
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of thermal conducting properties in epoxy thermoset systems using an aligned liquid-crystalline mesophase†
Thi En
Trinh
a and
Hyeonuk
Yeo
 *abcd
*abcd
aDepartment of Science Education, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea. E-mail: yeo@knu.ac.kr
bDepartment of Chemistry Education, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea
cDepartment of Nanoscience & Nanotechnology, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea
dDepartment of Pharmacy, Kyungpook National University, 80, Daehak-ro, Buk-gu, Daegu 41566, Republic of Korea
First published on 4th January 2024
Abstract
Liquid crystalline epoxy resins (LCERs) with high thermal conductivity (T/C) are an attractive solution that meet the thermal management requirements of electronic devices. The ordered microstructure derived from LCs can improve heat dissipation in the polymer network by reducing phonon scattering. Here a series of LCE systems named EB-n is successfully developed by connecting two biphenyl mesogens with several aliphatic spacers (n). The EB-n monomers with even spacers (n = 4, 6, 8) exhibit a wide nematic phase range between 160 °C and 230 °C, while EB-7 shows a weak mesophase only during heating, indicating that the spacer length controls the LC properties and molecular ordering. The curing reaction repairs the network using symmetric and asymmetric aromatic diamines capable of reacting in an aligned LC state. The resulting products exhibit high T/C with values ranging between 0.519 and 0.757 W m−1 K−1, which are about three times higher than those of conventional thermosets, and superior thermal and mechanical properties. Moreover, thermal infrared imaging demonstrates that their high T/C has an apparent effect on heat dissipation. These results suggest that the LCE system has many potential applications in the high-performance electric device industry.
Introduction
Thermoset materials have dominated the electronics market in recent years due to their outstanding features such as low weight, high mechanical strength, and compression resistance, in addition to strengths in composite manufacturing.1–3 Epoxy resins (ERs) are of particular interest to researchers and are widely used in fields such as electronics and aerospace engineering because of their special properties, such as easy curing, superior processability, high thermal resistance, good thermomechanical properties, and environmental stability.4–8 However, traditional ERs have an amorphous network structure, which makes them act as insulators with low thermal conductivity (T/C).9,10 In modern times, there is a growing need for thermal management systems and size minimization in high-performance electronic devices with integrated functions.11,12 To avoid malfunctions caused by heat accumulation, plastic parts must have high T/C to dissipate the heat generated during operation as rapidly as possible. This problem has attracted significant attention from researchers. This heat diffusion problem can be overcome by using composite materials that contain a polymer matrix and fillers, such as alumina, carbon fiber, and SiC, which can increase the thermal dissipation capacity of the materials.13–16 However, the T/C of the composites mainly depends on the amount of filler. Unfortunately, when the filler loading exceeds a certain content, phase separation occurs, resulting in increased interfacial resistance and a sudden decrease in material properties that do not meet application requirements.17,18 Therefore, the development of polymer matrices with high intrinsic T/C is necessary to improve the heat dissipation proficiency of such materials.19–21In organic polymers, heat transfer occurs through phonon vibration because of the absence of free electrons. The Debye equation expresses T/C values as follows: T/C = Cpvl/3.22 Here, Cp is the specific heat capacity per unit volume, v is the phonon velocity or sound velocity, and l is the mean free path of the phonon. For most single-chain polymers, Cp and v values are not significantly different. Therefore, to increase T/C, l needs to be enhanced. In polymer matrices with an amorphous structure, the value of l is ultra-low due to phonon scattering.23 Hence, increasing the crystallinity of the matrix by boosting the alignment of molecules is the best solution. From this point of view, previous studies have indicated that liquid crystalline epoxy resins (LCERs), which contain a rigid-rod backbone, retain their alignment in the cured products and LC states, resulting in a highly ordered structure that facilitates heat transfer in polymer networks by reducing phonon scattering and enhances thermal and mechanical properties.24–26 Particularly, LCERs based on biphenyl and phenyl benzoate have shown significant enhancement in thermal conducting properties and achieved T/C values that are five times higher than those of general-purpose ERs, although they were processed under fairly specific conditions.27–29 In this regard, most studies have confirmed that the possibility of breaking the T/C limit is quite high, but it requires special conditions such as very thin film states, molding under magnetic fields or high pressure, or the use of specific mixtures, which are difficult to use under general conditions and as composite materials with fillers.30–32
Our group has focused on developing polymeric materials with high T/C that can be used as bulk materials under normal conditions, and we believe that we have made significant progress in this area. For instance, we started with the simplest LCER, BP, and found that a suitable selection of curing agents and an efficient curing reaction play a crucial role in determining the physical properties of LCERs.33–37 Additionally, various LCERs with specific molecular structures that enhance mesogenic interactions have been reported.38–43 One such material is the twin mesogen-based bifunctional epoxy system, which has shown remarkable potential for improving T/C.41–43 As an extension of this strategy, the present study focuses on diversifying the spacer lengths in the polymers while retaining the simple biphenyl mesogens. Moreover, an LCER series containing an odd-numbered spacer system cured with two different kinds of curing agents has not yet been investigated. By maximizing self-assembly, the thermal and mechanical properties of the LCERs were investigated, and a pure thermoset resin system with high T/C values ranging between 0.519 and 0.757 W m−1 K−1 could be developed. As a result, highly functional bulk materials were obtained, which could be used without any special processing. These material development strategies and self-assembly maximization methods have significant implications in related fields and are expected to provide insights for the development of other high-performance heat dissipation materials.
Experimental
General
Chemical structures of the synthesized materials were determined using an NMR spectrometer (NMR, AVANCE III 500, Bruker). Their 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were obtained using CDCl3 as a solvent and tetramethylsilane (TMS) as an internal standard (taken as 0.0 ppm). FT-IR spectroscopy (FT/IR-4100, JASCO) was used to examine particular functional groups in the cured thermosets that could not be dissolved in common organic solvents. The molecular weights of the synthesized materials were determined using a mass spectrometer (MS, Xevo TQ-S micro, Waters). The thermal properties of monomers, curing mixtures, and cured samples were obtained using a DSC (DSC 25, TA Instruments) under N2 gas. Polarized optical microscopy (POM, BX53M, Olympus) and a precise hot stage (LINKAM, LTS420) were employed to observe the mesophase of the samples. Small amounts of compounds were sandwiched between two glass slides and preheated to form a homogeneous thin film on the glass surface. Then, the samples were heated and cooled repeatedly from room temperature to 250 °C at a rate of 1 °C min−1 to investigate the shift of birefringence. Moreover, POM was used to observe the formation of the mesophase during the curing process. To investigate the microstructures of cured specimens, X-ray diffraction analysis (XRD, EMPYREAN, Malvern Panalytical) was carried out in the 2θ range from 0° to 40°and a scan rate of 2° per minute. Thermogravimetric analysis (TGA, Q500, TA Instruments) was employed to investigate the thermal stability of the samples. The samples were put in an N2 gas environment and heated from room temperature to 1000 °C at a heating rate of 20 °C per minute. Thermomechanical properties of the cured samples were investigated using a DMA (DMA Q850, TA Instruments). The samples fabricated in rectangular shapes (40 × 5 × 1–2 mm) were heated from room temperature to 250 °C in an air atmosphere at a heating rate of 3 °C min−1 with a 1 Hz frequency. The tensile test was performed using a universal testing machine (UTM, AGS-X, Shimadzu) at a crosshead speed of 2 mm per minute. The thermal conductivity (T/C) was measured by the transient plane source method (TPS 3500, Hot Disk). All the sets of specimens were fabricated in a cylindrical round shape at a 10-mm radius and a 5-mm depth. The data were recorded repeatedly 3 times, and the average value was determined as the final data. To visualize the heat transferring ability of the cured samples, the change in color and temperature with the time of cured samples corresponding to infrared thermal images was recorded using a thermal imaging camera (IR camera, HT-02, HTI).Materials
Adipoyl chloride, sebacoyl chloride, and meta-chloroperoxybenzoic acid (mCPBA) were purchased from Daejung Chemical Company (Korea). Azelaoyl chloride, suberoyl chloride, and [1,1′-biphenyl]-4,4′-diol were purchased from TCI (Japan), and silica gel was obtained from Wako Chemical (Japan). Allyl bromide, 4,4′-diaminodiphenyl sulfone (DDS), and sulfanilamide (SSA) were obtained from Alfa Aesar (USA), and 4-allyloxy-4′-hydroxybiphenyl was synthesized and characterized according to a procedure reported in the literature.44 Common solvents and reagents were ordered from Duksan Chemical Company (Korea) and Daejung Chemical Company (Korea). All purchased chemicals were employed without further purification, and all reactions were conducted in an inert gas atmosphere.Synthesis of LCE monomers (EB-n)
LCE monomers were synthesized according to Scheme 1 in three steps, namely Williamson etherification, esterification, and epoxidation reaction. 4′-(Allyloxy)-[1,1′-biphenyl]-4-ol (A1) was easily synthesized through the one-side substitution of biphenol with allyl bromide.44 Subsequently, the remaining OH group was esterified by various diacid chlorides with different alkyl chain lengths to afford bis(4′-(allyloxy)-[1,1′-biphenyl]-4-yl) alkanedioate (EA-n, n = 4, 6, 7, 8-number carbon of aliphatic spacer). Thereafter, the vinyl groups were oxidized by mCPBA and converted to bifunctional epoxide monomers. All reactions proceeded as expected with appropriate yields. The final compounds, EB-n, were accurately characterized by 1H NMR, 13C NMR, FT-IR, and MS spectrometry, as shown in Fig. S1 and S2a (ESI†).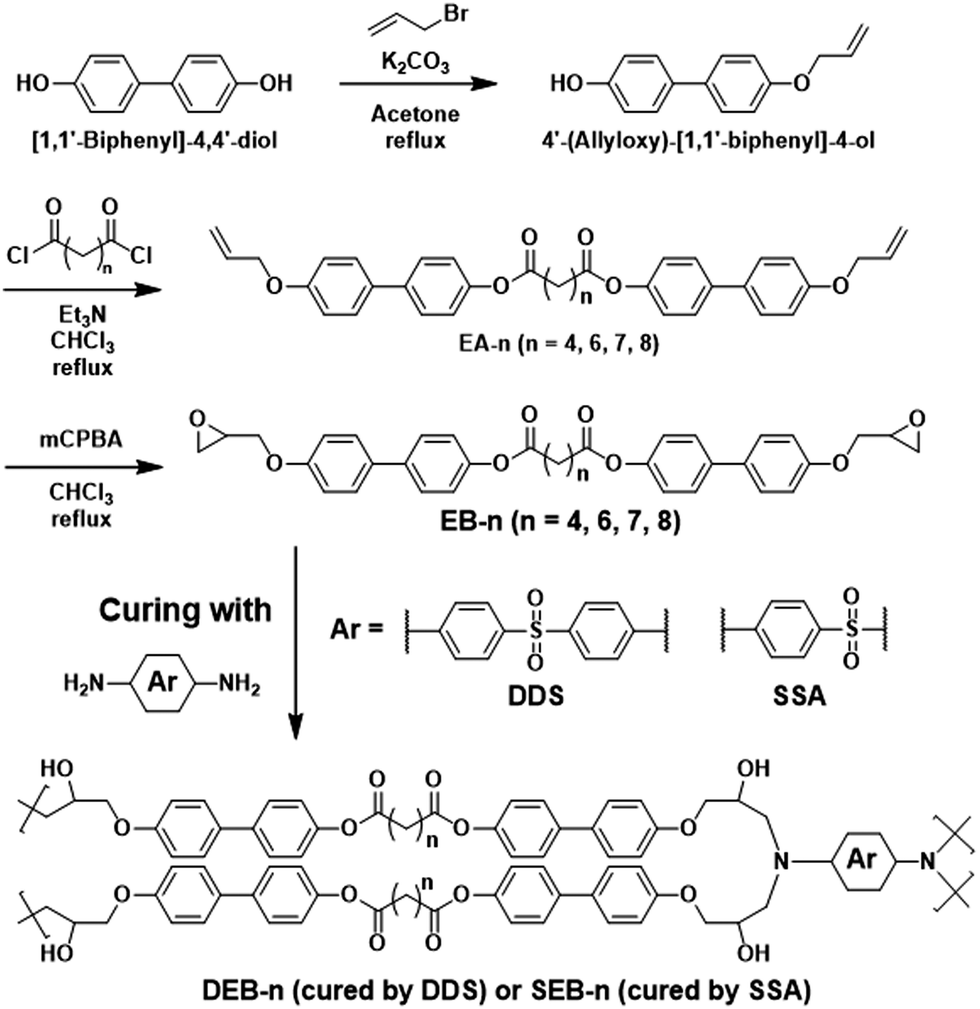 | ||
| Scheme 1 Chemical structures and synthetic scheme of bifunctional liquid crystalline epoxy monomers (EB-n), and their network structures (DEB-n and SEB-n) after curing with diamines (DDS and SSA). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) was used to purify A1 and the final compound was obtained as a white solid in 45.1% yield (5.10 g). 1H NMR (500 MHz, CDCl3): δ 7.45 (d, J = 9 Hz, 2Ar
:1, v/v) was used to purify A1 and the final compound was obtained as a white solid in 45.1% yield (5.10 g). 1H NMR (500 MHz, CDCl3): δ 7.45 (d, J = 9 Hz, 2Ar![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 7.42 (d, J = 8.5 Hz, 2Ar), 6.96 (d, J = 8.5 Hz, 2Ar), 6.87 (d, J = 9.0 Hz, 2Ar), 6.12–6.04 (ddt, J1 = 17.3 Hz, J2 = 10.5 Hz, J3 = 5.3 Hz, 1H, CH2
), 7.42 (d, J = 8.5 Hz, 2Ar), 6.96 (d, J = 8.5 Hz, 2Ar), 6.87 (d, J = 9.0 Hz, 2Ar), 6.12–6.04 (ddt, J1 = 17.3 Hz, J2 = 10.5 Hz, J3 = 5.3 Hz, 1H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O–), 5.44 (dq, J1 = 17.2 Hz, J2 = 1.6 Hz, 1H, C2CH–O–), 5.30 (dq, J1 = 10.4 Hz, J2 = 1.4 Hz, 1H, C2CH–O–), 4.71 (s, 1H, O), 4.57 (dt, J1 = 5.3 Hz, J2 = 1.6 Hz, 2H, –C2–O–Ar) ppm.
), 7.48 (d, J = 9.0 Hz, 4Ar), 7.14 (d, J = 8.5 Hz, 4Ar), 6.98 (d, J = 9 Hz, 4Ar), 6.08 (ddt, J1 = 17.3 Hz, J2 = 10.6 Hz, J3 = 5.3 Hz, 2H, CH2C–O–), 5.44 (dq, J1 = 17.2 Hz, J2 = 1.7 Hz, 2H, C2 = CH–O–), 5.31 (dq, J1 = 10.5 Hz, J2 =1.4 Hz, 2H, C2CH–O–), 4.58 (dt, J1 = 5.3 Hz, J2 =1.6 Hz, 4H, Ar–O–C2–), 2.63–2.70 (m, 4H, –CO–C2–), 1.95–1.89 (m, 4H, –COCH2–C2–) ppm. 13C NMR (125 MHz, CDCl3), δ 171.85 (–
C–O–), 5.44 (dq, J1 = 17.2 Hz, J2 = 1.6 Hz, 1H, C2CH–O–), 5.30 (dq, J1 = 10.4 Hz, J2 = 1.4 Hz, 1H, C2CH–O–), 4.71 (s, 1H, O), 4.57 (dt, J1 = 5.3 Hz, J2 = 1.6 Hz, 2H, –C2–O–Ar) ppm.
), 7.48 (d, J = 9.0 Hz, 4Ar), 7.14 (d, J = 8.5 Hz, 4Ar), 6.98 (d, J = 9 Hz, 4Ar), 6.08 (ddt, J1 = 17.3 Hz, J2 = 10.6 Hz, J3 = 5.3 Hz, 2H, CH2C–O–), 5.44 (dq, J1 = 17.2 Hz, J2 = 1.7 Hz, 2H, C2 = CH–O–), 5.31 (dq, J1 = 10.5 Hz, J2 =1.4 Hz, 2H, C2CH–O–), 4.58 (dt, J1 = 5.3 Hz, J2 =1.6 Hz, 4H, Ar–O–C2–), 2.63–2.70 (m, 4H, –CO–C2–), 1.95–1.89 (m, 4H, –COCH2–C2–) ppm. 13C NMR (125 MHz, CDCl3), δ 171.85 (–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O), 158.22 (–
O), 158.22 (–![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) –O–CH2–), 149.62 (––OCO–), 138.59, 133.08, 128.11, 127.71, 121.75, 115.06 (––), 133.23 (–HCH2), 117.76 (–CHH2), 68.90 (–H2–O), 34.02 (–OCO–H2–), 24.34 (–OCO–CH2–H2–) ppm. [EA–6, yield: 45.32%, white solid]: 1H NMR (500 MHz, CDCl3), δ 7.53 (d, J = 8.5 Hz, 4Ar), 7.48 (d, J = 9.0 Hz, 4Ar), 7.12 (d, J = 8.5 Hz, 4Ar), 6.98 (d, J = 9 Hz, 4Ar), 6.08 (ddt, J1 = 17.3 Hz, J2 = 10.5 Hz, J3 = 5.3 Hz, 2H, CH2C–O–), 5.44 (dq, J1 = 17.2 Hz, J2 = 1.6 Hz, 2H, C2CH–O–), 5.31 (dq, J1 = 10.5 Hz, J2 =1.4 Hz, 2H, C2CH–O–), 4.58 (dt, J1 = 5.3 Hz, J2 =1.6 Hz, 4H, Ar–O–C2–), 2.60 (t, J = 7.4 Hz, 4H, –CO–C2–), 1.79–1.84 (m, 4H, –CO–CH2–C2–), 1.55–1.48 (m, 4H, –CO–CH2–CH2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.32 (–O), 158.20 (––O–CH2–), 149.68 (––O–CO–), 138.50, 133.09, 128.10, 127.68, 121.76, 115.05 (––), 133.23 (–HCH2), 117.74 (–CHH2), 68.88 (–H2–O), 34.29 (–OCO–H2–), 28.73 (–OCO–CH2–CH2–H2–), 24.75 (–OCO–CH2–H2–) ppm. [EA-7, yield: 80.37%, white solid]: 1H NMR (500 MHz, CDCl3), δ 7.53 (d, J = 8.5 Hz, 4Ar), 7.47 (d, J = 8.5 Hz, 4Ar), 7.12 (d, J = 8.5 Hz, 4Ar), 6.98 (d, J = 8.5 Hz, 4Ar), 6.08 (ddt, J1 = 17.3 Hz, J2 = 10.5 Hz, J3 = 5.3 Hz, 2H, CH2C–O–), 5.44 (dq, J1 = 17.2 Hz, J2 = 1.6 Hz, 2H, C2CH–O–), 5.31 (dq, J1 = 10.5 Hz, J2 =1.4 Hz, 2H, C2CH–O–), 4.58 (dt, J1 = 5.3 Hz, J2 =1.5 Hz, 4H, Ar–O–C2–), 2.59 (t, J = 7.4 Hz, 4H, –CO–C2–), 1.79 (q, J = 7.3 Hz, 4H, –CO–CH2–C2–), 1.53–1.44 (m, 6H, –CO–CH2–CH2–C2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.30 (–O), 158.19 (––O–CH2–), 149.69 (––O–CO–), 138.47, 133.07, 128.08, 127.66, 121.77, 115.04 (––), 133.23 (–HCH2), 117.73 (–CHH2), 68.87 (–H2–O), 34.35 (–OCO–H2–), 28.87 (–CO–CH2–CH2–H2–, –CO–CH2–CH2–CH2–H2–), 24.85 (–CO–CH2–H2–) ppm. [EA-8, yield: 43.5%, white solid]: 1H NMR (500 MHz, CDCl3): δ 7.53 (d, J = 8.5 Hz, 4Ar), 7.48 (d, J = 8.5 Hz, 4Ar), 7.12 (d, J = 9.0 Hz, 4Ar), 6.98 (d, J = 8.5 Hz, 4Ar), 6.08 (ddt, J1 = 17.2 Hz, J2 = 10.5Hz, J3= 5.3 Hz, 2H, CH2C–O–), 5.44 (dq, J1 = 17.3 Hz, J2 = 1.7 Hz, 2H, C2CH–O–), 5.31 (dq, J1 = 10.5 Hz, J2 =1.4 Hz, 2H, C2CH–O–), 4.58 (dt, J1 = 5.3 Hz, J2 =1.5 Hz, 4H, Ar–O–C2–), 2.58 (t, J = 7.5 Hz, 4H, –CO–C2–),1.78 (q, J = 7.4 Hz, 4H, –CO–CH2–C2–), 1.46–1.38 (m, 8H, –CO–CH2–CH2–C2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.35 (–O), 158.19 (––O–CH2–), 149.71 (––O–CO–), 138.47, 133.10, 128.09, 127.67, 121.77, 115.04 (––), 133.23 (–HCH2), 117.14 (–CHH2), 68.87 (–H2–O), 34.39 (–OCO–H2–), 29.06 (–OCO–CH2–CH2–CH2–H2–), 29.02 (–OCO–CH2–CH2–H2–), 24.92 (–OCO–CH2–H2–) ppm.
–O–CH2–), 149.62 (––OCO–), 138.59, 133.08, 128.11, 127.71, 121.75, 115.06 (––), 133.23 (–HCH2), 117.76 (–CHH2), 68.90 (–H2–O), 34.02 (–OCO–H2–), 24.34 (–OCO–CH2–H2–) ppm. [EA–6, yield: 45.32%, white solid]: 1H NMR (500 MHz, CDCl3), δ 7.53 (d, J = 8.5 Hz, 4Ar), 7.48 (d, J = 9.0 Hz, 4Ar), 7.12 (d, J = 8.5 Hz, 4Ar), 6.98 (d, J = 9 Hz, 4Ar), 6.08 (ddt, J1 = 17.3 Hz, J2 = 10.5 Hz, J3 = 5.3 Hz, 2H, CH2C–O–), 5.44 (dq, J1 = 17.2 Hz, J2 = 1.6 Hz, 2H, C2CH–O–), 5.31 (dq, J1 = 10.5 Hz, J2 =1.4 Hz, 2H, C2CH–O–), 4.58 (dt, J1 = 5.3 Hz, J2 =1.6 Hz, 4H, Ar–O–C2–), 2.60 (t, J = 7.4 Hz, 4H, –CO–C2–), 1.79–1.84 (m, 4H, –CO–CH2–C2–), 1.55–1.48 (m, 4H, –CO–CH2–CH2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.32 (–O), 158.20 (––O–CH2–), 149.68 (––O–CO–), 138.50, 133.09, 128.10, 127.68, 121.76, 115.05 (––), 133.23 (–HCH2), 117.74 (–CHH2), 68.88 (–H2–O), 34.29 (–OCO–H2–), 28.73 (–OCO–CH2–CH2–H2–), 24.75 (–OCO–CH2–H2–) ppm. [EA-7, yield: 80.37%, white solid]: 1H NMR (500 MHz, CDCl3), δ 7.53 (d, J = 8.5 Hz, 4Ar), 7.47 (d, J = 8.5 Hz, 4Ar), 7.12 (d, J = 8.5 Hz, 4Ar), 6.98 (d, J = 8.5 Hz, 4Ar), 6.08 (ddt, J1 = 17.3 Hz, J2 = 10.5 Hz, J3 = 5.3 Hz, 2H, CH2C–O–), 5.44 (dq, J1 = 17.2 Hz, J2 = 1.6 Hz, 2H, C2CH–O–), 5.31 (dq, J1 = 10.5 Hz, J2 =1.4 Hz, 2H, C2CH–O–), 4.58 (dt, J1 = 5.3 Hz, J2 =1.5 Hz, 4H, Ar–O–C2–), 2.59 (t, J = 7.4 Hz, 4H, –CO–C2–), 1.79 (q, J = 7.3 Hz, 4H, –CO–CH2–C2–), 1.53–1.44 (m, 6H, –CO–CH2–CH2–C2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.30 (–O), 158.19 (––O–CH2–), 149.69 (––O–CO–), 138.47, 133.07, 128.08, 127.66, 121.77, 115.04 (––), 133.23 (–HCH2), 117.73 (–CHH2), 68.87 (–H2–O), 34.35 (–OCO–H2–), 28.87 (–CO–CH2–CH2–H2–, –CO–CH2–CH2–CH2–H2–), 24.85 (–CO–CH2–H2–) ppm. [EA-8, yield: 43.5%, white solid]: 1H NMR (500 MHz, CDCl3): δ 7.53 (d, J = 8.5 Hz, 4Ar), 7.48 (d, J = 8.5 Hz, 4Ar), 7.12 (d, J = 9.0 Hz, 4Ar), 6.98 (d, J = 8.5 Hz, 4Ar), 6.08 (ddt, J1 = 17.2 Hz, J2 = 10.5Hz, J3= 5.3 Hz, 2H, CH2C–O–), 5.44 (dq, J1 = 17.3 Hz, J2 = 1.7 Hz, 2H, C2CH–O–), 5.31 (dq, J1 = 10.5 Hz, J2 =1.4 Hz, 2H, C2CH–O–), 4.58 (dt, J1 = 5.3 Hz, J2 =1.5 Hz, 4H, Ar–O–C2–), 2.58 (t, J = 7.5 Hz, 4H, –CO–C2–),1.78 (q, J = 7.4 Hz, 4H, –CO–CH2–C2–), 1.46–1.38 (m, 8H, –CO–CH2–CH2–C2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.35 (–O), 158.19 (––O–CH2–), 149.71 (––O–CO–), 138.47, 133.10, 128.09, 127.67, 121.77, 115.04 (––), 133.23 (–HCH2), 117.14 (–CHH2), 68.87 (–H2–O), 34.39 (–OCO–H2–), 29.06 (–OCO–CH2–CH2–CH2–H2–), 29.02 (–OCO–CH2–CH2–H2–), 24.92 (–OCO–CH2–H2–) ppm.
Synthesis of bis(4′-(oxirane-2-ylmethoxy)-[1,1′-biphenyl]-4-yl) alkanedioate (EB-n). Bis(4′-(oxirane-2-ylmethoxy)-[1,1′-biphenyl]-4-yl) adipate (EB-4) was synthesized as per the following description as a representative example. In a system equipped with a temperature controller, reflux condenser, heating oil bath, and mechanical stirrer under inert gas, a mixture of EA-4 (2.00 g, 3.56 mmol), mCPBA (4.92 g, 28.48 mmol), and 100 ml anhydrous chloroform was introduced into a 250 ml round-bottom flask. The mixture was slowly heated to 65 °C and continuously stirred for 15 h at that temperature. After cooling to room temperature, the reaction solution was evaporated to remove a part of the solvent. The obtained mixture was then precipitated by methanol to obtain a crude product as a yellow solid. The compound was purified by column chromatography with an eluent of chloroform and ethyl acetate (13:1, v/v) to obtain the product, EB-4, as a white solid with a 60% yield (1.28 g). Bis(4′-(oxirane-2-ylmethoxy)-[1,1′-biphenyl]-4-yl) octanedioate
(EB-6), bis(4′-(oxirane-2-ylmethoxy)-[1,1′-biphenyl]-4-yl) nonanedioate (EB-7), and bis(4′-(oxirane-2-ylmethoxy)-[1,1′-biphenyl]-4-yl) decanedioate (EB-8) were synthesized using a similar method. [EB-4]:
1H NMR (500 MHz, CDCl3): δ 7.53 (d, J = 8.5 Hz, 4Ar), 7.49 (d, J = 9 Hz, 4Ar), 7.14 (d, J = 9.0 Hz, 4Ar), 6.99 (d, J = 8.5 Hz, 4Ar), 4.26 (dd, J1 = 11.0 Hz, J2 = 3.2 Hz, 2H, ArO–C2), 4.01 (dd, J1 = 11.0 Hz, J2 = 5.6 Hz, 2H, ArO–C2), 3.38 (ddt, J1 = 5.8 Hz, J2 = 4.1 Hz, J3 = 2.9 Hz, 2H, C of oxirane ring), 2.93 (dd, J1 = 4.9 Hz, J2 = 4.1 Hz, 2H, C2 of oxirane ring), 2.78 (dd, J1 = 4.9 Hz, J2 = 2.7 Hz, 2H, C2 of oxirane ring), 2.71–2.63 (m, 4H,–CO–C2–), 1.92 (q, J = 3.7 Hz, 4H, –CO–CH2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 171.82 (–O), 158.12 (––O–CH2–), 149.71 (––O–CO–), 138.48, 133.51, 128.20, 127.74, 121.78, 115.00 (––), 68.80 (–H2–O), 50.15 (H of oxirane ring), 44.73 (H2 of oxirane ring), 34.02 (–CO–H2–), 24.34 (–CO–CH2–H2–) ppm. MS (+ APCI) m/z: [M + Na]+ calcd for [C36H34O8 + Na]+, 617; found, m/z 617. [EB-6, yield: 62.0%, white solid]: 1H NMR (500 MHz, CDCl3): δ 7.53 (d, J = 9.0 Hz, 4Ar), 7.48 (d, J = 8.5 Hz, 4Ar), 7.12 (d, J = 8.5 Hz, 4Ar), 6.99 (d, J = 8.5 Hz, 4Ar), 4.26 (dd, J1 = 11.0 Hz, J2 = 3.2 Hz, 2H, ArO–C2), 4.01 (dd, J1 = 11.0 Hz, J2 = 5.6 Hz, 2H, ArO–C2), 3.38 (ddt, J1 = 5.8 Hz, J2 = 3.8 Hz, J3 = 2.7 Hz, 2H, C of oxirane ring), 2.93 (dd, J1 = 4.9 Hz, J2 = 4.1 Hz, 2H, C2 of oxirane ring), 2.78 (dd, J1 = 4.9 Hz, J2 =2.6 Hz, 2H, C2 of oxirane ring), 2.60 (t, J = 7.5 Hz, 4H,–CO–C2–), 1.55–1.48 (m, 8H, –CO–CH2–C2– C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.22 (–O), 158.11 (––O–CH2–), 149.77 (––O–CO–), 138.41, 133.54, 128.19, 127.72, 121.79, 114.99 (––), 68.88 (–H2–O), 50.15 (H of oxirane ring), 44.73 (H2 of oxirane ring), 34.02 (–CO–H2–), 28.74 (–CO–CH2–CH2–H2–), 24.75 (–CO–CH2–H2–) ppm. MS (+ APCI) m/z: [M + Na]+ calcd for [C38H38O8 + Na]+, 645; found, m/z 645. [EB-7, yield: 59.3%, white solid]: 1H NMR (500 MHz, CDCl3): δ 7.51 (d, J = 9.0 Hz, 4Ar), 7.48 (d, J = 9.0 Hz, 4Ar), 7.12 (d, J = 8.5 Hz, 4Ar), 6.98 (d, J = 8.5 Hz, 4Ar), 4.26 (dd, J1 = 11.1Hz, J2 = 3.2 Hz, 2H, ArO–C2), 4.00 (dd, J1 = 11.1 Hz, J2 = 5.6 Hz, 2H, ArO–C2), 3.38 (ddt, J1 = 5.7 Hz, J2 = 4.0 Hz, J3 = 2.8 Hz, 2H, C of oxirane ring), 2.93 (dd, J1 = 4.9 Hz, J2 = 4.1 Hz, 2H, C2 of oxirane ring), 2.78 (dd, J1 = 4.9 Hz, J2 = 2.6 Hz, 2H, C2 of oxirane ring), 2.59 (t, J = 7.5 Hz, 4H,–CO–C2–), 1.79 (q, J = 7.3 Hz, 4H, –CO–CH2–C2–), 1.51–1.42 (m, 6H, –CO–CH2–C2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.28 (–O), 158.10 (––O–CH2–), 149.79 (––O–CO–), 138.36, 133.52, 128.17, 127.70, 121.80, 114.98 (––), 68.87 (–H2–O), 50.14 (H of oxirane ring), 44.71 (H2 of oxirane ring), 34.36 (–CO–H2–), 28.87 (–CO–CH2–CH2–H2–, –CO–CH2–CH2–CH2–H2–), 24.86 (–CO–CH2–H2–) ppm. MS (+ APCI) m/z: [M + Na]+ calcd for [C39H40O8 + Na]+, 659; found, m/z 659. [EB-8, yield: 58.2%, white solid]: 1H NMR (500 MHz, CDCl3): δ 7.53 (d, J = 9.0 Hz, 4Ar), 7.48 (d, J = 9.0 Hz, 4Ar), 7.12 (d, J = 8.5 Hz, 4Ar), 6.99 (d, J = 9 Hz, 4Ar), 4.26 (dd, J1 = 11.0 Hz, J2 =3.2 Hz, 2H, ArO–C2), 4.01 (dd, J1 = 11.1 Hz, J2 =5.6 Hz, 2H, ArO–C2), 3.38 (ddt, J1 = 5.8 Hz, J2 = 3.8 Hz, J3 = 2.7 Hz, 2H, C of oxirane ring), 2.93 (dd, J1 = 4.9 Hz, J2 = 4.1 Hz, 2H, C2 of oxirane ring), 2.78 (dd, J1 = 4.9 Hz, J2 = 2.6 Hz, 2H, C2 of oxirane ring), 2.60 (t, J = 7.5 Hz, 4H, –CO–C2–), 1.78 (q, J = 7.5 Hz, 4H, –CO–CH2–C2–), 1.38–1.46 (m, 8H, –CO–CH2–CH2–C2–C2–) ppm. 13C NMR (125 MHz, CDCl3): δ 172.34 (–O), 158.10 (––O–CH2–), 149.80 (––O–CO–), 138.37, 133.55, 128.18, 127.71, 121.90, 121.80, 114.99 (––), 68.87 (–H2–O), 50.15 (H of oxirane ring), 44.72 (H2 of oxirane ring), 34.41 (–CO–H2–), 29.07 (–CO–CH2–CH2–CH2–H2–), 29.03 (–CO–CH2–CH2–H2–), 24.92 (–CO–CH2–H2–) ppm. MS (+ APCI) m/z: [M + Na]+ calcd for [C40H42O8 + Na]+, 673; found, m/z 673.
Preparation of curing mixtures and cured samples
To investigate the optimal conditions for preparing the cured samples, homogeneous mixtures of EB-n and DDS or SSA (named DEB-n or SEB-n, respectively) were prepared by mixing equal equivalent of a hydrogen atom in the amine group, and an epoxy group. Heating curves were obtained using a differential scanning calorimeter in the temperature range of 0 °C–300 °C. These curves were initially measured to find a suitable temperature for the curing reaction. Subsequently, isothermal heating at the optimal temperature was performed to determine the reaction time for each curing agent. Thereafter, the mixture was transferred to a rectangular parallelepiped or a round chip steel mold suitable for each measurement. DEB-n samples were cured at 182 °C for 2 h and SEB-n samples were cured at 173 °C for 3 h by hot compression to give cured DEB-n and SEB-n samples, respectively. Finally, all the samples were post-cured at 185 °C for 2 h to get the fully cured samples.Results and discussion
Design and synthesis of monomers
A series of monomers, EB-n, composed of two mesogens based on a biphenyl moiety terminated by epoxy functional groups and connected by two ester groups and an aliphatic spacer (n = 4, 6, 7, 8) was synthesized in three steps. In this reaction, [1,1′-biphenyl]-4,4′-diol was used as the starting material. The detailed synthetic process and molecular structures are shown in Scheme 1. For simple and efficient self-assembly, a molecular structure that can maximize intermolecular interactions was designed using the twin mesogen structure of biphenyl. In particular, the mesogens were connected with spacers of various lengths and four types of odd and even carbon numbers to enhance the intermolecular interaction. 4′-(Allyloxy)-[1,1′-biphenyl]-4-ol (A1) was easily synthesized through the one-side substitution of biphenol with allyl bromide.44 Subsequently, the remaining OH group was esterified by various diacid chlorides with different alkyl chain lengths to afford bis(4′-(allyloxy)-[1,1′-biphenyl]-4-yl) alkanedioate (EA-n). Thereafter, the vinyl groups were oxidized by mCPBA and converted to bifunctional epoxide monomers. All reactions proceeded as expected with appropriate yields. The final compounds, EB-n, were accurately characterized by 1H NMR, 13C NMR, FT-IR, and MS spectrometry, as shown in Fig. S1 and S2a (ESI†). Moreover, two series of cured products, DEB-n and SEB-n, were prepared by reacting EB-n with two different aromatic diamines, 4,4′-diaminodiphenyl sulfone (DDS) and sulfanilamide (SSA), respectively. These diamine curing agents were deemed suitable for this system after extensive thermal and optical investigation of the monomers.Mesophase of monomers
The phase transition behavior and thermal properties of the synthesized monomers were investigated by differential scanning calorimetry (DSC) and polarized optical microscopy (POM), as shown in Fig. 1(a) and (b), respectively. In addition, the mesomorphic properties are summarized in Fig. 1(c) and Table 1. Monomers with even number of carbons in the aliphatic chain, namely EB-4, EB-6, and EB-8, showed a typical Schlieren structure with a nematic LC regime. They also exhibited a wide temperature range of 160 °C–230 °C with slight differences depending on the chain length of the spacer for both heating and cooling processes. For all monomers, the changes in the starting temperature and LC region decreased with increasing alkyl chain length; however, this tendency was not observed for EB-7. During the heating cycle, EB-4 underwent a change from the crystalline state to the nematic state at 174 °C; the nematic state persisted until 230 °C before transforming into the isotropic state with an LC range around 56 °C. The same process was observed during the cooling cycle, where the temperature required to form the LC phase from the isotropic state was approximately 228 °C, and the LC range was approximately 65 °C. This phenomenon was also observed in EB-6 and EB-8, where the LC range was 43 °C–32 °C for the heating cycle and 41 °C–29 °C for the cooling cycle. This could be explained by the fact that the longer the flexible chain, the shorter the LC range. This was also caused by the flexibility of the carbon chain; the greater the number of methylene groups in a molecule, the faster it moves.42,43 As a result, EB-8 was more malleable than EB-6 and EB-4, leading to a lower starting temperature and a narrower LC range than that of EB-6 and EB-4. However, EB-7, which contained an odd number of carbon atoms, displayed different LC behaviors during both heating and cooling cycles. During the heating cycle, EB-7 showed an LC phase at a starting temperature of 155 °C, which was lower than that of even carbon monomers. Moreover, its LC phase was kept within a range of 20 °C before transforming into the isotropic state at 175 °C, which was also narrower than that of EB-8. The DSC curves of EB-7 clearly show that the enthalpy change of the endothermic peak was quite small at approximately 3.3 J g−1, indicating that the change from the LC phase to the isotropic state was quite fast. In the cooling cycle, two exothermic peaks of EB-7 appeared too close, and the enthalpy of the first peak from the isotropic state was quite low at 1.9 J g−1. As a result, the phase change from LC to the crystal state was so rapid leading to the LC phase region being narrower than the heating cycle. The differences in the mesophase between monomers with even and odd carbons in the flexible chain originated from dissimilarities in the molecular structure and the even–odd carbon chain effect.27,45,46 Overall, the mesomorphic properties of EB-n were significantly enhanced compared to epoxy monomers based on biphenyl mesogens.33–37,47,50 In addition, despite containing an aliphatic chain, the monomers were quite thermally stable, as shown in Fig. S3 and Table S1 (ESI†). All monomers except EB-7 exhibited a 5% weight loss at 291 °C during heating. Notably, EB-7 showed this weight loss at 350 °C.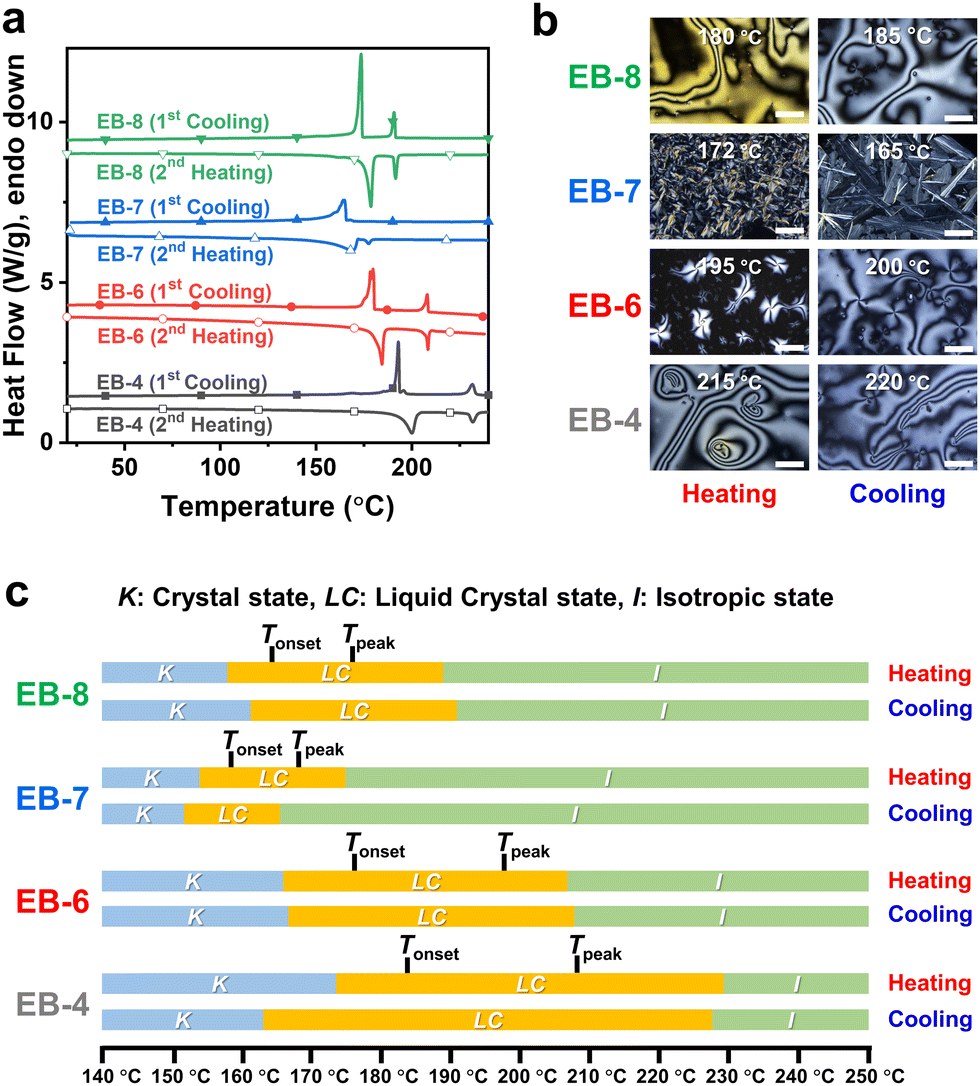 | ||
| Fig. 1 Liquid crystalline properties of the EB-n monomer series: (a) differential scanning calorimetry (DSC) curves of pure EB-n at a heating rate of 5 °C per minute in N2 gas. (b) Polarized microscopy (POM) images of EB-n (scale bar = 50 μm). (c) Illustrative diagrams of EB-n phase transition. | ||
| Sample | Heating cycle | Cooling cycle | ||||||
|---|---|---|---|---|---|---|---|---|
| K → LC [°C] | ΔH [J g−1] | LC → I [°C] | ΔH [J g−1] | I → LC [°C] | ΔH [J g−1] | LC → K [°C] | ΔH [J g−1] | |
| a Determined by DSC results of pure EB-n at a heating and cooling rate of 5 °C min−1. | ||||||||
| EB-4 | 174.0 | 54.7 | 230.0 | 13.1 | 228.5 | −14.1 | 163.4 | −54.0 |
| EB-6 | 166.4 | 53.2 | 207.5 | 11.7 | 208.6 | −13.4 | 167.2 | −51.4 |
| EB-7 | 154.7 | 58.1 | 175.7 | 3.3 | 166.0 | −1.9 | 152.0 | −60.1 |
| EB-8 | 158.3 | 73.0 | 190.4 | 13.9 | 191.5 | −15.2 | 162.0 | −72.0 |
Curing and self-assembly of cured products
To develop thermally conductive polymers with a self-aligned structure using cured EB-n LC, the curing behavior of the mixtures of EB-n with two aromatic curing agents (DDS and SSA) was investigated through DSC. The temperature-dependent and time-dependent heat flows at a specific temperature were thoroughly investigated. The DSC curves are presented in Fig. S4 (ESI†), and the values are summarized in Fig. 1(c) and Table 2. In this study, DDS and SSA were chosen as curing agents owing to the following specific reasons. Firstly, the LC region of the monomers falls within the 160 °C–230 °C range, which is a suitable curing temperature for aromatic diamines with electron-withdrawing substituents.35 Curing the LC monomers in this range would help retain molecular alignment in the final crosslinked products. Consequently, the polymer will have higher crystallinity, which in turn improves thermal conductivity by reducing phonon scattering when heat transfers through the polymer matrix.24,25 Previous studies have successfully demonstrated that the inclusion of biphenyl mesogens in curing monomers, utilizing DDS and SSA as curing agents, resulted in notable improvements in thermal conductivity and mechanical properties.34,38,47 The curing process of epoxy resin is widely recognized to involve three distinct stages of initial linear chain extension, followed by branching and ultimately crosslinking. In the case of LCERs, these stages significantly influence the alignment of the mesogenic units within the materials.35,49,50 Due to the temperature range of LCERs, sensitive control of suitable curing temperature is required during the initial stage, and it is essential to select a curing system with lower reactivity. DDS and SSA are often chosen as they exhibit reduced reactivity, primarily due to the electron-withdrawing sulfone unit present. This reduction in reactivity minimizes undesirable branching and crosslinking reactions at elevated temperatures. Specifically, the lowered reactivity between the primary amine and the oxirane ring helps prevent excessive heat generation and safeguards the integrity of the LC structure.48,51 Additionally, it is worth noting that DDS possesses a symmetrical structure, while SSA has an asymmetric structure. This distinction between the two curing agents presents an interesting opportunity for comparison and analysis within the system.| Sample | DEB-n series (DDS mixture) | Sample | SEB-n series (SSA mixture) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| T onset [°C] | T peak [°C] | ΔHc [J g−1] | ΔHd [J g−1] | T onset [°C] | T peak [°C] | ΔHc [J g−1] | ΔHd [J g−1] | ||
| a Starting temperature of the curing reaction determined from the DSC results of EB-n curing mixtures at a heating rate of 5 °C min−1. b Rate maximum temperature of the curing reaction determined from the DSC results of EB-n curing mixtures at a heating rate of 5 °C min−1. c Enthalpy change of the curing reaction determined from the DSC results of EB-n curing mixtures at a heating rate of 5 °C min−1. d Enthalpy change of the curing reaction determined from the isothermal DSC results of DEB-n at 182 °C and SEB-n at 173 °C. | |||||||||
| DEB-4 | 175.3 | 194.8 | 235.8 | 247.5 | SEB-4 | 162.6 | 171.0 | 206.3 | 219.7 |
| DEB-6 | 176.6 | 208.0 | 260.3 | 269.7 | SEB-6 | 165.0 | 171.0 | 234.8 | 243.2 |
| DEB-7 | 179.2 | 215.7 | 250.1 | 257.6 | SEB-7 | 172.7 | 183.6 | 285.5 | 305.9 |
| DEB-8 | 180.3 | 232.7 | 267.4 | 281.3 | SEB-8 | 172.9 | 184.5 | 349.6 | 368.1 |
From curing investigation by DSC analysis on curing mixtures prepared by mixing and grinding into a fine powder, all the curing mixtures had exothermic reaction temperature fall in the monomer LC region with the series of DEB-n from 175 °C to 270 °C and SEB from 163 °C to 260 °C and the values of the curing heat were over 200 J g−1. The onset curing temperature (Tonset) of DEB-n was 175 °C, which is 12 °C higher than that of the SEB-n system. This difference is not significant, but Tonset increases slightly with an increase in the chain length. This indicates that the curing rate is inversely proportional to the spacer length. In this regard, a mixture of EB-4 and the curing agent showed the fastest curing rate. The curing reaction of DEB-n showed a similar regime to a large difference between the Tonset and Tpeak values. Generally, derivatives with an even number of carbon atoms showed a higher difference between Tonset and Tpeak. This difference was approximately 20 °C for DEB-4, 30 °C for DEB-6, and 40 °C for DEB-8. In contrast, the temperature differences for the SEB-n derivatives varied. SEB-4 and SEB-6 showed a smaller difference between Tonset and Tpeak than SEB-7 and SEB-8. Notably, SEB-7 and SEB-8 displayed a small second exothermic peak in the curing reaction. The first and second peak temperatures of SEB-7 and SEB-8 were 183.6 °C and 246.4 °C and 184.5 °C and 252.2 °C, respectively. This observation is consistent with a previous report on the curing of epoxy monomers with SSA.38,52 The first amine group of SSA bonded directly with the phenyl group, which was more reactive than the second amine group directly attached to the strongly withdrawing sulfonyl group. As a result, it reacted first and corresponded to the first exothermic peak. The second exothermic peak was attributed to the second amine group, which required more energy to trigger the reaction. This phenomenon was not observed in shorter carbon chain derivatives (n = 4, 6) because the faster reaction rate of the first amine group was enough to ignite the reaction of the second amine group simultaneously, facilitated by the quick movement of smaller molecules.
From the isotherms measured at 182 °C for DEB-n and at 173 °C for SEB-n, the curing reactions of all derivatives were completed within an hour. Compounds with shorter carbon chains required less curing time for both DEB-n and SEB-n. For all systems, the heat of curing was not significantly different from that obtained from the temperature-dependent curves. Interestingly, the isothermal curves of SEB-7 and SEB-8 showed two distinguishable exothermic peaks, which is consistent with the dynamic curing investigation. Moreover, although faint, two peaks also appeared for DEB-7 and DEB-8. Presumably, such peaks were observed for long-chain systems with slow reaction rates. A similar phenomenon was observed in all systems. This may have occurred due to a special phase transition phenomenon, which was further investigated in this study.
To better understand the self-assembly during the curing reaction, POM was carried out at the same temperature at which the isotherms were obtained. This was done to investigate the mesomorphic properties of the DEB-n and SEB-n systems, as shown in Fig. 2(a) and (b), respectively. Fig. S5 and S6 (ESI†) show the images developed over time with specific mesophases. During curing, DEB-n samples with even carbon atoms displayed a smectic-like microcrystalline arrangement, while DEB-7 exhibited an amorphous structure with a black-out image under polarized light. In contrast, all SEB-n series showed mesomorphic crystalline structures, including the EB-7 system. Particularly, SEB-7 displayed a different mesophase structure compared to the other SEB-n systems; this structure was significantly different from that of DEB-7. This may have occurred because SEB-7 contains an odd number of carbon atoms; however, its structure is probably different from that of DEB-7 because of the difference in the resulting structure of the curing agent amine. EB-7 was confirmed to possess weak mesomorphic properties compared to other even-numbered EB-n systems; therefore, it is natural for DEB-7 to exhibit an ambiguous LC phase. However, despite having a shorter LC range in the heating cycle than other derivatives, SEB-7 could still form an LC structure with a suitable curing agent and temperature. Since the ordered structure of the cured product was different from that of the monomer, it was important to optimize the reaction conditions to maximize the self-assembly characteristics. As a result of the mesophase observation, it was confirmed that most DEB-n and SEB-n samples showed similar textures; however, those derived from EB-7 showed significantly different textures.45,46 This indicates that the self-assembly characteristics were different, and the differences in the properties of the cured product could be confirmed.37,38
 | ||
| Fig. 2 Microstructure analysis results of the cured samples: (a) and (b) mesophase images of DEB-n and SEB-n after the curing reaction, recorded by polarized microscopy (POM), respectively. (c) and (d) X-ray diffraction (XRD) charts of the DEB-n series and SEB-n series, respectively. | ||
Subsequently, bulk specimens were fabricated as shown in Fig. S7 (ESI†) by curing and self-assembly under the optimized conditions, as described above. The preparation was carried out on a heating press with round or rectangular steel molds by inserting the curing mixture. The sample dimensions were diversified to investigate specific properties. The DEB-n series initially reacted at 182 °C for 2 h, and the SEB-n series reacted at 173 °C for 3 h. The samples were then post-cured at 185 °C for 2 h. After curing, most of the samples turned opaque with an ivory white to brown color, except for DEB-7, which was slightly transparent with a brown color. This confirmed that the curing reaction of all SEB-n series samples and DEB-n series samples except for DEB-7 occurred in the LC phase, while a large portion of DEB-7 was cured in the isotropic state, resulting in a semi-transparent appearance of the final product. This observation was in good agreement with the POM observation results for the curing behavior. The changes in the chemical structure that occurred during the curing reaction were investigated using FT-IR spectroscopy, as shown in Fig. S2 (ESI†). As a major change, all the spectra showed the transformation of the epoxy group in the region around 920 cm−1 to the hydroxyl group in the region from 3300–3500 cm−1. This change is due to the epoxide ring opening reaction caused by the amine. In addition, it was determined that only the intended reaction was properly performed because all IR absorption bands changed reasonably after the formation of the cured network structure, as shown in Scheme 1.
To investigate the microstructure of the cured bulk samples, X-ray diffraction (XRD) was performed, as shown in Fig. 2(c) and (d). Except for DEB-7, all DEB-n samples showed a sharp peak at approximately 2θ = 20°, indicating the distance for the π–π interactions of the aromatic ring. This clearly demonstrates the highly crystalline microstructure of the cured sample. In contrast, DEB-7 showed a rather broad peak in the same region, indicating that cured DEB-7 was in an amorphous state. These results were consistent with the appearance of the samples. However, all SEB-n samples exhibited several sharp peaks near 2θ = 20° and small peaks in the region of 2θ < 10°, indicating a higher ordered crystalline structure than other DEB-n samples. Interestingly, SEB-7 and SEB-8 displayed distinguishable small peaks in the region of 2θ < 5°, indicating the smectic moiety in the microstructure. This may be attributed to the slow rate observed during the curing of the mixture, which lead to a more developed aligned structure. This structure differed from that of SEB-4 and SEB-6. To conclude, it was possible to successfully fabricate specimens with self-ordered microstructures.53
Thermal and mechanical properties
To examine the properties of the cured samples, several DSC measurements, dynamic mechanical analysis (DMA), tensile tests as mechanical property analysis, thermogravimetric analysis (TGA), and thermal conductivity (T/C) analysis were conducted. The data are summarized in Table 3. First, from the DSC results shown in Fig. S8 (ESI†), it was confirmed that the occurrence of additional reactions was not confirmed at all, and thus fully cured materials were obtained. Moreover, the glass transition temperatures (Tg,DSC) were confirmed in the range of 127 °C–154 °C for the DEB series and 120 °C–145 °C for the SEB-n series. At the same carbon number, DEB-n showed a slightly higher value than SEB-n. As the number of carbons increased, the tendency for Tg to decrease as per the increase in molecular fluidity was clearly confirmed. However, all the values were significantly higher than 100 °C, which was a fairly high level considering that EB-n contained aliphatic spacers.36 The glass transition was also investigated by DMA (Tg,DMA), as shown in Fig. 3. Although there was a slight difference between Tg,DSC and Tg,DMA, both methods showed the same consistency for DEB-n and SEB-n. All Tg,DMA values showed a uniform trend in both DEB-n and SEB-n series, with Tg increasing as the spacer chain length decreased. Generally, for samples with an even number of carbon atoms in the spacer (n = 4, 6, 8), DEB-n showed a higher Tg than SEB-n. However, this trend was reversed in the case of samples with an odd number of carbon atoms (n = 7). SEB-7 showed a higher Tg than DEB-7. This trend could be explained by the highly ordered crystalline structure of SEB-7, which plays a critical role in a further interaction in the resin matrix. Thus, in the heating process, more energy was required for molecular vibration resulting in enhancing the Tg.47,54| Sample | T g,DSC [°C] | T g,DMA [°C] | T d,5% [°C] | T d,10% [°C] | Thermal conductivityd [W m−1 K−1] | Crosslink densitye [mol cm−3] | Tensile strengthf [MPa] | Young's modulusf [MPa] | Elongation at breakf [%] | Density [g cm−3] | Sound velocityg [km s−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by DSC at a heating rate of 5 °C min−1. b Determined by DMA at a heating rate of 3 °C min−1 with 1 Hz frequency. c Determined by TGA at a heating rate of 20 °C min−1. d Measured using a HotDisk TPS 3500 thermal conductivity analyzer with accuracy over the whole range of thermal conductivities being within ±5% and the reproducibility being within ±2%.56 e Calculated by storage modulus curves at 200 °C. f Measured by the tensile test at room temperature with a crosshead speed of 2 mm min−1. g Calculated from the equation [sound velocity = (Young's modulus/density)1/2].57 | |||||||||||
| DEB-4 | 154.1 | 151.6 | 356.4 | 372.0 | 0.519 | 0.002068 | 62.7 ± 2.3 | 3025 ± 121 | 3.23 ± 0.13 | 1.26 | 1.55 |
| DEB-6 | 145.0 | 142.3 | 350.1 | 368.4 | 0.557 | 0.003526 | 61.8 ± 1.9 | 2950 ± 95 | 3.41 ± 0.11 | 1.18 | 1.58 |
| DEB-7 | 134.3 | 129.6 | 350.3 | 370.7 | 0.511 | 0.000261 | 58.8 ± 2.2 | 2050 ± 52 | 3.52 ± 0.09 | 1.17 | 1.32 |
| DEB-8 | 126.8 | 122.9 | 356.2 | 369.6 | 0.616 | 0.002973 | 60.8 ± 2.6 | 2175 ± 71 | 3.66 ± 0.12 | 1.11 | 1.4 |
| SEB-4 | 145.0 | 146.2 | 316.4 | 338.2 | 0.551 | 0.063194 | 57.4 ± 2.1 | 3300 ± 99 | 2.68 ± 0.08 | 1.35 | 1.56 |
| SEB-6 | 137.2 | 137.3 | 324.0 | 339.2 | 0.641 | 0.077308 | 56.4 ± 2.5 | 2900 ± 116 | 2.75 ± 0.11 | 1.26 | 1.52 |
| SEB-7 | 133.3 | 133.5 | 317.5 | 333.1 | 0.757 | 0.042652 | 58.1 ± 2.3 | 3450 ± 131 | 2.64 ± 0.1 | 1.25 | 1.66 |
| SEB-8 | 119.6 | 120.1 | 320.8 | 339.2 | 0.709 | 0.064833 | 55.1 ± 2.3 | 3075 ± 126 | 2.92 ± 0.12 | 1.22 | 1.59 |
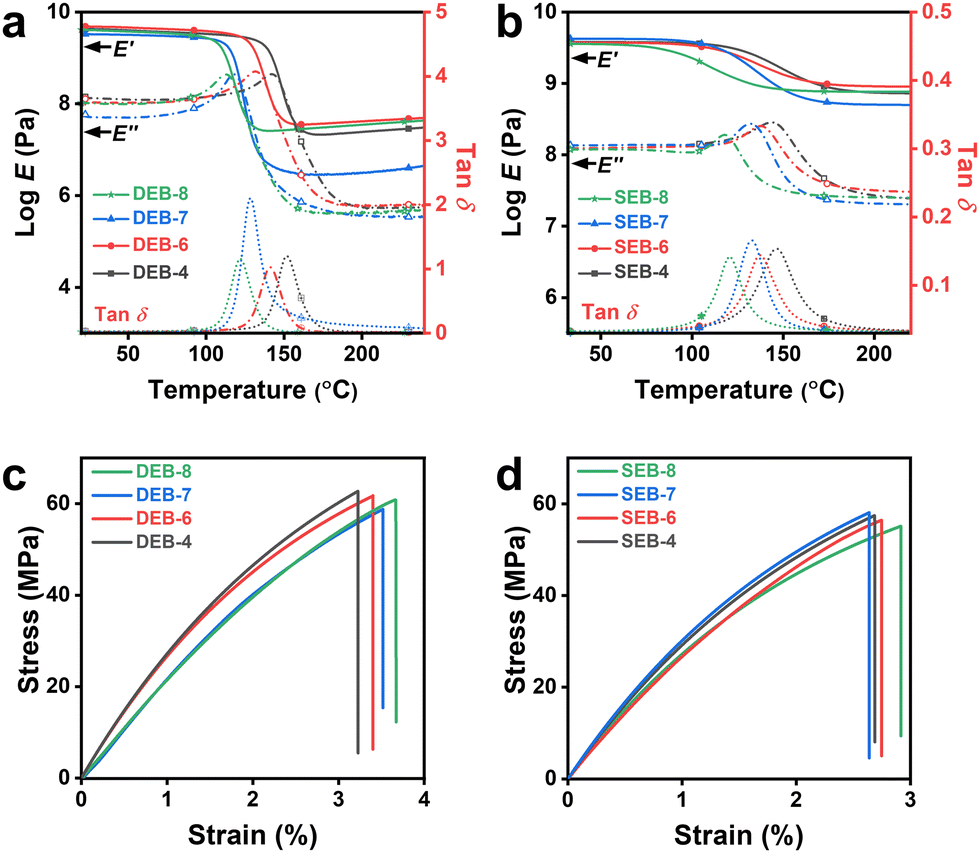 | ||
| Fig. 3 Mechanical properties of cured samples: (a) and (b) dynamic mechanical analysis (DMA) results at a frequency of 1 Hz at a heating rate of 3 °C min−1 in air. (c) and (d) Stress–strain curves for the DEB-n series and SEB-n series, respectively. | ||
The mechanical properties of the thermosets were also examined using DMA. It was evident that all cured samples exhibited good mechanical properties, with a high storage modulus (E′) of approximately 4.0 GPa. Although DEB-n showed a slightly higher E′ and loss modulus (E′′) than SEB-n in the glassy region, SEB-n had a higher modulus than DEB-n in the rubbery state. This feature was attributed to the nature of the curing agents. In the glassy state, DDS had one more aromatic ring than SSA, thereby strengthening the π–π interactions of the molecules. This made the polymer network more stable at Tg. Over Tg, the π–π interactions in DEB-n were weakened because the mobility of the molecule increased. In contrast, SSA had a shorter length between the crosslinked points than DDS, making the polymer network in SEB-n less flexible than DEB-n, leading to a lower reduction in E′. This became even clearer when looking at the crosslinking density (ν = E′/3RT, R: gas constant) calculated at 200 °C, where all the E′ curves were on the rubbery plateau.41 The υ values are listed in Table 3. SEB-n had higher υ values than DEB-n, and notably in both, only the odd number of spacers had a lower ν. There was a significant difference between the molecular packing of odd-numbered and even-numbered spacer systems. This effect led to a decrease in crystallinity, which was likely to cause large differences in moduli and crosslinking density. This accounted for the differences in the polymer structures of the two types of thermosets, leading to differences in their mechanical properties.
The crystallinity and the curing system also affect the mechanical properties of the cured samples. Fig. 3(c) and (d) show the typical stress–strain curves for the hard materials of DEB-n and SEB-n, respectively. It is clear that although DEB-n has a higher tensile strength and strain at break than SEB-n, both DEB-n and SEB-n possess a tensile strength over 55 MPa with even values of n (n = 4, 6, 8); samples exhibit a clear tendency with a decrease in strength and an increase in elongation deformation as n increases. In the DEB-n series, DEB-4 shows the highest strength of 62.7 MPa, while DEB-7 has the lowest value of 58.8 MPa. In the SEB-n series, SEB-7 has the highest tensile strength of 58.1 MPa, which is 0.7 MPa higher than that of DEB-4, and the lowest strain at 2.64%, which is approximately 0.04% lower than SEB-4. In both series, DEB-8 and SEB-8, which have the highest carbon number in the spacer length, exhibit the longest strains at 3.66% and 2.92%, respectively. The difference in tensile strength and strain at the break between the odd and even carbon numbers in flexible length is derived from dissimilarity in structure and crystallinity. While DEB-7 has an amorphous structure, SEB-7 displays a microcrystalline pattern that enhances the intermolecular forces but reduces the mobility of the molecules.55
The thermal decomposition of the cured samples was initially evaluated through TGA; the corresponding results are presented in Fig. S3 (ESI†), and the obtained data are summarized in Table 3. The DEB-n series exhibited a higher degree of thermal stability than the SEB-n series. Specifically, all DEB-n samples exhibited a 5% weight loss temperature (Td,5%) greater than 350 °C. However, the Td,5% of the SEB-n samples was approximately 30 °C lower. This disparity was likely due to the higher content of aromatic components in DEB-n relative to SEB-n. Overall, despite the presence of a long aliphatic chain in their structures, all the cured polymers exhibited excellent thermal stability, as evidenced by their 10% weight loss temperatures (Td,10%) all exceeding 330 °C.
Additionally, the T/C of the cured samples was assessed using two roundchip-shaped specimens with a diameter of 20 mm and a thickness of 4 mm. The T/C values are presented in Table 3. Notably, all cured samples exhibited high T/C values exceeding 0.5 W m−1 K−1. The values were considerably higher than those of common thermosets as well as other LC epoxy resins.36–43 In particular, the SEB-n series exhibited higher T/C than the DEB-n series. Although the T/C tendency was similar for both DEB-n and SEB-n with even carbon numbers (n = 4, 6, 8), there were slight differences in the n value of each series. T/C increased as the number of carbons increased.42 For instance, the T/C values of DEB-4, DEB-6, and DEB-8 were 0.519, 0.557, and 0.616 W m−1 K−1, respectively. Similarly, SEB-4, SEB-6, and SEB-8 exhibited T/C values of 0.551, 0.641, and 0.709 W m−1 K−1, respectively, which were around 0.1 W m−1 K−1 higher than the T/C values of the corresponding DEB-n samples. However, DEB-7 and SEB-7 showed different regimes compared to other derivatives with even n values.27 Interestingly, while DEB-7 had a lower T/C of 0.511 W m−1 K−1 compared to other DEB-n samples, SEB-7 had higher T/C of 0.757 W m−1 K−1 than other SEB-n samples. This deviation in the T/C between DEB-7 and SEB-7 could be attributed to the microstructure of the cured samples, as investigated by POM and XRD. DEB-7 had an amorphous structure, while SEB-7 exhibited a crystalline microstructure, which facilitated thermal conductivity in the polymer chains by reducing phonon scattering. Furthermore, the sound velocity (ν) derivative from Young's modulus (E) which is indirectly a representative of phonon velocity was calculated based on the equation57 (ν = (E/ρ)1/2, ρ = density) and is listed in Table 3. This calculation contributes to a better understanding of the influence of the material's structure on thermal conductivity. The sound velocity values of DEB-7 and SEB-7 show differences, with DEB-7 having a lower velocity (1.32 km s−1) compared to SEB-7 (1.66 km s−1). Although thermal conductivity depends on multiple parameters, there is not a direct and simple relationship between sound velocity and thermal conductivity. However, in general, higher sound velocity values tend to enhance thermal conductivity.58 It is evident that the spacer length (odd or even carbon numbers) influences the thermal conductivity of the cured samples based on the differences in the cured sample structure, which stems from the structural dissimilarity of EB-7 compared to EB-n (n = 4, 6, 8).27 This leads to variations in mesophase behavior, and under the same curing conditions, the final cured products differ. In the cured samples with even values of n (n = 4, 6, 8), the curing conditions play a key role in the formation of the final structure in the cross-linking matrix. At the same curing temperature, the curing reaction of mixtures containing monomers with longer spacer length is more favorable to utilize the LC region. This is because the slower movement of the monomer facilitates chain extension at the initial stage, leading to greater alignment in the final cured samples. The increased structural order reduces phonon scattering when heat is transmitted along the matrix. This explains why DEB-8 and SEB-8 have higher T/C values compared to DEB-6, DEB-4 and SEB-6, SEB-4 respectively. In contrast, in samples with an odd carbon number in the flexible chain (n = 7), the curing agent plays a crucial role in forming the crystalline structure, which significantly enhances heat transport efficiency through the matrix via the phonon vibration phenomenon. In this case, the SSA curing agent proves to be more effective than DDS.
Demonstration of the heat dissipation of thermally conductive materials is essential for their application and performance evaluation. Materials with high thermal conductivity conduct heat faster and become hotter or colder when placed in environments with different temperatures. This can be easily visualized using an infrared camera.41 In order to visualize the T/C of the cured specimens, thermal infrared images were captured during the heating and cooling processes, as illustrated in Fig. 4. For comparison, a commercial silicone rubber sample with a T/C of approximately 0.2 W m−1 K−1 was used as a reference. All samples with the same initial equilibrium temperature were placed on a temperature-fixed hot stage at 100 °C during the heating process. Pictures were taken every five seconds for 60 seconds, and the images and exact temperatures of each sample were collected, which have been presented in Fig. 4(a)–(c) and (g), (h). Notably, during contact with the heat source, the sample temperature initially increased rapidly and then gradually increased as it approached the equilibrium state, which facilitated interpretation. The temperatures of the DEB-n and SEB-n samples increased much faster than that of silicone rubber. After one minute of heating, the temperatures of DEB-n and SEB-n approached approximately 90 °C; however, the temperature of the reference stayed just below 80 °C. Moreover, a minor variation was observed for both the DEB-n and SEB-n series. At the same time, DEB-8 exhibited the highest temperature, followed by DEB-6 and DEB-4. DEB-7 possessed the lowest temperature. In the SEB-n series, SEB-7 showed the highest temperature changing rate, followed by SEB-8, SEB-6, and SEB-4. It was clear that SEB-7 heated up the fastest, and DEB-7 warmed up the slowest, which is consistent with their T/C values. In the cooling experiment, all samples were initially in a temperature-equilibrium state at 70 °C. The samples were then taken from the oven and placed on an insulated foam surface composed of polystyrene for cooling. The color images and temperatures of the samples were recorded until no color change occurred, as shown in Fig. 4(d)–(f) and (i), (j). The experimental conditions were designed to ensure that any environmental factors, which could impact sample heating diffusion, were equal, thereby allowing any differences in temperature decrease to be attributed solely to the intrinsic nature of the sample. Intuitively, the color-changing rate of all the cured specimens was much faster than that of the reference sample. Specifically, SEB-n exhibited a slightly higher changing rate than DEB-n. During the 4-min recording period, the temperature of all the cured samples decreased from 70 °C to approximately 20 °C. In contrast, the temperature of the reference sample only reduced to around 40 °C. This indicated that the LC thermosets had a much faster heat diffusion rate than common polymers. A phenomenon similar to the heating cycle was observed during the cooling process, where the sample temperature dropped rapidly in the first minute, gradually declined in the next two minutes, and decreased slowly in the last minute. In the series of samples with even n values (n = 4, 6, 8), the temperature reduction or the thermal spreading tendency decreased as n decreased. This tendency was consistent with the TC values, as well as the IR thermal imaging demonstration during the heating process. Notably, DEB-7 exhibited slightly lower heat transmission than other DEB-n samples at a higher temperature for 240 seconds, while SEB-7 showed better heat transmission than other SEB-n derivatives at a lower temperature after four minutes. This finding was consistent with the TC measurement and proved that the ordered structure of SEB-7 enhanced the thermal pathway more than the amorphous structure of DEB-7. Therefore, it was again confirmed that the formation of an ordered structure during the curing process significantly affected the thermal conducting properties even in the same LC material.
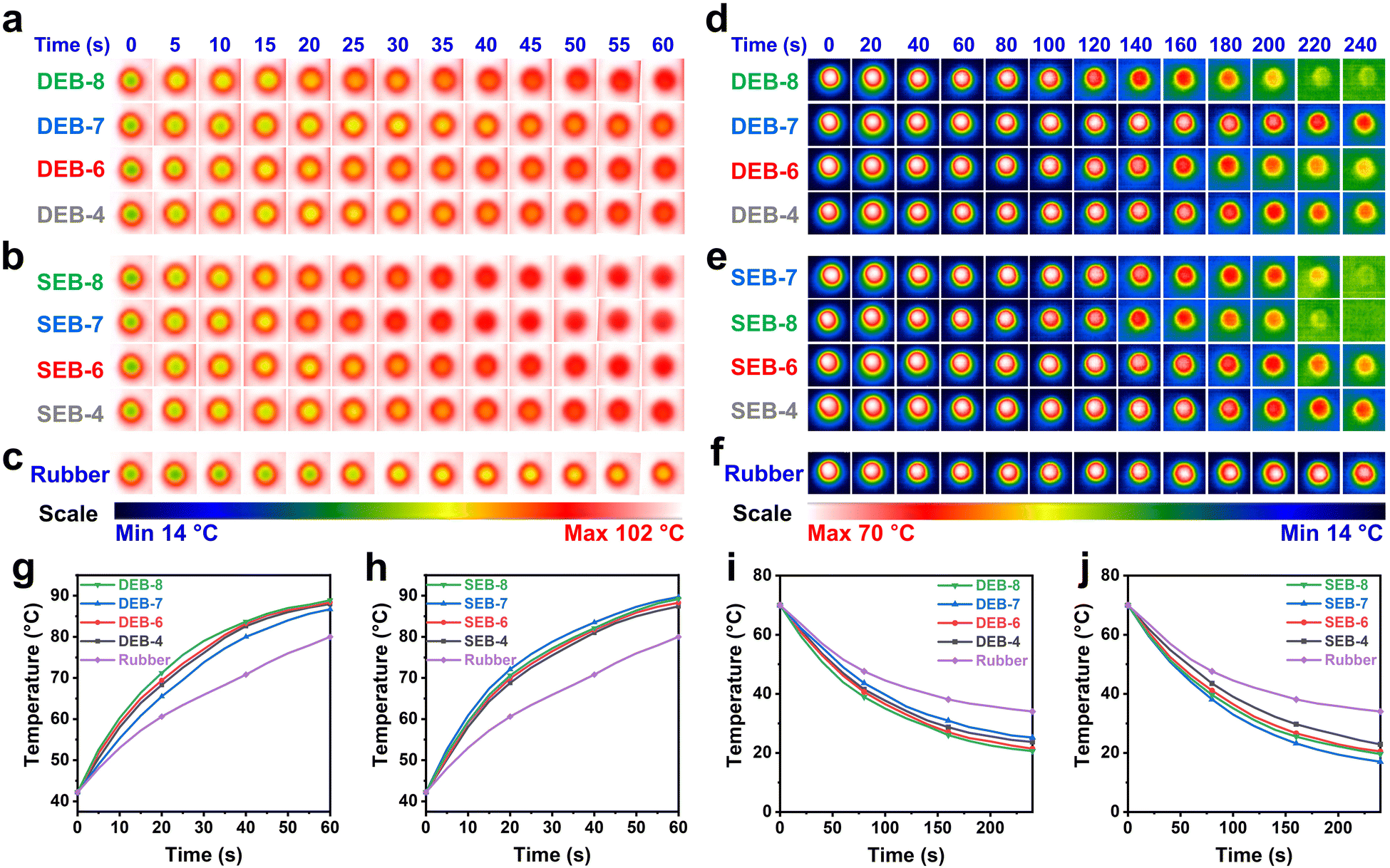 | ||
| Fig. 4 Heat conduction test results of the cured samples: (a)–(c) Temperature changes in DEB-n, SEB-n, and silicon rubber (reference) over time during the heating process observed using a thermal imaging camera, respectively. (d)–(f) Temperature changes of DEB-n, SEB-n, and silicone rubber over time during the cooling process observed using a thermal imaging camera, respectively. (g)–(j) Graph showing the temperature changes over time in both the heating and cooling processes. | ||
Conclusions
In this study, a bifunctional LCE series (EB-n, n = 4, 6, 7, 8) based on a biphenyl mesogen was synthesized and characterized. In this series, LCERs with even carbon numbers in the flexible chain (n = 4, 6, 8) displayed similar thermotropic LC behavior, with a typical nematic phase that spanned over the wide temperature range of 160 °C–230 °C. While there were slight variations among the derivatives, the LC region of the monomers was similar to the curing region of most aromatic amines. This finding represents a significant improvement in the monomer LC range compared to that of previous studies, which used epoxy monomers based on biphenyl mesogens with either no or short LC phases. However, EB-7 displayed a different mesophase behavior with a shorter LC region. This difference was consistent with the odd–even effect observed in the aliphatic carbon chains. The EB-n series was cured by two aromatic diamines (DDS and SSA) capable of curing in the LC range under the optimized conditions to enhance self-assembly properties. Notably, the cured samples showed high thermal stability, with 10% mass loss occurring at approximately 330 °C and a minimum Tg of 120 °C. The thermal resistance decreased slightly as the spacer length increased but showed a slightly exceptional value for n = 7, which contained an odd number of carbons. Moreover, the DEB-n series showed a slightly better thermal resistance than the SEB-n series, which was caused by their higher aromatic content with a slightly more regular structure of the symmetric amine. In addition, cured samples displayed good mechanical properties of a hard material with storage modulus values of approximately 4.0 GPa and a tensile strength more than 55 MPa with slight differences among derivatives. The ordered structures in the cured samples formed a larger crosslinked network by enhancing the thermal and mechanical properties. This was evidenced by DEB-7, which lacked alignment in its amorphous structure and exhibited a lower modulus as well as tensile strength than the other products. Furthermore, all cured products exhibited outstanding heat dissipation ability with T/C values higher than 0.5 W m−1 K−1, which were around 2.5 times higher than those of conventional epoxy-based systems, with slight variance among the derivatives depending on the spacer chain length. XRD analysis showed that the mesomorphic properties of SEB-n were more enhanced than those of DEB-n in the T/C investigation. Among them, SEB-7, which showed the most ordered structure, exhibited the highest T/C of 0.757 W m−1 K−1; this is one of the highest values reported among the bulk bifunctional epoxy systems developed so far. Heat dissipation tests demonstrated using IR thermal imaging also confirmed the good performance of the thermosets based on their high thermal conductivity. The high T/C of cured products originated from the formation of ordered LC structures during curing. Moreover, the curing reaction induces crosslinking, enhances microcrystallinity, and facilitates heat transfer pathways in the polymer matrix by reducing phonon scattering. These LC thermosets with high T/C and excellent thermal and mechanical properties have several potential applications in high-performance electronic devices.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2021R1I1A3042146) and the Ministry of Science and ICT (2022R1A4A1029580), and the Korea Evaluation Institute of Industrial Technology (KEIT) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (20011123).References
- Y. Sun, L. Wang, Y. Ni, H. Zhang, X. Cui, J. Li, Y. Zhu, J. Liu, S. Zhang, Y. Chen and M. Li, 3D printing of thermosets with diverse rheological and functional applicabilities, Nat. Commun., 2023, 14, 245 CrossRef CAS PubMed.
- L. Yang, Y. Li, M. Du, Y. He, Y. Lan, Q. Yin, F. Zhu and G. Chang, Force-Reversible and Energetic Indole-Mg-Indole Cation–π Interaction for Designing Toughened and Multifunctional High-Performance Thermosets, Adv. Funct. Mater., 2022, 32, 2111021 CrossRef CAS.
- X. Yang, M. Guo, J. Yan, L. Wang, C. Zhao, D. Xiang, H. Li, J. Lai, Z. Li, A. Ahmed, L. Sun and Y. Wu, A Repeatable Dual-Encryption Platform from Recyclable Thermosets with Self-Healing Ability and Shape Memory Effect, Adv. Funct. Mater., 2022, 32, 2205177 CrossRef CAS.
- J. C. Capricho, B. Fox and N. Hameed, Multifunctionality in Epoxy Resins, Polym. Rev., 2020, 60, 1–41 CrossRef CAS.
- J. Han, G. Du, W. Gao and H. Bai, An Anisotropically High Thermal Conductive Boron Nitride/Epoxy Composite Based on Nacre-Mimetic 3D Network, Adv. Funct. Mater., 2019, 29, 1900412 CrossRef.
- D.-W. Li, H.-Y. Wang, Y. Liu, D.-S. Wei and Z.-X. Zhao, Large-scale fabrication of durable and robust super-hydrophobic spray coatings with excellent repairable and anti-corrosion performance, Chem. Eng. J., 2019, 367, 169–179 CrossRef CAS.
- J. Liu, S. Wang, Y. Peng, J. Zhu, W. Zhao and X. Liu, Advances in sustainable thermosetting resins: From renewable feedstock to high performance and recyclability, Prog. Polym. Sci., 2021, 113, 101353 CrossRef CAS.
- J. Zhang, X. Mi, S. Chen, Z. Xu, D. Zhang, M. Miao and J. Wang, A bio-based hyperbranched flame retardant for epoxy resins, Chem. Eng. J., 2020, 381, 122719 CrossRef CAS.
- M. Bahrami, Z. Ranjbar, R. A. Khosroshahi and S. Ashhari, Investigating corrosion protection properties of epoxy thermal insulators through cyclic corrosion test, Prog. Org. Coat., 2017, 113, 25–30 CrossRef CAS.
- S. Nakamura, T. Fujii, S. Matsukawa, M. Katagiri and H. Fukuyama, Specific heat, thermal conductivity, and magnetic susceptibility of cyanate ester resins – An alternative to commonly used epoxy resins, Cryogenics, 2018, 95, 76–81 CrossRef CAS.
- I. Chowdhury, R. Prasher, K. Lofgreen, G. Chrysler, S. Narasimhan, R. Mahajan, D. Koester, R. Alley and R. Venkatasubramanian, On-chip cooling by superlattice-based thin-film thermoelectrics, Nat. Nanotechnol., 2009, 4, 235–238 CrossRef CAS PubMed.
- Y. Liu, R. Zheng and J. Li, High latent heat phase change materials (PCMs) with low melting temperature for thermal management and storage of electronic devices and power batteries: Critical review, Renewable Sustainable Energy Rev., 2022, 168, 112783 CrossRef.
- F. Guo, X. Shen, J. Zhou, D. Liu, Q. Zheng, J. Yang, B. Jia, A. K. T. Lau and J.-K. Kim, Highly Thermally Conductive Dielectric Nanocomposites with Synergistic Alignments of Graphene and Boron Nitride Nanosheets, Adv. Funct. Mater., 2020, 30, 1910826 CrossRef CAS.
- Z. Liu, J. Li and X. Liu, Novel Functionalized BN Nanosheets/Epoxy Composites with Advanced Thermal Conductivity and Mechanical Properties, ACS Appl. Mater. Interfaces, 2020, 12, 6503–6515 CrossRef CAS PubMed.
- Z. Wu, C. Xu, C. Ma, Z. Liu, H.-M. Cheng and W. Ren, Synergistic Effect of Aligned Graphene Nanosheets in Graphene Foam for High-Performance Thermally Conductive Composites, Adv. Mater., 2019, 31, 1900199 CrossRef PubMed.
- X. Xu, J. Zhou and J. Chen, Thermal Transport in Conductive Polymer–Based Materials, Adv. Funct. Mater., 2020, 30, 1904704 CrossRef CAS.
- H. Chen, V. V. Ginzburg, J. Yang, Y. Yang, W. Liu, Y. Huang, L. Du and B. Chen, Thermal conductivity of polymer-based composites: Fundamentals and applications, Prog. Polym. Sci., 2016, 59, 41–85 CrossRef CAS.
- S. Ahmed and F. R. Jones, A review of particulate reinforcement theories for polymer composites, J. Mater. Sci., 1990, 25, 4933–4942 CrossRef CAS.
- Y. Guo, Y. Zhou and Y. Xu, Engineering polymers with metal-like thermal conductivity—Present status and future perspectives, Polymer, 2021, 233, 124168 CrossRef CAS.
- Y. Liu, Y. Zhou and Y. Xu, State-of-the-art, opportunities, and challenges in bottom-up synthesis of polymers with high thermal conductivity, Polym. Chem., 2022, 13, 4462–4483 RSC.
- Y. Guo, K. Ruan, X. Shi, X. Yang and J. Gu, Factors affecting thermal conductivities of the polymers and polymer composites: A review, Compos. Sci. Technol., 2020, 193, 108134 CrossRef CAS.
- X. Xu, J. Chen, J. Zhou and B. Li, Thermal Conductivity of Polymers and Their Nanocomposites, Adv. Mater., 2018, 30, 1705544 CrossRef PubMed.
- V. Singh, T. L. Bougher, A. Weathers, Y. Cai, K. Bi, M. T. Pettes, S. A. McMenamin, W. Lv, D. P. Resler, T. R. Gattuso, D. H. Altman, K. H. Sandhage, L. Shi, A. Henry and B. A. Cola, High thermal conductivity of chain-oriented amorphous polythiophene, Nat. Nanotechnol., 2014, 9, 384–390 CrossRef CAS PubMed.
- M. Kisiel and B. Mossety-Leszczak, Development in liquid crystalline epoxy resins and composites – A review, Eur. Polym. J., 2020, 124, 109507 CrossRef CAS.
- K. Ruan, X. Zhong, X. Shi, J. Dang and J. Gu, Liquid crystal epoxy resins with high intrinsic thermal conductivities and their composites: A mini-review, Mater. Today Phys., 2021, 20, 100456 CrossRef CAS.
- J. M. McCracken, V. P. Tondiglia, A. D. Auguste, N. P. Godman, B. R. Donovan, B. N. Bagnall, H. E. Fowler, C. M. Baxter, V. Matavulj, J. D. Berrigan and T. J. White, Microstructured Photopolymerization of Liquid Crystalline Elastomers in Oxygen-Rich Environments, Adv. Funct. Mater., 2019, 29, 1903761 CrossRef.
- G. Lv, C. Shen, N. Shan, E. Jensen, X. Li, C. M. Evans and D. G. Cahill, Odd–even effect on the thermal conductivity of liquid crystalline epoxy resins, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2211151119 CrossRef CAS PubMed.
- Y. Lin, X. Huang, J. Chen and P. Jiang, Epoxy thermoset resins with high pristine thermal conductivity, High Voltage, 2017, 2, 139–146 CrossRef.
- M. Akatsuka and Y. Takezawa, Study of high thermal conductive epoxy resins containing controlled high-order structures, J. Appl. Polym. Sci., 2003, 89, 2464–2467 CrossRef CAS.
- S. Tanaka, F. Hojo, Y. Takezawa, K. Kanie and A. Muramatsu, Highly Oriented Liquid Crystalline Epoxy Film: Robust High Thermal-Conductive Ability, ACS Omega, 2018, 3, 3562–3570 CrossRef CAS PubMed.
- Y. Xu, X. Wang, J. Zhou, B. Song, Z. Jiang, E. M. Y. Lee, S. Huberman, K. K. Gleason and G. Chen, Molecular engineered conjugated polymer with high thermal conductivity, Sci. Adv., 2018, 4, eaar3031 CrossRef PubMed.
- G.-H. Kim, D. Lee, A. Shanker, L. Shao, M. S. Kwon, D. Gidley, J. Kim and K. P. Pipe, High thermal conductivity in amorphous polymer blends by engineered interchain interactions, Nat. Mater., 2015, 14, 295–300 CrossRef CAS PubMed.
- A. I. Olamilekan and H. Yeo, Curing Behavior of 4,4′-Diglycidyloxybiphenyl with p-Phenylene Diamine Derivatives, Macromol. Res., 2020, 28, 960–967 CrossRef CAS.
- H. Yeo, Curing kinetics of liquid crystalline 4,4′-Diglycidyloxybiphenyl epoxy cured with 4,4′-Diaminodiphenylsulfone, Polymer, 2018, 159, 6–11 CrossRef CAS.
- H. Yeo, Curing kinetics of liquid crystalline 4,4′-diglycidyloxybiphenyl epoxy with various diamines, Polymer, 2019, 168, 209–217 CrossRef CAS.
- H. Yeo, A. M. Islam, N.-H. You, S. Ahn, M. Goh, J. R. Hahn and S. G. Jang, Characteristic correlation between liquid crystalline epoxy and alumina filler on thermal conducting properties, Compos. Sci. Technol., 2017, 141, 99–105 CrossRef CAS.
- A. M. Islam, H. Lim, N.-H. You, S. Ahn, M. Goh, J. R. Hahn, H. Yeo and S. G. Jang, Enhanced Thermal Conductivity of Liquid Crystalline Epoxy Resin using Controlled Linear Polymerization, ACS Macro Lett., 2018, 7, 1180–1185 CrossRef CAS PubMed.
- M. M. Hossain, A. I. Olamilekan, H.-O. Jeong, H. Lim, Y.-K. Kim, H. Cho, H. D. Jeong, M. A. Islam, M. Goh, N.-H. You, M. J. Kim, S. Q. Choi, J. R. Hahn, H. Yeo and S. G. Jang, Diacetylene-Containing Dual-Functional Liquid Crystal Epoxy Resin: Strategic Phase Control for Topochemical Polymerization of Diacetylenes and Thermal Conductivity Enhancement, Macromolecules, 2022, 55, 4402–4410 CrossRef CAS.
- Y. Kim, H. Yeo, N.-H. You, S. G. Jang, S. Ahn, K.-U. Jeong, S. H. Lee and M. Goh, Highly thermal conductive resins formed from wide-temperature-range eutectic mixtures of liquid crystalline epoxies bearing diglycidyl moieties at the side positions, Polym. Chem., 2017, 8, 2806–2814 RSC.
- K. Ku, S. Choe and H. Yeo, Correlation of self-assembly and thermal conducting properties of PEG-backbone polyether with a room temperature mesophase, Mol. Syst. Des. Eng., 2022, 7, 520–527 RSC.
- K. Ku and H. Yeo, Phase-controllable topochemical polymerization of liquid crystalline epoxy according to spacer length, Polym. Chem., 2023, 14, 644–650 RSC.
- A. I. Olamilekan and H. Yeo, Thermal Conducting Thermosets Driven by Molecular Structurally Enhanced Mesogen Interactions, ACS Appl. Polym. Mater., 2021, 3, 4147–4155 CrossRef CAS.
- T. E. Trinh, K. Ku and H. Yeo, Reprocessable and Chemically Recyclable Hard Vitrimers Based on Liquid-Crystalline Epoxides, Adv. Mater., 2023, 35, 2209912 CrossRef CAS PubMed.
- J. S. Hu, K. Q. Wei, B. Y. Zhang and L. Q. Yang, Synthesis, structure and properties of new chiral liquid crystalline monomers and homopolysiloxanes containing menthyl groups, Liq. Cryst., 2008, 35, 925–935 CrossRef CAS.
- A. Jakli, Odd-even effects in liquid crystals, Liq. Cryst., 2022, 49, 1010–1019 CrossRef CAS.
- R. Saha, G. Babakhanova, Z. Parsouzi, M. Rajabi, P. Gyawali, C. Welch, G. H. Mehl, J. Gleeson, O. D. Lavrentovich, S. Sprunt and A. Jákli, Oligomeric odd–even effect in liquid crystals, Mater. Horiz., 2019, 6, 1905–1912 RSC.
- Y. Li, P. Badrinarayanan and M. R. Kessler, Liquid crystalline epoxy resin based on biphenyl mesogen: Thermal characterization, Polymer, 2013, 54, 3017–3025 CrossRef CAS.
- J. Y. Lee, J. Jang, S. S. Hwang, S. M. Hong and K. U. Kim, Synthesis and curing of liquid crystalline epoxy resins based on 4,4′-biphenol, Polymer, 1998, 39, 6121–6126 CrossRef CAS.
- H. Guo, M. Lu, L. Liang, K. Wu, D. Ma and W. Xue, Liquid Crystalline Epoxies with Lateral Substituents Showing a Low Dielectric Constant and High Thermal Conductivity, J. Electron. Mater., 2017, 46, 982–991 CrossRef CAS.
- H. Guo, J. Zheng, J. Gan, L. Liang, K. Wu and M. Lu, High thermal conductivity epoxies containing substituted biphenyl mesogenic, J. Mater. Sci.: Mater. Electron., 2016, 27, 2754–2759 CrossRef CAS.
- G. Chen, Q. Zhang, Z. Hu, S. Wang, K. Wu, J. Shi, L. Liang and M. Lu, Liquid crystalline epoxies bearing biphenyl ether and aromatic ester mesogenic units: Synthesis and thermal properties, J. Macromol. Sci., Part A: Pure Appl. Chem., 2019, 56, 484–495 CrossRef CAS.
- Y. Li and M. R. Kessler, Cure kinetics of liquid crystalline epoxy resins based on biphenyl mesogen, J. Therm. Anal. Calorim., 2014, 117, 481–488 CrossRef CAS.
- C. Ortiz, R. Kim, E. Rodighiero, C. K. Ober and E. J. Kramer, Deformation of a Polydomain, Liquid Crystalline Epoxy-Based Thermoset, Macromolecules, 1998, 31, 4074–4088 CrossRef CAS.
- C. Shen, Q. Zhao and C. M. Evans, Ion specific, odd–even glass transition temperatures and conductivities in precise network polymerized ionic liquids, Mol. Syst. Des. Eng., 2019, 4, 332–341 RSC.
- H. Memon, Y. Wei and C. Zhu, Correlating the thermomechanical properties of a novel bio-based epoxy vitrimer with its crosslink density, Mater. Today Commun., 2021, 29, 102814 CrossRef CAS.
- T. Log and S. Gustafsson, Transient plane source (TPS) technique for measuring thermal transport properties of building materials, Fire Mater., 1995, 19, 43–49 CrossRef CAS.
- P. D. Davidse, H. I. Waterman and J. B. Westerdijk, Sound velocity and young's modulus in polyethylene, J. Polym. Sci., 1962, 59, 389 CrossRef CAS.
- W. Li, S. Lin, B. Ge, J. Yang, W. Zhang and Y. Pei, Low sound velocity contributing to the high thermoelectric performance of Ag8SnSe6, Adv. Sci., 2016, 3, 1600196 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00585b |
| This journal is © The Royal Society of Chemistry 2024 |