
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophobic cyclodextrin dimer-assisted self-healing elastomer: movable crosslinks of pseudo-rotaxane with recyclable and separable functionality†
Shintaro
Kawano
 *a,
Kaito
Ichiwara
b,
Haruto
Taneichi
b,
Shieri
Hamada
b,
Yuki
Fujino
b,
Osamu
Shimomura
b and
Motohiro
Shizuma
a
*a,
Kaito
Ichiwara
b,
Haruto
Taneichi
b,
Shieri
Hamada
b,
Yuki
Fujino
b,
Osamu
Shimomura
b and
Motohiro
Shizuma
a
aResearch Division of Surfactant Group, Osaka Research Institute of Industrial Science and Technology (ORIST), Morinomiya, Joto-ku, Osaka 536-8553, Japan. E-mail: skawano@orist.jp
bDepartment of Applied Chemistry, Faculty of Engineering, Osaka Institute of Technology (OIT), Asahi-ku, Osaka 535-8585, Japan
First published on 24th July 2024
Abstract
We fabricated a movable cross-linked elastomer derived from a pseudo-rotaxane by utilizing a dimer structure, in which the ring molecules made a threaded structure onto a polymer backbone, by simple bulk polymerization. The pseudo-rotaxane cross-linked elastomer exhibited facile healing and good stretching corresponding to the stress relaxation, which can contribute to the increment of fracture energy. We separated the topologically cross-linked dimer compounds from the threaded polymer backbone compound, constituting a recyclable cross-linker for a circular economy as a sustainable material.
Self-healing polymers, in which defects can be repaired autonomously, are of great technological interest to extend the durable life of materials. Developing self-healing materials can contribute to a sustainable society because of the corresponding reduction in waste by recycling and restoration after damage. To date, one of the main approaches to self-healing systems is introducing reversible crosslinks of noncovalent bonds based on supramolecular chemistry. Various designs have reported use of hydrogen bonds,1,2 metal–ligand coordination bonds,3,4 and π–π stacking5,6 interactions as well as host–guest interactions.7,8 These types of reversible crosslinks can undergo repeated cleavage and reformation by regeneration of noncovalent bonds between the polymer main and/or side chains, resulting in healing through a chemical process. The other main approach is physical self-healing by dispersing stress at deformation and returning to the original state; deriving from the high toughness, elasticity, and self-restoration.9,10 The physical approach can prevent microcracks from cleaving covalent bonds during deformation. The most important physical healing has resulted from polymeric materials with movable cross-linkers (topological cross-linker), referring to cross-linkers that indirectly connect to polymers with a mechanically interlocked architecture such as a polyrotaxane (PRx).11 PRx is a macromolecule composed of ring molecules threaded by an axle polymer, with bulky stoppers at both ends. The composition of numerous PRx materials include macrocyclic oligosaccharide, α-cyclodextrin (α-CD) as the ring compound and poly(ethylene glycol) as the axle compound.12 These materials have been used as slide ring gels, in which figure-eight cross-linkers are interlocked topologically with bulky stoppers along the polymer chain.13 The crosslinks can freely slide along the backbone chains in a manner that disperses internal stress and minimizes local stress, similarly to pulleys. Recently, rotaxane/polyrotaxane structures have been introduced into the side chain against a methacrylate or an acrylate polymer main chain, affording movable cross-linked elastomers with higher fracture strain and stress.10,14,15 However, movable cross-linking based on PRx has been used only for incorporating physical self-healing. Because the molecules are mechanically interlocked with bulky stoppers, chemical self-healing does not occur, for example, on the interface of a completely cut bulk material.
To overcome this issue, various researchers have studied stretchable elastomers based on PRx materials, combining physical and chemical healing from slide-ring motion of ring molecules and continuous dissociation/recognition of dynamic bonds such as hydrogen bonds, obtained by group modification of the ring or polymer chain grafted onto the ring.16,17 However, these strategies introduce complications into synthesis. Another movable cross-linked elastomer has been prepared by bulk copolymerization between hydrophobic CD monomers and alkyl acrylate.18–20 In the case of ethyl acrylate (EA) and peracetylated γ-cyclodextrin (γ-CD) monomers, poly-ethyl acrylate (PEA) main chains thread γ-CD rings after polymerization. Although a ring molecule can slide a certain distance on the threaded main PEA chain in the system, another γ-CD ring molecule connected with the side chains of PEA functions as bulky stopper for the slide-ring behavior of a γ-CD molecule. Therefore, movable cross-linked elastomers consisting of the PRx systems have not been used for chemical self-healing to repair e.g. the cut surface of bulk materials. Accordingly, in this study, we focused on a movable cross-linker of a pseudo-PRx consisting of an eyeglass-shaped dimer inspired by a figure-eight; in which the CD ring molecules of both ends can be threaded onto polymer main chains in a manner that topologically links with the other polymer chain, providing a simple approach to realize chemical self-healing as well as physical healing of stress dispersal through slide-ring motion. In addition, the pseudo-PRx structure was separable in a manner that enabled collection of the movable cross-linker from the threaded structures of the polymer through extraction and precipitation, consistent with the need for a circular economy and reliably recycling sustainable materials.
To study the self-healing of an elastomer exhibiting a movable cross-linker, we utilized pseudo-rotaxane ring molecules at both ends connected by a linker molecule at the center, inspired by a figure-eight,11 enabling a pulley effect on the polymer main chain. We designed and synthesized an eyeglass-shaped macrocyclic dimer (D1) (Fig. 1A); consisting of a symmetrical structure of macrocyclic ring molecules, hydrophobic γ-CDs, covalently conjugated by a polyoxyethylene linker via the click reaction. In the case of the big cavity size of γ-CDs, we used a bulky substituent for arenesulfonylation to assess the selective C-6 position for mono-substitution on the primary face of the CD cavity21 (Scheme S1A, ESI†). In addition, purifying this compound was relatively easy by resin reverse-phase chromatography because of inhibition of further poly-substitutions on the C-6 position. In our case, we synthesized hydrophobic mono-6-substituted γ-CD by reaction of a monosulfonyl compound with sodium azide; next, we peracetylated the protected mono-6-azide CD (Scheme S1B and C†). We obtained the hydrophobic CD dimer, D1 by Cu-catalyzed 1,3-dipolar cycloaddition with a tetraethylene glycol-substituted dialkyne linker (Scheme S1D†), affording an eyeglass shape in which two rings of the both ends link with a flexible chain of the oligo-ethylene glycol chain [Fig. 1A (right)]. The ESI† shows experimental details and analytical data by 1H NMR and MS (Fig. S1–S15†), and are consistent with the proposed molecular structures.
 | ||
| Fig. 1 Chemical structure and schematic of AcO-γ-CD dimer (D1) (A) and synthetic route of PEA-D1 by bulk photo-polymerization of EA monomer with a movable cross-linker, D1 (B). | ||
We prepared acrylate-based elastomers using the movable crosslinker of the eyeglass-like dimer to clarify the stress dispersive effect and chemical self-healing, such as supramolecular recognition of the dynamic bonds derived from a threaded structure between the cross-linker and polymer main chains (Fig. 1B). We prepared the acrylate elastomers by bulk photo-polymerization of EA monomer containing a D1 movable cross-linker on the PTFE mold; the ESI shows details (Fig. S16†). We used 1,4-butanediol diacrylate (BDA) as a conventional chemical cross-linker for comparing the movable crosslinker. We also used a peracetylated γ-cyclodextrin; PAcO-γ-CD (M1; Fig. S11 and S12†) as a simple slide-ring PRx additive, but not linked with the other ring molecule to cross-link the threaded polymer chain as in the D1 dimer.
We characterized the threaded structure of the γ-CD ring molecule onto the PEA backbone by nuclear Overhauser effect (NOE) differential spectra of 1D NMR (Fig. S17†). Difference NOE can confirm that the object definitely threads the γ-CD ring of the movable cross-linker, rather than the mixing of both components. We also prepared a model compound by introducing a bulky long alkyl chain into the copolymer unit to function as a quasi-bulky stopper in 1H NMR.18 When we assigned the resonance field irradiation to specific 1H positions in which the γ-CD host-H3 (5.3 ppm) or H5 (3.6 ppm) was saturated, we detected the HA proton (1.6 ppm) corresponding to the PEA–CH2 backbone as a substantial correlation peak for each saturation in the nonirradiated differential NOE spectra. Thus, there was an inclusion complex between the γ-CD internal cavity (H3, H5) and the PEA main chain, indicating threading of the γ-CD host onto the PEA backbone.
We evaluated the mechanical properties PEA-D1 and PEA-M1 by tensile tests compared with chemically cross-linked PEA–BDA and linear PEA (without cross-linker). Fig. 2 shows the stress–strain curves of the elastomers, measured at a rate of 1 mm s−1. Table 1 shows the mechanical stress and strain at the elastic yield point, fracture strain at break, Young's modulus, and fracture energy. We calculated the fracture energy related to the material toughness from the integral of the stress–strain curve, representing the tensile test results. Prior to describing the effect of the movable cross-linker using D1, we evaluated the control linear PEA during the tensile tests. The stress of PEA was low because of the deformable plastic property (glass transition temperature Tg = −18 °C) although the fracture strain was extended to ca. 1100%. Furthermore, the chemically cross-linked elastomer (PEA–BDA-0.1) immediately fractured with a low strain of ca. 100%. This result might be attributable to chemically cross-linked covalent bonds, which cannot disperse the applied stress. However, the movable cross-linked elastomer PEA-D1 exhibited interesting mechanical properties from the tensile tests with varying quantities of D1 cross-linker. Whereas the elastic yield stress increased to ca. 380 kPa with increasing quantity of D1 from 0.1–0.5 mol%, the stress gradually decreasing after the elastic yield point. Moreover, the fracture strains of the D1 materials reached ca. 2300 and 2500% for added quantity of D1 at 0.5 and 0.2 mol%, respectively; which are much higher than that of chemically cross-linked PEA–BDA or linear PEA homopolymer. The Young's modulus also substantially increased (∼3.0 MPa) by increasing addition of D1 before reaching the elastic yield point. The gradual decrease of the stress for strain elongation, but no breaks at strain, might be owing to the stress relaxation of PEA-D1. In the study, we carried out tensile tests of PEA-M1, derived from only the threading material of the CD ring molecules but not cross-linking each PEA main chain, to compare the stress dissipation and relaxation effects. We did not observe a gradual decrease of stress until break although the elastic yield retained ca. 470% to 610% strain. This result might be attributable to concentrated placement of M1 axel molecules against the uniaxial extension, enabling constant stress.22 Thus, introducing a movable cross-linking unit D1 to bind each inter-polymer main chain and threading CD ring molecules facilitated stress relaxation as well as no bulky stopper for anchoring the threaded ring molecules, resulting in semi-permanent elongation in a manner that decreased the strain fracture. We confirmed this hypothesis by stress relaxation tests. Fig. S18† shows the process of relaxing the stress of movable cross-linked D1 materials and chemically cross-linked materials (PEA–BDA) yet keeping the strain at 100%. PEA-D1 exhibited a large stress relaxation over a longer time compared with PEA–BDA. At equilibrium in 1800 s, stress relaxation reached ca. 82%. PEA–BDA relaxed to ca. 30% stress and reached equilibrium in a shorter time compared with PEA-D1. In the tensile tests of relaxing the stress for PEA-M1, we observed constant stress relaxation even when using only threading materials without a linker at a 78% less relaxation rate than that of PEA-D1 upon maintaining the strain at 100%. The slight difference might be due to the sliding elasticity of the monomeric γ-CD ring molecules by entropic restoring force when the alignment entropy decreases with uniaxial deformation.23 More importantly, such specific stress relaxation of PEA-D1 might be derived from the movable cross-linked point. We increased the calculated fracture energy of PEA-D1 to ca. 25× higher than that of PEA–BDA by increasing the D1 content from 0.1 to 0.5 mol% (Table 1). These superior fracture energies might also result from the threading effect of a movable cross-linker of the D1 materials, which can efficiently dissipate the applied stress.
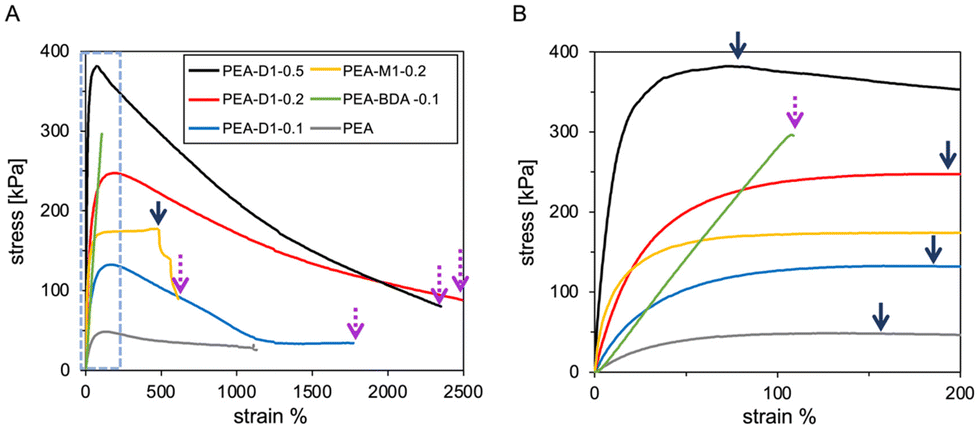 | ||
| Fig. 2 Stress–strain curves of PEA-D1, PEA-M1, PEA–BDA, and PEA by tensile tests at a rate of 1.0 mm s−1 (A) and enlarged view at comparatively low strains (dashed line) (B). The concentration of the cross-linker was 0.1 (for D1 and BDA), 0.2 (for D1 and M1), and 0.5 (for D1) mol%. Arrows indicate elastic fields (solid lines) and fracture points (dashed lines). | ||
| Elastic yield stress [kPa] | Strain% (for yield stress) | Young's modulus [MPa] | Fracture strain% | Fracture energy [MJ m−3] | |
|---|---|---|---|---|---|
| PEA-D1-0.5 | 382 | 74 | 3.0 | 2360 | 7.8 |
| PEA-D1-0.2 | 247 | 201 | 0.74 | 2500 | 6.6 |
| PEA-D1-0.1 | 133 | 190 | 0.43 | 1780 | 2.1 |
| PEA-M1-0.2 | 177 | 477 | 2.0 | 613 | 1.6 |
| PEA-BDA-0.1 | 298 | 106 | 0.29 | 106 | 0.32 |
| PEA | 49 | 160 | 0.14 | 1142 | 0.70 |
We evaluated the self-healing of the movable cross-linked PEA-D1 elastomer (Fig. 3A and Movie S1†). We cut the elastomer into two pieces with scissors and immediately reattached the two pieces with damaged cross sections. The recovered piece supported itself even after lifting by shaking with a tweezer, and then the piece adhered tightly as one piece without separation after pulling the opposite sides with a tweezer. However, the chemically cross-linked elastomer PEA–BDA did not adhere after the same experiment (Fig. S19†). Thus, self-healing of the movable cross-linked PEA-D1 elastomer is a result of the reversible reconstruction derived from the supramolecular pseudo-PRx structure, which can rapidly migrate to the most stable state on the fractured surface. It is not apparent whether the CD ring molecules would newly thread onto the axle PEA chain on the fractured surface. However, the pseudo-PRx cross-linked structure indicates that the dissociated structure after rupture by external stimuli might enable a return to the equilibrium complex structure (by the plastic flow generated by rotational motion of the CD rings around the PEA chain axis and the local slip-ring effect), resulting in self-healing. We evaluated the self-healing efficiency of the elastomer using tensile tests. We allowed self-healed pieces to stand at 25 °C and 80 °C for 24 h after contacting the cross sections of cut pieces. Herein, we defined the healing efficiency [(S1 or S2)/S0 × 100%] as the points of elastic yield (S0: original sample, S1: re-adhered sample at 25 °C, S2: re-adhered sample at 80 °C) before starting stress relaxation. The elastomers (PEA-D1-0.2) exhibited healing ratios of 80% and 85% at 25 °C and 80 °C, respectively (Fig. 3B). Whereas the elastic yield points were almost the same efficiency, the fracture strain was more effectively improved with increasing healing temperature. Thus, there were various binding interactions that did not clearly depend on the temperature within the polymer matrix, whereas the faster reconstruction and motion of the PEA chains were driven by higher temperature. Linear thermoplastic polymers such as homopolymer PEA also exhibit self-healing. However, because these rubbery elastomers exhibit a low Tg (<0 °C) and readily deform at room temperature, they are not mechanically stable polymers. More importantly, our movable cross-linked elastomers realized self-healing with self-supported and enhanced mechanical properties; potentially expanding the applicability of elastomers. The movable cross-linked elastomer PEA-D1, consisting of a pseudo-PRx structure with hydrophobic cyclodextrin dimer, has potential for separation and reuse because the PEA-D1 enables topological threading of D1 onto the PEA backbone without exhibiting a bulky stopper on the PEA chain ends. In addition to the ability to separate, recover, and reuse D1 (synthesized in multiple steps over time), application to reprocessing by recovery of PEA (a typical acrylate ester polymer) could contribute to a circular economy through a zero-waste process for these types of cross-linked polymer materials. We independently separated D1 (Mw: 4848), smaller than PEA (Mn: 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mw: 190000), by solvent dilution and precipitation. After we dissolved a used piece of PEA-D1 elastomer in tetrahydrofuran (THF) at a sufficiently diluted volume, followed by evaporating THF and drying in vacuo overnight, we precipitated only D1 by using diethyl ether from the PEA-D1 solid. We efficiently collected D1 from the used elastomer at 63% yield even after the first cycle. Gel permeation chromatography (GPC) measurements of the PEA-D1 mixture diluted in THF indicate complete separation at each size-excluded molecular fraction between PEA and D1 (Fig. 4A). In addition, we detected and fractionated the independent D1 compound after precipitation of D1 by using diethyl ether from the PEA-D1 solid without the PEA fraction (Fig. 4B). 1H NMR spectra of D1 also confirmed the high purity of D1 with a small quantity of PEA moiety (<1% compared with the starting mixture; Fig. S20†). We efficiently collected D1 at 63% yield even after the first cycle. Furthermore, we carried out zero-waste separable collection for PEA-D1 (by simple preparative GPC) by continuous separation utilizing the hierarchical molecular weight fraction; at 100000 and 4848 Da for PEA and D1, respectively. Thus, we independently separated PEA from D1 (Fig. S21†).
000, Mw: 190000), by solvent dilution and precipitation. After we dissolved a used piece of PEA-D1 elastomer in tetrahydrofuran (THF) at a sufficiently diluted volume, followed by evaporating THF and drying in vacuo overnight, we precipitated only D1 by using diethyl ether from the PEA-D1 solid. We efficiently collected D1 from the used elastomer at 63% yield even after the first cycle. Gel permeation chromatography (GPC) measurements of the PEA-D1 mixture diluted in THF indicate complete separation at each size-excluded molecular fraction between PEA and D1 (Fig. 4A). In addition, we detected and fractionated the independent D1 compound after precipitation of D1 by using diethyl ether from the PEA-D1 solid without the PEA fraction (Fig. 4B). 1H NMR spectra of D1 also confirmed the high purity of D1 with a small quantity of PEA moiety (<1% compared with the starting mixture; Fig. S20†). We efficiently collected D1 at 63% yield even after the first cycle. Furthermore, we carried out zero-waste separable collection for PEA-D1 (by simple preparative GPC) by continuous separation utilizing the hierarchical molecular weight fraction; at 100000 and 4848 Da for PEA and D1, respectively. Thus, we independently separated PEA from D1 (Fig. S21†).
 | ||
| Fig. 3 (A) Photographs of self-healing of PEA-D1-0.2 elastomer before and after healing: (1) and (2) we cut PEA-D1-0.2 into two pieces with scissors, (3) we reattached the two pieces with damaged cross sections, and (4) two pieces were sufficiently adhered to be pulled from opposite sides without breakage. (B) Stress–strain curves of original elastomer (S0: PEA-D1-0.2) and elastomers healed after heating at 25 °C (S1) and 80 °C (S2) by tensile tests; self-healing efficiency percentage = [(S1 or S2)/S0] × 100. | ||
 | ||
| Fig. 4 GPC analysis of PEA-D1-0.5 diluted in THF (upper) and product collected after separation by using diethyl ether (lower). | ||
Conclusions
In summary, we synthesized an eyeglass-shaped dimer (D1) to serve as a topological cross-linker and obtained movable cross-linked elastomer threading of D1 onto the polymer chain by bulk photo-polymerization of the EA monomer with D1. The mechanical properties of the obtained elastomer (PEA-D1) exhibited an improved elastic modulus, stretching, and toughness (even containing only 0.5 mol% D1 movable cross-linker) compared with that of linear PEA and covalently cross-linked PEA–BDA. The movable cross-linked elastomer derived pseudo-PRx structure without a bulky stopper exhibited rapid self-healing. The topological cross-linker (connected to a universal acrylate polymer) enabled separation and reuse by simple solvent dilution and precipitation, in addition to the durability of the self-healing materials, contributing a new paradigm for a circular economy and sustainable long-life materials.Author contributions
All authors discussed the results and contributed to the final manuscript.Data availability
The data supporting this article have been included as part of the ESI.†The data supporting this article have been included as part of the ESI as follow; ESI available: Detailed synthetic and experimental procedures; 1H NMR, COSY, 13C NMR, DEPT NMR, HMQC spectra, MALDI-TOF-MS, GPC traces for synthesized compounds; characterization of threaded inclusion complex structures from NOE 1H NMR spectra; characterization of stress relaxation of elastomer from tensile tests, self-healing property of PEA-BDA (chemically crosslinked elastomer), and recyclability for a PEA-D1 from 1H NMR spectra and GPC analysis (PDF); self-healing performance of a PEA-D1 elastomer (MP4). See ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Michael Scott Long, PhD, from Edanz for editing a draft of this manuscript. This work was supported by JSPS KAKENHI Grant Numbers JP18K14021.References
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334 CrossRef CAS PubMed.
- X. Wang, S. Zhan, Z. Lu, J. Li, X. Yang, Y. Qiao, Y. Men and J. Sun, Adv. Mater., 2020, 32, 2005759 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 44, 6717 RSC.
- J.-F. Mei, X.-Y. Jia, J.-C. Lai, Y. Sun, C.-H. Li, J.-H. Wu, Y. Cao, X.-Z. You and Z. Bao, Macromol. Rapid Commun., 2016, 37, 1667 CrossRef CAS PubMed.
- S. Nomimura, M. Osaki, J. Park, R. Ikura, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2019, 52, 2659 CrossRef CAS.
- J. Park, S. Murayama, M. Osaki, H. Yamaguchi, A. Harada, G. Matsuba and Y. Takashima, Adv. Mater., 2020, 32, 2002008 CrossRef CAS PubMed.
- M. Nakahata, S. Mori, Y. Takashima, H. Yamaguchi and A. Harada, Chem, 2016, 1, 766 CAS.
- K. Koyanagi, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2017, 50, 5695 CrossRef CAS.
- K. Mayumi, K. Ito and K. Kato, Polyrotaxane and Slide-Ring Materials, ed. J. Steed and P. Gale, Royal Society of Chemistry, Rondon, UK, 2015 Search PubMed.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485 CrossRef CAS.
- J. Sawada, D. Aoki, S. Uchida, H. Otsuka and T. Takata, ACS Macro Lett., 2015, 4, 598 CrossRef CAS PubMed.
- H. Gotoh, C. Liu, A. B. Imran, M. Hara, T. Seki, K. Mayumi, K. Ito and Y. Takeoka, Sci. Adv., 2018, 4, eaat7629 CrossRef CAS.
- R. Du, Z. Xu, C. Zhu, Y. Jiang, H. Yan, H. C. Wu, O. Vardoulis, Y. Cai, X. Zhu, Z. Bao, Q. Zhang and X. Jia, Adv. Funct. Mater., 2020, 30, 1907139 CrossRef CAS.
- Q. Jin, R. Du, H. Tang, Y. Zhao, W. Peng, Y. Li, J. Zhang, T. Zhu, X. Huang, D. Kong, Y. He, T. Bao, D. Kong, X. Wang, R. Wang, Q. Zhang and X. Jia, Angew. Chem., Int. Ed., 2023, 62, e202305282 CrossRef CAS PubMed.
- R. Ikura, J. Park, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, Macromolecules, 2019, 52, 6953 CrossRef CAS.
- Y. Kawai, J. Park, Y. Ishii, O. Urakawa, S. Murayama, R. Ikura, M. Osaki, Y. Ikemoto, H. Yamaguchi, A. Harada, T. Inoue, H. Washizu, G. Matsuba and Y. Takashima, NPG Asia Mater., 2022, 14, 32 CrossRef CAS.
- R. Ikura, S. Murayama, J. Park, Y. Ikemoto, M. Osaki, H. Yamaguchi, A. Harada, G. Matsuba and Y. Takashima, Mol. Syst. Des. Eng., 2022, 7, 733 RSC.
- R. Palin, S. J. A. Grove, A. B. Prosser and M.-Q. Zhang, Tetrahedron Lett., 2001, 42, 8897 CrossRef CAS.
- K. Kato, K. Nemoto, K. Mayumi, H. Yokoyama and K. Ito, ACS Appl. Mater. Interfaces, 2017, 9, 32436 CrossRef CAS.
- K. Mayumi, M. Tezuka, A. Bando and K. Ito, Soft Matter, 2012, 8, 8179 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic and experimental procedures; 1H NMR, COSY, 13C NMR, DEPT NMR, HMQC spectra, MALDI-TOF-MS, GPC traces for synthesized compounds; characterization of threaded inclusion complex structures from NOE 1H NMR spectra; characterization of stress relaxation of elastomer from tensile tests, self-healing property of PEA-BDA (chemically crosslinked elastomer), and recyclability for a PEA-D1 from 1H NMR spectra and GPC analysis (PDF); self-healing performance of a PEA-D1 elastomer (MP4). See DOI: https://doi.org/10.1039/d4lp00196f |
| This journal is © The Royal Society of Chemistry 2024 |