
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of polysulfide anions and molecular sulfur via coupling HPLC with ICP-MS†
Aleksei
Sadykov
ab,
Yannick P.
Stenzel
a,
Martin
Winter
ac,
Simon
Wiemers-Meyer
a and
Sascha
Nowak
 *a
*a
aMEET Battery Research Center, Institute of Physical Chemistry, University of Münster, Corrensstr. 46, 48149 Münster, Germany. E-mail: sascha.nowak@uni-muenster.de
bInternational Graduate School for Battery Chemistry, Characterization, Analysis, Recycling and Application (BACCARA), University of Münster, Corrensstr. 40, 48149 Münster, Germany
cHelmholtz Institute Münster, IEK-12, Forschungszentrum Jülich GmbH, Corrensstr. 46, 48149 Münster, Germany
First published on 22nd August 2024
Abstract
A novel method for the speciation and quantification of polysulfide anions and molecular sulfur in lithium polysulfide solutions in organic solvents is reported. The technique is based on hyphenation of high-performance liquid chromatography (HPLC) and inductively coupled plasma mass spectrometry (ICP-MS). A sector-field mass-spectrometer was utilized which made it possible to quantify various sulfur compounds without the need for single component standards and conduct the direct detection of the main isotope of sulfur regardless of interferents such as highly abundant 16O2. Key aspects of separation and sample preparation were considered which allowed complete separation of derivatized polysulfide anions. Gradual adjustment of essential parameters and hardware is described. Variation of plasma settings allowed for obtaining chromatograms with desired analyte peak shapes. The optimized method was applied for the quantification of various lithium polysulfide mixtures in organic solvents showing the accessibility of the corresponding polysulfide distributions with this technique.
1. Introduction
The formation of polysulfides, first described by Scheele,1 gained scientific interest in the 1960s during the development of spectroscopic methods. In his pioneering studies, Giggenbach2,3 reported qualitative observations on the polysulfide distribution both in aqueous and aprotic media based on the absorbance bands of different polysulfide species in solution. Since then, there have been several attempts to describe the distribution of polysulfides. Further interest in polysulfide distribution in organic media was caused by the development of lithium–sulfur (Li–S) batteries. As polysulfide ions are generated during operation of Li–S batteries, the so-called polysulfide shuttling causes significant issues during cycling.4 The reduction of shuttled polysulfide anions at the anode leads to Li corrosion and coulombic inefficiency. For example, X-ray photoelectron spectroscopy (XPS)5 and X-ray diffraction analysis (XRD)6 were applied to qualitatively monitor polysulfide formation during cycling of Li–S batteries. Being indispensable methods for in situ characterization of polysulfide distribution, these methods nevertheless are not suitable for investigation of liquid electrolytes.Nuclear magnetic resonance (NMR) spectroscopy was also reported as a tool for the analysis of polysulfide distribution.7 Since the only NMR-active sulfur nucleus 33S has a low gyromagnetic ratio (2.052 × 107 rad T−1 s−1) and low natural abundance (0.76%)8 it is necessary to detect polysulfide anions using derivatization (for example with alkyl halogenides) and then detect the nucleus with higher abundance. Nonetheless, the sensitivity of this method is not sufficient to detect polysulfides with higher chain lengths (7–8) which typically have smaller concentrations than shorter polysulfides.
Semi-quantitative analysis using ultraviolet-visible (UV-Vis) spectroscopy is possible when appropriate signal deconvolution is achieved.9 Even though this technique is a great tool for operando studies, for example, to analyze the discharge of Li–S batteries, the nature of the assumptions made to calculate the concentrations does not allow for obtaining accurate results. Also, the spectral assignment of polysulfide peaks may differ when different solvents or electrolytes are used.10
A next step in the speciation of polysulfides was taken when separation techniques such as liquid chromatography (LC) were used. In 2004 Kamyshny et al.11 showed that derivatized polysulfides can be separated in reversed-phase (RP) HPLC and detected using a UV-Vis detector. Following this, soft ionization mass-spectrometry techniques such as electrospray ionization (ESI)12 or atmospheric pressure chemical ionization (APCI)13 with suitable derivatization were applied for the detection of the species separated by HPLC. Although these methods offer high sensitivity (and in the case of non-destructive mass spectrometry they can confirm the structure of each eluted species), it is not possible to conduct quantification since no commercial standards are available for each species in the system.
The only MS method that allows full quantification of various species without using standards for each analyte is inductively coupled plasma-mass spectrometry (ICP-MS). Thanks to complete atomization in the ICP-unit, the detector response of a certain compound does not depend on its chemical structure but solely on the concentration of analyzed atoms. The detection of the sulfur main isotope (32S) requires the use of additional add-ons such as reaction cells due to the interference of highly abundant (16O2),14 with small adducts such as 32S16O+ being analyzed instead.
When an organic matrix is used, additional challenges may occur. Although the addition of small amounts of organic solvents may be beneficial for the ICP-MS signal intensity as some atoms can be measured interference-free (e.g., in the case of 80Se and 40Ar40Ar),15 decreasing the amount of Ar+-species mostly results in loss of signal intensity or instability of the plasma.16 In addition, existing methods of sulfur detection with ICP-MS face new difficulties due to further interference. For example, de Wolf et al.17 showed incapability of quadrupole ICP-MS equipped with a dynamic reaction cell to detect sulfur in an organic-rich matrix due to interference of 32S16O+ and 36Ar12C+. However, interference-free detection was possible using a sector-field (SF) ICP-MS in medium resolution mode, with 32S+ or 34S+ being directly detected using the mass analyzer.
The RP-HPLC-ICP-MS method is used for sulfur-containing biomolecules such as peptides and bioinorganic complexes.18,19 Due to the chemical nature of those molecules, eluents with low content of organics can be used, which do not extinguish the plasma, nor promote the deposition of elemental carbon on the hardware (cones). However, for many non-polar molecules stronger eluents must be used which in the case of reversed-phase chromatography means higher organic content.
The signal intensity, resolution, and detectability of sulfur in general depend on many parameters, such as eluent composition and flow, plasma power, and composition and flow of the gas mixtures introduced to the sample chamber. Nevertheless, literature data scarcely contain studies on the adjustment of plasma setup and little attention is paid to signal characteristics such as peak width and the signal-to-noise ratio which are to be considered as well.
In this study, a method for quantification of sulfur polyanions based on HPLC separation followed by ICP-MS detection on a sector-field device is presented. Method development and the optimization of parameters are described. The gradual adjustment of the setup settings may serve as a directive for other studies in the field of HPLC-ICP-MS separation.
2. Materials and methods
2.1. Chemicals and sample preparation
Deionized water from a Millipore Milli-Q water system, HPLC-grade methanol (Merck) and HPLC-grade acetonitrile (Merck) were used for preparation of eluents and dilution of samples. Lithium sulfide (99.9%, Alfa Aesar) and elemental sulfur (99.998%, Sigma-Aldrich) were used for the generation of lithium polysulfide distributions after vacuum drying for 12 h at room temperature. Additionally, ε-caprolactam (>98%, Fisher Scientific), acetamide (99%, Fisher Scientific), and anhydrous 1,3-dioxolane (DOL, 99.8%, Thermo Scientific) were used as solvent components for the dissolution of Li2Sn mixtures. Dimethyl sulfide (Me2S, >99%, Thermo Scientific), dimethyl disulfide (Me2S2, >99%, Merck), dimethyl trisulfide (Me2S3, >99%, TCI), and lithium bis(trifluoromethane)sulfonimide (LiTFSI, >99.95%, Sigma-Aldrich) were applied as qualitative and quantitative standards. Methyl trifluoromethanesulfonate (>98%, TCI) was used for the derivatization of the Li2Sn mixtures.For the generation of polysulfide distribution in organic electrolytes Li2S was mixed with S8 in various proportions to achieve a concentration of 0.11 M in a solvent containing ε-caprolactam, acetamide and 1,3-dioxolane in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 weight ratio. The chemicals were mixed in a glove box under an inert atmosphere (residual pressure of H2O and O2 < 1 ppm) and heated for 24 h at 50 °C to form dark green to dark red solutions (Fig. S1†) of lithium polysulfides with nominal composition ranging from Li2S3.8 to Li2S8.1 (for simplification in some parts of this paper the notations contain rounded indices, e.g. “Li2S4” denotes “Li2S3.8”). For analysis, 10 μL of sample solution was mixed with 10 μL methyl trifluoromethanesulfonate and diluted with 980 μL of acetonitrile. Qualitative standards were dissolved in methanol to obtain a 10 ppm sulfur concentration of LiTFSI, Me2S, Me2S2, and Me2S3 and a saturated solution of S8.
:1:3 weight ratio. The chemicals were mixed in a glove box under an inert atmosphere (residual pressure of H2O and O2 < 1 ppm) and heated for 24 h at 50 °C to form dark green to dark red solutions (Fig. S1†) of lithium polysulfides with nominal composition ranging from Li2S3.8 to Li2S8.1 (for simplification in some parts of this paper the notations contain rounded indices, e.g. “Li2S4” denotes “Li2S3.8”). For analysis, 10 μL of sample solution was mixed with 10 μL methyl trifluoromethanesulfonate and diluted with 980 μL of acetonitrile. Qualitative standards were dissolved in methanol to obtain a 10 ppm sulfur concentration of LiTFSI, Me2S, Me2S2, and Me2S3 and a saturated solution of S8.
2.2. Methods
For reversed-phase chromatographic separation, a Thermo Scientific Dionex UltiMate 3000 series was used equipped with a Hypersil GOLD C18 (50 × 2.1 mm, 1.9 μm) column. Thermo Element XR™ was employed for ICP-MS detection of the eluate.To achieve the best chromatographic separation and peak shape, an adjustment of parameters was conducted which will be described in the following sections. Chromatography signal intensity was measured internally using the ELEMENT software as the sum of all 20 channels that comprised the corresponding peak in the mass spectrum.
3. Results and discussion
Free polysulfide anions can transform into each other and organize a dynamic equilibrium:11 | (1) |
 | (2) |
After derivatization, e.g. with methyl triflate according to the equation:
| Sn2− + 2CF3SO3CH3 → CH3SnCH3 + 2CF3SO3−, | (3) |
polysulfide anions transform into organic molecules which can still disproportionate/degrade but significantly slower than the corresponding anionic species:
 | (4) |
Therefore, for chromatographic separation followed by any detection method, the derivatization of polysulfides has an advantage. As the derivatized species, e.g., dimethyl polysulfides, show lower dis- and comproportionation rates (conversion times ∼1 day for derivatized species20vs. ∼1 s for free polysulfides),11 the polysulfide distribution can be “frozen” and depicted within typical chromatographic measurement times. In this study, three derivatization agents were applied: methyl triflate, 4-(dimethylamino)benzoyl chloride, and benzyl chloride which were already described previously as derivatizing agents for polysulfide mixtures.7,12 Among all, methyl triflate was chosen due to its high reaction rate (see Table S1†) which was also confirmed in previous studies.11 Moreover, dimethyl polysulfides demonstrated single peaks in UV-Vis chromatograms which was not the case for 4-(dimethylamino)benzoylchloride.
First, HPLC separation was optimized to start with a defined sample introduction for the mass analyzer later. For testing the quality of separation three standard compounds were chosen, namely LiTFSI, dimethyl sulfide and dimethyl disulfide. Fig. 1 demonstrates LC-ICP-MS chromatograms of the standards acquired using water–methanol eluent with 55%, 65% and 75% (v/v) organic content, showing different elution behaviors depending on the fraction of methanol. LiTFSI being an ionic compound has no retention on the column in reversed-phase chromatography; therefore it elutes first. Furthermore, its retention time does not depend on the methanol fraction in the eluent. Dimethyl sulfides having a nonpolar nature elute later, with the retention time being longer for molecules containing longer sulfur chains. The percentage of organic solvent necessary for complete separation of the standards was 55%. A lower methanol fraction was not considered since it would make the separation time longer with no significant improvement. To make the separation time shorter it is possible to apply gradient LC separation instead of the isocratic one that was already demonstrated in the literature11 for derivatized polysulfides. Gradient separation is an HPLC technique which not only shortens the duration of the analysis but also ensures that retaining compounds (i.e., compounds with the highest affinity to the stationary phase) are washed out by applying the eluent with a higher content of strong solvent (methanol, acetonitrile, etc.). However, for quantification purposes, gradient separation may not be beneficial, as the detector response used for the quantification may vary at different eluent compositions. For example when analyzing the ICP-MS signal for 35ClH2+ ion, Vanhaecke and coworkers demonstrated a non-linear response depending on the methanol content in the eluent in a 15–90% (v/v) range and a linear response within the same range for acetonitrile.21 Even in the case of a clear linear dependence a correction does not seem favorable due to non-zero dwell volumes of the HPLC system and ICP-MS introduction system as well as non-zero equilibration times when changing between various eluent formulations. For the conditions applied in this work, the response of 32S+ shows a non-linear behavior with different methanol contents and the response factor (i.e., the ratio between the Me2S signal and its quantity) decreased by a factor of 100 when changing from 50% to 90% methanol content (Fig. 1b). This is the case because the plasma conditions were optimized for a 55% methanol percentage. Additionally, no improvement in the peak shape was observed (i.e., peak width remained the same) when a stronger eluent was used. Based on this, for the straightforward separation of the derivatized polysulfides the isocratic chromatography mode was applied.
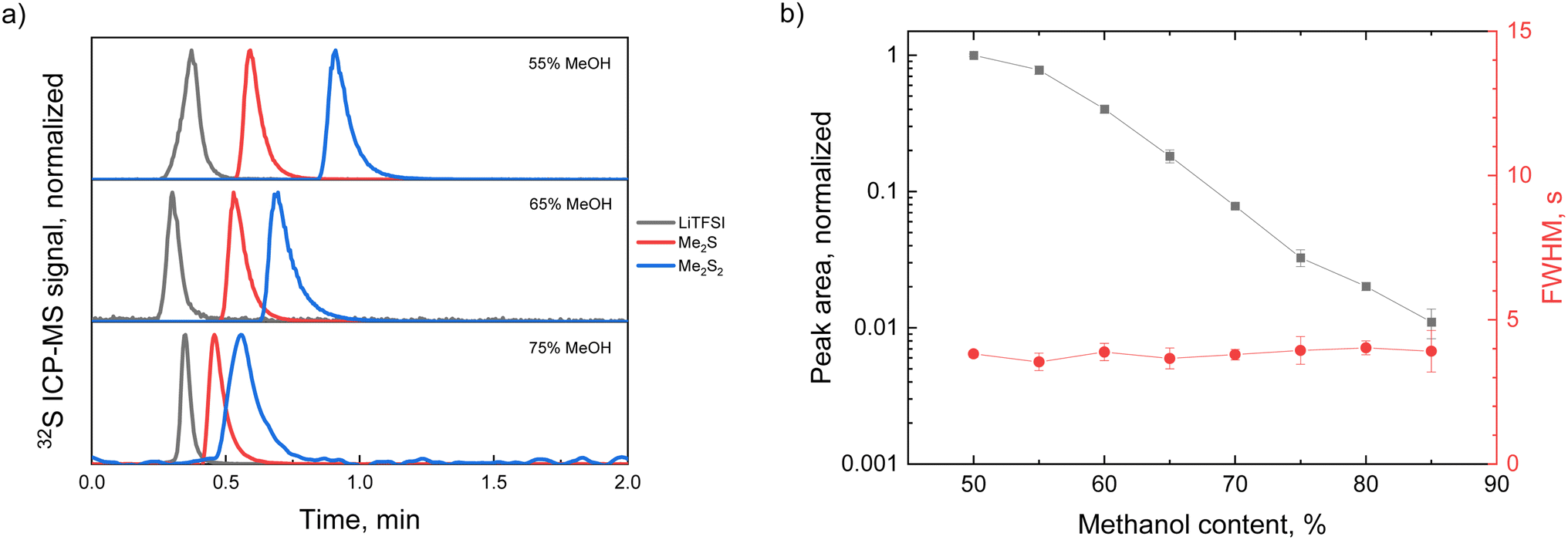 | ||
| Fig. 1 (a) LC-ICP-MS chromatograms of LiTFSI, Me2S and Me2S2 standards (separate injections) acquired using different methanol contents in the eluent (55%, 65% and 75%); (b) normalized response factor measured as peak area and the peak width (full width at half-maximum, FWHM) of the Me2S signal at various methanol contents in the eluent. The Ar sample gas flow value was 0.7 L min−1 and the eluent flow was 0.5 mL min−1. | ||
It is worth mentioning that response dependence on methanol content shown in Fig. 1b may have a different behavior when other gas parameters are applied in the sample introduction system. Fig. S2b† shows an increase in the 32S+ signal at higher methanol contents when oxygen was added to the sample gas to a small extent (7.6%). This may be due to the suppression of the 32S+ signal in the presence of oxygen in the plasma as an SO+ adduct is formed. However further addition of organics inhibits the formation of SO+ since O2 is consumed for the interaction with organic matter.
For a well-resolved chromatography acquisition not only a suitable eluent program is important but also a suitable post-column configuration. In the literature22 it was shown that peak shape depends on the sample introduction system used to transfer the liquid flow to the ICP unit. For example, the authors demonstrated peak broadening by a factor of 3.2 for a PFA-LC nebulizer in combination with various sample chambers. Fig. S3† shows chromatograms of dimethyl sulfide acquired under the aforementioned conditions using different sample chambers and a PFA-LC nebulizer. The peak width is increased by a factor of 2 when replacing a small volume cyclonic chamber with a conventional Scott spray chamber. The increased peak width is explained by an increased dispersion time when the solute/analyte is introduced to a larger chamber. In all cases the increase in the peak width is 1.7–5.5 s compared to the signal width without the spray chamber (1.8 s, UV-Vis signal on Fig. S3†). The broadening value is in good correlation with the amount of time the analyte spends in the spray chamber with adjusted sample gas flow (∼50 mL/0.7 L min−1 = ∼4 s). To keep the peak width as small as possible it was decided to use the sample chamber with the smallest volume.
Further enhancement of the chromatography signal can be achieved by varying the eluent flow and sample gas parameters. Thus, dimethyl sulfide was injected using conditions from Table 1 at two values of eluent flow and several values of Ar sample gas flow in the range of 0.5–1.0 L min−1 with or without admixed O2 (0–0.10 L min−1). Peak characteristics to calculate were peak full width at half-maximum (FWHM), signal-to-noise ratio (S/N), and sharpness (ratio of peak height to its FWHM). It should be noted that peak sharpness here equals the halved square root of the number of theoretical plates and therefore indicates the efficiency of the separation under applied conditions. Fig. 2 shows the results of this parameter adjustment. As can be seen from Fig. 2, a higher eluent flow of 0.5 mL min−1 is more favorable for the peak shape. In this case, chromatography peaks are sharper and the signal-to-noise ratio is at least two times larger than for lower eluent flow rates. It is worth noting that when recalculated to volume units (mL), the peak widths are smaller for low eluent flows which was also demonstrated in the literature.22 However, the peak sharpness (Fig. 2c) is still higher at 0.50 mL min−1 since higher eluent flow does not only result in lower peak width but also in increased signal intensity.
| Instrument | Thermo Scientific Dionex UltiMate 3000 Series |
| Binary pumps | HPG-3200RS |
| Autosampler | WPS-3000RS |
| Column oven | TCC-3000RS |
| UV-Vis detector | DAD-3000(RS) |
| Eluent | 55% methanol and 45% water (v/v) |
| Flow rate | 0.5 mL min−1 |
| Injection volume | 1.0 μL |
| Oven temperature | 40 °C |
| Column | Hypersil GOLD C18 50 × 2.1 mm, 1.9 μm |
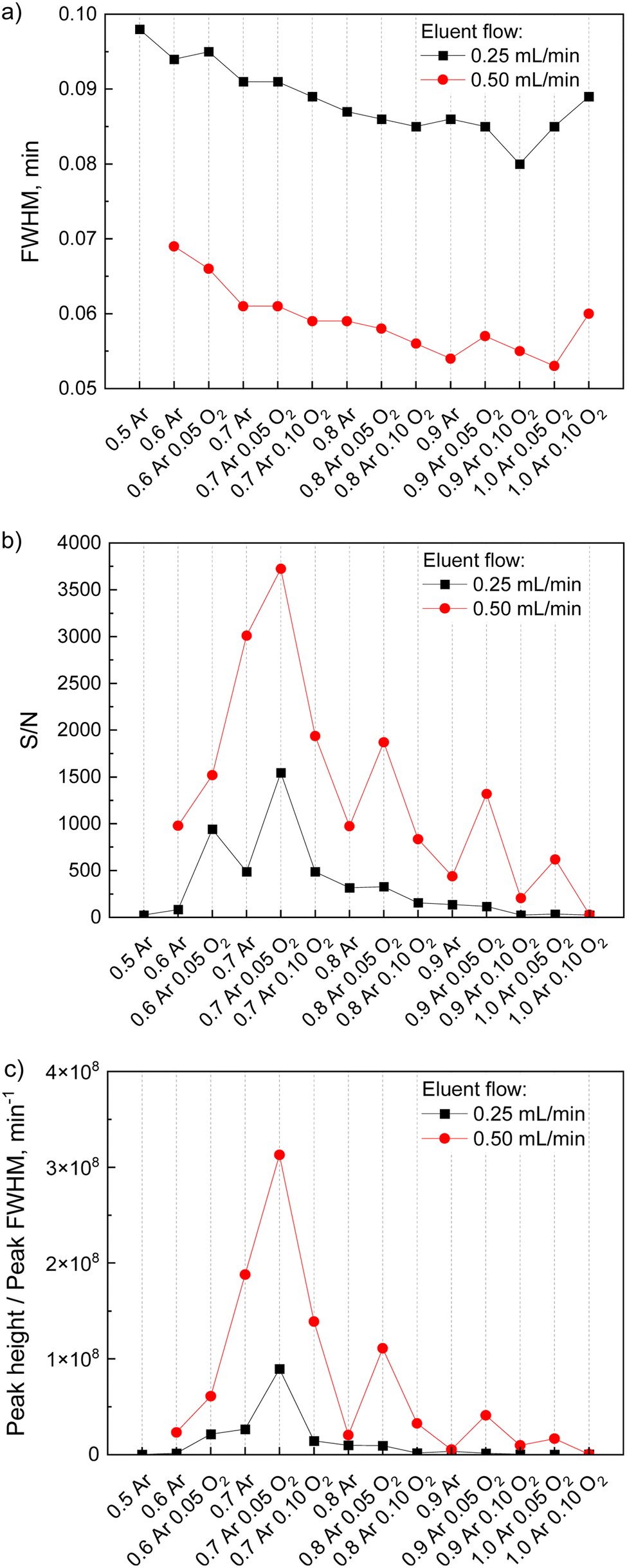 | ||
| Fig. 2 Peak width (a), signal-to-noise ratio (b) and sharpness (c) of the dimethyl sulfide signal collected in the HPLC-ICP-MS experiment for various eluent flows and sample gas flows with 55% methanol in the eluent. No chromatography peak is observed for 0.5 mL min−1 eluent flow with 0.5 L min−1 sample gas flow. | ||
As for different gas flows, a decreasing trend in peak width with Ar sample gas flow can be seen with a small increase for the highest Ar flows (Fig. 2a). Although varying gas settings do not lead to sufficient peak narrowing, other peak characteristics change drastically within the varied range. For instance, an increase in Ar flow from 0.6 to 0.7 L min−1 leads to a 3-fold increase in S/N and an 8-fold increase in peak sharpness, with the maximum observed at 0.7 L min−1 for both characteristics. The sharpness and the S/N could be further improved, when O2 was admixed to the sample gas. For both characteristics, an increase was observed when O2 flow within the sample gas was 0.05 L min−1, while further addition of O2 led to a drop in these parameters.
The variety of eluent flow-gas flow combinations is limited due to plasma instability at some values. For example, the plasma extinguished when oxygen was admixed to small Ar sample gas flows (0.5 L min−1 and lower) as high fractions of O2 destabilize the plasma. In contrast, a stable plasma flame at high Ar flows was only achieved with admixed O2 as it was needed to oxidize larger amount of organics in the mist.
Based on these evaluations, the final setup of the hyphenation was operated with 0.50 mL min−1 eluent flow (HPLC) and 0.7 L min−1 Ar sample gas flow (ICP), since it resulted in the best peak S/N and sharpness among all O2-free regimes. Although the addition of small amounts of oxygen led to an improved signal shape, it was not implemented further due to possible deviations in the gas flow controller of the mass-spectrometer at low gas flow values. The parameters optimized for signal collection and used for further experiments are presented in Table 2.
| Instrument | ELEMENT XR™ |
| Operation power | 1500 W (with shielded torch) |
| Cooling gas flow rate | 16.0 L min−1 |
| Auxiliary gas flow rate | 1.2 L min−1 |
| Sample gas | 0.70 L min−1 |
| Additional gas flow | 0.00 L min−1 |
| Sampling/skimmer cones | Platinum |
| Sample chamber | GE Twinnabar cyclonic (20 mL) |
| Sample chamber temperature | Room temperature |
| Torch | Quartz, 1.0 mm i.d. |
| Nebulizer | PFA-LC |
| Drain pump speed | 15 rpm |
| Detected elements | 32S and 7Li at medium resolution |
The optimized setup settings allow the separation of mixtures of dimethyl polysulfides (n = 1–9) with maximized intensity and signal-to-noise ratios. However, as derivatized mixtures of dimethyl sulfides contain large quantities of salt (lithium triflate, LiOTf), which elutes first, it can give an intense signal in the chromatogram and overlap following peaks. Therefore, the next adjustment of the separation was to optimize cut-off time of the flow diversion. Fig. 3 demonstrates the ICP-MS signal of a derivatized mixture with a nominal composition of Li2S3.8. As expected from the data in Fig. 1, 30 s cut-off time was sufficient to let all the charged non-retained compounds move to the waste and all organic species move to the ICP-MS part without losing their signals (which is demonstrated by a cut-off time value of 35 s). Moreover, for the retention time values smaller than 30 s, one can clearly see the overlap of the Me2S signal with that of the triflate anion.
 | ||
| Fig. 3 HPLC-ICP-MS chromatograms (32S) normalized to the signal of Me2S3 of a derivatized mixture with a nominal composition Li2S3.8 with a cut of the signal of LiOTf at 20, 30, and 35 s after injection as well as the chromatogram with no diverted flow (black solid line). | ||
Several mixtures with various S/Li ratios were prepared as described in Section 2.1. To achieve higher solubility of lithium polysulfides, highly polar organic compounds such as ε-caprolactam and acetamide were used in the solvent. The “eutectic” solvent with a similar composition was applied in the literature23 to prepare Li2Sn solutions with n in the range of 1–8; however in this work only 1,3-dioxolane was applied as the main solvent to dissolve highly polar ε-caprolactam and acetamide. Despite that using highly polar compounds increased the solvation of lithium cations and polysulfide anions and thus increased the solubility of lithium polysulfide, solutions of lithium polysulfides with a lower sulfur content were not investigated in this work. Fig. 4a shows chromatograms of the derivatized mixtures with nominal compositions from Li2S4 to Li2S8, as well as the chromatograms of some commercially available standards. The retention time of each derivatized species followed a logarithmic dependence on chain length (Fig. S4†) according to the literature.11 This made it possible to implicitly confirm the presence of Me2Sn with n = 4–8 as certified standards for these compounds are commercially unavailable. Although Kamyshny et al.11 demonstrated the possibility of detecting dimethyl nonasulfide, n > 9 was neglected since the retention time especially in an isocratic RP HPLC under the conditions used in this work would exceed the one hour mark (∼80 min).
 | ||
| Fig. 4 (a) HPLC-ICP-MS chromatograms (32S) of different Li2Sn mixtures after derivatization and comparison with the chromatograms of commercial standards: anionic, Me2S, Me2S2, Me2S3 and S8. Numbers (1–8) on the chromatogram of derivatized Li2S8.1 symbolize the corresponding chain length of dimethyl polysulfides (for clarity they are omitted for the rest of the chromatograms); (b) calculated mass fractions of each polysulfide species in the Li2Sn mixtures; (c) calculated molar fractions of each polysulfide species in the Li2Sn mixtures. For better visualization, the values in (a)–(c) are shown on the logarithmic scale. | ||
The chromatograms of the derivatized mixtures show the presence of polysulfide species of all chain lengths. However, peak areas for different polysulfides change when the nominal composition changes from Li2S4 to Li2S8. When converting peak areas to the mass/molar content of polysulfides in a mixture (Fig. 4b and c), trends for the fraction of each polysulfide over the nominal composition of lithium polysulfide solution can be observed. As anticipated, the mixtures with high S content show higher percentages of long chain polysulfides, S72− and S82−, and molecular sulfur S8. Surprisingly, for Li2S6 and Li2S8 mixtures the content of short polysulfides (S2− and S22−) is also higher than that for Li2S4 and Li2S5 mixtures. For example, the mass fraction of S2− amounts to 9% and 17% for Li2S6 and Li2S8 mixtures, respectively. The most abundant species of the considered polysulfide distributions was S42−; regardless of the S/Li ratio its mass content amounts to 32% for Li2S8 and 52% for Li2S4. It is worth noting that for this anion as well as for S32− the mass content decreases when the S/Li ratio increases. The data obtained are in good accordance with the literature: as it was derived from the standard Gibbs free energy of formation ΔfG°, the most stable species are the ones from tetra- to hexasulfide.11
The described species were not the only ones present in the chromatograms of derivatized samples. Unknown compound X1 with a retention time of 0.74 min might be the CH3S3˙ radical that was formed after the derivatization of the radical S3˙− which is present in polysulfide mixtures at high temperatures or at low concentrations.24 The repeated measurements of the same derivatized samples after a certain amount of time showed a decrease in the area of the mentioned chromatography peak over time (e.g., see Fig. 3, for which the chromatograms were recorded only 1 week after the derivatization) that may indirectly indicate the recombination of these radicals. Another possible assignment for this peak could be a compound formed during interaction of the solvent components with polysulfide anions. Anyway, further investigations are needed to clarify this question.
Additional metrics were determined based on the calibration with LiTFSI as a standard. The limit of detection was 1.5 ppm and the limit of quantification was 4.5 ppm evaluated with the 3-sigma and 10-sigma criterion, respectively. The limit of detection is notably higher than the value determined by Bluemlein25 with a similar HPLC-ICP-MS method (4.9 ppb). This deviation can be explained by the fact that the authors used 100%-water eluent, which again confirms strong dependency of the LC-ICP-MS signal response on the eluent composition (Fig. 1b). Nevertheless, the value determined in our work is lower than the one reported by Kamyshny11 in 90% methanol–water eluent with HPLC-UV/Vis (6 ppm for S32−).
4. Conclusion
A facile and straightforward quantification approach was developed for the separation and quantification of polysulfide anions and molecular sulfur in lithium polysulfide solutions in organic solvents, which is based on the coupling of HPLC with SF-ICP-MS. Since the separation was conducted in isocratic mode without counter-gradients or adjustment of gas end eluent parameters only, thermal control of the sample introduction system was not required as is usually the case in HPLC-ICP-MS studies. Moreover, no usage of oxygen was needed to maintain the stability of plasma.The optimization of parameters allowed the complete separation of derivatized polysulfide species and molecular sulfur to be achieved with the best performing ICP-MS detection. Despite quite high limits of detection and low time resolution, it was shown that adjusting gas and eluent flow parameters allows the signal-to-noise ratio to be enriched and a high peak sharpness to be reached.
The results of HPLC-ICP-MS analysis demonstrated that tetrasulfide was the most abundant species in Li2Sn mixtures for n = 4–8 with a mass fraction of 32–52% (in relation to all sulfur species). Also, there is a clear trend for increasing mass fraction of long chain polysulfides (S72− and S82−) and molecular sulfur S8 in the mixtures with high S content, with the same tendency being observed for the shortest polysulfide anions (S2− and S22−). Being a powerful tool for the quantification of polysulfide distributions the proposed method may shed brighter light on the electrochemical processes occurring during operation of Li–S batteries.
Author contributions
Aleksei Sadykov: conceptualization, methodology, investigation, validation, formal analysis, visualization, writing – original draft. Yannick P. Stenzel: methodology, validation, writing – review and editing. Martin Winter: supervision, writing – review & editing. Simon Wiemers-Meyer: supervision, writing – review & editing. Sascha Nowak: supervision, conceptualization, writing – review & editing.Conflicts of interest
The authors have no conflict of interest.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
The authors would like to acknowledge the Ministry for Culture and Science of North Rhine Westphalia for funding this work within the International Graduate School for Battery Chemistry, Characterization, Analysis, Recycling, and Application (BACCARA).References
- C. W. Scheele, Chemische Abhandlung von der Luft und dem Feuer, S. L. Crusius, Upsala und Leipzig, 1777 Search PubMed.
- W. Giggenbach, On the nature of the blue solutions of sulfur, J. Inorg. Nucl. Chem., 1968, 30, 3189–3201 CrossRef CAS.
- W. F. Giggenbach, Equilibriums involving polysulfide ions in aqueous sulfide solutions up to 240 deg, Inorg. Chem., 1974, 13, 1724–1730 CrossRef CAS.
- W. Ren, W. Ma, S. Zhang and B. Tang, Recent advances in shuttle effect inhibition for lithium sulfur batteries, Energy Storage Mater., 2019, 23, 707–732 CrossRef.
- Y.-S. Su, Y. Fu, T. Cochell and A. Manthiram, A strategic approach to recharging lithium-sulphur batteries for long cycle life, Nat. Commun., 2013, 4, 2985 CrossRef PubMed.
- S. Waluś, C. Barchasz, R. Bouchet, J.-C. Leprêtre, J.-F. Colin, J.-F. Martin, E. Elkaïm, C. Baehtz and F. Alloin, Lithium/Sulfur Batteries Upon Cycling: Structural Modifications and Species Quantification by In Situ and Operando X-Ray Diffraction Spectroscopy, Adv. Energy Mater., 2015, 5, 1500165 CrossRef.
- A. Kawase, S. Shirai, Y. Yamoto, R. Arakawa and T. Takata, Electrochemical reactions of lithium-sulfur batteries: an analytical study using the organic conversion technique, Phys. Chem. Chem. Phys., 2014, 16, 9344–9350 RSC.
- J. F. Hinton, Sulphur-33 NMR Spectroscopy, Annu. Rep. NMR Spectrosc., 1987, 19, 1–34 CrossRef CAS.
- M. U. M. Patel and R. Dominko, Application of in operando UV/Vis spectroscopy in lithium-sulfur batteries, ChemSusChem, 2014, 7, 2167–2175 CrossRef CAS PubMed.
- Q. He, Y. Gorlin, M. U. M. Patel, H. A. Gasteiger and Y.-C. Lu, Unraveling the Correlation between Solvent Properties and Sulfur Redox Behavior in Lithium-Sulfur Batteries, J. Electrochem. Soc., 2018, 165, A4027–A4033 CrossRef CAS.
- A. Kamyshny, A. Goifman, J. Gun, D. Rizkov and O. Lev, Equilibrium distribution of polysulfide ions in aqueous solutions at 25 degrees C: a new approach for the study of polysulfides' equilibria, Environ. Sci. Technol., 2004, 38, 6633–6644 CrossRef CAS PubMed.
- D. Zheng, D. Qu, X.-Q. Yang, X. Yu, H.-S. Lee and D. Qu, Quantitative and Qualitative Determination of Polysulfide Species in the Electrolyte of a Lithium–Sulfur Battery using HPLC ESI/MS with One-Step Derivatization, Adv. Energy Mater., 2015, 5, 1401888 CrossRef.
- S.-Y. Kim, R. Jang, J. Hyun, S. Kim, Y. C. Choi, S.-H. Park and Y. Y. Youn, Qualitative Determination of Polysulfide Species in a Lithium-Sulfur Battery by HR-LC-APCI-MSn with One-Step Derivatization, J. Am. Soc. Mass Spectrom., 2022, 33, 1653–1658 CrossRef CAS PubMed.
- J. Giner Martínez-Sierra, O. Galilea San Blas, J. M. Marchante Gayón and J. I. García Alonso, Sulfur analysis by inductively coupled plasma-mass spectrometry: A review, Spectrochim. Acta B Atom Spectrosc., 2015, 108, 35–52 CrossRef.
- J. Goossens, F. Vanhaecke, L. Moens and R. Dams, Elimination of interferences in the determination of arsenic and selenium in biological samples by inductively coupled plasma mass spectrometry, Anal. Chim. Acta, 1993, 280, 137–143 CrossRef CAS.
- S. Liu and D. Beauchemin, Effect of methanol and sodium dodecylsulfate on radial profiles of ion abundance in inductively coupled plasma mass spectrometry, Spectrochim. Acta B Atom Spectrosc., 2006, 61, 319–325 CrossRef.
- K. de Wolf, L. Balcaen, E. van de Walle, F. Cuyckens and F. Vanhaecke, A comparison between HPLC-dynamic reaction cell-ICP-MS and HPLC-sector field-ICP-MS for the detection of glutathione-trapped reactive drug metabolites using clozapine as a model compound, J. Anal. At. Spectrom., 2010, 25, 419 RSC.
- O. J. Lechtenfeld, B. P. Koch, W. Geibert, K.-U. Ludwichowski and G. Kattner, Inorganics in organics: quantification of organic phosphorus and sulfur and trace element speciation in natural organic matter using HPLC-ICPMS, Anal. Chem., 2011, 83, 8968–8974 CrossRef CAS PubMed.
- K. Bluemlein, E. M. Krupp and J. Feldmann, Advantages and limitations of a desolvation system coupled online to HPLC-ICPqMS/ES-MS for the quantitative determination of sulfur and arsenic in arseno-peptide complexes, J. Anal. At. Spectrom., 2009, 24, 108–113 RSC.
- U. Košir, I. Kralj Cigić, J. Markelj, S. Drvarič Talian and R. Dominko, Polysulfide species in various electrolytes of Li-S batteries – a chromatographic investigation, Electrochim. Acta, 2020, 363, 137227 CrossRef.
- B. Klencsár, L. Balcaen, F. Cuyckens, F. Lynen and F. Vanhaecke, Development and validation of a novel quantification approach for gradient elution reversed phase high-performance liquid chromatography coupled to tandem ICP-mass spectrometry (RP-HPLC-ICP-MS/MS) and its application to diclofenac and its related compounds, Anal. Chim. Acta, 2017, 974, 43–53 CrossRef PubMed.
- M. Bernardin, F. Bessueille-Barbier, A. Le Masle, C.-P. Lienemann and S. Heinisch, Suitable interface for coupling liquid chromatography to inductively coupled plasma-mass spectrometry for the analysis of organic matrices. 2 Comparison of Sample Introduction Systems, J. Chromatogr. A, 2019, 1603, 380–387 CrossRef CAS PubMed.
- Q. Cheng, W. Xu, S. Qin, S. Das, T. Jin, A. Li, A. C. Li, B. Qie, P. Yao, H. Zhai, C. Shi, X. Yong and Y. Yang, Full Dissolution of the Whole Lithium Sulfide Family (Li2 S8 to Li2 S) in a Safe Eutectic Solvent for Rechargeable Lithium-Sulfur Batteries, Angew. Chem., Int. Ed., 2019, 58, 5557–5561 CrossRef CAS PubMed.
- R. Steudel, in Elemental Sulfur und Sulfur-Rich Compounds II, ed. R. Steudel, Springer, Berlin, Heidelberg, 2003, pp. 127–152 Search PubMed.
- K. Bluemlein, A. Raab, A. A. Meharg, J. M. Charnock and J. Feldmann, Can we trust mass spectrometry for determination of arsenic peptides in plants: comparison of LC-ICP-MS and LC-ES-MS/ICP-MS with XANES/EXAFS in analysis of Thunbergia alata, Anal. Bioanal. Chem., 2008, 390, 1739–1751 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00231h |
| This journal is © The Royal Society of Chemistry 2024 |