
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCapturing of extracellular vesicles derived from single cells of Escherichia coli†
Fumiaki
Yokoyama
 ab,
André
Kling
a and
Petra S.
Dittrich
*a
ab,
André
Kling
a and
Petra S.
Dittrich
*a
aDepartment of Biosystems Science and Engineering, ETH Zurich, CH-4056 Basel, Switzerland. E-mail: petra.dittrich@bsse.ethz.ch
bThe University of Tokyo, Department of Physics, Tokyo 113-0033, Japan
First published on 12th February 2024
Abstract
Bacteria secrete extracellular vesicles (EVs), also referred to as bacterial membrane vesicles, which carry, among other compounds, lipids, nucleic acids and virulence factors. Recent studies highlight the role of EVs in the emergence of antibiotic resistance, e.g. as carrier and absorbent particles of the drug to protect the cells, or as a pathway to disseminate resistance elements. In this study, we are interested in characterizing the secretion of EVs at the single bacterial level to ultimately understand how cells respond to antibiotic treatment. We introduce a microfluidic device that enables culture of single bacterial cells and capture of EVs secreted from these individuals. The device incorporates parallel, narrow winding channels to trap single rod-shaped E. coli cells at their entrances. The daughter cells are immediately removed by continuous flow on the open side of the trap, so that the trap contains always only a single cell. Cells grew in these traps over 24 h with a doubling time of 25 minutes. Under antibiotic treatment, the doubling time did not change, but we observed small changes in the cell length of the trapped cells (decrease from 4.0 μm to 3.6 μm for 0 and 250 ng mL−1 polymyxin B, respectively), and cells stopped growing within hours, depending on the drug concentration. Compared to bulk culture, the results indicate a higher susceptibility of on-chip-cultured cells (250 ng mL−1vs. >500 ng mL−1 in bulk), which may be caused, among other reasons, by the space limitation in the cell trap and shear forces. During the culture, EVs secreted by the trapped cells entered the winding channel. We developed a procedure to selectively coat these channels with poly-L-lysine resulting in a positively charged surface, which enabled electrostatic capture of negatively charged EVs. Subsequently, the immobilized EVs were stained with a lipophilic dye and detected by fluorescence microscopy. Our findings confirm large variations of EV secretion among individual bacteria and indicate a relative high rate of EV secretion under antibiotic treatment. The proposed method can be extended to the detection of other secreted substances of interest and may facilitate the elucidation of unknown heterogeneities in bacteria.
Introduction
Cells from all biological kingdom secrete extracellular vesicles (EVs),1,2 which are lipid nanoparticles containing various biological cargos, including proteins, nucleic acids, and saccharides.3,4 EVs exhibit diverse sizes and compositions5–7 despite being derived from the same cell population, which can be attributed to the multiple secretion mechanisms in both eukaryotic and prokaryotic cells.4,8 It has been revealed that environmental fluctuations can also alter the production and components of EVs.9,10 EVs can be taken up by other cells in a nearby or distant environment, thus contributing to cell-to-cell communication.11,12In recent years, studies on the variation of secreted EVs at the single-cell level have become feasible, particularly for mammalian cells using various imaging methods13 as well as microfluidic devices.14–21 Microcompartments like microchambers, ring-shaped valves, and droplets have been employed to isolate single cells, enabling subsequent measurements of fluorescence protein-including EVs or protein cargos via immunostaining.21 These techniques have identified subpopulations of single-cell derived EVs, suggesting the heterogeneity of EV-based intercellular communications in mammalian cells.
While the secretion of EVs from mammalian cells has been extensively studied at both population and single-cell levels, investigating EVs secreted by bacterial cells could so far only be shown in bulk culture experiments. Various types of bacterial EVs like outer-membrane vesicles (OMVs), inner-membrane vesicle (IMVs), and outer-inner-membrane vesicles (OIMVs), have been reported recently,8,22 and heterogeneous EVs, in sizes, components, stiffness, etc., collected from a bulk culture23,24 may reflect both phenotypic variations of each secretory cell within a population and identical cells at different time points. To understand these heterogeneities of EV secretion at the single-cell and the single-vesicle level, sophisticated techniques are needed.25 However, detecting EVs from individual bacterial cells remain challenging due to the small size of the cells, motility, and rapid proliferation of bacterial cells like Escherichia coli. Besides, common molecular markers like proteins and surface saccharides within bacterial EVs have not yet been identified26 because bacterial EV cargos are heterogeneous not only with respect to molecular species but also to the secretion levels of the compound present in the EVs.3,24 Despite these challenges, EVs have gained considerable attention in recent years because of their roles in antibiotic tolerance, transmission of antibiotic resistance genes and potential use of vesicle pathways as a drug target.27–29 In particular, OMVs contribute to cell fitness by removing the unwanted cargo, i.e. EV formation could serve as a “garbage disposal” mechanism.27,30,31
In addition, the aspect of single-cell analysis for microbes should be highlighted. In microbiology, single-cell analysis by time-lapse microscopy typically means observing and tracking single cells within a growing population,32,33 while only few studies report true single-bacterial cell studies,34–36 and these do not provide long-term cell culture methods without neighboring cells like the ones done for yeast cells using microfluidic hydrodynamic traps.37–39 Although the most prevalent life form of bacteria in natural environments is thought to be a cellular community known as a biofilm,40 they can also survive in a single-cell state without neighboring cells as unicellular organisms and switch between these unicellular and multicellular states.41 Therefore, to fully comprehend bacterial life from unicellular to multicellular aspects, researchers may not only study single bacterial cells in a population but also completely isolated single cells.
In this study, we introduce a new microfluidic device, inspired by the mother machine,42,43 to achieve a culture of isolated, single bacterial cells and detect EVs secreted from these individual cells. We investigate cells treated and not treated with polymyxin B. This antibiotic compound was selected because it has been previously reported that it induces EV production in E. coli as part of an immediate defense strategy, where the EV membrane adsorbs polymyxin B to reduce its concentration and protect the cells.30 Our new method allows us to observe the growth patterns and EV secretion at the same time. This method has potential to open new vistas for elucidating unknown heterogeneities of antibiotic responses and detecting EV secretion from individual bacterial cells.
Experimental
Bacterial strains and culture conditions
For monitoring bacterial growth, E. coli K-12 MG1655 (pSEVA271-sfgfp) was used.42 A glycerol stock of this E. coli was streaked onto an LB plate containing 1.5% agar, incubated at 37 °C for 24 h and stored at 4 °C until use. A single colony on the plate was inoculated in LB with kanamycin (50 μg mL−1) and cultured at 200 rpm and 37 °C for 16 h using an Ecotron incubator (Infors, Bottmingen, Switzerland). Ten microliters of the pre-cultured medium were cultured in 1 mL of LB with kanamycin (50 μg mL−1) at 200 rpm and 37 °C approximately for 2 h until cells entered the exponential growth phase (the optical density at 600 nm, OD600, reached ∼0.3). This suspension was used for the single-cell culture in the microfluidic device. For bulk culture, 40 μL of the pre-cultured medium was diluted in 4 mL LB with kanamycin (50 μg mL−1) and cultured at 200 rpm and 37 °C for 16 h until cells entered the stationary growth phase (OD600 ∼ 7.0). The optical density was measured on a photometer (NanoPhotometer, Implen, Munich, Germany). For antibiotic treatment, we added polymyxin B (Sigma-Aldrich, St. Louis, MO, United States).Microfluidic device fabrication
Sketches of the PDMS-glass device and dimensions are displayed in the ESI,† Fig. S1. The protocol for the fabrication of the master mould is also included in the ESI.† Polydimethylsiloxane (PDMS) microfluidic devices were fabricated using a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio of Sylgard 184 silicone elastomer base and curing agent (Dowsil, Midland, MI, United States). These reagents were mixed well using a plastic spatula and degassed using a vacuum pump until visually bubble-free. Approximately 5 g of the mixture was poured onto the master mould and placed at 80 °C for 3 h. Afterwards, the cured PDMS was peeled off from the master mould and unnecessary parts of the PDMS block were cut off. Four inlets/outlets were punched using a 1.5 mm biopsy puncher (Integra LifeSciences, Princeton, NJ, United States). The surface of the device was cleaned using adhesive tape. A No. 1.5 microscopy glass slide (0.16–0.19 mm thick, Biosystems, Muttenz, Switzerland) was cleaned by washing with acetone, isopropanol, and water, then dried using nitrogen gas and a heater at 150 °C. The device and the glass slide were plasma-activated using a PDC-32G plasma cleaner (Harrick Plasma, Ithaca, NY, United States) at less than 0.9 mbar for approximately 1 min and bond together. The glass-bonded device was put on a heater at 150 °C for 5 min and stored at room temperature until use.
:1 mass ratio of Sylgard 184 silicone elastomer base and curing agent (Dowsil, Midland, MI, United States). These reagents were mixed well using a plastic spatula and degassed using a vacuum pump until visually bubble-free. Approximately 5 g of the mixture was poured onto the master mould and placed at 80 °C for 3 h. Afterwards, the cured PDMS was peeled off from the master mould and unnecessary parts of the PDMS block were cut off. Four inlets/outlets were punched using a 1.5 mm biopsy puncher (Integra LifeSciences, Princeton, NJ, United States). The surface of the device was cleaned using adhesive tape. A No. 1.5 microscopy glass slide (0.16–0.19 mm thick, Biosystems, Muttenz, Switzerland) was cleaned by washing with acetone, isopropanol, and water, then dried using nitrogen gas and a heater at 150 °C. The device and the glass slide were plasma-activated using a PDC-32G plasma cleaner (Harrick Plasma, Ithaca, NY, United States) at less than 0.9 mbar for approximately 1 min and bond together. The glass-bonded device was put on a heater at 150 °C for 5 min and stored at room temperature until use.
Two-patterned surface functionalization of the microfluidic device
0.01% (0.1 mg mL−1) poly-L-lysine (70000–150000 molecular weight, Sigma-Aldrich) was applied to a microfluidic device using a polytetrafluoroethylene (PTFE) tube (inner diameter 0.25 mm, outer diameter 1.59 mm, BGB, Orsa, Sweden, or PKM Konrad, Rotkreuz ZG, Switzerland) and 1 mL syringe (Becton, Dickinson and Company, Franklin Lakes, NJ, United States) from the lower-left inlet (Fig. S1A and S2A step 1†), and incubated at 20 °C for 1 h. Then, the air pressure was applied from the same inlet using the PTFE tube and syringe to remove the solution from the device (Fig. S2A step 2†). The device was heated at 80 °C for 19 h to evaporate the remaining poly-L-lysine solution. A top wide trench of the device was refilled with 10 mL of bovine serum albumin (BSA) by pipetting to the upper-left inlet, followed by flushing the solution with 20 mL of Dulbecco's phosphate-buffered saline (DPBS, KCl 0.2 g L−1, KH2PO4 0.2 g L−1, NaCl 8 g, Na2HPO4 1.15 g L−1, pH = 7.0–7.3, Thermo Fisher Scientific, Waltham, MA, United States) (Fig. S2A step 3†). The narrow channels of the device were filled with LB using the PTFE tube and syringe at the lower-left inlet and stored at 20 °C until use (less than 2 h) (Fig. S2A step 4†). For the visualization, 5 μg mL−1 streptavidin–Atto565 and 5 μg mL−1 biotin–fluorescein isothiocyanate (FITC) were used instead of poly-L-lysine and BSA at step 1 and 3, respectively (Fig. S2B and C†). Brightfield and confocal fluorescence images were taken with ×20 objective (0.75 numerical aperture), for FITC (emitter: ET525/50 m, Chroma Technology, Olching, Germany) using 5% laser power of 488 nm and 50 ms exposure time and for Atto565 (emitter: FF01-609/54, Semrock, DEX Health & Science, LLC Rochester, NY, USA) using 50% laser power of 561 nm and 1 s exposure time. For the visualization of the narrow channels in Fig. S2D,† the background of the overlaid image between Fig. S2B and C† was subtracted with a 50 pixel rolling ball radius in ImageJ/Fiji version 2.3.0/1.53f, then adjusted in brightness.
Time-lapse microscopy for growth measurement
For cell loading and culturing, the microfluidic device was connected to three three-way stop valves with tubing (Fig. S3A†). Cells in the exponential growth phase were pelleted by centrifugation at 6800 × g and at 20 °C for 5 min and washed twice in DPBS. One-thousand-times diluted cell suspension in a fresh medium was applied to the upper-left inlet with a 100 μL min−1 flow rate, while fresh medium with kanamycin (50 μg mL−1) and polymyxin B, when needed, was applied from the lower-right inlet with a 120 μL min−1 flow rate to keep the inlet non-contaminated (Fig. S3B†). This diluted culture supernatant contained 4.3 × 104 cells per mL calculated from colony forming units at the exponential growth phase (see the Experimental section in the ESI†). Once about ten cell traps among 38 ones in one microscopic view field were occupied by single cells in approximately 5 min, the flow containing cells was stopped, the three-way stop valve at the upper-left inlet for cell suspension was open to the outlet for waste, and the upper-right outlet used for waste in cell loading was closed with a three-way stop valve. This process allowed the fresh medium flow to direct to the narrow channels and then to the lower-left outlet (Fig. S3C†). The flow rate of the medium was set at 1 mL h−1 under 37 °C for the growth measurement to apply sufficient nutrition to the captured cells and simultaneously wash out daughter cells. Fluorescence images before and after valve opening and closing were taken by epi-fluorescence microscopy for excitation and optical filters and dichroic mirrors for the green fluorescent protein (GFP), as described right below, using 1 s exposure time to show the flow direction in Fig. S3.†The trapped cells were imaged in brightfield mode of a Nikon ECLIPSE Ti 2 inverted microscope (Nikon, Tokyo, Japan) with a light-emitting diode (LED) illumination system (CoolLED, Andover, UK), a motorized stage, a ×100 objective (1.49 numerical aperture, with immersion oil), and a Hamamatsu Orca Flash 4.0 V2 complementary metal-oxide-semiconductor (CMOS) camera with a sensor size of 2044 × 2048 pixels (Hamamatsu Photonics, Shizuoka, Japan). Fluorescence images of the cell traps were taken by epi-fluorescence microscopy mode using a Spectra X LED unit (Lumencor, Beaverton OR, United States) for excitation and optical filters and dichroic mirrors for GFP (exciter: ET470/40x, emitter: ET525/50, dichroic: T496lpxr, Chroma Technology) using 50 ms exposure time every 5 min for 24 h. Imaging was controlled by Visitron VisiView. For the acquisition of cell morphological parameters, the images were processed using ImageJ/Fiji version 2.3.0/1.53f. First, the scale was set using the known scale provided by Visitron VisiView. The background was subtracted with a 50 pixel rolling ball radius. Then, the threshold was automatically adjusted for the dark background with the default setting. By particle analysis, the fluorescence cells were fit to ellipses, and their major and minor length was measured as cell length and width, respectively. The doubling time was calculated from the cell length data.
Purification of extracellular vesicles by ultracentrifugation
EVs secreted from E. coli cells were collected from cultures at the exponential and the stationary growth phase, according to the previous method.44 The cells were pelleted by centrifugation at 6800 × g and 20 °C for 10 min. The supernatant was centrifuged at 13000 × g and 20 °C for 15 min to remove the remaining bacterial cells. The supernatant was filtered through a 0.2 μm pore polyethersulfone (PES) filter to remove the remaining debris. EVs were obtained by ultracentrifugation of the filtrate at 100000 × g (average centrifugal force) and 4 °C for 2 h with a centrifuge (SORVALL WX Ultra Series, Thermo Electron Corporation, Waltham, MA, United States). The pellets were ten times concentrated by resuspending in DPBS and used as EVs. The supernatants without EVs after ultracentrifugation were used as EV-free supernatants. These EVs and EV-free supernatants were kept at 4 °C for less than two days. Size and concentration of EVs were validated by nanoparticle tracking analysis (Experimental section in the ESI†).
Zeta potential of extracellular vesicles and poly-L-lysine
The zeta potential of EVs in DPBS and 0.01% poly-L-lysine (Sigma-Aldrich) was measured at 20 °C using a Zetasizer Nano dynamic light scattering analyzer (Malvern Panalytical, Worcestershire, UK). The distribution data were obtained by repeating up to 100 cycles.On-plate assay for extracellular vesicle detection
The surface of a 384-well black plate (Greiner, Kremsmünster, Austria) was incubated with 20 μL of 0.01% poly-L-lysine (Sigma-Aldrich) at 20 °C for 1 h, followed by washing twice with 50 μL of DPBS (Thermo Fisher Scientific). Twenty microliters of purified EVs or EV-free supernatants were applied to the wells and incubated at 37 °C for 24 h, followed by lipid-staining with 20 μL of 5 μg mL−1N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl)pyridinium dibromide (FM4-64, Thermo Fisher Scientific) at 20 °C for 30 min in the dark. The fluorescence intensity of FM4-64 was measured at 515/20 nm excitation and 635/20 nm emission wavelength using a Cytation 5 cell imaging multi-mode reader (BioTek, Winooski, VT, United States). A non-coated plate was used with EVs as a control.Extracellular vesicle detection by microscopy
After culture of cells in the microfluidic device for 24 h or applying purified EVs, 1 μg mL−1 FM4-64 (Thermo Fisher Scientific) in DPBS was applied to the device by flow at 1 mL h−1 for 30 min in the dark. Images of the narrow channels were taken on an inverted microscope as described above. Fluorescence images were taken at the bottom of the device using 50% laser power of 561 nm and 1 s exposure time (emitter: FF01-609/54, Semrock). For image analysis, each narrow channel excluding a cell trap region was selected by a rectangle as a region of interest (ROI), shown in Fig. 5B. We showed the total fluorescence signals of each ROI (summing up the fluorescence intensity of all pixels within the ROI). The value correlates to the fluorophores that diffused into the membrane, and we used it as a means of EV secretion. In this way, we simplify the data analysis, but it should be noted that we do not enumerate EVs.Results & discussion
Microfluidic device enabling capture of single E. coli cells
We designed a microfluidic device to capture an individual bacterial cell in a narrow channel. In contrast to the previous design,42 our device keeps the initially isolated cell only, while daughter cells are immediately washed out (Fig. 1A). We have named this modified mother machine device “the isolated mother machine”, or iMM. With the iMM, we can capture EVs secreted from single bacterial cells over extended periods longer than the division time of the cells. | ||
| Fig. 1 Design of the isolated mother machine (iMM) device for capturing bacterial cells and collecting EVs from individual cells. (A) The microfluidic device enables the isolation of individual bacteria and continuous removal of their daughter cells. The process of EV collection using the iMM device is as follows: step 1. Bacterial cells are loaded into the device through a wide trench by medium flow; step 2. A single bacterial cell is captured by a cell trap at the entrance of a narrow channel; step 3. During culture, daughter cells from the isolated cell are quickly removed from the cell trap by flow, so that the isolated single cell remains singular; step 4. EVs secreted from the single cell are carried towards the end of a narrow channel by flow; step 5. Staining of the EVs for detection. (B) Enlarged view of the narrow channels illustrating the cell trap and the winding structure for EV capturing. (C) Schematic side view of the narrow channel surface to immobilize EVs by electrostatic forces. The lipid-staining dye (FM4-64) integrates into the EV membrane and thereby increases the emission of fluorescence. All the sketches are not to scale. | ||
The iMM device has four inlets/outlets that are connected to two wider channels, referred here to wide trenches (Fig. 1 and S1A†). The trenches are 100 μm wide and 25 μm high and are connected to 1230 narrow (1.8 μm) and flat (0.85 μm) channels (Fig. S1B†). The design reported in the literature42 has a protrusion at the end of narrow channels to prevent isolated cells from escaping while allowing efficient and uniform flow of medium through the channel. Here, we have changed this design and included protrusions after 4 μm inside from the entrance of narrow channels to make space for one E. coli cell (Fig. S1C†). The height of the cell trap was designed to be slightly smaller (0.85 μm) than the E. coli cell width reported previously45 to ensure robust single cell trapping at high flow rates that are needed to prevent cell adhesion to the wide trench wall. In addition, the narrow channel following the trapped cell makes a turn (Fig. 1B), which prevents rod-shaped bacteria like E. coli from tilting or substantially deforming to escape the cell trap through the small channel.
To enable the slow flow of EVs secreted from single cells through the narrow channels, they are open at the end to the wide trench on the other side (Fig. 1B and S1C†). The wall surface of the narrow channels is rendered positively charged with poly-L-lysine to capture negatively charged EVs within the narrow channels. Following a common way for bacterial EV detection, membrane lipids of the captured EVs are stained with a lipophilic dye (Fig. 1C). Surfaces of the wide trench for medium flow were coated with BSA to prevent cell adhesion. This two-pattern modification was achieved by a sequential procedure shown in the brightfield microscopic images of Fig. S2A† and confirmed using fluorescence molecules in Fig. S2B and C.†
Growth dynamics of isolated E. coli cells
For cell loading and culturing, the microfluidic device was connected to three three-way stop valves with tubing (Fig. S3A†). The channel design depicted in Fig. S3B† was crucial to apply cells and medium while preventing contamination in the wide trench and the inlet for medium applying during cell loading. We introduced a diluted culture medium containing GFP-expressing E. coli at the exponential growth phase (4.3 × 104 cells per mL) and captured approximately 300 cells in total within 5 minutes, which relates to an efficiency (trapped/supplied cells) of ∼1.4%. Then, the medium was applied to the cells trapped in the narrow channels and collected in the outlets as indicated in Fig. S3C.† Remaining cells were flushed out by the continuous medium, preventing contamination of the cells growing in the device during culture (Fig. S4†). Under 37 °C and at 1 mL h−1 lysogeny broth (LB) flow, the isolated single cells grew toward the wide trench since the winding structure in the narrow channel prevent cells from growing to the opposite side (Fig. 2A). Then, they divided, producing daughter cells with a median doubling time of 25 min (Fig. 2B and C, Movie S1†) until we stopped the experiment after 24 hours. The doubling time was 4.5-min longer than the reported doubling time in bulk of LB medium at 37 °C.46 We speculate that the shear forces and the space constraint may influence the growth as reported before for other designs of the mother machine.45 We measured the cell length and width of each cell by fitting the fluorescence areas of the cells to ellipses, revealing oscillatory growth dynamics with length ranging from 2.5 to 7 μm and a constant cell width of approximately 1.2 μm during culture (Fig. 2D and S5A† and 2E and S5B,† respectively). Occasionally, a cell in this measurement stopped growing and expanded its cell width near the end of the measurement (Fig. S5 and Movie S2†). It is important to note that the measurement for cell length indicates that 33% of cell length of 6 μm elongated single cells was outside of the 4 μm cell trap, thus excluding EVs secreted from the outer body part from the analysis in this method. Some cells, in particular abnormally elongated cells, were washed out by the flow outside of the cell trap and detached at some points. In addition, daughter cells sometimes attached to the wall of narrow channels after cell division, preventing single-cell analysis. These data were removed from the analysis. Overall, the obtained growth patterns were similar to those observed in previous studies using the original mother machine,43,45 indicating that the trapped cells in our modified design grew well.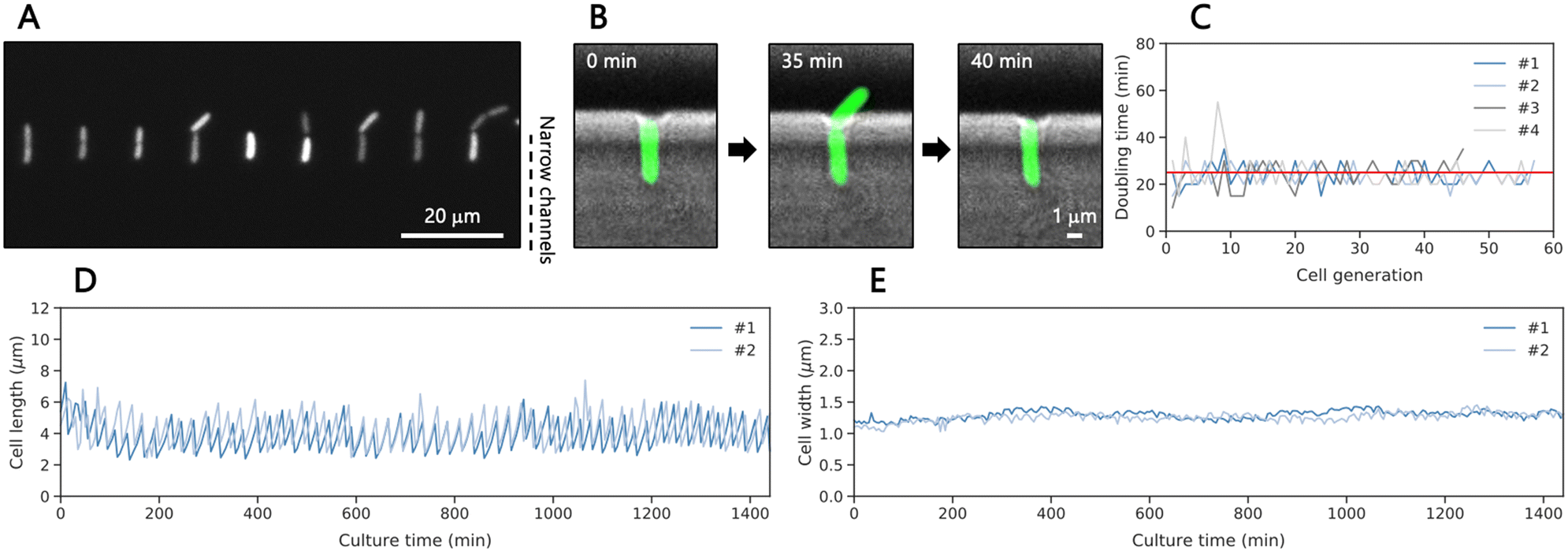 | ||
| Fig. 2 Growth dynamics of single cells of E. coli. (A) A representative image of GFP-expressing single E. coli cells isolated in cell traps on the iMM device. (B) Overlayed fluorescence and brightfield images of a cell depicting the removal of the daughter cell. The daughter cell is clearly visible 35 min after cell isolation and was removed by medium flow at 40 min. (C) Doubling times of single cells, calculated from the fluorescence images of four cells. The red line represents the median doubling time of the data at 25 min. Changes in the cell length (D) and width (E) of two individuals cells, obtained from the fluorescence images over 24 h. | ||
Growth dynamics of single cells of E. coli under antibiotic exposure
To investigate the antibiotic effects on the growth dynamics of isolated single bacterial cells, we compared untreated cells with cells exposed to the membrane-perturbing antibiotic polymyxin B. Although most cells in the absence of polymyxin B grew well for 24 h, until we stopped the observation, and some cells elongated at some points due to division deficiency as mentioned above (Fig. 3, S7, and Movie S3†). In the presence of polymyxin B at 50 and 250 ng mL−1, we observed that not all the cells grew over the full 24 h period (Fig. 3A, S7, and Movies S4 and S5†). Under these conditions, we observed a gradual loss of GFP fluorescence over time within cells, indicating cell death (Movie S5†). The dead cell, however, was still captured, as we stained the membrane with FM4-64 after 24-h culture (Fig. S8†), and therefore, the killing mechanism by polymyxin B did not cause complete membrane fragmentation of lysed cells. The concentration of 250 ng mL−1 is lower than expected from measurements in bulk culture (Fig. S6A†). The higher susceptibility to the antibiotics for the on-chip captured cells could be caused by additional stress on the cells due to space constraints and shear forces. In consequence, the duration of single cell observation under the antibiotic in our device is limited to the time until the cells stop growing. | ||
| Fig. 3 Growth dynamics of single E. coli cells under antibiotic exposure. (A) Time course of cell length dynamics at different concentrations of polymyxin B. The growth of captured single cells was monitored for up to 24 h. Each data line represents a single bacterial cell (additional examples in Fig. S7†). (B) Cell length of each single cell obtained from growth dynamics at different concentrations of polymyxin B. (C) Doubling time of each single cell obtained from the time course of growth dynamics. Each black line within the boxes represents the median, each lower and upper edges of the boxes represent the 25 and 75 percentiles of the data, respectively, and each error bar represents the maximum and minimum values of the box plots (n > 84 in B and 11 in C). The gray dots on the right side of the boxes represent each data point. The light blue graphs on the left side of the boxes represent each data distribution. *p < 0.01, **p < 0.05, and n.s.: no significant difference (two-sided Brunner–Munzel test). | ||
While the doubling time is almost not affected by the antibiotic treatment (Fig. 3C), we observed a small decrease of the cell length (Fig. 3B) with increasing polymyxin B concentration (4.0, 3.9, and 3.6 μm at 0, 50, and 250 ng mL−1 of polymyxin B, respectively, Fig. 3B). This is opposite as expected from bulk culture, where median values of cell length are 3.6 and 4.0 μm with and without polymyxin B at 250 ng mL−1, respectively (Fig. S6†). The bulk observation is consistent with a previous study,47 where increase in length (and volume) and decrease of surface area/volume is observed as response to an antibiotic. The difference between on-chip and bulk values may be caused again by shear forces.
Detection of extracellular vesicles on the microfluidic device
Since we wanted to capture EVs only in the winding channels, we first tested a suitable coating material and developed the coating procedure on-chip (Fig. S2†). Therefore, we used EVs purified from bulk culture. Measurements showed that EVs in DPBS have a zeta potential of −12.7 mV, while poly-L-lysine suspended in DPBS had a median zeta potential of −35.8 (Fig. 4A), indicating that poly-L-lysine-coated surface can electrostatically capture EVs. To validate the EV capture method, we modified the polystyrene surface of a multi-well plate using poly-L-lysine, then applied the purified EVs and performed lipid staining with a lipophilic dye, FM4-64. The median fluorescence intensity of the labeled EV lipid membrane was significantly higher on the coated surface (87.0 a.u.) than those on the non-coated surface (43.0 a.u.) and using EV-free supernatant on the coated surface (17.0 a.u.) (Fig. 4B). These results indicate that the combination of electrostatic force and lipid staining was a viable approach for EV detection in the narrow channels of the proposed microfluidic device.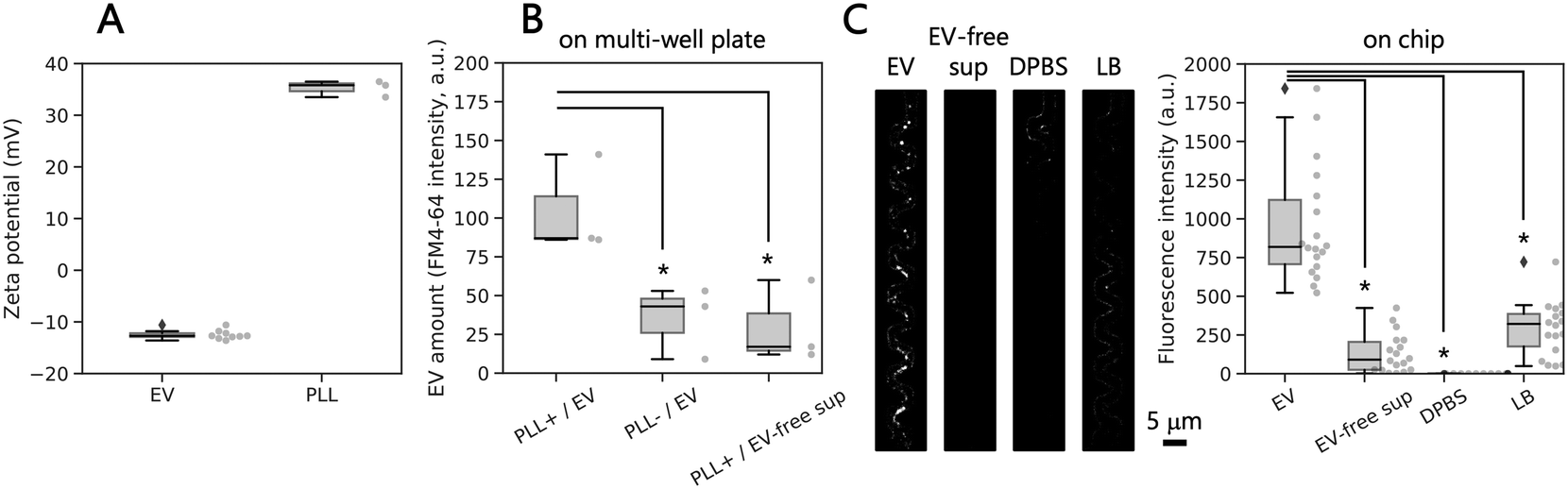 | ||
| Fig. 4 Capturing and detection of purified extracellular vesicles in the narrow channels of the microfluidic device. (A) Zeta potential of EVs and poly-L-lysine (PLL) in DPBS. (B) EV detection on a 386-well plate with a polystyrene surface modified with and without poly-L-lysine. As a control, EV-free supernatant (sup) was used as well. (C) Representative images of detected EVs on the microfluidic device by lipid-staining with FM4-64 (left) and the fluorescence intensity of EVs, EV-free supernatant, DPBS, and LB quantified with the images (right). Each black line within the boxes represents the median, each lower and upper edges of the boxes represent the 25 and 75 percentiles of the data, respectively, and each error bar represents the maximum and minimum values of the box plots (n = 3 or 9 in Fig. 4A and B, n = 18 in figure C). *p < 0.01 (two-sided Brunner–Munzel test). | ||
Next, to confirm successful capture of EVs in the iMM device, we introduced purified EVs into the device coated with poly-L-lysine. After incubation to allow EVs to be captured by the coated surface, we stained the membrane of the captured EVs using FM4-64 and observed fluorescence spots with diameters <1 μm by fluorescence microscopy on devices. In contrast, almost no fluorescence spots were observed when the device was incubated with EV-free supernatant, DPBS, or LB (Fig. 4C). Comparison of the fluorescence intensity in a region of interest along the narrow channels confirmed the higher fluorescence intensity for the channel exposed to EV suspension compared to the controls. These results indicate that the negatively charged EVs were electrostatically captured by the positively charged surface of the microfluidic device modified with poly-L-lysine and subsequently detected by lipid staining with a lipophilic dye and a microscope. Other negatively charged compounds could be captured on the surface as well, however, they should not be stained by the dye, therefore, they will not be detected by fluorescence microscopy.
Detection of EVs derived from single cells of E. coli
Next, we collected the EVs from individual E. coli cells with and without antibiotic treatment. At the cell trap, isolated single E. coli cells produced EVs by budding during culture (Fig. 5A). After culture for 24 h under all the conditions, FM4-64 was applied to the microfluidic device to stain both lipid membranes of the trapped cells and the captured EVs in the narrow channels. The winding structures, excluding cell traps of the narrow channels, were defined as a region of interest (ROI) (Fig. 5B). The fluorescence images (Fig. 5C) indicated that we could clearly harvest and detect EVs in the winding channels, where the cell trap was occupied by a bacterial cell. We observed that untreated cells produced a higher number of visible EVs than treated cells. For cells treated with a larger concentration of polymyxin B, the cells stopped growing, but remained in the trap what we confirmed after 24 h. We assume that EV production ceased once cell growth and division stopped, and therefore, we plotted the total fluorescence signal vs. growing time (Fig. 5D). The values of the EV intensity varied from cell to cell, even under the same culture conditions, and we observed low EV numbers in some cases. Additionally, we often found aggregated and chain-like structures, which have been reported before as pearling,48 confirmed by nanoparticle tracking analysis for EVs purified from bulk culture (Fig. S9 and Movie S6†). These structures hindered enumeration of EVs on our microscope. In the future, observation with super-resolution microscopy may support a more robust quantification of the EV production. | ||
| Fig. 5 Extracellular vesicle secretion of isolated single E. coli cells. (A) A representative image of an E. coli cell with membrane budding (white arrow). A fluorescence image of a GFP-expressing cell and a brightfield image of a narrow channel are overlayed. (B) A sketch of the narrow channel indicating the region of interest (ROI), where EVs are detected (not to scale). (C) Representative images of captured EVs after 24 h culture under treatment with polymyxin B at concentrations of 0, 50, and 250 ng mL−1, respectively. EVs appear as bright dots after staining with FM4-64. (D) Total fluorescence intensity of EVs plotted against growing time for different concentrations of polymyxin B (n = 4 for each condition). | ||
Finally, we compared these observations with results from bulk culture, where EVs were collected by centrifugation after 16 h of culture, when the cells were already in the stationary growth phase. Here, we conducted nanoparticle tracking analysis and found that cells treated with 250 ng mL−1 of polymyxin B secreted bigger EVs (Fig. S10†), and in accordance with a previous study30 the treated cells secreted a larger amount of EVs. This latter seems to be contradicting to the on-chip data. However, when we divide the obtained fluorescence signal by cell survival time on the device, we can confirm this enhanced secretion of EVs (Fig. S11†). Certainly, further studies with a larger cell number and measurements at more time points, ideally continuous monitoring of EV production, are required to confirm the findings.
Conclusions
The proposed iMM device has enabled the culture of individual bacterial cells for up to 24 hours, providing valuable insights into their growth and response to antibiotic treatment at the single-bacterial-cell level. In addition, due to local surface coating in the small-channel region of the device, we could capture EVs from individual cells. Our study confirmed the heterogeneity of EV secretion in bacteria. We also harvested EVs from bacteria under antibiotic treatment, however, bacteria died rapidly under treatment. In consequence, EV secretion stopped and accordingly, the period to collect EVs was short. Nevertheless, the proposed device has the potential to elucidate deeper characteristics in the secretion of EVs as well as proteins at the single-bacterial-cell level and may reveal different types of secretion mechanisms.49,50 Therefore, more specific staining is required; for example, one can capture and characterize EVs more specifically by implementation of immunoassays in the narrow channels. Instead of endpoint measurement, it would be interesting to monitor continuously the secretion of EVs, e.g. by employing surface plasmon resonance (SPR) spectroscopy.Future work will focus on optimizing the device to allow for higher throughput and more accurate statistics. The major hurdle to overcome is the loss of cells during the measurements. A better coating that more efficiently promotes bacteria adhesion could extend the duration of cells in the traps. Gentle squeezing of the cells in the traps by exposing pressure on the ceiling is another option to improve the microfluidic design. The device could be employed for other microbial cells, ideally non-motile species, as well as other secreted compounds such as virulence factors.
Author contributions
F. Y. designed the research, performed all experiments, and analyzed the data. A. K. fabricated master molds. F. Y and P. S. D. discussed the results and wrote the paper. All authors reviewed and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr. Voichita Mihali and Prof. Cornelia G. Palivan (University of Basel) for nanoparticle tracking analysis, Dr. Jonas M. Nikoloff (ETH Zurich) for help with microscopy, the team of Single-Cell Facility and of Clean Room Facility of the Department of Biosystems Science and Engineering (ETH Zurich) for their valuable help and advice, and F. Y. thanks Prof. David Juncker (McGill University) for discussion. We gratefully acknowledge the funding from the European Research Council (ERC Consolidator Grant No. 681587 to P. S. D.).References
- B. L. Deatherage and B. T. Cookson, Infect. Immun., 2012, 80, 1948–1957 CrossRef CAS PubMed.
- S. Gill, R. Catchpole and P. Forterre, FEMS Microbiol. Rev., 2018, 43, 273–303 CrossRef PubMed.
- C. Schwechheimer and M. J. Kuehn, Nat. Rev. Microbiol., 2015, 13, 605–619 CrossRef CAS PubMed.
- G. van Niel, G. D'Angelo and G. Raposo, Nat. Rev. Mol. Cell Biol., 2018, 19, 213–228 CrossRef CAS PubMed.
- E. Willms, C. Cabañas, I. Mäger, M. J. A. Wood and P. Vader, Front. Immunol., 2018, 9, 738 CrossRef PubMed.
- D. Jeong, M. J. Kim, Y. Park, J. Chung, H.-S. Kweon, N.-G. Kang, S. J. Hwang, S. H. Youn, B. K. Hwang and D. Kim, BMC Biol., 2022, 20, 270 CrossRef CAS PubMed.
- S. J. Biller, L. D. McDaniel, M. Breitbart, E. Rogers, J. H. Paul and S. W. Chisholm, ISME J., 2017, 11, 394–404 CrossRef CAS PubMed.
- M. Toyofuku, N. Nomura and L. Eberl, Nat. Rev. Microbiol., 2019, 17, 13–24 CrossRef CAS PubMed.
- S. Gebremedhn, A. Ali, A. Gad, R. Prochazka and D. Tesfaye, Front. Vet. Sci., 2020, 7, 602043 CrossRef PubMed.
- S. J. Biller, A. Coe, A. A. Arellano, K. Dooley, S. M. Silvestri, J. S. Gong, E. A. Yeager, J. W. Becker and S. W. Chisholm, Appl. Environ. Microbiol., 2023, 89, e00594-23 CrossRef PubMed.
- A. Chronopoulos and R. Kalluri, Oncogene, 2020, 39, 6951–6960 CrossRef CAS PubMed.
- S. Gurung, D. Perocheau, L. Touramanidou and J. Baruteau, Cell Commun. Signaling, 2021, 19, 47 CrossRef CAS PubMed.
- S. T.-Y. Chuo, J. C.-Y. Chien and C. P.-K. Lai, J. Biomed. Sci., 2018, 25, 91 CrossRef CAS PubMed.
- Y. Chiu, W. Cai, Y. V. Shih, I. Lian and Y. Lo, Small, 2016, 12, 3658–3666 CrossRef CAS PubMed.
- K. J. Son, A. Rahimian, D.-S. Shin, C. Siltanen, T. Patel and A. Revzin, Analyst, 2015, 141, 679–688 RSC.
- Y. Ji, D. Qi, L. Li, H. Su, X. Li, Y. Luo, B. Sun, F. Zhang, B. Lin, T. Liu and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5979–5984 CrossRef CAS PubMed.
- J. M. Nikoloff, M. A. Saucedo-Espinosa, A. Kling and P. S. Dittrich, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2106630118 CrossRef CAS PubMed.
- J. M. Nikoloff, M. A. Saucedo-Espinosa and P. S. Dittrich, Anal. Chem., 2023, 95, 1933–1939 CrossRef CAS PubMed.
- W. Cai, Y.-J. Chiu, V. Ramakrishnan, Y. Tsai, C. Chen and Y.-H. Lo, Lab Chip, 2018, 18, 3154–3162 RSC.
- K. Hattori, Y. Goda, M. Yamashita, Y. Yoshioka, R. Kojima and S. Ota, Anal. Chem., 2022, 94, 11209–11215 CrossRef CAS PubMed.
- L. Li, H. Su, Y. Ji, F. Zhu, J. Deng, X. Bai, H. Li, X. Liu, Y. Luo, B. Lin, T. Liu and Y. Lu, Adv. Sci., 2023, 10, 2301018 CrossRef CAS PubMed.
- M. Toyofuku, S. Schild, M. Kaparakis-Liaskos and L. Eberl, Nat. Rev. Microbiol., 2023, 21, 415–430 CrossRef CAS PubMed.
- Y. Kikuchi, N. Obana, M. Toyofuku, N. Kodera, T. Soma, T. Ando, Y. Fukumori, N. Nomura and A. Taoka, Nanoscale, 2020, 12, 7950–7959 RSC.
- P. D. Singorenko, V. Chang, A. Whitcombe, D. Simonov, J. Hong, A. Phillips, S. Swift and C. Blenkiron, J. Extracell. Vesicles, 2017, 6, 1324731 CrossRef PubMed.
- F. Zhu, Y. Ji, J. Deng, L. Li, X. Bai, X. Liu, B. Lin and Y. Lu, Chin. Chem. Lett., 2022, 33, 2893–2900 CrossRef CAS.
- J. Hong, P. Dauros-Singorenko, A. Whitcombe, L. Payne, C. Blenkiron, A. Phillips and S. Swift, J. Extracell. Vesicles, 2019, 8, 1632099 CrossRef CAS PubMed.
- C. R. MacNair and M.-W. Tan, Ann. N. Y. Acad. Sci., 2023, 1519, 63–73 CrossRef CAS PubMed.
- N. Krishnan, L. J. Kubiatowicz, M. Holay, J. Zhou, R. H. Fang and L. Zhang, Adv. Drug Delivery Rev., 2022, 185, 114294 CrossRef CAS PubMed.
- U. S. Kosgodage, P. Matewele, G. Mastroianni, I. Kraev, D. Brotherton, B. Awamaria, A. P. Nicholas, S. Lange and J. M. Inal, Front. Cell. Infect. Microbiol., 2019, 9, 227 CrossRef CAS PubMed.
- A. J. Manning and M. J. Kuehn, BMC Microbiol., 2011, 11, 258 CrossRef CAS PubMed.
- S. W. Kim, S. B. Park, S. P. Im, J. S. Lee, J. W. Jung, T. W. Gong, J. M. S. Lazarte, J. Kim, J.-S. Seo, J.-H. Kim, J.-W. Song, H. S. Jung, G. J. Kim, Y. J. Lee, S.-K. Lim and T. S. Jung, Sci. Rep., 2018, 8, 5402 CrossRef PubMed.
- Z. Ma, P. M. Chu, Y. Su, Y. Yu, H. Wen, X. Fu and S. Huang, Quant. Biol., 2019, 7, 171–181 CrossRef CAS.
- L. Xie, Q. Bao, A. Terada and M. Hosomi, Chem. Eng. J., 2016, 306, 1099–1108 CrossRef CAS.
- D. Choudhary, V. Lagage, K. R. Foster and S. Uphoff, Cell Rep., 2023, 42, 112168 CrossRef CAS PubMed.
- G. Pitruzzello, C. G. Baumann, S. Johnson and T. F. Krauss, Small Sci., 2022, 2, 2100123 CrossRef CAS.
- Y. Wakamoto, J. Ramsden and K. Yasuda, Analyst, 2005, 130, 311–317 RSC.
- E. A. Sarnoski, R. Song, E. Ertekin, N. Koonce and M. Acar, iScience, 2018, 7, 96–109 CrossRef CAS PubMed.
- M. C. Jo, W. Liu, L. Gu, W. Dang and L. Qin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9364–9369 CrossRef CAS PubMed.
- P. Liu, T. Z. Young and M. Acar, Cell Rep., 2015, 13, 634–644 CrossRef CAS PubMed.
- H.-C. Flemming and S. Wuertz, Nat. Rev. Microbiol, 2019, 17, 247–260 CrossRef CAS PubMed.
- S. Hooshangi and W. E. Bentley, Curr. Opin. Biotechnol., 2008, 19, 550–555 CrossRef CAS PubMed.
- Ö. Baltekin, A. Boucharin, E. Tano, D. I. Andersson and J. Elf, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9170–9175 CrossRef PubMed.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef CAS PubMed.
- F. Yokoyama, T. Imai, W. Aoki, M. Ueda, J. Kawamoto and T. Kurihara, Front. Microbiol., 2021, 12, 629023 CrossRef PubMed.
- D. Yang, A. D. Jennings, E. Borrego, S. T. Retterer and J. Männik, Front. Microbiol., 2018, 9, 871 CrossRef PubMed.
- S.-T. Liang, M. Ehrenberg, P. Dennis and H. Bremer, J. Mol. Biol., 1999, 288, 521–538 CrossRef CAS PubMed.
- L. K. Harris and J. A. Theriot, Cell, 2016, 165, 1479–1492 CrossRef CAS PubMed.
- M. Kaplan, G. Chreifi, L. A. Metskas, J. Liedtke, C. R. Wood, C. M. Oikonomou, W. J. Nicolas, P. Subramanian, L. A. Zacharoff, Y. Wang, Y.-W. Chang, M. Beeby, M. J. Dobro, Y. Zhu, M. J. McBride, A. Briegel, C. L. Shaffer and G. J. Jensen, eLife, 2021, 10, e73099 CrossRef CAS PubMed.
- E. R. Green and J. Mecsas, Microbiol. Spectrum, 2016, 4 DOI:10.1128/microbiolspec.vmbf-0012-2015.
- T. R. D. Costa, C. Felisberto-Rodrigues, A. Meir, M. S. Prevost, A. Redzej, M. Trokter and G. Waksman, Nat. Rev. Microbiol, 2015, 13, 343–359 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00707c |
| This journal is © The Royal Society of Chemistry 2024 |