
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding improved capacity retention at 4.3 V in modified single crystal Ni-rich NMC//graphite pouch cells at elevated temperature†
Galo J.
Páez Fajardo
 ah,
Meltiani
Belekoukia
ah,
Satish
Bolloju
a,
Eleni
Fiamegkou
a,
Ashok S.
Menon
ah,
Zachary
Ruff
bh,
Zonghao
Shen
ch,
Nickil
Shah
a,
Erik
Björklund
dh,
Mateusz Jan
Zuba
e,
Tien-Lin
Lee
f,
Pardeep K.
Thakur
f,
Robert S.
Weatherup
dh,
Ainara
Aguadero
cgh,
Melanie J.
Loveridge
a,
Clare P.
Grey
bh and
Louis F. J.
Piper
*ah
ah,
Meltiani
Belekoukia
ah,
Satish
Bolloju
a,
Eleni
Fiamegkou
a,
Ashok S.
Menon
ah,
Zachary
Ruff
bh,
Zonghao
Shen
ch,
Nickil
Shah
a,
Erik
Björklund
dh,
Mateusz Jan
Zuba
e,
Tien-Lin
Lee
f,
Pardeep K.
Thakur
f,
Robert S.
Weatherup
dh,
Ainara
Aguadero
cgh,
Melanie J.
Loveridge
a,
Clare P.
Grey
bh and
Louis F. J.
Piper
*ah
aWMG, University of Warwick, Coventry, CV4 7AL, UK. E-mail: louis.piper@warwick.ac.uk
bDepartment of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK
cDepartment of Materials, Imperial College London, Royal School of Mines, London SW7 2AZ, UK
dDepartment of Materials, University of Oxford, Oxford, England OX1 3PH, UK
eMaterials Engineering Research Facility, Energy Sciences Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
fDiamond Light Source Ltd., Harwell Science and Innovation Campus, Didcot, Oxfordshire OX1 3QR, UK
gCSIC, Instituto de Ciencia de Materiales de Madrid, C/Sor Juana Ines De la Cruz 3, 28049, Madrid, Spain
hThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot, UK
First published on 21st September 2023
Abstract
The capacity retention of commercially-sourced pouch cells with single crystal Al surface-doped Ni-rich cathodes (LiNi0.834Mn0.095Co0.071O2) is examined. The degradation-induced capacity fade becomes more pronounced as the upper-cut-off voltage (UCV) increases from 4.2 V to 4.3 V (vs. graphite) at a fixed cycling temperature (either 25 or 40 °C). However, cycles with 4.3 V UCV (slightly below the oxygen loss onset) show better capacity retention upon increasing the cycling temperature from 25 °C to 40 °C. Namely, after 500 cycles at 4.3 V UCV, cycling temperature at 40 °C retains 85.5% of the initial capacity while cycling at 25 °C shows 75.0% capacity retention. By employing a suite of electrochemical, X-ray spectroscopy and secondary ion mass spectrometry techniques, we attribute the temperature-induced improvement of the capacity retention at high UCV to the combined effects of Al surface-dopants, electrochemically resilient single crystal Ni-rich particles, and thermally-improved Li kinetics translating into better electrochemical performance. If cycling remains below the lattice oxygen loss onset, improved capacity retention in industrial cells should be achieved in single crystal Ni-rich cathodes with the appropriate choice of cycling parameter, particle quality, and particle surface dopants.
1 Introduction
Unlocking higher energy densities in Ni-rich lithium-ion batteries (LIB) is key for electric vehicles capable of higher range autonomy.1 A usual route to achieve higher densities on the material side resides on increasing the Ni content “x” in LiNixMnyCozO2 with x + y + z = 1. The increased Ni content renders more discharge capacity2,3 for the same voltage window, i.e. more lithium extraction for a given voltage cut-off. Among the Ni-rich cathode family members, LiNi0.8Mn0.1Co0.1O2 (NMC811) has become popular.4–9 However, the increased lithium extraction available with increasing Ni content means that degradation due to surface O loss is more pronounced.10 Namely, the gain in capacity with increased Ni contents comes in detriment of the cycle life with larger cycling voltage windows. Thus, understanding the mechanisms underlying the acceleration of degradation is crucial to improve cycle life with Ni-rich cathodes.Cycling with a potential window in the low voltage range is a usual approach to limit the unwanted acceleration degradation at high voltages (upper-cut-off voltage (UCV) >4.2 V). However, the characteristic Li diffusion drop at lower cutoff voltage11–13 represents kinetic limitations during Li reinsertion with their associated capacity loss according to Zhou et al.13 in a work on NMC811. Yet they show that cycling at an increased temperature (from 25 °C to 45 °C) avoids the low-voltage diffusion limitations across the cathode crystal structure and partially recovers the capacity loss associated with kinetic limitations. The slight temperature increment lowers the activation energy barrier in a divacancy diffusion model that could link with subtle preferentially-oriented thermal expansion of the NMC811 structure.14–16 However, there is a competing phenomenon besides the temperature-induced capacity recovery as sustaining long cycles at elevated temperature typically induces further electrolyte degradation.17 Therefore, it is imperative to design experimental tests to assess long-term cyclability effects of the competing forces between the capacity recovery and faster surface degradation at higher temperatures.
Cycling, including high voltage (V > 4.2), by fixing the lower-cut-off voltage (LCV) and increasing the upper-cut-off voltage (UCV) exhibits a degradation turning point at 4.2 UCV in Ni-rich materials. With UCV ≤ 4.2 V in 2.5–4.2 V potential windows, graphite solid-electrolyte interface (SEI) degradation leads the capacity retention decay revealed on some recent studies by Dose et al.18 on poly-crystalline (PC) Ni-rich NMC//graphite coin cells. Surface side reactions at the SEI alter the voltage potential of the graphite material, resulting in a profile shift (slippage) which differential voltage analysis (DVA) is sensitive to.18 Dose et al. also demonstrate slippage accounts for most of the capacity loss in 2.5–4.2 V up to a 300-cycle breaking point in a slippage capacity loss graph and suggest additional degradation sources principally associated with PC particle detachment contribute to extra capacity loss afterwards (>300 cycles).
The slippage cycling breaking point happens sooner (at nearly 100 cycles) with UCV = 4.3 V18 where surface sensitive X-ray spectroscopic evidence confirms some degree of O loss in the cycled cathode material of PC NMC811//graphite coin cells.19 However, with UCV at 4.4 V, O loss leads to exponentially larger gas evolution as early on as the first cycle revealed in studies of electrolyte chemical oxidation at the NMC811/LP57 electrolyte interface and showing 4.4 V UCV is the O loss onset.10,20–22 Thus, 4.3 V can be considered as a moderate O loss threshold; namely, few surface regions undergo an early O loss degradation whose effects accumulate and become statistically relevant with the number of cycles. Within the moderate O loss condition, any small source of O loss can be passivated by surface doping.23 Among the wide family of surface dopants, aluminum compounds as surface doping agents have successfully proven to be beneficial against HF/H2O attacks from electrolyte in a scavenger role of decomposed electrolyte species.17,24 However, it is unknown how the Al-scavenger role behaves in long-term cycle experiments.
This work expands both the studies of the temperature-induced capacity recovery and the surface Al-scavenger role, assessing the combined effects of both approaches in long-term cycles by exploring the electrochemical performance of Ni-rich cathodes during 500 cycles at 40 °C. We expect the long-term scavenger behavior of Al surface doping to delay the typical high-temperature induced degradation and to allow a more robust cathode-electrolyte interface (CEI) in cycling protocols with UCV up to the moderate O loss threshold (4.3 V). Thus, with the surface of the Ni-rich material being protected with the Al-based doping, the long-term high-temperature cycling should account for the low-voltage Li kinetics limitations delivering more capacity and better cycle life in the long-term.
We examine the capacity retention and degradation pathways in real cell formats with industry relevant mass loading and cell design. We employed commercially sourced dry pouch cells from Li-FUN Technology Corporation (LiFun) and studied with a range of cycling protocols and chemical analysis. The cathode of the cell is Al surface-doped LiNi0.834Mn0.095Co0.071O2 (Al-surface-doped-Ni-rich). The nominal cathode composition was found by inductively coupled plasma (ICP) within this study. The surface doping is a LiFun proprietary process with disclosure restriction. Material without the surface doping is not tested since the beneficial role has been largely proven elsewhere,17,24–26 and we focus on the Al effects in the long-term. Our cycling studies seek to understand degradation pathways at the moderate O loss (UCV at 4.3 V). We design a comprehensive study with an optimal combination of operating cycling temperature (25 and 40 °C) and UCV (4.2 V and 4.3 V) with a fixed LCV at 2.5 V to determine the origin of long-term degradation with high temperature and Al surface doping. LiFun cathode material morphology is Al-surface-doped-Ni-rich single crystals (SC). SCs can serve as a cleaner model for studying the effects of high voltage degradation at the particle surface during long-term cycling since this morphology shows reduced particle cracking22,27–29 compared to its polycrystalline counterparts.
We use synchrotron-based soft X-ray absorption spectroscopy (XAS) and hard X-ray photoelectron spectroscopy (HAXPES) with lab-based techniques (scanning electron microscopy (SEM), time-of-flight secondary ion mass spectrometry (ToF-SIMS), and inductively coupled plasma optical emission spectroscopy (ICP-OES)) to investigate the impact on electrochemical performance of combining the temperature-induced capacity recovery and the surface Al dopant scavenger role after 500 cycles. We observe higher initial capacity, negligible charge transfer impedance growth, and excellent capacity retention at 4.3 V cycling at 40 °C after 500 cycles. The simultaneous use of the single-crystal morphology, sacrificial surface agents, high quality commercial cells, and cycling protocols facilitate high-voltage cycling for long duration and highlight the importance of kinetic limitations of this chemistry.
2 Methodology
2.1 Full-cells
Al-surface-doped-Ni-rich//graphite pouch cells with double-sided electrode coating were obtained from Lifun technologies (402035-S204070C). Cells arrived without electrolyte and sealed with sufficient pouch cell material for degassing. The pouch cells consist of calendered double coated SC Ni-rich electrodes and graphite electrodes. The SC Al-surface-doped-Ni-rich cathode mass loading is 16.71 mg cm−2 with 95.5% active material and 76.96 cm2 of electrode active area and a C/20 practical specific capacity at 200 mAh g−1. The cathode electrode has a press density at 3.3 g cm−3. The graphite electrode has 11.3 mg cm−2 mass loading with 94.8% active material a C/20 practical specific capacity at 350 mAh g−1. The anode electrode has a press density at 1.5 g cm−3. LiFun manufactured cells with a N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio at 1.1747 at 4.3 V. Notice that all the LiFun cells in this work were manufactured with this N:P specification. Prior to electrolyte filling, cells were cut open and dried at 100 °C under vacuum overnight to remove residual moisture. The cells were then filled in a dry room (dew point of −45 °C) with 1.1 g of 1 M LiPF6 in 3:7 w/w ethylene carbonate – (EC)/ethyl methyl carbonate – (EMC) (LP57) with 2% vinylene carbonate – (VC) electrolyte solution (Solvionic).
:P ratio at 1.1747 at 4.3 V. Notice that all the LiFun cells in this work were manufactured with this N:P specification. Prior to electrolyte filling, cells were cut open and dried at 100 °C under vacuum overnight to remove residual moisture. The cells were then filled in a dry room (dew point of −45 °C) with 1.1 g of 1 M LiPF6 in 3:7 w/w ethylene carbonate – (EC)/ethyl methyl carbonate – (EMC) (LP57) with 2% vinylene carbonate – (VC) electrolyte solution (Solvionic).
Cells were held at 1.5 V for 20 hours in a climate chamber set at 40 °C to facilitate full wetting of the electrode. A Maccor 4000 series cycler was used for formation and charge–discharge cycling. Cells underwent two formation cycles at 40 °C using constant current (CC) at C/20 between 2.5 V and the designated upper cut-off voltage (UCV, 4.2 or 4.3 V). Following formation, cells were cut open in the dry room, degassed, and vacuum sealed. Subsequently, cells were placed in climate chambers set at 25 and 40 °C, and cycled from 2.5 V to the desired UCV (4.2 V or 4.3 V) for 500 cycles. Three cells per cycling condition were cycled to ensure reproducibility. A constant current – constant voltage (CCCV) mode with a fixed C-rate of C/3 was applied, limiting current down to C/30 during the constant voltage (CV) step for charge. Every 100 cycles, two additional diagnostic cycles CC at C/20 were performed.
Potentiostatic Electrochemical Impedance Spectroscopy (PEIS) was measured after formation and after 500 cycles using a Biologic VMP3 potentiostat. After a rest period at 2.5 V, cells were cycled to 3.8 V (CC C/20 charge) where EIS was performed at 3.8 V (CC C/20 charge) in a climate chamber set at 25 °C with an amplitude of 10 mV and a scanning frequency range between 100 kHz to 10 MHz.
Profiles of distribution of relaxation times were computed from experimental electrochemical impedance spectroscopy data with the DRTtool software30 available at DRTtool (https://sites.google.com/site/drttools/home).
Polarization cell analysis relates the changes in the cycle time average voltage during the cycling operation. Polarization is computed for each cycle and uses a voltage vs. time profile of a given cycle n for its calculation using the following equation:
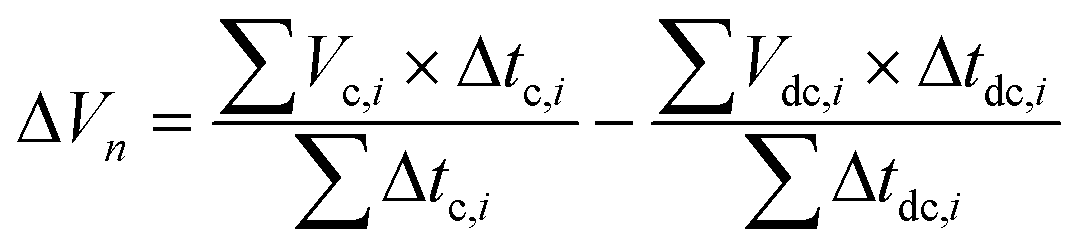 | (1) |
2.2 Electrode characterization
After cycling, cells were discharged to 2.5 V and disassembled in an argon glovebox (MBraun) (O2 < 1 ppm, H2O < 1 ppm) for subsequent characterization. The electrodes were washed with dimethyl carbonate solvent (DMC, anhydrous ≥99% purity, Sigma-Aldrich) in order to rinse off electrolyte residue and dried in the glovebox. Upon disassembly, electrodes were transferred from the glovebox in a proprietary airless transfer holder for particle morphology and elemental analysis.SEM images were obtained using a Carl Zeiss Sigma Field Emission Gun Scanning Electron Microscope (FEG-SEM) at 10 kV accelerating voltage in combination with an in-lens detector at working distances of approximately 5 mm.
Elemental analysis was performed using ICP-OES (Thermoscientific 7400 Duo). The SC Al-surface-doped-Ni-rich powder first was dissolved in concentrated, distilled hydrofluoric acid. The acid was then boiled off in a graphite heating block and collected for disposal. Subsequently, the remaining powder was dissolved in trace-element grade aqua regia. After complete digestion of the powder, the acid was again boiled off and the powder was dissolved in nitric acid and diluted for the ICP measurement. The concentration of a given element in the solutions was determined by comparing the emission of the sample solutions to a calibration line generated from a concentration series using a multi-elemental standard (VWR, Aristar) at each wavelength of interest. The emission wavelengths were selected such that there was no interference from other measured elements, elements in the standard, or the matrix solution (2% nitric acid). The composition of the powders was calculated by assuming that the molar fraction of nickel, manganese and cobalt was 1.
HAXPES measurements of core regions were performed at the beamline I09 at Diamond Light Source using photon energy at 2.2 keV and 6.6 keV with a pass energy of 200 eV corresponding to a 250 meV energy resolution. The binding energies of HAXPES regions were referenced to the so-called adventitious carbon (C 1s) peak at 284.5 eV, the most accepted value in the literature.31,32 Soft XAS in the total electron yield (TEY) mode also took place at the Diamond beamline I09. To account for beam irregularities, the absorption signal was divided by the signal from a reference Cu-coated mesh (I0) in the incident photon beam. For the O K-edge spectra, photon energies were calibrated using the characteristic Li2CO3 peak at 534 eV26,33 and scaled to the background. For the Mn and Ni L3-edge, photon energies were normalized and scaled to the Mn and Ni L3-edge peak at ∼643 eV34 and ∼855 eV,26,35 respectively.
ToF-SIMS (ToF-SIMS.5, IONTOF GmbH, Münster, Germany) was applied for the SEI and CEI chemical analysis. The analysis was performed with a 25 keV Bi+ primary beam within an analysis area of 100 μm × 100 μm and the cluster 10 nA Arn+ (n ≈ 1200) was applied for sputtering over an area of 300 μm × 300 μm. High current bunch mode (HCBM) of the primary beam with high mass resolution (≈10000) was chosen for the chemical analysis. All of the air-sensitive samples were mounted in an Ar-filled glovebox (O2 < 0.6 ppm, H2O < 0.6 ppm) and were transferred into the instrument within a vacuum suitcase. The vacuum transfer suitcase was opened only for load lock pressure lower than 3 × 105 mbar in order to minimise the influence gas.
3 Results and discussion
3.1 Thermally improved capacity retention
Fig. 1 shows the charging and discharging curves and the impact on the electrochemistry performance of the cells subjected to the four cycle conditions. Although cycling at higher UCV (4.3 V) at room temperature exhibits acceleration of degradation, increasing the cycling temperature to 40 °C results in enhanced capacity retention and higher specific capacity as highlighted by the green arrows in Fig. 1. Further analyses shown in Fig. 2 reveal the improvement of the cell performance at high UCV and cycling temperature. Table 1 shows a summary of the main results during cycling of Al-surface-doped-Ni-rich//graphite pouch cells. | ||
| Fig. 1 Voltage vs. specific capacity of the different cycling protocols. The grey lines are for cycle number 50, 100, 150, 200, 250, 300, 350, 400, and 450. | ||
 | ||
| Fig. 2 a) Coulombic efficiency, b) discharge capacity, and c) normalized discharge capacity of the LiFun SC Al-surface-doped-Ni-rich//graphite pouch cells at different cycling conditions. Short periods of power outage make 4.2 V 25 °C data slightly spread around 100 cycles, but statistical trends are not altered. | ||
| Temperature | |||
|---|---|---|---|
| 25 °C | 40 °C | ||
| UCV | 4.2 V | 180.66 mAh g−1 | 190.62 mAh g−1 |
| 91% | 92% | ||
| 4.3 V | 189.36 mAh g−1 | 199.49 mAh g−1 | |
| 74% | 85.5% | ||
The nominal specific capacity data (Fig. 2b) is normalized using the first-cycle specific capacity as a reference so that changes on degradation (slopes) are highlighted given a common reference (specific capacity of the first cycle) after normalization. After 500 cycles in the voltage window 2.5–4.2 V at 25 °C, the full cells (Fig. 2a) retain 91% of the initial C/3 capacity (180.66 mAh g−1) with the capacity fade being approximately linear (Fig. 2b). The observed performance reflects the industrial-like quality of the cells and outperforms other systems reported elsewhere.18,27,36,37
The cycling protocols with no thermally-improved Li kinetics (cycling temperature at 25 °C) shows the typical electrochemical behaviour upon increasing the UCV – larger initial discharge capacity with faster degradation. The initial discharge capacity of the 4.3 V UCV cycling protocol stands at 189.36 mAh g−1 (≈8.71 mAh g−1 above the 4.2 V UCV cycling protocol) as showed in Fig. 2b. However, the 8.71 mAh g−1 gain results in faster degradation, with steeper degradation at about 300 cycles, and retaining almost 74% of the initial capacity (17% lower than the 4.2 V UCV case) after 500 cycles (Fig. 2b). The gain in capacity and subsequent accelerated degradation upon UCV increase is commonly reported in the literature with SEI-based degradation governing up to 4.2 V UCV18 and a more cathode-leading degradation at 4.3 V UCV.18,38
Increasing the temperature from 25 °C to 40 °C (where the improved Li diffusion gets enabled) does not show sign of additional degradation in the 2.5–4.2 V cycling protocol, indicated by comparable slopes in Fig. 2c for both 25 and 40 °C cycling temperatures with the 4.2 V UCV. Yet the 40 °C choice provides a sustained 9.97 mAh g−1 additional capacity across the 500 cycles – Fig. 2b. The absence of additional degradation indicates how resilient the electrolyte is to the 40 °C cycling temperature, ruling out electrolyte thermal decomposition as the source of degradation in the 4.3 V UCV 40 °C cycling protocol, and rather suggesting chemical oxidation of the electrolyte as the possible source of additional degradation in the 4.3 V UCV 40 °C cycling condition.21
Higher temperature kinetic effects challenge the traditional view of cells decaying faster at higher temperature in the long-term. The higher temperature kinetic effects combined with wider potential windows from 4.2 UCV to 4.3 UCV and a fixed LCV at 2.5 V exhibit a remarkable role in the electrochemical performance of the Al-surface-doped-Ni-rich//graphite cells. Fig. 2b and c show that after 500 cycles with the 4.3 V UCV, the 40 °C cells perform remarkably better than the 25 °C ones with capacity retention being 85.5% and 74.0% of initial capacities at ≈200 and 189 mAh g−1, respectively. Accordingly, the capacity retention shows a significant improvement with profile slope tending to retain the linear nature of the baseline cycling protocol (4.2 V and 25 °C in Fig. 2b). The thermally-enhanced diffusion of Li ions, tested for a single cycle by Zhou et al.,13 translates into a long-term positive effects on the electrochemical performance with differentiated outcomes depending on the UCV choice. For the 4.2 V UCV case, we have a sustained additional capacity across the 500 cycles and no additional thermal degradation at the electrodes or electrolyte. For the 4.3 V UCV case, the moderate O loss impacts the electrochemical performance at the 25 °C cycling condition after 500 cycles while the 40 °C cycling condition results in more specific capacity and better retention. Possible degradation pathways can be associated with cathode surface degradation given a moderate chemical oxidation of the electrolyte due to cycling at the moderate O loss threshold. Degradation pathways can also be associated with cathode bulk degradation or with cathode particle morphology degradation. Cell resistance studies follow to further understand the observed behavior.
3.2 Thermally-improved kinetic during long-term cycling
Polarization is a measure of the overall Li-ion battery resistance. The resistance nature of the cell can be linked to ionic resistance (active material and electrolyte), charge transfer (active material – electrolyte interface), and diffusion.39 For a given cycle number, polarization is the difference of the time average of the voltage profile during charging and discharging as described in section 2. Thus an increasing polarization represents increasing electrochemically induced sources of resistance. Analyzing the evolution of cell polarization across the number of cycles provide a means to study the impact of the temperature and high UCV during long-term cycling.Fig. 3 shows the potential average (three cells) polarization evolution with the testing cycling protocols. For the 4.2 UCV case (Fig. 3), where section 3.1 demonstrates the absence of additional degradation for the 40 °C cycling temperature, high temperature cycling up to 200 cycles shows enhanced Li kinetics (less overall polarization) extending the proven single-cycle thermal benefit13 to the long term. Beyond 200 cycles, polarization remains statistically similar with the sustained additional capacity across the 500 cycles demonstrated in section 3.1. At 4.3 V UCV in Fig. 3, the differences are remarkable between cycling temperatures. We observe again a turning point at 200 cycles below which the 40 °C cycling condition exhibits lower polarization than even the 4.2 V 25 °C cycling protocol. The 4.3 V 25 °C polarization worsens with the number of cycles standing above of the rest of cycling protocols.
 | ||
| Fig. 3 Time average cell potential difference quantifies the amount of cell polarization. | ||
We hypothesize that at the cathode surface, the low 4.3 V 40 °C polarization corresponds to some surface regions with an early formation of Al–F–O passivating layers in the long term. Lebens-Higgins et al.17 show the Al-based scavenging surface benefit in a single cycle while our work suggests similar benefits prolong in the long term. An increased likelihood of electrolyte attacks stemming from instances of electrolyte chemical oxidation at the moderate O loss UCV threshold after 500 cycles could have driven the gradual Al–F–O passivating layer formation. At bulk, we hypothesize the 40 °C temperature during cycling might retain to some extent the crystal anisotropic expansion after cycles allowing more Li ions to cycle compared with the 25 °C case.
Following sections test these hypotheses with detailed analyses of the resistance components associated with the bulk or material interface kinetics to collate the polarization and electrochemical results and provide an understanding for the improved capacity retention at the 4.3 V 40 °C cycling condition after 500 cycles.
3.3 Bulk studies: crystal and particle degradation
The thermal impact on the cycle life differs with the choice of UCV according to section 3.1 with a sustained additional capacity (temperature-induced capacity recovery) and no signs of further degradation at the 4.2 V UCV while the 4.3 V UCV cycling protocol shows better capacity retention at 40 °C. Section 3.2 suggests temperature plays a relevant role in delaying or limiting polarization growth. We need to gain more insight into the thermally-induced effects on polarization components related with the SC particle and crystal structure level of the Al-surface-doped-Ni-rich material.Fig. 4 demonstrates how robust the bulk crystal structure is even after 500 cycles. The P4 dQ/dV peak has been typically used as a metric to explain capacity fade arising from high voltage structural instabilities associated with the structural H2/H3 phase transition with a direct relation between a decaying P4 peak with gradual emergence of the H2/H3 phase transition over cycles.40,41 However, ongoing debate cast doubts on the existence of the H2/H3 phase transition in NMC-811, similar to our Ni-rich material, since spectroscopic experiments by Märker et al.11 show a still decaying P4 peak even in the absence of the H2/H3 phase transition. Thus, P4 can rather be associated with a direct evolution of the c-lattice parameter of the crystal structure, as suggested by Kondrakov et al.,42 indexing the dQ/dV P4 peak signal to the cycling dynamics of the c-lattice parameter (derivative of the c-lattice parameter with respect to cell potential – dc/dV). Namely, a decaying dQ/dV P4 peak means a dc/dV → 0 which translates into a less responsive c lattice parameter during cycling. We refer dc/dV as the contraction dynamic of the c-lattice parameter.
 | ||
| Fig. 4 dQ/dV plot after 100 and 500 cycles of the full cell at a) 4.2 V and b) 4.3 V. Traditional peak assignment: P1 associated to the C6 → LiCx structural changes, P2 to the H1 → M transition, P3 to the M → H2 transition, and P4 to the H2 → H3 transition during cycling. Insets in both panels highlight the P4 evolution. | ||
At 4.2 V UCV (inset Fig. 4a), the 40 °C cycling protocol shows the same extent for the contraction dynamic of the c-lattice parameter with similar P4 peaks between 100 and 500 cycles. Conversely, the 25 °C cycling condition yields a decaying P4 peak between these two cycle points. These results indicate that in the 25 °C cycling condition, there is a relatively larger cathode bulk region whose c-lattice parameter does not collapse fully relative to other more redox-active bulk domains. The increase of less-redox active bulk regions is known as structural heterogeneity. A gradual deterioration of the contraction dynamic of the c lattice parameter has been reported previously43 where the gradual disappearing dc/dV parameter evolves in tandem with the emergence of increased bulk heterogeneity. In contrast, the 40 °C cycling protocol limits the increase of structural heterogeneity given the almost invariant P4 peak signal at 4.2 V UCV between cycle number 100 ad 500, showing a robust P4 peak to cycling.
At 4.3 V UCV (inset Fig. 4b), regardless of the cycling temperature, the 500 cycle P4 peak decays relative to the 100 cycle P4 peak, meaning the mild O loss threshold is significantly affecting the c lattice dynamics and in turn inducing heterogeneity at the crystal level. However, the 40 °C cycling condition shows advantages over the room temperature protocol with the 4.3 V UCV. The 40 °C P4 peak shows the five-hundred cycle P4 signal to be comparable to the one-hundred cycle P4 signal at 25 °C (i.e., comparable structural heterogeneity but at a longer cycling life with higher cycling temperature). Namely, the thermally-enhanced Li kinetics at 40 °C systematically delays the genesis of structural heterogeneity making the structure to show some level of electrochemically resilience. In conclusion, raising the cycling temperature avoids and delays the evolution of crystal heterogeneity in Al-surface-doped-Ni-rich at 4.2 V and 4.3 V respectively.
SEM studies on the Al-surface-doped-Ni-rich SC cathode particle (Fig. 5) show the lack of a particle heterogeneity evolution associated with redox dynamics. At a particle level, heterogeneity defines the visual particle changes, resulting from cycling and affecting Li diffusion. Literature reports particle slicing as the typical particle degradation mechanisms for SC that can lead to increased particle heterogeneity. According to Bi et al., particle slicing evolves at higher voltages (>4.2 V) as a cumulative effect of incomplete reversible lattice gliding during cycling.44 Particle slicing leads to internal moderate cracks. Neighboring planes along the 003 direction of the layered structure slide over each other when the cell reaches the UCV. The planes then does not fully recover their original morphological structure at the LCV of the cell. The partially recovered planes should induce inter-plane slicing and eventual micro-cracking formation in long-term cycling.
 | ||
| Fig. 5 a) SEM image on pristine SC Al-surface-doped-Ni-rich NMC cathode showing an average particle size in the order of micrometer. Primary particles are highlighted in light green. Inset table summarizes the ICP-OES results on the same material. Molar percentages are based on Mols of Ni + Mn + Co = 100%. b) SEM on electrodes from discharged cells showing cycled Al-surface-doped Ni-rich NMC at 2.5–4.3 V at 40 °C. Green arrows indicate surface stripes on the particles. Orange arrows mean clean crystal cuts from structural gliding. Red ovals show calendering induced cracking. | ||
The SEM imaging shows an average SC particle size at 2 μm with particles preserving their integrity and shape upon cycling to 4.3 V UCV at 40 °C, demonstrating their aversion to cycling degradation. The pristine and cycled particle (Fig. 5a and b respectively) show uneven cracks (fractures in red ovals) and surface stripes (green arrows) indicating these particle morphology defects do not arise from cycling but could rather be attributed to the calendering step.45 Conversely, the surface stripes in the orange arrows can indicate minor particle slicing. However, the amount of particle slicing is not sufficient to evolve into micro-cracking in most of the particles as negligible amounts of particle fracture and severe particle heterogeneity is observed for the 4.3 V 40 °C cycling protocol. We note that the Ni composition of the Al-surface-doped Ni-rich material exceeds the typical NMC811 Ni content at 83.4 per cent nickel, therefore the system at an UCV of 4.3 V is more delithiated (stressed) than conventional NMC811, yet these SC particles are robust to particle heterogeneity, meaning thermally-enhanced diffusion and the mild O loss threshold does not impose degradation at the particle level of the Al-surface-doped-Ni-rich material.
3.4 Surface studies: Al-surface dopants and impedance evolution
Even though high cycling temperature improves Li-ion kinetics resulting in partial recover of lost capacity, increasing the cycling temperature typically promotes faster electrolyte degradation at elevated temperature.17 Additionally, cycling protocols with UCV at the moderate O loss threshold further stress the Al-surface-doped-Ni-rich CEI stability. This section explores the long-term role of the Al-surface dopant in the competing performance effects between improved Li-ion kinetics, thermally-enhanced electrolyte degradation, and the moderate O loss threshold at the interface.The different cycling protocols induce an Al-surface-doped-Ni-rich interface with a resulting combination of impedance processes (electrolyte ohmic resistance process, the charge-transfer process, among others). These processes have their respective time scale given by their characteristic time constants (τ = RC) and associated relaxation frequencies 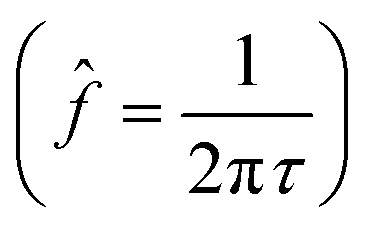 . Thus, distinguishing these different time constants unlocks an a priori knowledge of the relevant impedance processes associated with the cycling protocols.46
. Thus, distinguishing these different time constants unlocks an a priori knowledge of the relevant impedance processes associated with the cycling protocols.46
A model to study the time constants is the distribution of relaxation times (DRT).47Fig. 6 shows the DRT equivalent resistance values as a function of the different cycling protocols with the respective ![[f with combining circumflex]](https://www.rsc.org/images/entities/i_char_0066_0302.gif) .
.
 | ||
| Fig. 6 Distribution of relaxation time analyses of experimental EIS data after 500 cycles with the cycling protocols. The data is plotted versus the associated relaxation frequencies to equate regions where characteristics EIS arcs are expected for the different impedance processes. a) Highlights the changes in spectra as UCV changes from 4.2 V to 4.3 V at fixed 25 °C (red to yellow gradient) and the changes as the cycling temperature goes from 25 °C to 40 °C at a fixed 4.2 V UCV (red to brown gradient). b) Shows the temperature effects on impedance at a fixed 4.3 UCV. | ||
The DRT equivalent resistance at high frequency (105 Hz), associated with electrolyte diffusion processes, shows dependency to high UCV and cycling temperature in our cycling protocols as shown in Fig. 6a. In cycling protocols for the moderate O loss effects only (fixed 25 °C, UCV from 4.2 V to 4.3 V) and for the temperature-improved Li-ion kinetics only (fixed UCV at 4.2 V, temperature from 25 °C to 40 °C), the high frequency equivalent resistance increases from 0.04 to 0.08 Ω. This resistance increase favors the established view of a more electrochemically unstable electrolyte that can be driven by chemical oxidation at high UCV or minor thermal degradation. Indeed, the cycle protocol with both high UCV and high temperature further supports this statement with an equivalent resistance reaching 0.14 Ω in Fig. 6b.
However, the low frequency DRT peak (between 10−1 to 10−1 Hz), typically associated with ionic diffusion through the CEI,48,49 does not show signatures of a severely damaged interface despite of the cycling conditions with partial electrolyte decomposition. In cycling protocols for the moderate O loss effects only (fixed 25 °C, UCV from 4.2 V to 4.3 V), the equivalent low frequency resistance increases from 0.28 to 0.34 Ω in accordance with relatively higher electrolyte volatility at high voltages affecting the CEI. Conversely, the temperature-improved Li-ion kinetics testing protocol (fixed UCV at 4.2 V, temperature from 25 °C to 40 °C) improves the interface kinetics with a lower equivalent resistance at 0.26 Ω despite of the high frequency DRT peak showing increased instances of electrolyte decomposition. Similar trends are observed when we compare different cycling temperatures at higher UCV. The DRT findings suggest the Al-surface dopant plays a crucial role in long-term cycling allowing for a more stable interface even in the presence of increased surface attacks from cycling-induced electrolyte decomposition. We investigate further the CEI to elucidate the Al-based surface doping evolution with cycles.
The Al-surface-doping distribution in single crystal Ni-rich particles shows a gradient of concentration that decreases with the depth of the particle. ToF-SIMS in Fig. 7a shows Al-based species predominant at the surface with intensities in a ToF-SIMS point-to-point normalization analysis showing AlO− and AlO2− signal of one order of magnitude higher at shorter sputtering times. Longer sputtering times correspond to deeper depth probing.50,51 At sputtering time longer than 200 s, the transition metal (TM) values stabilize into a bulk-like stoichiometry (Fig. S1†). However, as the intensities of secondary ions are not only proportional to the concentrations but also affected by the matrix effects, a quantitative analysis on Al-dopants is not straightforward. HAXPES, (Fig. 7b), confirms the Al surface distribution is a Al3+ oxidation environment with a characteristic peak reflecting the Al 1s orbital at 1559.85 eV comparable to the Al 1s HAXPES peak in LiNi0.8Co0.15Al0.05O2.17
 | ||
| Fig. 7 a) ToF-SIMS on pristine SC Al-surface-doped-Ni-rich cathode. <TM> = Ni, Mn, or Co, distinguished by different dashes. b) Al 1s orbital from HAXPES experiments at two different photon energies with NCA reference for Al3+.17 c) ToF-SIMS on discharged cycled Al-surface-doped-Ni-rich MNC cathode after 500 cycles to investigate the Al–F reaction product from the Al scavenger surface effect. d) HAXPES at 6.6 keV on discharged electrodes of the SC Al-surface-doped-Ni-rich material showing fluorination of Al also an indicative of Al scavenger protection from electrolyte HF attack. | ||
Fig. 7c and d show the Al surface dopants undergo a fluorination process in cycling conditions which correspond with the higher instances of electrolyte decomposition according to the high frequency DRT peak in Fig. 6. The resulting high frequency DRT peak intensities from the cycling conditions share similar increasing trends to those of the AlF4− ToF-SIMS intensities Fig. 7 and 6.6 keV HAXPES Al–O–F Al 1s peak Fig. 7d. The Al scavenging role responds differently as a function of the cycling conditions that promote certain degree of electrolyte decomposition (UCV = 4.2 V, T = 40 °C) ≈ (UCV = 4.3 V, T = 25 °C) < (UCV = 4.3 V, T = 40 °C). With UCV at 4.3 V and cycling temperature at 40 °C, HAXPES shows increased fluorination in earlier cycle numbers allowing for the early protection of the interface resulting in a corresponding better polarization as observed in Fig. 3. As a result, ToF-SIMS and HAXPES respectively show byproducts of the aluminum fluorination process. In NCA cathodes, aluminum oxide components are known to promote a more stable CEI protecting the interface from decomposed electrolyte attacks.17,52–55 Al fluorination chemical pathways have been proposed56 for Al2O3 coating on cathode material:
| Al2O3 + 2HF → Al2O2F2 + H2O | (2) |
| Al2O2F2 + 2HF → Al2OF4 + H2O | (3) |
| Al2OF4 + 2HF → 2AlF3 + H2O | (4) |
3.5 Surface studies: evolution of the cathode electrolyte interface
In order to examine the long-term effect of the Al fluorination process on the Ni-rich cathode surface, we employed soft XAS in the total electron yield mode. O K-edge, Ni L3-edge, and Mn L3-edge (Fig. 8a–c, respectively) samples the cathode surface with secondary electrons emitted after the X-ray absorption. These electrons have a mean free path of several nanometers making the technique suitable to probe the cathode chemical surface structure, in particular the reduced ‘NiO’ surface layers.59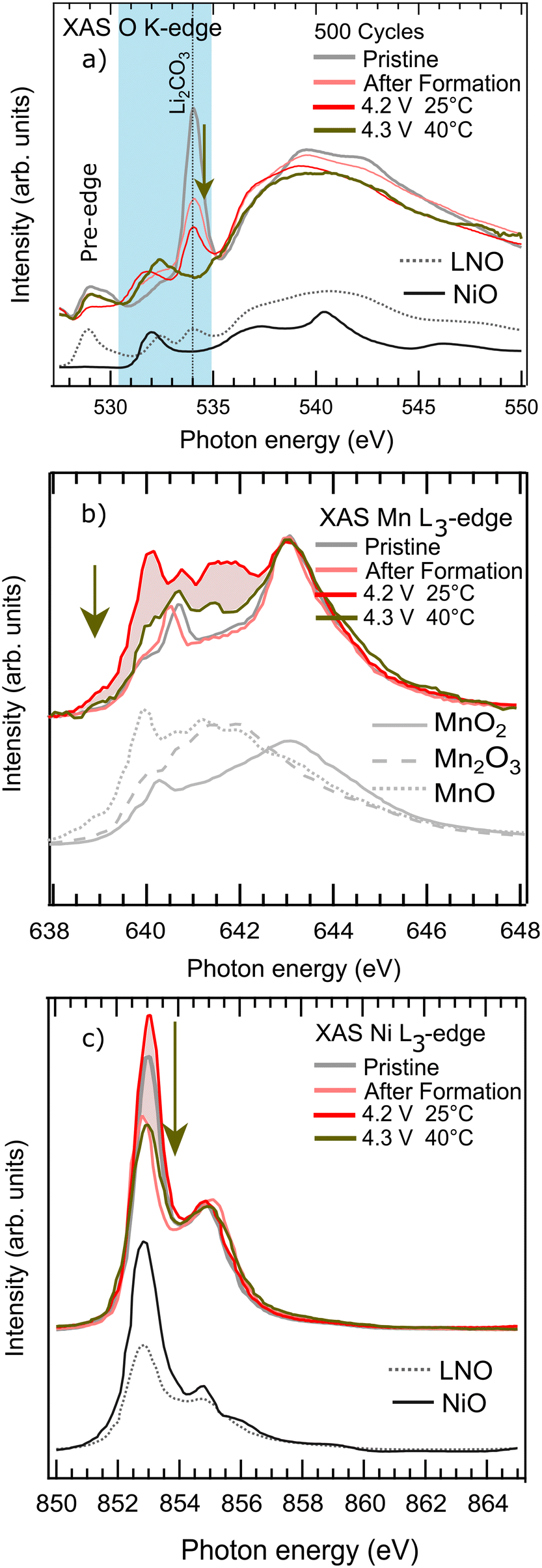 | ||
| Fig. 8 Soft XAS experiments on the SC Al-surface-doped-Ni-rich electrodes in the discharged state (2.5 V) after the formation cycle, and after 500 cycles in two representative cycling protocols (4.2 V 25 °C, and 4.3 V 40 °C). Pristine is the electrode as-received from LiFun. The energy regions are a) O K-edge, b) Mn L3-edge, and c) Ni L3-edge. Ni L3-edge and Mn L3-edge scans are normalized to peak at high energies to highlight variation at the cathode surface (shaded regions). | ||
The O K-edge, Ni L-edge, and Mn L-edge regions exhibit distinguishable photon energy domains where either the Ni-rich NMC electronic structure (cathode region) or cathode surface species electronic structure (surface region) dominates. It is worth mentioning spectral weight from these two regions might overlap because spectral contributions from the Ni-rich NMC material and its surface are not fully independent. However, by studying the O, Ni, and Mn XAS spectral relative changes at the surface domain using the cathode region peak as a normalization factor, we can characterize some components of the CEI as per Sallis et al.59 Sample data in Fig. 8 is in the discharged state (2.5 V) after the formation cycle and after 500 cycles with the baseline cycling protocol, and the 4.3 V 40 °C cycling protocol with more instances of some degree of electrolyte decomposition.
The energy domain 528–530.5 eV in the O K-edge corresponds to the Ni-rich NMC XAS cathode region due to the strong TM 3d–O 2p hybridization. The XAS cathode region can also be observed at 643 eV in the Mn L3-edge, and at 855 eV in the Ni L3-edge via the allowed dipole TM 2p → 3d absorption transition.
The XAS surface region of Ni-rich NMC material is highly sensitive to reactions with atmosphere (pristine) and electrolyte (cycled). As a result, the surface state can differ from bulk depending on exposure and cycling conditions. In the normally discharged (and pristine) state, the bulk is expected to exhibit Mn4+, Co3+, and Ni2+/3+. Thus, difference in the soft XAS TEY spectra reflects irreversible surface changes reflecting the cathode surface/(electrolyte) interface evolution. For example, these can include surface TM reduction due to lattice oxygen i.e. Ni(Mn,Co)O rock salt formation, Li2CO3 formation and leaching, and any other pathway leading to surface degradation. Here, the XAS surface region stands in the 531–535 eV range at the O K-edge, at 640 eV in the Mn L3-edge, and at 853 eV in the Ni L3-edge. The spectra related to the XAS surface region is highlighted in Fig. 8 as a guide to the eye for the TM surface evolution.
Meanwhile, the Mn L3 surface region (Fig. 8b) remains invariant to Li2CO3 decomposition. We conclude that the Li2CO3 leaching feature mainly affects the Ni ions rather than the Mn ions at the surface of our Al-surface-doped-Ni-rich material so becomes another Ni reduction source along with surface reconstruction. Conversely, Mn ions spectral changes are independent to Li2CO3 decomposition and hence to its leaching property, making Mn ions primarily sensitive to above-mentioned surface damage processes of the layered structure.
Interestingly, at the condition where the Al scavenger role activates (4.3 V UCV, 40 °C) as confirmed by DRT analysis, ToF-SIMS, and HAXPES, the Ni and Mn surface region massively decline relative to the intensities for the 4.2 V 25 °C case (large surface reconstruction). We also confirm this is a cleaner surface because we observe a spectral recovery of the pre-edge domain in the O K-edge to nearly back to the pristine cathode state with the added advantage of a dissolved Li2CO3 peak. Our work thus confirms the scavenger role of Al-based doping in restoring a surface where Ni and Mn surface reconstruction phases are absent and therefore further reduced oxidation states of the Ni and Mn ions are not observed. Additionally, surface contaminants have dissolved off the Al-surface-doped-Ni-rich material surface.
3.6 Surface studies: TM cross-talk
Increasing the UCV to 4.3 V and raising the temperature to 40 °C results in improved stability of surfaces. This finding is consistent with the DRT low frequency peak (cathode interface diffusion), indicating no significant charge-transfer impedance growth associated with the moderate O loss and temperature-induced electrolyte decomposition. As the UCV and temperature are raised, the decomposed Li2CO3 and the initial reduced surface metal species due to leaching are expected to be dissolved into the electrolyte. The TM dissolution in the electrolyte must appear as TM deposited at the anode, a process commonly referred as cross-talk. We note that this is different to the case for more stressed conditions where cross-talk is orders of magnitude larger in cycling protocols with full O loss degradation (V > 4.3 V). Additionally, note that the amount of TM dissolution on the graphite is significantly smaller than in polycrystalline NMC-811//graphite studies where pronounced capacity fade was observed in cycling with O loss.19 Given the good capacity retention, the level of cross-talk here is not sufficient to compromise the SEI. To quantify this, we employed HAXPES and ToF-SIMS studies on the anode graphite. The intensities of each TM species for different cells are compared with TOF-SIMS on the assumption that the major composition is graphite and it does not undergo significant chemical changes for different cells.Fig. 9 correlates TM deposition at different scales: particle (HAXPES) and electrode (ToF-SIMS). The cycling conditions progressively augments the initially-reduced Ni (and Mn) deposition (Fig. 9) on the graphite SEI layer in parallel to decomposition of Li2CO3 from the cathode (Fig. 8a). Ni 3p signal in Fig. 9a shows a maximum at 4.3 V UCV and 40 °C and links with the electrochemically-induced surface cleaning of decomposed Li2CO3. This correlation is similarly observed in the ToF-SIMS experiments depicted in Fig. 9b, where the 4.3 V 40 °C cycling protocol exhibits the highest Ni ionic intensity in comparison with the cells with the 4.2 V UCV and 25 °C cycling protocol. With 4.2 V UCV and 25 and 40 °C, only Mn ions (Mn+) present a tangible amount of signal while the signal intensities of Co and Ni ions are close to the detection limit level (ppm) of ToF-SIMS. Note that the Ni+, Co+, and Mn+ ions do not reflect the actual oxidation state of the TMs and instead result from ionization of the material surface after ionic bombarding with a 25 keV Bi+ beam in ToF-SIMS studies. However, TM XAS L-edge absorption experiments show that the TMs exist in a non-metallic oxidation state (i.e., TMn+, n > 0) (Fig. S2†) consistent with the literature.19 In conclusion, combining HAXPES and ToF-SIMS studies on the anode, we observe the SEI is being poisoned with islands of deposited TM which partially contribute to the capacity decay62 of the full cells.
 | ||
| Fig. 9 a) Li 1s HAXPES at 2.2 keV of cycled graphite in the discharge state with b) ToF-SIMS experiments on the same material to confirm HAXPES trends. c) Schematic explaining the solid-electrolyte-interface poisoning while Li2CO3 decomposes. TMn+ with n > 0 shows the TMs exist in a non-metallic oxidation state. | ||
Fig. 9c summarizes the mechanism: Li2CO3 formation induced TM reduction and its electrochemical decomposition releases the reduced species into the electrolyte where they deposit on the SEI. This study distinguishes surface reduction processes associated with Ni-rich NMC reacting with moisture and Li2CO3 from lattice O loss induced surface reduction at higher UCV. The former appears to result in some cross-talk but not electrochemically significant, whereas the latter is a relevant cause of degradation for Ni-rich cathode systems.
4 Conclusions
Cycling protocols accounting for a moderate O loss (below the O-loss release threshold) and for elevated temperatures (i.e. 40 °C) translate into improved lithium-ion kinetics and superior capacity retention only if single crystalline Ni-rich NMC morphologies are equipped with sacrificial surface dopants (e.g., Al). With the single-crystal particle morphology being resilient to the electrochemistry and surface sacrificial agent passivating surface attacks from decomposed electrolyte, increasing the cycling temperature improves particle and crystal homogeneity (less heterogeneity), resulting in improved Ni-rich NMC diffusion pathways for lithium ions, therefore capacities. In the case of the 4.3 V at 40 °C cycling protocol, our studies confirm that after 500 cycles the sacrificial surface agent delays surface degradation from decomposed electrolyte either from the moderate O loss threshold. In the bulk, high temperature cycling translates into improve c lattice dynamics and better Li kinetics. The observed degradation products link with stripping off of cathode surface species associated with Li2CO3 formation rather than surface phases resulting from the moderate NMC surface O loss.As a result, increasing the temperature from 25 to 40 °C results in improved specific capacity and capacity retention at 85.5% in contrast to a 74.0% capacity retention in the 4.3 UCV regime when surfaces are passivated. The benefit of increasing the temperature thus directly translates into improved energy density (E = V × Ah (Wh)) for lithium-ion batteries. Namely, more energy can be retrieved in every cycle, reducing the required number of charging cycles a potential user might face in the long term, so expanding the life-span of the cell.
Author contributions
Galo Páez Fajardo: writing - original draft, formal analysis, investigation, data curation, methodology. Meltiani Belekoukia: formal analysis, investigation, methodology, writing – review & editing. Satish Bolloju: writing - review & editing. Eleni Fiamegkou: formal analysis, investigation, writing – review & editing. Ashok S. Menon: investigation, writing – review & editing. Zachary Ruff: formal analysis, investigation, writing – review & editing, methodology. Zonghao Shen: formal analysis, investigation, writing – review & editing, methodology. Nickil Shah: writing – original draft, investigation, data curation. Erik Björklund: investigation. Mateusz Jan Zuba: investigation. Tien-Lin Lee: resources, methodology, writing – review & editing. Pardeep K. Thakur: resources, methodology. Robert S. Weatherup: resources, writing – review & editing, investigation. Ainara Aguadero: resources, writing – review & editing. Melanie J. Loveridge: resources, writing – review & editing. Clare P. Grey: funding acquisition. Louis F. J. Piper: funding acquisition, conceptualization, writing – review & editing, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ieuan Ellis and Paul Malliband from the Battery Scale Up facility at WMG for their assistance. This work is supported by the Faraday Institution under grant no. FIRG060, no. FIRG024, and no. FIRG061. We acknowledge Diamond Light Source for time on beamline I09 under proposal SI25355-5 and SI27494-2.Notes and references
- C. P. Grey and D. S. Hall, Nat. Commun., 2020, 11, 6279 CrossRef CAS PubMed.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- H.-H. Ryu, B. Namkoong, J.-H. Kim, I. Belharouak, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2021, 6, 2726–2734 CrossRef CAS.
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 CrossRef CAS.
- S. G. Booth, A. J. Nedoma, N. N. Anthonisamy, P. J. Baker, R. Boston, H. Bronstein, S. J. Clarke, E. J. Cussen, V. Daramalla, M. De Volder, S. E. Dutton, V. Falkowski, N. A. Fleck, H. S. Geddes, N. Gollapally, A. L. Goodwin, J. M. Griffin, A. R. Haworth, M. A. Hayward, S. Hull, B. J. Inkson, B. J. Johnston, Z. Lu, J. L. MacManus-Driscoll, X. M. De Irujo Labalde, I. McClelland, K. McCombie, B. Murdock, D. Nayak, S. Park, G. E. Pérez, C. J. Pickard, L. F. J. Piper, H. Y. Playford, S. Price, D. O. Scanlon, J. C. Stallard, N. Tapia-Ruiz, A. R. West, L. Wheatcroft, M. Wilson, L. Zhang, X. Zhi, B. Zhu and S. A. Cussen, APL Mater., 2021, 9, 109201 CrossRef CAS.
- M. Wentker, M. Greenwood and J. Leker, Energies, 2019, 12, 504 CrossRef CAS.
- Y. M. Alqahtani and Q. L. Williams, Materials, 2022, 15, 2146 CrossRef CAS PubMed.
- M. Shang, X. Chen and J. Niu, Cell Rep. Phys. Sci., 2022, 3, 100767 CrossRef CAS.
- M. Wood, J. Li, R. E. Ruther, Z. Du, E. C. Self, H. M. Meyer, C. Daniel, I. Belharouak and D. L. Wood, Energy Storage Mater., 2020, 24, 188–197 CrossRef.
- H. Zhang, H. Liu, L. F. J. Piper, M. S. Whittingham and G. Zhou, Chem. Rev., 2022, 122, 5641–5681 CrossRef CAS.
- K. Märker, P. J. Reeves, C. Xu, K. J. Griffith and C. P. Grey, Chem. Mater., 2019, 31, 2545–2554 CrossRef.
- K. Märker, C. Xu and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 17447–17456 CrossRef.
- H. Zhou, F. Xin, B. Pei and M. S. Whittingham, ACS Energy Lett., 2019, 4, 1902–1906 CrossRef CAS.
- A. Van der Ven and G. Ceder, J. Power Sources, 2001, 97–98, 529–531 CrossRef CAS.
- A. Van der Ven, J. Bhattacharya and A. A. Belak, Acc. Chem. Res., 2013, 46, 1216–1225 CrossRef CAS PubMed.
- E. Lee, S. Muhammad, T. Kim, H. Kim, W. Lee and W.-S. Yoon, Adv. Sci., 2020, 7, 1902413 CrossRef CAS.
- Z. W. Lebens-Higgins, D. M. Halat, N. V. Faenza, M. J. Wahila, M. Mascheck, T. Wiell, S. K. Eriksson, P. Palmgren, J. Rodriguez, F. Badway, N. Pereira, G. G. Amatucci, T.-L. Lee, C. P. Grey and L. F. J. Piper, Sci. Rep., 2019, 9, 17720 CrossRef PubMed.
- W. M. Dose, C. Xu, C. P. Grey and M. F. De Volder, Cell Rep. Phys. Sci., 2020, 1, 100253 CrossRef.
- E. Björklund, C. Xu, W. M. Dose, C. G. Sole, P. K. Thakur, T.- L. Lee, M. F. L. De Volder, C. P. Grey and R. S. Weatherup, Chem. Mater., 2022, 34, 2034–2048 CrossRef PubMed.
- W. M. Dose, W. Li, I. Temprano, C. A. O'Keefe, B. L. Mehdi, M. F. L. De Volder and C. P. Grey, ACS Energy Lett., 2022, 7, 3524–3530 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Phys. Chem. Lett., 2017, 8, 4820–4825 CrossRef CAS.
- J. Hu, L. Li, Y. Bi, J. Tao, J. Lochala, D. Liu, B. Wu, X. Cao, S. Chae, C. Wang and J. Xiao, Energy Storage Mater., 2022, 47, 195–202 CrossRef.
- A. S. Menon, N. Shah, J. A. Gott, E. Fiamegkou, M. J. W. Ogley, G. J. P. Fajardo, N. Vaenas, I. Ellis, P. Malliband, G. West, D. Walker, M. Loveridge and L. Piper, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-zs9kp.
- J. L. Tebbe, A. M. Holder and C. B. Musgrave, ACS Appl. Mater. Interfaces, 2015, 7, 24265–24278 CrossRef CAS.
- G. Kaur and B. D. Gates, J. Electrochem. Soc., 2022, 169, 043504 CrossRef CAS.
- Z. W. Lebens-Higgins, S. Sallis, N. V. Faenza, F. Badway, N. Pereira, D. M. Halat, M. Wahila, C. Schlueter, T.-L. Lee, W. Yang, C. P. Grey, G. G. Amatucci and L. F. J. Piper, Chem. Mater., 2018, 30, 958–969 CrossRef CAS.
- X. Fan, G. Hu, B. Zhang, X. Ou, J. Zhang, W. Zhao, H. Jia, L. Zou, P. Li and Y. Yang, Nano Energy, 2020, 70, 104450 CrossRef CAS.
- E. R. Logan, H. Hebecker, X. Ma, J. Quinn, Y. HyeJeong, S. Kumakura, J. Paulsen and J. R. Dahn, J. Electrochem. Soc., 2020, 167, 060530 CrossRef CAS.
- C.-H. Jo, N. Voronina and S.-T. Myung, Energy Storage Mater., 2022, 51, 568–587 CrossRef.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- X. Chen, X. Wang and D. Fang, Fullerenes, Nanotubes, Carbon Nanostruct., 2020, 28, 1048–1058 CrossRef CAS.
- G. Greczynski and L. Hultman, Prog. Mater. Sci., 2020, 107, 100591 CrossRef CAS.
- H. Kobayashi, S. Emura, Y. Arachi and K. Tatsumi, J. Power Sources, 2007, 174, 774–778 CrossRef CAS.
- W.-S. Yoon, M. Balasubramanian, K. Y. Chung, X.-Q. Yang, J. McBreen, C. P. Grey and D. A. Fischer, J. Am. Chem. Soc., 2005, 127, 17479–17487 CrossRef CAS.
- H. Wang, C. Y. Ralston, D. S. Patil, R. M. Jones, W. Gu, M. Verhagen, M. Adams, P. Ge, C. Riordan, C. A. Marganian, P. Mascharak, J. Kovacs, C. G. Miller, T. J. Collins, S. Brooker, P. D. Croucher, K. Wang, E. I. Stiefel and S. P. Cramer, J. Am. Chem. Soc., 2000, 122, 10544–10552 CrossRef CAS.
- J. Li, L. E. Downie, L. Ma, W. Qiu and J. R. Dahn, J. Electrochem. Soc., 2015, 162, A1401–A1408 CrossRef CAS.
- C. S. Yoon, K.-J. Park, U.-H. Kim, K. H. Kang, H.-H. Ryu and Y.-K. Sun, Chem. Mater., 2017, 29, 10436–10445 CrossRef CAS.
- W. Li, X. Liu, Q. Xie, Y. You, M. Chi and A. Manthiram, Chem. Mater., 2020, 32, 7796–7804 CrossRef CAS.
- J. Kang, B. Koo, S. Kang and H. Lee, Chem. Phys. Rev., 2021, 2, 041307 CrossRef CAS.
- J. Sun, X. Cao, H. Yang, P. He, M. A. Dato, J. Cabana and H. Zhou, Angew. Chem., Int. Ed., 2022, e202207225 CAS.
- N.-Y. Park, G.-T. Park, S.-B. Kim, W. Jung, B.-C. Park and Y.-K. Sun, ACS Energy Lett., 2022, 7, 2362–2369 CrossRef CAS.
- A. O. Kondrakov, H. Geßwein, K. Galdina, L. de Biasi, V. Meded, E. O. Filatova, G. Schumacher, W. Wenzel, P. Hartmann, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 24381–24388 CrossRef CAS.
- C. Xu, K. Märker, J. Lee, A. Mahadevegowda, P. J. Reeves, S. J. Day, M. F. Groh, S. P. Emge, C. Ducati, B. Layla Mehdi, C. C. Tang and C. P. Grey, Nat. Mater., 2021, 20, 84–92 CrossRef CAS.
- Y. Bi, J. Tao, Y. Wu, L. Li, Y. Xu, E. Hu, B. Wu, J. Hu, C. Wang, J.-G. Zhang, Y. Qi and J. Xiao, Science, 2020, 370, 1313–1317 CrossRef CAS PubMed.
- J. C. Stallard, S. Vema, D. S. Hall, A. R. Dennis, M. E. Penrod, C. P. Grey, V. S. Deshpande and N. A. Fleck, J. Electrochem. Soc., 2022, 169, 040511 CrossRef CAS.
- T. Paul, P. W. Chi, P. M. Wu and M. K. Wu, Sci. Rep., 2021, 11, 12624 CrossRef CAS PubMed.
- S. Wang, J. Zhang, O. Gharbi, V. Vivier, M. Gao and M. E. Orazem, Nat. Rev. Methods Primers, 2021, 1, 41 CrossRef CAS.
- N. Laszczynski, S. Solchenbach, H. A. Gasteiger and B. L. Lucht, J. Electrochem. Soc., 2019, 166, A1853 CrossRef CAS.
- J. A. Gilbert, J. Bareño, T. Spila, S. E. Trask, D. J. Miller, B. J. Polzin, A. N. Jansen and D. P. Abraham, J. Electrochem. Soc., 2016, 164, A6054 CrossRef.
- J. Brison, S. Muramoto and D. G. Castner, J. Phys. Chem. C Nanomater. Interfaces, 2010, 114, 5565–5573 CrossRef CAS.
- J. Ekar, P. Panjan, S. Drev and J. Kovăc, J. Am. Soc. Mass Spectrom., 2022, 33, 31–44 CrossRef CAS.
- M. Aykol, S. Kirklin and C. Wolverton, Adv. Energy Mater., 2014, 4, 1400690 CrossRef.
- L. David, K. Dahlberg, D. Mohanty, R. E. Ruther, A. Huq, M. Chi, S. J. An, C. Mao, D. M. King, L. Stevenson and D. L. Wood, ACS Appl. Energy Mater., 2019, 2, 1308–1313 CrossRef CAS.
- W. Zhu, X. Huang, T. Liu, Z. Xie, Y. Wang, K. Tian, L. Bu, H. Wang, L. Gao and J. Zhao, Coatings, 2019, 9, 92 CrossRef.
- Y. Tesfamhret, R. Younesi and E. J. Berg, J. Electrochem. Soc., 2022, 169, 010530 CrossRef CAS.
- S. Myung, K. Izumi, S. Komaba, Y. Sun, H. Yashiro and N. Kumagai, Chem. Mater., 2005, 17, 3695–3704 CrossRef CAS.
- L. Zou, J. Li, Z. Liu, G. Wang, A. Manthiram and C. Wang, Nat. Commun., 2019, 10, 3447 CrossRef PubMed.
- B. Han, B. Key, S. H. Lapidus, J. C. Garcia, H. Iddir, J. T. Vaughey and F. Dogan, ACS Appl. Mater. Interfaces, 2017, 9, 41291–41302 CrossRef CAS.
- S. Sallis, N. Pereira, P. Mukherjee, N. F. Quackenbush, N. Faenza, C. Schlueter, T.-L. Lee, W. L. Yang, F. Cosandey, G. G. Amatucci and L. F. J. Piper, Appl. Phys. Lett., 2016, 108, 263902 CrossRef.
- H. S. Liu, Z. R. Zhang, Z. L. Gong and Y. Yang, Electrochem. Solid-State Lett., 2004, 7, A190 CrossRef CAS.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132–A141 CrossRef CAS.
- N. V. Faenza, Z. W. Lebens-Higgins, P. Mukherjee, S. Sallis, N. Pereira, F. Badway, A. Halajko, G. Ceder, F. Cosandey, L. F. J. Piper and G. G. Amatucci, Langmuir, 2017, 33, 9333–9353 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ToF-SIMS relative intensities Al vs. Ni-rich NMC and XAS TM experiments on graphite to confirm TM oxidation states. See DOI: https://doi.org/10.1039/d3lf00093a |
| This journal is © The Royal Society of Chemistry 2024 |