
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering potential vascularization factors of on-chip co-cultured hiPSC-derived cerebral organoids†
Maneesha
Shaji
 a,
Atsushi
Tamada
b,
Kazuya
Fujimoto
a,
Keiko
Muguruma
*b,
Stanislav L.
Karsten
*a and
Ryuji
Yokokawa
*a
a,
Atsushi
Tamada
b,
Kazuya
Fujimoto
a,
Keiko
Muguruma
*b,
Stanislav L.
Karsten
*a and
Ryuji
Yokokawa
*a
aDepartment of Micro Engineering, Graduate School of Engineering, Kyoto University, Kyoto daigaku-Katsura, Nishikyo-ku, Kyoto – 615-8540, Japan. E-mail: karsten.stanislav.3i@kyoto-u.ac.jp; yokokawa.ryuji.8c@kyoto-u.ac.jp
bDepartment of iPS Cell Applied Medicine, Kansai Medical University, 2-5-1 Shin-machi, Hirakata City, Osaka – 573-1010, Japan. E-mail: muguruke@hirakata.kmu.ac.jp
First published on 29th January 2024
Abstract
The lack of functional vascular system in stem cell-derived cerebral organoids (COs) limits their utility in modeling developmental processes and disease pathologies. Unlike other organs, brain vascularization is poorly understood, which makes it particularly difficult to mimic in vitro. Although several attempts have been made to vascularize COs, complete vascularization leading to functional capillary network development has only been achieved via transplantation into a mouse brain. Understanding the cues governing neurovascular communication is therefore imperative for establishing an efficient in vitro system for vascularized cerebral organoids that can emulate human brain development. Here, we used a multidisciplinary approach combining microfluidics, organoids, and transcriptomics to identify molecular changes in angiogenic programs that impede the successful in vitro vascularization of human induced pluripotent stem cell (iPSC)-derived COs. First, we established a microfluidic cerebral organoid (CO)-vascular bed (VB) co-culture system and conducted transcriptome analysis on the outermost cell layer of COs cultured on the preformed VB. Results revealed coordinated regulation of multiple pro-angiogenic factors and their downstream targets. The VEGF-HIF1A-AKT network was identified as a central pathway involved in the angiogenic response of cerebral organoids to the preformed VB. Among the 324 regulated genes associated with angiogenesis, six transcripts represented significantly regulated growth factors with the capacity to influence angiogenic activity during co-culture. Subsequent on-chip experiments demonstrated the angiogenic and vasculogenic potential of cysteine-rich angiogenic inducer 61 (CYR61) and hepatoma-derived growth factor (HDGF) as potential enhancers of organoid vascularization. Our study provides the first global analysis of cerebral organoid response to three-dimensional microvasculature for in vitro vascularization.
Introduction
The recently discovered ability of cerebral organoids to emulate in vivo brain development makes them a powerful new tool for elucidating the mechanisms of human neurogenesis and various disease pathologies.1 Unfortunately, multiple limitations of current organoid technology prevent the realization of its full potential in both basic research and translational applications.2 Among them are laborious and costly production processes, lack of expected tissue-specific heterogeneity leading to the lack of functional repertoire, significant variability causing deviant behavior of phenotypes, and limited organoid life span (generally three months3) that prevent development of COs into fully functional mature organ tissue. The main reason is the lack of a vascular system, preventing both efficient oxygen and nutrient supply and simultaneous waste removal, and ultimately leading to hypoxia and necrosis.2,4Several strategies have been implemented to vascularize human brain organoids, including transplantation of organoids into an immunodeficient mouse brain,5 human ETS variant 2 induction,6 endothelialization of organoids,7,8 and fusion with vascular organoids.9–11 Although the growth of vessel-like networks in cerebral organoids has been reported, complete vascularization with functional and perfusable vasculature has only been achieved by transplantation.6 Therefore, understanding the molecular mechanisms that prevent efficient in vitro vascularization and identifying the extrinsic factors influencing this complex process are urgently needed.
Recent advances in microfluidic technology have facilitated the customization of microenvironments by accurately controlling mechanical, chemical, fluidic, and structural cues. It is now possible to mimic in vivo conditions by modeling vascularized tissue constructs in the presence of growth factors in vitro through microfluidic platforms (on-chip). Recent studies have demonstrated the successful vascularization of liver,12 adipose tissue,12 kidney,13 and tumor organoids.14,15 Although vascularization of brain organoids on chips has not yet been reported, such an approach may potentially be realized to increase flexibility and control of current microfluidic systems.
One possible in vitro vascularization approach is to embed organoids in a preformed vascular or capillary bed. Using this method, functional connections between the organoid and the preformed vascular network can be achieved by anastomosis.16 The formation of an efficient, functional, and interconnected vascular network between the organoid and the preformed vasculature depends on the successful initiation of angiogenesis, followed by lumenization, network formation, vascular remodeling, and pruning. Identifying the factors responsible for the above-mentioned events in an in vitro culture system will enable generation of stable and mature vasculature. Therefore, it is crucial to identify the factors involved in these processes.
Here, we integrated an on-chip organoid system with region-specific microdissection and transcriptomics to decipher the molecular mechanisms involved in early angiogenesis of human induced pluripotent stem cell (hiPSC)-derived COs. We developed a microfluidic platform to co-culture COs directly on a pre-formed VB generated by the self-assembly of human umbilical vein endothelial cells (HUVECs). To identify CO-specific expression changes, we used vacuum-assisted capillary-based microdissection and isolated the outer 200 μm thick organoid layer, excluding the central areas potentially affected by hypoxia and cell death, to dismiss tissue-reactive expression changes. We then performed whole transcriptome analysis and used DAVID functional annotation and GenCLiP3 literature search to identify key angiogenic factors altered at three distinct time points: initial vascular bed angiogenic response (day 28 or co-culture day 3/CC-D3), sprouting angiogenic response (day 31 or CC-D6), and inhibition of angiogenesis (day 35 or CC-D10).
Results
This study comprised of the following parts: on-chip co-culture system optimization, region-specific CO microdissection and microarray analysis, identification of critical angiogenesis factors and signaling pathways, and functional evaluation of identified extracellular factors with respect to VB formation and CO angiogenesis (Fig. 1A).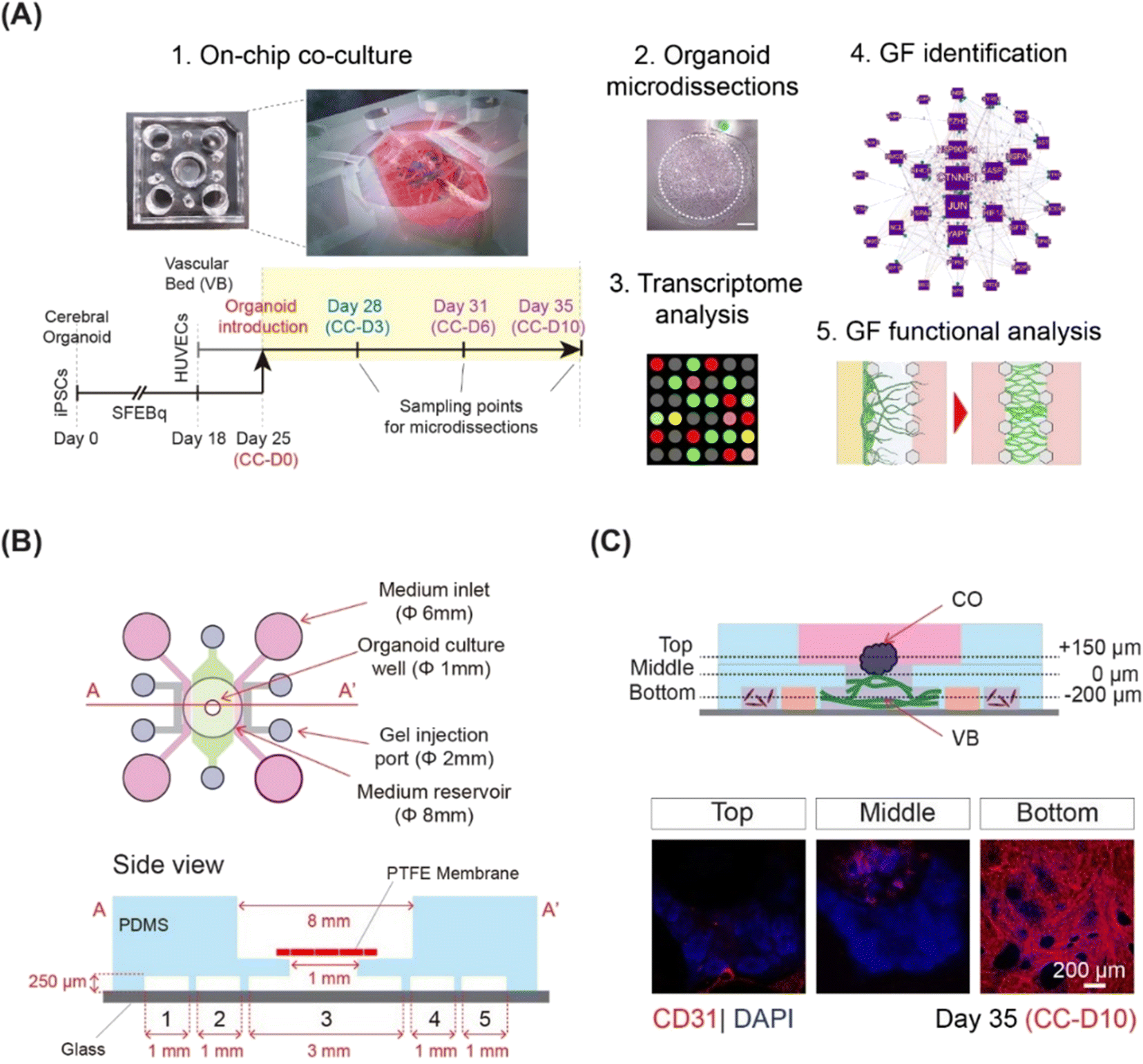 | ||
| Fig. 1 Co-culture of human iPSCs derived CO with 3D vasculature on a chip. (A) Schematic showing the overall workflow of the study. Day, CC-D, and GF represent organoid day, co-culture day, and growth factor, respectively. (B) The microfluidic chip used for the co-culture system. The schematic representation of the chip showing the device dimensions. The bottom layer consists of 5 channels: 1, 5 for hLFs, 3 for vascular bed and 2, 4 for media supply. The channels are separated by micropillars of width 100 μm and height 250 μm. (C) Whole mount immunostaining of the organoids on-chip at different z-sections (top, middle and bottom). Vasculature is stained by CD31 (shown in red) and nucleus is stained by DAPI (shown in blue). | ||
On-chip co-culture of cerebral organoids and 3D microvasculature
Here, we integrated microfluidics and organoid technology to develop an on-chip system to culture COs in direct contact with microvasculature. COs and the VB were grown separately and then co-cultured for 10 days. We employed a three-layer, five-channel, polydimethylsiloxane (PDMS)-based microfluidic device (Fig. 1B) with a removable membrane, as recently reported.17 Cerebral organoids derived from hiPSCs using the serum-free floating culture of embryoid body-like aggregates with quick aggregation (SFEBq) technique3 were co-cultured on a 3D perfusable VB created by self-organization of HUVECs (Fig. 1C and S1†). The differentiation of hiPSCs into cerebral organoids was confirmed using quantitative PCR for SOX1, FOXG1, EMX1, and PAX6 (Fig. S2A†).3 To exclude potential adverse effects of the mixed medium on organoid differentiation, we examined the expression of telencephalic markers FOXG1, EMX1, and PAX6 (Fig. S2B and C†).3The VB remained stable and perfusable after 10 days of co-culture (Fig. S3A and B†). We then investigated the effect of the vasculature on the growth and differentiation of CO. Immunostaining for neural progenitor markers SOX2 and FOXG1, neuroepithelial stem cell marker NESTIN, and differentiated neuron marker CTIP23 did not reveal any differences in the ventricular structures of COs grown with or without a pre-formed VB (Fig. S3C†). Furthermore, the CTIP2/SOX2 ratio, an indicator of overall neuron production during organoid maturation,18 significantly increased between days 25 and 35 for COs cultured with (p < 0.001) and without (p < 0.001) pre-formed VB (Fig. S4A and B†). No significant difference was observed in the CO sizes grown with and without VB (average diameter 1 mm, data not shown). Taken together, these observations suggest that the morphology and differentiation of organoids grown on a chip in direct contact with VB were not significantly different from those grown using the dish culturing protocol.
Because vasculature plays a role in tissue maintenance and survival through the exchange of nutrients and oxygen, we examined whether the co-culture of organoids on VB influenced the overall survival of the organoid. Expression of CASP3, an apoptotic marker, showed accumulation at the center of organoids without a VB, but exhibited a decreasing trend in COs co-cultured on a VB (Fig. S4C†). However, this decrease was not significant (Fig. S4D†).
Angiogenesis of hiPSC-derived cerebral organoids is inhibited by 10 days of co-culture
Using immunostaining, we monitored the progress and degree of vascularization during 10 days of co-culture. Sprout-like endothelial structures branching from the microvasculature towards the organoid on CC-D6 were observed at the interface between the CO and VB using confocal microscopy (Fig. 2A). To further investigate the extent of vascularization, we performed immunostaining assays using organoid cryosections on days 28 (CC-D3), 31 (CC-D6), and 35 (CC-D10) (Fig. 2B). After three days of co-culture (CC-D3), we observed that HUVECs partially covered the organoid, with a few cells migrating into the organoid (Fig. 2B). On CC-D6, sprouts were observed along the periphery of the organoid in the cryosection images (Fig. 2B), with a noticeable increase in the percentage of ECs with respect to the total number of cells (Fig. 2C). However, by CC-D10, no further sprouts were observed invading the organoids (Fig. 2B). Vascular structures were predominantly limited to the outermost layer of the organoids, with a significant reduction (p < 0.05) in the percentage of ECs (Fig. 2B and C) at CC-D10. Based on these observations, we conclude that CO vascularization was inhibited shortly after its initiation.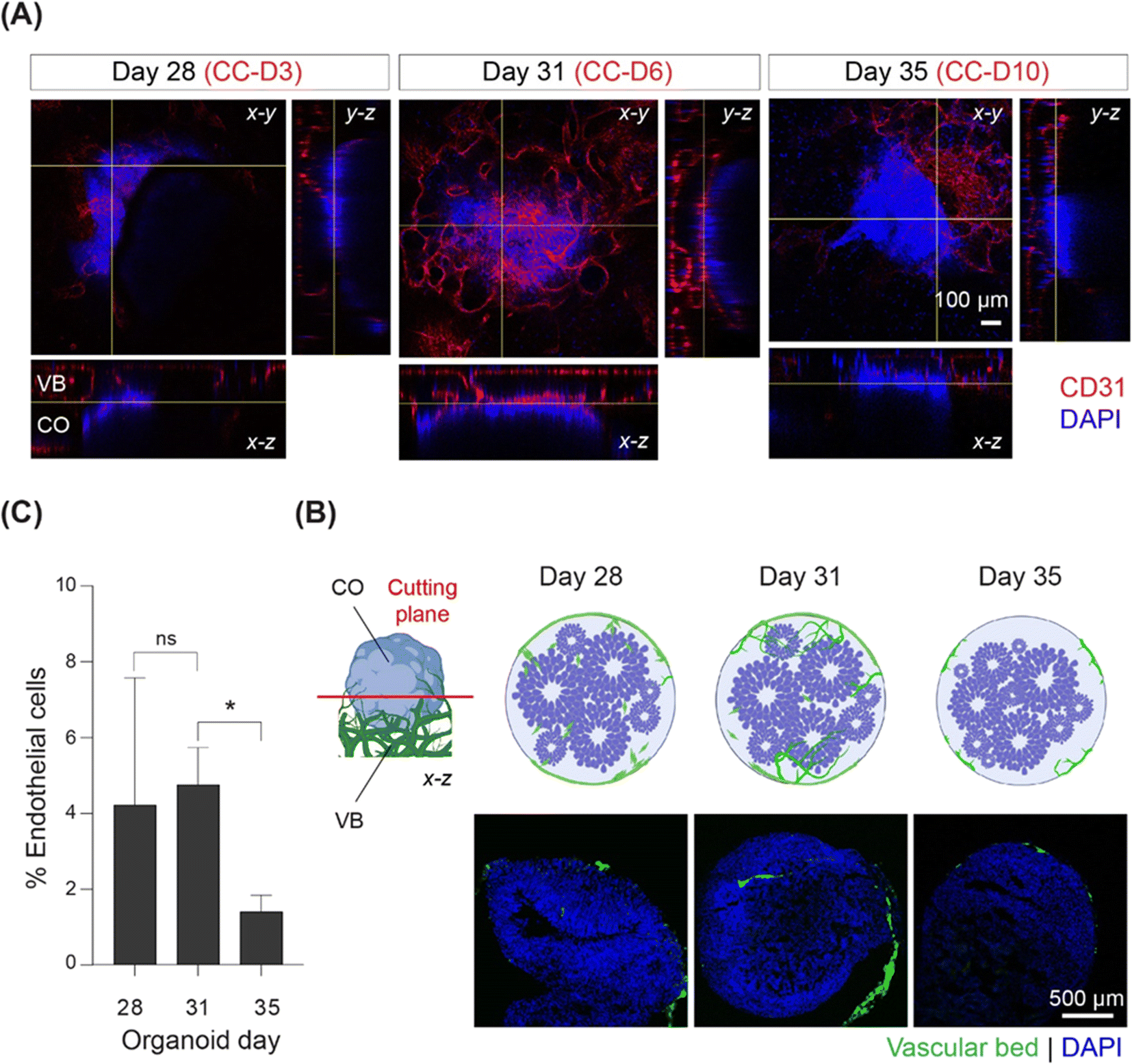 | ||
| Fig. 2 Co-culture of CO with 3D vasculature on a chip reveals initiation and subsequent inhibition of vascularization. (A) Whole mount immunostaining showing organoid and vasculature on-chip at CC-D 3, 6, and 10. The vasculature is stained by CD31, and nucleus by DAPI. The interface between cerebral organoid (CO) and vascular bed (VB) (approximately 260 μm from the glass bottom) is shown by yellow lines in x–z and x–y planes. (B) Schematic showing the sectioning (cutting) plane in red, CO (blue) in direct contact with GFP-positive VB (green) in the left side and the immunostained cryosections (right side) of COs at CC-D 3, 6, 10. (C) Percentage of endothelial cells relative to the total number of cells in the cryosections at day 28, 31, and 35, n = 6. | ||
Angiogenesis related genes are identified by on-chip cultured COs
Analysis of VB morphology after contact with COs showed sprouting of ECs by day 31 (Fig. 2), indicating probable angiogenic events in response to interaction with the outer organoid layer. However, organoid vascularization was notably inhibited by day 35 (Fig. 2). We hypothesized that signaling received from the cells at the periphery of the organoids may be the main driving mechanism guiding such in vitro angiogenic processes. Therefore, to identify the regulatory network of angiogenic factors involved in the initiation of the vascularization process, we conducted a whole-transcriptome analysis of the outermost cell layer of CO (200 μm thickness) using high-density oligonucleotide microarrays. This approach ensured that the transcriptome changes were detected only in the areas most in contact with the VB, and that the hypoxic/necrotic core of the cerebral organoid was excluded from the analysis (Fig. S5†). To capture the dynamics of the vascularization process, samples were collected on days 28 (CC-D3), 31 (CC-D6), and 35 (CC-D10) of the microarray experiment (Fig. 1A).This approach identified 1697 distinct known transcripts (long list), for which expression was significantly altered in at least one of the two comparisons (p ≤ 0.05). Sixty-six genes were differentially regulated at both time points: 715 on day 31 and 916 on day 35. After applying a higher significance level corresponding to the p value ≤0.01, the gene list was reduced to 314 known genes, with 270 and 40 genes regulated on days 31 and 35, respectively.
Gene ontology analysis using the DAVID functional annotation tool19,20 revealed 30 categories for various biological processes with at least 30% enrichment (EASE score = 0.05; Table S1†). The most prominent categories were related to vascular system and central nervous system (CNS) development, cell differentiation, and apoptosis. Interestingly, angiogenesis represented the most significantly enriched biological process (63% enrichment, p = 0.0002; Table S1†), followed by response to hypoxia (53%, p = 0.0005). Furthermore, DAVID functional annotation clustering using high stringency classification (EASE score = 0.01) of all known genes regulated at least one of the time points (n = 1697) revealed multiple overrepresented functional clusters, with a top cluster represented by vascular development and angiogenesis genes (109 genes, Fig. S6†).
Further search for angiogenesis related genes using GenCLIP3 analysis21 retrieved 287 hits showing co-occurrence of an official gene name with the keyword “angiogenesis” (Fig. S7, Table S2†). Among these genes, 42 were mentioned in at least 10 or more published studies, with the top three genes represented by VEGFA (↑d31), HIF1a (↓d35), and AKT1 (↑d31), the key elements of the vascular endothelial growth factor (VEGF) regulatory pathway. Combination of this group with the “angiogenesis” cluster identified in DAVID functional annotation clustering yielded a list of 332 known angiogenesis-related genes (Fig. S7, Table S2†). Given the well-known role of VEGFA in angiogenesis, we applied it as a keyword against all 332 genes and reduced the network to only VEGF-interacting genes (108 genes; Fig. 3A). This resulted in the identification of 43 genes directly regulated by VEGFA expression (Fig. 3B). Further stratification of the literature to determine the interaction of these genes with VEGFA resulted in the functional network depicted in Fig. 3.
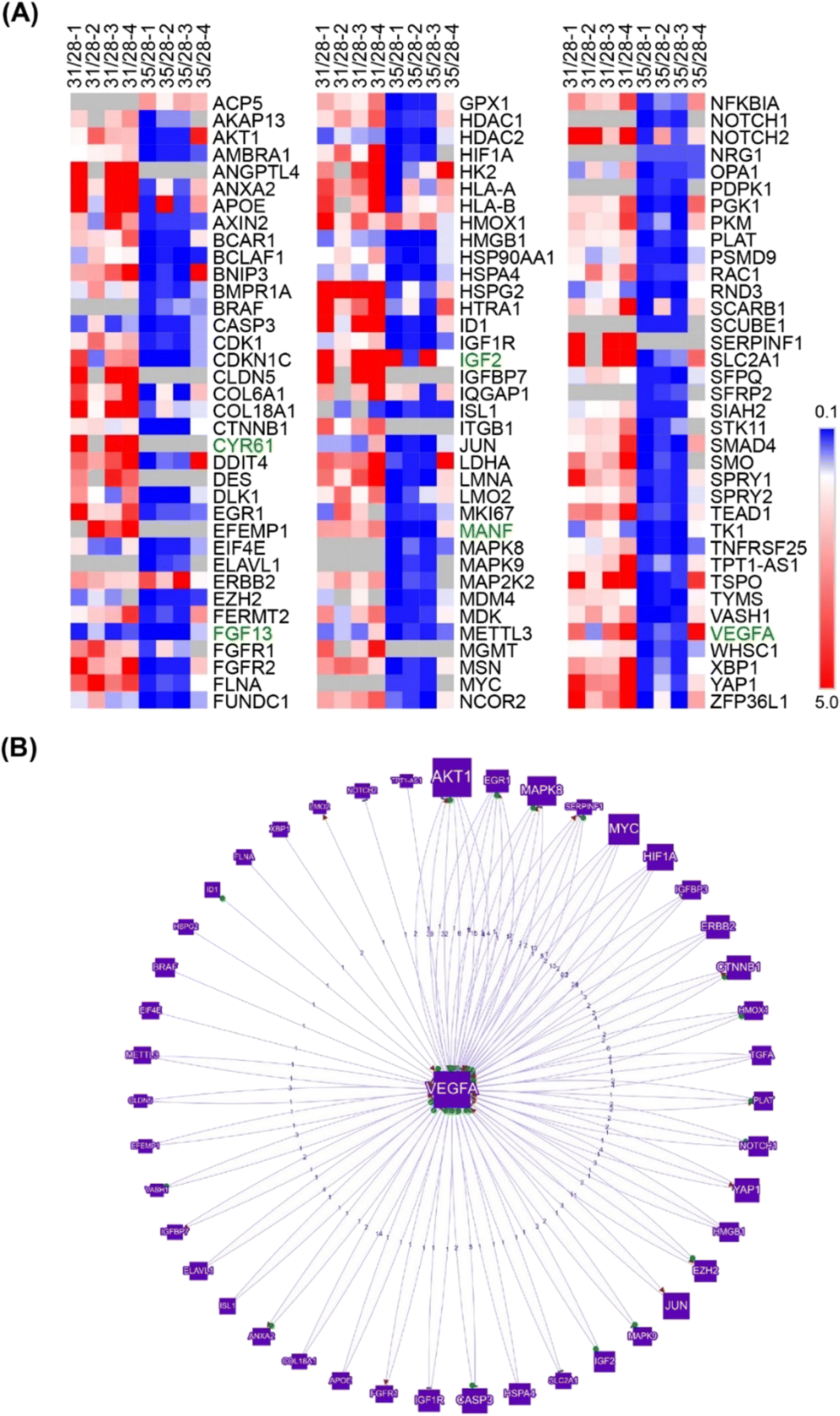 | ||
| Fig. 3 GenCLiP analysis reveals substantial dysregulation of VEGF pathway represented by direct interactors and downstream targets. (A) Genes involved in the VEGF pathway identified by DAVID functional annotation and GenCLiP3 literature search using “VEGF” as a keyword. (B) Genes directly interacting (regulators and downstream targets) with VEGFA. | ||
Key members of the VEGF signaling pathway agree with observed angiogenic processes
The VEGF pathway is a central regulatory mechanism in angiogenic process.22,23 One hundred and eight members of the VEGF/HIF1A signaling pathway, including 43 direct regulators and downstream targets of VEGFA, were identified in this study (Fig. 3, Table S2†). On day 31, the expression of VEGFA (↑d31) was elevated (Fig. 3), which agreed with the increased expression of multiple VEGF-activating factors on day 31 (Fig. 3 and 4; Text Data S1†). Likewise, decreased VEGFA expression at day 35 coincided with a decrease in the expression of multiple VEGF-activating genes and increased expression of VEGF inhibitors on day 31 (Fig. 4, Data S1†). Numerous targets activated by the VEGF pathway demonstrated changes in agreement with the activation of VEGFA on day 31 (Fig. 4, Text Data S1†).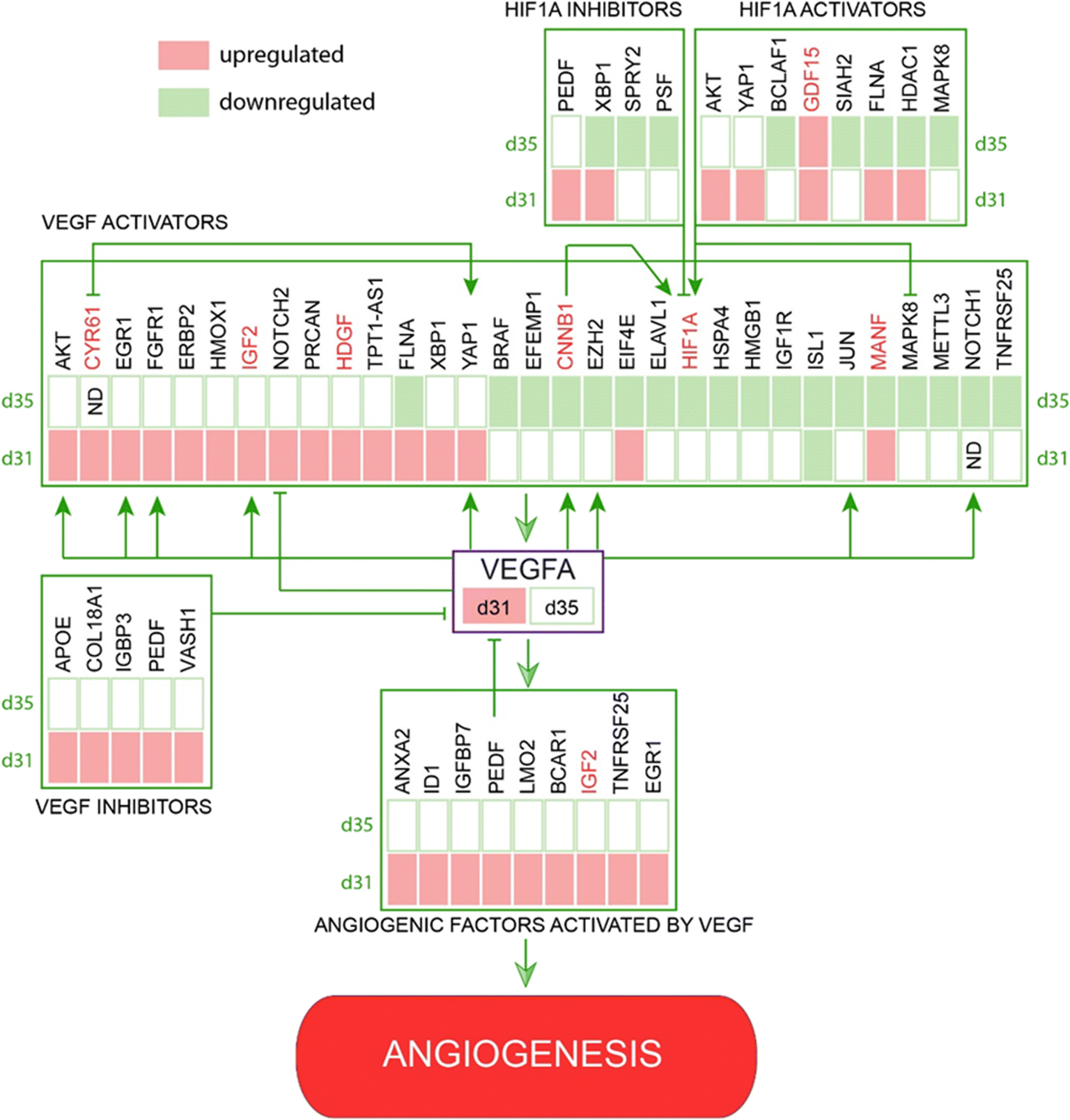 | ||
| Fig. 4 Identified network of co-regulated genes involved in VEGF controlled angiogenesis demonstrates regulation of VEGF pathway members and downstream targets at days 31 and 35. | ||
Other pro-angiogenic factors are downregulated after 10 days of co-culture
β-Catenin (CTNNB1; ↓d35), a central regulator of CNS angiogenesis acting on CNS vessels specifically,24 was significantly downregulated at day 35 (p < 0.05). Several activators of the WNT pathway were inhibited at day 35, including discs large MAGUK scaffold protein 125 (DLG1; ↑d31), DEAD-box helicase 526 (DDX5; ↓d35), heat shock protein HSP 90-beta27 (HSP90AB1; ↓d35), and secreted frizzled-related proteins 128–30 (sFRP1; ↓d31, ↓d35) (Table S1†).In addition, multiple pro-angiogenic factors not directly related to the VEGF or Wnt/β-catenin pathways were inhibited at day 35, including APC-stimulated guanine nucleotide exchange factor31,32 (ARHGEF4 or ASEF, ↑d31), cannabinoid receptor 133,34 (CNR1; ↓d35), cofilin 135 (CFL1; ↓d35), heterogeneous nuclear ribonucleoprotein K36 (hnRNPK; ↓d35), high mobility group box 137 (HMGB1; ↓d35), mitochondrial dynamin-like GTPase38 (OPA1; ↓d35), programmed cell death 539 (PDCD5; ↓d35), and YY1-associated factor 140,41 (YAF1 or HDAC2; ↓d35) (Table S1†). Similarly, some factors showed upregulation at day 31 during the peak of pro-angiogenic activity, with decreased expression by day 35. These included lamin A42 (LMNA; ↑d31), L-lactate dehydrogenase A chain43–45 (LDHA; ↑d31), and serine/threonine protein kinase 2646 (STK26 or MST4; ↑d31) (Text Data S2†).
Identified extracellular factors affect angiogenic sprouting
A thorough search for angiogenesis-related genes regulated under at least one of the conditions (d31/d28, d35/d28) identified six extracellular factors, including cysteine-rich angiogenic inducer 61, CYR61 (↑d31, d35-not detected), growth differentiation factor 15, GDF15 (↑d31), hepatoma-derived growth factor, HDGF (↑d31), hepatoma-derived growth factor-related protein-3, HDGFRP3 (↓d31, ↓d35), mesencephalic astrocyte-derived neurotrophic factor, MANF (↑d31), and secreted frizzled protein 1, sFRP1 (↓d35) (Fig. 4).We next hypothesized that angiogenesis-related extracellular regulatory factors have strong effects on the interaction between vascular cells and COs with regard to vascularization of the latter via angiogenic processes from the pre-formed vascular network.
To screen the identified growth factors for angiogenic activity, a three-channel microfluidic device was used (Fig. 5 and S8A†). In this angiogenesis assay (Fig. S8A†), ECs migrated through gel-forming sprouts in response to the growth factor gradient in the gel (Fig. 5A). The extent of sprout formation depended on the angiogenic activity of growth factors. Fig. S8B† shows the angiogenic activity of all the identified growth factors. CYR61 (100 ng ml−1) (p < 0.05) and HDGF (50 ng mL−1) treatment was associated with significantly higher activity within two days, with elongation of sprouts, interconnection of neighboring sprouts similar to intussusceptive microvascular growth, and creation of network-like structures. On day 10, CYR61 displayed superior angiogenic activity compared to that of HDGF (p < 0.05, Fig. 5B). Next, we tested the perfusability of the sprouts by flowing 70 kDa rhodamine dextran for both CYR61 and HDGF and observed the presence of dextran inside the sprouts, although leaky, confirming perfusable sprouts for both factors (Fig. 5C). The angiogenic activity measured by sprout coverage after 10 days of growth revealed negligible sprout formation for MANF and HDGFRP3 and no significant difference in sprout generation by FGF13 and sFRP1 when compared to the control (Fig. 5D and S8B†). In contrast, CYR61 and HDGF led to significantly higher angiogenic activity, with a maximum sprout coverage area of over 50% (p < 0.01) and 30% (p < 0.01), respectively (Fig. 5D).
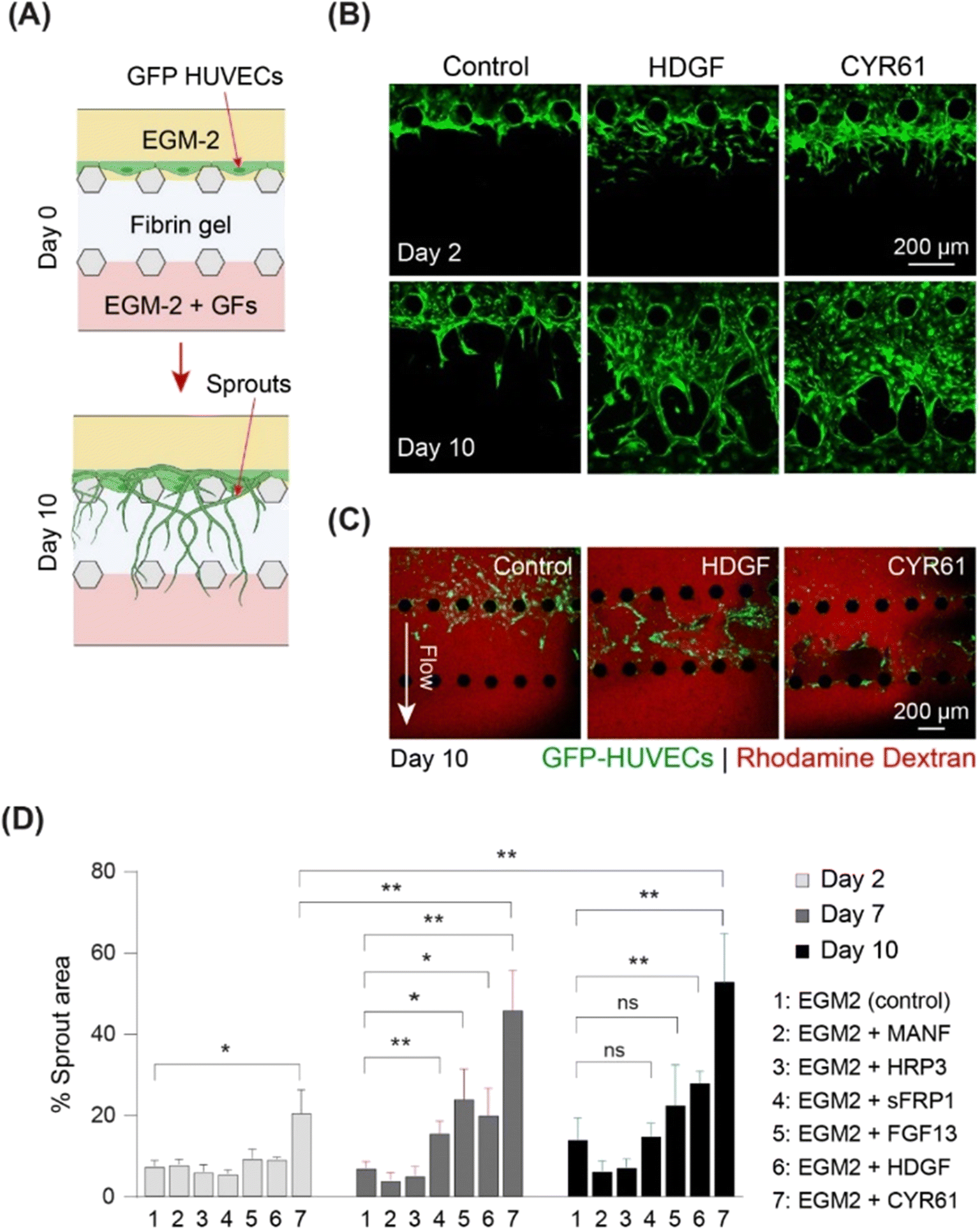 | ||
| Fig. 5 On-chip sprouting assay demonstrates strong pro-angiogenic effect of HDGF and CYR61. (A) Schematic of the angiogenesis assay. (B) Angiogenic sprouts (shown in green) at days 2 and 10 of culture in EGM2 only (control), EGM2 with HDGF, and EGM2 with CYR61. (C) Perfusability of rhodamine dextran inside angiogenic sprouts formed in the presence of EGM2 alone (control), EGM2 with CYR61, and EGM2 with HDGF at day 10. (D) Percentage area covered by sprouts at day 2, 7, and 10 of the angiogenesis assay, n = 6. | ||
Identified extracellular factors CYR61 and HDGF affect vascular network formation
To assess vasculogenic activity, we examined CYR61 and HDGF for their ability to promote the self-organization of ECs into complex network structures. We repurposed the same three-channel device (Fig. S8A†) by seeding HUVECs in the fibrin–collagen gel in the central channel and supplying growth factors from both sides (Fig. 6A). Vascular networks were formed in the presence of both CYR61 (100 ng mL−1) and HDGF (50 ng mL−1) within four days (Fig. 6B). After 10 days, CYR61-treated samples exhibited significantly more extensive vasculature (∼70%; p < 0.0001) than the control (∼25%). Similarly, HDGF-treated samples also exhibited significantly greater coverage than the control (∼60%; p < 0.001); however, the CYR61 vasculogenic effect was significantly stronger than that of HDGF (p < 0.0001; Fig. 6C). The generated vascular networks demonstrated significantly more branches for both CYR61 (p < 0.015) and HDGF (p < 0.05) than for the control (Fig. 6D). Furthermore, CYR61 treatment led to a markedly higher connectivity ratio in the vascular networks compared to control, although the difference was not statistically significant (Fig. S9A†). The perfusability of the vascular network was confirmed by injecting fluorescent rhodamine dextran (Fig. S9B†). From the results of both angiogenesis and vasculogenesis assays, we concluded that CYR61 was the best angiogenic enhancer, closely followed by HDGF. Therefore, we hypothesized that both CYR61 and HDGF could be putative modifiers of organoid vascularization.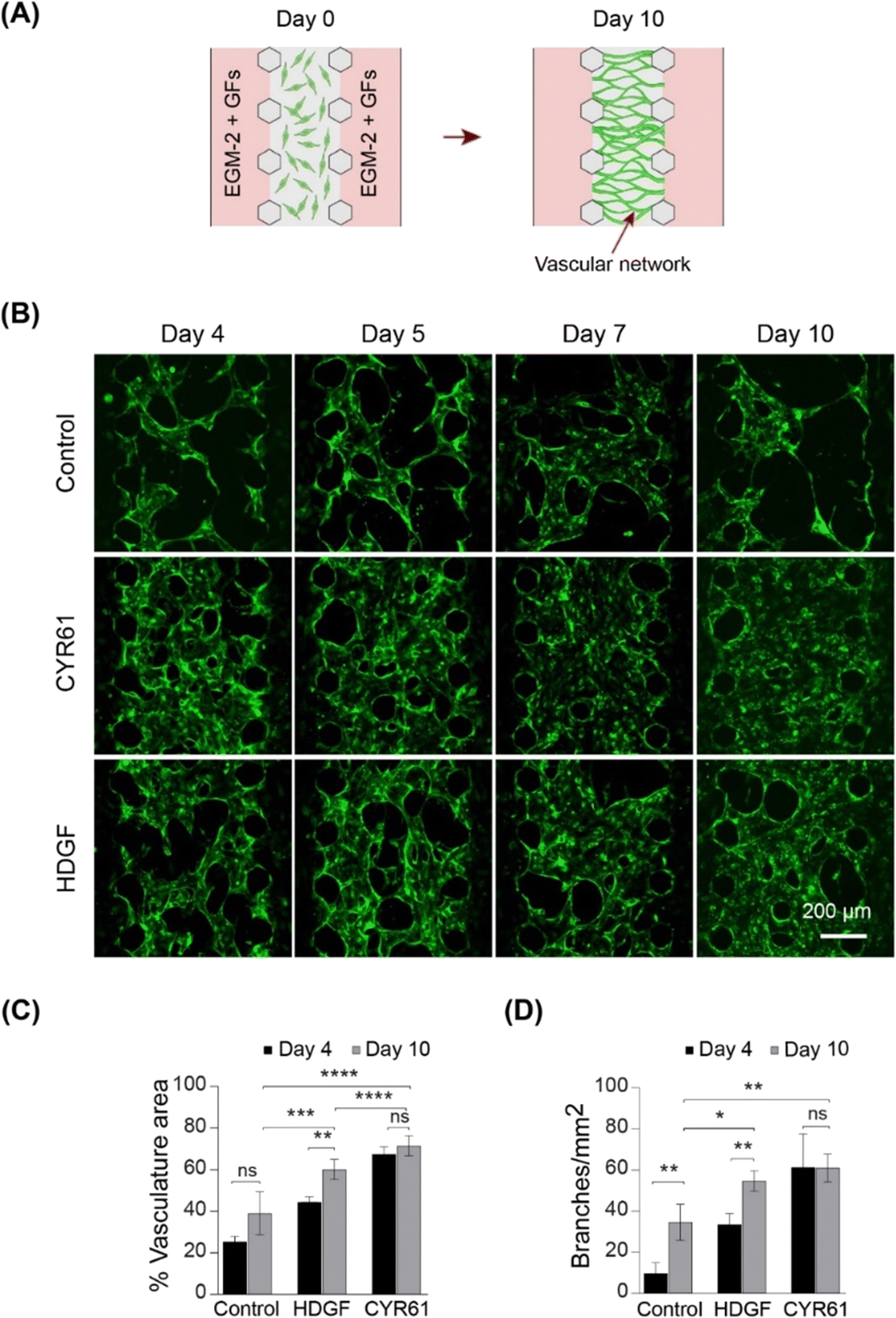 | ||
| Fig. 6 On-chip vasculogenesis assay demonstrates strong vasculogenic effect of HDGF and CYR61. (A) Schematic showing vasculogenesis assay. (B) Vascular networks formed at days 4, 5, 7 and 10 in EGM2 only (control), EGM2 with CYR61, and EGM2 with HDGF. (C) Percentage of vasculature area at days 4 and 10, under three different conditions (n = 4). (D) Branches per square millimeter at days 4 and 10, n = 4. | ||
CYR61 and HDGF show signs of improved angiogenesis and may stimulate organoid vascularization in vitro
Next, we investigated whether the addition of CYR61 and HDGF directly to the co-culture medium had significant effects on cerebral organoid vascularization. Both CYR61 (100 ng mL−1) and HDGF (50 ng mL−1) were added directly to the mixed medium (EC medium![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :organoid medium = 1:1), and organoids were grown in the on-chip co-culture system for 10 days (Fig. 7A). The sprout invasion was evaluated by both immunostaining of organoid sections and confocal microscopy. On CC-D10 (day 35), sprouts invading the ventricular structure (shown by CD31 staining) and many ECs migrating into the CO (shown by GFP-HUVECs) were observed in the co-cultures containing CYR61 (Fig. 7B, C and E and S9C†). In the case of HDGF, long vascular networks were observed inside the organoid in the sections (Fig. 7C and S9C†) as well as in the confocal images (Fig. 7F) and had higher percentage of vasculature are compared to CYR61 and control (Fig. S9C†). These data suggest that the co-culture of organoids in the presence of growth factors improved angiogenesis in the organoids and stimulated the organoid vascularization process in vitro.
:organoid medium = 1:1), and organoids were grown in the on-chip co-culture system for 10 days (Fig. 7A). The sprout invasion was evaluated by both immunostaining of organoid sections and confocal microscopy. On CC-D10 (day 35), sprouts invading the ventricular structure (shown by CD31 staining) and many ECs migrating into the CO (shown by GFP-HUVECs) were observed in the co-cultures containing CYR61 (Fig. 7B, C and E and S9C†). In the case of HDGF, long vascular networks were observed inside the organoid in the sections (Fig. 7C and S9C†) as well as in the confocal images (Fig. 7F) and had higher percentage of vasculature are compared to CYR61 and control (Fig. S9C†). These data suggest that the co-culture of organoids in the presence of growth factors improved angiogenesis in the organoids and stimulated the organoid vascularization process in vitro.
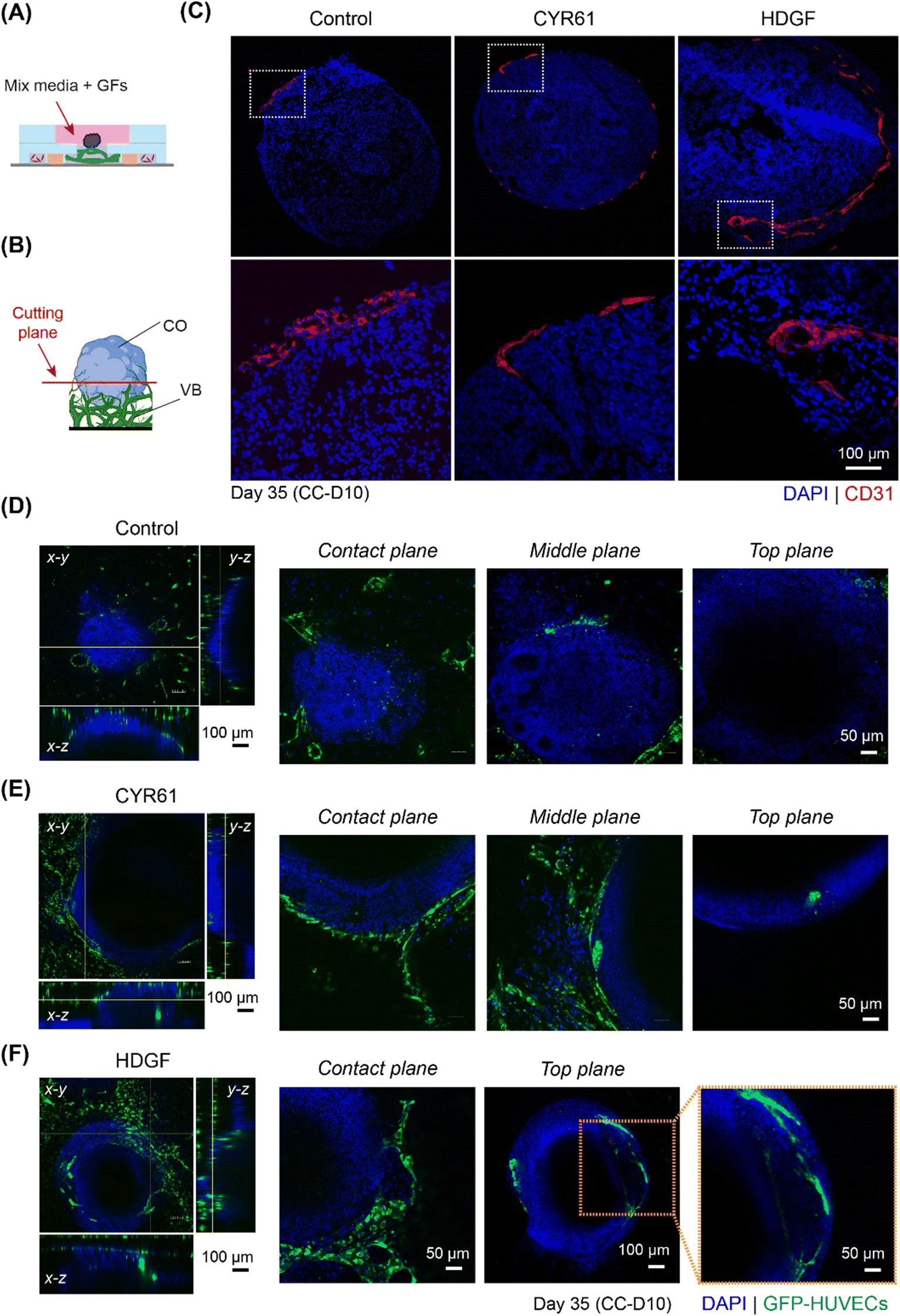 | ||
| Fig. 7 CYR61 and HDGF improve CO angiogenesis in vitro. (A) Schematic showing the CO co-culture with 3D vasculature in the presence of growth factors, CYR61 and HDGF. (B) Schematic showing the sectioning (cutting) plane in red, CO (blue) in direct contact with VB (green). (C) Immunostained cryosections of organoids co-cultured for 10 days on-chip in mixed medium (NDM/EGM2), mixed medium with CYR61, and mixed medium with HDGF. Vasculature is stained by CD31 (shown in red) and nuclei by DAPI (shown in blue). Lower panel shows the magnified images of the white dotted rectangular regions in the upper panel. (D–F) Confocal images showing the CO (blue) with VB (green) after 10 days of co-culture in (D) mixed medium alone/control, (E) mixed medium with CYR61 and (F) mixed medium with HDGF. COs are stained using DAPI. Images at the plane of contact of CO and VB (denoted by contact plane), plane inside the organoid co-culture well (denoted by middle plane) and plane above the co-culture well (denoted by top plane) are also shown. | ||
Discussion
Brain vascularization is achieved through complex interactions between the vascular and nervous systems. Understanding the cues of neurovascular communication is essential for building efficient long-term brain organoid systems that can be used to model developmental processes and determine disease mechanisms. Numerous molecular factors expressed by both vascular and nervous tissues have been identified, and their importance has been demonstrated both in vivo and in vitro.47 Their role is often beyond the control of angiogenic processes, having a strong impact on neurogenesis, neuronal migration, and general brain homeostasis. The formation of new blood vessels is not only triggered by the necessity to provide adequate nutrients and oxygen, but also directly controls cerebral developmental processes, including neurogenesis, gliogenesis, regionalization, and neuronal plasticity. In contrast, molecular factors exerted by neurons and surrounding cells regulate EC properties and their angiogenic potential.47 Recent single-cell-omics studies have convincingly demonstrated that organ development and tissue specification occur under the tight control of molecular factors produced by specific cells and are regulated in a complex temporo-spatial manner.18The human brain presents an additional dimension of complexity, guided by a sophisticated system of molecular gradients generated by various cell types. Its vascular system development utilizes mechanisms different from those of other organs and involves an additional structure, the blood brain barrier (BBB), composed of individual neurovascular units built of several cell types.48 The perineural vascular plexus (PNVP) is the first primitive vascular network to form in vivo around the neural tube and is mainly triggered by VEGFA expressed by the neural tube, which initiates a cascade of pro-angiogenic factors that further drive CNS vasculogenesis.49–51 The ingression of angiogenic sprouts from the PNVP produces the intraneuronal vascular plexus that develops in concert with migrating radial glial cells, ultimately forming the periventricular vascular plexus.47,52 ECs recruit pericytes, and the vascular system is covered by astrocyte processes connected to neurons forming the neurovascular unit, an essential element of the BBB.53 Further vascularization of the brain during development is primarily achieved through angiogenesis in a highly orchestrated process.48,54 Clearly, reproduction of such a process is difficult to achieve in conventional cell culture system.16,55
Reproducing the brain vascularization process in vitro is a monumental task that is yet to be successfully demonstrated.16 One reason is the inherent conflict in co-culturing dissimilar cell types controlled by extrinsic factors that impose direct, specific, and often mutually exclusive effects on different cell types. To preserve a specific brain organoid lineage, a particular type of culture medium and growth factors, some of which demonstrate inhibitory effects on microvasculature growth, must be used. Here, we applied a combination of growth culture conditions (NDM/EGM2) in a microfluidic system to simultaneously provide the required nutrients for both neurogenic CO specification and propagation of the vascular network. Our data demonstrated that the neuronal lineage of CO in this environment was preserved and that the preformed VB remained intact for at least 10 days (Fig. 2). The initial angiogenic response during co-culture correlates with the migratory behavior of endothelial cells (ECs) at the initiation of angiogenesis.56 Following the migration of ECs, angiogenesis proceeds via the formation of sprout-like structures, which then interconnect to form a vascular network, ultimately vascularizing the tissue.48 However, angiogenic processes observed after a few days in co-culture were quickly inhibited after day 7, which may have been in part due to changes in the molecular machinery evoked by COs on vascular cells (Fig. 2). A similar initiation of the angiogenic process was previously observed when COs were coated with ECs in Matrigel.7
We monitored global transcriptome changes in the outer layer of COs (200 μm) during critical time points and applied DAVID functional annotation in combination with a GenCLiP literature search to identify genes associated with angiogenesis and vascular development. We found that, while the expression of some pro-angiogenic factors was elevated at day 31, many activators of angiogenesis were downregulated at day 35, which may explain the overall inhibition of angiogenic processes by CC-D10 (Fig. 3 and 4).
This was particularly striking when focusing on the VEGFA (↑d31) regulatory network (Fig. 4). Angiogenesis is a multistage process of new blood vessel formation that involves many factors centered around VEGF signaling,57 which plays a crucial role in the guidance of endothelial tip cells and proliferation of stalk cells.47 The main source of VEGFA is neural progenitor cells49 and its activation occurs via physiological stimuli, such as hypoxia58 or activation of various pro-angiogenic factors.23 HIF1 is the main transcription factor in this group that triggers a cascade of pathways involved in the regulation of oxygen homeostasis and angiogenesis.59 It is a heterodimeric protein existing in two forms: HIF1a (↓d35) and HIF1b. Whereas HIF1b is constitutively expressed, HIF1a (↓d35) undergoes oxygen-dependent regulation.58,60 HIF1 is a positive regulator of VEGF expression and is critical for postnatal CNS angiogenesis.61,62 Our data indicate that, while the VEGFA network seemed to be activated at the early stage of co-culturing (day 31), many VEGFA-activating cues were suppressed at day 35 (Fig. 4).
The WNT/β-catenin signaling pathway is another major regulator of CNS-specific angiogenesis and is tightly connected to the HIF1/VEGF pathways.24 It is considered a main driver of sprouting angiogenesis and neurovascular unit formation.63 WNT-activating factors are expressed in CNS progenitors, stimulating the ingrowth and maturation of newly formed blood vessels.47 Transplantation studies indicate that extracellular signals from the brain parenchyma induce ECs to trigger CNS angiogenesis64,65 and is also regulated by the WNT pathway.24 Beta-catenin (CTNNB1; ↓d35), stabilized by WNT ligands, initiates the expression of specific genes involved in body pattern formation, neurogenesis, neurodegeneration, and vascular tissue development.66–68 Inhibition of WNT/β-catenin signaling in mice leads to severe defects in CNS angiogenesis, whereas non-CNS angiogenesis remain intact.24 Similar to VEGF pathway, beta-catenin (CTNNB1; ↓d35) was downregulated by day 35 being in agreement with inhibited CO angiogenic processes (Fig. 2).
In this study, we performed transcriptome analysis of the outer layer of COs to identify molecular changes in response to their crosstalk with 3D vasculature. Because of the close contact between the organoid and the VB, the collected outermost areas may have included endothelial cells. However, based on our estimates, the percentage of EC in the CO sections did not exceed 5% and 2% on days 31 and 35, respectively (Fig. 2C). Therefore, it is plausible that even if some ECs were accidentally captured during dissection, they would be insufficient to significantly alter the combined CO expression profile.
One way to promote vascularization process is to supply the culture media with certain angiogenic extracellular factors. In this study, we identified six growth factors, including CYR61 (↑d31, d35-not detected), GDF15 (↑d31), HDGF (↑d31), HDGFRP3 (↓d31, ↓d35), MANF (↑d31), and sFRP1 (↓d35) (Fig. 4). CYR61 (↑d31, d35-not detected), GDF15 (↑d31), HDGF (↑d31) and MANF (↑d31) are known pro-angiogenic factors involved in the VEGF/HIF1A signaling pathway. For example, CYR61 (↑d31, d35-ND) is an activator of the VEGF69–71 pathway (Fig. 4) that stimulates endothelial tip cell activity72 and plays an important role in neovascularization during embryonic development.73 HDGF (↑d31) is a secretory factor that promotes tumorigenesis, metastasis and enhances VEGF-dependent angiogenesis74–79 likely by stimulating the expression of HIF1A79 (Fig. 4). Similarly, increased levels of GDF15 (↑d31) and MANF (↑d31) promote angiogenesis via HIF1A80–82 and the VEGF pathway83 (Fig. 4). sFRP1 (↓d35) is a modulator of the Wnt/β-catenin pathway, which is known to stimulate angiogenesis and promote maturation of neovasculature in mesenchymal stem cells.29 The involvement of HDGFRP3 (↓d31, ↓d35), a neurotrophic factor that stimulates angiogenesis by promoting EC growth, migration, tube formation, and sprouting84 (Fig. 4), to a particular signaling pathway is unknown.
Our functional studies identified CYR61 (↑d31, d35-not detected) and HDGF (↑d31), as potential modifiers, that may improve CO vascularization (Fig. 5). While HDGF has been previously reported to be an important factor in vascular network formation, self-organization of ECs by CYR61 has not been previously demonstrated (Fig. 6). Moreover, both factors showed moderate improvement in CO-VB sprout formation within the entire co-culture period, (Fig. 7) compared to that in COs without growth factors (Fig. 2B and C), suggesting their possible functional involvement in CNS angiogenesis. However, in-depth investigations are required to determine the mechanisms of these factors and their roles in brain angiogenesis.
Particularly noteworthy is the potential influence of stromal cell quantity and origin on vasculogenesis. The brain vasculature contains specialized structures known as the BBB, which includes pericytes and other stromal cells. However, in this study, HUVECs and hLFs were used for the preparation of on-chip VB, which is a major limitation. Therefore, brain-specific microvascular ECs or iPSC-derived ECs could be used in future studies to generate a co-culture model closely resembling the developing human brain. In future studies, perfusion culture could be employed to improve the vascularization process. Additionally, single-cell sequencing or spatial omics analysis can be performed in the future for a better understanding of CO-specific vascularization process.
Materials and methods
Microfluidic devices: design and fabrication
To culture COs on a preformed VB, a three-layer microfluidic device was fabricated using a soft lithography process, as described previously.17 Briefly, the device comprised a top polydimethylsiloxane (PDMS) layer with gel injection ports, medium inlets, an organoid culture well, a PDMS middle layer with five parallel channels separated by micropillars (width = 100 μm, height = 250 μm), a removable polyester membrane coated with Sigmacote (Sigma) (pore size 0.4 μm; Sterlitech, Kent, WA, USA), and a glass bottom layer (Fig. 1B). To prepare the top layer, organoid culture wells (diameter: 8 mm), gel injection ports (diameter: 2 mm), and medium inlets (diameter: 6 mm) were punched out from a PDMS slab using biopsy punches (Sterile Dermal Biopsy Punch, Kai Industries, Tokyo, Japan) with corresponding diameters. Both PDMS layers were irreversibly air plasma bonded (40 s, 50 W, flow rate: 50 sccm; Femto Science, Hwaseong, Korea) and baked at 70 °C for 1 h.Angiogenesis and vasculogenesis assays were conducted using a three-channel microfluidic device with a width of 500 μm, separated by microposts (width 100 μm), and fabricated via standard photolithography using SU8 2100 (MicroChem, USA) on a silicon wafer with a feature height of 100 μm. The microfluidic device had two layers: a top PDMS layer featuring channel inlets of 2 mm, a medium reservoir of 6 mm, and a glass bottom layer (Fig. S8A†).
The PDMS portion of the device was bonded to a glass coverslip (24 mm × 24 mm, Matsunami Glass, Osaka, Japan) via plasma treatment. The bonding was completed by baking at 120 °C overnight. The devices were sterilized by UV treatment before the experiments.
For the five-channel device, the polyester membrane coated with Sigmacote was placed onto the organoid culture well, before gel injection.
Cell and cerebral organoid cultures
Control hiPSCs (201B7) were obtained from the RIKEN Bioresource Research Center. iPSCs were cultured under feeder-free conditions on iMatrix-511- (Nippi, Japan) coated plates with StemFit (AK02N, Ajinomoto, Japan) and maintained in an incubator under 5% CO2 at 37 °C. For passaging, iPSCs were harvested using 0.5× TryPLE Express (Thermo Fisher Scientific) and replated on iMatrix-511-coated dishes at a density of 1350 cells per cm2. Cerebral organoids were derived from hiPSCs using a previously reported method for serum-free floating culture of embryoid body-like aggregates with quick re-aggregation (SFEBq)3 (Fig. S2†).GFP-expressing HUVECs (GFP-HUVECs, Angio-Proteomie, Boston, MA, USA) were cultured in endothelial growth medium 2 (EGM2, Lonza, Basel, Switzerland), with GA1000 replaced with 1% penicillin and streptomycin (P/S, Thermo Fisher Scientific). Human lung fibroblasts (hLFs; Lonza) were cultured in fibroblast growth medium 2 (Lonza). Cells from passages four to five were used for the experiments. All cells were cultured in a humidified incubator at 5% CO2 and 37 °C.
Vascular bed preparation and perfusion assay
The cell seeding procedure is shown in Fig. S1.† Briefly, GFP-HUVECs and hLFs were prepared in EGM2 at concentration of 1.6 × 107 cells per mL and 1 × 107 cells per mL, respectively. A fibrin–collagen gel comprising fibrinogen (Sigma), collagen type I (Corning), and aprotinin (Sigma), was prepared on ice. The cell suspension was then mixed with fibrin–collagen gel solution at a ratio of 1:1. The concentrations of fibrinogen, collagen type I, and aprotinin in the final gel cell suspension were 2.5 mg mL−1, 0.2 mg mL−1, and 0.15 U mL−1, respectively. Thrombin (0.5 U mL−1, Sigma) was quickly added to the solution before injection into the five-channel device. Thirty microliters of HUVEC suspension (8 × 106 cells per mL) were injected into channel 3, and 20 μL hLF suspension (5 × 106 cells per mL) was injected into channels 1 and 5 (Fig. S1†). After polymerization of the gel at 37 °C for 15 min, EGM2 was introduced into channels 2 and 4. The porous polyester membrane was removed using tweezers. EGM2 was then introduced into organoid culture wells and medium reservoirs. The device was kept in a Petri dish covered with wet paper to prevent the evaporation of the media. After 2 d, GFP-HUVECs (5 × 106 cells in 20 μL EGM2) were introduced into channel 2 and incubated vertically at 37 °C for 30 min to allow the attachment of GFP-HUVECs to the side of the gel wall. The procedure was repeated for channel 4 to facilitate the opening of the constructed vascular network. The medium was changed every 2 days.
The perfusability of the vascular bed was tested on CC-D10 by introducing a fluorescent dye, 10 μl of 10 μM rhodamine B-conjugated dextran (70 kDa rhodamine dextran, Sigma) in DPBS, into channel 4 after removing EGM2 from the medium reservoirs. The pressure difference between channels 2 and 4 facilitated rapid perfusion of the dye through the vascular network, and images were obtained by confocal microscopy. The perfusability of the vascular network and angiogenic sprouts was tested on day 10 by introducing 10 μl of 10 μM rhodamine dextran into channel 1 after removing EGM2. The dye flowed through the center channel to channel 3, and images were captured.
Co-culture of hiPSC-derived cerebral organoids with preformed vasculature
For the on-chip co-culture system, the VB and COs were generated separately as described above. On day 25, multiple COs were introduced into the organoid culture well with a VB preformed on day 7, and then co-cultured for an additional 10 days (Fig. S1†) in mixed medium containing EGM2 and neural differentiation medium (NDM) in equal proportions. The NDM was prepared according to a previously published protocol.3 To test whether the mixed medium had any adverse effects on the cell growth in the co-culture experiments, COs were cultured in three types of media, NDM, EGM2, and mixed medium (NDM:EGM2 = 1:1), from day 25 to day 35 (Fig. S2B and C†). The medium was changed every 2 days.
Quantitative PCR
Total RNA was extracted from eight to ten COs for each condition using a Nucleospin RNA kit (MACHERY-NAGEL Inc., USA). cDNA was synthesized using PrimeScript RT Master Mix (Takara Bio Inc., Tokyo, Japan) and a thermal cycler (Bio-Rad Laboratories Inc., Tokyo, Japan). Quantitative PCR was performed using TB Green Premix Ex TaqII (Takara Bio Inc., Tokyo, Japan) and β-actin was used to normalize gene expression. The following primers were used: β-actin F: CAATGTGGCCGAGGACTTTG; β-actin-R: CATTCTCCTTAGAGAGAAGTGG; SOX1-F: GAACGCCTTCATGGTGTG; SOX1-R: CTGATCTCCGAGTTGTGCAT; FOXG1-F: AGAAGAACGGCAAGTACGAGA; FOXG1-R: CGGGTCCAGCATCCAGTAG; PAX6-F: TTCACATCTGGCTCCATGTT; PAX6-R: GGGTTGCATAGGCAGGTTAT; EMX1-F: GTCCGAGCAGAAGAAGAAGG; EMX1-R: AGTCATTGGAGGTGACATCG. The experiments were conducted in three independent replicates, each with five technical replicates.Brain organoid microdissection and RNA isolation
For gene expression experiments, day 28 (d28), day 31 (d31), and day 35 (d35) COs co-cultured on a VB were rapidly harvested, freshly frozen, cut into 15 μm sections using a cryostat (ThermoFisher Scientific, USA), and placed on non-charged glass slides. The 200 μm thick outer rims were collected from the serial sections of four independently cultured organoids (n = 4) for each time point using a capillary-based, vacuum-assisted cell and tissue acquisition system (UnipicK+, NeuroInDx, Inc., CA; Fig. S5†). On an average, at least 20 sections were microdissected per organoid. Total RNA was extracted from the collected tissues using the Qiagen RNeasy Micro kit, according to manufacturer protocol. The concentration and purity of the RNA were determined using a NanoDrop One/One spectrophotometer (Thermo Fisher Scientific, USA).Microarray experiment and data analysis
Twenty nanograms of isolated total RNA was used for T7-based amplification and labeling (Agilent Low RNA Input Linear Amplification Kit). The dye incorporation efficiency was detected using a NanoDrop One/One spectrophotometer (Thermo Fisher Scientific). Agilent whole human genome microarray comparisons of CO outer layers were performed for co-cultured organoids prior to detecting the vascular bed angiogenic response (d28, control), during visible angiogenic response (d31), and inhibited angiogenesis (d35), producing two independent comparison groups: d31 (angiogenesis) versus d28 (control) and d35 (inhibited angiogenesis) versus d28 (control) (Fig. 1). For each time point, four independently isolated RNA samples were used, producing four biological replicates. Eight microarray hybridizations were performed for each biological comparison, for a total of 16 microarrays, generating two groups of differentially expressed genes in the outer layers of organoids at d31/d28 (d31) and d35/d28 (d35) comparison points. The direction of expression for each gene in each comparison is indicated by up (↑) or down (↓) arrows in the text.Raw microarray data were acquired using an Agilent DNA Microarray scanner and processed with the accompanying Agilent Feature Extraction 10.5 Image Analysis software using default settings. Lowess normalized signal intensities with a >300 cut-off for the mean signal at each time point and >100 in each individual array were used to identify gene expression changes on d31 and d35 versus d28. To identify differential expression, the genes were tested against two conservative criteria: a ratio beyond the 99.5% confidence interval observed in homotypic comparisons,85 which corresponded to a ∼2-fold expression change; and a paired t-test yielding a p value less than 0.05. A Benjamini–Hochberg correction for multiple comparisons was performed. Identified differentially expressed genes were subjected to DAVID functional annotation analysis19,20 and a literature network search using GenCLiP321 for functional categories and specific “angiogenesis” factors. DAVID analysis was used to identify over-represented biological processes (EASE score = 0.05) and enriched clusters of genes for specific biological processes (EASE score = 0.01). The following parameters were used to identify enriched clusters: EASE score below 0.01, p-value below 0.05, adjusted for multiple comparisons using Benjamini–Hochberg correction, and at least 2-fold enrichment for each functional category.
Sprouting and vasculogenesis assays
For sprouting assay, fibrin–collagen gel consisting of 2.5 mg mL−1 fibrinogen (Sigma), 0.2 mg mL−1 collagen type I (Corning), and 0.15 U mL−1 aprotinin (Sigma) was prepared on ice. After adding 0.5 U mL−1 thrombin to the gel, the mixed solution was injected into channel 2 of the three-channel device (Fig. 5A and S8A†). The devices were incubated overnight for polymerization of the gel in a humidified chamber with 5% CO2 at 37 °C. GFP-HUVECs were prepared in EGM2 at a concentration of 1.6 × 107 cells per mL, and 20 μL of suspension was then added to channel 1 and incubated vertically at 37 °C to allow the attachment of HUVECs onto the gel wall for 20 min (denoted as day 0). EGM2, alone or with growth factors (EGM2 + GFs), was then added to channels 1 and 3, respectively, and cultured for 10 d (Fig. 5A). The medium was changed every day.For the vasculogenesis assay, 30 μL of GFP-HUVECs (8 × 106 cells per mL) in EGM2-based fibrin–collagen gel solution with thrombin, as described above, were injected into channel 2 of a three-channel device (Fig. 6A and S8A†) and incubated in a humidified chamber at 5% CO2 and 37 °C for 15 min to polymerize the gel. EGM2 + GFs was then added to channels 1 and 3 (day 0). After 2 d, 20 μL of GFP-HUVECS (5 × 106 cells per mL) was added to channel 1 and incubated vertically at 37 °C for 30 min. The same procedure was repeated for channel 3. EGM2 containing growth factors was then supplied to channels 1 and 3 (Fig. 6A) and cultured until day 10, with the media changed every day.
The following growth factors were purchased and reconstituted according to manufacturer guidelines: recombinant human CYR61 Fc chimera protein CF (CYR61; R&D Systems Inc.), hepatoma-derived growth factor recombinant protein (HDGF; ProSci, CA, USA), recombinant human FGF13 protein (FGF13; RayBiotech Life, Inc., GA), recombinant human sFRP1 protein CF (sFRP1; R&D Systems Inc.), recombinant human MANF protein CF (MANF; R&D Systems Inc.), and recombinant human HRP3 protein (HRP3; Abcam). CYR61, FGF13, sFRP1, MANF, and HRP3 were diluted in EGM2 to a final concentration of 100 ng mL−1; HDGF to a final concentration of 50 ng mL−1 for the experiments (Fig. 5 and 6). For the on-chip co-culture system, CYR61 and HDGF were added to the mixed medium (Fig. 7).
Immunohistochemistry
Organoids were washed twice with phosphate-buffered saline (DPBS) and then fixed in 4% paraformaldehyde (PFA) in DPBS on ice for 20 min and incubated overnight in 20% sucrose in PBS at 4 °C for cryoprotection. PFA-fixed or cryoprotected organoids were embedded and frozen in Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetek, Japan), serially sectioned at 10 μm thickness using a cryostat (Thermo Fisher Scientific) and mounted on glass slides (Matsunami). The tissue sections were washed in PBS to remove excess OCT compound, permeabilized with 0.3% Triton X-100 in PBS (DPBST) three times for 15 min each, blocked with 10% donkey serum (Jackson Immunoresearch) in DPBST for 1 h at room temperature, and incubated overnight with primary antibodies (Table S3†) in blocking solution prepared in 0.05% Tween 20 in DPBS (DPBT) at 4 °C. The sections were then washed three times with DPBT for 15 min each, incubated with secondary antibodies (Table S3†) and DAPI (#D3571, Thermo Fisher Scientific) diluted in blocking solution prepared in DPBT for 1 h at room temperature, and then washed thoroughly with DPBT. Tissue sections were then mounted in SlowFade Gold Antifade Reagent (Thermo Fisher Scientific) for imaging.86For whole-mount immunostaining, devices with co-cultured organoids on vasculature were washed twice in DPBS, fixed in 4% PFA for 20 min on ice, and washed twice in DPBS. The tissues were permeabilized with 1% Triton X-100 in DPBST (DPBST1) three times for 15 min each, blocked with 10% donkey serum in DPBST1 for 3 h at room temperature, and incubated with primary antibodies (Table S3†) in dilution buffer prepared in DPBST1 at 4 °C for 2 d. The devices were washed with DPBST1 three times for 20 min each and incubated for 1 h with secondary antibodies (Table S3†) and DAPI (D3571, Thermo Fisher) in DPBST1 at room temperature. The tissues were washed three times with DPBS for 30 min.
Image and statistical analysis
Images were captured using a confocal microscope (Olympus FV-3000, Tokyo, Japan). The immunostained images were binarized using ImageJ software and analyzed for the percentage area of at least four organoids and devices. To analyze the vascular network morphology, GFP images were acquired using a confocal microscope and post-processed to obtain the maximum intensity projected image. GFP images were filtered and binarized. The area fraction was then measured from the binary image using ImageJ software (NIH). The binary image was then skeletonized and analyzed for branches, junctions, and endpoints. The percent cover of vasculature area was determined by measuring the area fraction of the binary image. The average vessel diameter was calculated as follows:The number of branches per square millimeter was calculated by dividing the number of branches by the total area in square millimeters. The connectivity ratio was calculated by dividing the junctions by the endpoints.
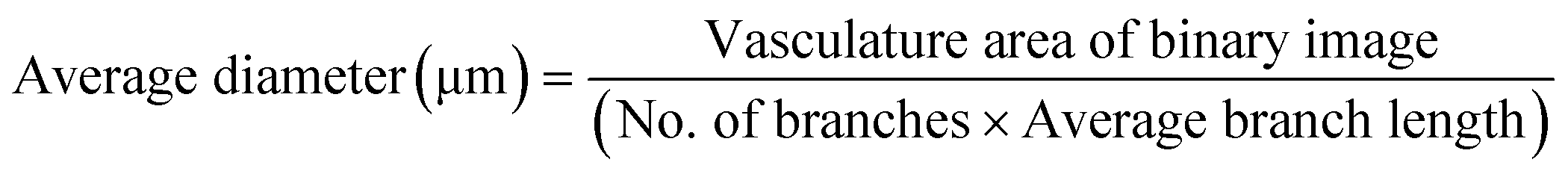
All data presented in this manuscript represent the mean ± SD, unless specified in the figure legends. Statistical tests were performed using the GraphPad Prism software (San Diego, CA, USA). Tukey's Multiple comparison test (one-way ANOVA) was used to determine statistical significance. The following symbols were used: * p < 0.05; ** p < 0.01; *** p < 0.001; *** p < 0.0001; and ns, no significance. Illustrations were prepared using BioRender and Adobe Illustrator.
Data availability statement
The datasets generated during and/or analysed during the current study are available in the NCBI GEO repository and are available from the corresponding author on reasonable request.Author contributions
SLK and RY conceived, developed, and mentored the project; KM provided the brain organoid culturing protocol and assisted in data interpretation; MS and SLK conceived and performed experiments; AT assisted with experiments; MS and SLK analyzed and interpreted the data; MS, AT, KF, KM, SLK, and RY wrote the manuscript. KF assisted in the fabrication of the microfluidic chip.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Microarray scanning was conducted at the Medical Research Support Center, Graduate School of Medicine, Kyoto University, and was supported by the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science, and Technology, Japan. The authors would like to thank Masatsugu Denawa for providing technical support; Aki Kubo, Shiho Morimoto, and Mayumi Moriwake for supporting fabrication, organoid, and cell maintenance; and Dr. Lili Kudo for critical reading of the manuscript. Microfabrication was supported by the Nanotechnology Hub, Kyoto University. This research was supported by Japan Agency for Medical Research and Development (AMED) grant numbers JP22be1004204 and JP17be0304205 and JSPS KAKENHI Grant Number JP 22K18319 (RY). KM was supported by NOVARTIS foundation for the Promotion of Science. M. S. was supported by the JICA FRIENDSHIP Program (No. D1956066).References
- A. Bhattacharya, W. W. Y. Choi, J. Muffat and Y. Li, J. Mol. Biol., 2022, 434, 167386 CrossRef.
- M. Hofer and M. P. Lutolf, Nat. Rev. Mater., 2021, 6, 402–420 CrossRef.
- T. Kadoshima, H. Sakaguchi, T. Nakano, M. Soen, S. Ando, M. Eiraku and Y. Sasai, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20284–20289 CrossRef.
- A. Bhaduri, M. G. Andrews, W. Mancia Leon, D. Jung, D. Shin, D. Allen, D. Jung, G. Schmunk, M. Haeussler, J. Salma, A. A. Pollen, T. J. Nowakowski and A. R. Kriegstein, Nature, 2020, 578, 142–148 CrossRef.
- A. A. Mansour, J. T. Gonçalves, C. W. Bloyd, H. Li, S. Fernandes, D. Quang, S. Johnston, S. L. Parylak, X. Jin and F. H. Gage, Nat. Biotechnol., 2018, 36, 432–441 CrossRef.
- B. Cakir, Y. Xiang, Y. Tanaka, M. H. Kural, M. Parent, Y. J. Kang, K. Chapeton, B. Patterson, Y. Yuan, C. S. He, M. S. B. Raredon, J. Dengelegi, K. Y. Kim, P. Sun, M. Zhong, S. Lee, P. Patra, F. Hyder, L. E. Niklason, S. H. Lee, Y. S. Yoon and I. H. Park, Nat. Methods, 2019, 16, 1169–1175 CrossRef PubMed.
- M. T. Pham, K. M. Pollock, M. D. Rose, W. A. Cary, H. R. Stewart, P. Zhou, J. A. Nolta and B. Waldau, NeuroReport, 2018, 29(7), 588–593 CrossRef.
- P. Wörsdörfer, N. Dalda, A. Kern, S. Krüger, N. Wagner, C. K. Kwok, E. Henke and S. Ergün, Sci. Rep., 2019, 9, 15663 CrossRef.
- M. G. Kook, S.-E. Lee, N. Shin, D. Kong, D.-H. Kim, M.-S. Kim, H. K. Kang, S. W. Choi and K.-S. Kang, Int. J. Stem Cells, 2022, 15, 85–94 CrossRef.
- F. Birey, J. Andersen, C. D. Makinson, S. Islam, W. Wei, N. Huber, H. C. Fan, K. R. C. Metzler, G. Panagiotakos, N. Thom, N. A. O'Rourke, L. M. Steinmetz, J. A. Bernstein, J. Hallmayer, J. R. Huguenard and S. P. Paşca, Nature, 2017, 545, 54–59 CrossRef PubMed.
- X.-Y. Sun, X.-C. Ju, Y. Li, P.-M. Zeng, J. Wu, Y.-Y. Zhou, L.-B. Shen, J. Dong, Y. Chen and Z.-G. Luo, eLife, 2022, 11, e76707 CrossRef.
- Y. Jin, J. Kim, J. S. Lee, S. Min, S. Kim, D.-H. Ahn, Y.-G. Kim and S.-W. Cho, Adv. Funct. Mater., 2018, 28, 1801954 CrossRef.
- K. A. Homan, N. Gupta, K. T. Kroll, D. B. Kolesky, M. Skylar-Scott, T. Miyoshi, D. Mau, M. T. Valerius, T. Ferrante, J. V. Bonventre, J. A. Lewis and R. Morizane, Nat. Methods, 2019, 16, 255–262 CrossRef.
- Y. Nashimoto, R. Okada, S. Hanada, Y. Arima, K. Nishiyama, T. Miura and R. Yokokawa, Biomaterials, 2020, 229, 119547 CrossRef PubMed.
- Y. Nashimoto, T. Hayashi, I. Kunita, A. Nakamasu, Y. Torisawa, M. Nakayama, H. Takigawa-Imamura, H. Kotera, K. Nishiyama, T. Miura and R. Yokokawa, Integr. Biol., 2017, 9, 506–518 CrossRef PubMed.
- S. Zhang, Z. Wan and R. D. Kamm, Lab Chip, 2021, 21, 473–488 RSC.
- Y. Kameda, S. Chuaychob, M. Tanaka, Y. Liu, R. Okada, K. Fujimoto, T. Nakamura and R. Yokokawa, Lab Chip, 2022, 22, 641–651 RSC.
- X. Qian, H. N. Nguyen, M. M. Song, C. Hadiono, S. C. Ogden, C. Hammack, B. Yao, G. R. Hamersky, F. Jacob, C. Zhong, K. Yoon, W. Jeang, L. Lin, Y. Li, J. Thakor, D. A. Berg, C. Zhang, E. Kang, M. Chickering, D. Nauen, C.-Y. Ho, Z. Wen, K. M. Christian, P.-Y. Shi, B. J. Maher, H. Wu, P. Jin, H. Tang, H. Song and G. Ming, Cell, 2016, 165, 1238–1254 CrossRef.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2009, 4, 44–57 CrossRef PubMed.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nucleic Acids Res., 2009, 37, 1–13 CrossRef PubMed.
- J.-H. Wang, L.-F. Zhao, H.-F. Wang, Y.-T. Wen, K.-K. Jiang, X.-M. Mao, Z.-Y. Zhou, K.-T. Yao, Q.-S. Geng, D. Guo and Z.-X. Huang, Bioinformatics, 2020, 36, 1973–1975 CrossRef.
- Y.-I. Chung, S.-K. Kim, Y.-K. Lee, S.-J. Park, K.-O. Cho, S. H. Yuk, G. Tae and Y. H. Kim, J. Controlled Release, 2010, 143, 282–289 CrossRef.
- R. S. Kerbel, N. Engl. J. Med., 2008, 358, 2039–2049 CrossRef.
- R. Daneman, D. Agalliu, L. Zhou, F. Kuhnert, C. J. Kuo and B. A. Barres, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 641–646 CrossRef PubMed.
- C. Cho, Y. Wang, P. M. Smallwood, J. Williams and J. Nathans, eLife, 2019, 8, e45542 CrossRef.
- C. Liu, L. Wang, Q. Jiang, J. Zhang, L. Zhu, L. Lin, H. Jiang, D. Lin, Y. Xiao, W. Fang and S. Guo, Front. Oncol., 2019, 9 Search PubMed.
- H. Wang, G. Deng, M. Ai, Z. Xu, T. Mou, J. Yu, H. Liu, S. Wang and G. Li, Oncogene, 2019, 38, 1489–1507 CrossRef PubMed.
- P. Dufourcq, T. Couffinhal, J. Ezan, L. Barandon, C. Moreau, D. Daret and C. Duplàa, Circulation, 2002, 106, 3097–3103 CrossRef PubMed.
- P. Dufourcq, L. Leroux, J. Ezan, B. Descamps, J.-M. D. Lamazière, P. Costet, C. Basoni, C. Moreau, U. Deutsch, T. Couffinhal and C. Duplàa, Am. J. Pathol., 2008, 172, 37–49 CrossRef.
- P. Dufourcq, B. Descamps, N. F. Tojais, L. Leroux, P. Oses, D. Daret, C. Moreau, J.-M. D. Lamazière, T. Couffinhal and C. Duplàa, Stem Cells, 2008, 26, 2991–3001 CrossRef.
- Y. Kawasaki, S. Tsuji, K. Muroya, S. Furukawa, Y. Shibata, M. Okuno, S. Ohwada and T. Akiyama, EMBO Rep., 2009, 10, 1355–1362 CrossRef.
- Y. Kawasaki, T. Jigami, S. Furukawa, M. Sagara, K. Echizen, Y. Shibata, R. Sato and T. Akiyama, J. Biol. Chem., 2010, 285, 1199–1207 CrossRef PubMed.
- A. Amlani, M. G. Hornick, K. Cooper, P. Prazad, R. Donovan and A. Gulati, Dev. Neurosci., 2017, 39, 498–506 CrossRef.
- S. Pisanti, P. Picardi, L. Prota, M. C. Proto, C. Laezza, P. G. McGuire, L. Morbidelli, P. Gazzerro, M. Ziche, A. Das and M. Bifulco, Blood, 2011, 117, 5541–5550 CrossRef PubMed.
- Y.-L. Xu, D.-B. Wang, Q.-F. Liu, Y.-H. Chen and Z. Yang, Hum. Reprod., 2010, 25, 2480–2488 CrossRef.
- Y. Chen, Y. Zeng, Z. Xiao, S. Chen, Y. Li, J. Zou and X. Zeng, J. Cell. Biochem., 2019, 120, 14296–14305 CrossRef CAS.
- W. Wang, H. Jiang, H. Zhu, H. Zhang, J. Gong, L. Zhang and Q. Ding, Oncol. Lett., 2013, 5, 884–888 CrossRef PubMed.
- S. Herkenne, O. Ek, M. Zamberlan, A. Pellattiero, M. Chergova, I. Chivite, E. Novotná, G. Rigoni, T. B. Fonseca, D. Samardzic, A. Agnellini, C. Bean, G. Di Benedetto, N. Tiso, F. Argenton, A. Viola, M. E. Soriano, M. Giacomello, E. Ziviani, G. Sales, M. Claret, M. Graupera and L. Scorrano, Cell Metab., 2020, 31, 987–1003.e8 CrossRef PubMed.
- H. Zhao, C. Peng, X. Lu, M. Guo, T. Yang, J. Zhou and Y. Hai, Am. J. Transl. Res., 2019, 11, 1116–1128 Search PubMed.
- M. Hulsurkar, Z. Li, Y. Zhang, X. Li, D. Zheng and W. Li, Oncogene, 2017, 36, 1525–1536 CrossRef PubMed.
- L. Hu, X. Lv, D. Li, W. Zhang, G. Ran, Q. Li and J. Hu, J. Cell. Mol. Med., 2021, 25, 2190–2202 CrossRef PubMed.
- N. Sayed, C. Liu, A. Mohamed, F. Himmati, J. Z. Zhang, S. Khanamiri, J. R. Moonen, A. Wnorowski, L. Cheng, J. W. Rhee, S. Gaddam, K. C. Wang, K. Sallam, J. H. Boyd, Y. J. Woo, M. Rabinovitch and J. C. Wu, Sci. Transl. Med., 2020, 12, eaax9276 CrossRef PubMed.
- H. Lin, R. Muramatsu, N. Maedera, H. Tsunematsu, M. Hamaguchi, Y. Koyama, M. Kuroda, K. Ono, M. Sawada and T. Yamashita, EBioMedicine, 2018, 27, 71–85 CrossRef PubMed.
- G. Parra-Bonilla, D. F. Alvarez, M. Alexeyev, A. Vasauskas and T. Stevens, PLoS One, 2013, 8, e75984 CrossRef.
- B. Yetkin-Arik, I. M. C. Vogels, P. Nowak-Sliwinska, A. Weiss, R. H. Houtkooper, C. J. F. Van Noorden, I. Klaassen and R. O. Schlingemann, Sci. Rep., 2019, 9, 12608 CrossRef.
- X. Zhang, L. Song, Y. Huang, S. Han, M. Hou and H. Li, Invest. Ophthalmol. Visual Sci., 2020, 61, 28 CrossRef.
- B. Peguera, M. Segarra and A. Acker-Palmer, Curr. Opin. Neurobiol., 2021, 69, 202–213 CrossRef PubMed.
- R. Milner, Cerebral Angiogenesis. Methods in Molecular Biology, Humana Press, New York, NY, 2014 Search PubMed.
- K. A. Hogan, C. A. Ambler, D. L. Chapman and V. L. Bautch, Development, 2004, 131, 1503–1513 CrossRef PubMed.
- J. M. James, C. Gewolb and V. L. Bautch, Development, 2009, 136, 833–841 CrossRef PubMed.
- J. M. James and Y. Mukouyama, Semin. Cell Dev. Biol., 2011, 22, 1019–1027 CrossRef PubMed.
- M. Tata and C. Ruhrberg, Neuronal Signaling, 2018, 2, NS20170139 CrossRef.
- M. Segarra, M. Rodriguez Aburto, J. Hefendehl and A. Acker-Palmer, Annu. Rev. Cell Dev. Biol., 2019, 35, 615–635 CrossRef.
- T. Wälchli, A. Wacker, K. Frei, L. Regli, M. E. Schwab, S. P. Hoerstrup, H. Gerhardt and B. Engelhardt, Neuron, 2015, 87, 271–296 CrossRef.
- H. J. Jeong, Z. Jimenez, K. Mukhambetiyar, M. Seo, J. W. Choi and T. E. Park, Tissue Eng. Regener. Med., 2020, 17, 747–757 CrossRef.
- H. M. Eilken and R. H. Adams, Curr. Opin. Cell Biol., 2010, 22, 617–625 CrossRef PubMed.
- P. Carmeliet, Nat. Med., 2000, 6, 389–395 CrossRef.
- A. Zimna and M. Kurpisz, BioMed Res. Int., 2015, 2015, 549412 Search PubMed.
- J. I. Bárdos and M. Ashcroft, Biochim. Biophys. Acta, Rev. Cancer, 2005, 1755, 107–120 CrossRef.
- A. Pezzuto and E. Carico, Curr. Mol. Med., 2018, 18, 343–351 CrossRef CAS PubMed.
- S. Tomita, M. Ueno, M. Sakamoto, Y. Kitahama, M. Ueki, N. Maekawa, H. Sakamoto, M. Gassmann, R. Kageyama, N. Ueda, F. J. Gonzalez and Y. Takahama, Mol. Cell. Biol., 2003, 23, 6739–6749 CrossRef PubMed.
- S. Zhang, B. Kim, X. Zhu, X. Gui, Y. Wang, Z. Lan, P. Prabhu, K. Fond, A. Wang and F. Guo, Nat. Commun., 2020, 11, 2027 CrossRef PubMed.
- C. Wu, J. Chen, C. Chen, W. Wang, L. Wen, K. Gao, X. Chen, S. Xiong, H. Zhao and S. Li, Sci. Rep., 2015, 5, 16151 CrossRef PubMed.
- P. A. Stewart and M. J. Wiley, Dev. Biol., 1981, 84, 183–192 CrossRef PubMed.
- R. C. Janzer and M. C. Raff, Nature, 1987, 325, 253–257 CrossRef PubMed.
- A. M. Goodwin and P. A. D'Amore, Angiogenesis, 2002, 5, 1–9 CrossRef PubMed.
- A. Cattelino, S. Liebner, R. Gallini, A. Zanetti, G. Balconi, A. Corsi, P. Bianco, H. Wolburg, R. Moore, B. Oreda, R. Kemler and E. Dejana, J. Cell Biol., 2003, 162, 1111–1122 CrossRef CAS PubMed.
- C. Y. Logan and R. Nusse, Annu. Rev. Cell Dev. Biol., 2004, 20, 781–810 CrossRef CAS.
- C.-C. Chen, F.-E. Mo and L. F. Lau, J. Biol. Chem., 2001, 276, 47329–47337 CrossRef.
- S. Ivkovic, B. S. Yoon, S. N. Popoff, F. F. Safadi, D. E. Libuda, R. C. Stephenson, A. Daluiski and K. M. Lyons, Development, 2003, 130, 2779–2791 CrossRef.
- A. N. Athanasopoulos, D. Schneider, T. Keiper, V. Alt, U. R. Pendurthi, U. M. Liegibel, U. Sommer, P. P. Nawroth, C. Kasperk and T. Chavakis, J. Biol. Chem., 2007, 282, 26746–26753 CrossRef.
- M.-H. Park, A. Kyung Kim, S. Manandhar, S.-Y. Oh, G.-H. Jang, L. Kang, D.-W. Lee, D. Y. Hyeon, S.-H. Lee, H. E. Lee, T.-L. Huh, S. H. Suh, D. Hwang, K. Byun, H.-C. Park and Y. M. Lee, eLife, 2019, 8, e46012 CrossRef PubMed.
- F. E. Mo, A. G. Muntean, C. C. Chen, D. B. Stolz, S. C. Watkins and L. F. Lau, Mol. Cell. Biol., 2002, 22, 8709–8720 CrossRef.
- C. Bao, J. Wang, W. Ma, X. Wang and Y. Cheng, Future Oncol., 2014, 10, 2675–2685 CrossRef.
- M. E. LeBlanc, W. Wang, X. Chen, Y. Ji, A. Shakya, C. Shen, C. Zhang, V. Gonzalez, M. Brewer, J.-X. Ma, R. Wen, F. Zhang and W. Li, Mol. Vision, 2016, 22, 374–386 CAS.
- A. D. Everett, J. V. Narron, T. Stoops, H. Nakamura and A. Tucker, Am. J. Physiol., 2004, 286, L1194–L1201 CAS.
- C. Thirant, E. Galan-Moya, L. G. Dubois, S. Pinte, P. Chafey, C. Broussard, P. Varlet, B. Devaux, F. Soncin, J. Gavard, M. Junier and H. Chneiweiss, Stem Cells, 2012, 30, 845–853 CrossRef PubMed.
- R. Eguchi and I. Wakabayashi, Oncol. Rep., 2020, 44, 14–28 Search PubMed.
- Y.-W. Lin, S.-T. Huang, J.-C. Wu, T.-H. Chu, S.-C. Huang, C.-C. Lee and M.-H. Tai, BMC Cancer, 2019, 19, 1083 CrossRef PubMed.
- S. Wang, M. Li, W. Zhang, H. Hua, N. Wang, J. Zhao, J. Ge, X. Jiang, Z. Zhang, D. Ye and C. Yang, Sci. Rep., 2017, 7, 9027 CrossRef.
- L. Wang, Y. Liu, W. Li and Z. Song, Clin. Res. Hepatol. Gastroenterol., 2017, 41, 408–414 CrossRef PubMed.
- H. Song, D. Yin and Z. Liu, Mol. Biol. Rep., 2012, 39, 4017–4022 CrossRef.
- B. Gao, J. Deng, X. Zhang, H. Sun, G. Jia, J. Li, K. Zhang, C. Wan, L. Wang, L.-J. Yan, Z. Cai and J. Ma, Neurosci. Lett., 2020, 715, 134657 CrossRef PubMed.
- M. E. LeBlanc, W. Wang, N. B. Caberoy, X. Chen, F. Guo, G. Alvarado, C. Shen, F. Wang, H. Wang, R. Chen, Z.-J. Liu, K. Webster and W. Li, PLoS One, 2015, 10, e0127904 CrossRef.
- S. L. Karsten, T.-K. Sang, L. T. Gehman, S. Chatterjee, J. Liu, G. M. Lawless, S. Sengupta, R. W. Berry, J. Pomakian, H. S. Oh, C. Schulz, K.-S. Hui, M. Wiedau-Pazos, H. V. Vinters, L. I. Binder, D. H. Geschwind and G. R. Jackson, Neuron, 2006, 51, 549–560 CrossRef.
- Y. Ishida, H. Kawakami, H. Kitajima, A. Nishiyama, Y. Sasai, H. Inoue and K. Muguruma, Cell Rep., 2016, 17, 1482–1490 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00930k |
| This journal is © The Royal Society of Chemistry 2024 |