
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructured-light-sheet imaging in an integrated optofluidic platform†
Petra
Paiè
 *ab,
Gianmaria
Calisesi
a,
Alessia
Candeo
ab,
Andrea
Comi
b,
Federico
Sala
b,
Francesco
Ceccarelli
b,
Ada
De Luigi
c,
Pietro
Veglianese
c,
Korbinian
Muhlberger
d,
Michael
Fokine
d,
Gianluca
Valentini
ab,
Roberto
Osellame
ba,
Mark
Neil
e,
Andrea
Bassi
ab and
Francesca
Bragheri
*b
*ab,
Gianmaria
Calisesi
a,
Alessia
Candeo
ab,
Andrea
Comi
b,
Federico
Sala
b,
Francesco
Ceccarelli
b,
Ada
De Luigi
c,
Pietro
Veglianese
c,
Korbinian
Muhlberger
d,
Michael
Fokine
d,
Gianluca
Valentini
ab,
Roberto
Osellame
ba,
Mark
Neil
e,
Andrea
Bassi
ab and
Francesca
Bragheri
*b
aDipartimento di Fisica, Politecnico di Milano, Piazza Leonardo da Vinci, 32, Milano, 20133, Italy. E-mail: petra.paie@polimi.it
bIstituto di Fotonica e Nanotecnologie, Consiglio Nazionale delle Ricerche, Piazza Leonardo da Vinci, 32, Milano, 20133, Italy. E-mail: francesca.bragheri@ifn.cnr.it
cIstituto di Ricerche Farmacologiche Mario Negri IRCCS, Via Mario Negri 2, Milano, 20156, Italy
dDepartment of Applied Physics, KTH Royal Institute of Technology, Roslagstullsbacken 21, Stockholm, 11421, Sweden
ePhysics Department, Imperial College London, Prince Consort Road, London, SW7 2BB, UK
First published on 3rd October 2023
Abstract
Heterogeneity investigation at the single-cell level reveals morphological and phenotypic characteristics in cell populations. In clinical research, heterogeneity has important implications in the correct detection and interpretation of prognostic markers and in the analysis of patient-derived material. Among single-cell analysis, imaging flow cytometry allows combining information retrieved by single cell images with the throughput of fluidic platforms. Nevertheless, these techniques might fail in a comprehensive heterogeneity evaluation because of limited image resolution and bidimensional analysis. Light sheet fluorescence microscopy opened new ways to study in 3D the complexity of cellular functionality in samples ranging from single-cells to micro-tissues, with remarkably fast acquisition and low photo-toxicity. In addition, structured illumination microscopy has been applied to single-cell studies enhancing the resolution of imaging beyond the conventional diffraction limit. The combination of these techniques in a microfluidic environment, which permits automatic sample delivery and translation, would allow exhaustive investigation of cellular heterogeneity with high throughput image acquisition at high resolution. Here we propose an integrated optofluidic platform capable of performing structured light sheet imaging flow cytometry (SLS-IFC). The system encompasses a multicolor directional coupler equipped with a thermo-optic phase shifter, cylindrical lenses and a microfluidic network to generate and shift a patterned light sheet within a microchannel. The absence of moving parts allows a stable alignment and an automated fluorescence signal acquisition during the sample flow. The platform enables 3D imaging of an entire cell in about 1 s with a resolution enhancement capable of revealing sub-cellular features and sub-diffraction limit details.
Introduction
Throughput is a fundamental property for an efficient investigation of cellular heterogeneity. Progresses in single-cell analysis combined with personalized medicine treatments are currently making gigantic steps, revolutionizing current laboratory procedures and clinical practices, but still striving for a throughput increase. Flow cytometry (FC) is undeniably the gold standard in terms of measured cells per second number, but it is characterized by poor information content, lacking spatial data acquisition. In recent years imaging flow cytometry (IFC) has gained increasing attention by combining image acquisition with a rapid and automated fluidic-assisted sample delivery through the detection channel.1 Different systems are now available both commercially (e.g. ImageStream, Luminex) and at prototype level in research laboratories, where advanced imaging techniques enabled improving the long-standing compromise between spatial and temporal resolution.2–6 Nevertheless, given the severe technical challenges in interfacing imaging setups with the IFC capillaries where the samples flow, these techniques have severely sacrificed image quality for throughput, being often characterized by low resolution and poor spatial information content (i.e. incompatibility with volumetric analysis).The availability of high-quality volumetric images of single cells combined with high throughput analysis would have a tremendous impact on cell heterogeneity investigation, constituting a new enabling method for addressing fundamental questions, identifying new markers for cellular health status, further advancing knowledge in cell cycles, and monitoring drug uptake.
Among different 3D imaging techniques, light sheet fluorescence microscopy (LSFM) is the one that best suits high throughput measurement, given its intrinsic fast dynamics based on non-invasive optical sectioning.7 Typical implementations use a cylindrical lens to focus the illumination light in one direction only, while the excited fluorescence is collected orthogonally by a microscope objective. In conventional setups, the specimens are manually positioned in a sample chamber, aligned in the optical system and automatically translated or rotated to allow for the acquisition of multiple image planes. By combining LSFM with microfluidics8 the samples can be automatically flown across the illumination region, thus overcoming limitations related to the sample mounting stages. This approach has been implemented by exploiting tubes,9 capillaries10 and microfluidic chips11–15 for the sample delivery. The system integration has been further increased by combining optical and fluidic elements on the same platform, by adding inclined micromirrors to reflect the incoming light sheet and project it orthogonally onto the sample4,16,17 or by embedding microlenses in the microfluidic chip to generate the light sheet.18–20 These approaches have been exploited to image samples of different nature and with different dimensions, from single cells to zebrafish embryos.12,19 While these implementations allow 3D and high throughput analysis at single cell level, image resolution is always subject to diffraction, thus severely limiting the ability to investigate subcellular features.
On the other hand, optical nanoscopy has emerged as a new field of research in the last decades21 because of its ability of overcoming the diffraction limit of classical fluorescence microscopy. Several techniques have already been developed to achieve super-resolution,22–25 which offer a huge resolution enhancement, down to a few nm, but at the expense of a long acquisition time in the range of several hours, thus hindering high throughput analysis. Structured illumination microscopy (SIM), based on the use of a patterned fluorescence excitation, is gaining increasing relevance for imaging beyond the diffraction limit26,27 because of its optimal compromise between temporal and spatial resolution as well as its compatibility with standard fluorescent labeling. Initially used for obtaining optical sectioning,28 SIM is now commonly and commercially used in microscopy offering a two-fold increase in spatial resolution over the diffraction limit. Combining SIM with microfluidics to perform super-resolution imaging of cells inside microchannels is desirable to increase its measurement throughput. In preliminary implementations the sample is flown either through an optical lattice,29 guaranteeing continuous sample delivery, or along a fixed illumination pattern as in standard epifluorescence SIM.30,31 The pattern generator has also been realized on-chip, using a counterpropagating beam approach to generate the required pattern, and integrated optical shifters to move it.32,33 Nevertheless, none of these systems is compatible with far-field 3D imaging. The combination of SIM and LSFM, i.e. structured light-sheet (SLS), has been presented by Chen et al.,34 enabling the imaging of molecular processes at sub-diffraction spatial scales. A structured light sheet generated by using a spatial light modulator and an annular mask has been launched through the light-sheet illumination objective and phase-shifted to allow SIM reconstruction and achieve resolution enhancement. The resulting interference pattern is bi-dimensional and cannot be rotated, thus resulting in an anisotropic resolution gain. This aspect, together with the technical complexity of the system has limited its implementation and the spread of this technique. Different methods have been proposed to achieve similar imaging performances. Chang et al. used the interference generated by two counter-propagating laser light sheets for patterned light-sheet generation.35 However, multiple illumination objectives are required for a symmetric resolution enhancement, leaving little space to handle the sample. A recent proposal allows for an isotropic enhancement in resolution by exploiting multidirectional structured illumination in oblique plane microscopy.36,37 A major flaw of these optical systems is the incompatibility with high throughput measurements as in IFC.
Here, we propose an automated platform for SLS-IFC, capable of performing 3D imaging of single cells flowing in a microfluidic channel with a resolution beyond the diffraction limit. The system, realized with femtosecond laser irradiation followed by chemical etching (FLICE) of glass substrates38 is an integrated optofluidic platform encompassing a directional coupler, a thermo-optic phase shifter and cylindrical lenses to generate and shift a patterned light sheet within a microchannel network. Meanwhile, the cells are flown in the channel and through the light plane, such that their fluorescence signal is automatically imaged performing dual-color SLS-IFC on-the-fly. Thanks to the resolution enhancement the system allows the recognition of sub-diffraction limit details such as vesicles within the volume of HeLa cells as well as nanoparticles, uptaken by the cells at different concentrations. The system enables the acquisition of an entire stack of 30 slices of a single cell in about 1.5 s with a capacity up to 40 cells per min.
Results
Platform design for SLS-IFC
In SIM microscopy, super-resolution is typically enabled by light modulation and the pattern periodicity defines the improvement in supported spatial frequencies. Moreover, the pattern translation over the sample is necessary for retrieving the sample information; having a 1D pattern, as in our case, entails the necessity of acquiring 3 raw images per plane, with optimal relative phase shifts of 0°, 120° and 240°.27To enable SLS on chip we have conceived an integrated sample and illumination platform, whose schematic design is reported in Fig. 1a. This device is composed of two separate glass components, connected by two polarization-maintaining (PM) fibers: a first purely optical chip and a second optofluidic device. The first one includes a symmetric directional coupler and a thermal phase shifter, and it is designed to split the light from a PM fiber pigtailed to the chip and to induce a rapid and on-demand phase shift between the two arms of the coupler. The second device is obtained by assembling two cylindrical microlenses and a microfluidic channel, as illustrated with a side-view in Fig. 1b. This is the component where SLS-IFC occurs. Each lens collects the beam from the corresponding fiber and focuses it in one direction only, generating a light sheet. The two light sheets, precisely tilted with respect to each other, overlap in correspondence with the microfluidic channel where the sample flows. The exact angle between the propagating beams during the assembly is set by custom-made lens holders. The interference between the beams generates a 1D modulation in the light sheet intensity which can be translated on demand by the thermal phase shifters. All the components are entirely fabricated by femtosecond laser micromachining (FLM), an enabling technique for 3D glass microstructuring38,39 that leads to permanent structural changes in the substrate by non-linear absorption processes. The extreme versatility of this fabrication method allows different types of material modification, including surface ablation, optical waveguide inscription and local enhancement of chemical etching selectivity. This last property combined with subsequent exposure of the substrate to an etchant solution can be used for glass micro-structuring as well as for fluidic channel formation. All these capabilities have been extensively used in the framework of this work for the fabrication of the different chip components, as detailed in Material and methods. The high level of integration that characterizes our system constitutes a major strength, as it enables advanced imaging capabilities together with device compactness, alignment stability and ease of use. Indeed, once the system is assembled it can be mounted as an add-on device on a standard fluorescence microscope, as shown in Fig. 1, upgrading it to a 3D super-resolved imaging flow cytometer, with only a few straightforward connections to external instrumentation and without requiring any specific end-user expertise.
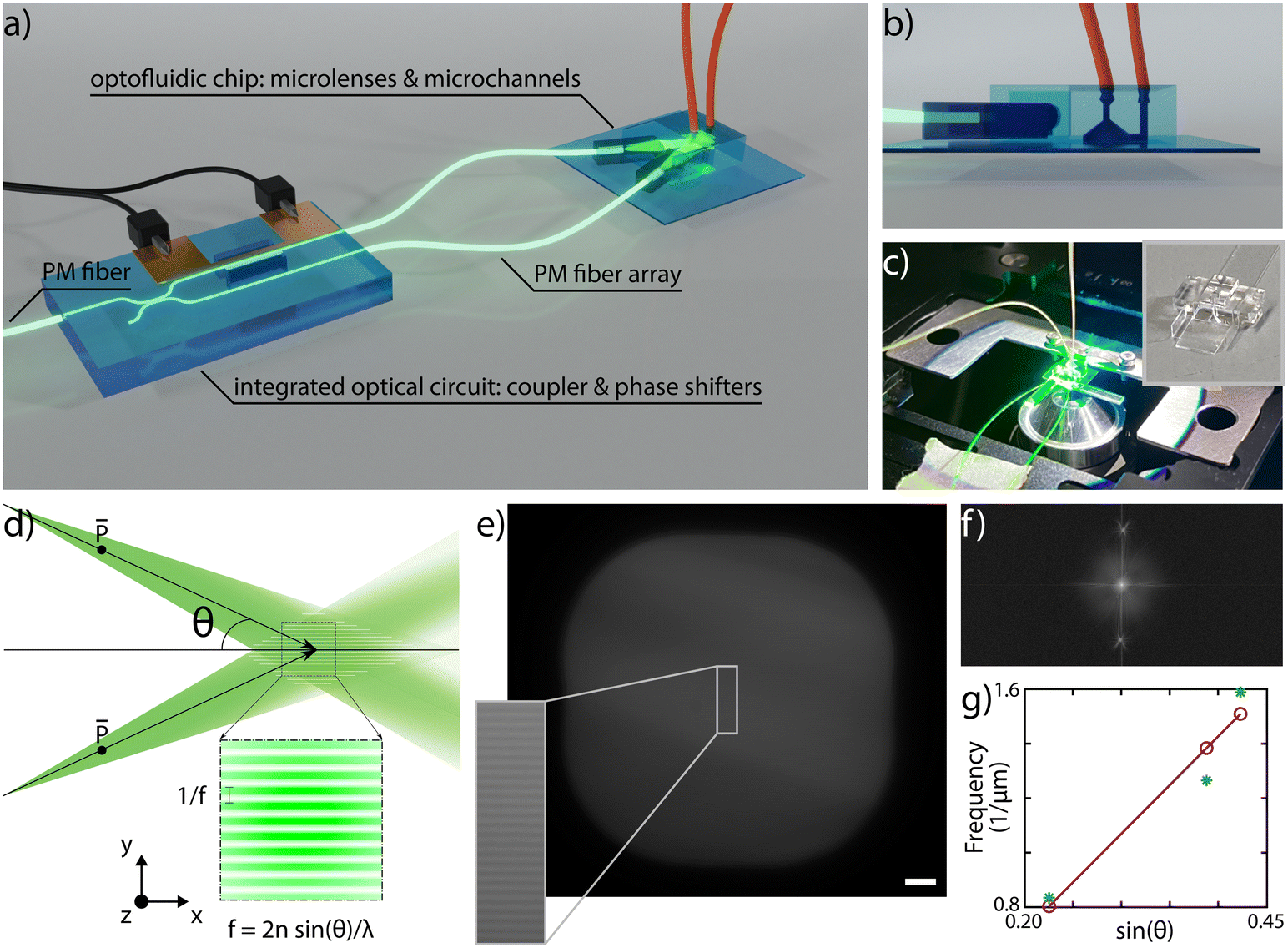 | ||
| Fig. 1 a) Schematic design of the system: a PM fiber is coupled to an integrated optical circuit designed to symmetrically split the beam and to induce an on-demand phase shift between the two outputs. The signal, collected by PM fibers is delivered to an optofluidic chip, where microlenses create two light sheets that overlap in correspondence with a microfluidic channel generating a patterned illumination plane. b) Detailed side view of the optofluidic device characterized by a U-shaped microchannel layout and a tapered design optimized to reduce aberrations in the detection path. c) Picture of the dual-color device mounted on a standard inverted microscope during its use. The device is clamped onto a custom-sample holder with the dimensions of a standard microscope slide. The inset shows a picture of the fabricated microchannel with a shelf-like structure on the front, which is designed to host the two microlenses. d) Principle used for patterned light generation. By overlapping two weakly diverging tilted beams an interference pattern occurs. The related periodicity depends on the semi-angle between the two beams, following the law reported at the panel bottom. e) Experimentally acquired on chip fluorescence emission of rhodamine and TDE solution when excited by the patterned light sheet. The detail of the modulation is highlighted. Given the linearity between fluorescence excitation and emission, the collected signal can be used to characterize the pattern properties. Scale bar is 10 μm. f) Fourier transform of the fluorescence signal reported in panel e) where two distinct peaks given by light modulation are clearly observable in the frequency space. g) Experimental verification of the frequency pattern dependence as a function of the angle reported in panel d) obtained by fabricating and analyzing three devices with different angles between the two lenses. | ||
On-chip pattern generation
The interference pattern of two non-collinear collimated beams has a frequency that directly depends on the semi-angle between the two: the larger the angle, the higher the pattern frequency and therefore the resolution enhancement, following the law illustrated in Fig. 1d. In our device there is a similar configuration, having two divergent light sheets, as determined by the fiber NA, that spatially overlap in correspondence of the sample channel. To demonstrate the capability of controlling the modulation frequency, we have assembled and experimentally characterized the pattern periodicity for three different devices having semi-angle values respectively of 13°, 23° and 25°. To do so, we have launched into the PM fiber a 561 nm laser source and filled the sample channel with a mixed fluorescent solution of Rhodamine diluted in water and 2.2′-thiodiethanol (TDE) in a concentration of 40% and 60%, respectively, to match the surrounding glass refractive index and to enable the pattern frequency analysis. Fig. 1e shows an image of the plane of the sample channel illuminated by the light sheets with an inset of the generated pattern. The Fourier transform of the image, Fig. 1f, highlights the presence of two precise peaks due to the pattern modulation in the frequency space, indicating that in a first approximation we can neglect the beam divergence over the channel cross-section. Measuring the peak location for each device allows us to plot the trend reported in Fig. 1g, showing a good agreement between experimental and expected results, highlighting the capability of controlling the fabrication and assembling steps, and the properties and performance of the integrated microscope. Moreover, for single-cell imaging, we used a custom analysis software that is able to automatically extract the pattern modulation frequency and measure the applied phase shifts. As a consequence, it is not mandatory for the pattern frequency to be uniform over the entire channel cross-section, but rather over selected smaller regions-of-interest containing single cells. Precise control of the polarization state is a fundamental requirement for SIM. Indeed, ensuring a parallel polarization of the two light sheets in correspondence of the sample channel, as indicated in Fig. 1d, enables maximizing the modulation contrast of the pattern. To achieve this, we have taken advantage of the birefringent axis of the optical waveguides, which is vertically aligned along the glass thickness plane and whose orientation can be controlled during the laser fabrication process.40 Therefore, aligning the slow axis of the PM fibers with the birefringent axis of the waveguide enables propagation of a linear polarization state throughout the entire optical circuit, from the first PM fiber until the microchannel. In this regard, it is important to control also the slow axis orientation of the fibers before gluing them to the microlenses, as discussed in the Methods section.SLS-IFC of single cells
We have experimentally validated our system by selecting the device with the highest modulation frequency, 1.6 × 106 m−1 at a wavelength of 561 nm, which corresponds to a pattern periodicity of 620 nm. This enables, in our setup, an increase of the supported spatial frequencies by up to 50% when using in the collection path a 40×, 1.15 NA microscope objective. The biological sample that we have analyzed on chip consists of a fixed population of HeLa cells fluorescently marked using Wheat Germ Agglutinin Alexa Fluor 594 (WGA 594) conjugate to label plasma membranes. We have diluted the sample in two different liquid suspensions at a concentration of 8 × 105 cells per ml, the first one made by mixing 1× PBS and TDE in order to match the glass refractive index, 1.45, and the second with 1× PBS only. In both cases, we have limited the acquisition area to a region of interest within the channel cross-section where the pattern is highly uniform. Similar image quality is demonstrated for both dilutions, see Fig. 2 for PBS only dilution and Fig. S1† for analysis with the index-matching solution. In addition, Fig. S2† presents analysis performed on a sample of fixed macrophages (raw 264.7 cell line) in PBS and Fig. S3† shows data acquired on a sample of fixed HeLa cells in TDE with phalloidin labelling.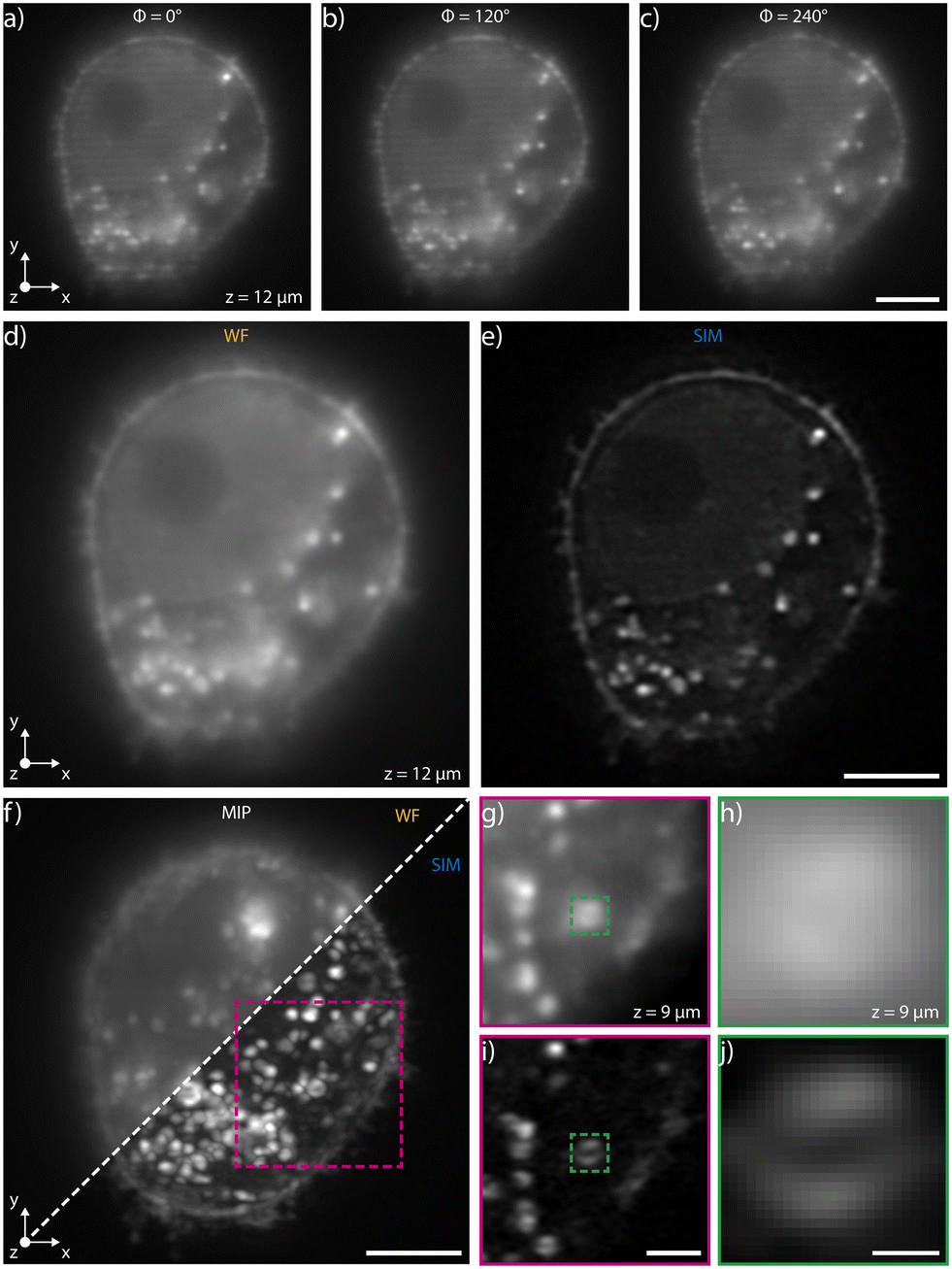 | ||
| Fig. 2 Acquisition of a fixed HeLa cell labelled with WGA 594 immersed in water during its flow through the patterned light sheet. a)–c) Raw images of a single plane for different phase shifts. Scale bar is 5 μm. d) Widefield image obtained by summing the three images at different phase shifts. The intensity uniformity highlights that the correct phase shift values have been applied to the chip. e) Corresponding super-resolved image obtained by post-processing the widefield images. Scale bar is 5 μm. f) Maximum intensity projections (MIPs) of the same cell obtained in widefield and in SIM imaging modality. The comparison highlights a significant quality improvement in the latter mode. Scale bar is 5 μm. g)–j) Resolution enhancement of the system evidenced by looking at a cellular vesicle in WF (g and h) and in SIM modality (i and j). Note that while the internal structure of the vesicle is not resolved in WF mode, its hollow feature is detectable with SIM. Scale bars are 2 μm (i) and 400 nm (j). | ||
High-precision pressure-driven pumps have been used to process the sample through the device at a flow rate of 13 nl min−1, which corresponds to a maximum cell velocity of about 10 μm s−1. In this condition, at a typical acquisition rate of 100 fps, the three modulated images required for SIM reconstruction are acquired within the depth of field and transverse resolution of the microscope avoiding motion artifacts (ESI† Notes). Image acquisition is performed with a custom software developed in Python that synchronizes the camera acquisition with the thermal phase shifters and with the laser (see Materials and methods). A Python module (napari-sim-processor) is used for enhanced resolution image reconstruction. Fig. 2a–c and ESI† Video S1 show the images of a single cellular plane, optically sectioned by the patterned light sheet, at the different phase steps necessary for image reconstruction, having nominal 0°, 120° and 240° relative phase shifts respectively. The sum of these three raw acquisitions, if the correct phase shifts are provided, gives a uniformly illuminated widefield (WF) image, as shown in Fig. 2d, S3a and b.† Post-processing the raw data enables retrieval of the corresponding super-resolved image for each plane, as shown in Fig. 2e. Fig. 2f directly compares the image quality obtained in wide-field and in SIM modality, highlighting the improved image quality of the latter imaging mode. Fig. 2g–j shows the detail of a fluorescent cellular vesicle, where it is possible to note how the WF imaging mode does not resolve its structure, while the SIM configuration shows fine inner details. In fact, the typical hollow structure of the vesicle could be appreciated only through SIM reconstruction. As expected, the resolution enhancement occurs along the pattern modulation direction (y axis in Fig. 2a). Additional examples of resolution enhancement are illustrated in Fig. S4 and in S5† in which nanobeads acquired profiles are analysed in 3D.
The 3D imaging capabilities of the SLS-IFC chip are illustrated, comparing wide-field and SIM volumetric image quality, in Fig. 3a–d, in ESI† Video S2, and in S6 and S7, showing 3D imaging of healthy cells and of cells treated to induce apoptosis, respectively. Note that SIM post-processing enables both background reduction to improve contrast and image resolution enhancement. Indeed, Fig. 3e–h reports WF and SIM images of a single z–y cellular plane and the plots of two intensity profiles along the indicated y and z direction. As expected, in the first case an increase in system resolution is observed, while in the second case a significant background reduction is demonstrated. The effect of sample movement due to drift and Brownian motion during sample scanning has been also considered. We have observed that no substantial artifacts are introduced in the reconstruction of a single plane of the sample. Moreover, the sample vibration and drift can be corrected by image registration in post-processing (see Fig. 3i–k and ESI† Notes for additional details) to improve the sample volume reconstruction. To better characterize the performance of our integrated device, we have taken advantage of hollow structures of the sample, like the subcellular vesicles. The throughput, the automation and the 3D imaging capability of this system have been fundamental for this analysis, enabling a rapid acquisition of hundreds of vesicles. Thanks to the microfluidic approach we can automatically image cells flowing in the sample channel, at a rate of up to 40 cells per min, depending on the concentration of the specimen. Vesicles are present in the entire cell volume, as highlighted in Fig. 4a and b where the MIP of a cell is shown respectively in widefield and SIM modalities. The higher image quality in SIM reconstructions allows the implementation of automated segmentation for vesicle recognition and identification, as depicted in Fig. 4c and visualized in ESI† Video S3. Through our segmentation pipeline implemented in napari, we can therefore locate multiple vesicles per sample by analyzing the different planes in the cell as it is optically sectioned. The result of this statistical analysis is reported in Fig. 4d, which compares the morphological properties of the same vesicles in WF and SIM configurations. The scatter plot displays the results obtained by imaging 65 HeLa cells, containing a total of 414 vesicles. Each point displays information from both the real and reciprocal space. On the y-axis of the graph the vesicle contrast is shown. For the SIM images this is calculated as the normalized intensity difference between the maximum and minimum intensity on a line parallel to the modulation direction, where the resolution enhancement is expected to be greatest. The line chosen for any given vesicle is the one that gives the largest contrast. The same points are then used to calculate the contrast in the WF image, as it may not exhibit a minimum in intensity for that vesicle. The higher the measured contrast, the better the capability of resolving the internal hollow structure of the vesicles. For each vesicle, the same intensity profile is analyzed also in the spectral domain and the result is reported on the x-axis of the same graph, where the weighted average of the measured spatial frequencies (weighted k) is reported. The higher this value, the better the corresponding image resolution. An example of this analysis is reported in Fig. 4e–h, for a single vesicle, both in WF and SIM modalities. The intensity profiles along the direction of the pattern modulation are compared in real and Fourier domains to evaluate the image contrast and the frequency bandwidth. From the scatter plot presented in Fig. 4d it is possible to observe that SIM and WF data form two well-separated clouds in the chosen space, with two distinct barycenters. The blue star, representing the average properties for vesicle images in SIM modality, has higher values on both axes with respect to the red circle, the center of the WF cloud. In particular, we measure an average increase of about 4.5 and 2.9 times, for the measured contrast and weighted k values, respectively. Therefore, SIM images are characterized by higher intensity contrast and larger frequency content, indicating a clear resolution enhancement.
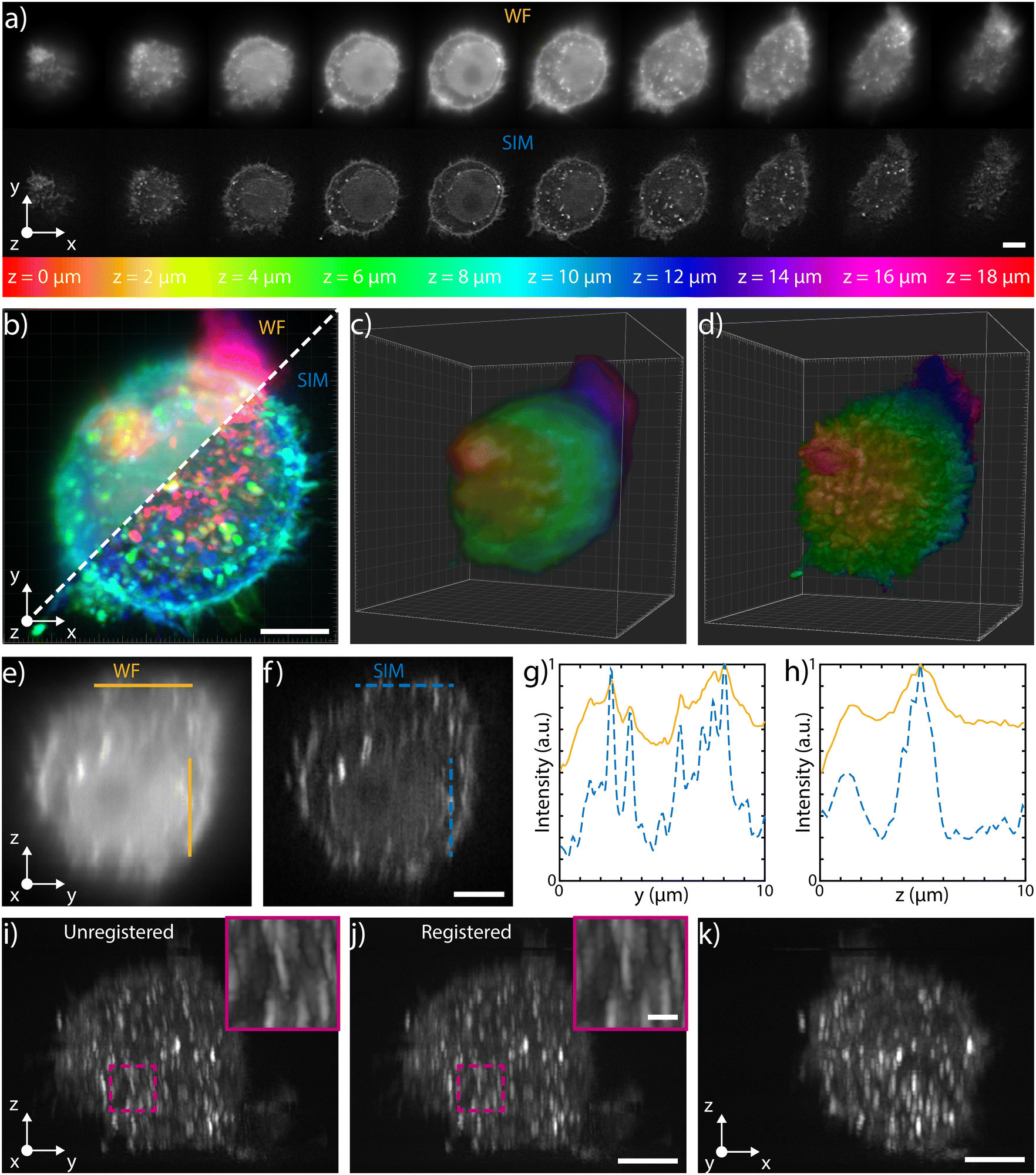 | ||
| Fig. 3 3D capabilities a) selected single planes, equally spaced of 2 μm, acquired during the flow in water of a fixed HeLa cell labeled with WGA 594 through the patterned light sheet. The top row shows the WF planes, while the bottom one depicts those reconstructed with SIM. The entire stack is illustrated in ESI† Fig. S6. b) MIP of the WF stack (top left) and of the SIM stack (bottom right), where the colors are representative of depth, following the depth color-coding shown at the bottom of panel a). Scale bar is 5 μm. c) and d) 3D reconstructions of the WF and of the SIM stacks, respectively (colors representative of depth). e) and f) y–z orthogonal projections of the WF and SIM stacks, respectively. Scale bar is 5 μm. g) Intensity profiles retrieved along the horizontal lines (y direction) shown in e) and f), highlighting the improvement obtained both in resolution and in contrast with the SIM reconstruction (blue dashed line) with respect to the WF (orange line). h) Intensity profiles retrieved along the vertical lines (z direction) shown in e) and f), highlighting the improvement obtained only in contrast with the SIM reconstruction (blue dashed line) with respect to the WF (orange line). i) Unregistered and j) registered MIP of the y–z orthogonal planes of the SIM stack, together with a zoomed-in detail of the corresponding magenta box. The comparison between the two demonstrates the possibility of correcting for possible translations of the cells while flowing through the channel. Scale bars are 5 μm and 1 μm for the main image and for the magenta box, respectively. k) x–z MIP of the SIM stack after registration. Scale bar is 5 μm. | ||
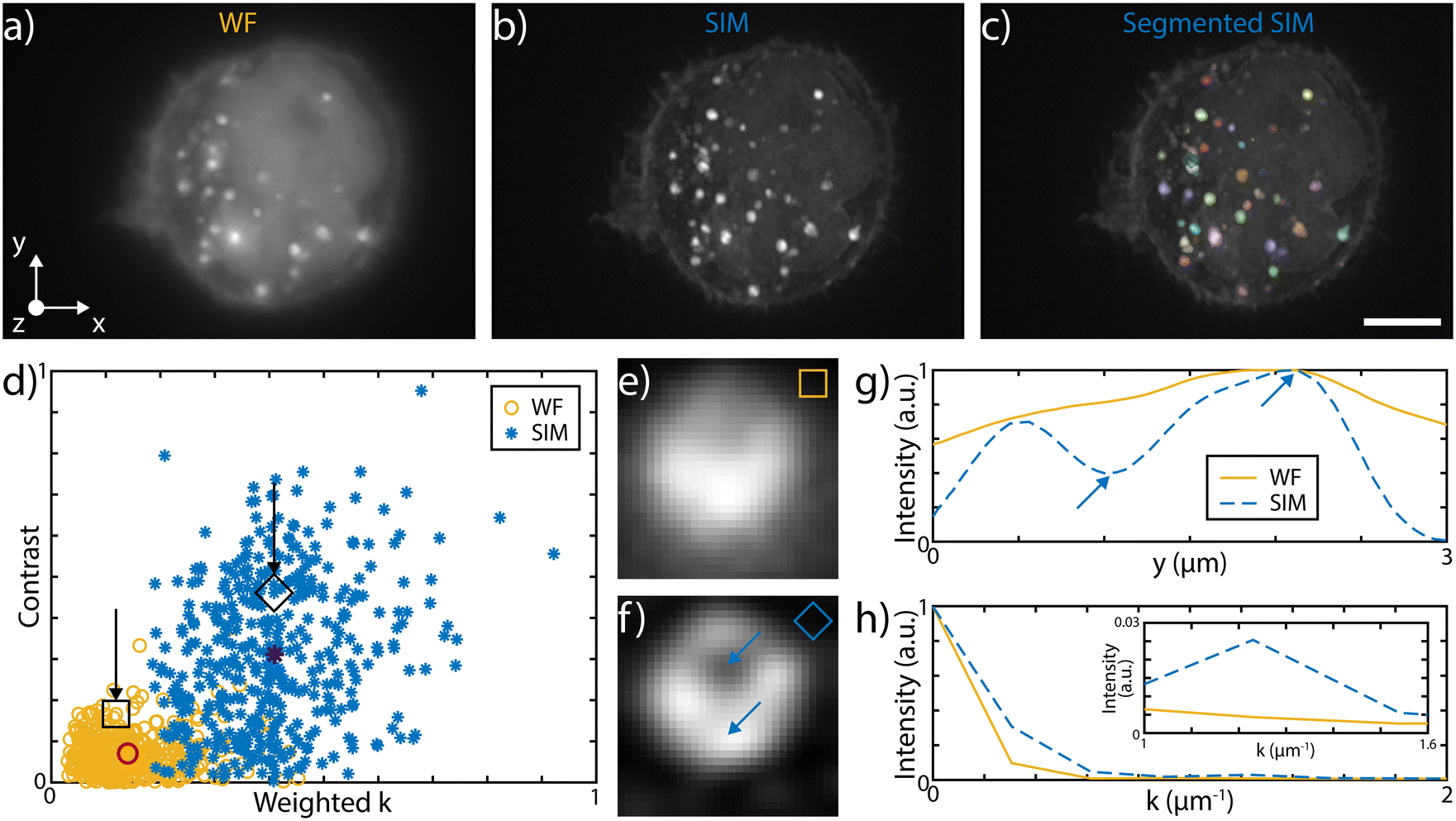 | ||
| Fig. 4 MIP of a fixed HeLa cell labeled with WGA 594 immersed in water in WF, panel a), and in SIM modality, panel b), with inner vesicles present in the entire volumes. c) Automated SIM segmentation, facilitated by increased image contrast, enables vesicle recognition and identification. The retrieved vesicles are subsequently post-processed for image quality analysis. Scale bar is 5 μm. d) Scatter plot of the cellular vesicles analysis in SIM and in WF mode. Both the intensity images and the corresponding Fourier transforms are analyzed. On the y-axis the contrast given by the normalized intensity difference between maximum and minimum values, due to the vesicle inner voids. On the scatter-plot x-axis the same intensity line is analyzed in the Fourier space and the weighted average of the measured spatial frequencies is reported. The average values for the two populations are represented by the red circle and by the dark blue star, for WF and SIM, respectively. The separation between the two populations highlights image quality improvement with SIM. Images of the same vesicle in WF and SIM mode are illustrated in panel e) and f), respectively. Void square and diamond locate the specific vesicle in the scatter plot, while blue arrows indicate the intensity minimum and maximum positions. g) Measured intensity profile for contrast analysis for both SIM and WF imaging mode. h) Fourier analysis of the intensity profile in the two imaging modalities used for weighted k measurement. The inset highlights intensity differences at high frequencies. | ||
Dual color imaging on chip
Multicolor imaging is key in fluorescence microscopy but its implementation on chip is not straightforward. Indeed, the wavelength-dependent behavior of certain optical components, such as the microlenses or the integrated beam-splitters, might significantly alter the properties of the patterned light sheet, degrading the final image quality. In order to achieve multicolor imaging, we have first investigated the chromatic aberrations of microlenses, using Zemax software for ray tracing simulations, as shown in Fig. S8.† We found that tuning the excitation wavelength from 561 nm to 488 nm mainly affects the focus position, which is moved closer to the lens as the wavelength decreases, with a maximum shift of 17 μm, at 488 nm. This light sheet alteration does not prevent its use for multicolor imaging. Indeed, if we consider an area of about 60 × 60 μm2 around the center of the sample channel, where the green beam is focused, the blue light sheet is a maximum of 15% thicker than the green. Indeed, the maximum blue waist was estimated to be about 1.6 μm while the green waist was 1.4 μm, guaranteeing good optical sectioning for cellular imaging at both excitation wavelengths. In our system, the major cause of chromatic dependence is given by the evanescent coupling of the integrated optical splitters that is highly sensitive to wavelength variation, which in turn affects the modulation contrast of the pattern. To address this aspect, we have modified the integrated circuit layout to produce close to 50–50 splitting at both 561 and 488 nm, by optimizing the coupling length of the splitter. To demonstrate dual color imaging performances, we processed fixed HeLa cells on-chip, previously incubated with polystyrene latex nanobeads (NPs) and with wheat germ agglutinin Alexa Fluor 488 conjugate (WGA 488). We have coupled to our device a blue and a green laser at 488 and 561 nm, respectively, and we have alternated the two sources in synchronism with the camera acquisition rate. The performance of the device in WF and SIM configuration are compared in Fig. 5 and in ESI† Video S4, which show 3D and multicolor imaging of a cell acquired by SLS-IFC. Cellular planes at increasing depth, together with single and dual color MIP are illustrated, comparing WF and SIM image quality and confirming the improvement in resolution and contrast at both colors. Moreover, the intensity profiles from a single cellular plane illustrate how, at both colors, it is possible to observe an increased resolution along the light sheet modulation direction as expected with SIM, Fig. 5f–i.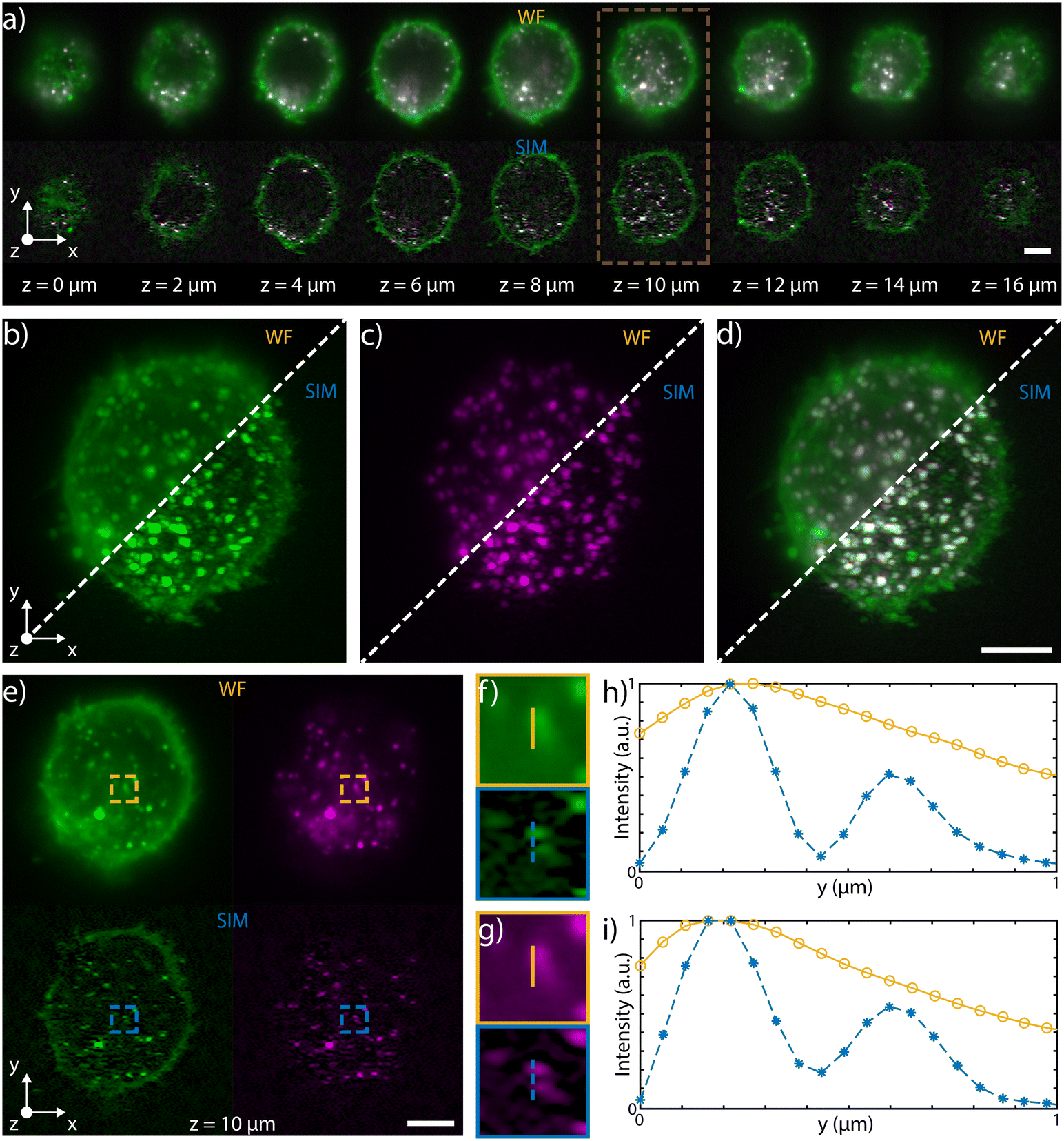 | ||
| Fig. 5 a) Selected single planes, equally spaced of 2 μm, acquired during the flow in water of a fixed HeLa cell, labelled with fluorescent red NPs and WGA 488, through the patterned light sheet. The top row shows the WF planes, while the bottom one depicts those reconstructed with SIM. Both top and bottom rows are composite images of the two acquired channels (green for excitation at 488 nm and emission at 520–550 nm, magenta for excitation at 561 nm and emission at 580–680 nm). b) and c) MIPs for the WF (top left) and of the SIM (bottom right) of the green and magenta channels, respectively. d) Composite image of the green and magenta channel of the WF (top left) and SIM (bottom right) MIPs. e) Single channel images of the plane highlighted by the brown box in panel a) (z = 10 μm). Top row displays the single-channel images for the WF, while the bottom row shows the correspondent SIM reconstructions. f) and g) Details of the singles plane images, corresponding to the boxes depicted on the green channel images and on the magenta ones, respectively. h) and i) Intensity profiles along the lines drawn on the detail images in f) and g) for the green channels and for the magenta ones, respectively. These latter graphs demonstrate the increase in resolution along the y direction of the SIM (blue dashed line) in comparison to the WF (orange line). For all WF and SIM images, a gamma correction of 0.5 was applied for visualisation purposes. Scale bar is always 5 μm. | ||
Discussion
SLS-IFC chip is an optofluidic platform capable of performing 3D imaging of single suspended cells with super-resolution in an automated fashion. The system is composed of two glass chips connected by polarization-maintaining fibers. The first device includes the optical components necessary to split the incoming light into two beams and to induce an on-demand phase-shift. Subsequently, the optofluidic platform uses the two beams to generate a light sheet, patterned by interference, to optically section the sample during its flow. For each slice, three images are acquired at different pattern shifts to obtain, after SIM algorithmic reconstruction, a 3D image of the cell with reduced background and enhanced contrast and resolution in single or dual color configuration. The implemented pattern allows up to a 50% increase in the image frequency support, therefore improving the transverse resolution from 300 nm available in our setup to 200 nm. The volumetric imaging capabilities of the system entail high information content acquisition per specimen, as demonstrated by the number of vesicles evaluated within each single cell with the support of automated segmentation and analysis software. Moreover, the fluidic-based sample delivery enables automatic and continuous sample processing. In particular, the SLS-IFC system is operated at a flow rate of 13 nl min−1, which enables the recording of 40 cells per min while maintaining an image acquisition rate of 100 fps over a given region of interest.In the present manuscript, we reported several patterns at different spatial frequencies, achieved by varying the angle between the two light sheets creating the pattern by interference, which in turn lead to different enhancements in resolution in one dimension. The system resolution could be further pushed by increasing the angle and ultimately by separating the beams by 180°, reaching the counter-propagating configuration. In this case, the pattern will not be directly visible and will be measurable only with cross-correlation methods, but the theoretical 2.4 enhancement in resolution would be reached. Further improvement with the addition of a third light sheet and orienting the three beams at 120° relative angles, gives a more uniform directional enhancement in resolution, as already foreseen in ref. 33 and 41.
The proposed system could be used in monitoring nanoparticles (NP) for drug delivery, in fact it provides information on both the amount of NP uptake by cells and on their position and possible accumulation. This information might be very useful also to study whether NP uptaken by cells are then released or if they remain accumulated, which might be potentially harmful for the cells themselves. The integrated system could be potentially used also in the study of chromatin within the cell nuclei. Indeed, chromatin 3D network has been efficiently visualized by SIM microscopes.42 Our system could be used to evaluate with high precision its abundance and location within the nuclei of large cell populations. This is an important information in the study of epigenetic role in cancer mutations and heterogeneity. In conclusion, we proved a new method for imaging flow cytometry capable of performing 3D and dual color super-resolved imaging of single cells diluted in a liquid suspension. This result has been achieved by taking advantage of the integrated approach that permits the necessary precision and stability in the alignment of the optical and fluidic components. We have presented the device layout, whose fabrication is enabled by advanced laser microfabrication techniques, and shown its successful validation with healthy and apoptotic HeLa cells as well as macrophages labelled with various fluorescent markers. The device that we have demonstrated here can be simply used as add-on to conventional brightfield microscopes, improving the measurement throughput as well as the imaging performances. These are two aspects often counterposed but both fundamental for targeting sample heterogeneity investigation at the single-cell level. Moreover, the integrated approach entails compactness and ease of use. Indeed, the complexity of the device is hidden from the final user who can operate the system without being trained in optics and photonics. We believe that this is an important aspect of our proposed method that could enable the widespread use of these advanced imaging techniques, which risk otherwise to be confined to specialized laboratories and facilities.
Materials and methods
Optical circuit fabrication
The optical circuit is fabricated by femtosecond laser micromachining (FLM) in a boro-aluminosilicate substrate (Corning EAGLE XG) using an Yb:KYW cavity-dumped mode-locked laser oscillator, with emission wavelength of 1030 nm, pulse duration of 300 fs, and a repetition rate of 1 MHz. A multi-scan irradiation process, followed by thermal annealing, has been optimized to obtain high-quality single-mode waveguides at both 488 and 561 nm. During the irradiation step, we used a 50×, 0.65 NA microscope objective to focus the beam into the substrate, which is translated by a high precision stage (FIBERGlide Aerotech 3D). The waveguides have been irradiated at a distance of 15 μm from the bottom surface of the glass substrate with 230 mW of laser power, 40 mm s−1 of scan velocity and 8 overlapped irradiations. Following a previously optimized annealing process up to 750 °C (ref. 43 and 44) we have obtained single mode waveguides with propagation losses of about 0.3 dB cm−1 and symmetric mode dimensions (FWHM) of about 1.8 × 1.9 μm2 at a wavelength of 488 nm and 2.7 × 2.8 μm2 at 561 nm. We have optimized the integrated coupler layout in terms of interaction length L and coupling distance d to perform a symmetric splitting of the incoming light power. We have first used L = 0.8 mm and d = 5 μm for the device working at 561 nm only (single-color device), while we have used L = 1.3 mm and d = 3.6 μm for the dual-color device. In these configurations, we have experimentally measured a splitting ratio of 48–52% for the single-color device and splitting ratios of 40–60 at 488 nm and 51–49% at 561 nm for the dual-color device, which guarantees fringe visibility of 97% and 99% for the blue and green light, respectively. The output distance between the two waveguides has been fixed to 127 μm to match the dimension of the fibers cladding. Fast thermal phase shifters (i.e. resistive microheaters) have been fabricated by following a previously optimized fabrication process:45 after a standard piranha cleaning bath, we have deposited 100 nm of gold, preceded by a 3 nm chromium adhesion layer, over the entire substrate surface using a magnetron sputtering process (Leybold LH Z400). Subsequently, thermal annealing has been used to increase both the stability and the reliability of the microheaters (1 h at 420 °C). Thermal phase shifters have been defined by removing the metal by laser irradiation. Each microheater is 3 mm long and 15 μm wide, corresponding to a resistance value of about 115 Ω. For local gold removal, the above-mentioned femtosecond laser has been used at 200 mW of average power, 2 mm s−1 scan velocity and focused on the glass surface using a 10×, 0.25 NA microscope objective. In addition, lateral trenches surrounding the microheater have been fabricated to reduce the thermal phase shifter response time, enabling system dynamics of about 1.5 ms, compatible with high acquisition rate cameras. For the fabrication of superficial trenches, before metal deposition the substrate was irradiated using multiple overlapped lines, 5 lines at each depth, from the surface to the trench bottom, at 800 mW of average laser power and 0.2 mm s−1 scan velocity. External connection to the driving electronics has been allowed by a male pin header bonded to the chip using an electrically conductive epoxy glue (CircuitWorks CW2400).Microchannel fabrication
The microfluidic channel is fabricated in a fused silica glass substrate by femtosecond laser irradiation followed by chemical etching (FLICE), a two-steps process constituted by laser irradiation followed by sample immersion in an aqueous solution of hydrofluoric acid (HF), that preferentially attacks irradiated regions, creating the microchannels. The laser source used is a commercial femtosecond laser system (Spectra-Physics Spirit 1040-16), with an emission wavelength of 1040 nm and 1 MHz repetition rate. The laser beam was frequency doubled and focused in a 2 mm thick fused silica substrate with a 50×, 0.6 NA microscope objective, the laser power was set to 260 mW, while the scan velocity was varied during the irradiation from 0.3 mm s−1 to 4 mm s−1 depending on the structure properties, such as depth and size. The layout of the microchannel is the same of our previous work,19 and it consists of a U-shaped microchannel, with inlet and outlet access holes for the tubes, and a tapered cone that is needed to match the collection objective numerical aperture in order to reduce the impact of aberrations due to the microchannel walls. The light sheets will intercept the channel just above the cone to avoid any beam deflection. In addition, to reduce the impact of aberrations that occur when imaging through rough surfaces as the ones fabricated by FLICE,46 the microchannel is not buried into the glass volume. The bottom surface is open and after etching it is sealed with a thin fused-silica coverslip, which guarantees high optical quality. The process has been optimized to prevent the UV curable glue (DELO Photobond GB345) used during the sealing process from entering the microchannel, thus keeping the bottom surface clear. In contrast to our previous design, in this work we used external microlenses that have to be aligned and glued in front of the sample channel. A “shelf”-like structure, having a base of 4.2 × 2.5 mm2, was realized in front of the channel, acting as a support surface for gluing the microlenses and providing a protection structure for them. Indeed the device was used in combination with a water immersion objective, thus it was mandatory to prevent water infiltration in front of the lenses. During the assembly process the lenses have been aligned and glued to the floor of this shelf. PEEK tubes for sample delivery were inserted in the inlet and outlet holes and glued to the device.Microlens fabrication
The microlenses fabricated in this work are obtained by shaping blocks of fused silica glasses into ferrule-like lenses using FLICE technique, as in ESI† Fig. S9. In contrast to our previous works,47 these microlenses are not directly buried in the microfluidic device substrate, having two major advantages: first, it enables to finely adjust the alignment of the two light sheets in the assembling phase, maximizing the pattern contrast. Second, having the focusing lens fabricated on an external surface enables further smoothing using CO2 laser polishing. For the irradiation process we have used the already mentioned commercial femtosecond laser (Spectra-Physics Spirit 1040-16), 260 mW of laser power and a scan velocity of 1 mm s−1. These components on one side have a hole for hosting fibers, while on the other side they have a curved profile designed to focus the light from the fiber along one axis. The cylindrical lens profile has been optimized using Zemax software for both single and dual color imaging. We have subsequently optimized the fabrication process, calibrating and compensating for the non-isotropic acid selectivity to match the theoretical lens profile, as in Fig. S9b and c.† Laser polishing was performed using a Synrad Firestar ti100HS CO2 laser operating at 10.6 μm and a Aerotech ALS130H-50 translation stage for scanning the beam across the sample. The beam with a diameter of 2 mm was focused onto the sample surface using a cylindrical ZnSe lens having a focal length of 127 mm, with the line focus aligned perpendicular to the scanning direction to ensure uniform heating across the surface. Optimum quality was achieved by scanning twice across the lens surface, in forward then backward direction, using approximately 20 W of laser power and a scanning speed of 0.1 mm s−1. Comparison of light sheet quality, before and after laser polishing is reported in Fig. S9d and e.† The measured beam waist was 1 μm, in good agreement with the theoretical one. Finally, tilted holders for the lenses are fabricated by FLICE and used, during the assembling phase, to control the relative angle between the lenses and therefore the pattern modulation frequency.Setup
In this work we used two different setups for image acquisition: a custom inverted microscope, with an Olympus LUMFLN60XW as collection objective, and a commercially available system, Nikon Ti2-U, with a Nikon 40×, 1.15 NA objective (MRD77410) with an additional magnification of 1.5× along the optical path. Both objectives are water immersion and are supplied by a correction collar. In both systems, we have filtered the excitation wavelength using a dual-band filter (Semrock FF01-512/630-25) and we have used a CMOS camera (Hamamatsu Orca Flash 4.0 V3) for image acquisition. Validation of the device on both these systems have been presented in the paper. As laser sources we used two Coherent OBIS lasers, at 488 and 561 nm, fiber coupled to two PM fibers, with the slow axis aligned parallel to the laser polarization state. A 488/561 PM laser combiner is used to launch both lasers into the single PM fiber pigtailed to the device input. Both the integrated optical component and the optofluidic one are mounted on the microscope stage, to which the output fibers are also secured to avoid any additional and undesired phase shifts during the measurement. The optofluidic component in particular is clamped to a custom sample holder with glass-slide dimensions, which can be easily mounted in the stage and aligned with respect to the collection objective.Sample preparation
HeLa cells and Raw 264.7 cells were grown in Dulbecco's modified Eagle medium (DMEM high glucose with pyruvate, Gibco Thermo Fisher Scientific) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS, Euroclone), 2 mM glutamine (Euroclone), and 1% (v/v) penicillin–streptomycin (Gibco Thermo Fisher Scientific) at 37 °C and 5% CO2 in a humidified atmosphere. For apoptosis induction, HeLa cells were treated with etoposide (50 μg ml−1) and, after incubation (24 hours), collected and resuspended in DPBS. For fluorescent nanoparticles labelling, cell lines were incubated with 2.5–25 μg ml−1 of polystyrene latex beads (aminated red 100 nm, Magsphere Inc.) for 60 min at 37 °C and 5% CO2 in a humidified atmosphere. After the treatment, the cells were washed with DPBS, detached with trypsin–EDTA (Gibco Thermo Fisher Scientific), and resuspended in DPBS. For labelling with fluorescent markers, cell lines were first subjected to centrifugation at 1000 rpm for 3 min at room temperature (RT) and fixed with 4% paraformaldehyde (Bio-Optica) for 15 min at RT. To label plasma membrane, fixed cell lines were incubated with wheat germ agglutinin Alexa Fluor 594 conjugate (Life Technologies) or with wheat germ agglutinin Alexa Fluor 488 conjugate (Life Technologies) for 30 min at RT. To label actin filaments (cytoskeleton), fixed cell lines were permeabilized in 0.1% Triton X-100 in DPBS for 15 min at RT, washed with DPBS and incubated with Alexa Fluor 568 Phalloidin (Life Technologies) for 30 min at RT. For two-colour labelling of cells, NPs-treated fixed cell lines were incubated with wheat germ agglutinin Alexa Fluor 488 conjugate (Life Technologies) for 30 min at RT. After incubation, cells were washed repeatedly (four times) and resuspended at 2–3 × 106 cells per ml.Image acquisition and real-time processing for single cells recording
The image acquisition with the CMOS camera was synchronized with the phase applied to the thermal shifters and with the lasers, in order to sequentially acquire frames with alternate illumination colors and subsequent phases. The camera was operated at 700px × 500px, corresponding to a field of view of 54 × 75 μm2 allowing acquisition rates up to 400 Hz, but the typical operation mode was at 50 to 200 Hz. The lasers and camera were triggered with two external digital signals generated by a digital-analog input–output (IO) board (National Instrument NI USB-6212) combined with a custom logic gate. The phases were controlled with the analog output port of the IO board, buffered through a high-speed voltage follower (Texas Instruments LMH6321) able to provide up to 300 mA to the resistive load. The latter circuit was operated in single-supply mode with a positive rail of 24 V. All hardware components were controlled by a custom-made Python program based on ScopeFoundry (https://www.scopefoundry.org/) open-source software. The pipeline for image acquisition and storage consisted of the following steps:– Images were captured sequentially, in batches of 6 consecutive frames consisting of 3 phases and 2 colors.
– The first image of the batch was processed in real-time to automatically segment the single (or multiple) cells in the field of view. Segmentation was based on Otsu's threshold and morphological operations. The cells were distinguished from other particles, possibly present in the field of view, based on the area of the segmented object.
– The segmentation was used to define a region of interest (ROI) around each cell. Only the ROIs were then saved on the hard disk. Typically, 20–100 batches, corresponding to different sections of the cells, were saved. The saving process continued until the cell, flowing away from the focus plane of the microscope, was no longer detected by the segmentation.
– The data were stored on the hard drive with a Hierarchical Data Format (HDF5) file, annotated with metadata that included all the hardware settings and measurement parameters.
Structured illumination microscopy reconstruction
We developed a Python module called napari-sim-processor for the reconstruction of SIM data within the napari ecosystem, (https://www.napari-hub.org/plugins/napari-sim-processor). The napari-sim-processor plugin is designed to work with raw SIM data acquired using a structured illumination microscope with standard and non-conventional illumination configurations. For the reconstruction of the data presented in this paper, the raw data consisted of a stack of 3 images of the samples acquired with a superimposed pattern with (expected) relative phases of 0°, 120° and 240°.The SIM reconstruction process consists of several steps. First, the raw SIM data are loaded into the plugin, a pre-calibration step is performed to determine the location of the carriers in the spatial frequency domain, their amplitudes, and phases. Pre-calibration is based on the cross-correlation in frequency space between low- and high-pass filtered replicas of the images (see Fig. S12†). Next, the plugin virtually extends the pupil of the optical detection system through spatial up-sampling of the data, followed by the separation and shifting of the different frequency bands in the image to their correct position, given by the spatial frequency of the detected illumination carriers. This process enables the reconstruction of extended-resolution images. The reconstruction also implements low spatial frequency suppression to remove out-of-focus signals and Wiener filtering to suppress the amplification of noise artifacts. The low spatial frequency suppression is achieved by applying a high-pass filter to the reconstructed image. The Wiener filter, on the other hand, uses an estimate of the power spectral density of the noise and to suppress noise artifacts in the reconstructed image. For comparison with the SIM reconstruction, pseudo-widefield reconstructions are created by taking the average value of the 3 raw images. The napari-sim-processor plugin is developed and maintained within the napari ecosystem, which provides a user-friendly interface for processing and analyzing imaging data. The plugin enables users to load SIM data, apply the reconstruction algorithms, and visualize the reconstructed images.
Data availability
All relevant data are presented in the paper and in ESI.† Additional data related to this paper may be requested from the authors.Author contributions
Conceptualization: PP, MN, AB, FB methodology: PP, FC, ADL, PV, KM, MF investigation: PP, GC, AC, AC, FS data analysis: PP, GC, AC, FC, AB supervision: PP, AB, FB writing – original draft: PP, FB writing – review and editing: PP, AC, FC, MF, GV, RO, AB, FB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thanks A. Zippo and S. D'Annunzio for providing cell samples for preliminary experiments. Financial support by the European Union through the H2020 FET Open project PROCHIP (Grant ID: 801336) is acknowledged.Notes and references
- P. Rees, H. Summers, A. Filby, A. Carpenter and M. Doan, Nat. Rev. Methods Primers, 2022, 2, 1–13 CrossRef PubMed.
- A. Rane, J. Rutkauskaite, A. deMello and S. Stavrakis, Chem, 2017, 3, 588–602 CAS.
- G. Holzner, B. Mateescu, D. van Leeuwen, G. Cereghetti, R. Dechant, S. Stavrakis and A. deMello, Cell Rep., 2021, 34, 108824 CrossRef CAS PubMed.
- T. Miura, H. Mikami, A. Isozaki, T. Ito, Y. Ozeki and K. Goda, Biomed. Opt. Express, 2018, 9, 3424 CrossRef CAS PubMed.
- H. Mikami, M. Kawaguchi, C. Huang, H. Matsumura, T. Sugimura, K. Huang, C. Lei, S. Ueno, T. Miura, T. Ito, K. Nagasawa, T. Maeno, H. Watarai, M. Yamagishi, S. Uemura, S. Ohnuki, Y. Ohya, H. Kurokawa, S. Matsusaka, C. Sun, Y. Ozeki and K. Goda, Nat. Commun., 2020, 11, 1162 CrossRef CAS PubMed.
- C. Lei, H. Kobayashi, Y. Wu, L. Ming, A. Isozaki, A. Yasumoto, H. Mikami, T. Ito, N. Nitta, T. Sugimura, M. Yamada, Y. Yatomi, D. Di Carlo, Y. Ozeki and K. Goda, Nat. Protoc., 2018, 13, 1603–1631 CrossRef CAS PubMed.
- J. Huisken, J. Swoger, F. Del Bene, J. Wittbrodt and E. Stelzer, Science, 2004, 305, 1007–1009 CrossRef CAS PubMed.
- E. J. Gualda, H. Pereira, G. G. Martins, R. Gardner and N. Moreno, Cytometry, Part A, 2017, 91, 144–151 CrossRef PubMed.
- E. J. Gualda, H. Pereira, T. Vale, M. F. Estrada, C. Brito and N. Moreno, Biomed. Opt. Express, 2015, 6, 4447–4456 CrossRef CAS PubMed.
- J. Wu, J. Li and R. Chan, Opt. Express, 2013, 21, 14474 CrossRef CAS PubMed.
- M. Ugawa and S. Ota, Biomed. Opt. Express, 2022, 13, 3647–3656 CrossRef CAS PubMed.
- R. Regmi, K. Mohan and P. Mondal, AIP Adv., 2014, 4, 097125 CrossRef.
- M. Ugawa and S. Ota, Small Sci., 2022, 2, 2100126 CrossRef CAS.
- H. Jiang, T. Zhu, H. Zhang, J. Nie, Z. Guan, C. Ho, S. Liu and P. Fei, Lab Chip, 2017, 17, 2193–2197 RSC.
- J. Son, B. Mandracchia, A. Silva Trenkle, G. A. Kwong and S. Jia, Lab Chip, 2023, 23, 624–630 RSC.
- M. B. M. Meddens, S. Liu, P. S. Finnegan, T. L. Edwards, C. D. James and K. A. Lidke, Biomed. Opt. Express, 2016, 7, 2219 CrossRef PubMed.
- R. Galland, G. Grenci, A. Aravind, V. Viasnoff, V. Studer and J. B. Sibarita, Nat. Methods, 2015, 12, 641–644 CrossRef CAS PubMed.
- P. Paie, F. Bragheri, A. Bassi and R. Osellame, Lab Chip, 2016, 16, 1556–1560 RSC.
- F. Sala, M. Castriotta, P. Paie, A. Farina, S. D'Annunzio, A. Zippo, R. Osellame, F. Bragheri and A. Bassi, Biomed. Opt. Express, 2020, 11, 4397–4407 CrossRef CAS PubMed.
- E. J. Vargas-Ordaz, S. Gorelick, H. M. York, B. Liu, M. L. Halls, S. Arumugam, A. Neild, A. de Marco and V. Cadarso, Lab Chip, 2021, 21, 2945–2954 RSC.
- S. Hell, Science, 2007, 316, 1153–1158 CrossRef CAS PubMed.
- S. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. Wolf Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS PubMed.
- S. Manley, J. M. Gillette, G.-H. Patterson, H. Shroff, H. F. Hess, E. Betzig and J. Lippincott-Schwartz, Nat. Methods, 2008, 5, 155–157 CrossRef CAS PubMed.
- R. Heintzmann and T. Huser, Chem. Rev., 2017, 117, 13890–13908 CrossRef CAS PubMed.
- M. G. Gustafsson, J. Microsc., 2000, 198, 82–87 CrossRef CAS PubMed.
- M. A. A. Neil, R. Juškaitis and T. Wilson, Opt. Lett., 1997, 22, 1905–1907 CrossRef CAS.
- F. Yu-Jui, H. Han-Yun, T. Sheng-Fang, W. Cheng-Hsuan, L. Chia-Ming, L. Yen-Ting, L. Chieh-Han, C. Shu-Wei and C. Bi-Chang, Lab Chip, 2021, 21, 344–354 RSC.
- C. Lu, N. C. Pegard and J. W. Fleischer, Appl. Phys. Lett., 2013, 102, 161115 CrossRef.
- B. Mandracchia, J. Son and S. Jia, Lab Chip, 2021, 21, 489–493 RSC.
- O. I. Helle, F. T. Dullo, M. Lahrberg, J. C. Tinguely, O. G. Helleso and B. S. Ahluwalia, Nat. Photonics, 2020, 14, 431–438 CrossRef CAS.
- M. Calvarese, P. Paie, A. Candeo, G. Calisesi, F. Ceccarelli, G. Valentini, R. Osellame, H. Gong, M. A. A. Neil, F. Bragheri and A. Bassi, Opt. Express, 2022, 30, 30246–30259 CrossRef PubMed.
- B. C. Chen, W. R. Legant, K. Wang, L. Shao, D. E. Milkie, M. W. Davidson, C. Janetopoulos, X. S. Wu, J. A. Hammer, Z. Liu, B. P. English, Y. Mimori-Kiyosue, D. P. Romero, A. T. Ritter, J. Lippincott-Schwartz, L. Fritz-Laylin, R. D. Mullins, D. M. Mitchell, J. N. Bembenek, A. C. Reymann, R. Bohme, S. W. Grill, J. T. Wang, G. Seydoux, U. Serdar Tulu, D. P. Kiehart and E. Betzig, Science, 2014, 346, 1257998 CrossRef PubMed.
- B. J. Chang, V. D. P. Meza and E. H. K. Stelzer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4869–4874 CrossRef CAS.
- B. Chen, B. J. Chang, P. Roudot, F. Zhou, E. Sapoznik, M. Marlar-Pavey, J. B. Hayes, P. T. Brown, C. W. Zeng, T. Lambert, J. R. Friedman, C. L. Zhang, D. T. Burnette, D. P. Shepherd, K. M. Dean and R. P. Fiolka, Nat. Methods, 2022, 19, 1419–1426 CrossRef CAS PubMed.
- C. Dunsby, Opt. Express, 2008, 16, 20306–20316 CrossRef CAS PubMed.
- R. Osellame, H. J. Hoekstra, G. Cerullo and M. Pollnau, Laser Photonics Rev., 2011, 5, 442–463 CrossRef CAS.
- R. R. Gattass and E. Mazur, Nat. Photonics, 2008, 2, 219–225 CrossRef CAS.
- G. Corrielli, A. Crespi, R. Geremia, R. Ramponi, L. Sansoni, A. Santinelli, P. Mataloni, F. Sciarrino and R. Osellame, Nat. Commun., 2014, 5, 4249 CrossRef CAS PubMed.
- H. Gong, W. Guo and M. A. Neil, Philos. Trans. R. Soc., A, 2021, 379, 20200162 CrossRef.
- L. Schermelleh, P. M. Carlton, S. Haase, L. Shao, L. Winoto, P. Kner, B. Burke, M. C. Cardoso, D. A. Agard, M. G. Gustafsson, H. Leonhardt and J. W. Sedat, Science, 2008, 320, 1332–1336 CrossRef CAS PubMed.
- G. Corrielli, S. Atzeni, S. Piacentini, I. Pitsios, A. Crespi and R. Osellame, Opt. Express, 2018, 26, 15101–15109 CrossRef CAS PubMed.
- A. Arriola, S. Gross, N. Jovanovic, N. Charles, P. G. Tuthill, S. M. Olaizola, A. Fuerbach and M. J. Withford, Opt. Express, 2013, 21, 2978–2986 CrossRef PubMed.
- M. Calvarese, P. Paie, F. Ceccarelli, F. Sala, A. Bassi, R. Osellame and F. Bragheri, Sci. Rep., 2022, 12, 239 CrossRef CAS PubMed.
- F. Sala, P. Paie, R. Martinez Vazquez, R. Osellame and F. Bragheri, Micromachines, 2021, 12, 180 CrossRef PubMed.
- R. Memeo, P. Paie, F. Sala, M. Castriotta, C. Guercio, T. Vaccari, R. Osellame, A. Bassi and F. Bragheri, J. Biophotonics, 2021, 14, e202000396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00639e |
| This journal is © The Royal Society of Chemistry 2024 |