
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic enhancement of seawater hydrogen generation via sulfur vacancy enriched and phases engineered CQD loaded CdS photocatalyst†
Bishal
Kumar Nahak
a and
Fan Gang
Tseng
 *abcde
*abcde
aDepartment of Engineering and System Science, National Tsing Hua University, Section 2, Kuang-Fu Rd, Hsinchu 30013, Taiwan, ROC. E-mail: fangang@ess.nthu.edu.tw; Fax: 886-3-5733054; Tel: 886-3-5715131 ext. 34270 Tel: 886-3-5733054
bInstitute of Nano Engineering and Microsystems, National Tsing Hua University, Hsinchu 30013, Taiwan, ROC
cDepartment Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan, ROC
dResearch Center for Applied Sciences, Academia Sinica, Taipei 11529, Taiwan, ROC
eFrontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing Hua University, Hsinchu 300, Taiwan, ROC
First published on 19th December 2023
Abstract
The use of photocatalytic water splitting, which involves generation of hydrogen (H2), is regarded as a possible remedy to counteract the obstacles in the use of fossil fuels and to confront the issues of the energy crisis and environmental contamination. Despite the advancements, there continues to be a scarcity of research on stable and efficient photocatalysts suitable for use in seawater. The present paper describes the process of creating a photocatalyst using a hydrothermal method which combines carbon quantum dots and cadmium sulfide (CdS) nanoparticles. The developed photocatalyst showed excellent photocatalytic H2 generation (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 450 μmol g−1 in 10 h) and dye degradation efficiency (97% in 60 min). Further, the H2 generation of bare CdS and CQD@CdS was also tested under seawater conditions where a longer lifetime of particle was observed up to 10 h. A comprehensive analysis was conducted to evaluate the photodegradation mechanism of Rhodamine B dye utilizing the CQD@CdS system. The results showed that the process kinetics can be depicted using a pseudo-second order reaction. The remarkable catalytic activity and outstanding durability of the CQD@CdS can be ascribed to the effective function of the CQDs, which augment the segregation of the photoinduced charge carriers and averts the occurrence of photocorrosion in seawater. Moreover, the CQDs also supported a higher specific surface area and phase transformation in CdS with a dominant hexagonal phase. All these synergistic effects on the properties by the CQD@CdS, aided in creation of a highly efficient photocatalyst that is suitable for applying to solar hydrogen generation.
450 μmol g−1 in 10 h) and dye degradation efficiency (97% in 60 min). Further, the H2 generation of bare CdS and CQD@CdS was also tested under seawater conditions where a longer lifetime of particle was observed up to 10 h. A comprehensive analysis was conducted to evaluate the photodegradation mechanism of Rhodamine B dye utilizing the CQD@CdS system. The results showed that the process kinetics can be depicted using a pseudo-second order reaction. The remarkable catalytic activity and outstanding durability of the CQD@CdS can be ascribed to the effective function of the CQDs, which augment the segregation of the photoinduced charge carriers and averts the occurrence of photocorrosion in seawater. Moreover, the CQDs also supported a higher specific surface area and phase transformation in CdS with a dominant hexagonal phase. All these synergistic effects on the properties by the CQD@CdS, aided in creation of a highly efficient photocatalyst that is suitable for applying to solar hydrogen generation.
1. Introduction
Hydrogen (H2) presently has an impressive position as one of the most viable and net zero carbon energy sources. Therefore, as the required global energy supply will continue to increase, the energy transition should be accompanied by efficient emerging technologies with worldwide availability.1 Converting solar radiation into H2 using a potent photocatalyst is regarded as a glimpse into the future for providing a sustainable resource to address energy concerns.2–6 Driven by natural photosynthesis process, production of H2via solar water splitting has gained the interest of researchers over the decades, dating all the way back to 1972, when TiO2 was first used for the electrochemical splitting of water in the presence of light.7–10 Given the potential for converting the renewable solar irradiation and plentiful natural seawater into chemical fuels, immense efforts have been made in devising new efficient photocatalysts, particularly those capable of utilizing visible light (420–700 nm)11–15 which accounts for approximately 43% of the solar spectrum.16Photocatalytic techniques are very effective, low-cost, and environmentally beneficial for remediation of various organic contaminants. Numerous semiconductors with and without alterations have been extensively studied as potential photocatalysts for the mineralization of organic compounds such as CeO2, Fe2O3, SnO2, TiO2, ZnO.11,16–22 However, the larger band gap of these oxide semiconductors, means that the catalytic activity is limited to the UV region. Another key challenge for using oxide photocatalysts is that the quick charge carrier recombination has to be dealt with ref. 24 and 25.
Chalcogenide semiconductors were regarded as promising candidates for visible light responsive (VLR) hydrogen generation due to their intrinsic merits which include its favorable band gap, fast light response and excellent thermal stability.25–27 Cadmium sulfide (CdS) is an ideal candidate for use as a VLR photocatalyst due to its narrow band gap of 2.4 eV. Because it has a small radius for the diffusion of photoinduced electrons and holes, it enables a bare CdS semiconductor to improve the solar light absorption and scattering capabilities, leading to a decrease of charge carrier recombination.28 However, under continuous exposure to light, CdS has shown a low resistance towards photo-corrosion. Various capping agents have been utilized to increase the stability of these particles in the nanodomain.29–31 This class of substances can act as a capping or passivating agent that decreases the energy required for the formation of new nuclei on a pre-existing structure by lowering the interfacial activation-energy.32–36 Among these different passivating agents, poly(vinylpyrrolidone) (PVD) gained much interest because of its water solubility, biocompatibility and because it is relatively harmless to the environment.
Strategies for surface treatment have been utilized to minimize the recombination of charge carriers and to lessen the photocorrosion of CdS. The prevailing approach involves the implementation of sacrificial reagents (SRs) such as formic acid, lactic acid, S2−, SO32−, that effectively scavenge the photo-induced holes. Another way to enhance the photocatalytic performance, is by creating cationic/anionic vacancies37 and using a co-catalyst such as TiO2,38,39 ZnO,40,41 MoS2,42,43 ZnS,44 Bi2WO6,45 g-C3N446 and precious metals.47 They may act as trap centers or electron acceptors for the photogenerated electron that help to reduce the recombination and boosts the CdS photocatalytic performance. However, these materials necessitate sophisticated, expensive processing equipment, use of harmful chemicals and at the same time they are not ecofriendly. Therefore, it is necessary to find an alternative co-catalyst that is ecofriendly, easy to synthesize and able to reduce charge carrier recombination.
Apart from these, some other alternatives have also been reported such as band-gap engineering and creating sulfur vacancies (SVs).23 Band-gap engineering plays a crucial role in achieving an appropriate phase, and facilitating the effective separation of charge carriers. Yu et al., reported cubic-phase CdS nanocrystals that were rich in sulfur, and which gave a hydrogen production rate of 680 μmol g−1 h−1.48 Similarly, Li et al., produced hexagonal CdS nanorods with an H2 generation efficiency of 740 μmol g−1 h−1.49 Some reports also claim that using multiple crystal phases or phase junctions in semiconductor crystals has been proven to enhance photocatalytic performance. Ai et al., synthesized a mixed phase CdS nanostructure which had a suitable bonding width that improved the H2 yield up to 4893 μmol g−1 h−1.50 Although the significance of these phase junctions was explored to some degree using the previously mentioned methods, they have not been given due consideration. Thus, additional investigations into the significance of phase junctions and the factors associated with them are still needed. Recently, anion vacancies have been one of the notable ways to create trap centers that capture the charge carriers. This has been quite common in narrow band gap semiconductors such as CdS, CdSe, SnS2, and ZnS. Conventionally, SVs are created by doping, etching, and controlling the ratio of reactants in the synthesis process. However, individually, none of these factors were successful in increasing the H2 generation efficiency to a great extent. Herein, a strategic increase of SVs was made with the addition of carbon quantum dots (CQDs) that helped in catalytic process. The presence of a synergistic effect between the mixed phase structure and SVs helped in lowering the photoinduced charge carrier recombination and provided a higher hydrogen evolution rate (HER).
The CQDs are a class of finely proportioned carbon-based nanomaterials that bear a resemblance to graphite particles. These carbonaceous materials are essential for facilitating the interaction between adsorbed O2 on the surface and photoinduced electrons, thus preventing charge carrier's recombination.51–53 The CQDs with a large specific Brunauer–Emmett–Teller (BET) area can be easily treated to anchor an array of functional groups, including –COOH, –OH, and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.54–56 Furthermore, the CQDs possess exceptional properties such as being excellent electron acceptors, having a unique electron reservoir, and they are also low-cost, non-toxic, and highly stable, making them a preferred cocatalyst in photocatalysis.57–59 Using graphene as a highly effective cocatalyst in CdS photocatalytic systems has been extensively studied. However, nanosized CQDs offer several advantages such as easy functionalization and the ability to generate a large number of reducing sites that enhance charge separation and give a greater H2 evolution.57 Although CQDs have low electrical conductivity, they are a cost-effective alternative for forming heterojunctions with CdS. In addition, CQDs offer a high surface area and a highly porous morphology that can effectively adsorb various organic pollutants.59 The functional groups present in CQDs can expertly interrupt the recombination process of the charge carriers in CdS.58
O.54–56 Furthermore, the CQDs possess exceptional properties such as being excellent electron acceptors, having a unique electron reservoir, and they are also low-cost, non-toxic, and highly stable, making them a preferred cocatalyst in photocatalysis.57–59 Using graphene as a highly effective cocatalyst in CdS photocatalytic systems has been extensively studied. However, nanosized CQDs offer several advantages such as easy functionalization and the ability to generate a large number of reducing sites that enhance charge separation and give a greater H2 evolution.57 Although CQDs have low electrical conductivity, they are a cost-effective alternative for forming heterojunctions with CdS. In addition, CQDs offer a high surface area and a highly porous morphology that can effectively adsorb various organic pollutants.59 The functional groups present in CQDs can expertly interrupt the recombination process of the charge carriers in CdS.58
Recently, various techniques have been developed to create a near-coaxial heterojunction by uniformly attaching conjugated carbon layers to the surface of CdS during the nucleation stage of nanoparticles. Lei et al., have published a study on the use of CdS nanoparticles combined with graphene quantum dots (GQDs) to boost the photocatalytic production of H2. In their study, the GQDs acted as electron acceptors which reduced the charge recombination.60 In a study by Guo et al.,61 a solvothermal method was utilized to create a photocatalyst consisting of activated carbon fibers and CdS, derived from coal. The resulting material demonstrated an exceptional VLR photocatalytic performance in degrading methylene blue dye. In all these cases the enhancement of the H2 evolution was claimed to be due to the presence of CQDs, where the CQDs acts as an electron acceptor that helps in reducing charge carrier recombination. However, as far as is known its influence on the change in phase transformation and creating sulfur-vacancies has not so far been reported. Herein, CQD-based CdS nanoparticles have been fabricated using a facial hydrothermal method. The main focus of this study is the design of a VLR photocatalyst with a high surface area and mixed phase structure that increases H2 evolution and stability of the pristine CdS. Furthermore, various characterizations have been performed to validate the properties of the sample. The developed photocatalyst was utilized for Rhodamine B (RhB) dye mineralization and H2 generation in normal water and seawater.
2. Experimental
2.1. Materials
All the chemicals were of analytical grade and used without further purification. Cadmium acetate (Cd(CH3COO)2), thiourea ((NH2)2CS), sodium hydroxide (NaOH), glucose (C6H12O6), urea (CO(NH2)2), methanol (MeOH) and triethylamine (TEA, N(CH2CH3)3) were purchased from Sigma-Aldrich (USA). Deionized (DI) water (resistivity: 18.5 MΩ cm) was used for all the analyses. The detailed characterization details given in ESI† (Text S1).2.2. Synthesis of the carbon quantum dots
A hydrothermal method was used to synthesize CQDs, with aqueous glucose as the reaction precursor. To begin with, 1 g of glucose and 50 mL of DI water was prepared at room temperature. The resulting solution was subjected to hydrothermal treatment at 200 °C for 12 h in a Teflon-lined stainless-steel autoclave. Upon cooling, the dark colored liquid was refined using a membrane with a pore diameter of 200 nm. It was then washed three times with ethanol to eliminate impurities, and centrifuged for 30 min at 10000 rpm to separate the carbon containing liquid from a heavy black precipitate. The CQD solution was used for further experiments. The detailed procedure is shown in Scheme 1.
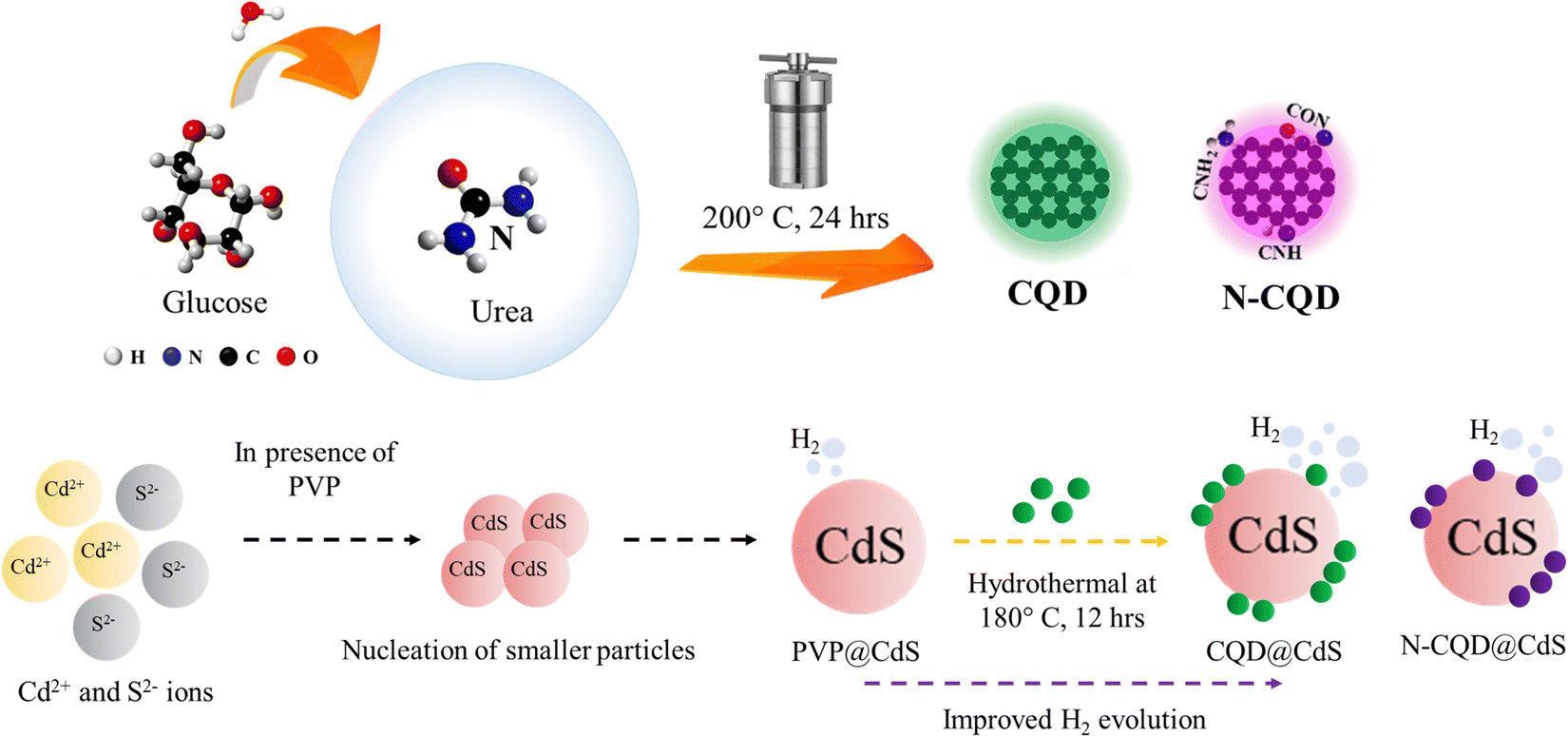 | ||
| Scheme 1 This shows the synthesis process of carbon quantum dots (CQDs) and the CQD integrated cadmium sulfide photocatalyst. | ||
Nitrogen doped CQD (N-CQDs) was synthesized with urea as the nitrogen source. Urea (0.5 g) was mixed with the initial reagent and the process was carried out similar way.
2.3. Synthesis of the CQD@CdS photocatalyst
Typically, cadmium and sulfur salts were mixed in 1:1 mole ratio during the synthesis process. First, an aqueous solution of salts was prepared using Cd(CH3COO)2 as the source of Cd2+ ions and (NH2)2CS for the S2− ions. CdS nanoparticles have higher tendency to get agglomerated in nucleation stage, thus an aqueous poly(vinylpyrrolidone) (PVP) solution have been used as capping agent that helps CdS to remain in nanodomain. The PVP solution was mixed with the cadmium salt solution under stirring condition, until it became transparent. Next, the (NH2)2CS solution was added dropwise to the previous solution under stirring and at a temperature of 75 °C. Finally, the pH of the solution was maintained at 11 using 2 N NaOH solution. As a result, a yellow colored solution was formed, which was transferred to a 100-mL Teflon-lined stainless-steel container and planed in autoclave at a temperature of 140 °C and left overnight. After the solution had cooled down to room temperature, the yellow colored precipitate was washed three times with DI water/ethanol to eliminate unwanted products. The particles were dried at 60 °C for 12 h, during which time the PVP@CdS formed (Scheme 1). Here, PVP@CdS has been used because bare CdS did not contain CQD.
The CQD@CdS samples were prepared by mixing the previously prepared CQDs from the initial CdS synthesis. In order to find optimal loading of CQD on CdS, different amounts (1 mL, 2 mL and 3 mL) of CQDs were used. Throughout the research 3 mL of CQD with CdS was used for different characterization and experimental purposes, due to its high performance (discussed in later sections). The rest of the procedure followed was similar to that used for PVP@CdS. Similarly, N-CQD@CdS was prepared using 3 mL of N-CQD at the reagent stage.
2.4. Photocatalytic experiment
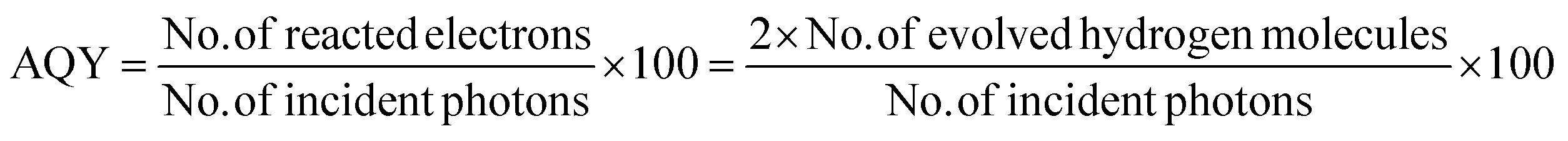 | (1) |
| Degradation efficiency (%) = (1 − C/C0) × 100 | (2) |
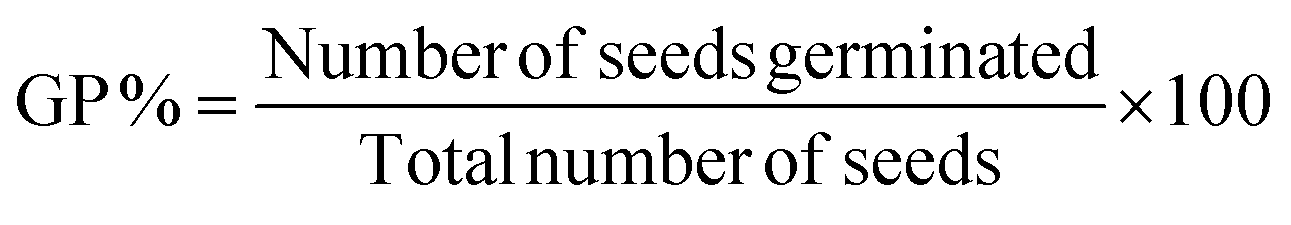 | (3) |
Next, length of the root and shoot after 6 d was measured by using the standard centimeter scale. The effect of the DW, untreated RhB solution, and treated RhB solution on the plant parameters was determined.
3. Results and characterizations
3.1. Structural and optical properties
To further verify the mixed phase structure of CQD@CdS, selected area (electron) diffraction (SAED) patterns and d-spacing analysis were done and their transmission electron microscopy (TEM) images are shown in Fig. 1b and c. The SAED rings for the respective lattice planes of the wurtzite phase (110, 101, 102, 112 and 004) and the zinc blende phase (200, 220) confirmed the co-existence of both phases. Some noticeable distortions were found on the phase junction region, and these were created due to the lattice strain and the structural defects in these nano-catalysts. The d-spacing for the hexagonal phase was found to be 3.17 Å and the cubic phase was 3.36 Å. The CQD doping on these mixed phase structures was also confirmed and the d-spacing was 2.1 Å. Also, by varying the CQD and CdS ratio there was no peak shift, whereas the intensity was decreased as shown in Fig. S8 (ESI†). These results were found to be similar with those reported in the literature Gogoi et al.65 It was found that hexagonal phase of CdS promoted high stability, low charge recombination and increased the catalytic action.66 The dominance of the hexagonal phase in any compound having a mixed phase elevated the overall photocatalytic efficiency.
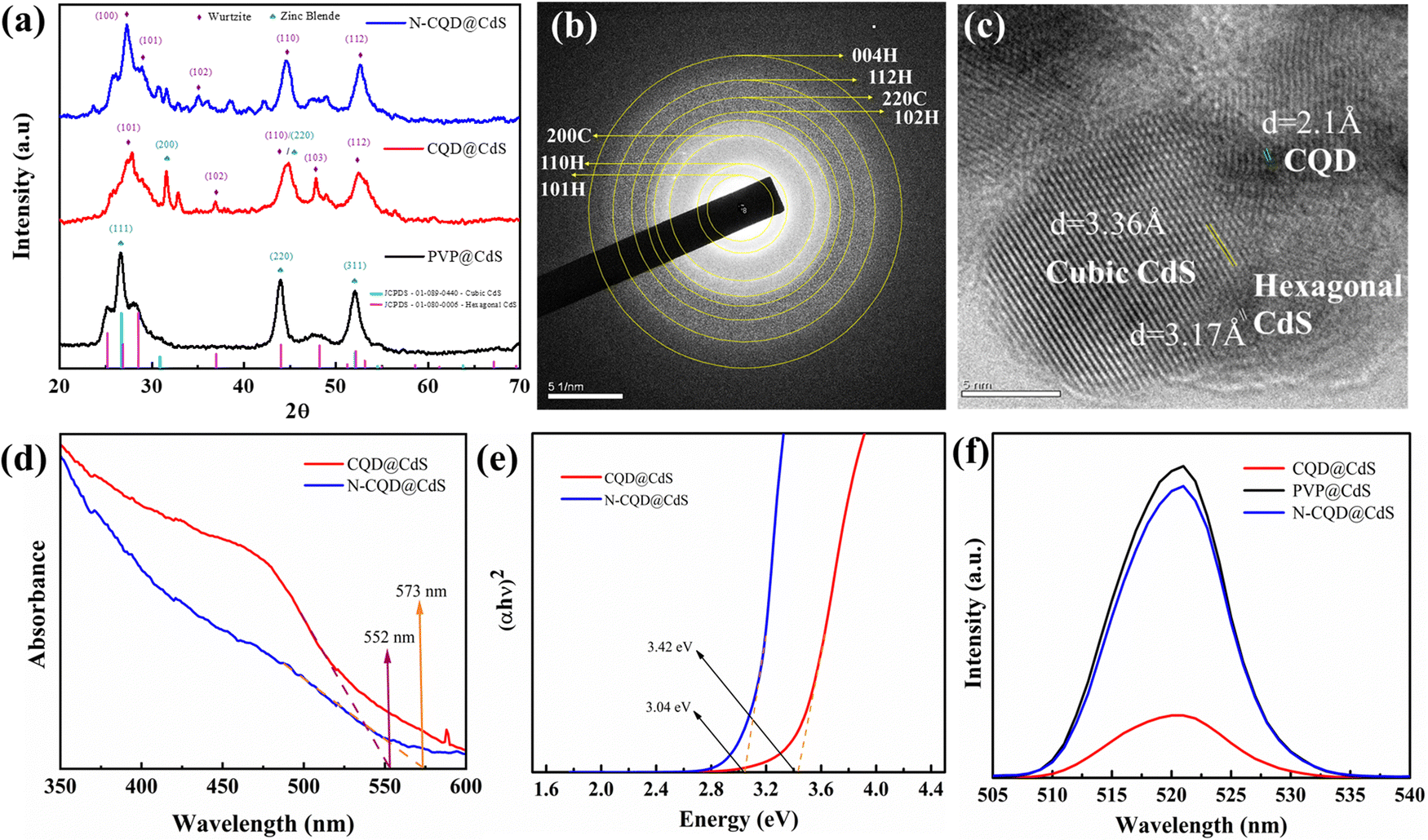 | ||
| Fig. 1 Shows the diffraction pattern (a), absorbance spectra (d), Tauc plot (e) and photoluminescence spectra (f) of all the CdS samples, and the SAED (b) and TEM lattice fringes (c) of the CQD@CdS samples. | ||
The average crystal size and microstrain stress were obtained as structural parameters from the XRD data. To estimate the average crystallite size of the CdS nanostructures, the Debye–Scherrer formula was employed as shown in eqn (4):
| D = kλ/βCosθ | (4) |
| ε = β/4tanθ | (5) |
The dislocation density was also calculated by using eqn (6):
| Δ = 1/D2 | (6) |
Nanostructures often have surface defects, which are caused by dislocations, vacancies and shear strain planes. These defects can act as traps for the photoinduced charge carriers and adsorption sites. When the electrons transfer to these adsorbed sites, it can prevent the recombination of photoexcited electron–hole pairs.67 Recent studies have shown that increasing the number of surface defects can enhance the charge separation and improve the catalytic activity.68 From Fig. S7 (ESI†), it was observed that CQD@CdS and N-CQD@CdS had a high average strain and dislocation density, and this can be due to its mixed phase and the presence of SVs confirmed by the X-ray photoelectron spectroscopy (XPS) and energy-dispersive X-ray spectroscopy (EDAX) analyses. Thus, helping the CQD@CdS to show better H2 evolution and better dye mineralization.
C bond which occurs due to the skeletal vibrations of the aromatic rings. The transmittance peaks at 2882 cm−1 and 1385 cm−1 correspond to the stretching vibrations of C–H. The stretching vibrations of the CO and –OH groups were confirmed from the peaks at 996 cm−1, 1142 cm−1 and 3350 cm−1, respectively.69 This confirmed that the synthesized CQDs are water soluble and are highly hydrophilic.
The bond interaction between PVP and CQD with CdS was studied and the results are shown in Fig. S1b (ESI†). The broad band centered at 3280 cm−1 was due to H–O–H bending vibrations of the water molecule. The characteristic bands located at 1654 cm−1, 1409 cm−1 and 2882 cm−1 were ascribed to the CO stretching modes, and C–H bonding of the PVP monomer. The presence of CQD on the CdS surface was confirmed by the bands at 1440 cm−1 and 1044 cm−1 that were assigned to the CC and C–N bonds, respectively. The occurrence of amine functional groups in the N-CQDs was confirmed by a slight peak at a wavenumber of 3117 cm−1. The N-CQDs were then coordinated to the surface of CdS by these –NH2 groups.64 A detailed description of the functional groups of each sample are shown in Table S1 (ESI†).
C bond with sp2 hybridization, whereas the peak at 300 nm was attributed to the n–π* transition of the carbonyl or amine groups present on the surface.71,72 The absorption spectrum in Fig. 1d demonstrates noticeable variations in the 550–600 nm wavelength range for the CQD loaded CdS samples. The “blue-shift” of λ from λ = 574 nm (for standard CdS samples) to 573 and 552 nm was observed in CdS when the N-CQD and CQD were introduced, respectively. This, established that the CQD@CdS and N-CQD@CdS particles had been successfully synthesized. The observed hypsochromic shifting in the synthesized CdS nanoparticles is believed to be associated with a slight reduction in particle size, which was consistent with the XRD results. This outcome also indicated that the decrease of absorption maximum was in the order of PVP@CdS > N-CQD@CdS > CQD@CdS. Specifically, PVP@CdS exhibited the strongest absorption (Fig. S2c, ESI†) in the visible light range at 574 nm. Whereas the N-CQD@CdS showed an absorption maximum at 573 nm, and the CQD@CdS showed a maximum at 552 nm as shown in Fig. 1d. Further, the corresponding optical band gap for the CdS samples were calculated and are shown in Table 1 and their respective Tauc plots are shown in Fig. 1e. The PVP@CdS possesses the smaller band gap of whereas the N-CQD@CdS and CQD@CdS have wide band gaps. The presence of CQD did not have a significant change in optical properties73 and this was also confirmed from other results reported in the literature that showed similar results to ours.65,74
| Sample name | Band gap (eV) | Absorption maxima (nm) | Size (nm) |
|---|---|---|---|
| PVP@CdS | 2.160 | 574 | 37 |
| CQD@CdS | 2.223 | 554 | 24 |
| N-CQD@CdS | 2.175 | 570 | 33 |
The average particle size (R) of the CdS nanoparticles was estimated using the Brus eqn (7):
| Eg = E + h2/8R2 [1/(me*) + 1/mh*] − 1.8e2/εR | (7) |
| Eg = 1240/λ | (8) |
3.2. Morphological properties
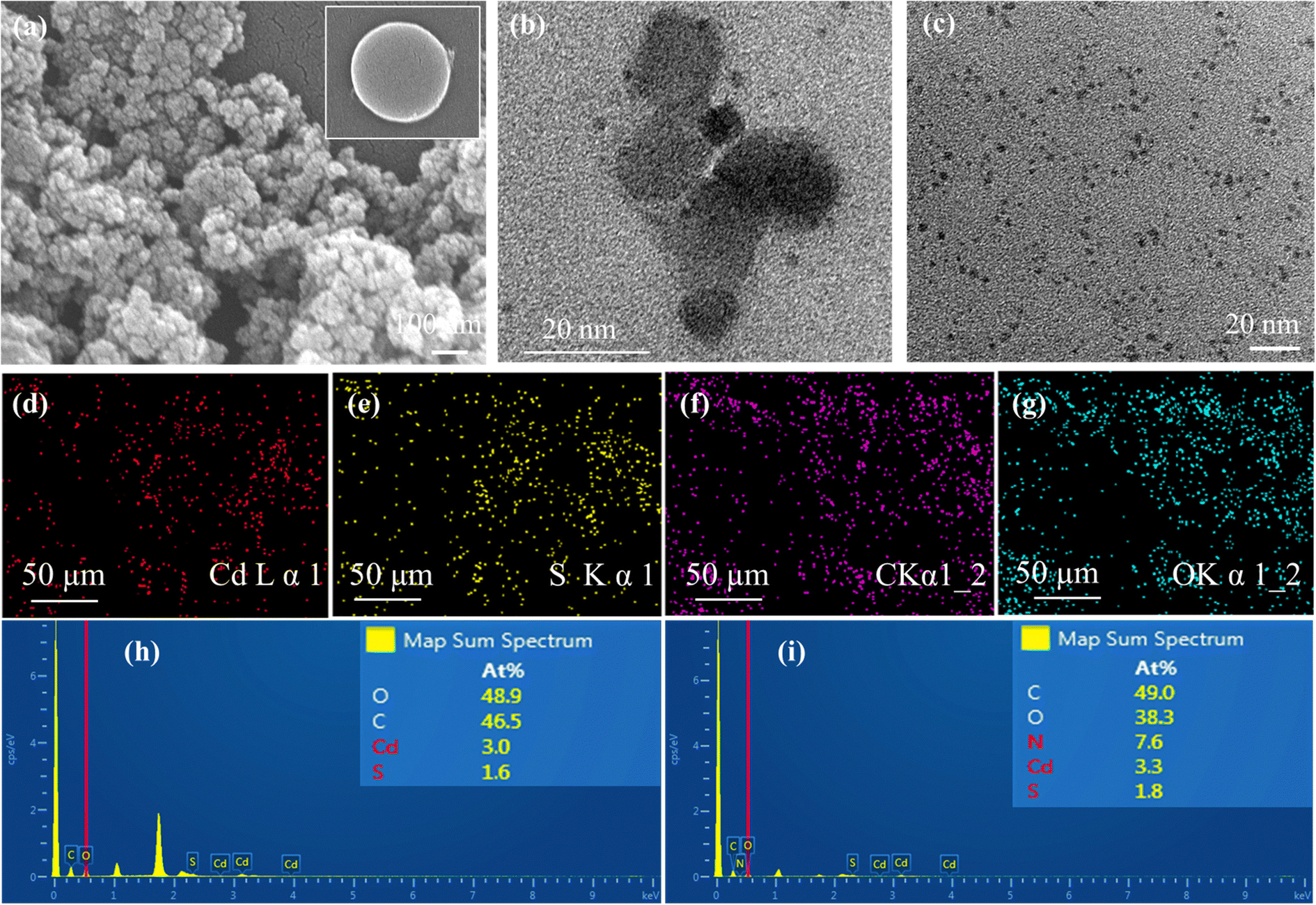 | ||
| Fig. 2 This shows (a) SEM and (b) TEM images of CQD@CdS, (c) the SEM image of CQD. (d)–(g) elemental mapping, and the EDAX spectra of (h) CQD@CdS and (i) N-CQD@CdS. | ||
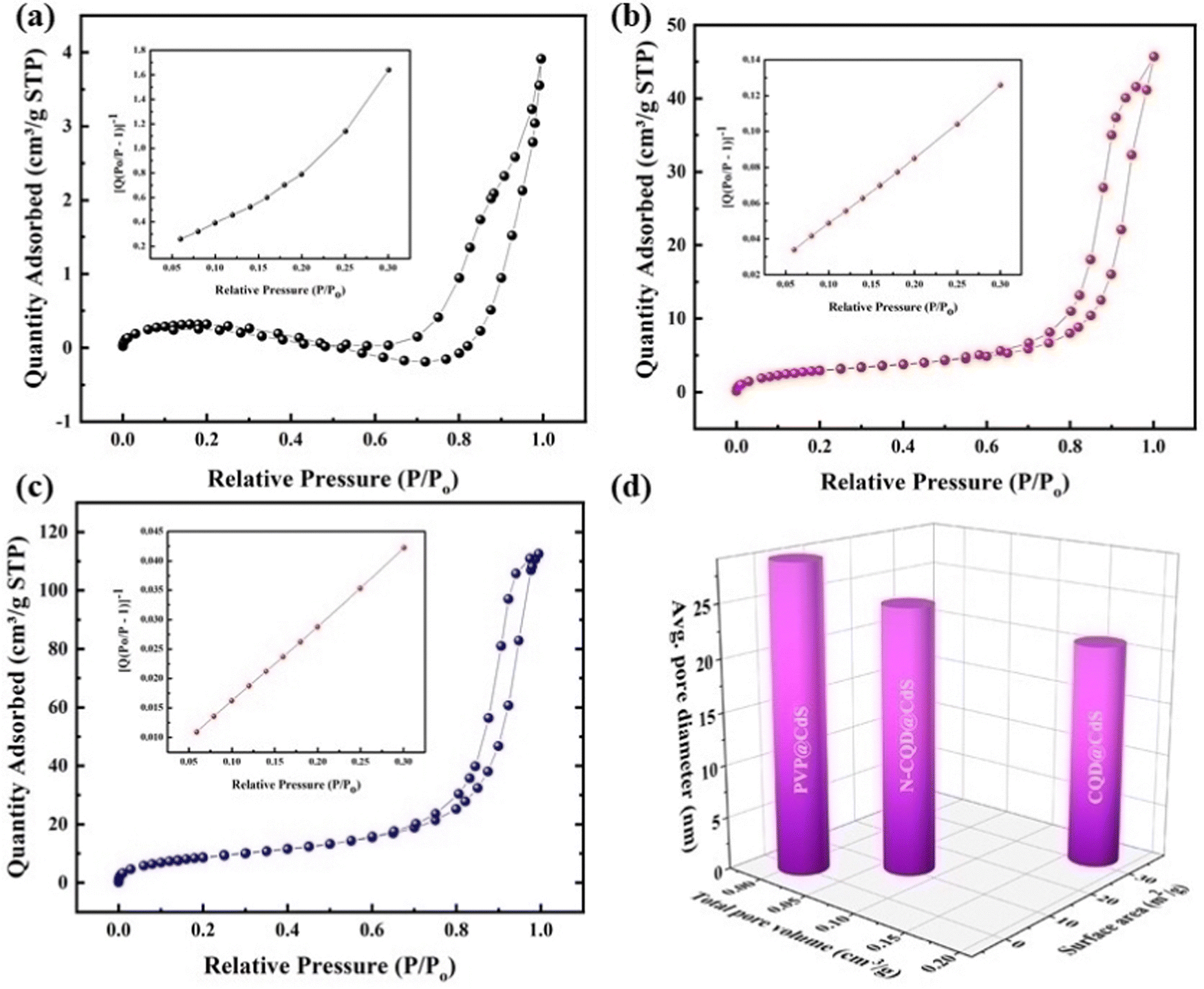 | ||
| Fig. 3 The BET absorption–desorption isotherms (a)–(c), and the pore size distribution and surface area (d) of the CdS sample. | ||
3.3. X-ray photoelectron spectroscopy (XPS)
The surface composition and oxidation states of the elements in the synthesized samples were confirmed by XPS analysis and the results are shown in Fig. 4. The full survey scans for the PVP@CdS, CQD@CdS and N-CQD@CdS spectra confirmed the presence of the elemental components: C, Cd, N, and S (Fig. S4, ESI†). The complete analysis of the high-resolution C 1s core level spectrum revealed intricate details, featuring distinct peaks indicative of various carbon functional groups. In particular, for PVP@CdS, the peaks centered at 284.39 eV, 286.09 eV, 287.95 eV (Fig. 4c) corresponded to –C–C/–CC, –C–O, and –CO, respectively. Similar functional groups were observed for CQD@CdS (284.51 eV, 285.85 eV, 287.36 eV, Fig. 4f) and N-CQD@CdS (284.38 eV, 285.36 eV, 287.01 eV, Fig. 4i). The full survey spectrum highlights a comparatively small carbon peak intensity in PVP@CdS, which confirmed the presence of CQD in later samples. The Cd 3d spectra (Fig. 4a, d and g) distinctly show the presence of two major peaks signifying the Cd2+ oxidation states in the samples.80 Furthermore, the deconvolution of the S 2p spectrum revealed doublet peaks for each sample: PVP@CdS (160.92 eV and 162.08 eV, Fig. 4b), CQD@CdS (160.49 eV and 161.68 eV, Fig. 4e) and N-CQD@CdS (160.49 eV and 161.61 eV, Fig. 4h) which were associated with S 2p1/2 and S 2p3/2, respectively.
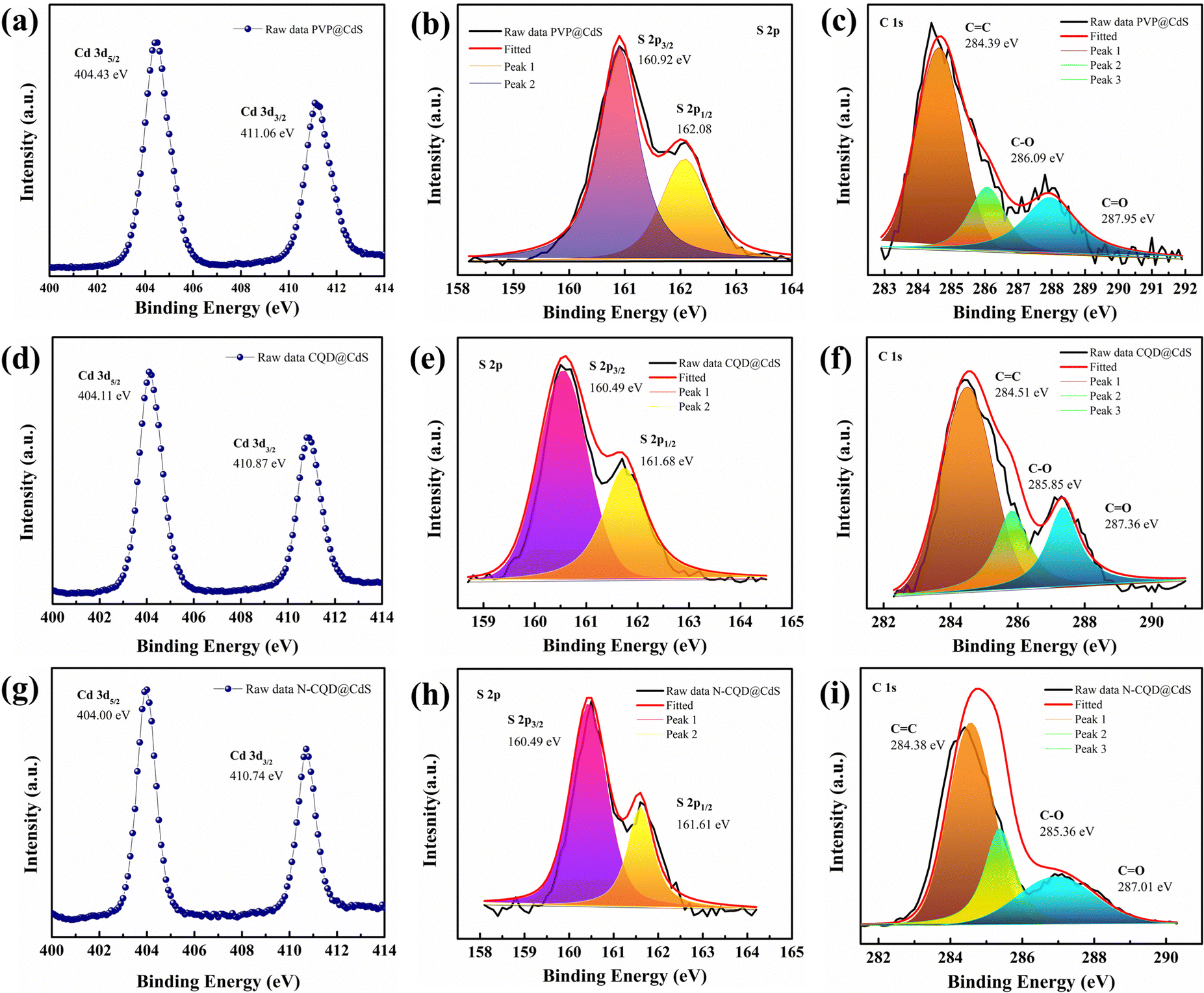 | ||
| Fig. 4 The XPS spectra of the PVP@CdS, CQD@CdS and N-CQD@CdS samples with their respective (a), (d) and (g) Cd 3d, (b), (e) and (h) S 2p, and (c), (f) and (i) C 1s elements. | ||
The Cd 3d and S 2p spectra of CQD@CdS and N-CQD@CdS showed a peak shift towards lower binding energies, when compared to bare PVP@CdS. This shift of energy levels indicated an enhancement in the electron density on CdS, which suggested there was a significant chemical interaction between the CQD and the CdS.63 The elemental analysis data are summarized in Table S4 (ESI†). Interestingly, the atomic percentage of sulfur in CdS became smaller with the use of CQD, when compared to that of bare PVP@CdS. This result indicated that the CQD supports the SV, thereby increasing the cadmium population. According to previous reports, the electron trap sites were identified in between the energy bands of 0.63–0.7 eV, i.e., below the conduction band (CB) of the bulk CdS, and these trap states in the crystal are assigned to SVs. Furthermore, the change in the Cd peak also supported the existence of a Cd surface with SVs81 and a decrease in the electronegative S atoms because of the lower binding energy shift occurred.82 This confirmed the presence of CdS with a deficiency in the S concentration that helped to increase the photocatalytic action.
Further, to confirm the XPS results for the SVs, electron paramagnetic resonance (EPR) analysis was performed as shown in Fig. S9 (ESI†). All the CdS samples showed resonance signals at g = 0.19. The weak signal for N-CQD@CdS prepared by a hydrothermal method involved low SVs. However, when the CQD was used instead of N-CQD, the EPR response also changed significantly. These results were also similar to the XPS elemental results shown in Table S4 (ESI†). Also, the SVs can enhance the free charge carrier, expose more active sites, and promote the mobility of the electrolyte ions, which was favorable for electrochemical activity.
4. Photocatalytic performance
4.1. Rhodamine B dye degradation
The assessment of photocatalytic efficiency for the CQD@CdS material was carried out using a comparative analysis with N-CQD@CdS, with RhB dye as the model target molecule.63 The degradation of RhB under visible light illumination was studied using a blank experiment, which showed minimal degradation. To assess the degradation efficiency of the photocatalysts, 25 mg of each catalyst was used for a 50 mg L−1 concentration of the RhB dye, and the decrease of the absorption peak was monitored under visible light conditions. The results in Fig. 5c indicate that the N-CQD@CdS achieved a degradation efficiency of 52%, whereas the CQD@CdS resulted in 97% degradation of RhB within 60 min. Fig. 5a and b illustrate the decline in RhB dye concentration over time periods of 115 min and 60 min for N-CQD@CdS and CQD@CdS, respectively. The N-CQD@CdS reached its saturation point after half of the time period, whereas the CQD@CdS continued to degrade the dye because of the strong adsorption of toxic components on catalyst surface causing the catalyst to be completely oxidized.83 To understand the kinetics and mechanism of the RhB dye mineralization with optimized catalytic loading, Langmuir–Hinshelwood and pseudo-second order kinetic models were utilized. The Langmuir–Hinshelwood model, which is also known as a pseudo-first order model is given by eqn (9):| ln(Ct/C0) = −Kappt | (9) |
| (1/Ct) − (1/C0) = Kappt | (10) |
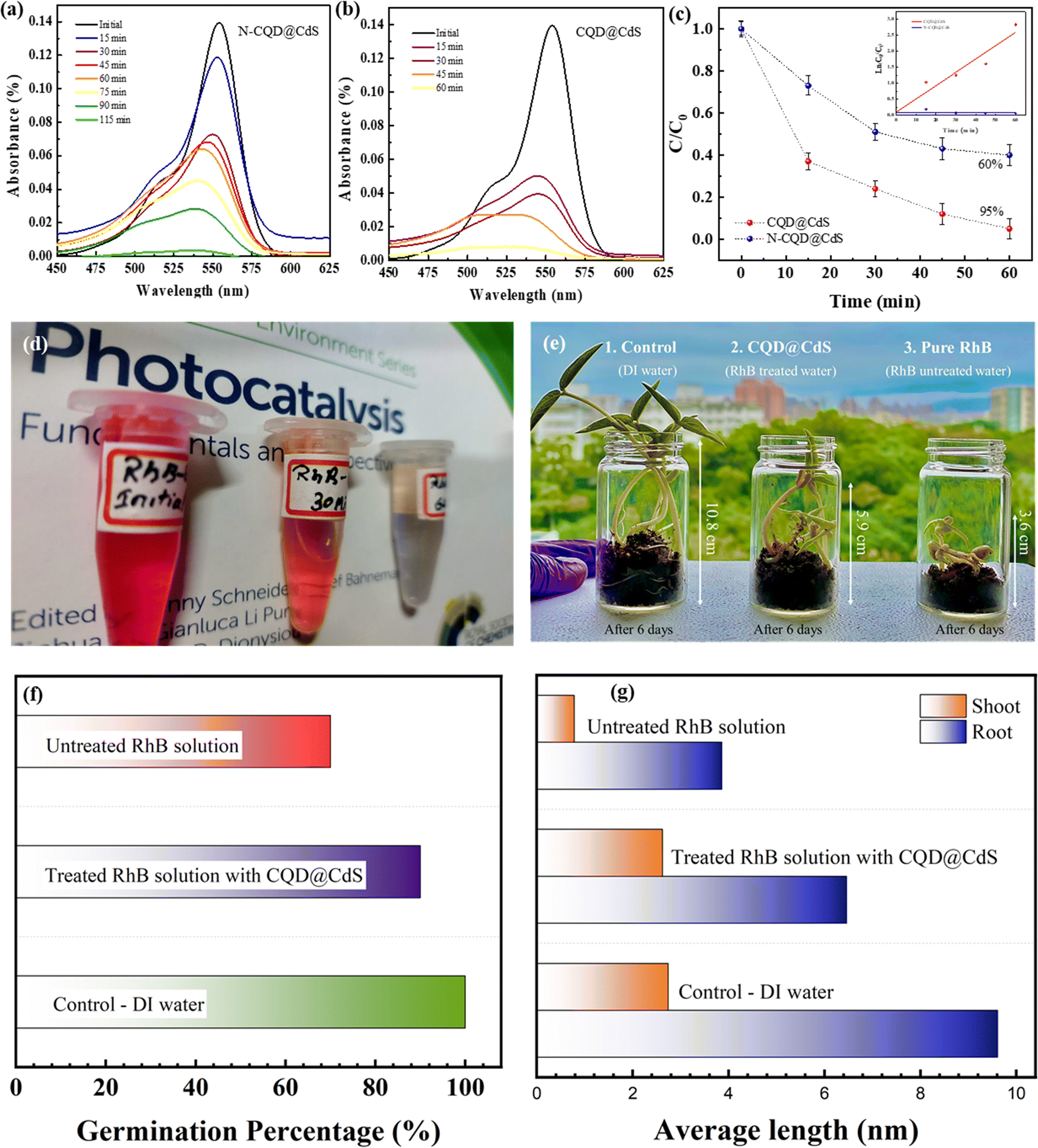 | ||
| Fig. 5 The Rhodamine B degradation absorbance spectra by N-CQD@CdS (a), and CQD@CdS samples (b). Dye degradation efficiency and pseudo-second order kinetic plot (c), change of dye over a 60 min time period (d), Vigna radiata growth rate profile with different sources of water (e) and growth rate (f), roots and shoots lengths (g). | ||
The linear fit between ln(1/Ct) against t is shown in Fig. 5d. Further, many previous reports have been done, and these show that with an increased amount of the pollutant, the degradation efficiency decreased which was due to a smaller number of active sites being available on the photocatalyst, which limited the hydroxyl and superoxide anion radicals generated.62
A comparative analysis of the degradation of the RhB dye using other reported materials is shown in Table S2 (ESI†). Bhavsar et al., synthesized activated carbon loaded CdS nanoflowers using a hydrothermal method that showed a degradation of 95% of RhB in 295 minutes.79 Similarly, Fan et al., reported a CdS heterostructure photocatalyst made by combining it with nonporous g-C3N4 nanoparticles and their combined effort degraded the RhB by 88.2% in 90 min.84 A unique Y-shaped CQD embedded on CdS nanorods was reported by Moniruzzaman et al., in which they successfully degraded 98% of RhB dye in 45 min.85 This higher efficiency was attributed to the Y-shape of the CQD that helped to increase the visible light absorption. In the research here, the CQD decorated the CdS nanostructures with surface defects which were synthesized by a hydrothermal process. Further, the dye degradation efficiency was checked with RhB, and degradation of 97% was achieved in 60 min. This improvement was attributed to the larger surface area of CdS which facilitated the superior adsorption of the dye. In addition, there was the presence of CQD with more active sites with functional groups (–COOH, –OH, –NO2) that interacted with the RhB molecules. This also increased the lifetime of the electron–hole charge carriers and enhanced their separation (Fig. 1f). Overall, the synergistic effects of adsorption and photocatalysis contributed to the improved degradation of RhB by the CQD@CdS composites.51,52 The ameliorated disintegration of RhB molecules by CQD@CdS composites was the collaborative outcome of adsorption and photocatalysis.
Further, a shift of absorbance was observed as the dye concentration gradually decreased (Fig. 5a and b). Similar findings were reported by Ahsaine et al.,86 who noted two possible photodegradation pathways for RhB: the cleavage of the complete conjugated chromophore and N-deethylation. If the degradation of RhB involves the cleavage of a conjugated chromophore, the peak intensity decreases while the peak position remains constant. However, if it follows the N-deethylation photodegradation pathway, the peak position gradually shifts to a lower wavelength (blue shift). In our study, it was observed that photodegradation of RhB follows a N-deethylation pathway, as shown by the main peak position shifting from 554 nm to 498 nm. The possible mechanism for this is shown in Fig. S10 (ESI†).
Further, the phytotoxicity experiments were carried out to evaluate the potential use of the pre-treated aqueous solution in the irrigation field, as well as the environmental impact of the release of the degrading solution. In fact, this technique can alleviate the burden of excessive underground water use, while encouraging the use of treated water to irrigate golf courses, parks, and gardens. Fig. 5e represents the phytotoxicity of untreated and treated RhB and DI solutions using a seed germination assay. The stubborn nature of RhB and DI was reflected in the fact that the untreated solution strongly inhibited seed germination in mung bean seeds (V. radiata) when compared to use of treated and distilled water solutions over 72 h. It was observed, that the pollutant molecule in the presence of a dye solution influenced plant length and seed germination. In seed germination, the percentage value when compared to control DI water (100%), and the RhB solution shows 70% and 82.2% degradation, respectively (Fig. 5f). These results clearly show that the toxicity of the degraded dye solution was within an acceptable range. Another significant length profiling study was also conducted (Fig. 5g), and this study also showed that the degraded dye solution was less toxic. These findings suggested that the photocatalytic degradation of RhB and DI aqueous solutions can reduce phytotoxicity. Hence, seed germination might be a suitable for the reuse of treated water.
4.2. Hydrogen generation
The overall water splitting (OWS) hydrogen generation of the developed photocatalyst were examined using a solar simulator with visible light irradiation (λ ≥ 420 nm). The photocatalysts were suspended in a solvent mixture containing TEA, MeOH together with DI water (1:1:10) and the evolved H2 was measured using a gas chromatography workstation. The TEA acted as a sacrificial electron donor and the MeOH was used to reduce the phase segregation between TEA and water.87Fig. 6a illustrates the sustained H2 production exhibited by all three photocatalysts over a period of 10 h, with their performance in the sequence of CQD@CdS > PVP@CdS-S > N-CQD@CdS. Remarkably, the CQD@CdS composites manifested a superior H2 evolution (3928 μmol g−1 h−1), which was nearly four-fold higher than that of the bare PVP@CdS (965 μmol g−1 h−1). This enhancement in H2 generation rate performance was attributed to the presence of CQD on the CdS surface as shown in Fig. 6b. However, a conspicuous decline in efficiency was observed in nitrogen doped CQD CdS samples, and a HER of 907 μmol g−1 h−1 was recorded. This can be attributed to the nitrogen impeding the active sites on the CQD surface, consequently diminishing its effective surface area (Fig. 3d), and this resulted in higher electron–hole recombination (Fig. 1f).
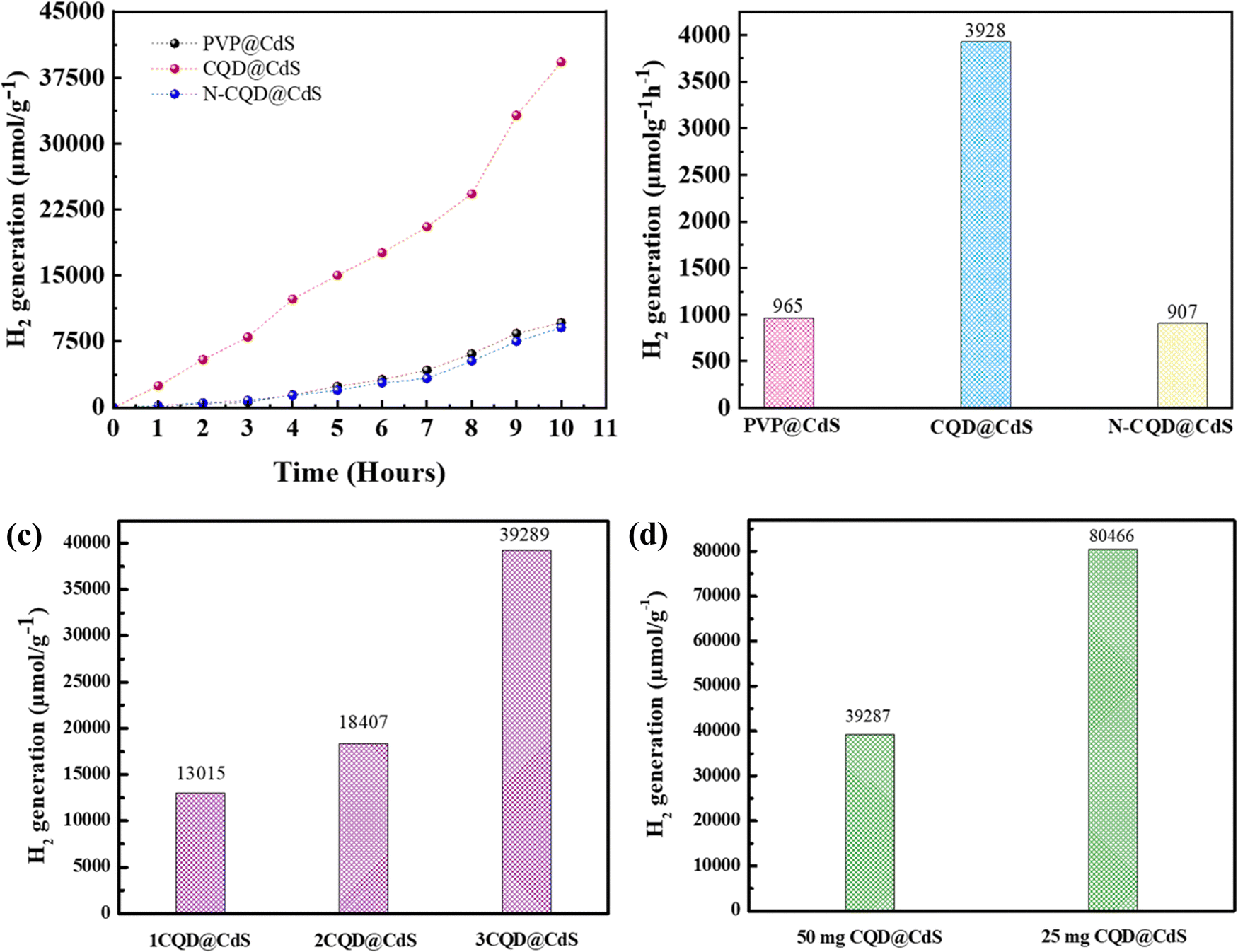 | ||
| Fig. 6 The time dependent photocatalytic hydrogen generation (a), the average hydrogen generation rate over 10 h by all the CdS samples (b). The hydrogen generation rate with varying ratios of the amounts of CQD (c), and the amounts of the CQD@CdS photocatalyst (d), used for reactions. | ||
Furthermore, the evaluation of the optimal loading of CQD (1 mL, 2 mL and 3 mL) on the CdS surface is shown in Fig. 6c. When 1 mL or 2 mL of CQD was used it showed a reduced H2 generation activity compared to when 3 mL of CQD was used. Conversely, an increase in the amount CQD, again resulted in a decrease in H2 generation as it can lead to blocking of the CdS surface. When the CQD amount was low, it was not able to absorb sufficient visible light for CdS. Therefore, maintaining an optimal amount of CQD is crucial to achieving the maximum enhancement in the photocatalytic performance. The AQY of all the three developed photocatalysts was calculated (eqn (1)), and the values were: PVP@CdS – 2.3%, N-CQD@CdS – 2.2% and CQD@CdS – 20.98%. The observed high AQY of CQD@CdS was due to the higher H2 evolution rate compared to other photocatalyst. These CQDs not only lower the overpotential for H2 generation in the CQD@CdS systems, but also serve as a consistent and easily approachable electron acceptor. Moreover, the CQD have a π-conjugated structure and oxygenated functional groups in its surface, which can easily interact with the CdS matrix.88 This interaction serves as an electronic buffer, enabling the continuous supply of electrons to the adjacent CdS semiconductor with appropriate band edge positioning, leading to H2 generation from H+.
287 μmol g−1 and 80466 μmol g−1, respectively, (Fig. 6d). At higher concentrations of photocatalyst, the catalytic system became turbid and this impeded the light irradiation.89 The reusability of the photocatalyst developed was examined using three consecutive cycles up to 20 h (Fig. S5, ESI†). Also, photocurrent analysis (Fig. S6, ESI†) of the CQD@CdS showed the long-term stability due to the CQD loading. It was observed that the H2 evolution remained almost constant for the first two cycles but was reduced in the third cycle, and this was possibly due to the oxidation of the radical scavenger during the redox reaction. Moreover, to verify the stability of the CQD@CdS, we measured the Cd2+ concentrations in the reaction solutions using ICP-MS and the results for this are shown in Table S5 (ESI†). An obvious increase of Cd2+ concentration was observed for PVP@CdS. However, in CQD@CdS, the concentration of Cd2+ was still low which supported its ability to hinder photocorrosion.
:1:10). A 1.5G solar simulator with a cut-off filter of λ > 420 nm was used as visible light source. The H2 evolution was monitored continuously every hour for 10 h (Fig. 7a) and the average H2 generation rates are shown in (Fig. 7b). Similar to the H2 evolution activity in DI water, also the trend here remained the same in the order of CQD@CdS > PVP@CdS > N-CQD@CdS. However, a decrease in the H2 generation rate could be seen in seawater. This was due to presence of additional salts and ions that reduced the charge carrier mobility. However, the catalyst developed showed a superior H2 generation rate compared to others reported in the literature. This shows that the use of CQD on the CdS surface together with its mixed phase structure and presence of SVs enhanced the stability of the photocatalyst.
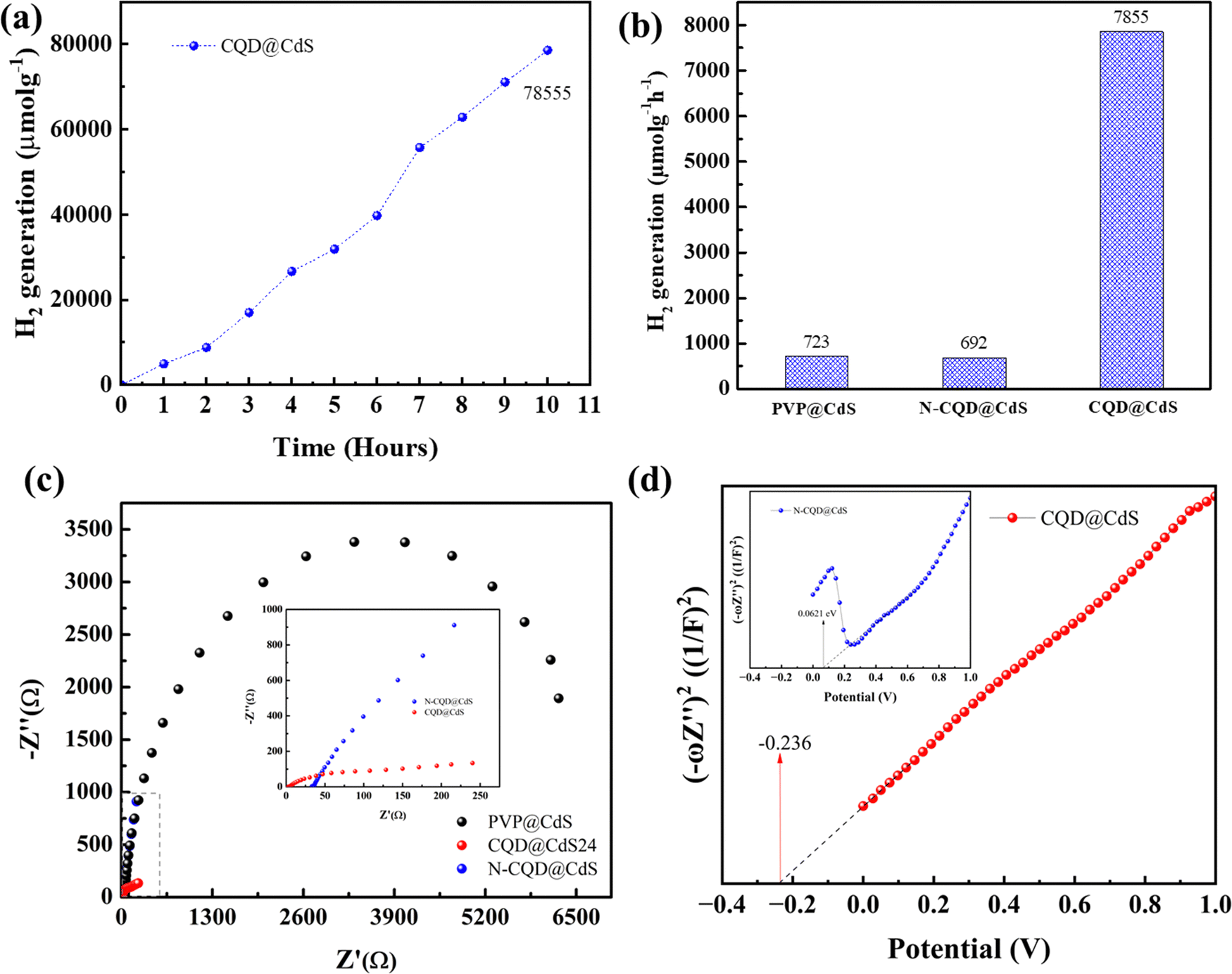 | ||
| Fig. 7 Hydrogen generation (a) using seawater with CQD@CdS for 10 hours. A comparison of the hydrogen generation rate using seawater (b), EIS spectra (c) and Mott–Schottky plot (d) for all the CdS samples. | ||
To elucidate the governing effect of CQD and N-CQD on photocatalytic H2 generation of CdS electrical impedance spectroscopy (EIS), Fig. 7c) and the Mott–Schottky (M–S, Fig. 7d) energy level diagram was studied. The EIS spectra of bare PVP@CdS, CQD@CdS and N-CQD@CdS under light irradiation are shown in Fig. 7c. The arc radius in the EIS plot represents the charge separation and transport resistance between the water and the photoelectrode. The arc radius of CQD@CdS was reduced when compared to those of N-CQD@CdS and bare PVP@CdS, that confirms their better, faster charge transport kinetics and which can be also seen in the photocurrent results (Fig. S6, ESI†) and the PL observations (Fig. 1f). The CQD@CdS shows the lowest resistance among all the developed photocatalysts that support its superior hydrogen generation activity. As is already known, resistance and conductivity are inversely proportional to each other, and the lower resistance of CQD@CdS signifies the increase in electrical conductivity which was due to the CQD electron acceptor on its surface.
The M–S plots of all the CdS samples were determined in a 0.2 M aqueous solution of Na2SO4 using eqn (11). From these plots, the flat band potential was calculated as the intersection point of the tangent line of the M–S curves and the x-axis (Fig. 7d). The flat band potentials of the samples were estimated to be −0.236 and 0.0621 vs. SCE for CQD@CdS and N-CQD@CdS, respectively. For efficient photocatalysis, the CB of CdS should more negative than its flat band potential, thus the final CB potential was converted to potential corresponding to a normal hydrogen electrode (ENHE) using eqn (12) as shown in Table 2 and Scheme 1. The obtained ECB values for CQD@CdS and N-CQD@CdS24 were 0.004 and 0.2337 vs. NHE, respectively.
| 1/C2 = 2 (NDeεε0)−1·(E − EFB − kT/e) | (11) |
| ENHE = ESCE + 0.24 V | (12) |
| ECB = EVB − Eg | (13) |
| Sample | E g (eV) | E FB (eV) | E CB (eV) | E VB (eV) |
|---|---|---|---|---|
| CQD@CdS | 2.26 | −0.236 | 0.004 | 2.264 |
| N-CQD@CdS | 2.20 | 0.0621 | 0.2337 | 2.433 |
5. Discussion
The photo-corrosion of CdS occurs mainly due to the oxidation of the sulfide ions by photogenerated holes as shown in eqn (14):| CdS + 2hVB+ → Cd2+ + S | (14) |
Tang et al.,90 demonstrated the curbing of the CdS photo-corrosion by applying an amorphous carbon layer. They also examined photodegradation and noted that the fortification of CdS in the presence of CQD, lead to an increase in the photocatalytic efficacy. Similarly, Zhu et al., synthesized a stable photocatalyst using carbon dots on CdS nanoparticles that generated an H2 rate of 2.55 μmol g−1 h−1 in absence of a SRs.58 Zero-dimensional carbon dots and a one-dimensional CdS nanowire heterostructure were reported by Chen et al.,13 which showed an H2 activity of 1633 μmol g−1. Zhu et al.,56 revealed the shape dependent photocatalytic H2 generation of the CdS nanostructures with CDs loaded on it. The study showed that the CDs/CdS nanosheets showed the highest H2 generation rate of 260 μmol g−1 h−1. Previously carbon loaded CdS nanostructures have been reported for use in H2 generation however, the efficiency was still found to be low. In this research, the CQD@CdS showed superior H2 generation rate of 8046 μmol g−1 h−1. This was due to CdS having mixed phase structure with SVs that help in increasing the catalytic reaction. Also, the CQD on the surface of CdS acts to capture electrons from the CB of CdS and shields CdS from reactive species, particularly OH radicals/intermediate species S− to S2−. In addition, holes were captured by SR which prevents the photo-corrosion of CQD@CdS.91 A comparison has been shown in Table S3 (ESI†).
Based on the previously mentioned findings, a feasible mechanism for the water splitting action of the CQD@CdS nanocatalysts using a visible light source is proposed in Scheme 2. Upon exposure to light, on the surface of catalyst it excites the electrons to move from the valence band (VB) to the CB leaving holes in the VB of the mixed phase CdS. Usually, these electron–hole pairs have a tendency to recombine with each other. However, due to the phase junction created between the cubic and hexagonal phases, which serves as an energy transition area to minimize energy loss and generates an internal electric field that separates the electrons and holes. These distinct band structures of phases cause the electrons to migrate from the hexagonal to the cubic phase (lower CB edge) and the holes move towards the hexagonal phase (higher VB edge).50 Here some of the electrons get trapped in defect states (Sv) created due to SVs that reduce the probability of the electrons and holes recombining.92 Because the CQDs are a great source for electron acceptors,74 they attract electrons from these trap states that reduce the H+ ions to produce H2 gas. Furthermore, the holes in the VB reacts with TEA to form H2O and CO2.
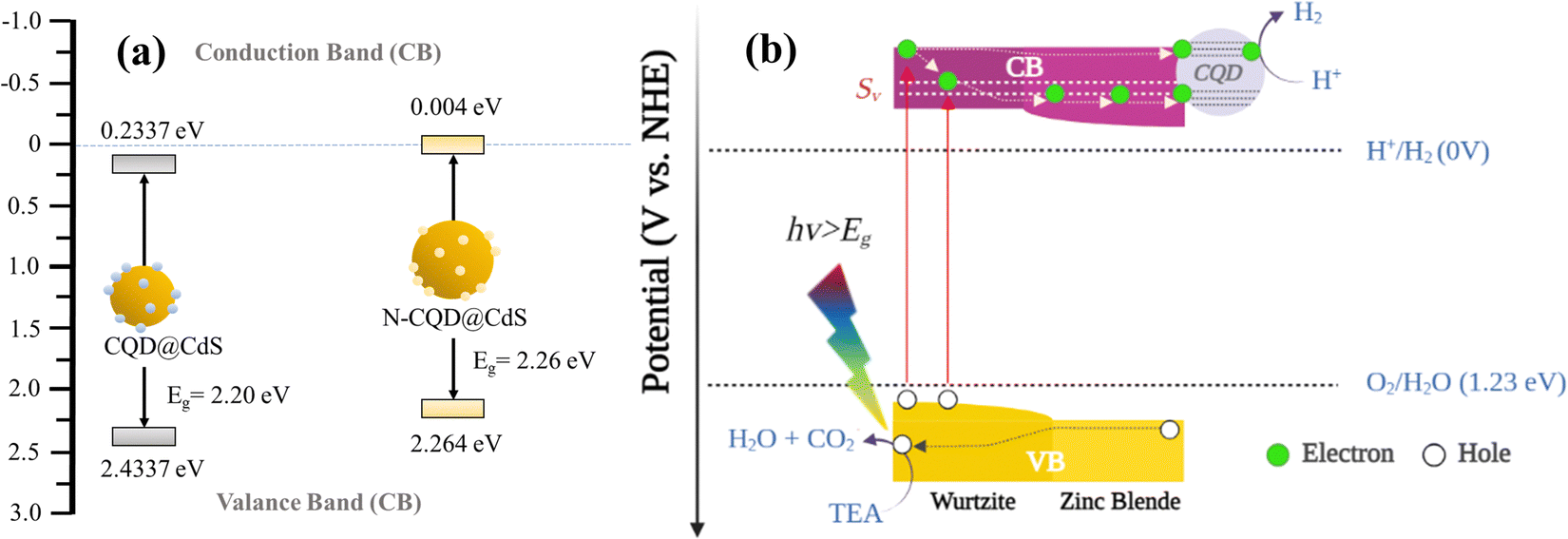 | ||
| Scheme 2 The band positions of CQD@CdS and N-CQD@CdS photocatalyst (a). The photocatalytic hydrogen generation mechanism (b) for phase engineered, sulfur vacancy enriched, and CQD loaded CdS nanostructures. | ||
In some cases it has been observed that the CQDs loading increases the absorption spectrum of the material thereby increasing the catalytic efficiency.93 However, in the present study there has been no significant change in absorbance observed (Fig. 1d) with the introduction of CQD, and this indicates the role of the CQD in catalytic system. Here, CQD did not behave as a photosensitizer, instead it acted as a buffer zone to store electrons, thus supporting the photoinduced charge separation94 and giving an outstanding HER performance.58
A sustainable way for the production of green H2 was evaluated in this work. The present renewable H2 generation systems pricing is at least two-fold more expensive than H2 generated from fossil fuels. This research provides potential insights that could decrease electrolyzer costs, replaces the traditional Pt/Ir catalyst, and adopts a new generation of cost-effective carbon-semiconductor catalysts for superior H2 evolution activity. Until today, most work with “renewable H2” has focused on using clean water, however, we have successfully demonstrated utilization of seawater with minimum loss in H2 evolution rate. It is expected that further understanding of the chemistry of these technologies may bring solutions to address the concerns of water issues in H2 production in near future.
Conclusion
In summary, carbon quantum dots as an efficient cocatalyst have been used with cadmium sulfide nanoparticles which form an effective photocatalyst for hydrogen generation and water treatment. The CQD coupled with CdS exhibited an excellent hydrogen generation rate of 80450 μmol g−1 and Rhodamine B dye degradation was 97% in 60 min. This was due to synergistic effect of the mixed phase, sulfur vacancies and the porous structure of CQD@CdS. The CQD not only successfully separated the photoinduced charge carriers but it also helped in increasing the lifetime of the CdS in seawater conditions up to 30 hours. The apparent quantum yield of the CQD@CdS photocatalyst developed was reported to be up to 20.07%. It is hoped that this research can provide a reliable approach to devising cost-effective and high-performance hybrid photocatalysts, that can effectively support for industrial applications.
Author contributions
Bishal Kumar Nahak drafted the manuscript, designed and performed the experiments, and analyzed the data, Fan-Gang Tseng designed the experiments, supervised the research, and reviewed and modified the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Satish Bolloju from Department of Chemical Engineering, National Taiwan University for use of the X-ray diffraction facility; the Department of Chemical Engineering and Engineering and System Science, National Tsing Hua University for allowing use their of their scanning electron microscope and transmission electron microscope; the Department of Materials Science Engineering, National Yang Ming Chiao Tung University for help with the X-ray photoelectron auger spectroscopy; the Instrumentation Center at National Tsing Hua University for their support with the electron paramagnetic resonance and induction-coupled plasma mass spectroscopy. The authors are thankful for the financial support for this work from the Ministry of Science and Technology, Taiwan (MOST 110-2634-F-007-023-MY3), and the Ministry of Education, Taiwan, Frontier Research Centre on Fundamental and Applied Sciences of Matters (MOE-111QR001I5).References
- B. K. Nahak, S. Preetam, D. Sharma, S. K. Shukla, M. Syväjärvi, D.-C. Toncu and A. Tiwari, Renewable Sustainable Energy Rev., 2022, 161, 112393 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Titanium Dioxide-Based Nanomaterials for Photocatalytic Fuel Generations, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- N. Z. Muradov and T. N. Veziroğlu, Int. J. Hydrogen Energy, 2008, 33, 6804–6839 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Solar Water Splitting Cells, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- C. Acar, I. Dincer and C. Zamfirescu, Int. J. Energy Res., 2014, 38, 1903–1920 CrossRef CAS.
- H. Ahmad, S. K. Kamarudin, L. J. Minggu and M. Kassim, Renewable Sustainable Energy Rev., 2015, 43, 599–610 CrossRef CAS.
- J. Gong, S. B. Darling and F. You, Energy Environ. Sci., 2015, 8, 1953–1968 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- C. Gomes Silva, R. Juárez, T. Marino, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 595–602 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- G. Di, Z. Zhu, Q. Dai, H. Zhang, X. Shen, Y. Qiu, Y. Huang, J. Yu, D. Yin and S. Küppers, Chem. Eng. J., 2020, 379, 122296 CrossRef CAS.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- T. Arshad, S. A. Khan, M. Faisal, Z. Shah, K. Akhtar, A. M. Asiri, A. A. Ismail, B. G. Alhogbi and S. B. Khan, J. Mol. Liq., 2017, 241, 20–26 CrossRef CAS.
- Y. Qiao, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Chem. Commun., 2019, 55, 4067–4070 RSC.
- H. M. Lwin, W. Zhan, S. Song, F. Jia and J. Zhou, Chem. Phys. Lett., 2019, 736, 136806 CrossRef CAS.
- A. M. Al-Hamdi, U. Rinner and M. Sillanpää, Process Saf. Environ. Prot., 2017, 107, 190–205 CrossRef CAS.
- P. K. Labhane and G. H. Sonawane, Inorg. Chem. Commun., 2020, 113, 107809 CrossRef CAS.
- Y. Jia, Y. Ma, L. Zhu, J. Dong and Y. Lin, Chem. Phys., 2019, 521, 1–4 CrossRef CAS.
- R. Roshan, B. K. Nahak, D. Mahata, P. Yadav, S. Panda, S. Patra, S. S. Mahato, A. Tiwari and S. Mahata, Energy Convers. Manage., 2023, 283, 116917 CrossRef CAS.
- N. K. Elumalai, C. Vijila, R. Jose, A. Uddin and S. Ramakrishna, Mater. Renew. Sustainable Energy, 2015, 4, 11 CrossRef.
- Y. Xu, Y. Huang and B. Zhang, Inorg. Chem. Front., 2016, 3, 591–615 RSC.
- M. Tayyab, Y. Liu, Z. Liu, Z. Xu, W. Yue, L. Zhou, J. Lei and J. Zhang, Chem. Eng. J., 2023, 455, 140601 CrossRef CAS.
- S. A. Ali and T. Ahmad, Int. J. Hydrogen Energy, 2022, 47, 29255–29283 CrossRef CAS.
- P. Garg, P. Bhauriyal, A. Mahata, K. S. Rawat and B. Pathak, Chem. Phys. Chem., 2019, 383–391 CrossRef CAS PubMed.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jäckel, J. Feldmann, F. Würthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862–869 CrossRef CAS.
- B. Nahak, R. Roshan, N. Jhariya, S. Bolloju, B. Pandit, S. Mahato and S. Mahata, Surf. Interfaces, 2023, 42, 103351 CrossRef CAS.
- S. Panda, A. Sahu, A. Patra, S. Panda, B. K. Nahak, B. N. Patra, S. S. Mahato and S. Mahata, Mater. Today: Proc., 2021, 47, 1197–1202 CAS.
- Y. Xia, K. D. Gilroy, H.-C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- H. Wang, P. Fang, Z. Chen and S. Wang, Appl. Surf. Sci., 2007, 253, 8495–8499 CrossRef CAS.
- M. Pattabi, B. Saraswathi Amma and K. Manzoor, Mater. Res. Bull., 2007, 42, 828–835 CrossRef CAS.
- T. Thongtem, A. Phuruangrat and S. Thongtem, Ceram. Int., 2009, 35, 2817–2822 CrossRef CAS.
- D. Hao, C. Liu, X. Xu, M. Kianinia, I. Aharonovich, X. Bai, X. Liu, Z. Chen, W. Wei, G. Jia and B.-J. Ni, New J. Chem., 2020, 44, 20651–20658 RSC.
- Q. Wang, S. Zhu, S. Zhao, C. Li, R. Wang, D. Cao and G. Liu, Fuel, 2022, 322, 124163 CrossRef CAS.
- B. K. Nahak, D. Mahata, N. Jhariya, P. Yadav, S. Panda, S. S. Sahu, K. Swatishree, A. P. Khedulkar, S. Bolloju, B. Pandit, S. S. Mahato and S. Mahata, Ceram. Int., 2023, 49, 32104–32115 CrossRef CAS.
- Q. Lin, S. Liang, J. Wang, R. Zhang, G. Liu and X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 3093–3102 CrossRef CAS.
- X. Li, J. Wang, J. Zhang, C. Zhao, Y. Wu and Y. He, J. Colloid Interface Sci., 2022, 607, 412–422 CrossRef CAS PubMed.
- C. Liu, Y. Xiao, W. Wan, Y. Wei, Y. Cao, L. Hong, Y. Wang, J. Chen, Q. Zhang and H. Jing, Appl. Catal., B, 2023, 325, 122394 CrossRef CAS.
- L. Zhang, X. Jiang, Z. Jin and N. Tsubaki, J. Mater. Chem. A, 2022, 10, 10715–10728 RSC.
- H. Sun, Z. Xiao, Z. Zhao, S. Zhai and Q. An, Appl. Surf. Sci., 2023, 611, 155631 CrossRef CAS.
- Y. Su, X. Xu, R. Li, X. Luo, H. Yao, S. Fang, K. Peter Homewood, Z. Huang, Y. Gao and X. Chen, Chem. Eng. J., 2022, 429, 132241 CrossRef CAS.
- X. Bai, X. Wang, X. Lu, Y. Liang, J. Li, L. Wu, H. Li, Q. Hao, B.-J. Ni and C. Wang, J. Hazard. Mater., 2020, 398, 122897 CrossRef CAS PubMed.
- Z. Jin, T. Wei, F. Li, Q. Zhang and L. Xu, New J. Chem., 2020, 44, 3471–3477 RSC.
- H. Yu, W. Zhong, X. Huang, P. Wang and J. Yu, ACS Sustainable Chem. Eng., 2018, 6, 5513–5523 CrossRef CAS.
- K. Li, M. Han, R. Chen, S.-L. Li, S.-L. Xie, C. Mao, X. Bu, X.-L. Cao, L.-Z. Dong, P. Feng and Y.-Q. Lan, Adv. Mater., 2016, 28, 8906–8911 CrossRef CAS PubMed.
- Z. Ai, G. Zhao, Y. Zhong, Y. Shao, B. Huang, Y. Wu and X. Hao, Appl. Catal., B, 2018, 221, 179–186 CrossRef CAS.
- I. Velo-Gala, J. J. López-Peñalver, M. Sánchez-Polo and J. Rivera-Utrilla, Appl. Catal., B, 2017, 207, 412–423 CrossRef CAS.
- K. S. Bhavsar, P. K. Labhane, R. B. Dhake and G. H. Sonawane, Inorg. Chem. Commun., 2019, 104, 134–144 CrossRef CAS.
- J. Ge, Y. Zhang and S.-J. Park, Materials, 2019, 12, 1916 CrossRef CAS PubMed.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- H. Ming, Z. Ma, Y. Liu, K. Pan, H. Yu, F. Wang and Z. Kang, Dalton Trans., 2012, 41, 9526–9531 RSC.
- C. Zhu, Y. Fu, C. Liu, Y. Liu, L. Hu, J. Liu, I. Bello, H. Li, N. Liu, S. Guo, H. Huang, Y. Lifshitz, S.-T. Lee and Z. Kang, Adv. Mater., 2017, 29, 1701399 CrossRef PubMed.
- Y. Wang, X. Liu, J. Liu, B. Han, X. Hu, F. Yang, Z. Xu, Y. Li, S. Jia, Z. Li and Y. Zhao, Angew. Chem., 2018, 130, 5867–5873 CrossRef.
- C. Zhu, C. Liu, Y. Zhou, Y. Fu, S. Guo, H. Li, S. Zhao, H. Huang, Y. Liu and Z. Kang, Appl. Catal., B, 2017, 216, 114–121 CrossRef CAS.
- S. Sharma, V. Dutta, P. Singh, P. Raizada, A. Rahmani-Sani, A. Hosseini-Bandegharaei and V. K. Thakur, J. Cleaner Prod., 2019, 228, 755–769 CrossRef CAS.
- Y. Lei, C. Yang, J. Hou, F. Wang, S. Min, X. Ma, Z. Jin, J. Xu, G. Lu and K.-W. Huang, Appl. Catal., B, 2017, 216, 59–69 CrossRef CAS.
- J. Guo, M. Guo, D. Jia, X. Song and F. Tong, Chem. Phys. Lett., 2016, 659, 66–69 CrossRef CAS.
- S. P. Smrithi, N. Kottam, A. Narula, G. M. Madhu, R. Mohammed and R. Agilan, J. Colloid Interface Sci., 2022, 627, 956–968 CrossRef CAS PubMed.
- C. Tang, Y. Zhang, J. Han, Z. Tian, L. Chen and J. Chen, Environ. Pollut., 2020, 262, 114246 CrossRef CAS PubMed.
- C. Zhu, C. Liu, Y. Fu, J. Gao, H. Huang, Y. Liu and Z. Kang, Appl. Catal., B, 2019, 242, 178–185 CrossRef CAS.
- D. Gogoi, R. Koyani, A. K. Golder and N. R. Peela, Sol. Energy, 2020, 208, 966–977 CrossRef CAS.
- Y.-J. Yuan, D. Chen, Z.-T. Yu and Z.-G. Zou, J. Mater. Chem. A, 2018, 6, 11606–11630 RSC.
- H. Arandiyan, S. S. Mofarah, C. C. Sorrell, E. Doustkhah, B. Sajjadi, D. Hao, Y. Wang, H. Sun, B.-J. Ni, M. Rezaei, Z. Shao and T. Maschmeyer, Chem. Soc. Rev., 2021, 50, 10116–10211 RSC.
- M. Kong, Y. Li, X. Chen, T. Tian, P. Fang, F. Zheng and X. Zhao, J. Am. Chem. Soc., 2011, 133, 16414–16417 CrossRef CAS PubMed.
- H. Zhang, H. Wang, Y. Wang and B. Xin, Appl. Surf. Sci., 2020, 512, 145751 CrossRef CAS.
- C. Carrillo-Carrión, S. Cárdenas, B. M. Simonet and M. Valcárcel, Chem. Commun., 2009, 5214–5226 RSC.
- H. Li, W. Kong, J. Liu, N. Liu, H. Huang, Y. Liu and Z. Kang, Carbon, 2015, 91, 66–75 CrossRef CAS.
- A. Sharma, T. Gadly, S. Neogy, S. K. Ghosh and M. Kumbhakar, J. Phys. Chem. Lett., 2017, 8, 1044–1052 CrossRef CAS PubMed.
- M. Ma, Y. Liu, Y. Wei, D. Hao, W. Wei and B.-J. Ni, J. Environ. Manage., 2021, 294, 113046 CrossRef CAS PubMed.
- M. Wang, Z. Wang, B. Zhang, W. Jiang, X. Bao, H. Cheng, Z. Zheng, P. Wang, Y. Liu, M.-H. Whangbo, Y. Li, Y. Dai and B. Huang, ACS Catal., 2020, 10, 13031–13039 CrossRef CAS.
- A.-M. Alam, B.-Y. Park, Z. Khan Ghouri, M. Park and H.-Y. Kim, Green Chem., 2015, 17, 3791–3797 RSC.
- B. K. Nahak, R. Roshan, S. D. Roy, A. Patra, A. Sahu, S. Panda, S. Panda, S. S. Mahato and S. Mahata, J. Mater. Sci.: Mater. Electron., 2022, 33, 15191–15208 CrossRef CAS.
- A. Sachdev and P. Gopinath, Analyst, 2015, 140, 4260–4269 RSC.
- Y. Pei, R. Pei, X. Liang, Y. Wang, L. Liu, H. Chen and J. Liang, Sci. Rep., 2016, 6, 21551 CrossRef CAS PubMed.
- K. S. Bhavsar, P. K. Labhane, R. B. Dhake and G. H. Sonawane, Chem. Phys. Lett., 2020, 744, 137202 CrossRef CAS.
- L. Wu, J. C. Yu and X. Fu, J. Mol. Catal. A: Chem., 2006, 244, 25–32 CrossRef CAS.
- X. Bai, X. Wang, T. Jia, L. Guo, D. Hao, Z. Zhang, L. Wu, X. Zhang, H. Yang, Y. Gong, J. Li and H. Li, Appl. Catal., B, 2022, 310, 121302 CrossRef CAS.
- A. Veamatahau, B. Jiang, T. Seifert, S. Makuta, K. Latham, M. Kanehara, T. Teranishi and Y. Tachibana, Phys. Chem. Chem. Phys., 2015, 17, 2850–2858 RSC.
- W. Zou, B. Gao, Y. S. Ok and L. Dong, Chemosphere, 2019, 218, 845–859 CrossRef CAS.
- Q. Fan, Y. Huang, C. Zhang, J. Liu, L. Piao, Y. Yu, S. Zuo and B. Li, Catal. Today, 2016, 264, 250–256 CrossRef CAS.
- M. Moniruzzaman and J. Kim, Appl. Surf. Sci., 2021, 552, 149372 CrossRef CAS.
- H. Ait Ahsaine, M. Ezahri, A. Benlhachemi, B. Bakiz, S. Villain, F. Guinneton and J.-R. Gavarri, Ceram. Int., 2016, 42, 8552–8558 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- W. Shi, F. Guo, M. Li, Y. Shi and Y. Tang, Sep. Purif. Technol., 2019, 212, 142–149 CrossRef CAS.
- N. N. Yunus, F. Hamzah, M. S. So’aib and J. Krishnan, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 206, 012092 Search PubMed.
- Y. Tang, X. Hu and C. Liu, Phys. Chem. Chem. Phys., 2014, 16, 25321–25329 RSC.
- S. Feizpoor, S. Rahim Pouran and A. Habibi-Yangjeh, Mater. Sci. Semicond. Process., 2023, 162, 107444 CrossRef CAS.
- J. Shi, J. Zhang, Z. Cui, S. Chu, Y. Wang and Z. Zou, Dalton Trans., 2022, 51, 5841–5858 RSC.
- M. Sabri, A. Habibi-Yangjeh, S. Rahim Pouran and C. Wang, Catal. Rev., 2023, 65, 118–173 CrossRef CAS.
- A. Habibi-Yangjeh and K. Pournemati, Crit. Rev. Environ. Sci. Technol., 2023, 1–31 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00741c |
| This journal is © The Royal Society of Chemistry 2024 |