
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multiplexed, allele-specific recombinase polymerase amplification assay with lateral flow readout for sickle cell disease detection†
Megan M.
Chang‡
 a,
Mary E.
Natoli
a,
Alexis F.
Wilkinson
a,
Venée N.
Tubman
bc,
Gladstone E.
Airewele
bc and
Rebecca R.
Richards-Kortum
*a
a,
Mary E.
Natoli
a,
Alexis F.
Wilkinson
a,
Venée N.
Tubman
bc,
Gladstone E.
Airewele
bc and
Rebecca R.
Richards-Kortum
*a
aDepartment of Bioengineering, Rice University, Houston, TX, USA. E-mail: rkortum@rice.edu
bTexas Children's Cancer and Hematology Centers, Houston, TX, USA
cBaylor College of Medicine, Houston, TX, USA
First published on 25th July 2024
Abstract
Isothermal nucleic acid amplification tests have the potential to improve disease diagnosis at the point of care, but it remains challenging to develop multiplexed tests that can detect ≥3 targets or to detect point mutations that may cause disease. These capabilities are critical to enabling informed clinical decision-making for many applications, such as sickle cell disease (SCD). To address this, we describe the development of a multiplexed allele-specific recombinase polymerase amplification (RPA) assay with lateral flow readout. We first characterize the specificity of RPA using primer design strategies employed in PCR to achieve point mutation detection, and demonstrate the utility of these strategies in achieving selective isothermal amplification and detection of genomic DNA encoding for the healthy βA globin allele, or genomic DNA containing point mutations encoding for pathologic βS and βC globin alleles, which are responsible for most sickle cell disorders. We then optimize reaction conditions to achieve multiplexed amplification and identification of the three alleles in a single reaction. Finally, we perform a small pilot study with 20 extracted genomic DNA samples from SCD patients and healthy volunteers – of the 13 samples with valid results, the assay demonstrated 100% sensitivity and 100% specificity for detecting pathologic alleles, and an overall accuracy of 92.3% for genotype prediction. This multiplexed assay is rapid, minimally instrumented, and when combined with point-of-care sample preparation, could enable DNA-based diagnosis of SCD in low-resource settings. The strategies reported here could be applied to other challenges, such as detection of mutations that confer drug resistance.
Introduction
Nucleic acid amplification tests (NAATs) employing polymerase chain reaction (PCR) are the gold standard diagnostic for many diseases. However, PCR requires specialized thermocycling equipment and highly trained personnel, limiting its feasibility in point-of-care (POC) testing. Isothermal NAATs have the potential to improve diagnostic medicine at the POC due to their simplified instrumentation and user requirements, but the utility of isothermal NAATs is often limited due to the difficulty of incorporating three or more targets into a single test, and the lack of tests to distinguish targets with clinically significant point mutations.Multiplexed isothermal NAATs are needed to detect multiple targets in a single test to decrease costs, increase clinical utility, and reduce diagnostic turnaround time.1 Multiplexed tests can be a key part of syndromic panels to screen for several pathogens responsible for similar symptoms or can provide important treatment information, such as identifying the presence of one or more drug-resistance mutations.1 Of existing isothermal amplification methods, recombinase polymerase amplification (RPA) is considered the most readily multiplexable as it only requires two to three primers per target;2 however, there are limited reports describing RPA assays that detect more than two targets in minimally instrumented formats that have sufficient sensitivity or are suitable for use in resource-limited settings due to the challenges of biochemical multiplexing.3–7
In addition, point mutations are important biomarkers that can impact clinical decision-making for many diseases. For example, sickle cell disease (SCD) is a life-threatening inherited blood disorder caused by one or more point mutations in the human β-globin gene (HBB) that affects over 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 newborns around the world each year, with over 90% of these births occurring in low- and middle-income countries (LMICs).8,9 It is characterized by the presence of at least one βS globin allele, and a second pathologic HBB variant allele that leads to the predominant formation of hemoglobin S (HbS).8 Abnormal polymerization of HbS can lead to severe anemia, chronic organ damage, and increased susceptibility to infections.10 Multiple strategies have been developed to resolve single nucleotide differences following amplification with RPA11–19 that overcome the limitations of the low operating temperature of RPA and its high tolerance to mismatches.20–23 However, post-amplification processing increases time-to-result and may lead to workspace contamination with amplified DNA.24,25 Moreover, many of the above approaches require heating steps at widely different temperatures from RPA's operating temperature of 37–42 °C,11–16 which increases the complexity of instrumentation required and limits the translation of these methods to resource-limited settings.
000 newborns around the world each year, with over 90% of these births occurring in low- and middle-income countries (LMICs).8,9 It is characterized by the presence of at least one βS globin allele, and a second pathologic HBB variant allele that leads to the predominant formation of hemoglobin S (HbS).8 Abnormal polymerization of HbS can lead to severe anemia, chronic organ damage, and increased susceptibility to infections.10 Multiple strategies have been developed to resolve single nucleotide differences following amplification with RPA11–19 that overcome the limitations of the low operating temperature of RPA and its high tolerance to mismatches.20–23 However, post-amplification processing increases time-to-result and may lead to workspace contamination with amplified DNA.24,25 Moreover, many of the above approaches require heating steps at widely different temperatures from RPA's operating temperature of 37–42 °C,11–16 which increases the complexity of instrumentation required and limits the translation of these methods to resource-limited settings.
There have been some reports of point mutation detection within a single round of RPA using specificity-enhancing strategies such as amplification refractory mutation system (ARMS) primer design,26–28 zip nucleic acids,29 peptide nucleic acids,26,30 and locked nucleic acids.31 However, these reported methods detect products with gel electrophoresis,26,27 electrochemical detection,28 or fluorescence,29–31 all of which require bulky or expensive peripheral equipment. Further, multiplexed point mutation detection with RPA in a single tube remains challenging and has required physical separation of primers, either through separate tubes27,31 or separate electrodes.28 We previously developed a novel RPA-based assay for SCD detection that differentiates DNA containing the most common pathologic point mutation in the β-globin gene, βS, from DNA containing the healthy βA in less than 30 minutes with high sensitivity and specificity.31 However, the assay relies on detection of fluorescent products, which requires dedicated optical instrumentation, and separate reactions for each point mutation of interest, increasing reagent costs and user complexity.
To streamline the assay workflow and reduce instrumentation requirements, here we report a multiplexed allele-specific RPA assay that is compatible with lateral flow readout to detect the healthy β-globin gene, βA, and the two most common pathologic point mutations, βS and βC. We optimized the assay for differentiation of genomic DNA from the following clinically relevant genotypes: AA (healthy), AS (sickle cell trait), SS (sickle cell anemia), and SC (hemoglobin SC disease), of which SS and SC are considered to have SCD and require clinical management. Finally, we validated the test performance with genomic DNA extracted from 20 whole blood samples from both patients with SCD and healthy volunteers.
Experimental section
Clean reaction setup
Amplification reaction setup was conducted in a biosafety cabinet inside a separate room designated for pre-amplification activities. Another dedicated biosafety cabinet was used for all DNA extraction and target preparation. All biosafety cabinets were regularly decontaminated with UV light, bleach, and RNAse Away. Single-use nuclease-free water aliquots were used for both reaction set-up and target dilution. Post-amplification analysis of amplicons was conducted in a space physically separated from pre-amplification activities to prevent workspace contamination with amplified DNA. Different lab coats were used in each lab space (sample preparation, pre-amplification, post-amplification) to further reduce the risks of environmental contamination with amplicons.Target preparation and storage
DNA associated with clinically relevant genotypes (AA, AS, SS, SC, and CC) for use in assay design and optimization was obtained from multiple sources. Mixed gender human genomic DNA was purchased from Promega (G3041) for genotype AA. Human genomic DNA was purchased from the Coriell Institute for Medical Research (Camden, NJ) for genotypes AS (NA20838), SS (NA16265), and SC (NA16266). Full-length genomic DNA targets were quantified using Agilent's Genomic DNA ScreenTape assay on the 4200 TapeStation system according to manufacturer's instructions.To represent genotype CC, a synthetic gBlock containing a 499-bp sequence of the β-globin gene (NCBI Gene ID: 3043) containing the βC allele was ordered from Integrated DNA Technologies (Coralville, Iowa, USA), as full length genomic DNA with genotype CC was unavailable to purchase. CC gBlock DNA was quantified in qPCR using TapeStation-quantified genomic DNA as standards. 20 μL qPCR reactions were prepared with PowerUP SYBR Green PCR Master Mix (Thermo Fisher Scientific), 1 μL each of forward and reverse primers at 10 μM working concentration (Table 1; KM29 and KM38), and 5 μL of sample. Reactions were incubated and monitored with a Bio-Rad CFX96 using the following thermocycling protocol: 50 °C UDG activation for 2 minutes, 95 °C polymerase activation for 2 minutes, followed by 40 cycles of denaturation at 95 °C for 15 seconds and annealing and extension at 60 °C for 1 minute.
| Primer name | Assay | Primer sequence (5′ → 3′) |
|---|---|---|
| KM29 | qPCR | GGTTGGCCAATCTACTCCCAGG |
| KM38 | qPCR | TGGTCTCCTTAAACCTGTCTTG |
| fP-A | RPA | /5DiGN/AGGGCAGTAACGGCAGACTTCTCC+TC |
| fP-S | RPA | /56-FAM/AGGGCAGTAACGGCAGACTTCTGC+A |
| fP-C | RPA | /56-TAMN/AGGGCAGTAACGGCAGACTTCTCAT+T |
| Probe | RPA | /5BiosG/GCTTACATTTGCTTCTGACACAACTGTGTTC |
| AC/idSp/AGCAACCTCAAACAG/3SpC3/ | ||
| rP | RPA | GGGCAGAGCCATCTATTGCTTACATTTGCTTCT |
Single-use aliquots of DNA associated with all target genotypes were prepared in 1× TE buffer and stored at −80 °C. Prior to each experiment, target DNA was diluted in nuclease-free water.
Primer screens
All allele-specific forward primer candidates (Tables S1–S3†) were screened in 50 μL TwistAmp nfo RPA reactions according to manufacturer's instructions. Primers and probe that amplified a 115 base pair region of the β-globin gene were adapted from previous work.31 Specifically, the probe was modified according to RPA nfo guidelines to incorporate an antigenic label to allow for lateral flow detection,33 and forward primer candidates were designed to incorporate locked nucleic acids or primer-template mismatches within the last three nucleotides to achieve selective amplification; the βA and βC allele-specific primers were extended by one nucleotide such that the 3′ end of the primers were positioned on the βC point mutation site. Primers and probe were prepared at 10 μM working concentrations in 1X TE buffer. Each 50 μL reaction contained one nfo enzyme pellet, and 37.5 μL of a master mix containing 29.5 μL rehydration buffer, 2.1 μL antigenically labelled forward primer candidate, 2.1 μL reverse primer, 0.6 μL fluorescein-labelled probe, and 3.2 μL of nuclease-free water. 10 μL of target containing either nuclease-free water of 104 input copies of genomic DNA was added, and 2.5 μL of 280 mM magnesium acetate catalyst was added to tube caps. Reactions were initiated with a quick centrifuge, then incubated for 20 minutes at 39 °C on a T8-ISO (Axxin Pty Ltd., Australia); a 2 mm stainless steel ball bearing (Simply Bearings Ltd., Manchester, UK) was included with each reaction to provide continual mixing during incubation. After amplification, products were immediately diluted 1:50 in running buffer (Milenia, Giessen, Germany), and 10 μL of diluted product was added to the sample pad of commercially available lateral flow strips (HybriDetect, Milenia). Strips were then placed upright into 100 μL of running buffer and allowed to develop for at least 3 minutes before scanning on a flatbed scanner (Epson Perfection V550 Photo).
Singleplex RPA
Primers that were able to selectively amplify target DNA containing either the healthy βA allele or the point mutation of interest in the βS or βC allele were chosen for further validation and designed to contain unique antigenic labels compatible with multiplexed amplification and lateral flow detection (Table 1).6 To conserve nfo enzyme pellets and reduce per-test cost, small-volume 10 μL singleplex RPA reactions were prepared by assembling 37.5 μL of master mix as described above. Each lyophilized nfo enzyme pellet was rehydrated with 37.5 μL of master mix, of which 7.5 μL was aliquoted into individual PCR tubes. 2 μL of target was added, and 0.5 μL of 280 mM magnesium acetate was added to the caps. Reactions were initiated with a quick spin to combine all reagents, vortexed briefly and spun again before incubating for 20 minutes on a heat block set to 39 °C. After four minutes, all reactions were removed from the heat block and vortexed, then briefly centrifuged before replacing on the heat block for the remaining 16 minutes of incubation.34 Following amplification, reactions were immediately diluted with 90 μL of 1× PBST to stop the reaction. The entire 100 μL volume of diluted RPA amplicons was pipetted onto the end of a custom lateral flow strip, and followed with 100 μL of 1× PBST as a running buffer. Strips were allowed to flow for ten minutes, then scanned on a flatbed scanner.Multiplex RPA
All RPA primers and the probe listed in Table 1 were prepared at 20 μM working concentrations in 1× TE buffer to assemble multiplexed RPA reactions. Each lyophilized nfo enzyme pellet was rehydrated with 37.5 μL containing 29.5 μL of rehydration buffer, 1.05 μL unmodified reverse primer, 0.5 μL biotinylated probe, 0.481 μL fP-S, 0.656 μL fP-C, 0.613 μL of fP-A, 1 μL of Tte UvrD helicase (New England Biolabs), and 3.7 μL nuclease-free water. 10 μL multiplex RPA reactions contained 7.5 μL of the rehydrated enzyme pellet mixture, 2 μL of target, and 0.5 μL of 280 mM magnesium acetate in the caps, such that the final concentrations were 420 nM unmodified reverse primer, 200 nM biotinylated probe, 192.5 nM fP-S, 245 nM fP-A, 262.5 nM fP-C, and 0.4 ng of Tte UvrD helicase per reaction. Reactions were initiated with a quick spin to combine all reagents, vortexed briefly and spun again before incubating for 20 minutes on a heat block set to 39 °C, with a brief vortex and centrifuge step after four minutes of incubation. After amplification, amplicons were detected on custom lateral flow strips as described above.Clinical samples: genomic DNA extraction and testing
De-identified venous blood samples from patients in the Sickle Cell Program at Texas Children's Hospital were collected under a protocol reviewed and approved by the Institutional Review Boards at Rice University and Baylor College of Medicine. Genotypes, as determined by HPLC, were provided with samples. Venous blood samples from healthy volunteers were obtained under Rice University IRB Protocol 2017-303; these samples were presumed to have genotype AA. All blood samples were collected into EDTA anticoagulant tubes and stored at 4 °C until testing. Genomic DNA was extracted from samples using the Qiagen DNA Micro Kit (56304) or the Qiagen DNeasy Blood & Tissue Kit (69504) according to manufacturer's instructions, with a final elution step into either 50 or 100 μL of buffer AE. DNA was stored at −20 °C until use. Upon thawing, samples were input undiluted into the multiplexed reaction containing 168 nM of fP-S, 269.5 nM fP-A, 262.5 nM fP-C, and 0.4 ng of Tte UvrD helicase per reaction. DNA concentration of each sample was also measured using a NanoDrop (ND-1000). Samples with discordant results were amplified with PCR and submitted for Sanger sequencing (Azenta Life Sciences, Burlington, MA).Lateral flow assays
Multiplexed lateral flow strips (Fig. S1†) were adapted from ref. 25 and were fabricated and stored at room temperature in a desiccated foil bag until use. All paper components were cut to size with a laser cutter (Universal Laser Systems VLS 3.60, Scottsdale, AZ) prior to assembly.Anti-FITC (ab19224, abcam, Cambridge, MA), anti-digoxigenin (ab64509, abcam), and biotinylated goat anti-mouse IgG (B7401, Sigma-Aldrich) were prepared as previously described.3 Briefly, antibodies were diluted to concentrations of 0.5 mg mL−1, 2.5 mg mL−1 and 1 mg mL−1 in a buffer containing 5% methanol and 2% sucrose in 100 mM sodium bicarbonate. Anti-TAMRA (MA1041, ThermoFisher) was used undiluted at 1 mg mL−1. A sciFLEXARRAYER S3 (Scienion, Berlin, Germany) was used to deposit approximately 18 μg, 9 μg, 112.5 μg and 4.5 μg of anti-TAMRA, anti-FITC, anti-digoxigenin, and biotinylated anti-mouse IgG, respectively, in four parallel lines on a 31 mm × 250 mm backed sheet of nitrocellulose (Unistart CN95, Sartorius, Goeingen, Germany). The first test line was printed 14.5 mm from the proximal edge, and subsequent test lines printed three mm apart.
Conjugate pads were prepared by diluting streptavidin-coated gold nanoshells (GSIR150, Nanocomposix, San Diego, CA) in a three to five ratio with 5% BSA in 1X PBST. Two mL were pipetted onto a 16 mm × 250 mm glass fiber pad (Grade 8980, Ahlstrom, Mt Holly Springs, PA), then lyophilized without freezing for at least 24 hours in a freeze-drying system (LabConco FreeZone 12, Kansas City, MO). After lyophilization, the conjugate pad was then used for lateral flow strip assembly.
Lateral flow strips (Fig. S1†) were assembled on an 80 mm backing card (MIBA-010, Diagnostics Consulting Network, Carlsbad, CA). The covertape between the kiss cuts at 17 mm and 43 mm was removed, and the nitrocellulose membrane was aligned. Then, a 19 mm × 250 mm cellulose wicking pad (CFSP223000, Millipore) was placed at the distal edge of the nitrocellulose membrane, with a two mm overlap. The conjugate pad was placed at the proximal end of the nitrocellulose membrane, also with a two mm overlap. Finally, a 27 mm × 250 mm glass fiber sample pad (Ahlstrom Grade 8951) was placed at the proximal end, overlapping three mm with the conjugate pad with a six mm overhang. The assembled card was cut into five mm wide strips using an A-Point Digital guillotine cutter (Arista Biologicals, Allentown, PA, USA).
Image analysis
Signal-to-background ratios (SBR) at the test and control lines were calculated using a custom image analysis program in MATLAB. Raw RGB images were converted to grayscale and inverted, and the average pixel intensity values across the width of the strip were computed to create a mean intensity line profile along the length of the strip. For custom lateral flow strips containing three test lines, the signal at the A, S, and C test lines was found by searching for the highest peaks within a 15-pixel windows centered 20, 40, and 60 pixels from the control line peak, respectively; if no peaks were found, then the median pixel value of the mean intensity line profile in each window was taken. The background was calculated by averaging the mean intensity line profile at pixels centered between the test line search regions, (i.e. located 10, 30, and 50 pixels away from the control line location) to form one overall background signal value. The SBR at each test line was then calculated as the ratio of the test line signal to that of the background. For Milenia strips used during primer screens, the SBR was calculated similarly; the signal at the test line was found by searching for the highest peak within a 15-pixel window centered 60 pixels from the control line peak, the background signal was the average of the highest peaks in the inverted mean intensity line profile, and the SBR was calculated by dividing the test line signal over the background signal.Data analysis
To set positivity thresholds for the singleplex allele-specific RPA reactions, the mean SBR of no-target controls at the test line of interest was calculated, and the positivity threshold was set three standard deviations above the mean. For the optimized multiplexed allele-specific RPA, the positivity threshold for A, S, or C lines was set by averaging the SBR at the test line of interest of no-target controls and samples containing off-target alleles, adding three standard deviations to the mean, and increasing the resultant value by ten percent.The positivity thresholds for the A, S, and C lines in the multiplex reaction were then used to determine the genotype for each clinical sample according to the following algorithm (Fig. S2†): 1) if only the signal at the A test line exceeded the A positivity threshold, then the sample was assigned genotype AA; 2) if the signal at both the A and S test lines exceeded the corresponding positivity thresholds, then the sample was assigned genotype AS; 3) if only the signal at the S test line exceeded its positivity threshold, then the sample was assigned genotype SS; 4) if the signals at both the S and C test lines exceeded the corresponding positivity thresholds, or if the signal at all three test lines exceeded their thresholds, the sample was assigned genotype SC; 5) if the signal at all test lines was below the corresponding positivity threshold, the test was considered invalid, as it is expected that all individuals tested will have at least one of the three targeted β-globin alleles, and the absence of test lines likely indicates that sufficient amplification did not occur.
To evaluate whether the mean DNA concentration was significantly different for clinical samples that produced valid and invalid results, a one-tailed unpaired t-test assuming unequal variances was performed (GraphPad Prism, version 9.5.1).
Results and discussion
Primer screens
To facilitate LFA detection, we incorporated a 5′ antigenic label to the previously published allele-specific primer for the βS gene and modified the probe according to RPA nfo guidelines to allow for direct amplicon detection on commercially available lateral flow strips.33 However, the primer could no longer differentiate between the βA and the βS alleles in this adapted format (Fig. 1, fP-S-4). To overcome this limitation, we screened primers listed in Table S1,† and found that the primer demonstrating the best specificity had a locked nucleic acid on the 3′ end of the primer at the point mutation site, and an additional strategic mismatch on the third base from the 3′ end (fP-S). The difference in specificity observed between the RPA nfo and the RPA exo kit is likely due to the 3′ to 5′ exonuclease activity.22,30 Both the LNA and the ARMS mutation were found to be necessary to suppress off-target amplification of the βA allele – the lack of an LNA on the point mutation site (fP-S-1) and the lack of an additional mismatch (fP-S-3) resulted in amplification of both the βA and the βS alleles, but more than one LNA in the last three bases of the primer resulted in markedly reduced amplification efficiency of the βS allele (fP-S-2, fP-S-5, fP-S-6).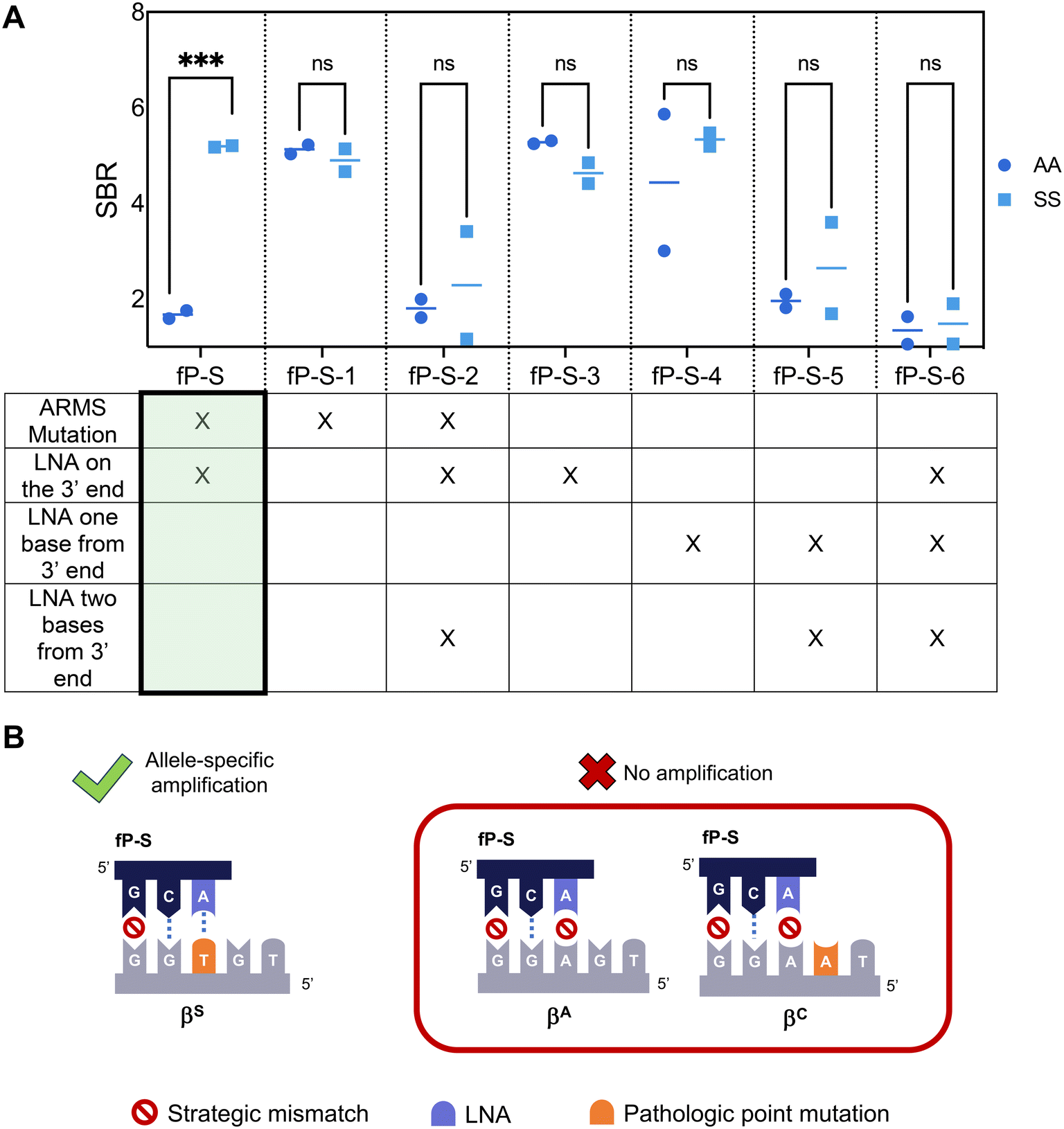 | ||
| Fig. 1 βS Allele-Specific Forward Primer Design and Screening. (A) Allele-specific forward primers for the βS allele were designed to incorporate specificity-enhancing modifications listed in the table. Primers were subsequently screened using 10000 input copies of either AA or SS target. Signal-to-background ratios of the resultant lateral flow assays are graphed. Individual replicates are represented by a circle, and horizontal lines represent the mean (n = 2). fP-S, containing an LNA on the point mutation site and a strategic ARMS mismatch, is highlighted as it was the only combination to have a statistically significant difference in signal formation between the two targets (p = 0.0006, two-tailed unpaired t-test). (B) Schematic of specificity enhancing modifications incorporated into fP-S that achieve allele-specific amplification of DNA encoding for the βS globin allele. | ||
We then adapted the allele-specific primer design to selectively amplify the βC allele and confirmed that the specificity enhancing modifications (strategic mismatch on third base and LNA on the mutation site) were enough to suppress amplification of both the βS and the βA alleles while maintaining amplification of the βC allele (Fig. S3†). Finally, we sought to re-design our previously published βA allele primer such that off-target amplification of both the βS and the βC alleles was suppressed. We extended the length of the βA primer such that the 3′ end was positioned on the βC point mutation and applied design strategies to suppress both βS and βC allele amplification – the final primer designs are listed in Table S3.† We then screened these βA primer candidates using target DNA with genotypes AA, SS, and CC (Fig. 2A).
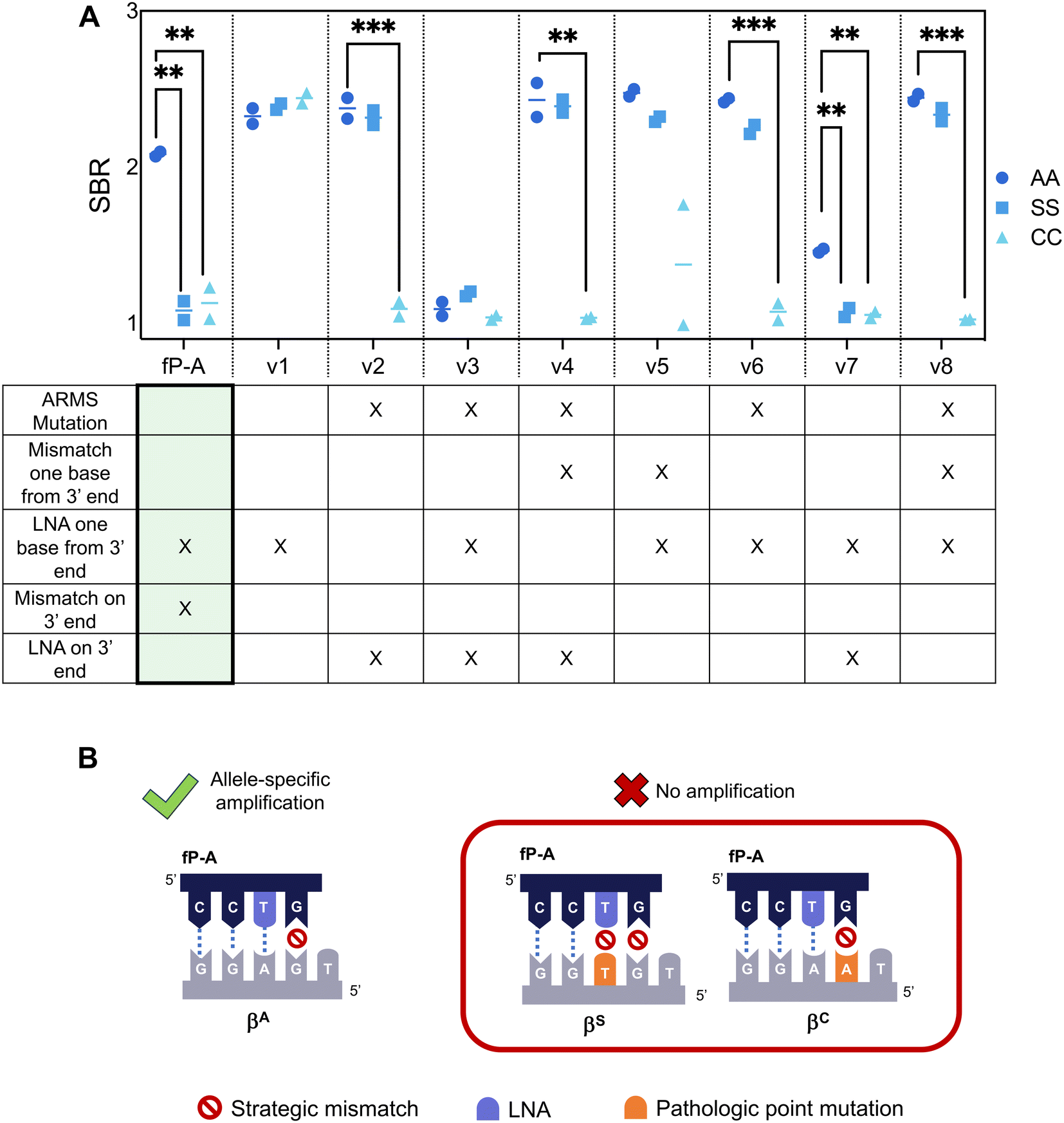 | ||
| Fig. 2 βA Allele-Specific Forward Primer Design and Screening. (A) Allele-specific forward primers for the βA allele were designed to incorporate specificity-enhancing modifications listed in the table. Primers were subsequently screened using 10000 input copies of AA, SS, or CC target. Signal-to-background ratios of the resultant lateral flow assays are graphed. Individual replicates are represented by a circle, and horizontal lines represent the mean (n = 2). fP-A, containing an LNA one base from the 3′ end and a strategic mismatch on the 3′ end (highlighted) had a statistically significant difference in signal formation between target containing only the βA globin allele and off-targets containing either the βS globin allele (p = 0.004) or the βC globin allele (p = 0.004), while maintaining strong signal formation of the target allele. **p < 0.01; ***p < 0.001; ****p < 0.0001; significance determined for each primer pair by a one-way ANOVA with post-hoc Tukey's test. (B) Schematic of specificity enhancing modifications incorporated into fP-A that achieve allele-specific amplification of DNA encoding for the βA globin allele. | ||
Results show that previously successful primer designs for allele-specific amplification, such as the use of an LNA on the penultimate base (A-v1) or the combination of an LNA on the 3′ end with an additional mismatch on the third base from the 3′ end (A-v2), were unable to suppress both off-target βS and βC amplification. In fact, many of the primer designs screened were unable to successfully suppress βS amplification; this is perhaps because the βS mutation site was located at the penultimate nucleotide position of the βA primer, and RPA has been shown to be tolerant to mismatches on the interior of primers.20–22 We found that it was necessary to include specificity enhancing modifications on both the penultimate and ultimate base to suppress both βS and βC allele amplification (fP-A, A-v7). fP-A was chosen as the best primer for allele-specificity given its ability to suppress both off-target alleles while maintaining strong amplification of the target βA globin allele; A-v7, while successful at suppressing amplification for off-target alleles, had reduced amplification efficiency of the target βA allele, likely due to the proximity of two LNAs in the 3′ end of the primer.
Singleplexed allele-specific RPA
We next sought to translate the allele-specific assays to a custom lateral flow strip that would allow simultaneous visualization of all three allele-specific products. We incorporated a unique antigenic tag on each allele-specific primer and fabricated lateral flow strips that captured products at separate test lines (Fig. 3A), then validated that the translated amplification and lateral flow detection assays were able to maintain allele specificity and correctly differentiate purchased genomic DNA for the following clinically relevant genotypes: healthy (AA), trait (AS), sickle cell anemia (SS), and hemoglobin SC disease (SC). | ||
| Fig. 3 Assay overview and singleplex performance. (A) Schematic of amplification (left) and lateral flow detection (right) of allele-specific amplification products. Each allele-specific forward primer contains a distinct antigenic label that allows for lateral flow capture and visualization of allele-specific products at separate test lines on a custom lateral flow assay. (B) Scans of representative lateral flow strips following amplification of 10000 input copies of DNA per reaction from purchased DNA samples with clinically relevant genotypes AA, AS, SS, or SC. DNA was amplified using each of the allele-specific primers in singleplex reactions and detected on custom lateral flow strips. (C) Signal-to-background ratio (SBR) of test lines for each condition shown in (B). Horizontal line represents the mean SBR for three replicates of DNA or the mean SBR for four negative replicates containing only water. Only genotypes containing the allele of interest result in signal formation above the positivity threshold (dashed line) at the corresponding test line of interest. NTC: no-target control. | ||
In a singleplex format, the allele-specific reactions only yield signal at the test line of interest for genotypes containing the target allele (Fig. 3B and C). Interestingly, the signal at the C test line is lower than the signal at either the S of A test lines, which is likely a result of the increased flow rate in the proximal portion of the strip that allows less reaction time for antigen and antibody binding, whereas flow is slower at the more distal S and A test lines, allowing for more reaction time to form stronger signal.35,36
Optimized multiplexed allele-specific RPA
To further simplify assay set-up, we next sought to multiplex the three allele-specific amplification assays into a single reaction. We optimized primer concentrations (Fig. S4 and S5†), magnesium concentration (Fig. S6†), reaction additives (Fig. S7†), and reaction temperature (Fig. S8†). We then evaluated the performance of the optimized multiplexed reaction with purchased DNA of both homozygous (AA, SS) and heterozygous (AS, SC) genotypes of interest in triplicate. The resultant lateral flow strips are shown in Fig. 4A. | ||
| Fig. 4 Multiplexed allele-specific RPA can genotype the β-globin gene in a single round of amplification. (A) Lateral flow strips and (B) corresponding signal-to-background ratios at each test line of purchased genomic DNA amplified and detected with the optimized multiplexed assay in triplicate. Genotypes AA, AS, SS, and SC are visually distinguishable, and samples containing the allele of interest result in SBRs above the positivity threshold (dashed lines) at the corresponding test lines of interest. NTC: no-target control. | ||
Faint non-specific signal formation is observed at the A test line even when the βA allele is not present in the multiplexed reaction, especially for SC samples, despite extensive optimization of the multiplexed reaction. This is likely due to concentration dependent specificity, where fP-A is unable to maintain allele specificity as millions of copies of off-target amplicons are generated. Thus, SC target was excluded when setting the positivity threshold for the A line. Using the combination of signals formed on the lateral flow strips that cross the corresponding threshold for positivity, the optimized multiplexed assay could successfully differentiate DNA from healthy individuals (AA), DNA from patients with sickle cell trait (AS), and DNA from patients with sickle cell anemia (SS) or hemoglobin SC disease (SC) (Fig. 4B). To increase usability of the assay in the field and enable definitive and consistent genotype predictions, the allele-specific positivity thresholds could be programmed into a low-cost lateral flow reader to automate strip interpretation.
Performance of multiplexed assay with clinical samples
Finally, we evaluated the performance of the multiplexed amplification and detection assay with genomic DNA extracted from whole blood samples from both sickle cell patients and healthy volunteers using the workflow outlined in Fig. 5A. A total of 20 samples were tested (seven AA samples, seven SS samples, and six SC samples), of which 13 had valid results by the algorithm in Fig. S2;† seven samples were considered invalid as they did not have any test lines above the corresponding positivity thresholds, indicating amplification was not sufficient to produce detectable amplicons. Lateral flow strips and SBRs for each clinical sample are shown in Fig. S9.†Fig. 5B shows a confusion matrix of the genotype predictions from the multiplexed assay (left) compared to the genotypes reported by HPLC (top). Of the 13 tests that produced valid results, four AA samples were correctly genotyped as AA, three SS samples were correctly genotyped as SS and one was incorrectly genotyped as AS, and all five SC samples were correctly genotyped as SC. The misidentification of a patient with SS genotype as sickle cell trait (AS) could delay critical care. The discordant sample was submitted for Sanger sequencing to identify if the sample was contaminated by amplicons in the laboratory workspace during DNA purification; however, sequencing confirmed the genotype identified by HPLC. Thus, the multiplexed assay's overall accuracy at genotype prediction for valid samples was 92.3%, with a sensitivity for SCD detection of 89% and a specificity of 100%. | ||
| Fig. 5 Testing workflow and performance of multiplexed assay with clinical samples. (A) Overview of the workflow used to test clinical samples. (B) Confusion matrix of test results predicted by the multiplexed assay (left) compared to the genotypes reported by the reference standard HPLC (top). (C) DNA concentration (ng μL−1) as measured by NanoDrop of clinical samples that produced invalid and valid test results. The mean DNA concentration in samples that produced invalid test results was not significantly different than the mean DNA concentration for clinical samples that produced valid test results (p = 0.1102), as determined by a one-tailed unpaired t-test assuming unequal variances. (D) Allele sensitivity and specificity within the multiplexed reaction was calculated based on samples that generated valid results. | ||
Next, we compared the DNA concentration for samples that produced invalid results to that for samples that produced valid results, as we hypothesized invalid results were produced when the amount of input DNA was below the multiplexed assay limit of detection. We found that the mean DNA concentration of samples that produced invalid results was not significantly lower than the mean DNA concentration of samples that produced valid results (Fig. 5C; p = 0.1102, significance determined using a one-tailed unpaired t-test assuming unequal variances). The one SC sample that had an invalid test result had an input DNA concentration of 18.2 ng μL−1; however two SC samples with similar input concentrations of 16.7 and 18.4 ng μL−1 had valid test results. Similarly, the three SS samples that produced invalid results had DNA concentrations between 17.5 and 42.3 ng μL−1 (Fig. S8†), but one SS sample with a DNA concentration in that range of 33.5 ng μL−1 produced a valid test result. These results are likely due to some stochastic variability near the multiplexed assay's limit of detection. However, the invalid AA samples tended to have higher DNA concentrations of 31–62.5 ng μL−1. A larger validation with clinical samples is needed to better understand the relationship between input DNA concentration and assay performance.
Finally, we calculated the sensitivity and specificity of the multiplexed assay to identify the presence of the βA, βS, and βC alleles based on samples that produced valid results (Fig. 5D). For the βA allele, the assay correctly identified four of four valid AA samples as having the βA allele, and correctly predicted eight of nine SCD samples as negative for the βA allele, resulting in a sensitivity of 100% and a specificity of 90% for βA. Of the nine SCD samples that produced valid results, all nine were correctly identified as having the βS allele present, and all four AA samples were correctly classified as not having the βS allele, leading to a sensitivity and specificity of 100% for the βS allele. Similarly, the assay correctly identified the presence of the βC allele in five of five valid SC samples, and correctly did not identify the βC in eight AA and SS samples; thus, the multiplexed assay has a 100% sensitivity and 100% specificity at βC allele detection.
Conclusion
We successfully developed a novel, multiplexed, allele-specific recombinase polymerase amplification assay with lateral flow detection that can detect pathologic β-globin alleles, βS and βC, in a single round of isothermal amplification. The results of the multiplexed amplification and detection assay can be used to predict a patient's genotype, and thus differentiate patients with sickle cell disease that require treatment (SS and SC) from patients without a sickling mutation (AA) or patients with sickle cell trait who do not require treatment (AS). When validated in a pilot set of extracted genomic DNA from individuals with AA, SS and SC genotypes, the assay demonstrated an overall genotype prediction accuracy of 92.3%, with 100% sensitivity and 100% specificity at detecting pathologic alleles βS and βC in samples that produced valid test results.Following additional optimization and validation, the test developed here could meet the urgent need for a rapid, low-cost, and easy-to-use diagnostic for SCD suitable for use in all patients.37 Most children born in LMICs die of severe anemia or bacterial infections before age five, often without having received a diagnosis8,38,39 due to the high cost and complexity of conventional diagnostic methods for SCD. Although there are several commercially available tests to reduce the cost and infrastructure requirements associated with SCD diagnosis, including lateral flow immunoassays such as SickleScan™ and HemotypeSC™,40–45 and paper-based electrophoresis devices,46–48 protein-based diagnostics generally cannot be used reliably on patients without a prior diagnosis of SCD who have recently received a transfusion of red blood cells, as transfusions contain globin proteins from the donor,49–51 leading individuals with SCD to be misdiagnosed as having sickle cell trait. This is a significant limitation that delays diagnosis and critical life-saving care for many SCD patients who are emergently transfused without a prior diagnosis.50,51 In contrast, the DNA-based diagnostic developed here circumvents the challenges of protein-based testing by identifying the underlying genetic cause of SCD, and could enable more timely diagnosis for potential SCD patients who have recently undergone a blood transfusion.
The work presented here adds to the growing body of literature of using RPA to detect point mutations in a single round of isothermal amplification,26,27,29–31,52 and further elucidates the impacts of locked nucleic acids and strategic mismatches as specificity enhancing strategies in RPA. Moreover, to our knowledge, it is the first report of an assay to achieve multiplexed point mutation detection in a single round of RPA that is compatible with lateral flow readout, which simplifies the workflow and reduces the cost associated with multiple reactions and expensive readout equipment. In addition, the assay described significantly reduces instrumentation requirements of previous reports by coupling RPA with lateral flow detection. Overall, the test achieves an estimated per-test materials cost of <$7 USD (Table S4†), of which $5 USD is due to the use of pre-functionalized gold nanoshells. With an alternative reporter such as gold nanoparticles,3 and additional cost reductions expected with at-scale manufacturing, the developed test could meet the price threshold for a diagnostic SCD test determined by previous cost analyses.53
The assay developed here has some limitations that should be addressed in future work. In its current format, the test still requires manual transfer of amplicons to the sample pad of the lateral flow strip, which could lead to workspace contamination with amplified DNA. Integration into a manufacturable, self-contained disposable could mitigate the risks of such contamination. In addition, point-of-care sample preparation strategies, such as alkaline lysis with sodium hydroxide,31 enzymatic lysis with achromopeptidase,24,25,54 or heat lysis,18 need to be integrated with the assay to make it more suitable for use in low-resource settings. Moreover, additional optimization to improve assay specificity, genotype accuracy, and enable consistent and definitive visual interpretation is needed; in the assay's current form, a low-cost lateral flow reader would be required for genotype prediction. Finally, a validation study with a larger set of clinical samples, especially in samples with genotype AS, is needed to better understand the assay's ability to differentiate SS patients that require treatment from AS patients that do not.
In conclusion, we developed a multiplexed RPA assay with lateral flow readout that is capable of identifying human genomic DNA containing point mutations encoding for pathologic βS and βC globins in a single round of isothermal amplification, and can be used to determine a person's genotype. We validated its performance with extracted genomic DNA from clinical samples and demonstrated strong genotype prediction agreement with a clinical reference test. With incorporation of sample preparation strategies, additional assay optimization, and a larger clinical evaluation, the developed test could enable DNA-based diagnosis of sickle cell disease for patients in low-resource settings.
Data availability statement
The data supporting the findings of this study are available within the paper and its ESI† files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.Author contributions
Conceptualization: MMC, MEN, RRK. Investigation: MMC, MEN. Methodology: MMC, AFW, MEN. Visualization: MMC, RRK. Supervision: VNT, GEA, RRK. Writing- original draft: MMC, RRK. Writing – review & editing: MMC, AFW, MEN, VNT, GEA, RRK.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Research in this paper was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Numbers F31HL154614 and R01HL166413. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to thank Dr. Meaghan Bond for helpful discussions and feedback, and gratefully acknowledge Tripti Halder and Dr. Meaghan Bond for their assistance with clinical samples.References
- A. Deshpande and P. S. White, Multiplexed nucleic acid-based assays for molecular diagnostics of human disease, Expert Rev. Mol. Diagn., 2012, 12(6), 645–659 CrossRef CAS PubMed.
- O. Mayboroda, I. Katakis and C. K. O'Sullivan, Multiplexed isothermal nucleic acid amplification, Anal. Biochem., 2018, 545, 20–30 CrossRef CAS PubMed.
- Z. Crannell, A. Castellanos-Gonzalez, G. Nair, R. Mejia, A. C. White and R. Richards-Kortum, Multiplexed Recombinase Polymerase Amplification Assay To Detect Intestinal Protozoa, Anal. Chem., 2016, 88(3), 1610–1616 CrossRef CAS PubMed.
- B. Ma, J. Li, K. Chen, X. Yu, C. Sun and M. Zhang, Multiplex recombinase polymerase amplification assay for the simultaneous detection of three foodborne pathogens in seafood, Foods, 2020, 9(3), 278 CrossRef CAS.
- C. Lu, J. Wang, L. Pan, X. Gu, W. Lu and D. Chen, et al., Rapid detection of multiple resistance genes to last-resort antibiotics in Enterobacteriaceae pathogens by recombinase polymerase amplification combined with lateral flow dipstick, Front. Microbiol., 2023, 13, 1062577 CrossRef PubMed.
- J. Li, N. M. Pollak and J. Macdonald, Multiplex detection of nucleic acids using recombinase polymerase amplification and a molecular colorimetric 7-segment display, ACS Omega, 2019, 4(7), 11388–11396 CrossRef CAS PubMed.
- A. V. Ivanov, I. V. Safenkova, A. V. Zherdev and B. B. Dzantiev, Multiplex Assay of Viruses Integrating Recombinase Polymerase Amplification, Barcode-Anti-Barcode Pairs, Blocking Anti-Primers, and Lateral Flow Assay, Anal. Chem., 2021, 93(40), 13641–13650 CrossRef CAS PubMed.
- G. J. Kato, F. B. Piel, C. D. Reid, M. H. Gaston, K. Ohene-Frempong and L. Krishnamurti, et al., Sickle cell disease, Nat. Rev. Dis. Primers, 2018, 4(1), 1–22 Search PubMed.
- F. B. Piel, A. P. Patil, R. E. Howes, O. A. Nyangiri, P. W. Gething and M. Dewi, et al., Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates, Lancet., 2013, 381(9861), 142–151 CrossRef PubMed.
- H. F. Bunn, Pathogenesis and treatment of sickle cell disease, N. Engl. J. Med., 1997, 337(11), 762–769 CrossRef CAS PubMed.
- M. E. Natoli, B. A. Rohrman, C. De Santiago, G. U. van Zyl and R. R. Richards-Kortum, Paper-based detection of HIV-1 drug resistance using isothermal amplification and an oligonucleotide ligation assay, Anal. Biochem., 2018, 544, 64–71 CrossRef CAS PubMed.
- E. S. Yamanaka, L. A. Tortajada-Genaro and A. Maquieira, Low-cost genotyping method based on allele-specific recombinase polymerase amplification and colorimetric microarray detection, Microchim. Acta, 2017, 184(5), 1453–1462 CrossRef CAS.
- S. Martorell, S. Palanca, A. Maquieira and L. A. Tortajada-Genaro, Blocked recombinase polymerase amplification for mutation analysis of PIK3CA gene, Anal. Biochem., 2018, 544, 49–56 CrossRef CAS PubMed.
- A. Lázaro, Á. Maquieira and L. A. Tortajada-Genaro, Discrimination of Single-Nucleotide Variants Based on an Allele-Specific Hybridization Chain Reaction and Smartphone Detection, ACS Sensors., 2022, 7(3), 758–765 CrossRef PubMed.
- A. S. de Olazarra, D. L. Cortade and S. X. Wang, From saliva to SNP: non-invasive, point-of-care genotyping for precision medicine applications using recombinase polymerase amplification and giant magnetoresistive nanosensors, Lab Chip, 2022, 22(11), 2131–2144 RSC.
- S. Martorell, Á. Maquieira and L. A. Tortajada-Genaro, A genosensor for detecting single-point mutations in dendron chips after blocked recombinase polymerase amplification, Analyst, 2022, 147(10), 2180–2188 RSC.
- P. Thakur, P. Gupta, N. Bhargava, R. Soni, N. Varma Gottumukkala and S. G. Goswami, et al., A Simple, Cost-Effective, and Extraction-Free Molecular Diagnostic Test for Sickle Cell Disease Using a Noninvasive Buccal Swab Specimen for a Limited-Resource Setting, Diagnostics., 2022, 12(7), 1765 CrossRef CAS PubMed.
- M. Ortiz, M. Jauset-Rubio, D. Kodr, A. Simonova, M. Hocek and C. K. O'Sullivan, Solid-phase recombinase polymerase amplification using ferrocene-labelled dNTPs for electrochemical detection of single nucleotide polymorphisms, Biosens. Bioelectron., 2022, 198, 113825 CrossRef CAS PubMed.
- B. Y. C. C. Ng, E. J. H. H. Wee, K. Woods, W. Anderson, F. Antaw and H. Z. H. H. Tsang, et al., Isothermal Point Mutation Detection: Toward a First-Pass Screening Strategy for Multidrug-Resistant Tuberculosis, Anal. Chem., 2017, 89(17), 9017–9022 CrossRef CAS PubMed.
- D. S. Boyle, D. A. Lehman, L. Lillis, D. Peterson, M. Singhal and N. Armes, et al., Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification, MBio, 2013, 4(2), e00135-13 CrossRef PubMed.
- R. K. Daher, G. Stewart, M. Boissinot, D. K. Boudreau and M. G. Bergeron, Influence of sequence mismatches on the specificity of recombinase polymerase amplification technology, Mol. Cell. Probes, 2015, 29(2), 116–121 CrossRef CAS PubMed.
- M. Higgins, O. W. Stringer, D. Ward, J. Andrews, M. S. Forrest and S. Campino, et al., Characterizing the Impact of Primer-Template Mismatches on Recombinase Polymerase Amplification, J. Mol. Diagn., 2022, 24(11), 1207–1216 CrossRef CAS PubMed.
- C. P. Mancuso, Z.-X. Lu, J. Qian, S. A. Boswell and M. Springer, A Semi-Quantitative Isothermal Diagnostic Assay Utilizing Competitive Amplification, Anal. Chem., 2021, 93(27), 9541–9548 CrossRef CAS.
- K. A. Kundrod, M. Barra, A. Wilkinson, C. A. Smith, M. E. Natoli and M. M. Chang, et al., An integrated isothermal nucleic acid amplification test to detect HPV16 and HPV18 DNA in resource-limited settings, Sci. Transl. Med., 2023, 15(701), eabn4768 CrossRef CAS PubMed.
- M. M. Chang, A. Ma, E. N. Novak, M. Barra, K. A. Kundrod and J. R. Montealegre, et al., A novel tailed primer nucleic acid test for detection of HPV 16, 18 and 45 DNA at the point of care, Sci. Rep., 2023, 13(1), 20397 CrossRef PubMed.
- Y. Liu, T. Lei, Z. Liu, Y. Kuang, J. Lyu and Q. Wang, A novel technique to detect EGFR mutations in lung cancer, Int. J. Mol. Sci., 2016, 17(5), 792 CrossRef PubMed.
- N. Singpanomchai, Y. Akeda, K. Tomono, A. Tamaru, P. Santanirand and P. Ratthawongjirakul, Rapid detection of multidrug-resistant tuberculosis based on allele-specific recombinase polymerase amplification and colorimetric detection, PLoS One, 2021, 16(6), e0253235 CrossRef CAS PubMed.
- M. Ortiz, M. Jauset-Rubio, O. Trummer, I. Foessl, D. Kodr and J. L. Acero, et al., Generic Platform for the Multiplexed Targeted Electrochemical Detection of Osteoporosis-Associated Single Nucleotide Polymorphisms Using Recombinase Polymerase Solid-Phase Primer Elongation and Ferrocene-Modified Nucleoside Triphosphates, ACS Cent. Sci., 2023, 9(8), 1591–1602 CrossRef CAS PubMed.
- L. Zhang, J. Peng, J. Chen, L. Xu, Y. Zhang and Y. Li, et al., Highly Sensitive Detection of Low-Abundance BRAF V600E Mutation in Fine-Needle Aspiration Samples by Zip Recombinase Polymerase Amplification, Anal. Chem., 2021, 93(13), 5621–5628 CrossRef CAS PubMed.
- S. Duan, G. Li, X. Li, C. Chen, T. Yan and F. Qiu, et al., A probe-directed recombinase amplification assay for detection of MTHFR A1298C polymorphism associated with congenital heart disease, BioTechniques, 2018, 64(5), 211–217 CrossRef CAS PubMed.
- M. E. Natoli, M. M. Chang, K. A. Kundrod, J. B. Coole, G. E. Airewele and V. N. Tubman, et al., Allele-Specific Recombinase Polymerase Amplification to Detect Sickle Cell Disease in Low-Resource Settings, Anal. Chem., 2021, 93(11), 4832–4840 CrossRef CAS PubMed.
- R. K. Saiki, D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi and G. T. Horn, et al., Primer-Directed Enzymatic Amplification of DNA with a Thermostable DNA Polymerase, Science, 1988, 239(4839), 487–491 CrossRef CAS PubMed.
- TwistDx. TwistAmp® DNA Amplification Kits Assay Design Manual [Internet].
- L. Lillis, J. Siverson, A. Lee, J. Cantera, M. Parker and O. Piepenburg, et al., Factors influencing Recombinase polymerase amplification (RPA) assay outcomes at point of care, Mol. Cell. Probes, 2016, 30(2), 74–78 CrossRef CAS PubMed.
- E. I. Newsham, E. A. Phillips, H. Ma, M. M. Chang, S. T. Wereley and J. C. Linnes, Characterization of wax valving and μPIV analysis of microscale flow in paper-fluidic devices for improved modeling and design, Lab Chip, 2022, 22(14), 2741–2752 RSC.
- D. Gasperino, T. Baughman, H. V. Hsieh, D. Bell and B. H. Weigl, Improving Lateral Flow Assay Performance Using Computational Modeling, Annu. Rev. Anal. Chem., 2018, 11(1), 219–244 CrossRef PubMed.
- D. Dexter and P. T. McGann, Saving lives through early diagnosis: the promise and role of point of care testing for sickle cell disease, Br. J. Haematol., 2022, 196(1), 63–69 CrossRef PubMed.
- F. B. Piel, S. I. Hay, S. Gupta, D. J. Weatherall and T. N. Williams, Global Burden of Sickle Cell Anaemia in Children under Five, 2010-2050: Modelling Based on Demographics, Excess Mortality, and Interventions. Osrin D., editor, PLoS Med., 2013, 10(7), e1001484 CrossRef PubMed.
- J. Mburu and I. Odame, Sickle cell disease: Reducing the global disease burden, Int. J. Lab. Hematol., 2019, 41(S1), 82–88 CrossRef PubMed.
- M. M. Nwegbu, H. A. Isa, B. B. Nwankwo, C. C. Okeke, J. Edet-Offong and N. O. Akinola, et al., Preliminary Evaluation of a Point-of-Care Testing Device (SickleSCAN TM) in Screening for Sickle Cell Disease, Hemoglobin, 2017, 41(2), 77–82 CrossRef CAS PubMed.
- A. Y. Segbena, A. Guindo, R. Buono, I. Kueviakoe, D. A. Diallo and G. Guernec, et al., Diagnostic accuracy in field conditions of the sickle SCAN® rapid test for sickle cell disease among children and adults in two West African settings: the DREPATEST study, BMC Hematol., 2018, 18, 26 CrossRef PubMed.
- M. B. Mukherjee, R. B. Colah, P. R. Mehta, N. Shinde, D. Jain and S. Desai, et al., Multicenter Evaluation of HemoTypeSC as a Point-of-Care Sickle Cell Disease Rapid Diagnostic Test for Newborns and Adults Across India, Am. J. Clin. Pathol., 2020, 153(1), 82–87 CrossRef CAS PubMed.
- O. Nnodu, H. Isa, M. Nwegbu, C. Ohiaeri, S. Adegoke and R. Chianumba, et al., HemoTypeSC, a low-cost point-of-care testing device for sickle cell disease: Promises and challenges, Blood Cells, Mol., Dis., 2019, 78, 22–28 CrossRef PubMed.
- R. Nankanja, S. Kadhumbula, A. Tagoola, M. Geisberg, E. Serrao and S. Balyegyusa, HemoTypeSC Demonstrates >99% Field Accuracy in a Sickle Cell Disease Screening Initiative in Children of Southeastern Uganda, Am. J. Hematol., 2019, 94(6), E164–E166 CrossRef PubMed.
- C. Steele, A. Sinski, J. Asibey, M.-D. D. Hardy-Dessources, G. Elana and C. Brennan, et al., Point-of-care screening for sickle cell disease in low-resource settings: A multi-center evaluation of HemoTypeSC, a novel rapid test, Am J Hematol., 2019, 94(1), 39–45 CrossRef PubMed.
- M. N. Hasan, A. Fraiwan, R. An, Y. Alapan, R. Ung and A. Akkus, et al., Paper-based microchip electrophoresis for point-of-care hemoglobin testing, Analyst, 2020, 145(7), 2525–2542 RSC.
- R. An, Y. Man, S. Iram, E. Kucukal, M. N. Hasan and Y. Huang, et al., Point-of-care microchip electrophoresis for integrated anemia and hemoglobin variant testing, Lab Chip, 2021, 21(20), 3863–3875 RSC.
- K. Qua, S. M. Swiatkowski, U. A. Gurkan and C. M. Pelfrey, A retrospective case study of successful translational research: Gazelle Hb variant point-of-care diagnostic device for sickle cell disease, J. Clin. Transl. Res., 2021, 5(1), e207 Search PubMed.
- J. Michlitsch, M. Azimi, C. Hoppe, M. C. Walters, B. Lubin and F. Lorey, et al., Newborn screening for hemoglobinopathies in California, Pediatr. Blood Cancer, 2009, 52(4), 486–490 CrossRef PubMed.
- W. Reed, P. A. Lane, F. Lorey, J. Bojanowski, M. Glass and R. R. Louie, et al., Sickle-cell disease not identified by newborn screening because of prior transfusion, J. Pediatr., 2000, 136(2), 248–250 CrossRef CAS PubMed.
- L. R. Smart, E. E. Ambrose, K. C. Raphael, A. Hokororo, E. Kamugisha and E. A. Tyburski, et al., Simultaneous point-of-care detection of anemia and sickle cell disease in Tanzania: the RAPID study, Ann. Hematol., 2018, 97(2), 239–246 CrossRef PubMed.
- M. Natoli, M. Chang, K. Kundrod, J. Coole, G. Airewele and V. N. Tubman, et al., Allele-specific recombinase polymerase amplification for real-time detection of sickle cell anaemia in low-resource settings: evaluation of an isothermal nucleic acid amplification test to detect the βS globin point mutation in paediatric patients, Lancet Global Health, 2021, 9, S13 CrossRef.
- M. Mvundura, C. Kiyaga, M. Metzler, C. Kamya, J. M. Lim and C. Maiteki-Sebuguzi, et al., Cost for sickle cell disease screening using isoelectric focusing with dried blood spot samples and estimation of price thresholds for a point-of-care test in Uganda, J. Blood Med., 2019, 10, 59–67 CrossRef PubMed.
- C. A. Smith, M. M. Chang, K. A. Kundrod, E. N. Novak, S. G. Parra and L. López, et al., A low-cost, paper-based hybrid capture assay to detect high-risk HPV DNA for cervical cancer screening in low-resource settings, Lab Chip, 2023, 23(3), 451–465 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI included with submission. See DOI: https://doi.org/10.1039/d4lc00281d |
| ‡ Current affiliation: Department of Bioengineering, University of Washington, Seattle, WA, USA. |
| This journal is © The Royal Society of Chemistry 2024 |