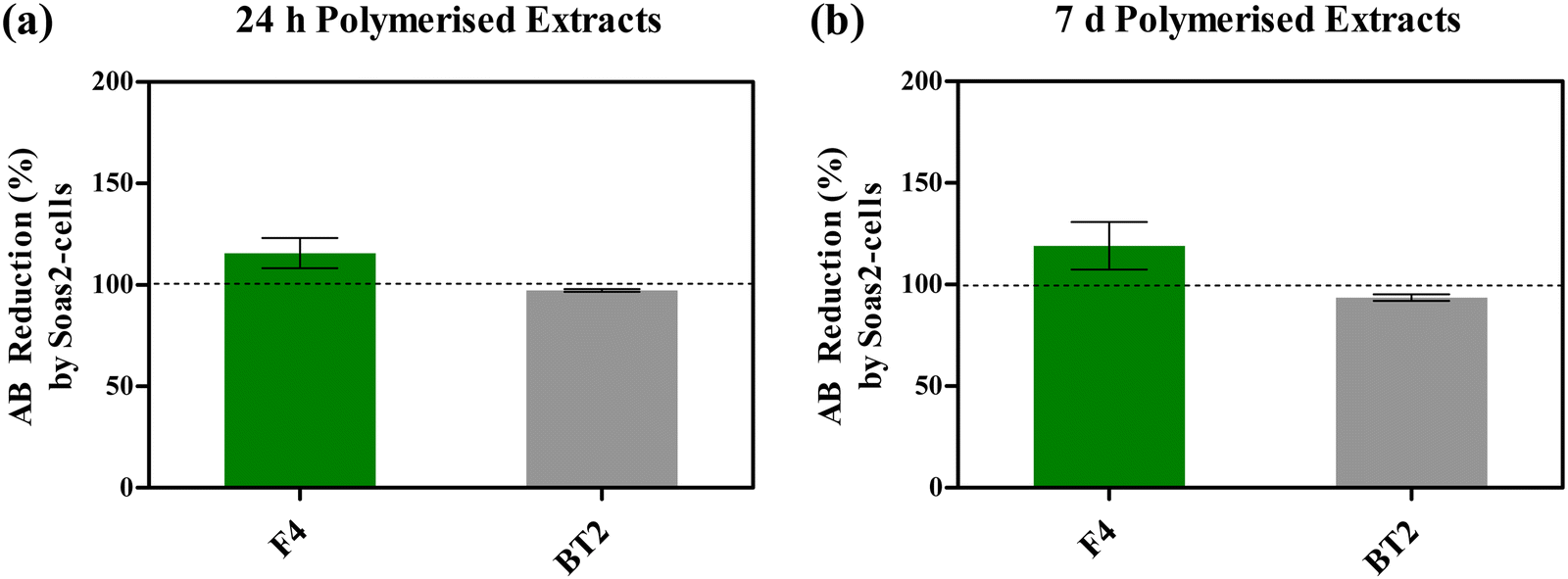 | ||
| Fig. 5 Reduction of AB into resorufin (%) by Saos-2 cells treated with 24 h (a) and 7 d extracts (b) of photo-cured formulations, in supplemented DMEM. Dashed lines represent untreated groups (control). Columns and error bars represent mean and standard deviation, respectively. | ||
On the other hand, Saos-2 cells exhibited slightly lower cytocompatibility when exposed to photocured BT2 media extracts, with relative resorufin production of 97.26% and 93.63% following treatment with 24 h and 7 d media extracts (Fig. 5).
3.5 Saos-2 cells attachment
Fig. 6 depicts SEM images of Saos-2 cells following 24 h incubation on photocured disc surfaces of F4 and BT2 systems. Thermanox® coverslips were used as positive controls. Generally, cells appear to have attached with polygonal morphology generally associated with Saos-2 cell lines.69 On both Thermanox® (Fig. 6(a)) and BT2 surfaces (Fig. 6(b)), cell attachments have taken place in small isolated clusters with most cells appearing round, and typically associated with initial phases of substrate adhesion.66,70 The most extensive cell adhesion and proliferation of Saos-2 cells can be visualised on F4 specimens (Fig. 6(c)), where under the same incubating conditions substrate surfaces have been completely covered with cells of pronounced surface anchorages. Significant increases in adhesive structures such as cytoplasmic projections (filopodia) extending beyond lamellipodia can be clearly observed.47,66 | ||
| Fig. 6 SEM images of Saos-2 cells attachment on (a) a Thermanox coverslip, (b) bisGMA/TEGDMA (BT2), and (c) the PLLA/PCF-DM (F4) surface, following 48 h incubation in supplemented media. | ||
4. Discussion
Generally, the characterisation of PLLA-DM and PCF-DM monomers via1H NMR and GPC (Fig. 2 and 3; ESI,† S1B) revealed reproducibility in terms of chemical structure, MW and PDI involving different batches. On average, the methacrylate contents of newly synthesised PLLA-DM and PCF-DM monomers were 2.16 and 1.67 meq g−1, respectively. The fumarate group in PCF-DM has been shown to participate in free radical polymerisation and facilitate crosslinking.27,51 Unlike previous findings,27,43,51,52 the new monomers consist of relatively small chains with narrow PDI. As a result, final monomers were viscous fluids at room temperature. This served to aide blending (with complete miscibility), and handling requirements during formulation development. It is also worth noting that, both PLLA-DM and PCF-DM monomers were not incorporated with stabilisers such as hydroquinone (HQ), that are generally included in commercial products to preserve shelf-life and avoid premature polymerisation.71The use of CQ as a photosensitiser was to avoid the safety implications associated with UV irradiation involving type I photosensitisers, especially when extended periods of photocuring are required in a clinical setup. The use of (tertiary) amines as a co-initiator accelerated the reaction rates by utilising α-aminoalkyl radicals produced during the hydrogen abstraction process. These radicals are highly effective and predominantly responsible for initiating polymerisation reactions72,73 compared with the cetyl radical produced by CQ alone. Previous studies have shown that aliphatic amines namely DMAEMA have good-to-comparable polymerisation efficiency with relatively better cytocompatibility than their aromatic counterparts such as EDMAB.74
Relatively lower DC (%) at 6 mm curing depth involving high initial viscosities formulations (F4 and F5, Fig. 4; ESI,† S1B), might be attributed to the mobility of propagating species such that the free radical entrapment may have reached a diffusion-controlled reaction sooner than those with relatively lower initial viscosities (i.e., F1 and F2, Fig. 4; ESI,† S1B). A rapid increase in local viscosity of crosslinking networks may have increased the termination rate constant to supersede propagation, thus causing DC to plateau due to the early onset of vitrification.75,76 Both bisGMA and TEGDMA monomers used in present study had a narrow PDI, with bisGMA containing two aromatic rings per monomer. In contrast, both PLLA-DM and PCF-DM are aliphatic oligomers with an array of smaller and larger chains. Consequently, the general improvement in the degree of conversion of PLLA-DM and PCF-DM formulations may be attributed to the increased diffusion of smaller propagating chains through larger chains within the crosslinking network, which effectively created new growth centres. Equally, the use of BT2 was to serve as a suitable comparison mixture, particularly for F4. Since the use of bisGMA and TEGDMA in orthopaedics (bone augmentation adhesives) and restorative dentistry (resin composite tooth restoratives) are widely documented.4,13,77,78
The lower DC120 (%) observed at 0.5 mm compared with thicker samples was likely a result of different local viscosities caused as a result of heat generated during polymerisation. A larger surface area to volume ratio resulted in a higher rate of heat loss during polymerisation, which had less impact on reducing local viscosity and network formation. The effect of temperature on reaction rates and polymerisation was less noticeable at 6 mm thickness, where differences in the rate of polymerisation within surface layers may have affected both heat and mass transfer rates in deeper layers.
PLA is generally regarded as a brittle material with insufficient fracture toughness (∼5% strain at breaking) for load bearing applications.79,80 Whereas, PCL is flexible with rubber-like properties, and a relatively higher fracture toughness with low strength.81 The intrinsic physical nature of PLLA-DM with higher Tg and stiffness than PCF-DM, serve to explain the relative increase in elastic modulus and overall strength characteristics of formulations as the PCF content was decreased (Fig. 3). During deformation, it is thought that stress transfer between PLLA and PCF component within the polymer matrix, was mediated through chemical (covalent) bonding formed during photo-crosslinking, which may have been reinforced by the vinyl double bond of fumarate participating in free-radical polymerisation.27,51 However, this toughening effect induced by PCL segments may have been reduced below 20% weight fraction, as characterised by somewhat brittle fracture observed for F5 specimens. Chain mobility and relaxation were likely to be more associated with higher PLLA segments which exhibited less deformation per given stress unit (MPa) than PCL, thus leading to decline in elastic modulus and tensile strength (Fig. 3(a)). In agreement with previous studies, Zhao et al.,80 and Douglas et al.,81 reported tensile strengths of ∼50 and 43 MPa for extruded solids consisting of 90/10 and 80/20 wt% PLA/PCL, respectively. Similarly, Malinowski,82 reported respective tensile and flexural strengths of ∼55 and 80 MPa, for an injection-moulded PLA/PCL (80/20 wt%) blend, and higher flexural modulus with increasing PLA content.
An array of elastic moduli and yield strengths displayed by F1-5 blends (4.4 ± 0.5 to 11.7 ± 2.5 GPa) in the current study, served to validate the tuneability criterion of the system. The elastic modulus of human cortical bone is around 18 GPa (compression), but varies greatly between 4 to 23 GPa due to factors such as ageing, mineral content, porosity and anatomical location. Likewise, trabecular bone reportedly has an elastic modulus of around 400 MPa in the longitudinal direction, with a load bearing capacity and ultimate tensile strength of around 50 and 8 MPa under compression and tension, respectively.83–85 Consequently, the tuneability and mechanical properties of PLLA/PCF-DM blends presented herein would help to develop polymer systems that avoid stress-shielding effects caused due to inadequate stress transfer loading involving conventional bone repair and restoration interventions.
The current study was designed to assess cytocompatibility of monomers and formulations, both in cured and uncured format under a clinically relevant setting. The use of F4 as the ‘suitable blend’ for biological studies was based on several observations. Firstly, the modulus of elasticity and UTS of F4 were comparable to that of F5 (Fig. 3), without significant effect on polymerisation dynamics and in turn DC (%) Both of which are largely dictated by initial viscosities and subsequent onset of vitrification mentioned above. Secondly, F4 had a relatively better flow behaviour than F5 (Fig. 4; ESI,† S1B). This served to meet key material handling requirements and extended working time prior to setting.
The increased resorufin production by Saos-2 cells following treatment with neat (unpolymerised) PLLA-DM, PCF-DM and F4 might be attributed to cell growth and possibly cell proliferation, discussed below. The microenvironment of normal fracture healing is known to be acidic, due to elevated levels of lactic acid. Such acidic conditions lead to increased osteoblast activities.86 It is proposed that L-lactic acid (lactate) may have been converted to pyruvate by lactate dehydrogenase. Pyruvate is subsequently converted into acetyl CoA by pyruvate dehydrogenase complex, which enters the Krebs cycle.87,88 Benoit et al.,89 tested osteoblast cells encapsulated in a PEG-lactic acid based dimethacrylate hydrogel and reported enhanced metabolic activity, cell proliferation, ALP production and mineralisation as a result of increased release of lactic acid during degradation. Further, ε-caprolactone segments may have been degraded by esterases into hydroxycaproic acid, which are taken up by cells to enter 2-β oxidation pathway to yield 3 acetyl coenzyme A (Acetyl CoA) molecules, which enter the Krebs cycle to subsequently generate ATP.87 Similarly, degraded fumarate (−OOC–C2CH2–COO−) and succinate (−OOC–C2H4–COO−) segments can be metabolised by fumarase and succinate dehydrogenase complex to generate more ATP molecules in the cycle.
In contrast, the cyto- and genotoxicity of Bis-GMA/TEGDMA monomers, on a variety of cell types, have been widely documented.11,20,78,91–93 Beside delaying cell cycles, these monomers also affect cell proliferation and survival.92 Significant reduction in metabolic activity of Saos-2 cells following exposure to TEGDMA media extracts (Fig. 4) might be attributed to its potency towards penetrating cellular membranes and interacting with intracellular molecules and structures. Studies have shown that TEGDMA causes depletion in intracellular glutathione (GSH) antioxidant, thus making cells vulnerable to oxidative damage, cell cycle delays and possibly cell death. Relatively constant metabolic activity of cells treated with neat bisGMA extracts can be assigned to its hydrophobic nature as opposed to TEGDMA, where oxygen atoms present in ethylene glycol segments make TEGDMA molecules more reactive. Regardless, there still remain concerns over DNA damage and the ability of bisGMA to degrade to bisphenol A.
Since PCL and PLA are generally hydrophobic and lack bioactivity in neat forms, it is possible that the presence of hydrophilic segments as such as O—C4H8—O (precursor to succinic acid) and fumarate, may have altered surface free energy and in turn facilitated ECM production to promote cell attachment and spreading involving transmembrane receptor proteins and extracellular matrix (ECM).66 Significantly higher levels of cell attachment observed on F4 surfaces (Fig. 6(c)) might be attributed to local acidity induced by polymer segments, since osteoblast cells favour acidic microenvironments.89 Chemical moieties (e.g., carboxyl groups) present on copolymer surfaces may have served as cell recognition sites to favour higher levels of ECM production. El-Amin et al.,89 reported consistently higher levels of osteoblastic cell adhesion on poly(lactic-co-glycolic acid) (PLGA) matrix surfaces compared with neat PLA and positive control (polystyrene surfaces). Production of ECM components such as collagen, fibrinogen, laminin and vitronectin were also reportedly increased. In particular, osteonectin and osteopontin were found both intra- and extracellularly on the PLGA polymer. The authors further reported a more extensive actin cytoskeletal network of osteoblasts, cultured on copolymer surfaces. Higher osteopontin and collagen type I gene expression by osteoblasts, are also reported by Benoit et al.,89 involving PEG-lactic acid based dimethacrylate hydrogels, and Yin et al.,88 involving dopamine modified PLLA surfaces.
The work presented here has covered the design and development of several formulations, with particular emphasis on F4 PLLA-DM/PCF-DM, (80/20 wt/wt%) composition. The research findings have shown that it is possible to develop a photocurable, flowable polyester-based monomers, using conventional ROP and polycondensation routes, with enhanced mechanical properties and cytocompatibility suitable for bone repair and augmentation involving load bearing applications. From practical standpoint, the new polymer system appears to offer relatively safer alternative to PMMA (bone cement) and resin-matrix systems used to prepare resin-based composites (RBCs) in restorative dentistry. Nonetheless, it is proposed that further physico-chemical and in vivo studies are still needed to truly assess osteogenesis capacity, long-term biocompatibility, degradation, and post-degradation mechanical properties in a more clinically-relevant settings.
5. Conclusion
Formulation development and subsequent characterisation revealed that PLLA-DM/PCF-DM monomer formulations are photocurable up to several millimetres depth using conventional light curing technologies and photoinitiating chemistries. Polymer mixtures can be tailored to achieve required mechanical properties without significant compromise of other physicochemical properties such as DC (%) and unpolymerised flow behaviour. Initial biological studies, in terms of cell attachment and cytocompatibility assay revealed biological interactions with promising signs of material-induced biological development. The present work has provided a solid platform for future studies on these novel monomers towards achieving a workable and potentially functional design for regenerative biomaterial for a given bone application.Data availability
The current research endeavour is subjected to on-going patent applications with priority dated 31st July 2019. All data supporting this study are present within the paper.Conflicts of interest
The authors declare no competing interests.Acknowledgements
AS Hamidi gratefully acknowledges financial support from EPSRC through a studentship from the Physical Sciences for Health Centre for Doctoral Training (EP/L016346/1). The authors further acknowledge Specific Polymers, Montpellier, France for their support in supplying monomer consumables.References
- GrandViewResearch. Orthopedic Implants Market Analysis, By Application (Spinal fusion, Long bone, Foot & Ankle, Craniomaxillofacial, Joint replacement, Dental), And Segment Forecasts To 2024, in Medical Devices. 2016.
- A. Hasan, B. Byambaa, M. Morshed, M. I. Cheikh and R. A. Shakoor, et al., Advances in osteobiologic materials for bone substitutes, J. Tissue Eng. Regener. Med., 2018, 12(6), 1448–1468 CrossRef CAS.
- GrandViewResearch. Bone Grafts And Substitutes Market Size, Share & Trends Analysis Report By Material Type (Allograft, Synthetic), By Application Type (Spinal Fusion, Craniomaxillofacial, Long Bone), By Region, And Segment Forecasts, 2019–2026. 2019. p. 126.
- K. O. Böker, K. Richter, K. Jäckle, S. Taheri and I. Grunwald, et al., Current State of Bone Adhesives-Necessities and Hurdles, Materials, 2019, 12(23), 3975 CrossRef PubMed.
- P. A. Leggat, D. R. Smith and U. Kedjarune, Surgical applications of cyanoacrylate adhesives: A review of toxicity, Aust. N. Z. J. Surg., 2007, 77(4), 209–213 CrossRef.
- S. Gheduzzi, J. Webb and A. Miles, Mechanical characteristics of three percutaneous vertebroplasty biomaterials, J. Mater. Sci.: Mater. Med., 2006, 17, 421–426 CrossRef CAS PubMed.
- Heraeus-Medical. Osteopal Family. Online. 2020. Available from the URL: https://www.heraeus.com/en/hme/products_solutions_heraeus_medical/spinal_augmentation/osteopal_vertebral_compression_fractures.html.
- Heraeus-Medical. Palacos Family. Online. 2020. Available from the URL: https://www.heraeus.com/en/hme/products_solutions_heraeus_medical/arthrosis/palacos_bone_cements.html.
- Heraeus-Medical. Copal G + C – Bone cement with genamicin and clindamycin. Online. 2020. Available from the URL: https://www.heraeus.com/en/hme/products_solutions_heraeus_medical/overview_products_indication/copal_gc_antibiotics.html.
- Stryker. SpinePlex bone cement. Online. 2020. Available from the URL: https://www.stryker.com/us/en/interventional-spine/products/spineplex-cement.html.
- A. L. McSweeney, B. G. Zani, R. Baird, J. R. L. Stanley and A. Hayward, et al., Biocompatibility, bone healing, and safety evaluation in rabbits with an IlluminOss bone stabilization system, J. Orthop. Res., 2017, 35(10), 2181–2190 CrossRef CAS PubMed.
- PRLOG. Swiss Ozics Group Announces CE Mark for the CompO6 (TM) Bone Reinforcement Composite. Ozics Group, 2011. Online. 2020. Available from the URL: https://www.prlog.org/11712867-swiss-ozics-group-announces-ce-mark-for-the-compo6-tm-bone-reinforcement-composite.html.
- Stryker. Costoss bone augmentation material. Online. 2020. Available from the URL: https://www.stryker.com/us/en/interventional-spine/products/cortoss-bone-augmentation-material.html.
- T. Juvonen, J.-P. Nuutinen, A. P. Koistinen, H. Kröger and R. Lappalainen, Biomechanical evaluation of bone screw fixation with a novel bone cement, Biomed. Eng. Online, 2015, 14, 74 CrossRef PubMed.
- R. C. R. Gergely, K. S. Toohey, M. E. Jones, S. R. Small and M. E. Berend, Towards the optimization of the preparation procedures of PMMA bone cement, J. Orthop. Res., 2016, 34(6), 915–923 CrossRef CAS.
- S. Kurata, K. Morishita, T. Kawase and K. Umemoto, Cytotoxic effects of acrylic acid, methacrylic acid, their corresponding saturated carboxylic acids, HEMA, and hydroquinone on fibroblasts derived from human pulp, Dent. Mater. J., 2012, 31(2), 219–225 CrossRef CAS PubMed.
- P. L. Lai, L. H. Chen, W. J. Chen and I. M. Chu, Chemical and physical properties of bone cement for vertebroplasty, Biomed. J., 2013, 36(4), 162–167 CrossRef.
- J. Palussière, J. Berge, A. Gangi, A. Cotten and A. Pasco, et al., Clinical results of an open prospective study of a bis-GMA composite in percutaneous vertebral augmentation, Eur. Spine J., 2005, 14(10), 982–991 CrossRef.
- J. G. Teeguarden and S. Hanson-Drury, A systematic review of Bisphenol A “low dose” studies in the context of human exposure: a case for establishing standards for reporting “low-dose” effects of chemicals, Food Chem. Toxicol., 2013, 62, 935–948 CrossRef CAS.
- S. Krifka, G. Spagnuolo, G. Schmalz and H. Schweikl, A review of adaptive mechanisms in cell responses towards oxidative stress caused by dental resin monomers, Biomaterials, 2013, 34(19), 4555–4563 CrossRef CAS.
- P. H. Long, Medical devices in orthopedic applications, Toxicol. Pathol., 2008, 36(1), 85–91 CrossRef CAS PubMed.
- J. P. Fisher, D. Dean and A. G. Mikos, Photocrosslinking characteristics and mechanical properties of diethyl fumarate/poly(propylene fumarate) biomaterials, Biomaterials, 2002, 23(22), 4333–4343 CrossRef CAS PubMed.
- M. D. Timmer, C. G. Ambrose and A. G. Mikos, In vitro degradation of polymeric networks of poly(propylene fumarate) and the crosslinking macromer poly(propylene fumarate)-diacrylate, Biomaterials, 2003, 24(4), 571–577 CrossRef CAS PubMed.
- E. Jabbari, S. Wang, L. Lu, J. A. Gruetzmacher and S. Ameenuddin, Synthesis, material properties, and biocompatibility of a novel self-cross-linkable poly(caprolactone fumarate) as an injectable tissue engineering scaffold, Biomacromolecules, 2005, 6(5), 2503–2511 CrossRef CAS PubMed.
- S. Wang, L. Lu, J. A. Gruetzmacher, B. L. Currier and M. J. Yaszemski, Synthesis and characterizations of biodegradable and crosslinkable poly(ε-caprolactone fumarate), poly(ethylene glycol fumarate), and their amphiphilic copolymer, Biomaterials, 2006, 27(6), 832–841 CrossRef CAS PubMed.
- K.-W. Lee, S. Wang, M. J. Yaszemski and L. Lu, Physical properties and cellular responses to crosslinkable poly(propylene fumarate)/hydroxyapatite nanocomposites, Biomaterials, 2008, 29(19), 2839–2848 CrossRef CAS PubMed.
- D. L. Alge, J. Bennett, T. Treasure, S. Voytik-Harbin and W. S. Goebel, et al., Poly(propylene fumarate) reinforced dicalcium phosphate dihydrate cement composites for bone tissue engineering, J. Biomed. Mater. Res., Part A, 2012, 100(7), 1792–1802 CrossRef PubMed.
- X. Liu, I. A. L. Miller, B. E. Waletzki, M. J. Yaszemski and L. Lu, Novel biodegradable poly(propylene fumarate)-co-poly(L-lactic acid) porous scaffolds fabricated by phase separation for tissue engineering applications, RSC Adv., 2015, 5(27), 21301–21309 RSC.
- L. Wang, Da-G. Guo, H. Zhu and L. Xie, Light emitting diodes (LEDs) encapsulation of polymer composites based on poly(propylene fumarate) crosslinked with poly(propylene fumarate)-diacrylate, RSC Adv., 2015, 5(65), 52888–52895 RSC.
- A. Hughes, H. Tai, A. Tochwin and W. Wang, Biodegradable and Biocompatible PDLLA-PEG1k-PDLLA Diacrylate Macromers: Synthesis, Characterisation and Preparation of Soluble Hyperbranched Polymers and Crosslinked Hydrogels, Processes, 2017, 5, 18 CrossRef.
- P. Chocholata, V. Kulda and V. Babuska, Fabrication of Scaffolds for Bone-Tissue Regeneration, Materials, 2019, 12(4), 568 CrossRef CAS PubMed.
- P. A. Gunatillake and R. Adhikari, Biodegradable synthetic polymers for tissue engineering, Eur. Cells Mater., 2003, 5, 1–16 CrossRef CAS ; discussion 16.
- S. H. Hsu, K.-C. Hung and C.-W. Chen, Biodegradable polymer scaffolds, J. Mater. Chem. B, 2016, 4(47), 7493–7505 RSC.
- K. Masutani and Y. Kimura, Chapter 1: PLA Synthesis. From the Monomer to the Polymer, in Poly(lactic acid) Science and Technology: Processing, Properties, Additives and Applications, The Royal Society of Chemistry, 2015, pp.1–36 Search PubMed.
- T. Maharana, B. Mohanty and Y. S. Negi, Melt–solid polycondensation of lactic acid and its biodegradability, Prog. Polym. Sci., 2009, 34(1), 99–124 CrossRef CAS.
- B. van Bochove and D. W. Grijpma, Photo-crosslinked synthetic biodegradable polymer networks for biomedical applications, J. Biomater. Sci., Polym. Ed., 2019, 30(2), 77–106 CrossRef CAS.
- S. Wei, J.-X. Ma, L. Xu, X.-S. Gu and X.-L. Ma, Biodegradable materials for bone defect repair, Mil. Med. Res., 2020, 7(1), 54, DOI:10.1186/s40779-020-00280-6.
- C. Zhang, in Biodegradable Polyesters: Synthesis, Properties, Applications, ed. S. Fakirov, Wiley-VCH Verlag GmbH & Co.KGaA, 1st edn, 2015 Search PubMed.
- M. E. Broz, D. L. VanderHart and N. R. Washburn, Structure and mechanical properties of poly(D,L-lactic acid)/poly(ε-caprolactone) blends, Biomaterials, 2003, 24(23), 4181–4190 CrossRef CAS.
- B. Laycock, M. Nikolić, J. M. Colwell, E. Gauthier and P. Halley, et al., Lifetime prediction of biodegradable polymers, Prog. Polym. Sci., 2017, 71, 144–189 CrossRef CAS.
- R. Shi, J. Xue, M. He, D. Chen and L. Zhang, et al., Structure, physical properties, biocompatibility and in vitro/vivo degradation behavior of anti-infective polycaprolactone-based electrospun membranes for guided tissue/bone regeneration, Polym. Degrad. Stab., 2014, 109, 293–306 CrossRef CAS.
- K. Shinno, M. Miyamoto, Y. Kimura, Y. Hirai and H. Yoshitome, Solid-State Postpolymerization of L-Lactide Promoted by Crystallization of Product Polymer: An Effective Method for Reduction of Remaining Monomer, Macromolecules, 1997, 30(21), 6438–6444 CrossRef CAS.
- Z. Wang, L. Yu, M. Ding, H. Tan and J. Li, et al., Preparation and rapid degradation of nontoxic biodegradable polyurethanes based on poly(lactic acid)–poly(ethylene glycol)–poly(lactic acid) and L-lysine diisocyanate, Polym. Chem., 2011, 2(3), 601–607 RSC.
- S. Kaihara, S. Matsumura, A. G. Mikos and J. P. Fisher, Synthesis of poly(L-lactide) and polyglycolide by ring-opening polymerization, Nat. Protoc., 2007, 2(11), 2767–2771 CrossRef CAS PubMed.
- S. He, M. D. Timmer, M. J. Yaszemski, A. W. Yasko and P. S. Engel, et al., Synthesis of biodegradable poly(propylene fumarate) networks with poly(propylene fumarate)–diacrylate macromers as crosslinking agents and characterization of their degradation products, Polymer, 2001, 42(3), 1251–1260 CrossRef CAS.
- M. B. Runge, H. Wang, R. J. Spinner, A. J. Windebank and M. J. Yaszemski, Reformulating polycaprolactone fumarate to eliminate toxic diethylene glycol: effects of polymeric branching and autoclave sterilization on material properties, Acta Biomater., 2012, 8(1), 133–143 CrossRef CAS PubMed.
- Sigma-Aldrich. Polymer Analysis by NMR. 2020. Online. Available from the URL: https://www.sigmaaldrich.com/technical-documents/articles/material-matters/polymer-analysis-by.html.
- Bacher, A. Infrared Spectroscopy, 2002. Online. Available from the URL: https://www.chem.ucla.edu/~bacher/spectrocopy/IR1.html.
- P. P. Albuquerque, A. D. Moreira, R. R. Moraes, L. M. Cavalcante and L. F. Schneider, Color stability, conversion, water sorption and solubility of dental composites formulated with different photoinitiator systems, J. Dent., 2013, 41, 67–72 CrossRef PubMed.
- U. Hansen, P. Zioupos, R. Simpson, J. D. Currey and D. Hynd, The Effect of Strain Rate on the Mechanical Properties of Human Cortical Bone, J. Biomech. Eng., 2008, 130, 110–118 CrossRef PubMed.
- R. Zange, Y. Li and T. Kissel, Biocompatibility testing of ABA triblock copolymers consisting of poly(L-lactic-co-glycolic acid) A blocks attached to a central poly(ethylene oxide) B block under in vitro conditions using different L929 mouse fibroblasts cell culture models, J. Controlled Release, 1998, 56, 249–258 CrossRef CAS PubMed.
- G. S. Sittampalam, Assay guidance manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD, 2004, pp. 263–267 Search PubMed.
- Thermofisher. Protocol for Passaging Adherent Cells, 2016. Online. Available from the URL: https://www.thermofisher.com/uk/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/subculturing-adherent-cells.html.
- H.-J. Hausser and R. E. Brenner, Phenotypic instability of Saos-2 cells in long-term culture, Biochem. Biophys. Res. Commun., 2005, 333(1), 216–222 CrossRef CAS.
- Invitrogen Corporation. AlamarBlue Assay manual (PI-DAL1025/1100Rev 1.0). 2002. Online. Available from the URL: https://tools.invitrogen.com/content/sfs/manuals/PIDAL1025-1100_TI%20alamarBlue%20Rev%201.1.pdf.
- E. A. Abou Neel, G. Palmer, J. C. Knowles, V. Salih and A. M. Young, Chemical, modulus and cell attachment studies of reactive calcium phosphate filler-containing fast photo-curing, surface-degrading, polymeric bone adhesives, Acta Biomater., 2010, 6, 2695–2703 CrossRef CAS PubMed.
- G. Longo, C. Ioannidu, A. Scotto d'Abusco, F. Superti and C. Misiano, et al., Improving Osteoblast Response In Vitro by a Nanostructured Thin Film with Titanium Carbide and Titanium Oxides Clustered around Graphitic Carbon, PLoS One, 2016, 11, e0152566 CrossRef PubMed.
- J.-B. Zeng, Y.-D. Li, Q.-Y. Zhu, K.-K. Yang and X.-L. Wang, et al., A novel biodegradable multiblock poly(ester urethane) containing poly(L-lactic acid) and poly(butylene succinate) blocks, Polymer, 2009, 50(5), 1178–1186 CrossRef CAS.
- A. S. Karikari, W. F. Edwards, J. B. Mecham and T. E. Long, et al., Influence of Peripheral Hydrogen Bonding on the Mechanical Properties of Photo-Cross-Linked Star-Shaped Poly(D,L-lactide) Networks, Biomacromolecules, 2005, 6(5), 2866–2874 CrossRef CAS PubMed.
- C. Pautke, M. Schieker, T. Tischer, A. Kolk and P. Neth, et al., Characterization of osteosarcoma cell lines MG-63, Saos-2 and U-2 OS in comparison to human osteoblasts, Anticancer Res., 2004, 24(6), 3743–3748 CAS.
- N. Ayobian-Markazi, T. Fourootan and M. J. Kharazifar, Comparison of cell viability and morphology of a human osteoblast-like cell line (SaOS-2) seeded on various bone substitute materials: An in vitro study, Dent. Res. J., 2012, 9(1), 86–92 CrossRef CAS.
- X. Zhao, I. Olsen, H. Li, K. Gellynck and P. G. Buxton, et al., Reactive calcium-phosphate-containing poly(ester-co-ether) methacrylate bone adhesives: Chemical, mechanical and biological considerations, Acta Biomater., 2010, 6(3), 845–855 CrossRef CAS PubMed.
- J. Jakubiak, X. Allonas, J. P. Fouassier, A. Sionkowska and E. Andrzejewska, et al., Camphorquinone–amines photoinitating systems for the initiation of free radical polymerization, 2003, vol. 44, pp.5219–5226 Search PubMed.
- T. Y. Kwon, R. Bagheri, Y. K. Kim, K. H. Kim and M. F. Burrow, et al., Cure mechanisms in materials for use in esthetic dentistry, J. Investig. Clin. Dent., 2012, 3(1), 3–16 CrossRef PubMed.
- A. S. Hamidi, M. A. Hadi and W. M. Palin, Alternative co-Initiators for photocurable dental resins: Polymerisation, quantum yield of conversion and cytotoxicity, Dent. Mater., 2022, 38(8), 1330–1343 CrossRef CAS PubMed.
- N. Davidenko, O. García and R. Sastre, Photopolymerization kinetics of dimethacrylate-based light-cured dental resins, J. Appl. Polym. Sci., 2005, 97(3), 1016–1023 CrossRef CAS.
- J. G. Leprince, W. M. Palin, M. A. Hadi, J. Devaux and G. Leloup, Progress in dimethacrylate-based dental composite technology and curing efficiency, Dent. Mater., 2013, 29(2), 139–156 CrossRef CAS PubMed.
- A. M. Ballo, I. Cekic-Nagas, G. Ergun, L. Lassila and A. Palmquist, et al., Osseointegration of fiber-reinforced composite implants: Histological and ultrastructural observations, Dent. Mater., 2014, 30(12), 384–395 CrossRef PubMed.
- N. J. Lin and S. Lin-Gibson, Osteoblast response to dimethacrylate composites varying in composition, conversion and roughness using a combinatorial approach, Biomaterials, 2009, 30(27), 4480–4487 CrossRef CAS PubMed.
- L. Wang, J. Qiu and E. Sakai, Microstructures and mechanical properties of polylactic acid prepared by a cold rolling process, J. Mater. Process. Technol., 2016, 232, 184–194 CrossRef CAS.
- H. Zhao and G. Zhao, Mechanical and thermal properties of conventional and microcellular injection molded poly (lactic acid)/poly (ε-caprolactone) blends, J. Mech. Behav. Biomed. Mater., 2016, 53, 59–67 CrossRef CAS.
- P. Douglas, A. B. Albadarin, A. H. Al-Muhtaseb, C. Mangwandi and G. M. Walker, Thermo-mechanical properties of poly ε-caprolactone/poly l-lactic acid blends: Addition of nalidixic acid and polyethylene glycol additives, J. Mech. Behav. Biomed. Mater., 2015, 45, 154–165 CrossRef CAS.
- R. Malinowski, Mechanical properties of PLA/PCL blends crosslinked by electron beam and TAIC additive, Chem. Phys. Lett., 2016, 662, 91–96 CrossRef.
- R. A. Stockwell, The Mechanical Properties of Biological Materials, J. Anat., 1981, 133(Pt 1), 99–100 Search PubMed.
- E. Novitskaya, P.-Y. Chen, E. Hamed, J. Li, V. A. Lubarda, I. Jasiuk and J. McKittrick, Recent advances on the measurement and calculation of the elastic moduli of cortical and trabecular bone: A review, Theor. Appl. Mech., 2011, 38(3), 209–297 CrossRef.
- N. H. Hart, S. Nimphius, T. Rantalainen, A. Ireland and A. Siafarikas, et al., Mechanical basis of bone strength: influence of bone material, bone structure and muscle action, J. Musculoskeletal Neuronal Interact., 2017, 17(3), 114–139 CAS.
- J. A. Spector, B. J. Mehrara, J. A. Greenwald, P. B. Saadeh and D. S. Steinbrech, J.M. Osteoblast expression of vascular endothelial growth factor is modulated by the extracellular microenvironment, Am. J. Physiol. Cell Physiol., 2001, 280, C72–C80 CrossRef CAS PubMed.
- C. C. Hardin and J. A. Knopp, 23.1Pathway, in Biochemistry – Essential Concepts, Oxford University Press, 2012, ISBN: 978-0-19-976562-1 Search PubMed.
- A. Kumari, Chapter 1 – Glycolysis. Sweet Biochemistry, Academic Press, 2018, p.1–5. ISBN: 978-0-12-814453-4 Search PubMed.
- S. W. Danielle, A. R. D. Benoit and S. A. Kristi, Manipulations in Hydrogel Degradation Behavior Enhance Osteoblast Function and Mineralized Tissue Formation, Tissue Eng., 2006, 12(6), 1663–1673 CrossRef PubMed.
- E. Malikmammadov, T. E. Tanir, A. Kiziltay, V. Hasirci and N. Hasirci, PCL and PCL-based materials in biomedical applications, J. Biomater. Sci., Polym. Ed., 2018, 29(7–9), 863–893 CrossRef CAS.
- C. A. Martins, G. Leyhausen, W. Geurtsen and J. Volk, Intracellular glutathione: A main factor in TEGDMA-induced cytotoxicity?, Dent. Mater., 2012, 28, 442–448 CrossRef CAS.
- N. J. Walters, W. Xia, V. Salih, P. F. Ashley and A. M. Young, Poly(propylene glycol) and urethane dimethacrylates improve conversion of dental composites and reveal complexity of cytocompatibility testing, Dent. Mater., 2016, 32(2), 264–277 CrossRef CAS PubMed.
- H. Schweikl, A. Hartmann, K.-A. Hiller, G. Spagnuolo and C. Bolaya, et al., Inhibition of TEGDMA and HEMA-induced genotoxicity and cell cycle arrest by N-acetylcysteine, Dent. Mater., 2007, 23, 688–695 CrossRef CAS PubMed.
- G. Spagnuolo, K. Galler, G. Schmalz, C. Cosentino, S. Rengo and H. Schweikl, Inhibition of phosphatidylinositol 3-kinase amplifies TEGDMA-induced apoptosis in primary human pulp cells, J. Dent. Res., 2004, 83, 703–707 CrossRef CAS PubMed.
- H. Schweikl, G. Schmalz and K. Rackebrandt, The mutagenic activity of unpolymerized resin monomers in Salmonella typhimurium and V79 cells, Mutat. Res., 1998, 415, 119–130 CAS.
- A. Abdal-hay, N. T. Raveendran, B. Fournier and S. Ivanovski, et al., Fabrication of biocompatible and bioabsorbable polycaprolactone/magnesium hydroxide 3D printed scaffolds: Degradation and in vitro osteoblasts interactions, Composites, Part B, 2020, 197, 108158 CrossRef CAS.
- H.-M. Yin, C. Mao, W. Liu, Y.-H. Liu and Y. Ren, et al., Nanotopographical polymeric surface with mussel-inspired decoration to enhance osteoblast differentiation, Appl. Surf. Sci., 2019, 481, 987–993 CrossRef CAS.
- S. F. El-Amin, H. H. Lu, Y. Khan, J. Burems and J. Mitchell, et al., Extracellular matrix production by human osteoblasts cultured on biodegradable polymers applicable for tissue engineering, Biomaterials, 2003, 24(7), 1213–1221 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01049j |
| This journal is © The Royal Society of Chemistry 2024 |