
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave-assisted extraction, separation, and chromogenic detection of laced marijuana for presumptive point-of-interdiction testing†
Killian C.
O'Connell
 *a,
Mariana B.
Almeida
bh,
Renna L.
Nouwairi
a,
Emmet T.
Costen
c,
Nicola K.
Lawless
de,
Maura E.
Charette
f,
Brennan M.
Stewart
g,
Suzana L.
Nixdorf
h and
James P.
Landers
ai
*a,
Mariana B.
Almeida
bh,
Renna L.
Nouwairi
a,
Emmet T.
Costen
c,
Nicola K.
Lawless
de,
Maura E.
Charette
f,
Brennan M.
Stewart
g,
Suzana L.
Nixdorf
h and
James P.
Landers
ai
aDepartment of Chemistry, University of Virginia, Charlottesville, Virginia 22904, USA. E-mail: kco4yh@virginia.edu
bChemical Institute of São Carlos, University of São Paulo, São Paulo 05001, Brazil
cAerospace Structural Research Corporation, Milford, Connecticut, 06460 USA
dDepartment of Biology, University of Virginia, Charlottesville, Virginia 22904, USA
eDepartment of Cognitive Science, University of Virginia, Charlottesville, Virginia 22904, USA
fDepartment of Biochemistry, University of Virginia, Charlottesville, Virginia 22904, USA
gDepartment of Biomedical Engineering, University of Virginia, Charlottesville, Virginia 22904, USA
hDepartment of Chemistry, Londrina State University, Londrina, Paraná 86057, Brazil
iMechanical and Aerospace Engineering, University of Virginia, Charlottesville, Virginia 22904, USA
First published on 20th August 2024
Abstract
Presumptive drug screening enables timely procurement of search and arrest warrants and represents a crucial first step in crime scene analysis. Screening also reduces the burden on forensic laboratories which often face insurmountable backlogs. In most scenarios, on-site presumptive drug screening relies on chemical field tests for initial identification. However, even when used appropriately, these test kits remain limited to subjective colorimetric analysis, produce false positive or negative results with excessive sample quantities, and are known to cross-react with numerous innocuous substances. Previous efforts to develop microfluidic devices that incorporate these chromogenic indicator reagents address only a few of the many challenges associated with these kits. This is especially true for samples where the drug of interest is present as a lacing agent. This work describes the development of a centrifugal microfluidic device capable of integrating facile sample preparation, by way of a 3D printed snap-on cartridge amenable to microwave assisted extraction, followed by chromatographic separation and chromogenic detection on-disc. As cannabis is among the most widely used controlled substance worldwide, and displays strong interference with these indicator reagents, mock samples of laced marijuana are used for a proof-of-concept demonstration. Post extraction, the microdevice completes high throughput metering just prior to simultaneous reaction with four of the most commonly employed microchemical tests, followed by objective image analysis in CIELAB (a device-independent color model). Separation and recovery of a representative controlled substance with 93% efficiency is achieved. Correct identification, according to hierarchical cluster analysis, of three illicit drugs (e.g., heroin, phencyclidine, and cocaine) in artificially laced samples is also demonstrated on-disc. The cost effective microdevice is capable of complete automation post-extraction, with a total analysis time (including extraction) of <8 min. Finally, sample consumption is minimized, thereby preventing the complete destruction of forensic evidence.
Introduction
Cannabis sativa is not only the most consumed drug worldwide, with an estimated 219 million users in 2021 alone, it is also the most frequent choice among polydrug users.1,2 Although cannabis is most often used in conjunction with licit drugs such as tobacco and alcohol, illicit narcotic use remains widespread. For example, 90–98% of people who take 3,4-methylenedioxymethamphetamine (MDMA), a synthetic opioid colloquially referred to as ecstasy, also reported concomitant use of cannabis.3 This can be due to a desire to enhance the hallucinatory effect of cannabis (e.g., using lysergic acid or phencyclidine) or to use cannabis to modulate the negative side effects of stimulants (e.g., cocaine, methamphetamine, or Adderall). The health repercussions associated with paring cannabis with additional drugs can be serious. For instance, combining marijuana with hallucinogens can lead to memory impairment and increase the risks associated with mood disorders,3 while combining it with stimulants raises the potential for heart attack and stroke.4 The psychobiological impact of long term polydrug use are complex and not well understood. However, chronic use has been implicated in cumulative neurobiological impairments. Although nation-wide legalization of recreational marijuana appears imminent within the United States and elsewhere, the popularity of intentional lacing presents serious risks for inadvertent overdose and accidents.5 The manufacture and consumption of the top lacing agents for cannabis, including cocaine, synthetic opioids, and amphetamines remains in the tens of millions, with qualitative assessments suggesting an increase in use.6–8 Drug screening, either at the roadside, workplace, or in harm reduction facilities is intended to minimize these risks by preventing operation of potentially lethal machinery while under the influence. Unfortunately, the unreliable nature of the most commonly employed presumptive screening tests is well-documented.9–12There are several methods available for on-site screening of illicit drugs of interest, ranging from portable analytical instrumentation to more rudimentary chromatographic or bio/chemical test kits. Portable instrumentation, although highly discriminatory and capable of discerning new substances of concern, are expensive (5–50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 USD) and require substantial technical training to operate.13 Beyond the initial investment in acquisition are continuing expenses for reagents and periodic servicing. For forensic laboratories that do not have adequate funding or industry partnerships, these devices (portable or not) may be entirely unattainable. Alternatives include thin layer chromatography, immunoassays, and microchemical “spot” tests. Although these tests are far more affordable (100–500 USD), require no electricity, and involve minimal training, they are destructive and incapable of identifying previously uncharacterized substances with accuracy.
000 USD) and require substantial technical training to operate.13 Beyond the initial investment in acquisition are continuing expenses for reagents and periodic servicing. For forensic laboratories that do not have adequate funding or industry partnerships, these devices (portable or not) may be entirely unattainable. Alternatives include thin layer chromatography, immunoassays, and microchemical “spot” tests. Although these tests are far more affordable (100–500 USD), require no electricity, and involve minimal training, they are destructive and incapable of identifying previously uncharacterized substances with accuracy.
Despite these drawbacks, commercially available colorimetric test kits remain well-established worldwide, comprising the bulk of chemical evidence submitted to investigative crime labs each year.14 Progress has been made in addressing some of the inherent downsides to these tests, mainly the subjectivity of assessing color responses.15–17 Yet, despite advancements toward more objective measures of chromogenic results, these techniques are not wholly capable of addressing the inherent cross-reactivity of these indicator reagents.18 Advanced software methods have attempted to compensate for these scenarios with some success.14 However, they perform best when there is some preconception of sample composition (i.e., selection of the correct testing agent) or when mixed samples result in only slight changes relative to expected outcomes. However, without objective analysis, actual color results observed by an analyst can be influenced not only by differences in visual perception capabilities (i.e., color blindness) or environmental conditions (e.g., flashing police cruiser lights, inclement weather, or background landscape), but also by drug concentration, drug salt or free base form, identity of salt counterion, and contaminants.19
Previous narcotic detection microdevices have almost exclusively targeted either liquid samples (e.g., body fluids or wastewater) or dissolvable ‘unknown white powders’.17,20–24 Although highly relevant, these testing platforms are not readily adaptable to more complex solid samples, such as marijuana. The underlying choice for these sample matrices is due, in part, to the limited sample preparation required prior to analysis. However, there is an extensive array of solid sample matrices (e.g., skeletal remains25 or hair26) within the field of forensics which currently require extensive, complex extraction procedures. In a variety of instances, closed vessel microwave assisted extraction (MAE) has been demonstrated to be a rapid, streamlined technique providing higher extraction yields and greater sensitivity for target detection, be it a synthetic or biological compound.27,28 Despite the numerous advantages of closed-vessel MAE compared to conventional extraction procedures, including a reduced risk of sample contamination and degradation, its integration within microfluidic platforms remains limited.
Finally, aside from the health hazards associated with combining illicit drugs, unambiguous identification is parament for enaction of fair and equal justice under the law. There are approximately 1.5 million drug-related arrests per year in the United States alone, with many of those arrested identifying as African American.11,29,30 Even a small error rate in test results equates to an enormous number of false convictions annually.31 As a moratorium on the reliance of these tests is unlikely to pass in the near future, a strategy for addressing their key failure points is critical.32 These deficiencies include the potential for cross reactions, addition of inappropriate sample quantities, subjective color analysis, and manual record keeping. Importantly, the method of redress must maintain simplicity, rapidity, and cost efficiency if it is to be realistically adopted. As will be discussed further, although the microfluidic platform designed for this application slightly increases the time-to-result due to the additional sample preparation steps, the massively parallel reaction procedure should not only make up for this lost time, but also mitigate the potential for compounding user error.17 New benefits, such as replicate testing and partially non-destructive, high-throughput sample extraction through MAE are also realized, in addition to objective identification. Electronic recording of results also simplifies evidence preservation and efficient data cataloguing.
Materials and methods
Materials
3 mil ChemFilm fluorinated ethylene propylene type FS was generously provided by Saint-Gobain (Worcester, MA, USA), 1.5 mm clear poly(methylmethacrylate) (PMMA) 8560K171 was purchased from McMaster-Carr (Aurora, OH, USA), TRANS NS 4 MIL polyethylene terephthalate (PET) was purchased from Film Source, Inc. (Maryland Heights, MO, USA), Tokyo Film Service Co., LTD. 75 μm Lumirror X30 black PET was purchased from Toray Industries, Inc. (Tokyo, JP), both 50.8 μm Arclad® 797039 heat activated adhesive (HAA) and 141 μm Arcare® 90106 double-sided pressure sensitive adhesive (PSA) were purchased from Adhesives Research Inc. (Glen Rock, PA, USA), Thermal Seal TS-RT2RR-100 single-sided pressure sensitive adhesive (ssPSA) was purchased from Excel Scientific, Inc. (Victorville, CA, USA), 81158 Permatex Black Silicone Adhesive Sealant was purchased from http://amazon.com (Bellevue, WA, USA), RS-F2-HTAM-02 (High Temp Resin) was purchased from FormLabs (Somerville, MA, USA), WHA1825047 glass microfiber filter paper grade GF/F and WHA1001090 qualitative filter paper grade 1 were both purchased from Whatman® GE Healthcare (Little Chalfont, UK), 66883-U 47 mm Empore™ SPE C18 Disks were purchased from MilliporeSigma (St. Louis, MO, USA), R12030B SiliaFlash Irregular Silica Gel P60 were purchased from SiliCycle (Quebec City, Quebec, Canada), polyethylene (PE) fast PCR tube strips were purchased from Eppendorf (Hamburg, GE), Lichen Cottage UFPT-F-10 polypropylene pipette tips were purchased from http://amazon.com/, Inc. (Bellevue, WA, USA), 15–45 μm pore size porous PE was generously provided by Porex Corporation (Fairburn, GA, USA).Reagents
98% sulfuric acid (H2SO4) certified ACS Plus, 38% hydrochloric acid (HCl) certified ACS Plus, glacial acetic acid (CH3COOH) certified ACS, citric acid monohydrate ACS certified, L-ascorbic acid reagent grade, 37% formaldehyde (v/v formaldehyde/water) (CH2O) ACS certified, 100% acetone (CH3)2CO certified ACS, sodium hydroxide (NaOH), cobalt thiocyanate and ammonium metavanadate were all purchased from Thermo Fisher Scientific (Waltham, MA, USA), both Fast Green FCF and tartrazine were purchased from MilliporeSigma (St. Louis, MO, USA), electrophoresis grade bromophenol blue (BPB) was purchased from Fisher Biotec (Wembley, WA, AU), D9805 Fast Blue B was purchased from MilliporeSigma (St. Louis, MO, USA). All NIK® Presumptive Drug Test Pouch Kits were purchased from Arrowhead Forensics (Lenexa, Kansas, USA). All controlled substances, including: A-007 (±)-amphetamine (amphetamine), M-023 (±)-methamphetamine methamphetamine), H-038 diacetylmorphine (heroin), C-006 Codeine, M-013 (±)-3,4-methylenedioxymethamphetamine (MDMA), P-007 phencyclidine (PCP), C-008 cocaine, C-045 cannabidiol, C-046 cannabinol, and T-005 (−)-Δ9-Tetrahydrocannabinol (THC) were purchased from Cerilliant Analytical Reference Standards (Round Rock, TX). Unless stated otherwise, all water used was Milli-Q Reference grade (MilliporeSigma, Burlington, MA, USA). 10023-LL blackberry sage loose leaf tea (blend of Sri Lankan black tea with blackberry flavor and organic sage) was purchased from Happy Lucky's Teahouse (Fort Collins, CO, USA).Software
Principal Component Analysis and Agglomerative Hierarchical Clustering were performed within XLSTAT version 2022.4.1 for Windows, XLSTAT Statistical Software, Addinsoft Inc., New York, NY USA, http://www.xlstat.com. Surface Response Modelling was performed using Statistica version 14.0.1 for Windows, Tibco, Palo Alto, CA, USA, http://www.tibco.com. All image analysis was performed using the open source software Fiji.33 All other statistical analysis, including ANOVA and confidence interval calculations, were performed using GraphPad Prism version 9.4.1 for Windows, GraphPad Software, San Diego, California USA, http://www.graphpad.com. Real time flow rate videos were analysed within Adobe Premier Pro Version 22.6.2 (Build 2) for Windows, Adobe Creative Cloud and Acrobat, San Jose, California USA, http://www.adobe.com. Microdevice designs were created within AutoCAD version 2018 for Windows, AutoDesk Inc., San Francisco, CA, USA, http://www.autodesk.com. 3D printed components were designed in SolidWorks version 2022 for Windows, Vèlizy-Villacoublay, France, EU, Dassault Systèmes, http://www.3ds.com and prepared for 3D printing using PreForm version 2.16.0 for Windows, Somerville, Massachusetts USA, http://www.formlabs.com.Preparation of chromogenic detection reagents
Chromogenic detection reagents were prepared according to the National Institute of Justice (NIJ) Standard for Color Test Reagents/Kits for Preliminary Identification of Drugs of Abuse (Fig. S1†).34 Briefly, the Marquis reagent was prepared as a 1% (v/v) solution of 37% (v/v) formaldehyde in 98% (v/v) H2SO4. The Mandelin reagent was prepared as a 1% (w/v) solution of ammonium metavanadate in 98% (v/v) H2SO4. The Mecke reagent was prepared as a 1% (w/v) solution of selenious acid in 98% (v/v) H2SO4. The Scott reagent was prepared as a 20% (w/v) solution of cobalt(III) thiocyanate in 10% acetic acid.The optimal testing solution for Fast Blue B (FBB) salt was found to be 0.2% (w/v) dissolved in CH3OH added to Whatman 1 filter paper pre-treated with [1 M] NaOH (Fig. S10†).
Screening of illicit drugs of interest across all chromogenic detection reagents
1 mg mL−1 drug standards were vacufuged to remove storage solvents, then reconstituted to 10 mg mL−1 in CH3OH. 2.5 mm diameter GF/F microfiber filters were arranged on a clean borosilicate glass slide then placed within a custom photo studio box. The studio box was lined with reflective material on all sides, with the exception of the bottom, which provided a matte white background. Once assembled, the photo box included only two slots: one allowing access for a white light source and the second for placement of a smartphone camera for image capture. The imaging box provided consistent lighting conditions and camera focal distances throughout all imaging procedures. Immediately prior to sample standard addition, 2.0 μL of the indicator reagent was deposited per GF/F punch, followed by the addition of 1.0 μL of either CH3OH (negative control) or a drug standard. Chromogenic responses recorded using a smartphone and an image capture sequence of 30, 60, 120, and 180 sec post drug standard addition. All images taken after 180 sec were converted to a CIELAB stack within Fiji. A standardized ROI selection across all replicates was used to extract mean L*a*b* values, then organized within either a 3 × 27 matrix per indicator reagent (Fig. 1A–D), or a 12 × 27 matrix for the combined analysis (Fig. 1E). A Pearson's correlation principal component analysis (PCA) was separately performed for all five matrices using XLSTAT statistical software. Qualitative supplementary variables were provided for the calculation of 95% confidence ellipses according to the six drug classes. All observations were standardized prior to PCA performance to avoid variable weighting between the different L*a*b* stack scales. | ||
| Fig. 1 Comparison of chromogenic response for pure vs. mixed illicit drug samples (n = 3). PCA analysis using pixel values from the CIELAB color model for both pure drug standards of D-amphetamine (amphetamine), D-methamphetamine (methamphetamine), diacetylmorphine (heroin), codeine, 3,4-methylenedioxymethamphetamine (MDMA), phencyclidine (PCP), cocaine, and Δ9-tetrahydrocannabinol (Δ9-THC) as well as 1:1 mixtures with Δ9-THC after reaction with the A. marquis, B. Mandelin, C. Mecke, and D. Scott commercial indicator reagents. E. PCA factor plane combining all CIELAB chemometric results across all indicator reagents. Centroids represent the 95% confidence interval, according to chi-square analysis, for specified drug classes. Insets display example images of actual spot test results used for subsequent image analysis. The top row “(−)” are the results from pure standards while the bottom row “(+)” are with 1:1 mixtures with Δ9-THC. Blank samples contain only solvent. | ||
Dynamic micro solid phase extraction optimization of illicit drugs of interest
A calibration curve of codeine drug standard reacted with the Marquis reagent was performed according to the procedure outlined in the previous section (Fig. 2C). Codeine concentrations ranging from 0–10 mg mL−1, in 2.5 mg mL−1 increments with an additional 1 mg mL−1 test point, were evaluated across four replicates. In contrast to the previous procedure, image analysis was performed within the HSB color model to quantify increasing mean saturation values. | ||
| Fig. 2 Selection of pH adjusting excipient and optimal heating time for lacing agents (mean ± SD). A. Plot of percent of ionized species expected in a sample after pH adjustment, assuming a pKa of 8 (light green) or 10 (dark green), representing the lowest and highest pKa of the illicit drugs evaluated. B. pKa value scale for all illicit drugs tested. Highlights within structure indicate location of positive charge for amine salt formation. The log(P) coefficients for each molecule are provided below their individual pKa values (the pKa for amphetamine and MDMA are identical). C. Calibration curve of saturation values measured in the HSB color model for codeine reacted with the Marquis reagent (R2 = 0.9886). Shading along the curve represents the 95% confidence interval around the line of best fit. Insets above the curve show example results used for subsequent image analysis. D. Heatmap results of separation efficiency for codeine in various pH-modifying extraction solvents, after partitioning with octyldecyl silane (C18) modified silica particles. The concentration of codeine in the organic vs. aqueous phase was calculated according to the previous calibration curve. A negative control containing only solvent was used as a reference for each liquid phase. A neutral pH positive control, with no applied heat (room temperature “rt” versus heat “Δ”), was used as a reference for all codeine containing samples. E. A multiple comparisons ANOVA was performed to assess whether any result (positive–negative) differed significantly from its respective negative control (α = 0.01). Numbers correspond accordingly: 1) H2O (rt), 2) 3% HCl in H2O (rt), 3) 3% HCl in H2O (Δ), 4) 3% HCl in (CH3)2CO (Δ), 5) 10% citric in H2O (Δ), 6) 10% ascorbic in H2O (Δ), and 7) 10% acetic in H2O (Δ). The only two values which were found to not be significantly different from the negative control are highlighted (light green) (p-values of 0.5962 and0.8238 for 3 and 4, respectively). | ||
All acidified extraction solvents were prepared on the day of testing. The pH of each solution was verified using Hydrion pH paper. The Marquis reagent and codeine standards were prepared as previously described, with codeine reconstituted in acidified extraction solvent to a final concentration of 10 mg mL−1. Once reconstituted, samples were placed within a 90 °C hot water bath for 5 min before being removed and allowed to equilibrate back to room temperature.
2.5 mg of C18-functionalized SPE microparticles were distributed among separate PCR tubes, with the microparticle mass matching the amount contained within the microdevice PMMA μColumns (Fig. S4Ai†). Prior to the introduction of extracted drug standards, the SPE microparticles were conditioned as follows: 40 μL of CH3OH was added to the microparticles and briefly mixed. The dispersed microparticles were then centrifuged at 3000 RPM for 10 sec, followed by removal of the supernatant. This same process was repeated with MQ H2O. A reconstituted codeine sample was then transferred to the microparticles and mixed briefly to ensure resuspension. Samples were then centrifuged at 3000 RPM for 10 sec, followed by collection of the aqueous supernatant. Next, 5 μL of CH3OH was added to the microparticles, mixed briefly, centrifuged, and the organic supernatant collected. The aqueous supernatant was vacufuged to a 5 μL final volume in order to normalize the comparison in saturation values between the organic and aqueous phases. Samples were tested as described previously, with image analysis performed in the HSB color model to quantify differences in mean saturation values. These values were then used to calculated codeine concentration in each phase according to a previously generated calibration curve (Fig. 2C).
Real time flow rate analysis
Simplified 12-plex microdevices with SPE μColumns were fabricated according to the PCL method. Sample input chambers were designed with equidistant rastered markings to quantify mock aqueous sample drainage over time through each μColumn. Real time monitoring of sample drainage was performed using a custom built stroboscopic spin system, composed of a DC brushless motor (EFLM1400 890 kV E-flite Park 450 Brushless Outrunner Motor; RC Visions, Huntington Beach, CA, USA), high intensity white light LED ring (Everbright 70 mm White 60 SMD COB LED Headlight; Amazon, Bellevue, WA, USA), and high speed video camera (MotionBLITZ EoSens® mini high-speed CMOS recording camera; Mikrotron-GmbH, Unterschleißheim, Germany), with an attached telephoto lens (TV ZOOM LENS G6X16 16–100 mm 1:1.9 1′′ macro; Mikrotron-GmbH). A custom breadboard (Propeller P8X32A-M44; Propeller Inc., Rockland, CA, USA) and software program (Propeller 1 Software for Windows; Parallax Inc. Rocklin, CA, USA) synced the motor spin frequency with the LED stroboscope by way of a photointerrupting optical switch (TT Electronics/Optek Technology; Woking, UK) which provided programmable system control through a simple user interface.
Spin frequencies ranging from 3000–5000 RPM, at intervals of 500 RPM, were monitored in real time after the addition of 100 μL of 0.1% (v/v) Allura Red Ac in MQ H2O. Flow rates were calculated based on correlating the fluid draining behavior with the theoretical volume between each rastered marking (calculated within AutoCAD) (Fig. 3A). Prior to mock aqueous sample introduction, each SPE membrane was conditioned with 80 μL of CH3OH spun through the μColumn at 4000 RPM for 50 sec. 100 μL of MQ H2O was then spun through the μColumn at 4000 RPM for 95 sec. The channel to the column conditioning waste chamber was then closed via laser-based occlusion, and the main valve to the sample collection chamber opened via laser ablation. All video files were imported into Adobe Premier Pro for frame-by-frame analysis.
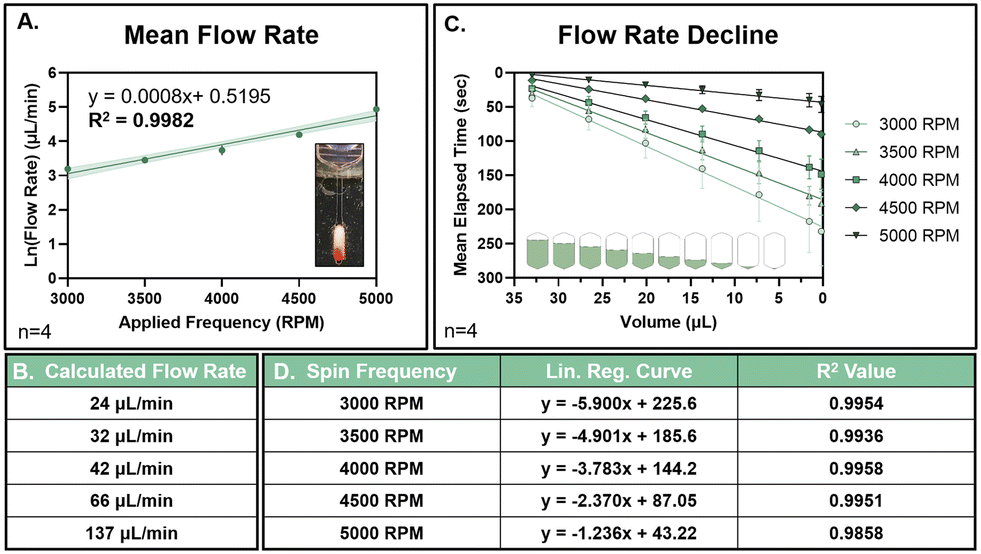 | ||
| Fig. 3 Flow rate characterization of integrated μColumn (mean ± SD). Flow rates through the integrated μColumn were evaluated according to high-speed video monitoring. A. The average flow rate per applied spin frequency was fit on a semi-log plot with a linear regression curve (R2 = 0.9982) across a frequency range of 3000–5000 RPM at 500 RPM intervals. Shading around the curve represent the 95% confidence interval around the line of best fit. B. The average calculated flow rate (μL min−1) per applied frequency is provided below. C. Real-time flow rate results per spin frequency applied. Results are converted from the average drain time per rastered demarcation to the corresponding mean elapsed time and predicted volume. At a volume of 0 μL, the sample input chamber has fully drained. D. Real time flow rate results were fit with a linear regression curve, with the corresponding equations and R2 values per applied frequency, provided in the table below. | ||
Stereolithographic 3D printing of sample preparation cartridges
Sample preparation cartridges (Fig. 4D) were fabricated using a Formlabs Form 3B+ stereolithography (SLA) 3D printer with Formlabs High Temp Resin 1. Components were printed in adaptive mode at 25 μm resolution using full support rafts. Upon print completion, components are rinsed in an IPA bath for 6 min, dried using compressed air, then cured for 120 min at 80 °C under UV exposure. A final cure step at 160 °C for 180 min, without UV exposure, ensured maximum heat resistance (≤238 °C), according to the manufacturer's recommendations. The lid and sample holder were held together using either a laser-cut PSA ring (room temperature use) or a manually applied silicone adhesive sealant (high temperature use). Solid samples were retained within the sample holder using a laser-cut filter composed of heat-resistant single-sided PSA. After addition of the extraction solvent, the fluidic port was transiently covered by an inert (PTFE) membrane attached using a single-sided PSA ring. PET tabs on either side of the membrane allowed for easy removal post dielectric heating. | ||
| Fig. 4 Performance characterization of 3D printed sample preparation cartridges. A. Calibration curve correlating the number of auto-selected pixels to known volumes of dyed aqueous solution. Shading along the curve represents the 95% confidence interval around the line of best fit. B. Percent volume recovery from sample preparation cartridges attached to a simplified microdevice. Recovered volumes were calculated according to the previously established calibration curve. Four distinct sample compositions and pre-treatment methods were compared (all contained 140 μL of dyed aqueous solution), including liquid-only “L”, liquid with a solid herbaceous component “S” (no heat step), and their heated counterparts “ΔL” and “ΔLS”, respectively. A multiple Comparison's ANOVA was performed to determine whether the recovery from any sample composition or pre-treatment method differed significantly from the liquid only domain (α = 0.01). C. Measured internal temperature immediately after exposure to dielectric heating. D. SolidWorks rendering (top) of sample preparation cartridge exposing internal features pertinent to the strategy used for internal temperature measurements. Actual image (bottom) of sample preparation cartridge used for internal temperature studies. The removable PTFE membrane cover of the fluidic port (added to prevent vapor escape during heating) is shown covering the entrance to the thermocouple insertion point. | ||
Characterization of the sample preparation cartridge
A calibration curve correlating pixel area to a known volume of [100 mM] tartrazine solution, located within a recovery chamber on-disc, was first generated with input volumes ranging from 50–140 μL, in increments of 10 μL (Fig. 4A). Tartrazine solution was first pipetted into a chamber located 26 mm away from the centre of disc rotation, matching the distance of the sample preparation cartridge distance from center. A laser valve between the input and recovery chamber was included to account for the small volume loss anticipated in the final microdevice design. After laser ablation of the valve, the disc was spun at 3000 RPM for 30 sec to recover the volume, before finally scanning the disc on a desktop scanner. Image analysis was carried out in Fiji, according to a previously published method.33,35 This process was repeated for sample preparation cartridges subjected to four separate treatment methods (Fig. 4B). In the first iteration, 140 μL of tartrazine solution was added to four separate sample preparation cartridges, sealed, then affixed to the recovery disc after a 100 sec waiting period. In the second iteration, this procedure was repeated with the addition of 30 mg of tea leaves prior to the addition of 140 μL of tartrazine solution. In the third iteration, no solid sample was included. Instead, all four cartridges were placed within the microwave (12 cm from the center of the rotating glass tray). All cartridges were then heated at 1100 W for 100 sec before immediate removal and disc attachment. In the final iteration, this process was repeated with the inclusion of 30 mg of tea leaves. Microdevice operation and image analysis was performed as previously outlined, using the previously established calibration curve to calculate volume recovery based on the measured pixel area (Fig. 4A). A modification to the published thresholding values had to be implemented for heated solid samples, as extracted compounds from the tea darkened the recovered tartrazine solution. The new optimal values were found to be Y: 50–255, U: 0–100, and V: 130–255.The internal temperature of the sample preparation cartridges was measured using a type-T thermocouple inserted through the fluidic exit port immediately after irradiation for 100, 200 or 300 sec (Fig. 4C and D). As described previously, 140 μL of MQ H2O was pipetted into the cartridge, sealed, and the fluidic exit port transiently protected using a PTFE membrane. All cartridges were placed 12 cm from the center of rotation.
Extraction optimization using response surface morphology
Simplified 3D printed sample preparation cartridges were designed to mimic the finalized microdevice sample preparation cartridges with regard to heat transfer and volume capacity (Fig. S6A†). Replicate samples were placed within three identical, isolated chambers joined only by a bottom plate. Lids with an identical fluidic port were transiently sealed using a PTFE membrane and heat resistant ssPSA. All heat-resistant simplified cartridges were 3D printed and cured according to the previously described method. All simplified cartridges were placed 12 mm from the center of rotation within the microwave during dielectric heating. Simulated SPE μColumns were prepared by laser-cutting the Empore™ PLM at 4% power, 8% speed. The surface area (8.03 mm2) of the PLM cut-outs matched the surface area of the on-disc μColumns. Each PLM was then placed within a pipette tip containing a 2.5 mm diameter porous PE frit to support the membrane, then nested within a collection vial. Prior to sample addition, each simulated μColumn was conditioned with 80 μL of CH3OH followed by 100 μL of MQ H2O.Several factors known to influence extraction were studied using response surface morphology (RSM) Box–Wilson central composite design (CCD) modelling (Table 1). These factors included pH (0–2), temperature (0–40.5 °C), and total incubation time (0–400 sec). One illicit drug of interest (codeine) and indicator reagent (the Marquis reagent) were used throughout the study. The CCD model included three levels of factorial design: low (−1), medium (0), and high (+1). The model was defined by the equation 2k + 2k + Cp, where k equates to the number of factors (3) and Cp the number of center points (5). The CV for each level was set to ±1.68 with an associated p-value of 0.10. 19 individual experiments were performed in triplicate. The microwave time required to reach a specified temperature, based on a previous calibration curve, was subtracted from the total incubation time. Two experimental conditions could not be fully accommodated with this design. For experiments 3 and 7, the specified total incubation time was less than the time required to heat to the target temperature. In these two instances, the time difference was 19 sec, representing an 8% error.
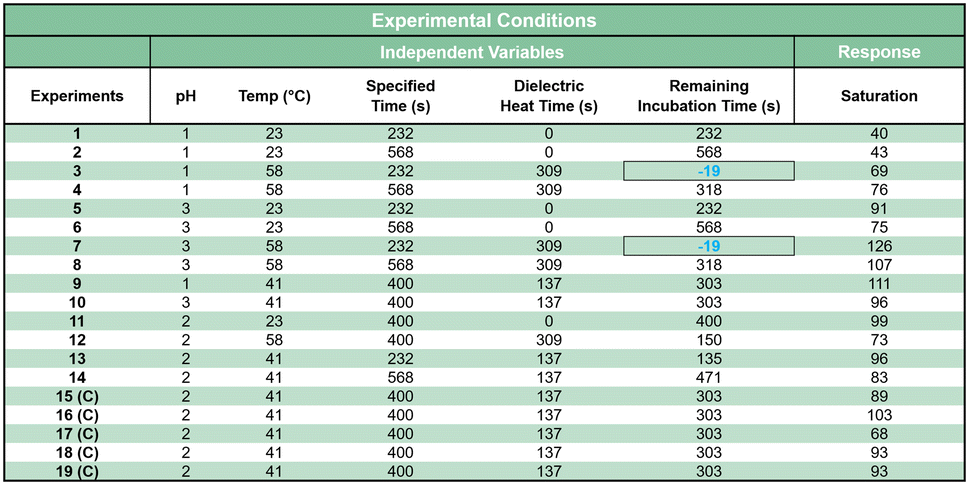
|
The Marquis reagent and acidified extraction solvents were prepared just prior to use. Codeine standards were prepared as described previously, then reconstituted to 1 mg mL−1 in CH3OH. 57 GF/F microfiber filters (2.5 mm diameter) were arranged on a clean borosilicate glass slide, pretreated with the Marquis reagent, then placed within the photo studio box. Chromogenic responses were recorded using a smartphone 180 sec after drug standard addition. Image analysis was performed within the HSB color model, to provide mean saturation values for RSM modelling (Fig. 5).
 | ||
| Fig. 5 CCD for extraction yield optimization. Response surface plots from a central composite design describe the ionization efficiency of a codeine standard based on the interaction between A. temperature (Δ) vs. pH, B. Time (θ) vs. pH, and C. Time vs. temperature. D. Pareto chart of standardized effects displays the impact of each screening factor (Δ, θ, and pH), modelled either linearly (L) or quadratically (Q), and their significance. The individual extraction parameters for each of the 19 conditions are provided in Table 1. | ||
Acidified extraction solvent and codeine standards were combined in each replicate sample preparation cartridge, covered with the 3D printed lid, heated, and incubated for the allotted time before transferal to a previously conditioned SPE μColumn. Collected samples were vacufuged to dryness then reconstituted to 10 mg mL−1 in CH3OH before deposition onto pretreated GF/F microfiber filters for image capture. Samples with complex chromogenic responses were eliminated from consideration within the model (Fig. S6B–D†).
Characterization of on-disc metering and mixing
A calibration curve (Fig. 6A), for quantifying volume delivery to the microdevice detection chambers, was established using simplified fluidic architecture consisting of an inlet and outlet channel on either side of a chamber matching the diameter of the intended detection wells. Image analysis was performed within the CIELAB color model in Fiji, with threshold parameters optimized (L*: 0–176, a*: 44–110, b*: 0–255) to select only green pixels.36 From this, a correlation between the number of green pixels to known pipetted volumes (ranging from 0.2–1.0 μL, in 0.1 μL increments) of 100% (v/v) Fast Green FCF dye was generated. | ||
| Fig. 6 Characterization of automated metering and sample mixing (mean ± SD). A. Calibration curve (R2 = 0.9816) correlating the number of pixels to known input volumes of dyed aqueous sample, with a volume range between 0.2–1.0 μL in 0.2 μL intervals. Shading around the curve represent the 95% confidence interval around the line of best fit. Examples of auto-selection during image analysis are displayed above the curve. B. Plot of average volume of metered and delivered aqueous samples per domain, according to image analysis. Actual volumes were calculated using the previously generated calibration curve. A multiple comparisons ANOVA was performed to determine whether any of the mean volumes differed significantly across domains on the simplified centrifugal microdevice (α = 0.01). C. Assessment of the mixing behavior between the metered aqueous sample with a stored reagent. Detection wells containing either tartrazine (yellow) or bromophenol blue (BPB) (blue) were measured according to their hue values as a reference. Hue measurements were repeated after the delivery of BPB to stored tartrazine (green). Insets display example images of detection wells used for analysis. Histograms display the mean pixel count per hue value across the surface of all replicate detection wells. D. Measured effect of variable input volumes on metering. Metered delivery volumes were compared after on-disc recovery from sample preparation cartridges loaded with volumes ranging from 50–125 μL. A multiple comparisons ANOVA was performed to determine whether any of the mean volumes differed significantly across input volumes (α = 0.01). | ||
An evaluation of mixing potential, between reagents pre-stored within each detection well and delivered sample aliquots, was performed by way of image analysis with aqueous dyes. 2 μL of either [100 mM] tartrazine solution or 100% (v/v) bromophenol blue (BPB) dye were added separately to 12 detection wells containing 2.5 mm diameter GF/F microfiber filters, affixed to the bottom layer of a simplified microdevice, then scanned after 180 sec using a desktop scanner. Next, 2 μL of [100 mM] tartrazine solution was added to a new set of detection wells, as previously described, before the microdevice was attached to the custom micromechanical control system. 100 μL of 100% (v/v) BPB solution was then added to the sample inlet, followed by on-disc metering and eventual delivery to the pretreated detection wells. The microdevice was scanned 180 sec after mock sample delivery and each detection well analysed for hue within the HSB color model in Fiji.
Microdevice fabrication and operation
Print-cut-laminate (PCL) centrifugal microfluidic devices layers and 2.5 mm diameter reagent storage microencapsulation wells were fabricated as described in previous publications37,38 with three modifications. Prior to lamination, both the laser-cut absorbent pad (Fig. 7A2.ii), composed of Whatman 1 filter paper, and the laser-cut functionalized membrane (Fig. 7A2.iii), composed of Empore™ C18 solid phase extraction (SPE) Disk material, were placed within their allotted cavities after aligning the main microdevice layers. Next, two additional passes through the laminator were added (total of six passes), followed by manually pressing on either edge of the SPE membrane border with flathead forceps to ensure a tight seal. A 0.1 mm overlap of the optically dense PET layer (Fig. 7A4.iii) accounted for the slight dimensional reduction of the SPE membrane during laser ablation, while a 0.2 mm overlap of the HSA layers (Fig. 7A4.ii and 4.iv) above and below the SPE membrane served to prevent liquid penetration between the interleaved microdevice layers throughout use. Fluid flow within the device was actuated using a custom-built micromechanical system with integrated laser valving, as described in previous publications (Fig. S9†).36,39 Laser valving was automated by entering the optimized control parameters, including: laser power, laser exposure time, laser z-height, laser distance from the center of rotation (CoR), number of valves, and valve angles (Fig. S9†).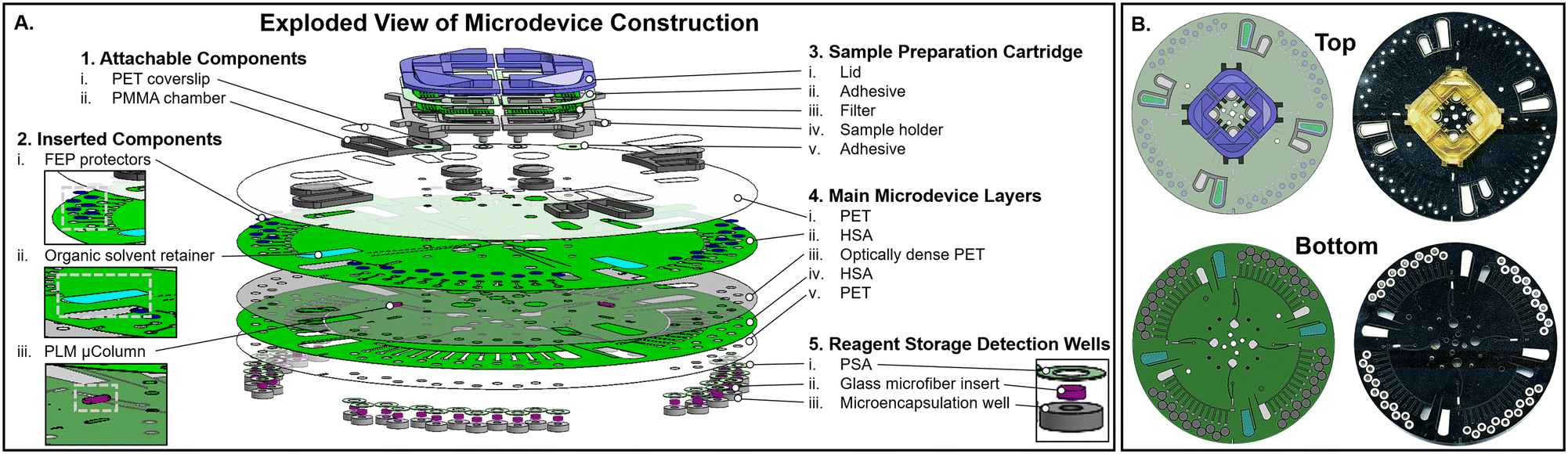 | ||
| Fig. 7 Centrifugal microfluidic device construction. A. SolidWorks rendering of four domain microdevice (exploded isometric view). 4. Each disc is composed of five main layers, fabricated according to the print, cut, and laminate (PCL) method. i. Layer 1 seals the underlying layers while also containing fluidic access ports and vents. ii. Layers 2 and iv. 4 accommodate the fluidic architecture in addition to bonding all other layers in direct contact. iii. Layer 3 acts as a programmable barrier, whereby brief laser irradiation allows for either highly localized laser ablation of layer 3 or channel occlusion in layers 2 and 4. Ablation results in a new access portal for fluid to traverse between the encompassing layers. v. Layer 5 acts as the final sealing layer of the adjoining fluidic architecture, as well as providing the fluidic exit ports. All internal layers, namely layers 2–4, contain the 2. insertable components including the PTFE corrosive vapor protective covers, the absorbent pad for organic solvent waste, and particle loaded membrane (PLM) micro-column (μColumn) for μSPE. 1. Attachable components provide additional volume capacity for select chambers within the device. 3. Initial sample preparation is performed in the attachable 3D printed cartridge, capable of holding up to 30 mg of solid herbaceous material and 100 μL of extraction solvent. Chambers are resistant to elevated temperatures for dielectric heating, easily attachable to each microdevice domain for a one-step sample interface and allow efficient passage of liquid extract while retaining solid matter. 5. Highly corrosive chromogenic reagents are stored within the detection wells according to a previously defined method. Each domain on the disc incorporates twelve detection wells. B. Top and bottom SolidWorks rendering (left) and scanned images (right) of fully fabricated 132 mm diameter centrifugal microdevice. | ||
Detection of laced illicit drugs of interest in contrived marijuana samples on-disc
30 mg of loose-leaf tea were separately spiked with 6 mg of a cannabinoid mixture containing a 1:2:0.5 ratio of Δ9-tetrahydrocannabinol, cannabidiol, and cannabinol (equating to 1.71 mg Δ9-THC, 3.44 mg CBD, and 0.85 mg CBN) were laced with either 5 mg heroin, phencyclidine, or cocaine standard. Samples were allowed to dry, protected from light, prior to the addition of either 5 mg heroin, cocaine, or PCP drug standard. An equivalent volume of CH3OH was added to one sample as a negative control. All samples were dried, protected from light, just prior to microwave assisted extraction. Sample preparation cartridges, and PCL microdevices with integrated SPE membranes, were fabricated according to the previously described methods. 2.5 mm diameter GF/F microfiber filters were loaded into acid storage wells38 pretreated with either the Marquis, Mandelin, Mecke, or Scott reagent. All wells were immediately attached to the bottom of the microdevice via PSA rings. Device operation proceeded as follows (Fig. 8): μColumns were pre-conditioned using 80 μL of CH3OH followed by 100 μL of MQ H2O at 4000 RPM for 50 and 95 sec, respectively. All conditioning reagents were directed toward the conditioning waste chamber. This chamber was closed off using laser-based occlusion and the main valve opened using laser ablation (Fig. S8†). Next, each contrived sample was loaded into a sample preparation cartridge then sealed with a silicone adhesive before the addition of 150 μL of 10% (v/v) CH3COOH. The sample preparation cartridge fluidic port was transiently sealed with a PTFE membrane prior to dielectric heating at 1100 W for 137 sec. After heating, the cartridges were immediately removed from the microwave, the PTFE membrane peeled off, and the cartridge manually pressed onto the microfluidic device top surface. Sample preparation cartridges were automatically aligned using alignment pegs, while PSA rings on the microdevice surface provided a water-tight seal. The fully assembled microdevice was loaded onto the micromechanical platform (Fig. S9†) and extracted sample passed through the conditioned μColumn via spinning at 5000 RPM for 80 sec. During this stage, solid sample components remained within the sample preparation cartridge while any extracted cannabinoids were retained within the μColumn. Sample eluent was metered across twelve separate chambers, while excess extract directed toward a secondary containment chamber. Valves below each metering chamber were opened upon executing a programmable laser valving sequence. A final spin step at 4000 RPM for 30 sec transferred all metered aliquots to the detection chambers containing pre-loaded indicator reagents. After 180 sec, the disc was scanned using a desktop scanner. Images of the detection wells were converted to a L*a*b* stack in the CIELAB color model within Fiji. A standardized ROI across all wells was used to extract mean L*a*b* values, then organized within a 12 × 39 matrix for combined reagent analysis in comparison to the previously established chemometric database. A Pearson's correlation principal component analysis (PCA) was performed using XLSTAT statistical software. Qualitative supplementary variables were provided for the calculation of 95% confidence ellipses according to the six drug classes. All observations were standardized prior to PCA performance to avoid variable weighting between the different L*a*b* stack scales. Finally, agglomerative hierarchical clustering (AHC) analysis was performed in XLSTAT, whereby the similarity index was calculated according to the Pearson correlation coefficients, and the agglomeration method based on weighted pair-group averages.
 | ||
| Fig. 8 Centrifugal microdevice protocol A. Schematic of single domain design, with major architectural features labelled. B. Sequence for on-disc sample processing. Initially, the μColumn is conditioned with the column conditioning (CC) reagents: i. a methanol wash followed by aqueous equilibration with ii. Water. Excess CC reagents flow to the designated CC waste chamber. iii. The channel to CC waste is then closed, and the main valve opened, via laser irradiation. iv. The solubilized sample, which has separately undergone MAE within the sample preparation (SP) cartridge is manually loaded onto the disc. v. The sample is subsequently passed through the μColumn, removing interfering cannabinoids, and metered into twelve 1 μL aliquots. Any excess sample is localized within the sample recovery chamber. vi. All valves leading from the metering chambers are opened, followed by a final spin step which delivers the extract into the detection chambers. vii. Any illicit drug present is reacted with the stored detection reagents (Marquis, Mandelin, Mecke, and Scott, n = 3). Finally, an image of the detection chambers is captured and any chromogenic response is noted via objective image analysis. | ||
Results and discussion
Among the indicator reagents available in the Narcotics Identification Kit (NIK®) polytesting system, both the Marquis and Scott reagents (Fig. S1†) are employed to a far greater extent than any other. This, according to an independent analysis performed by the United Nations Office on Drugs and Crime (UNODC) International Collaborative Exercise (ICE) of 181 laboratories in 67 countries.40 The reagents for these tests are provided in sealed glass ampoules contained within plastic pouches. To perform a test, the sample is dropped within a pouch, resealed, and the glass ampoule manually shattered. Some tests require consecutive breakages of multiple ampoules within the pouch, followed by intermittent shaking, before review of the final color change, if any. Some compounds must be tested further, sample amount allowing, in a cascade of tests to narrow down its presumed identity. The results from these tests have been standardized for use within the National Institutes of Justice (NIJ) Standard 0604.018 (ref. 34) as well as United Nations (UN) Manual.41 The Optical Society of America (OSA) also provides specifications for nuanced color interpretation based on the differentiation of more than 1600 Munsell colors.42 Given the difficulty in consulting the Munsell color chart in practice, in-field identification is more commonly made in reference to pre-printed swatches on the disposable pouches.13 Chemical interference from mixed samples during testing can result in several outcomes, either a false negative, false positive, or an inconclusive response is possible. False negatives occur most often when illicit material is disguised to mask detection, thereby facilitate trafficking. A prime example of this is black cocaine.43 More concerningly however, is the occurrence of false positives. Numerous examples have now been documented demonstrating incorrect identification with the commercially available NIK® test kits in the presence of innocuous substances.12,44,45 A study of the Duquenois–Levine field test kit, marketed for the identification of marijuana, found that patchouli, spearmint, and eucalyptus all resulted in a positive test result. Finally, chromogenic responses that do not match any anticipated result may indicate one or both of two possibilities: a new synthetic compound or a mixed sample.Compounding the inherent limitations of these chemical reactions, actual color results observed by an analyst may also be influenced by differences in visual perception capabilities (i.e., color blindness) or through environmental contributions (e.g., street lighting compared to sunlight). In comparison to visual inspection, digital image analysis can offer a sensitive and objective alternative for chemical identification, assuming images are taken under consistent conditions. However, standard practices are not in place for automated, objective image analysis. Custom software has been developed, in the form of smartphone applications such as ColorAssist® and Colorimeter®, to capture and evaluate image data within the red green blue (RGB) color model, with one study demonstrating good correlation across applications relative to the Munsell color chart.14 However, it was found that changes in the camera model, focusing distance, enablement of flash, or unintended tinting limited full correlation between results. Although the RGB color model is perhaps the most well-known, neither the values within each channel nor the combination of all channels translate intuitively to human perception. The Munsell color chart was the first color model to separate perceived color into the three properties of hue, intensity, and brightness (HSB) in order to map them into perceptually uniform, independent dimensions.46 Yet critically, both RGB and HSB are device dependent color models.47 Whereby separate devices will detect or reproduce a given RGB or HSB value differently. Therefore, without the use of a device independent model, the variances observed between applications would only serve to exacerbate the challenges faced from chromogenic abnormalities due to mixed composition samples.
Finally, whether or not an indicator reagent possesses an intrinsic color prior to any reaction (e.g., the Scott and Mandelin reagents) may also confound straightforward detection strategies across reagents when relying upon single-channel image analysis23 (Fig. S2†). Therefore, simultaneous detection across reagents and drug classes, assuming no preconception of the illicit drug of interest, necessitates an agnostic yet robust identification method. Two approaches in combination showed promise for attaining this objective. The first was the use of a device independent color model, CIELAB, which is defined relative to the Commission Internationale de l'Éclairage (CIE) standard observer in place of any particular monitor or printer.48 As with RGB and HSB, CIELAB is quantized using three values, with a* and b* representing chromaticity in the green-red and blue-yellow opponent systems respectively, and L* representing luminosity. Although all smartphone photodetectors employ three-channel RGB sensors, necessitating camera model and ambient lighting consistency, translation of these RGB indices to their corresponding CIELAB coordinates enables precise color communication across devices for accurate analysis, evidence preservation, and image reproduction. Second, in order to effectively navigate the differences attributed to indicator reagents with or without an inherent color, a combinatorial approach across channels was employed using principal component analysis (PCA). Importantly, unlike direct 3D scatter plots of raw data values,14 PCA provides non-correlated factors that can be used for logistic regression modelling or discriminant analysis.49
Chemometric image analysis of illicit drugs of interest
To determine whether a multivariate approach would enable discernment between separate drugs or drug classes, an initial screening across four indicator reagents (Marquis, Mandelin, Mecke, and Scott) against seven controlled substances (D-amphetamine, D-methamphetamine, diacetylmorphine (heroin), codeine, 3,4-methylenedioxymethamphetamine (MDMA), phencyclidine (PCP), cocaine, and Δ9-THC) was performed (Fig. 1A). Alongside each of the pure drug standards, a negative control containing only solvent was included. Mixed samples containing Δ9-tetrahydrocannabinol (Δ9-THC), the main psychoactive component in Cannabis sativa, were also assessed to determine the cannabinoid's potential to cross react with each indicator reagent. The results from these tests were analysed using the CIELAB color model, and the values for L*, a*, and b* used for PCA according to a Pearson's correlation matrix. Factors for the observation plane were selected to maximize the descriptive potential of the data, with a cumulative variability of at least 85%. Scatter plots of the chosen factors were then plotted with the inclusion of 95% confidence ellipses per supplementary variable (i.e., drug classification) (Fig. 1A–D, top row). This process was repeated for the 1:1 mixed samples with Δ9-THC (Fig. 1A–D, bottom row). Finally, all observations across the four reagents were combined into a single matrix for PCA (Fig. 1E). From these results, clear groupings between the separate drug classes, and in many cases individual drugs, were observable for each indicator reagent. As not all compounds react with a unique color, or at all, to each indicator, some degree of overlap between individual drugs (according to class) or with the negative control was anticipated. For example, neither cocaine nor PCP is expected to provide any chromogenic response with the Marquis reagent (Fig. S1†). As such, the tight grouping of the factor coordinates for these two compounds around the negative control values match expectation (Fig. 1A, top row). The combination of all four reagents in the final PCA demonstrated the additive discriminatory capabilities attainable with multivariate analysis. Conspicuously, only Δ9-THC was observed to partially overlap a 95% confidence ellipse outside of its drug class, partially mimicking the responses of amphetamines. Further, upon the addition of Δ9-THC to each standard, discrimination between the tested compounds essentially failed across all reagents. For those compounds that did remain distinct, their response was altered and no longer aligned with the originally calculated centroid, precluding accurate identification of the correct drug class. Notably, interference from Δ9-THC did not result in uniform masking of the original color. Instead, unique color combinations prevented identification of even the interfering agent itself. It is worth noting that these chromogenic responses were the result of 1:1 mixtures. However, in a more realistic scenario, the illicit drug of interest would be present in much lower quantities as a lacing agent. From these results, it was apparent that a widely inclusive sample preparation method, capable of removing marijuana and its associated cannabinoids, was necessary prior to presumptive screening with these reagents.
Sample extraction optimization and microdevice integration
Due to the high lipophilicity of cannabinoids, (octyl)decyl (C18) silane bonded silica would provide a method for high-capacity entrapment and removal within an aqueous mobile phase. However, as many of the illicit compounds of interest are present in neutral form (i.e., weak bases), their solubility within water alone would be inadequate for efficient recovery.53,54 Salt formation is commonly employed to enhance aqueous solubility as well as the dissolution rate of pharmaceutical type compounds. Although solution pH has been previously determined to be the most critical factor determining ionization status, particularly with regard to avoiding disproportionation,55 the influence of raised temperature and distinctive acidic excipients in extraction efficiency have also been found to promote the conversion to free acid form.56–58 Much of this effect may be attributable to the reduction in solvent surface tension and viscosity at elevated temperatures, which improve both wetting behaviour and solid matrix penetration. The specific pKa and logP values for the various compounds under investigation are provided in Fig. 2B. Among the possible cannabinoid constituents within a sample of marijuana, Δ9-THC is known to degrade readily in acidic solutions according to first-order kinetics. Both Δ8-THC and cannabinol (CBN) represent the primary degradation products in this process, alongside lower quantities of other less well-characterized compounds.59,60 Critically, each of these degradation products remain highly lipophilic59 (Fig. 2B). Even Δ9-THC metabolites, such as Δ9-THC–COOH, remain best extracted with a C18-functionalized sorbent.61
To determine the ability to effectively ionize the various primary (amphetamine), secondary (MDMA and methamphetamine), and tertiary amines (remaining compounds, excluding Δ9-THC), an initial proof-of-concept screening of several acidic excipients was performed to assess their relative impact on the separation efficiency of codeine from an organic (methanol) to an aqueous phase. Codeine was chosen as a representative molecule given the abundance of tertiary amines among the illicit drugs, alongside its uncomplicated image analysis for the creation of a standard curve (Fig. 2C). Samples of a codeine drug standard were rehydrated in five separate acidic solvents followed by the addition of C18 particles for dispersive μSPE. Prior to μSPE, a subset of these samples were subjected to elevated temperature. After brief dispersive mixing with the functionalized particles, the aqueous supernatant was removed and tested in reference to an organic supernatant. The effect of each acidic excipient was compared to a sample at neutral pH, while the effect of heat was compared between two otherwise identical preparations of 3% (v/v) hydrochloric acid (HCl). One extraction solution was prepared with acetone. A heatmap, comparing the calculated concentration of partitioned codeine for each set of conditions, is shown in Fig. 2D. A Multiple Comparisons ANOVA was performed to assess whether any result differed significantly from its respective negative control (α = 0.01) (Fig. 2E). From this, the effect of lowering the pH was immediately evident between water and 3% (v/v) HCl at 22 °C. For water alone, close to 90% of codeine remained within the organic phase, compared to less than 25% at a pH of 1–2. Without heat, the recovery of codeine in either phase remained markedly low. This is surmised to be due to ineffectual recovery from the polyethylene tube walls, later tested by adding Marquis reagent directly to the emptied tube and observing a strong color in response. Heating of the same solution raised the partition of codeine to the aqueous phase from <30% to >70%. As anticipated, the inclusion of a small percentage of acetone improved partitioning to the aqueous phase to ∼100%. The greater than 100% recovery is attributed to the removal of residual codeine from the polyethylene tube walls, a phenomenon present even for the calibration curve. Of the weak organic acids tested, separate effects from each counterion were discernible, as recovery between equal concentrations of ascorbic (pH 6–7), citric (pH 1), and acetic acid (pH 2) did not correlate well with pH, while the >90% recovery with acetic acid outperformed that of HCl despite identical pH values. This may be due, in part, to the relative abundance and solubility between the dissociated and undissociated forms of weak acids.62 From the ANOVA test between means, only two conditions, 3% (v/v) HCl at elevated temperature with and without acetone, were found to display codeine concentrations within the organic phase that were not significantly different from the negative control, implying highly efficient removal from this phase (p-values of 0.5962 and 0.8238, respectively, α-level: 0.01). Of the other conditions evaluated, only 10% (v/v) acetic acid came close to this efficiency, with a barely significant difference between means (p-value: 0.0410, α-level: 0.01). Based on these results, 10% (v/v) acetic acid was chosen for its superior ionization capabilities as well as its widespread compatibility across indicator reagents.63 Despite its even higher recovery, 3% (v/v) HCl with acetone was not selected due to its lower stability as a reagent and the incompatibility of acetone with many of the materials used for microfluidic device construction.
Ultimately, the flow rate through the PLM is influenced by several factors, including the applied external pressure head of the incoming fluid,68 the radius of the membrane's largest pores (rmax), the resistive pressure due to the surface tension of the mobile phase (γL), and the interfacial surface tension between the mobile and stationary phases (γwL). The resistance to flow can be determined based on the required liquid entry pressure (LEP) calculated according to eqn (1),69,70
 | (1) |
Real time monitoring of a 100 μL mock aqueous sample was performed using a high-speed video camera and custom stroboscope. From an analysis of the fluid draining behavior (Fig. S5†), a correlation between the average flow rate for a given spin frequency (ranging between 3000–5000 RPM, in increments of 500 RPM) was determined (Fig. 3A), with the calculated flow rates provided in the accompanying table (Fig. 3B). The average flow rate was found to be highly predictable based on the applied spin frequency, with an R2 value of 0.9982. The decline in the real time flow rate observed over time (Fig. 3C and D), per frequency, was also found to be highly consistent. This decline is due to the decreasing pressure head applied by the shrinking liquid column above the membrane. In contrast to previous studies with larger volumes and swellable fibers,72 the decline in real time flow rate was best fit according to a linear regression curve in place of an exponential decay function. If necessary, this behaviour allows for the facile prediction of flow rate decay over time at a specific frequency, offering a means of maintaining an optimal flow rate through linear, real-time adjustment in the applied spin frequency.
Unfortunately, integrated microextraction strategies, for solid samples that are not readily dissolvable, remain particularly challenging within the field of microfluidics. Given the vast range of possible solid samples derived from the environmental, clinical, forensic, and agricultural industries, this represents an enormous loss in potential for providing timely, point-of-need analysis. However, dielectric heating (i.e., microwave irradiation) is uniquely situated to help address the challenges associated with heating distinct states of matter for microdevice processing. This is due, in part, to forgoing the requirement for physical contact with a heating element, enabling flexible microdevice construction. Capitalizing on this, 3D printed sample preparation cartridges were designed to accept and retain 30 mg of an herbaceous sample, along with 50–150 μL of aqueous extraction solvent. The cartridge structure was engineered for high volumetric recovery and facile attachment to the centrifugal microdevice surface, using only a pressure sensitive adhesive. Small pegs enabled automatic alignment with the fluidic port entrance, preventing leaking. Beyond removal of the adhesive coverings and simply pressing the cartridges to the disc, no other steps were necessary for a robust world-to-chip interface. Furthermore, single or multiple cartridges may be simultaneously heated using a conventional microwave oven. By separating sample processing from the main microdevice structure, avoidance of heating thermally labile on-board reagents was achieved.38
Volumetric recovery from the 3D printed cartridges was first assessed by correlating known input volumes of a colored solution to pixel count, in reference to a calibration curve (Fig. 4A), according to a previously published method.35 Mock aqueous samples were then deposited within replicate sample cartridges (Fig. 4D), followed by centrifugal recovery on-disc. Several mock sample processing procedures were then compared: either with or without the inclusion of an herbaceous solid sample, as well as with or without microwave irradiation (Fig. 4B). No significant difference in recovery was observed from an unheated, liquid-only mock sample except for instances where heat facilitated liquid absorption into the porous solid material (ANOVA comparison of means, p-values: 0.9715, 0.9715, and 0.0079, α-level: 0.01). Despite this, under the conditions tested, volume recovery dropped by less than 10%. As temperature was expected to play a crucial role in microextraction efficiency57 based on prior assessments (Fig. 2), a correlation between irradiation time at constant power versus liquid temperature was measured (Fig. 4C). A linear response (R2 value of 0.9993) between microwave exposure and measured temperature was observed, implying tight control over the applied temperature was possible.
:1 mixtures with Δ9-THC (Fig. 1A). Several parameters and their interactions were studied, including pH, temperature, and incubation time (Table 1). HCl was used for pH modulation in this study in order to test a larger range of pH values, some of which were not achievable using acetic acid. The CCD model included three levels of factorial design: low (−1), medium (0) and high (+1), which were useful for obtaining response surfaces and desirability functions. The model was defined by the equation 2k + 2k + Cp, where k represented the number of factors and Cp is the number of center points. In this case, k and Cp were set to 3 and 5, respectively. 19 experiments (n = 3) were performed, with 5 experiments in the center of the design. The control variables within the model were selected to ensure the experimental conditions fell within the previously tested temperature and time studies performed using the 3D-printed sample preparation cartridges (Fig. 4C). One caveat to this study was the dynamic temperature profile throughout the incubation period, due to the dielectric heating method. In order to achieve the specified temperature, microwave irradiation was applied according to the previously generated calibration curve (Fig. 4C). Microwave irradiation also initiated the start of the incubation period. Once the target temperature was attained, dielectric heating ceased. Any additional incubation time remaining for the experimental condition took place in the absence of active heating. Therefore, in order to mimic the temperature decay curve within a sample cartridge, simplified 3D printed containers were designed to closely mimic the relevant cartridge dimensions (Fig. S6†), in place of standard laboratory containers. In spite of these efforts, two experimental conditions could not be fully accommodated with this design (Table 1, highlighted in blue). For 2 out of the 19 experiments, the specified total incubation period was less than the time required to heat to the target temperature. In these two instances, the time difference was 19 sec, representing an 8% error. This error level was considered an acceptable compromise in favour of reaching the target temperature.
The response surfaces obtained for codeine extraction revealed that higher saturation values, and therefore more efficient sample extraction, were obtained for pH values of ∼2. Furthermore, neither the incubation time, temperature, or combination thereof were found to significantly influence the extraction efficiency (based on an ANOVA, the R2 obtained was only 0.6092) (Fig. 5A–C). This finding was reiterated according to the Pareto chart of standardized effects (Fig. 5D), where only the pH reached a significance factor of p < 0.05. Altogether, these results implied that the pH-modulated MAE extraction method was highly robust and essentially insensitive to changes in incubation time and temperature. This is highly advantageous for in-field operation, where exact temperature and incubation periods may be more difficult to apply precisely, in contrast to the use of a standardized extraction solvent. According to the predicted values and desirability model (Fig. S7†), the maximum response (i.e., highest extraction efficiency) was reached using a pH of 2 at 40 °C for 400 seconds. These optimized conditions were later applied for extraction of contrived, laced marijuana samples on-disc.
Centrifugal microdevice design and operation
Reagent to sample ratios were separately evaluated, with a ratio of either 1:2 or 1:3 found to give consistent results with the appropriate color changes across all reagents. A sample volume of 1.0 μL was selected for each on-disc reaction, minimizing the total required extracted sample volume to 12 μL (3 replicates for each indicator reagent), orders of magnitude below the minimum recoverable volume (Fig. 6B). Any remaining extract was retained within a separate collection chamber, making later recovery possible for confirmatory analysis. According to a recent method,36 small volume metering chambers were integrated within the fluidic architecture and evaluated using a simplified centrifugal microdevice for both accuracy and consistency. A calibration curve of known input volumes (between 0.2–1.0 μL) to pixel count was first generated (Fig. 6A), according to a previously published method.35 Delivered sample volumes to the on-disc detection chambers (Fig. 6B), across three separate domains, were next assessed and found to be both highly accurate to the intended volume as well as self-consistent according to a multiple comparisons ANOVA (p-values of 0.8997, 0.7699, and 0.7699, α-level: 0.01). However, delivery of the correct sample volume, while essential, does not account for one of the major challenges associated with microfluidics: adequate mixing. A secondary study was performed to determine the extent to which an ultra-low volume sample would interact with a stored reagent contained inside a microencapsulation well (Fig. 6C). Image analysis of detection chambers loaded with glass microfiber membranes, containing either yellow or blue dye, were used to establish reference hue values for either a simulated indicator reagent alone (yellow) with on-disc metered and delivered mock sample (blue). Both the average hue values, as well as their homogeneity across the detection chamber, led to the conclusion that even mixing was occurring despite the low volumes due to the interplay between centrifugal actuation and capillary wicking throughout the membrane. The skew of the mixed reagent hue values toward the yellow end of the spectrum was anticipated given the sample to stored reagent volume ratio (1:3). Finally, the sensitivity of the metering consistency was challenged across several sample input volumes, ranging from 50–125 μL (Fig. 6D). This was intended to simulate the likely possibility of variable volume recovery from a solid sample during extraction. Despite the nearly 3-fold difference in sample volume, the metered and delivered volume was not found to appreciably alter, according to an ANOVA comparison of means (p-values of 0.8666, 0.3196, and 0.8143 for 75, 100, and 125 μL relative to 50 μL, α-level: 0.01). From this, it was concluded that both the microdevice fluid dynamics, in addition to the sample preparation method, represented robust strategies for use in-field.
Contrived marijuana samples containing 6 mg of Δ9-THC, cannabidiol (CBD), and CBN79 (at a 1:2:0.5 ratio, respectively) were previously laced with either methanol (as a negative control) or 5 mg of cocaine, PCP, or heroin80–82 and extracted via the optimized MAE parameters within separate 3D printed sample preparation cartridges. Each cartridge was attached to one of four domains on the otherwise fully assembled centrifugal microdevice, then subjected to a partially automated protocol (Fig. S8†) for sample metering and reaction with the pre-stored indicator reagents. The microdevice was then scanned using a desktop scanner, and the CIELAB values measured in Fiji. All results were compiled in a 39 × 12 matrix alongside the CIELAB values from Fig. 1, which acted as a chemometric reference database. PCA, according to a Pearson's correlation matrix, was performed with factors for the observation plane selected for maximal cumulative variance (at least 68%). Scatter plots of the chosen factors were then plotted with the inclusion of 95% confidence ellipses per supplementary variable (i.e., drug classification) (Fig. 9A). Additionally, agglomerative hierarchal clustering (AHC), based on Pearson's correlation coefficient for similarity, was also performed (Fig. 9B). From these results, each replicate within the four contrived samples mapped within the 95% confidence ellipse of the anticipated drug class. From the AHC, five clusters were identified, which aligned closely with the PCA clusters. Two differences between the analysis methods were observed, whereby Δ9-THC was placed within a cluster separate from the psychostimulant drug class (containing amphetamine and methamphetamine) and MDMA was included with the opiates cluster containing heroin and codeine (Fig. S11†).
 | ||
| Fig. 9 Identification of lacing agents. Contrived marijuana samples (composed of loose-leaf tea spiked with a cannabinoid mixture at a 1:2:0.5 ratio of Δ9-THC, CBN, and CBN, respectively) were laced with either 5 mg heroin, phencyclidine, or cocaine standard. One contrived sample was laced only with solvent to serve as a negative control. A. PCA factor plane, pooling all CIELAB results across all four indicator reagents (n = 3), relative to the chemometric database of pure illicit drug standards (Fig. 1). Centroids represent the 95% confidence interval, according to chi-square analysis, for specified drug classes. B. AHC analysis of all contrived samples, including negative controls (negative), cocaine (C), PCP (P), and heroin (H) laced samples (bolded) alongside the chemometric database F1 and F2 values. Clusters 1–5 are color coded according to the PCA drug classes. | ||
Conclusions
The combination of a 3D printed sample preparation cartridge, fully integrated centrifugal microfluidic disc, and objective image analysis using a device-independent color model allowed for rapid (<8 min) processing of a traditionally challenging forensic sample (i.e., mixed composition solid sample) in a facile manner. Total sample consumption was minimized and sample extraction largely non-destructive, with both the original sample as well as excess extracted sample reserved for later confirmatory testing (e.g., HPLC-MS or HPLC-GC). Identification of an illicit drug using microchemical detection reagents, which remain decidedly favoured by law enforcement officers, was improved through several mechanisms, the most important of which was accuracy via the removal of known interferents coupled to objective image analysis. In addition, simultaneous testing with multiple screening reagents removed the possibility for compounding error during standard hierarchical testing strategies. In a proof-of-concept illustration, the correct identification of three separate illicit drugs of interest (or their drug class) on-disc was demonstrated from contrived forensic samples, relative to a chemometric database of pure standards.The explosive growth of the legal marijuana industry still faces significant challenges with contamination from a wide range of compounds,83 including heavy metals,84,85 pesticides,86,87 and mycotoxins.88 In 2023 alone, federal health authorities received thousands of reports of heavy metal poisonings and fungal infections due to marijuana use.89 The integration of a unique MAE compatible sample preparation cartridge, coupled to functionalized membranes integrated within a centrifugal microfluidic device, offers an intentionally adaptable format for processing traditionally challenging samples. With respect to marijuana alone, the microdevice may be tailored for applications in the realm of consumer safety in place of law enforcement. Adaptation for these various targets should be achievable through selection of the appropriate SPE membrane, with many options available commercially, and the substitution of pertinent detection reagents.90 Screening of several compounds simultaneously is also possible within a single domain, just as the four colorimetric detection reagents were incorporated side-by-side. Another prospect is segmenting one domain per target class, offering advanced parallelization and greatly limiting sample consumption. Likewise, detection of drugs of abuse from solid samples, such as hair and nails, are essential for toxicological analysis in clarifying the cause of death related to overdoses, addictions, accidents, or injuries.75 The customized detection wells employed within this microdevice are only necessary for instances where 1) microdevice materials are incompatible with the desired indicator reagents, 2) the reagents are thermally sensitive and incompatible with the lamination procedure, 3) retention of liquid reagents is preferred, or 4) high volume capacity (either for preconcentration of detection reagents or for reception of larger processed sample volumes) is needed.38 Otherwise, cellulose microfiber filters with desiccated reagents, previously demonstrated with this microdevice format,15 may be directly incorporated within the main microdevice layers and sealed via lamination.
Although both the sample cartridges and microdevice offer serious potential for portability, in-field operation of MAE in particular would require the use of a portable microwave,16,91 in addition to a custom micromechanical system combining fluid actuation and laser valving. Although portable and fully automated centrifugal microfluidic platforms have been previously demonstrated, dedicated engineering would be required to achieve portable MAE. In this proof-of-concept demonstration, the assay may be readily performed within a laboratory, requiring minimal bench space and only a household microwave.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. C. O. conceived the presented idea, performed data curation, formal analysis, investigation, and project administration, as well as developed methodologies, acquired funds, created illustrations, and composed writing. M. B. A. performed formal analysis and investigation, developed methodologies, acquired funds, as well as assisted with illustrations and writing. R. L. N. performed formal analysis and investigation, developed methodologies, and assisted with illustrations and writing. E. T. C. performed all SolidWorks design creation and assisted with formal analysis as well as illustrations. N. K. L. assisted in investigation and formal analysis. M. E. C. assisted with AutoCAD design work as well as performed formal analysis. B. M. S. assisted in investigation and formal analysis. Both S. L. N. and J. P. L. acquired funds as well as supervised conceptualization and writing. All authors read and approved the completed manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Assistant Professor Michelle Peace from the Department of Forensic Science at Virginia Commonwealth University (VCU) for her helpful feedback during initial project conceptualization as well as Professors Nigel Kent and Andrew Kinman from the School of Mechanical & Manufacturing Engineering at Dublin City University (DCU) for their kindness and incredible patience while troubleshooting mechanical malfunctions. The authors would also like to thank Isaac Buell and Ryan Gibisier from the UVA Department of Mechanical Engineering for their assistance in customizing the high-speed video platform for flow rate analysis. Finally, M. B. A. would like to thank CAPES-PDSE (Edital 19/2016, process 88881.133648/2016-01) for their Ph.D. scholarship. This work was funded in part by the University of Virginia Graduate School of Arts & Science (GSASC).References
- Polydrug Use Among Cannabis Users, https://cannabissupport.com.au/polydrug-use-among-cannabis-users/, (accessed October 24, 2022) Search PubMed.
- World Drug Report 2023, http://www.unodc.org/unodc/en/data-and-analysis/world-drug-report-2023.html, (accessed August 5, 2023) Search PubMed.
- A. C. Parrott, R. M. Milani, E. Gouzoulis-Mayfrank and J. Daumann, J. Neural Transm., 2007, 114, 959–968 CrossRef CAS PubMed.
- R. W. Foltin, M. W. Fischman, J. J. Pedroso and G. D. Pearlson, Pharmacol., Biochem. Behav., 1987, 28, 459–464 CrossRef CAS PubMed.
- K. Schaeffer, 6 facts about Americans and marijuana, https://www.pewresearch.org/fact-tank/2021/04/26/facts-about-marijuana/, (accessed October 25, 2022) Search PubMed.
- World Drug Report 2022, http://www.unodc.org/unodc/en/data-and-analysis/world-drug-report-2022.html, (accessed October 25, 2022) Search PubMed.
- R. J. Peters, M. Williams, M. W. Ross, J. Atkinson and S. A. McCurdy, J. Drug Educ., 2008, 38, 285–295 CrossRef PubMed.
- C. R. Gilbert, M. Baram and N. C. Cavarocchi, Tex. Heart Inst. J., 2013, 40, 64–67 Search PubMed.
- R. Gabrielson and T. Sanders, The New York Times, 2016 Search PubMed.
- R. Gabrielson, Since We Reported on Flawed Roadside Drug Tests, Five More Convictions Have Been Overturned, https://www.propublica.org/article/since-we-reported-on-flawed-roadside-drug-tests-five-more-convictions-have-been-overturned, (accessed October 25, 2022) Search PubMed.
- K. Phelps, Pleading Guilty to Innocence: How Faulty Field Tests Provide False Evidence of Guilt, Roger Williams University Law Review, 2019, vol. 24, iss. 1, p. 7. Available at: https://docs.rwu.edu/rwu_LR/vol24/iss1/7 Search PubMed.
- Y. Tsumura, T. Mitome and S. Kimoto, Forensic Sci. Int., 2006, 155, 158–164 CrossRef PubMed.
- L. Harper, J. Powell and E. M. Pijl, Harm Reduct. J., 2017, 14, 52 CrossRef PubMed.
- K. M. Elkins, A. C. Weghorst, A. A. Quinn and S. Acharya, Drug Test. Anal., 2017, 9, 306–310 CrossRef CAS PubMed.
- S. T. Krauss, T. P. Remcho, S. M. Lipes, R. Aranda IV, H. P. Maynard III, N. Shukla, J. Li, R. E. Tontarski Jr. and J. P. Landers, Anal. Chem., 2016, 88(17), 8689–8697 CrossRef CAS PubMed.
- S. Krauss, PhD, University of Virginia, 2018 Search PubMed.
- T.-L. E. Lockwood, T. Leong, S. L. Bliese, A. Helmke, A. Richard, G. Merga, J. Rorabeck and M. Lieberman, J. Forensic Sci., 2020, 65, 1289–1297 CrossRef CAS PubMed.
- C. Darsigny, M. Leblanc-Couture and I. Desgagné-Penix, Austin J. Forensic Sci. Criminal., 2018, 5(1), 1074 Search PubMed.
- C. L. O'Neal, D. J. Crouch and A. A. Fatah, Forensic Sci. Int., 2000, 109, 189–201 CrossRef PubMed.
- G. Musile, L. Wang, J. Bottoms, F. Tagliaro and B. McCord, Anal. Methods, 2015, 7, 8025 RSC.
- M. A. Alhefeiti, J. Barker and I. Shah, Molecules, 2021, 26, 3291 CrossRef CAS PubMed.
- N. A. Abdelshafi, J. Bell, K. Rurack and R. J. Schneider, Drug Test. Anal., 2019, 11, 492–500 CrossRef CAS PubMed.
- S. T. Krauss, M. S. Woolf, K. C. Hadley, N. M. Collins, A. Q. Nauman and J. P. Landers, Sens. Actuators, B, 2019, 284, 704–710 CrossRef CAS.
- H. I. S. Kafeenah, R. Osman and N. K. A. Bakar, RSC Adv., 2018, 8, 40358–40368 RSC.
- N. A. Desrosiers, C. C. Betit and J. H. Watterson, Forensic Sci. Int., 2009, 188, 23–30 CrossRef CAS PubMed.
- I. Álvarez, A. M. Bermejo, M. J. Tabernero, P. Fernández, P. Cabarcos and P. López, Anal. Bioanal. Chem., 2009, 393, 1345–1350 CrossRef PubMed.
- C. Sparr Eskilsson and E. Björklund, J. Chromatogr. A, 2000, 902, 227–250 CrossRef CAS PubMed.
- F. Taglia, L. Wang, C. H. Setser, N. Fernández-Tejero, B. R. McCord and S. B. Lee, Forensic Sci. Int. Synergy, 2022, 5, 100291 CrossRef PubMed.
- R. Subramanian, L. Digard, M. Washington II and S. Sorage, In the Shadows: A Review of the Research on Plea Bargaining, Vera Institute of Justice, New York, 2020 Search PubMed.
- S. R. Gross, M. Possley and K. Stephens, Race and Wrongful Convictions in the United States, The National Registry of Exonerations, Newkirk Center for Science and Society, 2017 Search PubMed.
- R. R. Foundation, Arrests and the Criminal Legal System, https://www.drugpolicyfacts.org/chapter/crime_arrests, (accessed October 25, 2022) Search PubMed.
- 9780966080223, https://www.abebooks.com/9780966080223/False-Positives-Equal-Justice-John-096608022X/plp, (accessed October 25, 2022).
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- NIJ (National Institute of Justice) Standard for Color Test Reagents/Kits for Preliminary Identification of Drugs of Abuse | Office of Justice Programs, https://www.ojp.gov/ncjrs/virtual-library/abstracts/nij-national-institute-justice-standard-color-test-reagentskits, (accessed October 21, 2022) Search PubMed.
- M. S. Woolf, L. M. Dignan, A. T. Scott and J. P. Landers, Nat. Protoc., 2021, 16, 218–238 CrossRef CAS PubMed.
- R. L. Nouwairi, C. K. Jones, M. E. Charette, E. Holmquist, Z. Golabek and J. P. Landers, Anal. Chem. Search PubMed submission under revision.
- B. L. Thompson, Y. Ouyang, G. R. M. Duarte, E. Carrilho, S. T. Krauss and J. P. Landers, Nat. Protoc., 2015, 10, 875–886 CrossRef PubMed.
- K. O'Connell, N. Lawless, B. Stewart and J. Landers, Lab Chip, 2022, 22, 2549–2565 RSC.
- M. S. Woolf, L. M. Dignan, H. M. Lewis, C. J. Tomley, A. Q. Nauman and J. P. Landers, Lab Chip, 2020, 20, 1426–1440 RSC.
- M. Philp and S. Fu, Drug Test. Anal., 2018, 10, 95–108 CrossRef CAS PubMed.
- Rapid Testing Methods of Drugs of Abuse, http://www.unodc.org/unodc/en/scientists/rapid-testing-methods-of-drugs-of-abuse_new.html, (accessed October 25, 2022) Search PubMed.
- S. M. Newhall, D. Nickerson and D. B. Judd, J. Opt. Soc. Am., 1943, 33, 385–418 CrossRef.
- M. Binette and P. Pilon, Microgram J., 2013, 10(1), 8–11 Search PubMed.
- J. Kelly, K. Addanki and O. Bagasra, Open Forensic Sci. J., 2012, 5, 4–8 CrossRef.
- D. J. Symonsbergen, M. J. Kangas, M. Perez and A. E. Holmes, Forensic Sci., 2018, 2, 57 Search PubMed.
- The early development of the Munsell system - Kuehni - 2002 - Color Research & Application - Wiley Online Library, https://onlinelibrary.wiley.com/doi/10.1002/col.10002, (accessed October 26, 2022) Search PubMed.
- N. Ibraheem, M. Hasan, R. Z. Khan and P. Mishra, ARPN Journal of Science and Technology, 2012, 2(3), 265–275 Search PubMed.
- A. Zeileis, K. Hornik and P. Murrell, Comput. Stat. Data Anal., 2009, 53, 3259–3270 CrossRef.
- Principal Component Analysis (PCA), https://www.xlstat.com/en/solutions/features/principal-component-analysis-pca, (accessed October 26, 2022) Search PubMed.
- Laced Weed, https://www.drugrehab.com/addiction/drugs/marijuana/laced-weed/, (accessed October 24, 2022) Search PubMed.
- Synthetic cannabinoids, https://www.cdc.gov/nceh/hsb/chemicals/sc/default.html, (accessed October 27, 2022) Search PubMed.
- Drug Scheduling, https://www.dea.gov/drug-information/drug-scheduling, (accessed October 27, 2022) Search PubMed.
- R. Gulaboski, M. N. D. S. Cordeiro, N. Milhazes, J. Garrido, F. Borges, M. Jorge, C. M. Pereira, I. Bogeski, A. H. Morales, B. Naumoski and A. F. Silva, Anal. Biochem., 2007, 361, 236–243 CrossRef CAS PubMed.
- C. A. S. Bergström and P. Larsson, Int. J. Pharm., 2018, 540, 185–193 CrossRef PubMed.
- Y.-L. Hsieh and L. S. Taylor, Pharm. Res., 2015, 32, 549–561 CrossRef CAS PubMed.
- E. Skierka and M. Sadowska, Food Chem., 2007, 105, 1302–1306 CrossRef CAS.
- J. van der Merwe, J. Steenekamp, D. Steyn and J. Hamman, Pharmaceutics, 2020, 12, 393 CrossRef CAS PubMed.
- N. P. A. Christensen, J. Rantanen, C. Cornett and L. S. Taylor, Eur. J. Pharm. Biopharm., 2012, 82, 410–416 CrossRef CAS PubMed.
- E. R. Garrett and C. A. Hunt, J. Pharm. Sci., 1974, 63, 1056–1064 CrossRef CAS PubMed.
- M. A. Huestis, Chem. Biodiversity, 2007, 4, 1770–1804 CrossRef CAS PubMed.
- Extraction of THC Metabolites in Urine using the Empore™ Membrane C18 Extraction Cartridge, https://www.cdsanalytical.com/single-post/extraction-of-thc-metabolites-in-urine-using-the-empore-membrane-c18-extraction-cartridge, (accessed October 23, 2022) Search PubMed.
- 17.5, https://chem.libretexts.org/Bookshelves/General_Chemistry/Map%3A_Chemistry_-_The_Central_Science_(Brown_et_al.)/17%3A_Additional_Aspects_of_Aqueous_Equilibria/17.05%3A_Factors_that_Affect_Solubility, (accessed October 24, 2022).
- [PDF] Technical Note A Study of Acids Used for the Acidified Cobalt Thiocyanate Test for Cocaine Base | Semantic Scholar, https://www.semanticscholar.org/paper/Technical-Note-A-Study-of-Acids-Used-for-the-Cobalt-Deakin/7baf753f7c5e92fe1a7a03d9b1b854e24eb17bf6, (accessed October 25, 2022) Search PubMed.
- M. Miró, W. Frenzel, J. M. Estela and V. Cerdà, Analyst, 2001, 126, 1740–1746 RSC.
- H. Lingeman and S. J. F. Hoekstra-Oussoren, J. Chromatogr. B: Biomed. Sci. Appl., 1997, 689, 221–237 CrossRef CAS PubMed.
- Empore™ SPE Disks matrix active group C18, diam. 47 mm, pk of 20 | Sigma-Aldrich, http://www.sigmaaldrich.com/, (accessed October 9, 2022) Search PubMed.
- C. Erger and T. C. Schmidt, TrAC, Trends Anal. Chem., 2014, 61, 74–82 CrossRef CAS.
- J. Ducrée, S. Haeberle, S. Lutz, S. Pausch, F. von Stetten and R. Zengerle, J. Micromech. Microeng., 2007, 17, S103–S115 CrossRef.
- C. A. Cramers, J. A. Rijks and C. P. M. Schutjes, Chromatographia, 1981, 14, 439–444 CrossRef CAS.
- M. C. García-Payo, M. A. Izquierdo-Gil and C. Fernández-Pineda, J. Colloid Interface Sci., 2000, 230, 420–431 CrossRef PubMed.
- Wettability Characterization | Life Science Research | MilliporeSigma, https://www.emdmillipore.com/US/en/life-science-research/chromatography-sample-preparation/membrane-learning-center/Wettability-Characterization/lp2b.qB.f7IAAAFM20p88eJt,nav, (accessed October 3, 2022) Search PubMed.
- M. S. Woolf, L. M. Dignan, S. M. Karas, H. M. Lewis, K. C. Hadley, A. Q. Nauman, M. A. Gates-Hollingsworth, D. P. AuCoin, H. R. Green, G. M. Geise and J. P. Landers, Micromachines, 2022, 13, 487 CrossRef PubMed.
- V. Cavalloro, E. Martino, P. Linciano, S. Collina, V. Cavalloro, E. Martino, P. Linciano and S. Collina, Microwave-Assisted Solid Extraction from Natural Matrices, IntechOpen, 2021 Search PubMed.
- A. Liazid, M. Palma, J. Brigui and C. G. Barroso, J. Chromatogr. A, 2007, 1140, 29–34 CrossRef CAS PubMed.
- P. Fernández, S. Seoane, C. Vázquez, A. M. Bermejo, A. M. Carro and R. A. Lorenzo, Anal. Bioanal. Chem., 2011, 401, 2177 CrossRef PubMed.
- A. Namera, S. Yamamoto, T. Saito, S. Miyazaki, H. Oikawa, A. Nakamoto and M. Nagao, J. Sep. Sci., 2011, 34, 2232–2239 CrossRef CAS PubMed.
- About synthetic cannabinoids | NCEH | CDC, https://www.cdc.gov/nceh/hsb/chemicals/sc/About.html, (accessed February 2, 2023) Search PubMed.
- K2/Spice - Drug Fact Sheet, https://www.dea.gov/documents/2020/2020-06/2020-06-05/k2spice-drug-fact-sheet, (accessed February 2, 2023) Search PubMed.
- E. M. Mudge, S. J. Murch and P. N. Brown, Anal. Bioanal. Chem., 2017, 409, 3153–3163 CrossRef CAS PubMed.
- L. R. Drake and P. J. H. Scott, ACS Chem. Neurosci., 2018, 9, 2358–2372 CrossRef CAS PubMed.
- J. L. Bertron, M. Seto and C. W. Lindsley, ACS Chem. Neurosci., 2018, 9, 2459–2474 CrossRef CAS PubMed.
- J. Mella-Raipán, J. Romero-Parra and G. Recabarren-Gajardo, ACS Chem. Neurosci., 2020, 11, 3905–3927 CrossRef PubMed.
- What Lab Tests Reveal About Cannabis in New York's Illegal Dispensaries - The New York Times, https://www.nytimes.com/2022/12/01/nyregion/cannabis-bacteria-pesticides-illegal-dispensary.html, (accessed June 19, 2023) Search PubMed.
- L. Bengyella, M. Kuddus, P. Mukherjee, D. J. Fonmboh and J. E. Kaminski, Toxin Rev., 2021, 0, 1–11 CAS.
- S. Magazine and B. Handwerk, Modern Marijuana Is Often Laced With Heavy Metals and Fungus, https://www.smithsonianmag.com/science-nature/modern-marijuana-more-potent-often-laced-heavy-metals-and-fungus-180954696/, (accessed October 25, 2022) Search PubMed.
- D. V. Pinkhasova, L. E. Jameson, K. D. Conrow, M. P. Simeone, A. P. Davis, T. C. Wiegers, C. J. Mattingly and M. C. K. Leung, Curr. Res. Toxicol., 2021, 2, 140–148 CrossRef PubMed.
- Z. Montoya, M. Conroy, B. D. Vanden Heuvel, C. S. Pauli and S.-H. Park, Front. Pharmacol., 2020, 11, 571832 CrossRef CAS PubMed.
- Legalizing marijuana and the new science of weed (video), https://www.eurekalert.org/news-releases/735205, (accessed October 24, 2022) Search PubMed.
- For Marijuana Users, Even Legalization Doesn't Guarantee Safety, https://www.msn.com/en-us/health/other/for-marijuana-users-even-legalization-doesn-t-guarantee-safety/ar-BB1izG5m, (accessed February 28, 2024) Search PubMed.
- L. Yang, X. Ye, X. Li, Z. Huang, F. Chen, W. Yang and Z. Wang, Anal. Chim. Acta, 2022, 1207, 339815 CrossRef CAS PubMed.
- A. C. Scott, PhD, University of Virginia, 2020 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00223g |
| This journal is © The Royal Society of Chemistry 2024 |