
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonomeric and oligomeric amyloid-β cause distinct Alzheimer's disease pathophysiological characteristics in astrocytes in human glymphatics-on-chip models†
Aria R.
Yslas
 ,
Rena
Park
,
Nozomi
Nishimura
and
Esak
Lee
*
,
Rena
Park
,
Nozomi
Nishimura
and
Esak
Lee
*
Nancy E. and Peter C. Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: el767@cornell.edu
First published on 16th July 2024
Abstract
Alzheimer's disease (AD) is marked by the aggregation of extracellular amyloid-β (Aβ) and astrocyte dysfunction. For Aβ oligomers or aggregates to be formed, there must be Aβ monomers present; however, the roles of monomeric Aβ (mAβ) and oligomeric Aβ (oAβ) in astrocyte pathogenesis are poorly understood. We cultured astrocytes in a brain-mimicking three-dimensional (3D) extracellular matrix and revealed that both mAβ and oAβ caused astrocytic atrophy and hyper-reactivity, but showed distinct Ca2+ changes in astrocytes. This 3D culture evolved into a microfluidic glymphatics-on-chip model containing astrocytes and endothelial cells with the interstitial fluid (ISF). The glymphatics-on-chip model not only reproduced the astrocytic atrophy, hyper-reactivity, and Ca2+ changes induced by mAβ and oAβ, but recapitulated that the components of the dystrophin-associated complex (DAC) and aquaporin-4 (AQP4) were properly maintained by the ISF, and dysregulated by mAβ and oAβ. Collectively, mAβ and oAβ cause distinct AD pathophysiological characteristics in the astrocytes.
Introduction
Alzheimer's disease (AD) is a progressive brain disorder, characterized by memory loss and cognitive decline, which remains the predominant cause of dementia in the elderly.1 The pathological hallmarks of AD encompass extracellular amyloid plaques, constituted of amyloid-β (Aβ) peptides and intracellular neurofibrillary tangles derived from hyperphosphorylated tau protein.2 While both amyloid-β isoforms, Aβ40 and Aβ42, are present in the brain, Aβ42 tends to form neurotoxic oligomeric aggregates, and plays the predominant role over Aβ40 in AD pathogenesis, influencing synaptic functions, instigating oxidative and inflammatory responses, and leading to the hallmark symptoms of AD.3–5The formation of Aβ42 aggregates or oligomers in AD is based on the presence of monomeric Aβ42 in the brain parenchyma and its accumulation under the dysregulation of the brain's waste-clearance machinery such as the glymphatic system.6 In a normal glymphatic system, dictated by convective fluid dynamics, cerebrospinal fluid (CSF) penetrates periarterial spaces, interacting and mobilizing waste throughout the brain parenchyma. This waste-laden fluid forms interstitial fluid (ISF), drains into the perivenous spaces, and is eventually cleared via the meningeal lymphatics.7 In AD, the glymphatic system can be compromised when the system is influenced by Aβ42 and diminishes in its functionality. Although the Aβ42 monomers serve as a basic building block of the Aβ42 oligomers, the distinct roles of monomeric Aβ42 and oligomeric Aβ42 in glymphatic dysfunction and AD pathogenesis are unclear.
Astrocytes are one of the main cell types in the aforementioned waste-clearing glymphatic system, where the end feet of astrocytes are localized at the perivascular spaces (PVS), including the periarterial and perivenous spaces, in the brain parenchyma.8–10 Aquaporin-4 (AQP4) is a water channel expressed on these astrocytic endfeet at the PVS, which enables the flow of water molecules, especially when expressed on the endfeet rather than throughout the cell body (spatial polarization), thereby centralizing their role in the brain's waste clearance mechanism.11,12 Recent investigations have highlighted the pivotal role of the dystrophin-associated complex (DAC) within astrocytes in the AQP4 regulation.13 Integral for anchoring AQP4 to the plasma membrane of astrocytes, the DAC, through its interaction with laminin, modulates intracellular calcium (Ca2+) homeostasis and AQP4 polarization, which is frequently dysregulated in AD pathology.13 To our knowledge, the distinct roles of monomeric and oligomeric Aβ42 in DAC regulation, Ca2+ homeostasis, and AQP4 polarization in astrocytes are unknown.
While in vivo models are widely used in the AD and glymphatic fields, and the knowledge and insight obtained from the animal models are invaluable, they often bear intrinsic limitations, given the complexity of the model systems with tightly coupled biological and biophysical factors, which has hampered a deeper mechanistic understanding of the multifactorial neurodegenerative diseases such as AD. Two-dimensional (2D) cell culture is an easily controllable system, but it does not recapitulate the 3D structure of the glymphatic system and cannot introduce ISF across the system. To address these challenges, here we cultured astrocytes in a brain-mimicking three-dimensional (3D) extracellular matrix and the 3D culture model evolved into a microfluidic glymphatics-on-chip containing human primary astrocytes and human primary endothelial cells with the ISF. These models provided a unique opportunity to delve into the effects of monomeric and oligomeric Aβ on the function of astrocytes in the glymphatic contexts, which include astrocytic atrophy, hyper-reactivity, Ca2+ changes, and the regulation of the components of the dystrophin-associated complex (DAC) and AQP4, a hallmark of glymphatic function.
Experimental
Cell culture
The human astrocytes (ScienCell #1800) were cultured in complete ScienCell astrocyte media (#1801) on cell-culture-treated polystyrene. The d-HMVEC-BlNeos were cultured in complete EBM-2 with EGM-2 MV Single Quots (Lonza) on cell-culture-treated polystyrene. Both cell types received media changes every three days and were incubated in a standard mammalian cell culture incubator of 37 °C, 95% humidity, and 5% CO2.Amyloid-β preparation
Amyloid-β42 monomers and oligomers were prepared as established in Stine et al.,14 starting with Beta-Amyloid (1–42) HFIP (rPeptide, A-1163-2). Briefly, the dried peptide film was dissolved in dimethylsulfoxide (DMSO, Sigma-Aldrich) to make a 5 mM solution. The solution was briefly vortexed and then pulsed in a microcentrifuge before sonicating for 10 minutes in a bath sonicator. For the unaggregated/monomeric Aβ, ice-cold H2O was added to create a 100 μM solution. It was briefly vortexed for about 15 seconds and then used. For the aggregated/oligomeric Aβ, cold phenol-free F-12/DMEM cell culture media (Gibco) was added to the 5 mM Aβ DMSO solution to make 100 μM Aβ solution and kept ice-cold. It was then briefly vortexed for 15 seconds and then incubated at 4 °C for 24 hours before use. The solutions were then characterized via native Western Blot (WB). 5 μg of each suspension was loaded with water and a Laemmli SDS non-reducing buffer (Thermo Scientific Chemicals) for SDS-page. The final membrane was stained with the same amyloid-β antibody listed in Table 1 (Invitrogen), but at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 concentration prior to imaging with an Amersham Western Blot Imager. The characterization results can be seen in Fig. S1.†
:1000 concentration prior to imaging with an Amersham Western Blot Imager. The characterization results can be seen in Fig. S1.†
| Antibody | Dilution ratio | Vendor & catalog # |
|---|---|---|
| AQP4, rb pAb | 1:100 |
Invitrogen, #PA5-53234 |
| Nestin (SP103), rb mAb | 1:100 |
Thermofisher, #MA5-16421 |
| NFAT1 (25A10.D6.D2), m mAb | 1:100 |
Abcam, #ab2722 |
| CD31 (JC70A), m mAb | 1:100 |
Dako, #M0823 |
| GFAP, g pAb | 1:100 |
Abcam, #ab53554 |
| DAPI | 1:500 |
Thermo Scientific, #62248 |
| Phalloidin, Alexa Fluor 647 | 1:200 |
Invitrogen, #A22287 |
| Secondary antibodies | Invitrogen | |
| Dnk, anti-rb IgG, Alexa Fluor 647 | 1:500 |
#A-31573 |
| Dnk, anti-g IgG, Alexa Fluor 568 | #A-11057 | |
| Dnk, anti-m IgG Alexa Fluor 488 | #A-21202 |
3D cell-suspended ECM plate model
The well plates were 8-well glass-bottomed from MatTek Life Sciences. Before adding the ECM with the suspended astrocytes, the glass was treated with oxygen plasma, 0.01% poly-L-lysine (Sigma-Aldrich), and 1% glutaraldehyde (EMS) to reduce hydrophobicity. The well plates were washed with distilled water overnight and treated with UV before 200 μL of cell-suspended ECM was added to each well. The ECM consisted of 2.5 mg mL−1 collagen 1 (Corning), 1.25 mg mL−1 thiol-treated hyaluronic acid (HA, HyStem kit from Advanced Biomatrix), 0.26 mg mL−1 human plasma fibronectin (Sigma-Aldrich), 10× phosphate-buffered saline (PBS, Gibco), 1 N sodium hydroxide (NaOH, EMS), and astrocyte media containing human astrocytes with a final concentration of 1 million cells per mL. For experiments containing amyloid-β, they were also included in the ECM for a final concentration of 1 μM. After incubating the ECM in the devices in the cell culture incubator for one hour, astrocyte media was added. After one day of static culture, the plates were placed on a rocker in the cell culture incubator.Chip model
Devices were assembled as established in Soden et al.15 In summary, the pattern of the mold was created by silicon photolithography. Polydimethylsiloxane (PDMS) was mixed in a 10:1 ratio with its curing agent (included in Sylgard kit) and degassed via vacuum before being poured onto the molds and baked at 80 °C overnight. The mold was detached and shaped using razors and biopsy punches before being treated with oxygen plasma to attach to the 1.5 glass coverslips (EMS) and cured at 80 °C overnight. Following assembly, the devices were oxygen plasma treated and sequentially treated with 0.01% poly-L-lysine (Sigma-Aldrich), 1% glutaraldehyde (EMS), and overnight distilled water wash to reduce the hydrophobicity of the PDMS. Blunt-end acupuncture needles of diameter 0.25 μm (OHM Hwato) were soaked in ethanol and treated with 1% bovine serum albumin (BSA) in PBS before being loaded into the devices. The devices were then dried and UV sterilized before adding the astrocyte-rich ECM (same components as 3D cell-suspended ECM), and the chips were incubated overnight in a static cell culture incubator. The needles were removed, and the devices were sealed with vacuum grease (EMS) the next day, received fresh astrocyte media, and incubated on a gravity rocker overnight. On the following day, the BECs were seeded at 500000 cells per mL in complete microvascular endothelial media through the media reservoirs and suspended both upside down and right-side up before being placed on the gravity rocker for a shear and radial stress of ∼4 dyne per cm2 and ∼2 dyne per cm2, respectively. Interstitial fluid flow was induced via hydrostatic pressure differences in all devices excluding the no ISF control. 200 μL of complete microvascular endothelial cell media was added to the left media reservoirs while only 20 μL was added to each of the right media reservoirs. The reservoirs were allowed to reach hydrostatic equilibrium for 16 hours before downstream processing (either fixation for immunofluorescence or RNA isolation via TRIzol).
Fluo4-AM calcium imaging
Fluo-4 AM calcium imaging was prepared using Fluo-4 AM from Thermo Fisher Scientific following their specific protocol. Briefly, a loading solution was created by adding 50 μL of the ∼860 μM Fluo-4 AM/Pluronic® F-127 prepared solution to 14.3 mL of calcium-free Hank's buffered saline solution (HBSS, Gibco). Each well of the well-plate was washed with 200 μL HBSS before loading 200 μL of loading solution was added to each well and incubated in the dark at room temperature for ∼60 minutes. The solution was removed, and the wells were washed with 200 μL HBSS before imaging. Imaging was performed using a Lecia DMi8 confocal light microscope with an accompanying stage incubator from Oko-Lab. Each sample was imaged for 5 minutes, and signal intensity as well as signal shape parameters were measured blindly via ImageJ.Immunofluorescence imaging and image analysis
Both well plates and chips were prepared for immunofluorescence imaging the same way after the fixation step. Each sample was fixed with 4% paraformaldehyde (PFA, EMS) in PBS solution for the well-plate (astrocyte only) samples and 4% PFA in EBM-2 for the co-culture chips for 30–40 minutes and then washed thrice with PBS before being soaked in PBS at least overnight at 4 °C. Triton X (Sigma-Aldrich) was dissolved in PBS to create a 0.3% PBST solution. Each sample had its PBS aspirated before adding the PBST solution for 45–60 minutes at room temperature while shaking. The next steps were each performed with overnight incubation at 4 °C on a shaker on sequential days. The samples were then blocked in 3% BSA in PBS before receiving their primary antibodies via 3% BSA solution. After primary staining, the samples were washed with PBS before receiving secondary antibodies and DAPI (Thermo Scientific, 62248). The samples were washed one final time before imaging with a Leica DMi8 confocal light microscope. The antibodies used and their concentration are listed in Table 1.RT-qPCR
The cells suspended in ECM were collected from the chip devices. RNAs from these cells were isolated via TRIzol. The RNA yield and purity were measured using absorbance at 260 nm and 280 nm via a SpectraDrop microplate and microplate reader. Reverse transcription was performed using the TaqMan Reverse Transcription Reagents (Applied Biosystems by Thermo Fisher Scientific), random hexamers, and the isolated RNA in a Bio-Rad T100 Thermal Cycler. qPCR was performed using the GoTaq Green Master Mix (Promega), the cDNA, and custom primers (listed in Table 2) in a Bio-Rad CFX96 system. Data was retrieved via the Bio-Rad CFX Manager software with each sample run in triplicate and averaged before data analysis.| Gene | Forward primer | Reverse primer |
|---|---|---|
| AQP4 | GCCATCATTGGAGCAGGAATCC | ACTCAACCAGGAGACCATGACC |
| ARNT | CTGTCATCCTGAAGACCAGCAG | CTGGTTCTCATCCAGAGCCATTC |
| DAG1 | CCACAGTGGTTGGCATTCCTGA | CCAAGATGGCAAAGCCTCCTTC |
| DMD | GCTCAACCATCGATTTGCAGCC | TTCAGCCTCCAGTGGTTCAAGC |
| DTNA | CGGCTTGATGAAGAACACAGGC | GATGTCAGGAGCACTTCTCTGC |
| KCNIP4 | GTACGCTCAGAACAGCACCAAG | CCATCTCCAGTTCATCTTCCACG |
| MLC1 | CGCCAACATTCTGGACGAAGTG | GCCTGAAACTGAGTCATCCACG |
| SLC1A2 | TGCCAACAGAGGACATCAGCCT | CAGCTCAGACTTGGAGAGGTGA |
| SLC4A4 | GGAAAGCCAAGTCCTACCACGA | TACCAGCAATCAGGTCGTGCCT |
| SNTA1 | CCAGGACATCAAGCAGATTGGC | GAGACAAGTAGAGGAGCAGTTCC |
| STMN1 | AGAACCGAGAGGCACAAATGGC | TCTCGTCAGCAGGGTCTTTGGA |
Statistics
Student t-tests, one-way ANOVAs, and two-way ANOVAs were performed using Microsoft Excel and GraphPad Prism for the data represented in bar graphs (molecular marker and Ca2+ readouts). RT-qPCR results (represented in heat maps) were quantified using the Livak method using three technical replicates and pooled biological samples of all replicates. The Livak method was performed by hand using Microsoft Excel. *P < 0.05 was the threshold for statistical significance. ** stands for P < 0.01, *** stands for P < 0.001, and **** stands for P < 0.0001.Results
Both monomeric Aβ and oligomeric Aβ cause astrocytic atrophy and hyper-reactivity seen in Alzheimer's disease
We created the brain-mimicking extracellular matrix (ECM) hydrogels composed of collagen I (2.5 mg mL−1), hyaluronan (HA, 1.25 mg mL−1), and fibronectin (0.26 mg mL−1) and seeded human primary astrocytes inside the 3D ECM (Fig. 1A). To test the effects of the monomeric Aβ and oligomeric Aβ (the mass characterization of each form can be seen in Fig. S1†) on astrocytes, we seeded astrocytes in the ECM with either no Aβ42, 1 μM monomeric Aβ42, or 1 μM oligomeric Aβ42, and cultured them for three days. Then, we fixed the cells, and immunostained them with phalloidin (F-actin staining), anti-nestin antibodies, and DAPI (Fig. 1B–D). Actin was used to visualize the cytoskeletal morphology of the astrocytes and nestin was used to see astrocyte reactivity. The astrocytes exposed to either monomeric Aβ or oligomeric Aβ experienced atrophy, as quantified by decreased cell areas (Fig. 1E), lower actin signals (Fig. 1F), and increased cell roundness (Fig. 1G). Atrophy is a phenotype experienced by astrocytes in AD, signifying a morphological match to our pathophysiology of interest. Further characterization of the astrocyte reactivity was performed by quantifying the signal of nestin (Fig. 1H). The astrocytes exposed to either monomeric Aβ or oligomeric Aβ showed significantly higher expression of nestin compared to the control group. There were no statistically significant differences between the two Aβ groups in the degree of atrophy or reactivity. Collectively, our data shows that both monomeric Aβ and oligomeric Aβ cause astrocytic atrophy and hyper-reactivity just like pathological astrocytes in AD.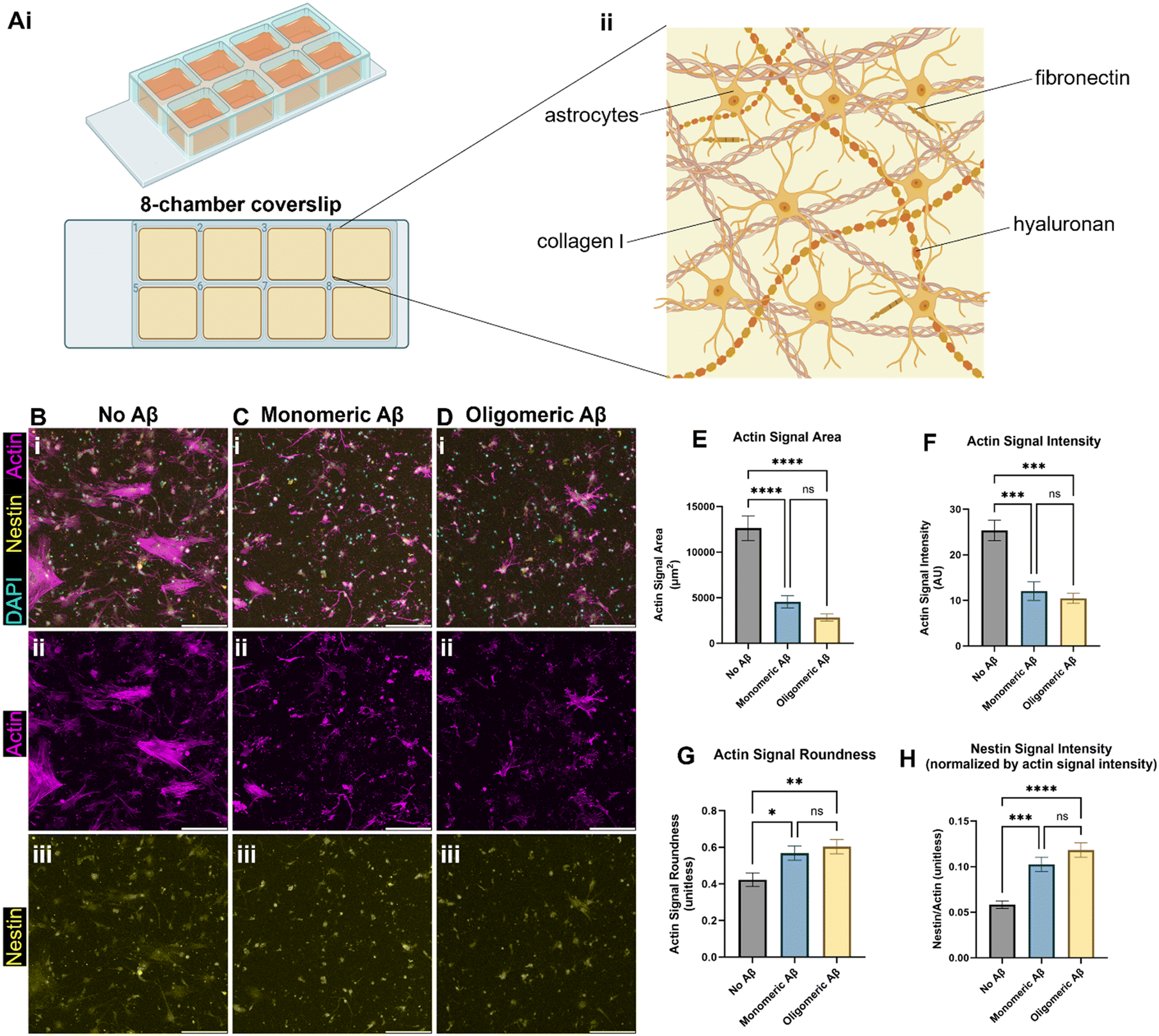 | ||
| Fig. 1 Astrocytic atrophy and reactivity changes in astrocytes exposed to monomeric and oligomeric Aβ. (A) A schematic of our initial 3D culture of astrocytes. (Ai) Human astrocytes were cultured in 3D ECM in chambers on a glass coverslip, (Aii) with brain-mimicking ECM components including collagen I, hyaluronan, and fibronectin. (B) Immunostaining astrocytes with phalloidin (F-actin staining), anti-nestin antibodies, and DAPI. The astrocytes were exposed to either no Aβ (B), monomeric (C), or oligomeric Aβ (D). (E) Changes in actin signal area per cell by monomeric or oligomeric Aβ. (F) Changes in total actin signal intensity by monomeric or oligomeric Aβ. (G) Changes in roundness by monomeric or oligomeric Aβ. (H) Changes in astrocytic reactivity by monomeric or oligomeric Aβ, as measured by nestin signal intensity relative to actin signal intensity. Scale bars (B–D) = 200 μm. * (p < 0.05), ** (p < 0.01), *** (p < 0.001), and **** (p < 0.0001) indicate statistical significance. ns = not significant. | ||
Monomeric Aβ and oligomeric Aβ cause different pathologies related to calcium ion homeostasis and transport in astrocytes
We next investigated alterations in functional metrics such as changes in calcium ion (Ca2+) homeostasis and transport in astrocytes. Altered calcium concentrations are one of the hallmarks of astrocytic responses, such as astrocytic reactivity, which further affect downstream cellular processes.16 Current literature states that in vivo mouse models of AD show increased intracellular Ca2+ and increased Ca2+ transient waves, so we explored a nuclear factor activated by calcineurin (NFAT) that is involved in the calcium signaling system and Ca2+ homeostasis. Calcium signaling activates the phosphatase calcineurin and induces the movement of NFAT into the nucleus.17 We showed that the NFAT1 signal was highly increased when monomeric or oligomeric Aβ was introduced to astrocytes, and white-colored DAPI signals overlapped with NFAT showing NFAT nuclear translocation in the presence of monomeric or oligomeric Aβ (Fig. 2A). When we quantified NFAT1 signal normalized by nuclear signal (DAPI), astrocytes exposed to mAβ had a significant increase in NFAT1 expression compared to the no Aβ control astrocytes (Fig. 2B). Oligomeric Aβ affected NFAT, but the change was not significant.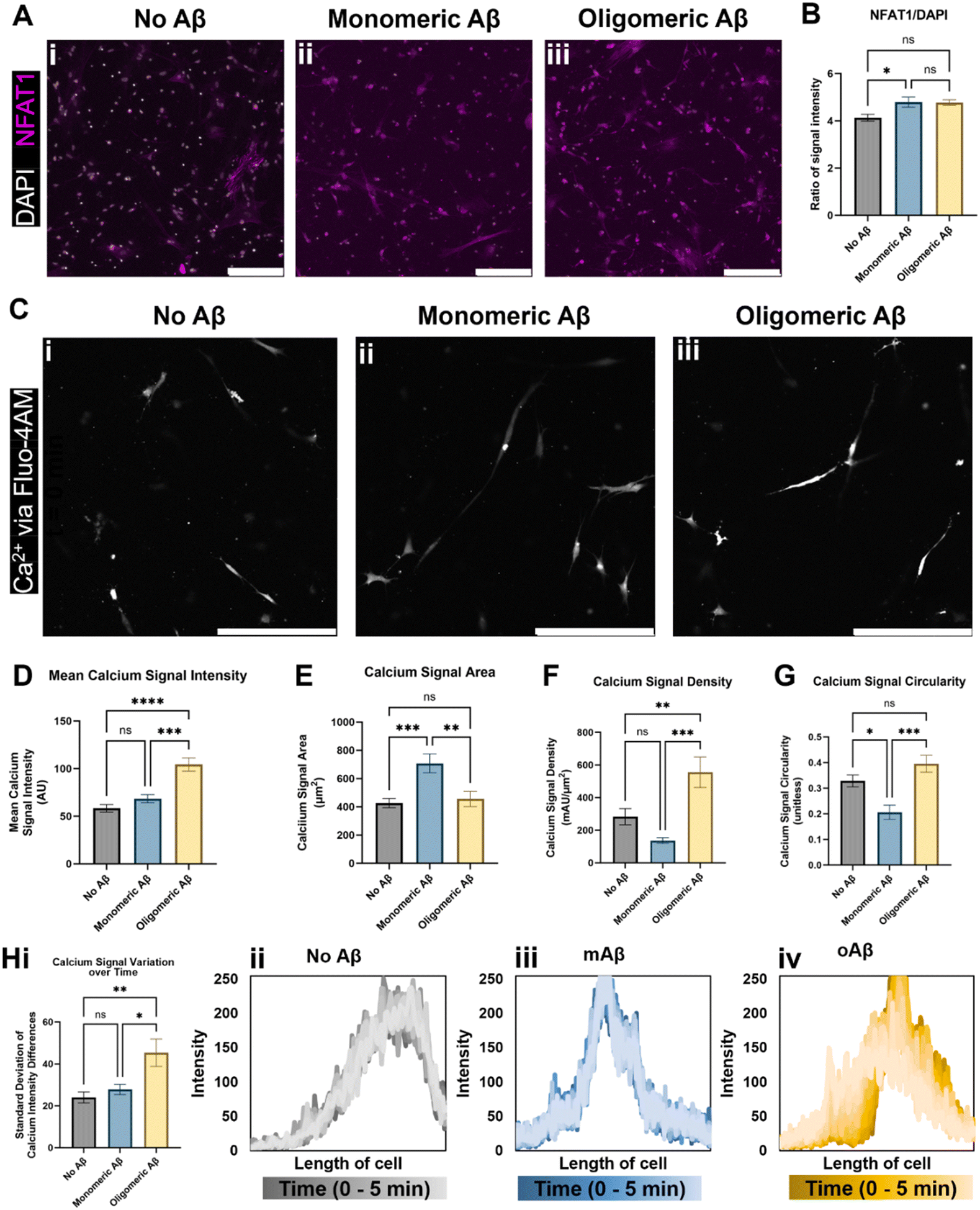 | ||
| Fig. 2 Intracellular Ca2+ changes in astrocytes exposed to monomeric and oligomeric Aβ. Immunofluorescent imaging of the astrocytes (A) indicated an increase in transcription factor NFAT1 (nuclear factor of activated T-cells 1) relative to DAPI signal in astrocytes exposed to (Aii) monomeric Aβ compared to (Aiii) oligomeric Aβ and (Ai) no Aβ exposure. (B) Despite having similar values, astrocytes cultured with oligomeric Aβ did not experience a statistically significant increase in NFAT1, a component of the calcium/calcineurin signaling pathway, while astrocytes cultured with monomeric Aβ did. (C) Live cell calcium ion (Ca2+) imaging revealed intracellular calcium differences in the three groups of astrocytes (Ci–iii). (D) Notably, astrocytes exposed to oligomeric Aβ had a strong increase (∼2-fold) in intracellular Ca2+ signal compared to both other astrocyte conditions. The (E) area, (F) signal intensity relative to signal area, and signal shape parameters of (G) circularity were quantified. In all three quantifications (E–G), astrocytes exposed to monomeric Aβ experienced differences compared to the other conditions. (E) Astrocytes exposed to mAβ had larger Ca2+ signal areas, which led to (F) much lower signal density compared to oAβ astrocytes. The (G) circularity of Ca2+ signals in the mAβ was statistically lower than the other conditions, indicative of a more branched signal pattern, perhaps throughout the entire cell instead of focused in one specific location. (H) Astrocytes exposed to oligomeric Aβ also experienced greater transients in intracellular Ca2+ compared to the other conditions as measured by variation in the signal intensity across the cell over time. The summary figure is shown in Hi while representative traces of the Ca2+ signal across the length of cells are shown in Hii–iv. All traces are shown in Fig. S2,† and the time progression is shown by dark to light color progression (darkest color is time = 0 minutes and lightest color is time = 5 minutes). Both increased intracellular Ca2+ concentration and increased Ca2+ transients are known to be found in AD. Scale bars (A and C) = 200 μm. * (p < 0.05), ** (p < 0.01), *** (p < 0.001), and **** (p < 0.0001) indicate statistical significance. ns = not significant. | ||
To examine Ca2+ fluctuation in the cells in real-time and investigate Ca2+ waves, we used Fluo4-AM to measure the Ca2+ signal in live/unfixed samples in our 3D culture for five minutes each. Representative images from the center of the time period (Fig. 2C) were used to quantify the calcium signal intensity (Fig. 2D), calcium signal area (Fig. 2E), calcium signal density (Fig. 2F), and calcium signal shape via circularity (Fig. 2G). The mAβ group showed the same intensity as the control group, but a greater Ca2+ signal area than either other group, indicating a decrease in calcium signal density throughout the cell body (Fig. 2D–F). The mAβ treated astrocytes also showed a decrease in circularity in the calcium signal compared to either group (Fig. 2G), implying that the Ca2+ is spread more throughout the cell body rather than focused in a certain area. By contrast, the oAβ group experienced a very different pathological change with no statistically significant change in signal area (Fig. 2E) or shape/circularity (Fig. 2G) compared to the control astrocytes without Aβ; however, the astrocytes exposed to oAβ showed a much higher intensity in Ca2+ signal, leading to a signal density statistically significantly higher than that of the control astrocytes (Fig. 2D–F). When the calcium signal over time was measured for calcium intensity differences (fluctuations) (Fig. 2H, i), only the astrocytes exposed to oAβ showed an increase in Ca2+ transient waves (Fig. 2H, iv) compared to those exposed to either no Aβ (Fig. 2H, ii) or mAβ (Fig. 2H, iii). All traces, beyond the representative ones found in Fig. 2H, can be found in Fig. S2.† Taken together, the astrocytes exposed to oAβ matched existing AD in vivo Ca2+ pathophysiology of increased intracellular Ca2+ with increased Ca2+ transient waves, while astrocytes exposed to mAβ showed intermediate pathophysiology in Ca2+ homeostasis before formal AD (as marked by Aβ oligomers) and during the over-aggregation of Aβ42.
Glymphatics-on-chip with interstitial fluid flow properly regulates DAC and AQP4
Although we examined the atrophy, reactivity, and intracellular Ca2+ changes in astrocytes exposed to monomeric and oligomeric Aβ (Fig. 1 and 2), the previous 3D model did not have blood endothelium nor the interstitial fluid (ISF). Glymphatics-on-chip devices were assembled as established in our previous study15 (Fig. 3A). The device has two parallel, cylindrical hollow channels embedded in the brain-mimicking ECM hydrogel that contains astrocytes. The hollow channels were seeded with endothelial cells by introducing endothelial cell suspension to the circular media reservoirs to model the glymphatic system of arterial and venous endothelium surrounded by astrocytes (Fig. 3A, i). We then introduced ISF into the device by adding excess media to the reservoirs connected to the left side channel, driving a convective ISF flow to the right side channel (Fig. 3A, ii). The device was immunostained with anti-CD31 and anti-GFAP antibodies to visualize 3D-engineered blood vessels and astrocytes in the ECM (Fig. 3B). Enlarged CD31 images show the rudimentary engineered blood vessel (Fig. 3C). Collectively, the device models the glymphatic system of arterial and venous endothelium surrounded by astrocytes, experiencing ISF flow from the arterial to venous endothelium (Fig. 3D).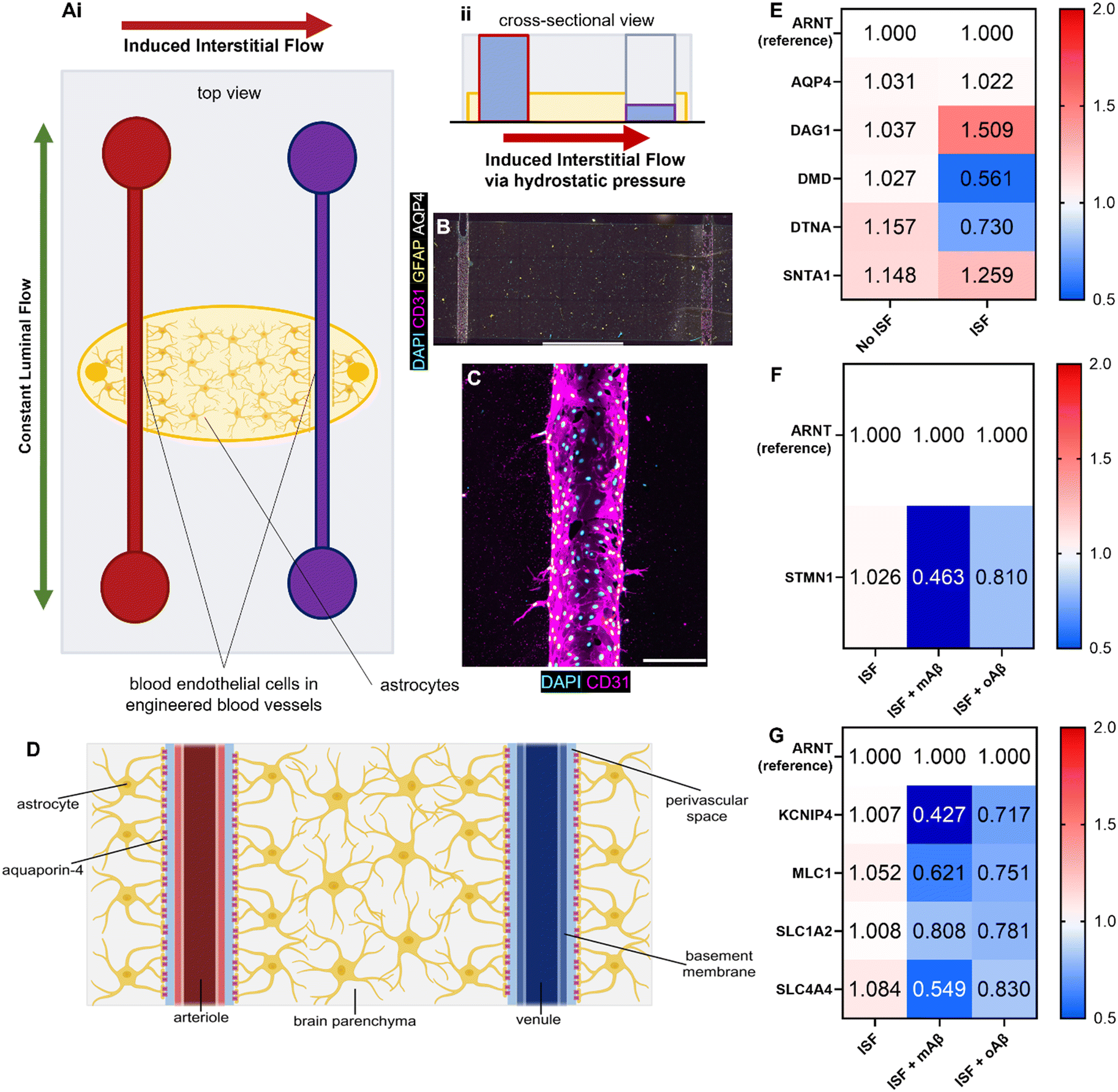 | ||
| Fig. 3 Glymphatics-on-chip model with RT-qPCR results. (Ai) The top view of the glymphatics-on-chip model illustrates the two directionalities of microfluidic flow. Luminal fluid flow (green arrow) is bidirectional throughout the chip. Convective interstitial fluid flow (red arrow) is induced from the left channel to the right channel, mirroring the periarterial to perivenous directionality of the glymphatic system. (Aii) The glymphatics-on-chip model's cross-sectional view illustrates how the interstitial fluid flow is induced via hydrostatic pressure differences in the media reservoirs. (B) Immunofluorescence microscopy image of the microfluidic glymphatics-on-chip model. It has two channels seeded with human primary blood endothelial cells (BECs, expressing CD31) embedded in a biomimetic ECM reservoir laden with human primary astrocytes (expressing GFAP throughout and AQP4 at their endfeet). Amyloid-β can be seeded into the ECM, drug treatment can be perfused via media through the two channels, and interstitial fluid flow can be induced from one channel to the other. (C) An immunofluorescence microscopy image of the 3D engineered blood vessel in the glymphatics-on-chip. (D) The gross structure of the glymphatic system, which mirrors our glymphatics-on-chip model. (E) RT-qPCR results illustrating different regulations of the DAC components after our device was exposed to 16 hours of inducible ISF compared to no ISF. ARNT, a reference gene; AQP4 (aquaporin-4), the water pore anchored by the complex; DAG1 (dystroglycan 1), one of the transmembrane and extracellular components of the complex; DMD (dystrophin), the protein that links the transmembrane components to the intracellular cytoskeleton; DTNA (dystrobrevin), a protein that binds to dystrophin and allows the formation of the dystrobrevin/syntrophin triplet; SNTA1 (α-1-syntrophin), a protein that binds to both dystrophin and dystrobrevin and is involved in signal coordination for multiple molecules. (F) RT-qPCR results illustrating different regulations of the cytoskeletal component, STMMN1, a protein involved in destabilizing microtubules, in the presence of ISF with or without mAβ or oAβ. (G) RT-qPCR results illustrating different regulations of the ion channel and transporters in the presence of ISF with or without mAβ or oAβ. KCNIP4, a voltage-gated K+ channel-interacting protein 4; MLC1, a membrane transport protein expressed in astrocytes; SLC1A2, a glutamate transporter; SLC4A4, a sodium bicarbonate transporter. Scale bars (B) = 2 mm, (C) = 200 μm. | ||
Aquaporin-4 (AQP4) is a critical component of the glymphatic system and is believed to be involved in the convective ISF flow responsible for brain waste clearance. The dystrophin-associated complex (DAC) anchors AQP4 to the cell membrane and is believed to be involved in AQP4 polarization, so before introducing Aβ to our devices, we explored how the presence or absence of inducible ISF affected the expression of DAC components via RT-qPCR with ARNT (aryl hydrocarbon receptor nuclear translocator) expression as reference (Fig. 3E). Specifically, we measured the expression of AQP4 and the following DAC components: dystroglycan-1 (DAG1), one of the transmembrane and extracellular components of the complex; dystrophin (DMD), the protein that links the transmembrane components to the intracellular cytoskeleton; dystrobrevin (DTNA), a protein that binds to dystrophin and allows the formation of the dystrobrevin/syntrophin triplet; α-1-syntrophin (SNTA1), a protein that binds to both dystrophin and dystrobrevin and is involved in signal coordination for multiple molecules.18 Notably, we saw trends of upregulation in DAG1, no major change in AQP4 or SNTA1, and 2-fold downregulation of DMD as well as lesser downregulation of DTNA. Taken together, our device recapitulated that the components of the dystrophin-associated complex (DAC) and AQP4 were properly maintained by the ISF, compared to no flow controls, though not statistically significantly (Fig. S4A†).
Glymphatics-on-chip with interstitial fluid flow shows the effects of mAβ and oAβ on astrocytic atrophy and ion channels in AD
We next treated monomeric and oligomeric Aβ (mAβ and oAβ) in the glymphatic-on-chip model to examine the roles of mAβ and oAβ in AD. We first examined STMN1 (stathmin 1), a ubiquitous cytosolic phosphoprotein that is involved in the regulation of the microtubule filament system of the cell cytoskeleton (Fig. 3F) given our finding that mAβ and oAβ caused astrocytic atrophy in Fig. 1. With ARNT as the reference between groups, we showed a trend of lower expression of STMN1 in the oAβ group compared to the control and statistically significantly lower STMN1 expression in the mAβ group compared to the control (Fig. 3F, statistics in Fig. S4B†), matching known astrocytic atrophy in AD, specifically between no AD and early stage AD ex vivo human samples.19Based on the findings in Fig. 2 that astrocytes exposed to oAβ and mAβ matched existing AD in vivo Ca2+ pathophysiology, other ions transport elements were measured via RT-qPCR of the cells in the microfluidic glymphatics-on-chip device immediately following 16 hours of ISF with or without mAβ and oAβ. With ARNT as the reference between groups, a voltage-gated K+ channel-interacting protein 4 (KCNIP4), a membrane transport protein expressed in astrocytes (MLC1), a glutamate transporter (SLC1A2), and sodium bicarbonate transporter (SLC4A4) expression levels were measured (Fig. 3G). All transport components showed downregulation trends compared to the healthy control. As with the Ca2+ signal, distinct differences compared to either the control group or the other Aβ group were seen in all ion transporters except MLC1 and SLC1A2, both of which still showed downregulation compared to the no Aβ control. KCNIP4, which is responsible for K+ homeostasis/transport, was downregulated in both conditions with over 2-fold downregulation in the mAβ condition while the oAβ astrocytes still had ∼1.5-fold downregulation compared to the no Aβ control. The glutamate and sodium bicarbonate transporters SLC1A2 and SLC4A4 showed different trends between the mAβ and oAβ groups. SLC1A2 was mildly downregulated in both Aβ groups. SLC4A4 was downregulated in both groups, but it was markedly more downregulated in the mAβ group than in the oAβ group. Statistics for this work can be found in Fig. S4C.† This work illustrates that monomeric Aβ may play a stronger role in many ion transporters and other transporters, but that both oligomeric and monomeric Aβ are responsible for some of the accepted ion dyshomeostasis of in vivo mouse and ex vivo human samples in AD, but in separate yet similar ways. Taken together, glymphatics-on-chip with interstitial fluid flow shows the effects of mAβ and oAβ on astrocytic atrophy and ion channels in AD.
Glymphatics-on-chip with interstitial fluid flow shows the effects of mAβ and oAβ on AQP4 polarization and DAC regulation
AQP4 is a critical component of the glymphatic system and is also known to be spatially depolarized in AD. We investigated the effects of mAβ and oAβ on AQP4 expression, including polarization, and GFAP expression using our glymphatics-on-chip system. The glymphatic system under flow was treated with mAβ and oAβ for three days. We then fixed the devices and performed immunostaining (Fig. 4A): staining for nuclei (DAPI), endothelial cells (CD31) to visualize our 3D engineered BVs, glial fibrillar acidic protein (GFAP) for some astrocytes, and AQP4. In the control group without Aβ, we saw more AQP4 polarization in the venous-side endothelium compared to the arterial-side endothelium (Fig. 4A, i). Strikingly, treatment with monomeric Aβ diminished the AQP4 spatial distribution as PVS polarizational directionality, by increasing AQP4 expression on the arterial side and decreasing AQP4 expression on the venous side (Fig. 4A, ii). Treatment with oligomeric Aβ overall decreased the expression of AQP4 on both sides and further eliminated the AQP4 polarization (Fig. 4A, iii). To perform polarization quantification, each image was split into five equal-sized portions, or “quintiles”, labeled 1–5 from left to right (Fig. 4B). In this system, portions 1 and 5 should be considered periarterial and perivenous, respectively, which is ideally where we would hope to see AQP4 signal in the polarized condition. By comparing the AQP4 signal in each quintile of the image to the total AQP4 signal, we could quantify the AQP4 polarization in each condition (Fig. 4C). Total depolarization would show a value of 20% (as indicated by the dashed line) from each quintile (100% ÷ 5 portions = 20% each portion), illustrating equal expression across the device. AQP4 polarization was seen in the ISF no Aβ control, with some depolarization in the mAβ group and nearly full depolarization (no polarization) in the oAβ group, implying that both monomeric and oligomeric Aβ play a role in AQP4 depolarization seen in AD. GFAP signal polarization was also quantified (Fig. 4D), but all three groups illustrated GFAP polarization, supporting the recent trend in the field to prove astrocyte pathology beyond simple GFAP staining. Total area fraction of both AQP4 (Fig. S3A†) and GFAP (Fig. S3B†) was compared between all three groups. There were no statistically significant differences between any of the groups, though the ratio of AQP4 total area fraction to GFAP total area fraction was greater in the group without Aβ compared to the group with mAβ and there were no statistically significant differences between the group with oAβ compared to either of the other groups (Fig. S3C†). This implies that the depolarization seen in the mAβ group is not simply due to lower expression of AQP4, but lower expression may play a role in the depolarization when exposed to oAβ. Either way, it invites further investigation.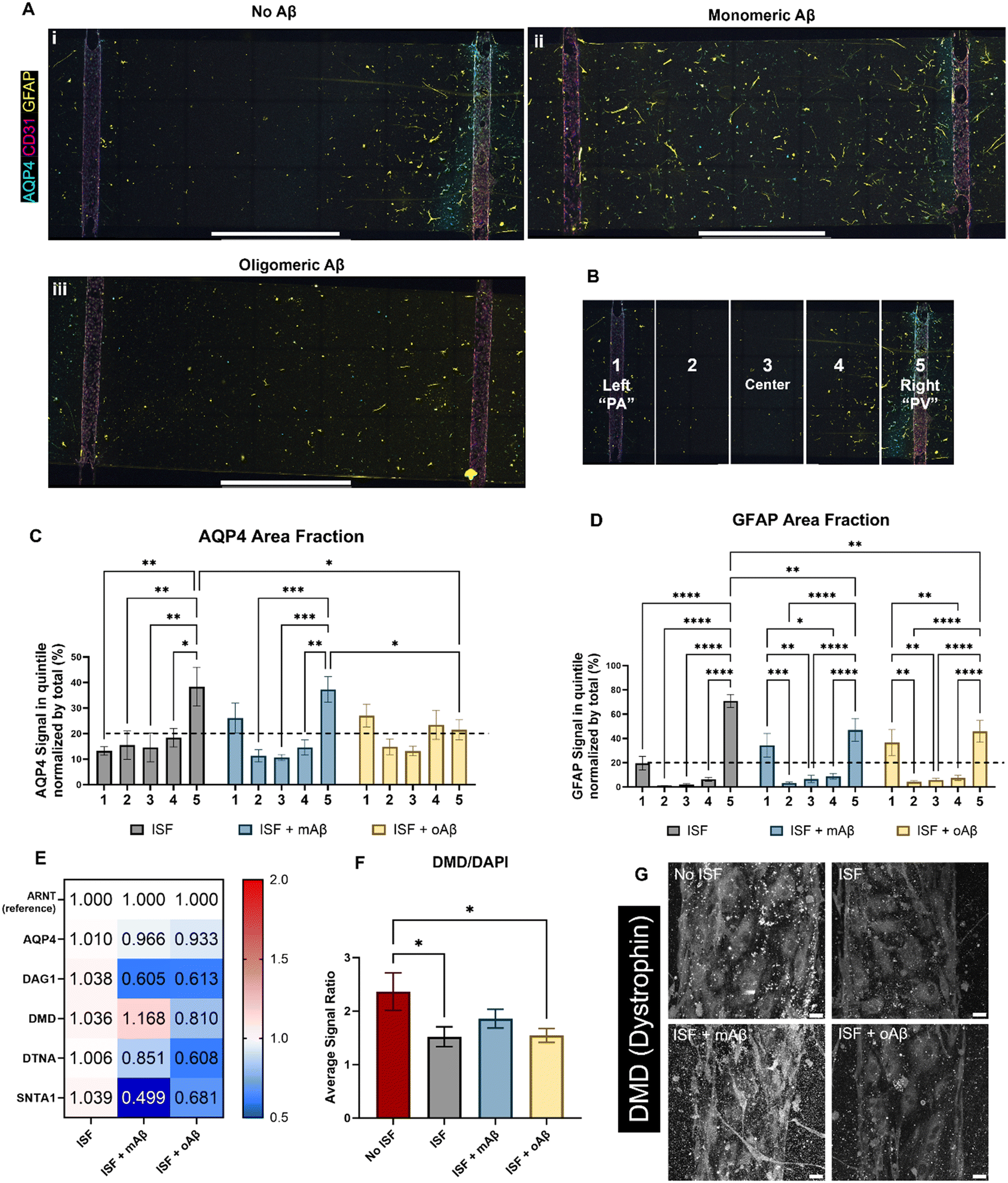 | ||
| Fig. 4 Glymphatics-on-chip system reveals spatial protein expression and RNA expression differences as a co-culture system. Following 16 hours of induced interstitial flow in the co-culture glymphatics-on-chip system, the samples were either prepared for immunofluorescent microscopy (A) or used for RT-qPCR (E). (A) Qualitative analysis of the IF images seems to show AQP4 polarization, especially to the right channel (“PV”) in the no Aβ control (Ai). Distinct GFAP and CD31 signals indicate that the astrocytes and BECs, respectively, maintained their identity in the co-culture devices across all treatment groups (Ai–iii). (B) For quantification, each image was split into five equal-width portions and labeled 1–5, left to right. Portions 1 and 5 are considered perivascular. (C) Quantitative analysis of the AQP4 area fractions of the ECM reservoir shows AQP4 polarization in the ISF no Aβ control group while values closer to 20% (dashed line) across all portions, indicating depolarization, were found in the two groups exposed to Aβ. (D) GFAP expression patterns were fairly consistent across all groups, with polarization in all groups. (E) RT-qPCR results of AQP4 and the DAC components, including DAG1 (dystroglycan), DMD (dystrophin), DTNA (dystrobrevin), and SNTA1 (α-1-syntrophin) with ARNT as the reference gene/protein. (F) Quantitative analysis of DMD (dystrophin) expression via IF imaging, normalized by DAPI expression in each image. (G) Representative images of DMD expression in the three ISF groups and the no ISF control. Scale bars (A) = 2 mm, (G) = 25 μm. * (p < 0.05), ** (p < 0.01), *** (p < 0.001), and **** (p < 0.0001) indicate statistical significance. ns = not significant. | ||
For mechanistic analysis, we performed RT-qPCR with the devices comparing the expression of different components of the DAC (Fig. 4E), with ARNT as the reference gene. In general, more dramatic differences were observed in the mAβ group than the oAβ group, but both showed important trends compared to the no Aβ control ISF group (statistics in Fig. S4D†). Despite the control and mAβ groups showing higher polarization of AQP4 than the oAβ group, both Aβ groups showed little to no downregulation in AQP4. DAG1 was nearly 2-fold downregulated in both Aβ groups. DMD was slightly upregulated in the mAβ group and slightly downregulated in the oAβ group. DTNA, like DAG1, was downregulated in both groups compared to the no Aβ ISF control but had a greater downregulation in the oAβ group compared than the mAβ group. SNTA1, however, was 2-fold and statistically significantly more downregulated in the mAβ group than the control, though both were downregulated compared to the no Aβ control ISF group. Interestingly, the DAC component expression changes in the mAβ + ISF group compared to ISF were nearly perfectly inverted of the DAC component expression changes in the ISF group compared to the no ISF control, with the exception of DTNA, while the oAβ group simply had trends of downregulation for every DAC component compared to the no Aβ ISF control. Finally, we stained for DMD expression in the vascular space of all three ISF groups and the no ISF control (Fig. 4F and G). While the mAβ group did not demonstrate statistically significantly more DMD than the ISF control or the oAβ group, it was the only group that did not have statistically significantly less DMD than the no ISF control, implying that it had similar levels of DMD to the no ISF control. Much like the ion homeostasis and transporter data, astrocytes exposed to either mAβ and oAβ both showed distinct AD pathologies different from the control and the other Aβ group, demonstrating that the AD pathophysiology largely attributed to Aβ plaques may be more complicated than simply the presence or absence of oligomeric Aβ. Taken together, glymphatics-on-chip with interstitial fluid flow shows the effects of mAβ and oAβ on AQP4 polarization and DAC regulation.
Discussion
After our previous work designing a preliminary glymphatics-on-chip model,15 we made some key improvements to the device to ensure more consistency between chips and enhanced biomimicry. These changes included changing the type of human blood endothelial cells used, reducing the concentration of human astrocytes suspended in our ECM, and changing the composition of our ECM by removing Matrigel, adding fibronectin, and changing the concentration of hyaluronan to better mimic natural human brain tissue. The previous model included Matrigel, which contains growth factors of unknown amounts and types since it is sourced from murine sarcoma. The Matrigel may promote angiogenesis, which leads to leaks and compromises the tight endothelium characteristic of cortical blood vessels, so we removed the Matrigel. Tunesi et al. created a brain ECM hydrogel with 1.2 mg mL−1 and 2.5 mg mL−1 collagen and HA, respectively.20 This inspired us to greatly increase the HA content, nearly doubling it from 0.645 mg mL−1 to 1.2 mg mL−1. We decided against adjusting the collagen since proper vessel morphology is a high priority in this model of perivascular space, and 2.5 mg ml−1 collagen has always supported our endothelium well in the models produced by our lab. We also included fibronectin in one of the new ECM formulations for two reasons. First and foremost, fibronectin is a key component of the brain ECM for its neuroprotective role.21 Secondly, from our own research, fibronectin tightens junctions,22 which could help provide a tight endothelium in our model. While our model is not a blood–brain–barrier (BBB) model, a fibronectin-rich ECM is paramount to forming the basement membrane of blood vessels in the brain and a key component of the BBB.23 Lam et al. state that the brain ECM is largely composed of glycosaminoglycans (like HA) with lower levels of fibrillar proteins (like collagen),24 so increasing the HA content also promotes the creation of a biomimetic ECM.With a more reliable model, we began characterizing the differences between healthy and AD-like pathology via the addition of a human recombinant Aβ42 in our model as monomeric or oligomeric Aβ42. Peng et al. demonstrated differences between monomeric Aβ40 and oligomeric Aβ42 in their in vivo mouse models.25 However, we cannot compare monomeric Aβ40 and oligomeric Aβ42 fairly because the Aβ40 monomer is chemically different from the Aβ42 monomer. Since most in vivo models showed soluble forms of Aβ42 namely monomers, and oligomers, are the most pathological in AD, but could not distinguish the effects of the monomeric and oligomeric Aβ42, we decided to include either monomeric or oligomeric Aβ42 at the same sub-lethal concentration in our study. Current studies suggest that hexamers and dodecamers of Aβ42 are the most neurotoxic isoforms of Aβ, but soluble Aβ typically exists as a combination of isoforms with continuous aggregation and degradation. Aβ monomers and small oligomers have natural structures primarily focused on α-helices with conversions to β-sheets at the dodecamer level as the Aβ begins to form fibrils. Aβ42 tetramers have a flexible bending structure that allows the addition of monomers or dimers to form ring-like pentamers and planar hexagonal hexamers, which can further stack to create the dodecamer and larger structures.26 Unfortunately, most research on the effects of different Aβ isoforms has been focused on their interactions with neurons rather than glial cells like the astrocytes we have in our model. With that said, Aβ dimers have been shown to accumulate in the lipid rafts of cell membranes27 and have been associated with memory loss and impairments.28 Aβ trimers can trigger aberrant cell signaling, reducing electrical activity in synapses,29 with their very strong ability to attenuate long-term potentiation in neurons via glutamate release by astrocytes.30 Aβ oligomer internalization, in neurons, can lead to decreased cell viability through mediating calcium imbalance31 and other systemic dysfunction. Aβ oligomer internalization may occur through a variety of pathways with astrocytes, including when astrocytes attempt to clear Aβ via phagocytosis32 or if Aβ induces membrane perforation in astrocytes, as it does in neuronal and mitochondrial membranes.33 With monomeric and oligomeric Aβ exhibiting similar fibril structures, we decided to investigate both forms in our study.
First, we validated the astrocyte pathology in either of our Aβ models following standards set through the consensus statement by Escartin et al.34 and assayed AD-like astrocyte pathology beyond morphology through a variety of functional readouts in our in vitro models. Astrocytic atrophy and cytoskeletal dysregulation are known phenomena in AD astrocytes,34 including the downregulation of stathmin 1,19 a protein involved in cytoskeletal regulation. Via phalloidin signal intensity and shape parameter quantification (Fig. 1E–G), nestin signal quantification (Fig. 1H), and stathmin 1 protein expression via RT-qPCR (Fig. 3F), we characterized that the cytoskeleton of astrocytes exposed to either mAβ or oAβ both demonstrated AD pathology. This validates our model and further suggests that the morphological and cytoskeletal changes seen in AD astrocytes depend more on the mere presence of Aβ42 rather than specifically oligomers of Aβ42.
Monomeric and oligomeric Aβ exposure produced distinct changes in molecular markers and functional readouts related to ions and other small molecules and their transporters. With Ca2+, mAβ matched AD with NFAT1 increase (Fig. 2B), but oAβ matched AD with increased intracellular calcium concentration (Fig. 2D and F) and transients (Fig. 2H) according to the AD parameters stated in the consensus statement by Escartin et al.34 mAβ astrocytes showed a potentially different phenotype not previously recorded in AD literature in which there was a widespread calcium signal rather than the more centralized signal seen in either the no Aβ control or the oAβ exposed astrocytes (Fig. 2E and G) without the significant calcium signal fluctuations (Fig. 2H). Now, a downside of our model is that there is not a temporal component to fully characterize the transition from astrocytes exposed primarily to mAβ to astrocytes primarily exposed to oAβ, but the widespread intracellular Ca2+ signal seen in astrocytes exposed to mAβ may be an intermediate pathological state before the traditional changes in Ca2+ seen in AD in vivo models and our in vitro astrocytes exposed to oAβ. The differences seen in proteins related to the transport of other ions and small molecules also brought some interesting differences between the two Aβ groups. For example, KCNIP4, which is a voltage-gated K+ ion channel-interacting protein and is therefore responsible for K+ homeostasis/transport, was downregulated in both Aβ groups but much more so in the mAβ condition (Fig. 3G), matching early stage AD ex vivo human samples compared to no AD samples;19 this effect from mAβ may potentially support the hypothesis that over-accumulation of monomeric Aβ42 leads to toxic Aβ42 plaques, which are the primary hallmarks of AD staging. However, some Aβ readouts seemed less intuitive when compared to post-mortem AD patient samples. SLC1A2 was upregulated in late AD compared to early AD in ex vivo human brain samples,19 so our data showing SLC1A2 slight downregulation in astrocytes exposed to either Aβ species category compared to control astrocytes (Fig. 3G) supports further discussion of the best way to characterize AD stages in patients and questions our belief that only Aβ oligomers and plaques are responsible for even late-stage AD pathology.
The AQP4 and DAC data also showed intriguing differences between the two Aβ groups. Similarly to the Ca2+ data that potentially implied an intermediate state in the astrocytes exposed to mAβ compared to known AD pathophysiology demonstrated by astrocytes exposed to oAβ, AQP4 depolarization, a well-characterized phenomenon in both human AD patient post-mortem samples35 and in vivo mouse models,36 was seen only in the astrocytes exposed to oAβ (Fig. 4C) while a potential precursor, changes in DAC expression, was seen stronger in astrocytes exposed to mAβ (Fig. 4E). This statement must include the caveats that our work (1) does not prove temporal transitions and (2) did not investigate—and therefore prove—DAC involvement in AQP4 depolarization via knockout, knockdown, or pharmaceutical inhibition, so saying that decreased DAC protein expression precedes AQP4 depolarization is simply a hypothesis. Further work would need to be performed to support this hypothesis. Work by Hablitz et al. has shown that DAG1, DTNA, and AQP4 were upregulated during greater AQP4 polarization and associated with the portion of the circadian rhythm during sleep in mice.13 Within our model, only one of those components (DAG1) was also upregulated after 16 hours of induced ISF compared to the no ISF control chips, but interestingly, SNTA was another component with a trend of upregulation (Fig. 3E). The primary downregulated component was DMD, though DTNA also illustrated a trend of downregulation. These results were nearly an inversion of the results seen in the Aβ conditions (Fig. 4E), especially in mAβ + ISF group compared to no Aβ + ISF control. DAG1, DTNA, and SNTA1 were all downregulated in both Aβ conditions, and SNTA1 was the component we saw downregulated the most in both Aβ conditions, with statistical significance in the mAβ condition. SNTA1 is an adaptor protein, and AQP4 anchors to the membrane through the connection of its carboxyl terminus to SNTA1.37 Mouse studies have shown that the knockdown of α-syntrophin results in decreased bidirectional water flow in the brain37 and decreased polarization of AQP4 to the endfeet of astrocytes in the perivascular space.38 α-Syntrophin was also found to be the central organizer of the astrocyte dystrophin complex since its presence was found to be required for the proper localization of dystrophin.38 The single component upregulated in any Aβ condition was dystrophin (DMD), which was slightly upregulated in only the astrocytes exposed to mAβ. Additionally, DMD protein was statistically significantly more expressed in the no ISF control than the ISF and ISF + oAβ conditions, but with no statistically significant difference with the ISF + mAβ condition (Fig. 4F). Most studies of dystrophin have been focused on its role in Duchenne muscular dystrophy, a disease of progressive muscular degeneration with 1/3 of patients experiencing cognitive deficits caused by mutations in the DMD gene. Lange et al. used human induced pluripotent stem cells (iPSCs) of Duchenne muscular dystrophy patients to create 2D and 3D astrocyte in vitro cultures and found that the mutated DMD astrocytes had behavioral and metabolic defects compared to control astrocytes.39 Their work paired with our current findings encourages further research into the involvement of DMD expression and mutations in AQP4 polarization and AD.
The work contained in this article demonstrates that our model is a promising model for highly detailed and mechanistic work in the glymphatic system and AD pathology, with all results summarized from both the preliminary suspension model and the organ-on-chip ISF model in Table 3. Like many other in vitro models, it has numerous advantages and disadvantages over the traditional in vivo models. Working directly with human biology rather than murine analogs, easy access for visualization via microscopy, and the ability to modify the model in very specific ways are some of the most apparent advantages. The disadvantages are mentioned in the limitations paragraph below. Some of these disadvantages could be addressed in future models, but our current model still served its purpose by investigating changes in astrocytes and the perivascular expression of AQP4 in the glymphatic system despite its simplicity.
| AD characteristic | mAβ | oAβ |
|---|---|---|
| Cytoskeletal changes: atrophy, nestin, & stathmin 1 | Matched AD in suspension and ISF models | Matched AD, particularly in suspension model |
| Ca2+: NFAT, increased intracellular concentration, increased transient waves | NFAT matched AD, intermediate Ca2+ (both in suspension model) | Ca2+ matched AD (suspension model) |
| Ion & molecular transport | Matched early AD pathology (ISF model) | Only slight changes (ISF model) |
| AQP4 depolarization | No change (ISF model) | Matched AD (ISF model) |
| DAC downregulation | Matched no ISF (ISF model) | Slightly matched no ISF (ISF model) |
Limitations of the study
As mentioned above, in vitro models have some inherent advantages and disadvantages compared to other model types, and our model is no exception. Disadvantages of our model include that the model is manufactured rather than naturally formed and that it simplifies the complexities of the human brain, missing key components such as neurons, blood circulation, CSF influx, and any immune system to respond to inflammation. It uses a mixture of sources for the ECM components, including rat tail, so despite the use of human cells and human Aβ, it is not entirely human. The BECs used in our model are dermally sourced rather than from human brain tissue. However, this may be a minor issue since primary organ-specific BECs lose their organ-specificity after expansion.40 As addressed in the discussion section, our study investigates the differences between monomeric and oligomeric Aβ42. It does not include Aβ40, which is a key source of monomeric Aβ in vivo, and it does not include a temporal component to visualize how/if the transition of Aβ42 from monomers to oligomers, including a mixture of the two forms, affects astrocytes or the glymphatic system. Finally, it bears to be repeated that our work implying DAC involvement in AQP4 depolarization was not proven via knockout, knockdown, or pharmaceutical inhibition, so saying that decreased DAC protein expression precedes AQP4 depolarization is simply a hypothesis to be further investigated in future work.Conclusions
Our study demonstrates the utility of our biomimetic 3D in vitro models, including a microfluidic glymphatics-on-chip model, by allowing characterizing and mechanistic studies into the specific effects caused by either monomeric or oligomeric Aβ on astrocytes and the polarization of AQP4 and downregulation of DAC components in the glymphatic system. Additionally, our work demonstrated an upregulation of DAC components in the presence of interstitial fluid flow, a feat currently only possible in an in vitro system rather than existing models and samples. Our model is validated by data from both in vivo mouse models and post-mortem samples from human Alzheimer's disease patients, and it provides a new tool in the study of both healthy glymphatic function and glymphatic dysfunction in Alzheimer's disease.Data availability
All source data are available in the main text or the supplementary materials. Additional data are available from the corresponding authors upon reasonable request.Author contributions
A. R. Y. designed and performed most of the experiments as well as wrote the draft of the manuscript, excluding the introduction, and created the figs. R. P. assisted in the experiments, wrote the draft of the introduction of the manuscript, and reviewed the manuscript. E. L. advised A. R. Y. and R. P. in experimental design and performance and provided funding. E. L. and N. N. gave a final review of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
National Science Foundation Graduate Research Fellowship Program for Aria R. Yslas (DGE-2139899), Cornell Engineering Learning Initiatives Student Grant Program for Rena Park, Mong Junior Cornell Neurotech Fellowship 2020, National Institute of Health grants (AI166772, HL165135). PDMS chip molds are manufactured at the Cornell CNF by Renhao Lu, a current Lee Lab member.Notes and references
- L. S. Schneider, Psychopharmacol. Bull., 2023, 53, 8–65 Search PubMed.
- V. J. De-Paula, M. Radanovic, B. S. Diniz and O. V. Forlenza, Subcell. Biochem., 2012, 65, 329–352 CrossRef CAS PubMed.
- Y. Huang, R. Potter, W. Sigurdson, A. Santacruz, S. Shih, Y. E. Ju, T. Kasten, J. C. Morris, M. Mintun, S. Duntley and R. J. Bateman, Arch. Neurol., 2012, 69, 51–58 CrossRef PubMed.
- T. Iwatsubo, A. Odaka, N. Suzuki, H. Mizusawa, N. Nukina and Y. Ihara, Neuron, 1994, 13, 45–53 CrossRef CAS.
- U. Sehar, P. Rawat, A. P. Reddy, J. Kopel and P. H. Reddy, Int. J. Mol. Sci., 2022, 23, 12924 CrossRef CAS PubMed.
- R. Hussain, U. Graham, A. Elder and M. Nedergaard, Trends Neurosci., 2023, 46, 901–911 CrossRef CAS.
- A. Louveau, B. A. Plog, S. Antila, K. Alitalo, M. Nedergaard and J. Kipnis, J. Clin. Invest., 2017, 127, 3210–3219 CrossRef.
- J. J. Iliff, M. Wang, Y. Liao, B. A. Plogg, W. Peng, G. A. Gundersen, H. Benveniste, G. E. Vates, R. Deane, S. A. Goldman, E. A. Nagelhus and M. Nedergaard, Sci. Transl. Med., 2012, 4, 147ra111 Search PubMed.
- B. T. Kress, J. J. Iliff, M. Xia, M. Wang, H. S. Wei, D. Zeppenfeld, L. Xie, H. Kang, Q. Xu, J. A. Liew, B. A. Plog, F. Ding, R. Deane and M. Nedergaard, Ann. Neurol., 2014, 76, 845–861 CrossRef CAS PubMed.
- H. Mestre, L. M. Hablitz, A. L. Xavier, W. Feng, W. Zou, T. Pu, H. Monai, G. Murlidharan, R. M. Castellanos Rivera, M. J. Simon, M. M. Pike, V. Pla, T. Du, B. T. Kress, X. Wang, B. A. Plog, A. S. Thrane, I. Lundgaard, Y. Abe, M. Yasui, J. H. Thomas, M. Xiao, H. Hirase, A. Asokan, J. J. Iliff and M. Nedergaard, eLife, 2018, 7, e40070 CrossRef.
- S. Mader and L. Brimberg, Cells, 2019, 8, 90 CrossRef CAS PubMed.
- E. A. Nagelhus and O. P. Ottersen, Physiol. Rev., 2013, 93, 1543–1562 CrossRef CAS PubMed.
- L. M. Hablitz, V. Pla, M. Giannetto, H. S. Vinitsky, F. F. Staeger, T. Metcalfe, R. Nguyen, A. Benrais and M. Nedergaard, Nat. Commun., 2020, 11, 4411 CrossRef CAS PubMed.
- W. B. Stine, L. Jungbauer, C. Yu and M. J. LaDu, Methods Mol. Biol., 2011, 670, 13–32 CrossRef CAS PubMed.
- P. A. Soden, A. R. Henderson and E. Lee, Adv. Biol., 2022, 6, e2200027 CrossRef.
- C. Agulhon, J. Petravicz, A. B. McMullen, E. J. Sweger, S. K. Minton, S. R. Taves, K. B. Casper, T. A. Fiacco and K. D. McCarthy, Neuron, 2008, 59, 932–946 CrossRef CAS PubMed.
- G. R. Crabtree and E. N. Olson, Cell, 2002, 109(Issue 2, Suppl 1), S67–S79 CrossRef CAS PubMed.
- Q. Q. Gao and E. M. McNally, Compr. Physiol., 2015, 5, 1223–1239 Search PubMed.
- H. Mathys, J. Davila-Velderrain, Z. Peng, F. Gao, S. Mohammadi, J. Z. Young, M. Menon, L. He, F. Abdurrob, X. Jiang, A. J. Martorell, R. M. Ransohoff, B. P. Hafler, D. A. Bennett, M. Kellis and L. H. Tsai, Nature, 2019, 570, 332–337 CrossRef CAS PubMed.
- M. Tunesi, L. Izzo, I. Raimondi, D. Albani and C. Giordano, J. Tissue Eng., 2020, 11, 2041731420945633 Search PubMed.
- J. Wang, L. Yin and Z. Chen, Neural Regener. Res., 2013, 8, 376–382 Search PubMed.
- A. R. Henderson, I. S. Ilan and E. Lee, Microcirculation, 2021, 28, e12730 CrossRef CAS.
- K. M. Baeten and K. Akassoglou, Dev. Neurobiol., 2011, 71, 1018–1039 CrossRef CAS PubMed.
- D. Lam, H. A. Enright, J. Cadena, S. K. G. Peters, A. P. Sales, J. J. Osburn, D. A. Soscia, K. S. Kulp, E. K. Wheeler and N. O. Fischer, Sci. Rep., 2019, 9, 4159 CrossRef PubMed.
- W. Peng, T. M. Achariyar, B. Li, Y. Liao, H. Mestre, E. Hitomi, S. Regan, T. Kasper, S. Peng, F. Ding, H. Benveniste, M. Nedergaard and R. Deane, Neurobiol. Dis., 2016, 93, 215–225 CrossRef CAS PubMed.
- H. Yang, J. Li, X. Li, L. Ma, M. Hou, H. Zhou and R. Zhou, Front. Mol. Neurosci., 2022, 15, 927530 CrossRef CAS PubMed.
- T. Kawarabayashi, M. Shoji, L. H. Younkin, L. Wen-Lang, D. W. Dickson, T. Murakami, E. Matsubara, K. Abe, K. H. Ashe and S. G. Younkin, J. Neurosci., 2004, 24, 3801–3809 CrossRef CAS.
- G. M. Shankar, S. Li, T. H. Mehta, A. Garcia-Munoz, N. E. Shepardson, I. Smith, F. M. Brett, M. A. Farrell, M. J. Rowan, C. A. Lemere, C. M. Regan, D. M. Walsh, B. L. Sabatini and D. J. Selkoe, Nat. Med., 2008, 14, 837–842 CrossRef CAS PubMed.
- M. Townsend, G. M. Shankar, T. Mehta, D. M. Walsh and D. J. Selkoe, J. Physiol., 2006, 572, 477–492 CrossRef CAS.
- M. Talantova, S. Sanz-Blasco, X. Zhang, P. Xia, M. W. Akhtar, S. Okamoto, G. Dziewczapolski, T. Nakamura, G. Cao, A. E. Pratt, Y. J. Kang, S. Tu, E. Molokanova, S. R. McKercher, S. A. Hires, H. Sason, D. G. Stouffer, M. W. Buczynski, J. P. Solomon, S. Michael, E. T. Powers, J. W. Kelly, A. Roberts, G. Tong, T. Fang-Newmeyer, J. Parker, E. A. Holland, D. Zhang, N. Nakanishi, H. S. Chen, H. Wolosker, Y. Wang, L. H. Parsons, R. Ambasudhan, E. Masliah, S. F. Heinemann, J. C. Pina-Crespo and S. A. Lipton, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2518–E2527 CrossRef CAS.
- R. Resende, E. Ferreiro, C. Pereira and C. Resende de Oliveira, Neuroscience, 2008, 155, 725–737 CrossRef CAS PubMed.
- R. G. Nagele, M. R. D'Andrea, H. Lee, V. Venkataraman and H. Y. Wang, Brain Res., 2003, 971, 197–209 CrossRef CAS PubMed.
- T. L. Lau, J. D. Gehman, J. D. Wade, K. Perez, C. L. Masters, K. J. Barnham and F. Separovic, Biochim. Biophys. Acta, 2007, 1768, 2400–2408 CrossRef CAS PubMed.
- C. Escartin, E. Galea, A. Lakatos, J. P. O'Callaghan, G. C. Petzold, A. Serrano-Pozo, C. Steinhauser, A. Volterra, G. Carmignoto, A. Agarwal, N. J. Allen, A. Araque, L. Barbeito, A. Barzilai, D. E. Bergles, G. Bonvento, A. M. Butt, W. T. Chen, M. Cohen-Salmon, C. Cunningham, B. Deneen, B. De Strooper, B. Diaz-Castro, C. Farina, M. Freeman, V. Gallo, J. E. Goldman, S. A. Goldman, M. Gotz, A. Gutierrez, P. G. Haydon, D. H. Heiland, E. M. Hol, M. G. Holt, M. Iino, K. V. Kastanenka, H. Kettenmann, B. S. Khakh, S. Koizumi, C. J. Lee, S. A. Liddelow, B. A. MacVicar, P. Magistretti, A. Messing, A. Mishra, A. V. Molofsky, K. K. Murai, C. M. Norris, S. Okada, S. H. R. Oliet, J. F. Oliveira, A. Panatier, V. Parpura, M. Pekna, M. Pekny, L. Pellerin, G. Perea, B. G. Perez-Nievas, F. W. Pfrieger, K. E. Poskanzer, F. J. Quintana, R. M. Ransohoff, M. Riquelme-Perez, S. Robel, C. R. Rose, J. D. Rothstein, N. Rouach, D. H. Rowitch, A. Semyanov, S. Sirko, H. Sontheimer, R. A. Swanson, J. Vitorica, I. B. Wanner, L. B. Wood, J. Wu, B. Zheng, E. R. Zimmer, R. Zorec, M. V. Sofroniew and A. Verkhratsky, Nat. Neurosci., 2021, 24, 312–325 CrossRef CAS.
- D. M. Zeppenfeld, M. Simon, J. D. Haswell, D. D'Abreo, C. Murchison, J. F. Quinn, M. R. Grafe, R. L. Woltjer, J. Kaye and J. J. Iliff, JAMA Neurol., 2017, 74, 91–99 CrossRef PubMed.
- D. M. Wilcock, M. P. Vitek and C. A. Colton, Neuroscience, 2009, 159, 1055–1069 CrossRef CAS PubMed.
- M. Amiry-Moghaddam, T. Otsuka, P. D. Hurn, R. J. Traystman, F. M. Haug, S. C. Froehner, M. E. Adams, J. D. Neely, P. Agre, O. P. Ottersen and A. Bhardwaj, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2106–2111 CrossRef CAS.
- A. D. Bragg, M. Amiry-Moghaddam, O. P. Ottersen, M. E. Adams and S. C. Froehner, Glia, 2006, 53, 879–890 CrossRef PubMed.
- J. Lange, O. Gillham, R. Alkharji, S. Eaton, G. Ferrari, M. Madej, M. Flower, F. S. Tedesco, F. Muntoni and P. Ferretti, Glia, 2022, 70, 466–490 CrossRef CAS.
- R. Marcu, Y. J. Choi, J. Xue, C. L. Fortin, Y. Wang, R. J. Nagao, J. Xu, J. W. MacDonald, T. K. Bammler, C. E. Murry, K. Muczynski, K. R. Stevens, J. Himmelfarb, S. M. Schwartz and Y. Zheng, iScience, 2018, 4, 20–35 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00287c |
| This journal is © The Royal Society of Chemistry 2024 |