
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBacterial cellulose nanoparticles as a sustainable drug delivery platform for protein-based therapeutics†
Gabrielle N.
Balistreri
ac,
Ian R.
Campbell
b,
Xinqi
Li
b,
Julia
Amorim
e,
Shuai
Zhang
 abd,
Elizabeth
Nance
ac and
Eleftheria
Roumeli
*ab
abd,
Elizabeth
Nance
ac and
Eleftheria
Roumeli
*ab
aMolecular Engineering & Sciences Institute, University of Washington, Seattle, WA 98195, USA. E-mail: eroumeli@uw.edu
bDepartment of Materials Science and Engineering, University of Washington, Seattle, WA 98195, USA
cDepartment of Chemical Engineering, University of Washington, Seattle, WA 98195, USA
dPhysical Sciences Division, Pacific Northwest National Laboratory, Richland, WA99354, USA
eDepartment of Biotechnology, Federal Rural University of Pernambuco, Recife PE 52171-900, Brazil
First published on 13th December 2023
Abstract
Sustainable nanomedicine is an emerging field aiming to address the challenges of scalability, reproducibility, thermal stability, and excessive waste generation for nanotherapeutic manufacturing. Bacterial cellulose (BC) fibers have the potential to overcome these challenges and be a versatile drug delivery platform. Here we report the development of BC nanoparticles (BCNPs) for sustainable drug delivery applications motivated by the material's biodegradability upon environmental disposal and biocompatibility, which are important properties for nanomedicine applications. In addition, BCNPs formulation has a reduced environmental impact, an overall eco-friendly life cycle, and can be implemented following green engineering principles. In this study, we fabricated BCNPs grown in a kombucha medium in agitated and aerated conditions for 24 h and size separated using centrifugation and polysorbate 80 as a surfactant. The produced particles are approximately 100 nm in diameter and have a slightly negative zeta-potential and predominantly amorphous morphology. We also investigated the growth of BC fibers after 1, 3, and 5 days and evaluated the BC's time-dependent physicochemical properties using X-ray diffraction, Fourier transform infrared spectroscopy, and thermogravimetric analysis. We report primarily amorphous BCNPs obtained after culturing for 1 day, while longer culture duration leads to larger BC particles comprised of fibers with increasing degree of crystallinity. Moreover, we show BCNPs are thermally stable up to 90 °C. We performed proof of concept studies to show drug loading capability by incorporating bovine serum albumin (BSA) as a model drug and quantified sustained release of BSA. These results further motivate the use of BCNPs as a promising nanotherapeutic platform.
1. Introduction
Within the pharmaceutical industry, nanomedicine is a rising sector where manufacturing is a major issue. By 2030, the global nanomedicine market is projected to be a $427-billion-dollar industry driven by continued investment in the development of novel drug delivery systems and an increasing demand for safe and affordable therapeutics.1 The state of the art materials being used for these applications are synthetic polymers derived from nonrenewable petroleum resources such as poly(glycolic acid) (PGA) and poly(lactic-co-glycolic acid) (PLGA) and lipids.2–4 Advantages of synthetic polymers and lipids are the tunability of mechanical properties and degradation kinetics, batch reproducibility, and consistency.2,5 However, not only are these synthetic feedstocks non-renewable, but they often require use of organic solvents during their synthesis and processing which then need to be disposed of appropriately.6 These solvent disposal processes create further environmental challenges, particularly in terms of generating waste streams associated with undesirable environmental impacts of the therapeutic products.7 The field of sustainable nanomedicine aims to circumvent these harmful effects of traditional nanotherapeutics by eliminating solvent use and adopting low-waste and green engineering methodologies.Cellulose, the most abundant renewable natural polymer, has been studied in the nanomedicine industry for biomedical and drug delivery applications.8 It is a biocompatible polymer that is naturally excreted as waste from the human body, yet exhibits biodegradation upon environmental disposal, which enables a less harmful end-of-life fate compared to non-biodegradable alternatives. Most studies utilize plant biomass-sourced cellulose where there is a need for extraction which requires solvent processing and significant energy consumption for mechanical defibrillation and chemical extraction.9 Alternatively, cellulose can be produced as an extracellular product by some bacterial species in specific culturing conditions.8,10 The key benefit of bacterial cellulose (BC) is that it is the only product of that biosynthesis process and therefore there is no need to extract and chemically treat the resulting biomass to isolate cellulose fibers.
However, the large-scale production of BC has been a long-standing challenge. Key limitations include the cost of materials, with growth media accounting for about 50–65% of the total cost,11 the energy expenditure required to maintain optimal growth conditions12 and the often limited and inconsistent yield, which consequently restrict product reproducibility.13 To address these challenges, prior studies have proposed various strategies. For instance, industrial applications employing low-temperature microbial cultures have been explored to reduce energy consumption.12,14 Furthermore, there has been research into utilizing agricultural waste, such as fruit peels and juices,12 wastewater,15 or other low-cost carbon sources, such as glycerol remaining from biodiesel production11 to reduce material costs. Additionally, co-cultures consisting of acetic acid bacteria and lactic acid bacteria have shown promise in enhancing cellulose yield and improving the limitations of batch to batch variability.12,13
BC has also been produced in an aqueous and low-cost sugar-based medium, known as a kombucha, from certain bacterial strains co-cultured with yeast species.16 BC grown in such co-cultures has several advantages: (1) it is produced in mild conditions with no pressure, specific gas flows, or sterility required, which are distinct advantages over single strain cultures; (2) BC production uses a metabolic, fossil-free process, where the only product of the bacterial cultures is BC, (3) the co-culture creates an environment that restricts the proliferation of certain contaminants, especially fungi17 and (4) it is produced at a commercial scale.16,18,19 For these reasons, BC is an emerging biopolymer platform used in engineering and biomedical applications.16
To date, BC has primarily been used in the drug delivery field for transdermal applications, where BC films are prepared for use in antimicrobial wound healing and dressing, and face and eye masks for anti-aging and scarred skin rejuvenation.20–22 Spherical BC particles with diameters in the 0.5–1.0 mm range have also previously been reported to enable a controlled drug release and exhibit antibacterial properties for tissue engineering and drug delivery applications.8,16,22–25 Despite the drug delivery potential demonstrated by BC microparticles, their application for long-circulation times and deep tissue penetration is limited due to their size. For injectable drug delivery systems, a nanoscale BC particle would provide longer circulation times, improved bioavailability and biodistribution, and greater targeted delivery through increased tissue penetration and cell specific uptake. Therefore, here we aim to create BC in nanoparticle form, by controlling the growth of cellulose, to leverage the opportunity to expand the utilization of BC as a drug delivery platform.
In order to grow BC nanoparticles, we must understand that the BC growth occurs as self-assembled cellulose chains extracellularly extruded from certain types of bacteria such as Gluconacetobacter xylinus, formerly known as Acetobacter xylinum, a Gram negative bacterium.9 The cellulose chains are arranged in a parallel configurations stabilized by intermolecular bonding (hydrogen bonding, van der Waals interactions) forming elemental fibrils which subsequently bundle to form fibers.9 Elemental BC fibrils from Gluconacetobacter xylinus have high crystallinity (up to 80%) with a cross-sectional thickness of 6–10 nm. Elementary fibrils bundle to form larger fibers with reported thicknesses ranging from 30–80 nm and lengths up to several microns.9 Depending on the culture conditions, the yield, morphology, structure, crystallinity, and degree of polymerization of the BC fibers can be altered and controlled.26 Incubation in static conditions enables the formation of a layered 3D-network of BC fibers, called a pellicle, in which the interlayer connectivity is facilitated by entangled, randomly oriented BC fibers. Agitated culture conditions, on the other hand, utilize centrifugal forces to bundle the growing cellulose fibers into spherical-like particles. In both static and agitated culture conditions, the high degree of crystallinity, high fiber aspect ratio, and the absence of other polymers as extracellular components of the produced pellicle or spherical particle, serve as key advantages for the produced BC as compared to plant-derived cellulose.
Here, we leverage agitated culture conditions to develop BC nanoparticles (BCNPs) and discuss their performance as a drug delivery platform. To bridge the gap between laboratory-scale production and scaled-up manufacturing to make BCNPs a more feasible solution for widespread applications in nanomedicine, we use an industrially scalable bacterial-yeast co-culture platform (Symbiotic Cultures of Bacteria and Yeast, SCOBY). To demonstrate the drug loading capacity of BCNPs, we used bovine serum albumin (BSA) as a model drug. BSA is a low cost, biodegradable, and biocompatible protein therapeutic with a high aqueous solubility and can be easily surface modified.27,28 Our results aim to serve as the foundation for producing BCNPs to be used as sustainable natural polymer carriers for delivery of protein therapeutics.
2. Materials and methods
2.1. BCNPs preparation, endotoxin removal, and size separation
BCNPs were prepared in agitated and aerated conditions in a kombucha medium on an orbital plate shaker at 150 rpm and 30 °C (Fig. 1A). The tea media was prepared with 1 L of nitrogen sourced black tea (Lipton Classic – black tea bags, Englewood Cliffs, NJ, USA) steeped for 5 min in hot deionized (DI) water, 200 g white pure cane sugar (Domino Sugar, Yonkers, NY, USA), and 20 mL apple cider vinegar (Bragg Organic, Santa Barbara, CA, USA). SCOBY were purchased from Joshua Tree Kombucha (Joshua Tree, CA, USA), and its contents were separated by membrane (pellicle) and liquid, both containing microbial co-cultures (Fig. S1†). 23 mL of tea media and 2 mL SCOBY liquid were added to a 250 mL Erlenmeyer flask and placed on a plate shaker for 24 h. Agitation enables the synthesized cellulose fibers to take a spherical shape, rather than pellicle form.16,18 At 24 h, 0.1 M sodium hydroxide solution (NaOH, Thermo Fisher, Waltham, MA, USA) is titrated into the kombucha media to a slight excess to terminate the growth of cellulose.16,18 The BCNPs were harvested from media with vacuum filtration and cleaned from excess media and NaOH via dialysis tubing made of cellulose ester (MW cutoff: 3 kDa, Spectrum Laboratories, Inc., San Francisco, CA, USA) for 24 h (Fig. 1B). A 100 K centrifugal tube filter centrifuged at 4000 rpm for 25 min was used to separate BCNPs from solution.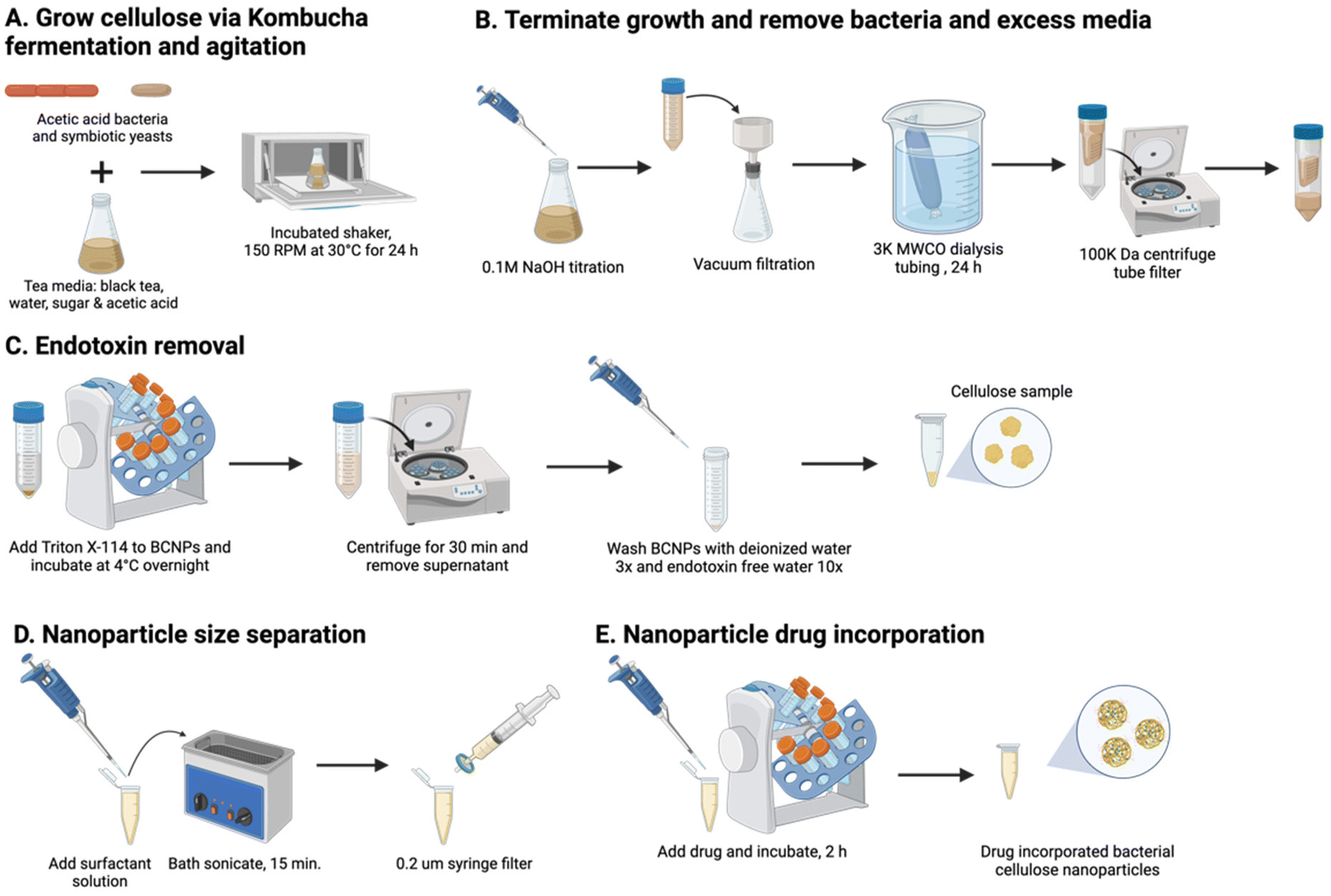 | ||
| Fig. 1 BCNPs synthesis and preparation for drug loading applications: (A) growth process of BC in a kombucha medium and agitated for 24 h, (B) termination of growth and isolation of BCNPs, (C) endotoxin removal from BCNPs, (D) application of surfactant solution and particle size separation, and (E) drug incorporation process. Created with Biorender.com. | ||
2.2. Endotoxin removal and endotoxin quantification assay for BCNPs
To remove the endotoxins from BCNPs (Fig. 1C), 25 mL of 0.1% Triton X-114 (TX-114, Thermo Fisher, Waltham, MA, USA) were added to the BCNPs and incubated overnight at 4 °C on a RotoBot R4045 standardized sized tube rotator (Stellar Scientific, Baltimore, MD, USA). The BCNPs and TX-114 solution were centrifuged at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rcf for 30 min, collected, and washed with 1.5 mL of DI water three times at 15000 rpm for 30 min. The collected BCNPs were next washed with 1.5 mL of endotoxin-free ultra-pure water (Sigma-Aldrich, St Louis, MO, USA) 10 times at 15000 rpm for 30 min. To quantify the endotoxin units per milliliter (EU mL−1), a Pierce™ Chromogenic Endotoxin Quant Kit (Thermo Fisher, Waltham, MA, USA) measured the supernatant after the last wash step. Prior to assay analysis, the supernatant was filtered with a 200 nm syringe filter, dilute 1:50 in endotoxin free water, and filtered with a 200 nm syringe filter.
000 rcf for 30 min, collected, and washed with 1.5 mL of DI water three times at 15000 rpm for 30 min. The collected BCNPs were next washed with 1.5 mL of endotoxin-free ultra-pure water (Sigma-Aldrich, St Louis, MO, USA) 10 times at 15000 rpm for 30 min. To quantify the endotoxin units per milliliter (EU mL−1), a Pierce™ Chromogenic Endotoxin Quant Kit (Thermo Fisher, Waltham, MA, USA) measured the supernatant after the last wash step. Prior to assay analysis, the supernatant was filtered with a 200 nm syringe filter, dilute 1:50 in endotoxin free water, and filtered with a 200 nm syringe filter.
2.3. BCNPs size separation
To size separate further, 1 mL BCNPs suspended in water were centrifuged and the supernatant was removed. The BCNPs were bath sonicated with 500 μL of 0.1 M polysorbate 80 surfactant solution (P80, Thermo Fisher, Waltham, MA, USA) and size filtered with a 200 nm syringe filter until further use (Fig. 1D).2.4. BCNPs size and zeta potential characterization
The particle size was measured by nanoparticle tracking analysis (NTA, NanoSight Malvern PANalytical, Salisbury, UK) at 23.9–24.1 °C (n = 5) and zeta potential (ζ-potential) was determined using a ζ-potential analyzer (NanoSizer Zeta Series, Malvern Instruments, Malvern, UK) at 37 °C. Samples were diluted to a 1:100 ratio (sample to DI water) from a batch concentration of ∼5 mg mL−1. To accurately measure the ζ-potential, 5 μL of 10 mM sodium chloride (NaCl, Sigma-Aldrich, St Louis, MO, USA) was added to the dilute samples.
2.5. Scanning transmission electron microscopy (STEM)
To prepare BCNPs for STEM, the samples were diluted to 1:200 ratio (sample to DI water) from batch concentration. BCNPs were adsorbed on a 200-mesh carbon film grid (Fisher Scientific, Waltham, MA, USA), negatively stained with uranyl acetate replacement electron microscopy stain (UAR-EMS, Electron Microscopy Sciences, Hatfield, PA, USA), and then washed in DI water. Samples were prepared and imaged on the Tecnai G2 F20 SuperTwin TEM (Field Electron and Ion Company, Hillsboro, OR, USA).
2.6. BC particle 3-day and 5-day preparation
BC particles were grown for 3-day (3d) and 5-day (5d) timepoints in a kombucha medium and in agitated and aerated conditions, as mentioned in section 2.1. At the 3d and 5d timepoints, the BC particles were separated from media via gravity filtration and then washed with 0.1 M NaOH and water to terminate the cellulose growth. Particles were placed in dialysis tubing (MW cutoff: 3 kDa, Spectrum Laboratories, Inc., San Francisco, CA, USA) for 24 h to fully remove excess media and NaOH, and then were left suspended in water.2.7. Light microscopy
A Motic Panthera TEC-BF light microscope (Motic, Kowloon Bay, Hong Kong) equipped with a 5× objective was used to conduct particle size measurements of the 3d and 5d BC particles (n = 20). The Feret's particle diameter was measured for 3d and 5d particles (n = 20).2.8. BC growth curve measurement
To measure the mass of BC particles over time, the collected particles were gravity filtered and weighed before and after air-drying (n = 3 technical replicates).2.9. Scanning electron microscopy (SEM)
The particles were air-dried individually on a silicon wafer (University Wafer, Inc., Boca Raton, FL, USA) and placed on an aluminum stub (Ted Pella, Inc., Redding, CA, USA) with a piece of carbon tape (Ted Pella, Inc., Redding, CA, USA). The samples were then sputter coated with platinum on a Coater Leica-ACE600 (Leica Microsystems, Wetzlar, Germany) and imaged on a Thermo-Apreo-S SEM (Thermo Fisher Scientific, Waltham, MA, USA).2.10. Atomic force microscopy (AFM)
Particles were ultrasonicated using a Fisher Scientific Model 505 probe sonicator (Thermo Fisher, Waltham, MA, USA) and diluted to an approximate weight percent of 0.1% with Mili-Q water (EMD Millipore, Burlington, MA). 60 μL of the sample solution were subsequently dropped onto a freshly cleaved mica (Ted Pella, Redding, CA), and naturally dried in a clean and dry environment, facilitating the adsorption of the samples onto the mica substrate. The samples were imaged on a Dimension Icon AFM (Bruker Corporation, Billerica, MA) using tapping mode. The Multi75-G AFM probe (Budget Sensors, Sofia, Bulgaria) with a force constant of 3.0 N m−1 and a resonance frequency of 75 kHz was utilized for the imaging. The offline data processing was done with NanoScope Analysis (Bruker Corporation, Billerica, MA). To measure the fiber thickness (n = 27–70 measurements from 3–5 technical replicates), height measurements were conducted and adjusted by comparing points on the same fiber, subtracting overlaps when present, to estimate the closest approximation to the true value, with a preference for non-stacking areas.2.11. BCNPs chemical and physical characterization
To prepare BCNPs for Fourier Transform Infrared Spectroscopy (FTIR), X-ray diffraction (XRD), and thermal gravimetric analysis (TGA), the samples were lyophilized using Labconco FreeZone 2.5 Plus 2.5 Liter Cascade Benchtop Freeze Dry System (Labconco Corporation, Kansas City, Missouri, USA). Samples were analyzed using a ThermoNicolet iS10 FT-IR instrument (Thermo Fisher, Waltham, MA, USA) where spectra were collected in the 4000 to 400 cm−1 range with 20 scans and 4 cm−1 resolution. XRD was analyzed on a D8 Advance XRD (Bruker Co., Billerica, MA, USA) using a Cu/Kα radiation source wavelength of 1.5418 Å, accelerating voltage of 40 kV, and filament emission of 40 mA. The degree of crystallinity, Xc, was calculated using Segal's method (eqn (1)).29Ic is the crystalline peak intensity (at ∼22.8°) and Ia is the amorphous peak intensity (at ∼18°) after the background signal is subtracted. The apparent crystallite size, D, is also calculated for a specific plane orientation (2θ) using the Scherrer equation (eqn (2)) where λ is the wavelength of incident radiation, k is a dimensionless shape factor set to 1, and β is the full width at half maximum intensity of the peak corresponding to the crystal orientation in question.16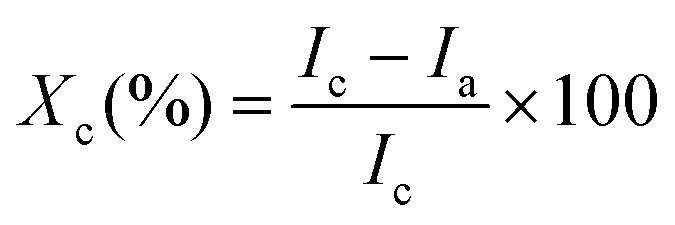 | (1) |
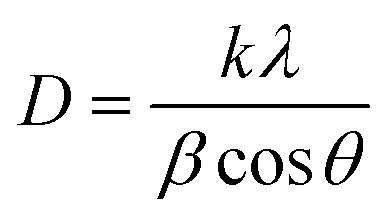 | (2) |
TGA was performed on a TA Discovery TGA 550 instrument (TA Instruments, New Castle, DE, USA) from 25 to 1000 °C at a heating rate of 10 °C mL−1, and under nitrogen atmosphere of 25 mL min−1 to prevent thermoxidative degradation.
2.12. Drug incorporation with BCNPs
BSA (Thermo Fisher, Waltham, MA, USA) was the model drug incorporated into BCNPs (Fig. 1E). Phosphate buffer solutions (PBS, Thermo Fisher) containing 0.25 mg mL−1, 0.5 mg mL−1, and 1.0 mg mL−1 BSA, respectively, were prepared. 1 mL of each solution was added to 0.5 mL of size separated BCNPs with P80 surfactant (BCNPs/P80-BSA at a concentration of 10 mg mL−1). Thus, the 3 prepared BCNPs/P80-BSA solutions contained 4.8, 9.1 and 16.7% (w/w) BSA, respectively. The BCNPs/P80-BSA solutions were incubated for 2 h at ambient temperature on an orbital rotator mixer and collected and washed in DI twice at 15000 rpm for 25 min via centrifugation. The supernatant was collected from each wash and combined for BSA quantification using bicinchoninic acid (BCA) protein assay kit (Pierce BCA Protein Kit, Thermo Fisher, Waltham, MA, USA).
2.13. Bicinchoninic acid assay to quantify BSA in BCNPs
The supernatant samples from the 3 prepared BCNPs/P80-BSA solutions were added to a 96-well microplate containing the BCA standard solutions and BCA reagents from the BCA assay kit. The BCA assay was measured using Ultraviolet–visible (UV-vis) spectrometry (BioTek Synergy H1 Microplate Reader, Fisher Scientific, Waltham, MA, USA) at 562 nm. A calibration curve was created by measuring the absorbances of BCA standard solutions and known BSA concentrations. The supernatant of non-BSA incorporated BCNPs served as a control. The BSA drug loading (DL) and encapsulation efficiency (EE) of the BCNPs were calculated using the following equations:30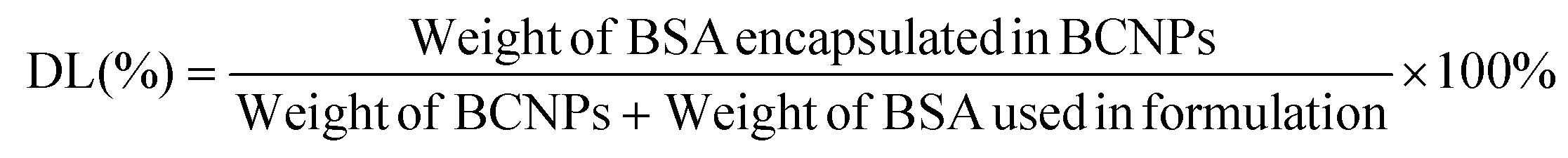 | (3) |
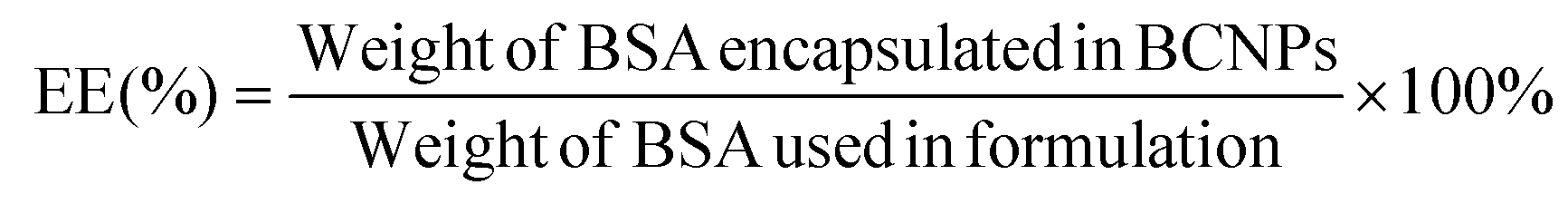 | (4) |
The particle size and ζ-potential were measured as described in section 2.2. From the NTA results, the polydispersity index (PDI) was calculated using the following equation where σ is the standard deviation of the mean particle diameter and d is the mean particle diameter:31
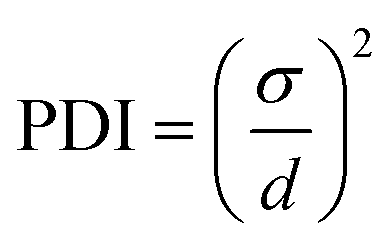 | (5) |
2.14. In vitro drug release profile
BCNPs/P80-BSA were resuspended in 1 mL of phosphate buffer solution (PBS) and placed in dialysis tubing (MW cutoff: 3 kDa, Spectrum Laboratories, Inc., San Francisco, CA, USA). The dialysis membrane was submerged in 20 mL of pH 4.5 PBS containing 1% P80 surfactant solution (Thermo Fisher, Waltham, MA, USA), ratio 100:1, in a 250 mL Erlenmeyer flask and placed on an incubated plate shaker at 90 rpm and 37 °C.30 At the designated timepoints, the PBS/1% P80 solution with BSA was collected and fresh PBS/1% P80 was replaced for the next time point. BSA released at each timepoint was quantified using a BCA assay measured on UV-Vis spectrometry. The percent of BSA released by BCNPs at each timepoint was calculated using the following equation:30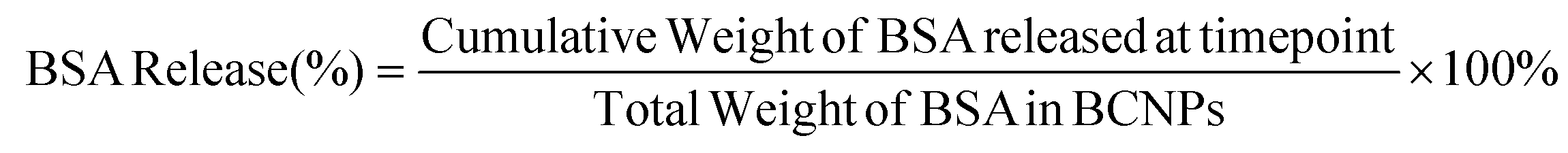 | (6) |
3. Results and discussion
3.1. BCNPs growth and morphological observations
BCNPs were prepared by bacteria co-cultured with yeast cells (see Methods section 2.1). Photographs of the culture flasks taken every 24 h until day 5 show that early-stage formation of BC can be visually observed from the 2-day timepoint when distinct particles have started to form (Fig. 2A). Further aggregation of the observed particles becomes apparent at 4-day and 5-day as macroscopic particles are formed. The average particle diameter was 206.9 ± 56.2 nm, 1.04 ± 0.187 mm, and 1.535 ± 0.644 mm for the BC 1 day (1d, Fig. 2B), 3d, and 5d particles respectively (Fig. S3†). It is noted that for 1d the average particle diameter was measured for n = 12 particles where particles that appeared aggregated, such as the three particles in Fig. 2B, were measured individually. For 3d and 5d, the average diameter is measured for n > 20 particles. The micrographs show that all collected particles (1d, 3d, and 5d) are comprised of randomly oriented BC fibers that form a porous nanofibrillar network as an extracellular matrix. The SEM images of the macroscopic 3d and 5d particles show the distinct BC fiber bundles that form large sheets as the particles are dried on the imaging substrate (Fig. 2C). The BC fiber bundles observed in Fig. 2C extend further than the field of view, suggesting that they are at least tens of microns in length. High-magnification AFM images (Fig. 2D) showed the cellulose fiber morphology and revealed stark differences with increasing growth time. In BC 1d images, only a limited number of individual fibers, comprised of fibrils with a thickness of 3.358 (±1.684) nm, are present. The fibers are arranged in a mesh-like network (Fig. 2Di). After 3d and 5d, the average thickness of elementary fibrils was measured to be 7.395 (±3.595) nm and 12.182 (±4.057) nm respectively. Analysis of the measured fibril thicknesses (Fig. 2E) shows that both the average fibril thickness and the variation in fibril thickness increase with time. Therefore, the elemental fibrils available at a given time have increasingly larger thickness distributions as growth time increases. Unlike the mesh-like network observed for 1d BC, the fibrils observed for 3d (Fig. 2Dii) and 5d (Fig. 2Diii) BC are assembled into fibers of increasing thickness. At both 3d and 5d, fibers can be observed that appear to be directly formed from intertwined elementary fibrils. There are also fibers present, especially at 5d (Fig. 2Diii), that are formed from bundles of these intermediate fibers, demonstrating the true hierarchical nature of BC. The size of these fiber bundles increases from 15.481 (±2.835) to 19.834 (±2.946) nm between 3d and 5d. These results collectively show that over the 5-days of monitored culture, BC fibrils increase their thickness and intertwine to form thicker and longer fibers which ultimately aggregate to form spherical-like particles. The diameter of these particles increases from a few hundreds of nanometers (1d) to a few millimeters. It's possible that for short growth times (1d) shear forces associated with agitation prevent the formation of organized fibers above a critical threshold that would promote hierarchical fiber formation, and instead, the distinct mesh-like network morphology is favored. However, given sufficient time (3d and 5d), it is apparent that the BC will preferentially assemble, as expected, to form the hierarchical fibers of increasing magnitude. Thus, our insights shed light into the different fiber arrangements obtained for short (1d) versus longer (3d–5d) culture durations.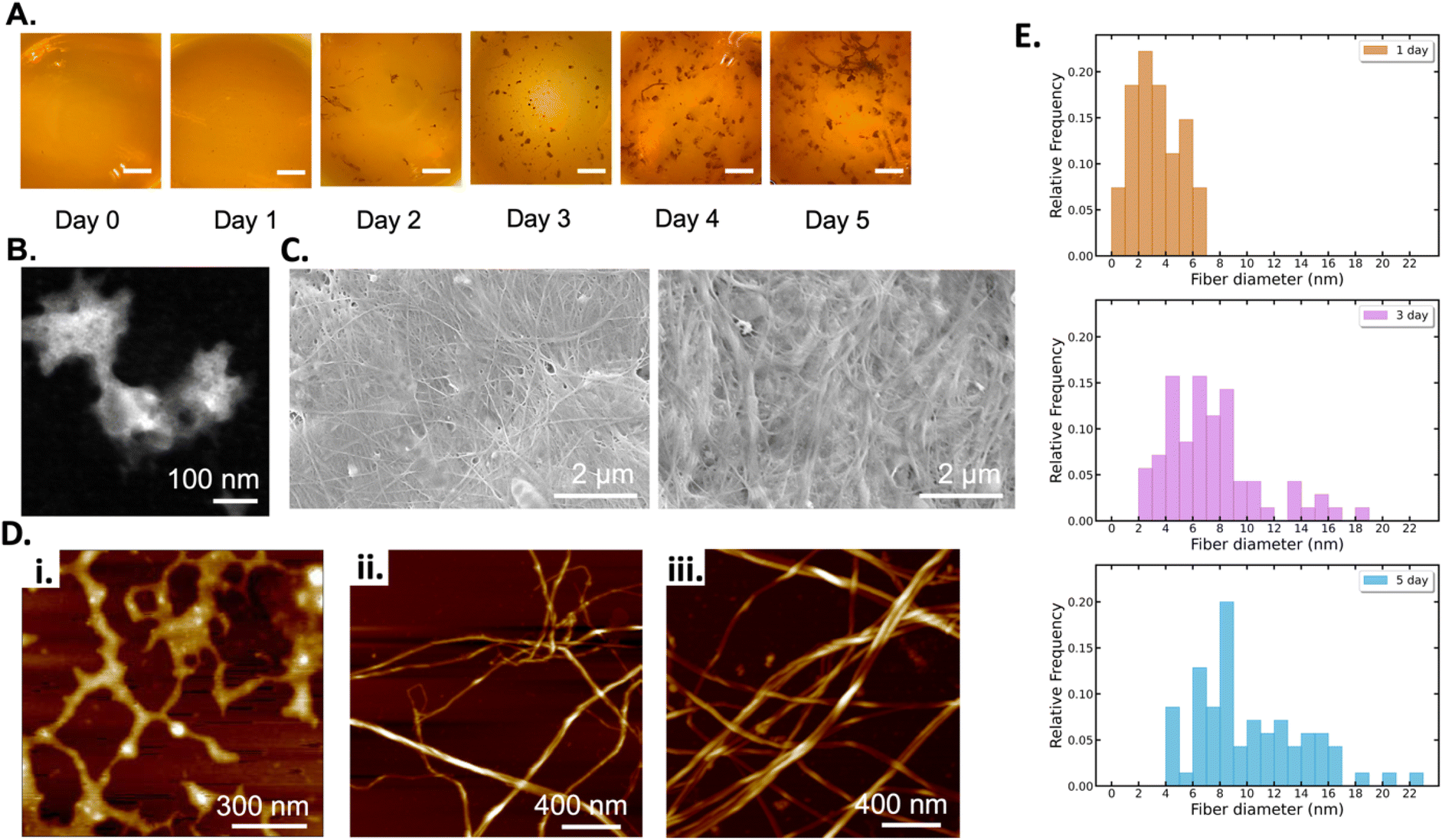 | ||
| Fig. 2 BC growth at timepoints: 1d, 3d, and 5d: (A) Photographs of the culturing flasks at 24 h timepoints until 5-day (all scale bars are 5 mm). (B) STEM image of BC 1d, (C) SEM images of BC 3d and 5d, (D) AFM images of BC (i) 1d, (ii) 3d and (iii) 5d, and (E) histogram of the fibril diameter distribution for 1d, 3d, and 5d measured in the AFM data. | ||
The BC yield data (wet and dry mass) over the growth period of 5 days are presented in Fig. 3. We fit the measured data with a logistic growth model in which biomass (M) is plotted as a function of time (t):
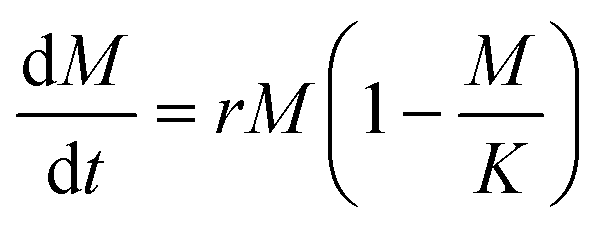 | (7) |
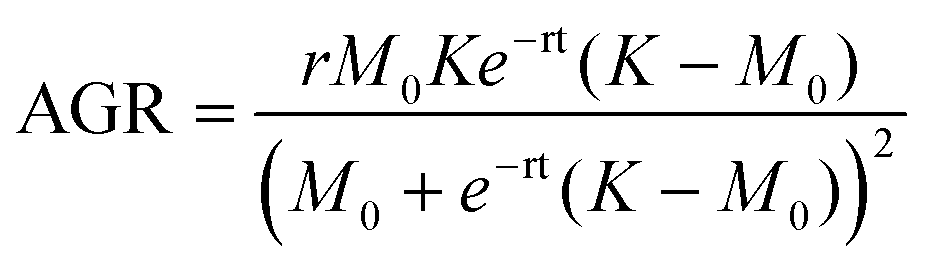 | (8) |
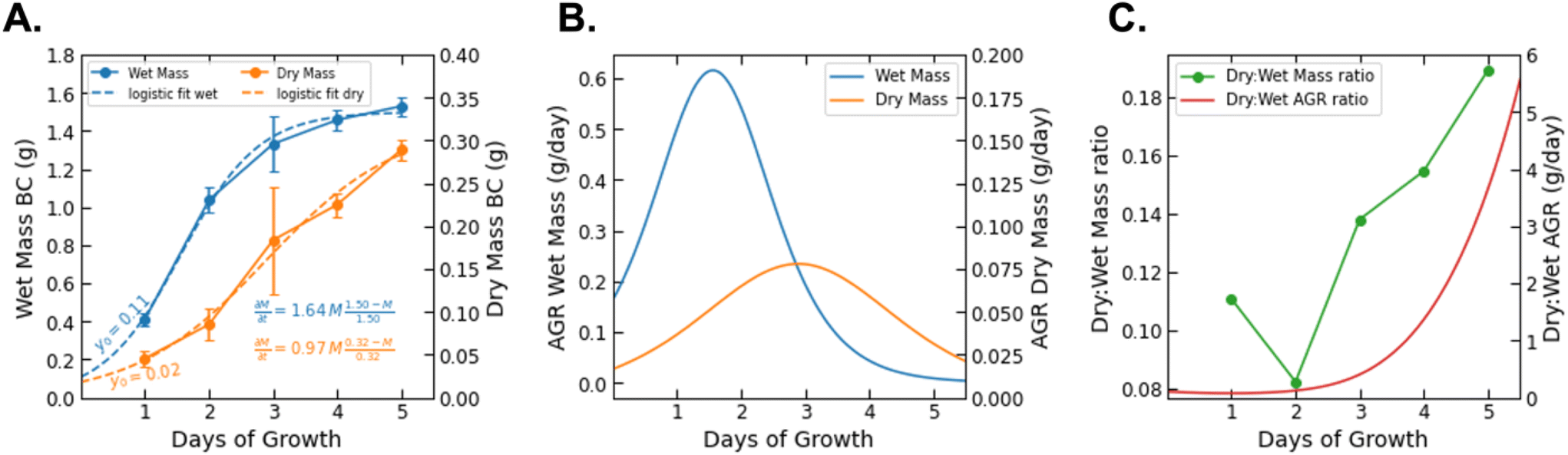 | ||
| Fig. 3 (A) The growth of BC (grams) wet mass and dry mass as a function of growth time (days) with logistic growth fit curves and corresponding equations. (B) The AGR of wet and dry BC mass plotted as a function of time on opposing axes. (C) The ratio of dry to wet mass (dry:wet mass) at each recorded time point plotted against the ratio of the dry AGR to the wet AGR (dry:wet AGR) at all times 0–6 days. | ||
While data was only collected at 24 h increments, the logistic fit extrapolates the mass growth, and thus the AGR, to time = 0. The growth rate parameter (r) observed for dry BC (r = 0.97) is greater than that previously reported for BC grown in a static culture (r = 0.55).32 This is consistent with additional findings that BC production can be increased by agitation for some bacteria species. It should be noted that the logistic fit for dry mass growth projects the maximum value of biomass to be 0.32 g. However, it is difficult to determine if the AGR has truly passed its peak based on the raw data.26 The mass measurements show that the wet biomass AGR starts from approximately 0.2 and is maximized at 0.6 g per day between days 1 and 2. From that point on it is projected to decrease and level off after day 4. On the other hand, the dry biomass growth rate shows a symmetric curve peaking at 0.2 g per day on day 3. Between day 3 and 4 of growth, the ratio between the AGR of dry and wet mass (dry:wet) surpasses 1.0 and increases nonlinearly until data collection ended (Fig. 3C). We note that the ratio between the dry and wet mass also began to increase significantly between day 2 and the end of the experiment. This difference may be related to the water holding capacity of BC. After a critical point (∼2 days), it's possible that the interfibrillar density of growing BC particles increases more rapidly than particle volume, thereby decreasing available volume for water holding.33 This would explain the increase of dry:wet mass and AGR ratios with increasing growth time. The water holding capacity of BC is also reflected by the intense fiber network paired with a swelling behavior from its porosity and high tensile strength that demonstrate BC's biological and mechanical properties.34 The reported deviations, both in terms of yield and in terms of fiber thickness, reflect the inherent variability of the material and the chosen culture conditions.
3.2. Structure and physical properties of BCNPs
The structural and thermal properties of BCNPs were compared to BC particles grown for 3d and 5d. XRD, FTIR, and TGA were used to identify the crystallinity, chemical bonding, and thermal behavior of the BC particles. XRD patterns of the BCNPs grown for 1d (Fig. 4A) show broad, indistinct peaks at 18–19° and 30–35° 2θ. The lack of sharp, salient peaks in the diffraction pattern indicates that while some nanocrystalline regimes may exist, the BCNPs are predominantly amorphous.35,36 The formation of nanocrystalline cellulose is more evident for particles grown for 3d and 5d as the characteristic peaks at ∼15° 2θ (corresponding to (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and (110) Iβ planes) and 22.7° 2θ (corresponding to (200) Iβ plane) are detected.37,38 Cellulose Iβ diffraction planes are used herein to denote specific peaks although likely both Iα and Iβ polymorphs coexist. In the absence of high temperatures, the alkaline conditions used to purify bacteria cellulose particles are not harsh enough to significantly reduce the Iα content.9 The degree of crystallinity calculated for BCNPs, and the BC particles grown for 3d and 5d are 37.8%, 76.6%, and 81.5%, respectively. The crystallite sizes for the (200) crystallographic planes are 3.29 and 4.80 nm respectively for the 3d and 5d particles, indicating that both crystallinity and crystallite size increase with time. The BCNPs do not exhibit crystals with the (200) orientation and so they have been excluded from the crystallite size analysis. However, the broad nature of the peaks in the diffraction pattern for BCNPs suggest crystallite sizes smaller than those observed for 3d and 5d BC particles. Therefore, our results show that primarily amorphous cellulose nanoparticles are obtained after 1d, while the particles harvested after 3d and 5d of culture are comprised of nanocrystalline BC microfibers. The fact that predominantly amorphous BCNPs can be obtained is key to their applicability as a drug delivery system, as amorphous carriers promote the bioavailability of the drug, and increase the solubility and the dissolution rate of a drug.39,40
0) and (110) Iβ planes) and 22.7° 2θ (corresponding to (200) Iβ plane) are detected.37,38 Cellulose Iβ diffraction planes are used herein to denote specific peaks although likely both Iα and Iβ polymorphs coexist. In the absence of high temperatures, the alkaline conditions used to purify bacteria cellulose particles are not harsh enough to significantly reduce the Iα content.9 The degree of crystallinity calculated for BCNPs, and the BC particles grown for 3d and 5d are 37.8%, 76.6%, and 81.5%, respectively. The crystallite sizes for the (200) crystallographic planes are 3.29 and 4.80 nm respectively for the 3d and 5d particles, indicating that both crystallinity and crystallite size increase with time. The BCNPs do not exhibit crystals with the (200) orientation and so they have been excluded from the crystallite size analysis. However, the broad nature of the peaks in the diffraction pattern for BCNPs suggest crystallite sizes smaller than those observed for 3d and 5d BC particles. Therefore, our results show that primarily amorphous cellulose nanoparticles are obtained after 1d, while the particles harvested after 3d and 5d of culture are comprised of nanocrystalline BC microfibers. The fact that predominantly amorphous BCNPs can be obtained is key to their applicability as a drug delivery system, as amorphous carriers promote the bioavailability of the drug, and increase the solubility and the dissolution rate of a drug.39,40
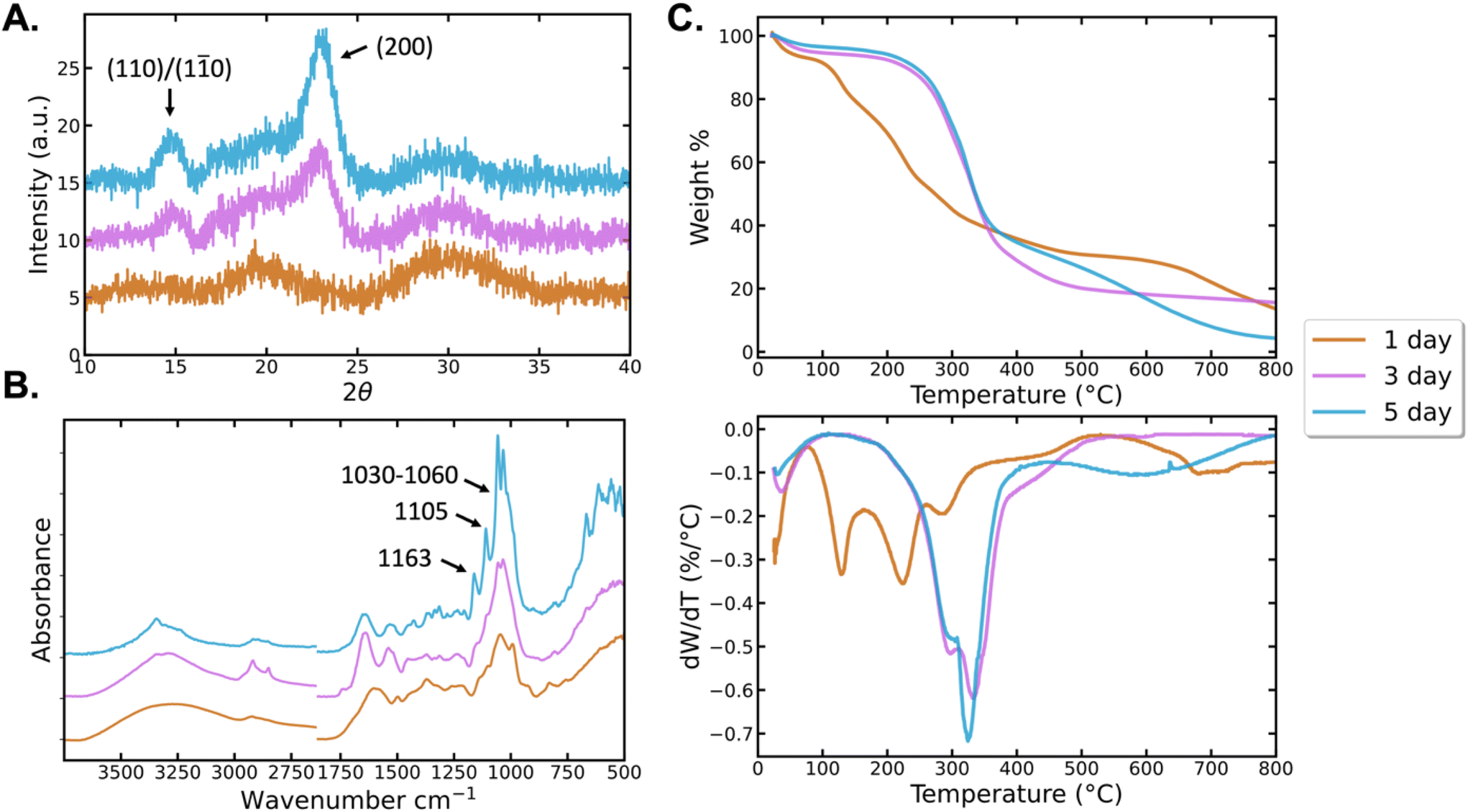 | ||
| Fig. 4 Characterization of BC structure and thermal properties: (A) XRD showing the 24 h BCNPs are amorphous, the 3d and 5d particles have small amounts of cellulose I nanocrystals (B) FTIR spectra to identify cellulose functional groups, and (C) TGA and DTGA profiles show the thermal degradation for each sample. All results include the samples BCNPs, BC 3d, and BC 5d. | ||
We next assess the FTIR spectra of the BC samples (Fig. 4B). The broad peak between 3000–3500 cm−1 is attributed to O–H stretching vibrations.11,41 In this region, the BCNPs spectrum is broader and more uniform than the spectra of the 3d and 5d particles. The 3d particle O–H band, while also broader than the 5d, has more salient features. The O–H band of the 5d particles is the narrowest and most defined of the BC samples. These differences in the O–H band suggest that the ratio of intermolecular and intramolecular hydrogen bonding between surface hydroxyl groups varies between samples. The peak-narrowing as a function of growth time for BC particles may be the result of increasing homogeneity of the physical and chemical environment as fibrillar order increases over time.29,30 The XRD spectra support this hypothesis as crystallinity was found to increase with growth time (Fig. 4A). Peaks between 2920–2922 cm−1 represent C–H stretching and vibrations, while 2897 cm−1 represents asymmetric –CH2 stretching and 2855 cm−1 represents symmetric –CH3 stretching.42,43 1440 cm−1 identifies –CH2 bending and 1300 cm−1 identifies C–H bending.44 The peaks at 1163 cm−1, 1030–1060 cm−1 and 1105 cm−1 correspond to C–O–C stretch of the β (1–4) glycosidic bonds, C–O stretch and C–O–C in-plane stretch of the glucose ring in cellulose, respectively.44,45 These signature peaks are observed clearly in the 5d particles, and are broader, yet still distinguishable in the 3d particles. The spectra of the 24 h BCNPs shows the peaks at 1030–1060 cm−1 and ∼1105 cm−1 broader than all other samples, while the 1160 cm−1 peak appears as a broad shoulder downshifted to ∼1140 cm−1. The increasing intensity, area, and sharpness of C–O and glycosidic bond peaks with longer culture times suggests that the cellulose fibers have a higher number of those skeletal bonds (i.e. higher molecular weight). Lastly, the broad peak around 1650 cm−1 is associated with the O–H vibrations of the adsorbed water.46
The TGA data for BC allows assessment of the thermal degradation behavior of our materials (Fig. 4C). The BCNPs show a ∼10% mass loss immediately upon heating, which is associated with water evaporation. The 3d and 5d BC particles have lower amount of withheld water (∼5%) that evaporates more gradually from room temperature to about 100 °C. Across all studied samples, there is no thermal degradation observed before 90 °C. The thermal decomposition of cellulose gives rise to the mass loss steps observed between 190–400 °C for the BC particles grown for 3 and 5d and 90–350 °C for the BCNPs.47 Differences in the crystal size, molecular weight, and molecular weight dispersity of cellulose give rise to the large differences in the thermal degradation profiles.48 The thermal stability is indicated by the initiation of thermal decomposition and not the temperature range that the decomposition spans, or the temperature at which the maximum decomposition rate occurs. The T5% (temperature at which 5% of the sample mass has been lost) for 3d BC and 5d BC are approximately 80 °C and 180 °C respectively, while T10% is ∼230 °C for 3d BC and 245 °C for 5d BC (Table 1). Therefore, the thermal stability of 5d BC particles is higher than that of 3d BC. In addition, the maximum degradation rates (DTG peaks) are achieved between 290 °C and 330 °C for the BC particles. The DTG peaks for BCNPs appear at ∼140 °C, 230 °C and 290 °C. Lastly, the molecular weight distribution can be associated with the slope of the weight loss curve or the intensity of the DTG peak, as a uniformity in molecular weight causes a larger share of the molecules to degrade at a similar temperature. Large slope or high DTG peak intensity indicate tight molecular weight distribution or relative homogeneity in molecular weight. We can therefore infer that the BC particles have more uniform molecular weight dispersity than the BCNPs. The higher molecular weight (as inferred from the FTIR spectra), tighter molecular weight distribution, and similar crystal size (as shown from XRD analysis) of the BC 3d and 5d particles justify their higher thermal stability, tighter decomposition temperature window, and higher temperatures of maximum degradation rate compared to the BCNPs. The shorter cellulose chains for BCNPs will result in a larger number of chain-ends per sample mass, compared to higher molecular weight 3d and 5d BC particles. Chain-ends serve as thermal degradation initiation points, justifying the lower thermal stability of BCNPs.49 Thus, the mass loss profiles confirm that BCNPs are the particles with the smallest crystal size and lowest molecular weight, while culturing for longer times leads to cellulose fibers with higher molecular weight. Moreover, the TGA confirmed that the thermal stability of the BC samples is stable across a range of temperatures relevant for shipping and storage conditions.
| Sample | X c (%) | D (nm) | DTG (°C) | T 5% (°C) | T 10% (°C) |
|---|---|---|---|---|---|
| 1d | 37.82 | N/A | 128.92, 223.22, 282.53 | 46.16 | 110.09 |
| 3d | 76.58 | 3.29 | 297.08, 333.91 | 81.28 | 228.78 |
| 5d | 81.47 | 4.80 | 304.64, 324.31 | 178.05 | 243.16 |
3.3. BSA drug loading and encapsulation efficiency in BCNPs
To prepare the BCNPs for the drug loading process, we sonicated the collected nanoparticles with P80 surfactant solution (see section 2 Materials and methods). The measured BCNPs concentration was 8.35 × 107 ± 1.18 × 107 particles mL−1 with an average particle size of 103.0 ± 1.5 nm (Fig. 5A, n = 5). We note that the NTA data report the size of BCNPs formed by using P80 as a surfactant to prevent the BCNP aggregation compared to the STEM image (Fig. 2B) of BCNPs prepared without size separation and P80 surfactant solution. To assess the long-term stability of BCNPs, we measured the particle size after storage in room temperature for 2 weeks, 5 months, and 7 months (Table S1†). The stored particles have negligible changes in their size up to 5 months of storage, without requiring temperature-controlled storage conditions. | ||
| Fig. 5 (A) Hydrodynamic size of BCNPs at concentration of 8.35 × 107 ± 1.18 × 107 particles mL−1 and measured on nanoparticle tracking analysis. The red region indicates peak intensity and standard deviation; (B) STEM of BCNPs at 1 μm scale bar, and (C) STEM of a BCNP at 100 nm scale bar. | ||
To observe if the fibrillar morphology of BCNPs is maintained after treatment with the surfactant solution, we conducted STEM. The collected images of BCNPs (Fig. 5B and C and Fig. S2†) reveal that the fibrillar structure of the nanoparticles was retained. Moreover, the treated and dried BCNPs had a particulate shape most resembling an elliptical morphology with an average particle size 478.9 ± 129.6 nm. This size is about 4 times larger than the NTA-measured particle size reflecting the effects of drying on the imaging substrate and possible particle–particle association therein.
In addition to size, another key desirable property for nanoparticles in biomedical applications is a near-neutral or slightly negative surface charge as it minimizes ionically-driven cellular membrane disruption.50 To achieve a desired surface charge, this is dependent on the therapeutic application where the administration route and target would be known. Here, we measured the average ζ-potential of the BCNPs treated with the surfactant solution to be slightly negative, −14.1 ± 4.2 mV (n = 3). The endotoxin units per milliliter (EU mL−1) of the post processed BCNPs and prior to drug incorporation were 0.46 EU mL−1 (n = 3). Our measured value is below the FDA regulation limit of 0.50 EU mL−1 for medical devices containing bacteriostatic water for injection.51,52
Given the nanoscale size, surface charge, high surface area, and nanofibrillar (mesh-like) morphology of the amorphous BCNPs, we intend to evaluate their applicability as a natural polymer platform for drug delivery applications.16,53,54 Different concentrations of BSA were incorporated into BCNPs/P80 at three relative targeted drug loadings (DL). High encapsulation efficiency (EE) around 90% was achieved for all target loadings. Incorporating BSA into the BCNP/P80 increased the particle size by 2.5-fold, to approximately 250 nm, with similar or more negative ζ-potentials compared to non-BSA containing BCNPs/P80 (Table 2).
| Mass of BCNPs (mg) | Mass of BSA (mg) | Target DL (% w/w) | DL ± STD (% w/w) | EE ± STD (% w/w) | Particle size ± STD (nm) | PDI | ζ-Potential ± STD (mV) |
|---|---|---|---|---|---|---|---|
| 5 | 0.25 | 4.76 | 4.31 ± 0.01 | 89.99 ± 0.31 | 256.9 ± 8.3 | 0.0010 | −11.9 ± 8.7 |
| 5 | 0.50 | 9.09 | 8.34 ± 0.03 | 90.99 ± 0.36 | 255.0 ± 5.4 | 0.0004 | −26.2 ± 4.7 |
| 5 | 1.0 | 16.67 | 15.46 ± 0.02 | 91.42 ± 0.12 | 231.7 ± 3.5 | 0.0002 | −18.7 ± 9.4 |
3.4. BSA drug release profile from BCNPs
The STEM images of the 8.34% DL sample showed similar particle shape to non-DL BCNPs/P80 and a slight increase in size due to the BSA incorporation (Fig. 6A). The drug release assay showed a burst release of BSA at 2 h, with a significant burst release of 8.9% for the 4.31% DL samples. We report data both in % w/w (Fig. 6) and in μg (Fig. S6†). In fact, within the first 4 h, the 4.31% DL samples release 30% of the total amount of BSA that they ultimately release over the entire 168 h period. After that initial burst release, these samples demonstrate a sustained release profile. At the end of the 168 h testing period, the 4.31% DL samples released cumulatively 55.3% of BSA, which corresponds to 126.2 μg in total, out of 229.5 μg initially loaded. The 8.34% DL samples showed a low release with a sustained profile. After the initial burst release of 3.9%, an average of 2.5% (11.3 μg) of BSA were released every 24 h compared to the 4.31% DL samples where the average release was of 5.5% (12.6 μg). Cumulatively, the 8.34% DL samples released 24.7% of BSA, corresponding to 113 μg out of 459.1 μg initially loaded, which is 45% less than the 4.31% DL samples. The 15.46% DL samples showed small quantitative changes in the release at later time points, indicated by a flattened curve (Fig. 6B). Over the 168 h period, these samples had a cumulative release of 8.7%. Notably, only 11.6% of this total release was observed within the first 48 h. Subsequently, an average of 10% of the total release, which corresponds to 10.2 μg was released every 24 hours thereafter. At the end of the release study window, the BSA remaining associated with BCNPs is 44.7 ± 18.5, 75.3 ± 10.5 and 91.4 ± 8.1% w/w for the 4.76, 8.34 and 15.46% DL, respectively. Therefore, we note the highest cumulative release and lowest amount of bound BSA in the 4.31% DL samples and hypothesize that the majority of BSA incorporated was surface associated. The 8.34% and 15.46% DL samples may have provided a higher BSA concentration gradient driving the BSA into the material core, seen similarly in a BC-based microsphere study.23 As the BSA-BCNP association is stronger, these samples have a lower release, compared to the sample with minimum BSA loading.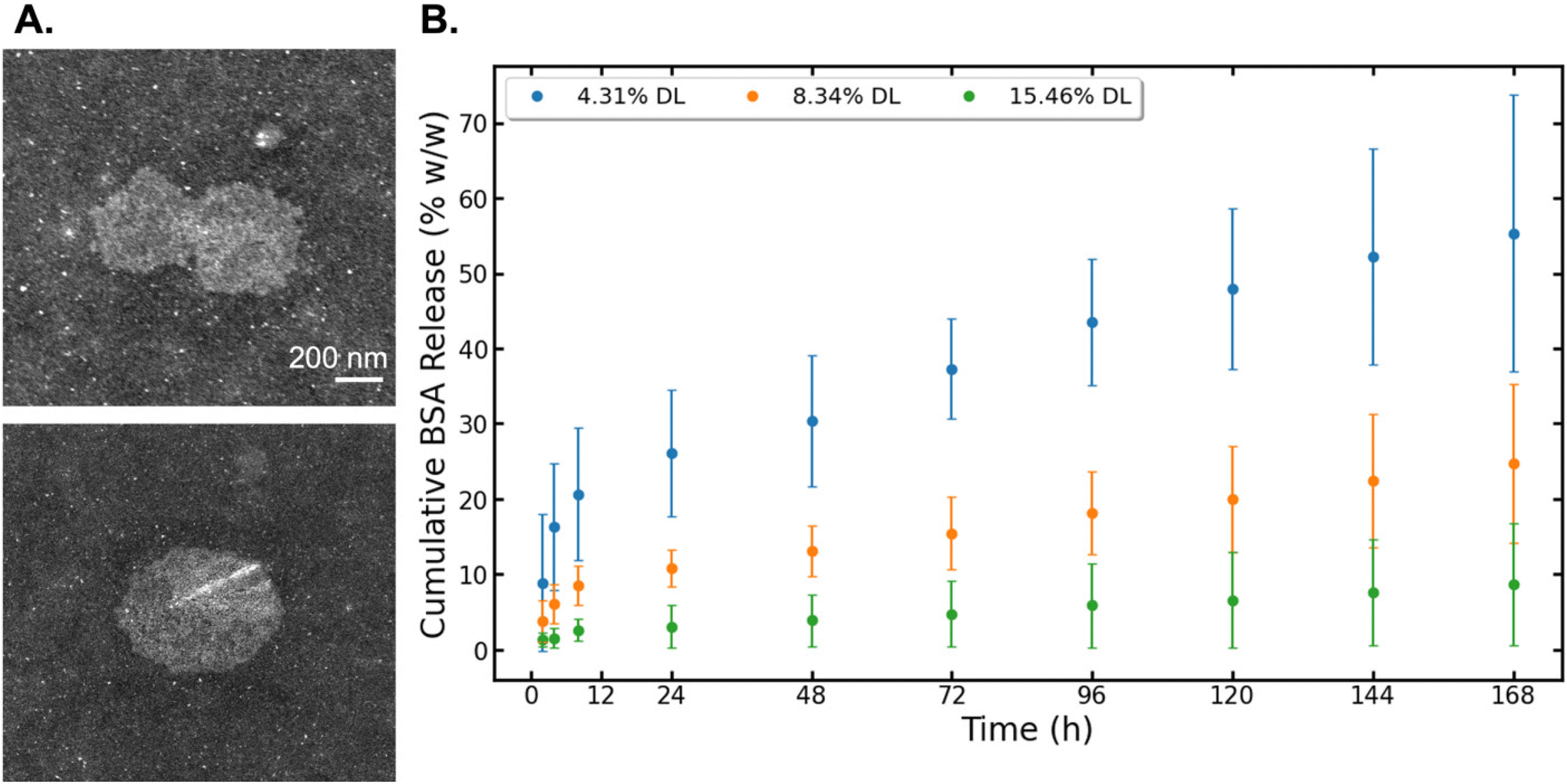 | ||
| Fig. 6 (A) STEM images of 8.34% DL BSA-BCNPs/P80. Scale bar set to 200 nm. (B) Results for cumulative BSA drug release (% w/w) from BCNPs/P80, calculated using eqn (6). | ||
Drug loading of BSA into BCNPs/P80 showed a strong binding affinity16 with a sustained release profile. We used physiologically relevant conditions to simulate BSA release. Our results align with prior studies loading BSA into BC microparticles, prepared using a bottom-up approach via the culturing of porous BC-based spheres and adsorbing BSA.23 BC-BSA microparticle studies by Lin et al.55 and Zhang et al.23 showed a strong electrostatic interaction between BC and BSA, where BSA adsorbing at the inner pore surface of the BC sphere was defined by a first-order release model, indicating a controlled drug release of BSA. BSA is one representative drug that can be incorporated into BC for therapeutic delivery, but many other drugs of interest have been incorporated into macroscale or micro-BC platforms.56,57 A recent study loaded a BC film with ibuprofen, a low solubility drug, and propranolol hydrochloride, a highly soluble drug.21 Both drugs showed immediate release with freeze-dried and air-dried BC film preparations, yet the freeze-drying method showed a sustained release for soluble drugs while air drying was optimal for DL at low drug concentrations.21 Another study used a top-down approach to mill a BC film into microparticles to drug load cloxacillin and cefuroxime sodium salts, adequate model drugs for antimicrobial activity in wound healing and facial scrubs.22 All methods showed an immediate drug release of approximately 85% in the initial 30 min while demonstrating good antibacterial activity.22 Additional examples of alternative therapeutics include benzalkonium chloride, an antimicrobial surfactant used in BC wound dressing, curcumin/polysorbate 20 used in cellulose nanoparticles, and cloxacillin and cefuroxime sodium salts in BC particles for wound dressing and medicated cosmetics.22,58,59 Overall our results contribute to the potential of cellulose-based materials to be used as platforms for drug delivery.
4. Conclusion
This work describes the preparation and characterization of BCNPs as a drug delivery platform. We describe a facile fabrication with minimal processing steps to minimize the inherent variability in the yield and properties of the produced BCNPs. The use of BCNPs serves as a sustainable alternative to the existing synthetic polymer and lipid nanoparticles used in drug delivery in that BCNPs are (1) bioderived (produced entirely from biomass), (2) the only product of the culturing process, (3) processed using minimal non-organic solvent and (4) do not require high heat or pressure to be produced.Our results show that BCNPs produced after 24 h of bacterial culture and size separated with P80 surfactant solution have an average size of approximately 103 nm, slightly negative zeta-potential, and are comprised of predominantly amorphous and entangled cellulose fibrils forming a mesh-like structure. Longer culture duration produces large BC particles that reach a diameter of approximately 1000–1500 μm. These particles are comprised of longer and thicker cellulose fibers which themselves comprise of bundled fibrils that have a higher crystallinity and MW compared to the nanoparticles harvested after 24 h. Nanoparticles can diffuse and distribute more readily in tissue compared to bulk BC; therefore, BCNPs offer numerous advantages over BC microparticles and BC pellicles for drug delivery. TGA data showed BCNPs are thermally stable until 90 °C. Further stability tests showed no changes in particle size upon storage at room temperature for up to 5 months, suggesting that a cold-chain storage and distribution are not required to ensure BCNP stability. BCNPs are biodegradable upon disposal in the environment and can be produced as an alternative and tunable drug delivery therapeutic to treat disease while having a reduced environmental impact through use of green engineering. Future optimization of the BCNP culturing conditions could enable higher production yields, minimize waste upon scale up, and improve the carbon footprint of the manufacturing process of nanoparticle-based drug delivery platforms.
Author contributions
Gabrielle N. Balistreri: Conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization. Ian R. Campbell: Methodology, data analysis, writing – review & editing. Xinqi Li: Julia Amorim: Investigation, methodology, writing – review & editing. investigation, methodology, writing – review & editing. Shuai Zhang: Methodology, writing – review & editing. Elizabeth Nance: Conceptualization, resources, writing – review & editing, supervision, project administration, funding acquisition. Eleftheria Roumeli: Conceptualization, resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Jeremy Filteau for assisting with BCA assay and endotoxin detection assay training, Ellen Lavoie for assisting with TEM sample preparations and instrument training at the MAF, and Dr Paul Grandgeorge for assisting with figure visualization. Part of this work was conducted at the Molecular Analysis Facility, a National Nanotechnology Coordinated Infrastructure (NNCI) site at the University of Washington, which is supported in part by funds from the National Science Foundation (awards NNCI-2025489, NNCI-1542101), the Molecular Engineering & Sciences Institute, and the Clean Energy Institute. The AFM experiments were supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences through the Center for the Science of Synthesis Across Scales, an Energy Frontier Research Center at the University of Washington, under Award DE-SC0019288. I. R. C. acknowledges funding from the National Science Foundation Graduate Research Fellowship under Grant No. DGE-2140004.References
- Nanomedicine Market Size Worth $427.18 Billion By 2030, https://www.grandviewresearch.com/press-release/global-nanomedicine-market.
- S. Bhatia, Natural Polymer Drug Delivery Systems, 2016 Search PubMed.
- S. Grund, M. Bauer and D. Fischer, Adv. Eng. Mater., 2011, 13, B61–B87 CrossRef.
- L. M. Ickenstein and P. Garidel, Expert Opin. Drug Delivery, 2019, 16, 1205–1226 CrossRef CAS PubMed.
- P. Ghasemiyeh and S. Mohammadi-Samani, Res. Pharm. Sci., 2018, 13, 303 Search PubMed.
- W. R. Roy, in Handbook of Solvents, 2014, ch. 3, pp. 1203–1253 Search PubMed.
- M. J. Savelski, S. C. Slater, P. V. Tozzi and C. M. Wisniewski, Clean Technol. Environ. Policy, 2017, 19, 2467–2477 CrossRef CAS.
- M. M. Abeer, M. C. Mohd Amin and C. Martin, J. Pharm. Pharmacol., 2014, 66, 1047–1061 CrossRef CAS PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- F. Wahid, L. H. Huang, X. Q. Zhao, W. C. Li, Y. Y. Wang, S. R. Jia and C. Zhong, Biotechnol. Adv., 2021, 53, 107856 CrossRef CAS PubMed.
- A. Vazquez, M. L. Foresti, P. Cerrutti and M. Galvagno, J. Polym. Environ., 2013, 21, 545–554 CrossRef CAS.
- C. Zhong, Front. Bioeng. Biotechnol., 2020, 8, 605374 CrossRef PubMed.
- J. Wood, C. Van Der Gast, D. Rivett, J. Verran and J. Redfern, Front. Bioeng. Biotechnol., 2022, 10, 561 Search PubMed.
- Z. Xiao, L. Zhang, Z. L. Li, J. Zhong, D. Chen and H. Huang, Environ. Res., 2021, 196, 110392 CrossRef CAS PubMed.
- H. Zhao, J. Xia, J. Wang, X. Yan, C. Wang, T. Lei, M. Xian and H. Zhang, Biotechnol. Biotechnol. Equip., 2018, 32, 350–356 CrossRef CAS.
- F. Yoshinaga, N. Tonouchi and K. Watanabe, Biosci., Biotechnol., Biochem., 1997, 61, 219–224 CrossRef CAS.
- D. Laavanya, S. Shirkole and P. Balasubramanian, J. Cleaner Prod., 2021, 295, 126454 CrossRef CAS.
- W. F. Dudman, J. Gen. Microbiol., 1960, 22, 25–39 CrossRef CAS PubMed.
- N. Petersen and P. Gatenholm, Appl. Microbiol. Biotechnol., 2011, 91, 1277–1286 CrossRef CAS PubMed.
- S. Mohammadi, F. Jabbari and V. Babaeipour, Int. J. Biol. Macromol., 2023, 242, 124955 CrossRef CAS PubMed.
- C. Jantarat, P. Muenraya, S. Srivaro, A. Nawakitrangsan and K. Promsornpason, RSC Adv., 2021, 11, 37354–37365 RSC.
- H. Ullah, M. Badshah, A. Correia, F. Wahid, H. A. Santos and T. Khan, Curr. Pharm. Des., 2019, 25, 3692–3701 CrossRef CAS PubMed.
- W. Zhang, X. C. Wang, J. J. Wang and L. L. Zhang, Int. J. Biol. Macromol., 2019, 140, 196–205 CrossRef CAS PubMed.
- Y. Hu and J. M. Catchmark, Biomacromolecules, 2010, 11, 1727–1734 CrossRef CAS PubMed.
- M. Jovic, P. Ingale, D. Radjenovic and V. Lakshminarayanan, Startups Developing Nanocellulose Products in 2020, 2020 Search PubMed.
- P. Singhsa, R. Narain and H. Manuspiya, Cellulose, 2018, 25, 1571–1581 CrossRef CAS.
- C. Y. Wong, J. Martinez, H. Al-Salami and C. R. Dass, Anal. Bioanal. Chem., 2018, 410, 6991–7006 CrossRef CAS PubMed.
- J. Wang and B. Zhang, Curr. Med. Chem., 2018, 25, 2938–2953 CrossRef CAS PubMed.
- E. J. Foster, R. J. Moon, U. P. Agarwal, M. J. Bortner, J. Bras, C.-E. Sandra, K. J. Chan, M. J. D. Clift, E. D. Cranston, S. J. Eichhorn, D. M. Fox, W. Y. Hamad, L. Heux, B. Jean, M. Korey, W. Nieh, K. J. Ong, M. S. Reid, S. Renneckar, R. Roberts, J. A. Shatkin, J. Simonsen, K. Stinson-Bagby, N. Wanasekara and J. Youngblood, Chem. Soc. Rev., 2018, 47, 2609–2679 RSC.
- A. Joseph, T. Wood, C.-C. Chen, K. Corry, J. M. Snyder, S. E. Juul, P. Parikh and E. Nance, Nano Res., 2018, 11, 5670–5688 CrossRef CAS.
- N. Raval, R. Maheshwari, D. Kalyane, S. R. Youngren-Ortiz, M. B. Chougule and R. K. Tekade, in Basic Fundamentals of Drug Delivery, 2019, pp. 369–400, DOI:10.1016/b978-0-12-817909-3.00010-8.
- M. Hornung, M. Ludwig, A. M. Gerrard and H. P. Schmauder, Eng. Life Sci., 2006, 6, 537–545 CrossRef CAS.
- M. Konopacki, B. Grygorcewicz, M. Kordas, P. Ossowicz-Rupniewska, A. Nowak, M. Perużyńska and R. Rakoczy, Biochem. Eng. J., 2022, 182, 108432 CrossRef CAS.
- M. Badshah, H. Ullah, S. A. Khan, J. K. Park and T. Khan, Cellulose, 2017, 24, 5041–5052 CrossRef CAS.
- W. Yao, Y. Weng and J. M. Catchmark, Cellulose, 2020, 27, 5563–5579 CrossRef CAS.
- C. Aulin, S. Ahola, P. Josefsson, T. Nishino, Y. Hirose, M. Österberg and L. Wågberg, Langmuir, 2009, 25, 7675–7685 CrossRef CAS PubMed.
- T. G. Fawcett, C. E. Crowder, S. N. Kabekkodu, F. Needham, J. A. Kaduk, T. N. Blanton, V. Petkov, E. Bucher and R. Shpanchenko, Powder Diffr., 2013, 28, 18–31 CrossRef CAS.
- A. D. French, Cellulose, 2014, 21, 885–896 CrossRef CAS.
- A. M. Stewart and M. E. Grass, Mol. Pharm., 2020, 17, 180–189 CrossRef CAS PubMed.
- B. C. Hancock and G. Zografi, J. Pharm. Sci., 1997, 86, 1–12 CrossRef CAS PubMed.
- C. M. Lee, J. D. Kubicki, B. Fan, L. Zhong, M. C. Jarvis and S. H. Kim, J. Phys. Chem. B, 2015, 119, 15138–15149 CrossRef CAS PubMed.
- V. V. Revin, A. V. Dolganov, E. V. Liyaskina, N. B. Nazarova, A. V. Balandina, A. A. Devyataeva and V. D. Revin, Polymers, 2021, 13, 1422 CrossRef CAS PubMed.
- E. Erbas Kiziltas, A. Kiziltas, M. Blumentritt and D. J. Gardner, Carbohydr. Polym., 2015, 129, 148–155 CrossRef CAS PubMed.
- N. Halib, Sains Malays., 2012, 41, 211 Search PubMed.
- O. Asanarong, V. Minh Quan, S. Boonrungsiman and P. Sukyai, Eur. Polym. J., 2021, 143, 110224 CrossRef CAS.
- S. Cichosz and A. Masek, Materials, 2020, 13, 4573 CrossRef CAS PubMed.
- F. Jiang and Y. L. Hsieh, Carbohydr. Polym., 2013, 95, 32–40 CrossRef CAS PubMed.
- O. M. Vanderfleet, M. S. Reid, J. Bras, L. Heux, J. Godoy-Vargas, M. K. R. Panga and E. D. Cranston, Cellulose, 2019, 26, 507–528 CrossRef CAS.
- K. Watanabe, M. Tabuchi, Y. Morinaga and F. Yoshinaga, Cellulose, 1998, 5, 187–200 CrossRef CAS.
- J. D. Clogston and A. K. Patri, in Methods in Molecular Biology, Humana Press, 2011, pp. 63–70, DOI:10.1007/978-1-60327-198-1_6.
- U. S. Food & Drug Administration, Baterial Endotoxins/Pyrogens, 1985, 9 Search PubMed https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-technical-guides/bacterial-endotoxinspyrogens .
- U. S. Food & Drug Administration, Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers, 2012, 11 Search PubMed https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-pyrogen-and-endotoxins-testing-questions-and-answers .
- S. Gorgieva, Processes, 2020, 8, 624 CrossRef CAS.
- C. A. Rosas-Casarez, Int. J. Adv. Comput. Sci. Appl., 2014, 4, 226 Search PubMed.
- Q. Lin, Y. Zheng, G. Wang, X. Shi, T. Zhang, J. Yu and J. Sun, Int. J. Biol. Macromol., 2015, 73, 264–269 CrossRef CAS PubMed.
- Y. L. Tan and H. K. Ho, Drug Discovery Today, 2018, 23, 1108–1114 CrossRef CAS PubMed.
- M. T. Paolo Caraceni and M. E. Bonavita, Blood Transfus., 2013, s25 Search PubMed.
- R. K. S. Bhavana, V. Mohite and S. V. Patil, Cellul. Chem. Technol., 2016, 50, 223 Search PubMed.
- Y. C. Ching, T. M. S. U. Gunathilake, C. H. Chuah, K. Y. Ching, R. Singh and N.-S. Liou, Cellulose, 2019, 26, 5467–5481 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00184a |
| This journal is © The Royal Society of Chemistry 2024 |