
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-precision low-level Cd isotopic analysis using MC-ICP-MS and its application to marine samples from Terra Nova Bay (Antarctica)†
Maria Alessia
Vecchio
 ab,
Lana
Abou-Zeid
b,
Marco
Grotti
a and
Frank
Vanhaecke
*b
ab,
Lana
Abou-Zeid
b,
Marco
Grotti
a and
Frank
Vanhaecke
*b
aDepartment of Chemistry and Industrial Chemistry, University of Genoa, Via Dodecaneso 31, 16146 Genoa, Italy
bAtomic & Mass Spectrometry – A&MS Research Group, Department of Chemistry, Ghent University, Campus Sterre, Krijgslaan 281-S12, 9000 Ghent, Belgium. E-mail: frank.vanhaecke@ugent.be
First published on 12th September 2024
Abstract
This study presents the development, validation and use of an approach for precise and accurate cadmium (Cd) isotopic analysis at low concentration levels using multi-collector inductively coupled plasma-mass spectrometry (MC-ICP-MS). The MC-ICP-MS unit used was equipped with a standard sample introduction system, thus using an ICP operated under wet plasma conditions, and Faraday cup amplifiers equipped with a 1013 Ω resistor. One-column anion exchange chromatography allowed isolation of Cd with 80–99% recovery and absence of an effect of potential on-column fractionation on the isotope ratio results was demonstrated. Use of both an internal (using Ag) and an external (measured in a sample-standard bracketing sequence) standard was relied on for correction of the bias introduced by instrumental mass discrimination. A long-term precision of 0.09‰ (2SD) for δ114/110Cd was achieved at a Cd concentration of 10 ng mL−1. The method developed was validated by analysing NIST SRM 2711a (Montana soil) and NRC TORT-3 (Lobster hepatopancreas) reference materials, yielding results consistent with literature values. Subsequently, the method was applied to two Antarctic marine organisms Adamussium colbecki and Trematomus bernacchii, collected during both the 1990s and 2020s, to investigate its potential for identifying changes in the biogeochemical cycle of Cd over time and reveal natural or anthropogenic sources. A preferential uptake of the lighter Cd isotopes in both species was observed, indicated by negative δ114/110Cd values ranging between −0.24 and −0.09‰. This finding is consistent with previous studies that have reported Cd fractionation during its uptake by marine organisms, with a preference for the lighter isotopes. No significant differences in δ114/110Cd values were observed between organs of the same species or between the same species collected in the different decades, suggesting minimal Cd isotope fractionation during internal transfer and consistent Cd sources over time. Comparison with literature data suggests that the Cd source in Antarctic biota may be predominantly of natural origin, with δ114/110Cd values indicating isotopically heavier Cd than that found in Cd-polluted areas. However, further Cd isotope ratio data from various Antarctic sample types are necessary to further evaluate the Cd sources in marine samples.
1 Introduction
Cadmium (Cd) is a toxic element found naturally in the Earth's crust at trace concentration levels (∼0.1 μg g−1), with higher values in Zn, Pb and Cu ores.1 Its concentration in the environment is increased due to anthropogenic activities such as ore mining,2 smelting/refining,3,4 industrial production of nickel–cadmium batteries,5 waste incineration, use of phosphate fertilisers and coal combustion.6Cadmium can be associated with atmospheric aerosol and transported over long distances, eventually reaching the Antarctic continent.7 Next to the deposition of these wind-transported soil particles via wet processes,8 also natural processes, including submarine volcanic activity9 and direct emissions from sea ice,10 can contribute to the occurrence of Cd in the Antarctic marine environment.
Biota of Antarctic marine ecosystems, such as the bivalve Adamussium colbecki (Antarctic scallop) and the fish Trematomus bernacchii (emerald rockcod), are used as proxies for studying the biogeochemistry of Cd.11 These organisms, living in the Southern Ocean, can accumulate much higher levels of Cd compared to their counterparts in regions more influenced by human activities. This occurs because Cd acts as a substitute for the essential element Zn, for which the input from the icy continent is low.12 Therefore, bioaccumulation and biomagnification of Cd in the Southern Ocean occur as a natural phenomenon.13 However, relatively high Cd levels between 100 to 200 μg g−1 have been reported in the digestive gland of the bivalve mollusc Adamussium colbecki, raising the question whether all of this Cd is indeed from natural origin.12,14,15 Confirming that such high Cd accumulation is solely due to natural processes is challenging when monitoring Cd concentrations only, highlighting the need for deeper and complementary information to reveal the sources of Cd in Antarctic environments.3
Cd possesses eight stable isotopes, covering a wide mass range from 106 to 116 amu. As with all other elements with ≥2 isotopes, it shows natural variation in its isotopic composition owing to mass-dependent isotope fractionation, as a result of its different isotopes participating in physical processes and biogeochemical reactions to slightly different extents.16 Examining variation in Cd isotopic composition showed to be a powerful tool to investigate the biogeochemical cycle of this element and to reveal the sources of anthropogenic Cd pollution in the environment.3,17,18
Research into stable isotope fractionation of Cd dates back to the 1970s with pioneering work carried out by Rosman and collaborators,19–21 revealing pronounced variations from 0.6‰ to 1.5‰ per amu in extraterrestrial materials (meteorites).20,21 Subsequently, Cd isotope fractionation was also evidenced in terrestrial materials, such as seawater, geological and environmental samples, by several authors.22 For instance, Lacan et al.22 found that phytoplankton at the surface of seawater preferentially absorbs the lighter Cd isotopes, making surface seawater isotopically heavy. In contrast, decomposition and release of sinking Cd-containing particulates govern the shape of Cd isotopic profiles in the deeper ocean.23 Moreover, the water mass circulation plays an important role in the redistribution of Cd and its isotopes, while deep seawater remains lighter due to mixing and nutrient regeneration.22–25 Birch leaves showed δ114/110Cd variation up to 1‰ during root uptake and translocation.26 Cd isotopic analysis was also relied on to reveal and distinguish between natural and anthropogenic sources of Cd in the environment. Some authors demonstrated that industrial activities may affect the Cd isotopic composition. Cd deposited around a Pb–Zn refinery was shown to be isotopically lighter than the corresponding slag.3 Others observed significant Cd isotope fractionation between the final products and the raw materials.27 The δ114/110Cd values ranged from −2.3‰ in Pb refinement waste to +5.86‰ in coal combustion slag, highlighting the method's usefulness in tracking Cd pollution. Shiel et al. used this approach to identify the sources of Cd in bivalves from different regions, revealing varying natural and anthropogenic contributions in Atlantic and Mediterranean bivalves.5,17,28
Multi-collector inductively coupled plasma-mass spectrometry (MC-ICP-MS) is the technique of choice for measuring Cd isotope ratios as it offers high precision owing to the simultaneous monitoring of the ion beams of interest, enabling very small differences in the isotopic composition of the target element to be revealed and quantified.29–31 However, for also assuring accuracy, (i) the bias introduced by instrumental mass discrimination needs to be adequately corrected for and (ii) potential spectral and non-spectral interferences need to be addressed.
For Cd, the instrumental mass bias is typically corrected for by means of (i) external correction with the standard measured in a sample-standard bracketing (SSB) approach,26,32 (ii) external correction (SSB) combined with the use of Ag as an internal standard (IS) in a double correction approach or (iii) a double-spike approach.24,25,31,33–38 Spectral interferences affecting the isotopic analysis may be caused by the occurrence of elements with isobaric nuclides, such as Pd, Sn and In, and polyatomic interferences such as argide and oxide ions of elements such as Mo, Zn and Ge. Non-spectral interferences caused by the co-presence of matrix elements can introduce errors in the mass bias correction. To overcome spectral and non-spectral interferences, Cd is typically isolated from matrix elements through chemical purification usually achieved by single- or multiple-stage ion exchange chromatography, with the use of AG-MP-1M, AG1-X8 and/or Eichrom TRU Spec resins.4,23,26,29,31,34,36,37,39–45
Precise and accurate isotopic analysis of Cd in Antarctic biota samples is challenging due to the low Cd concentration present in such samples, leading to Cd concentration in the solutions finally analysed typically in the 20–300 ng mL−1 range.46 In addition, many studies report on-column Cd fractionation during the chromatographic isolation, especially when low Cd amounts are loaded into the column. This artificial fractionation is typically corrected through the use of a double-spike approach.23,31,34,38,40,41,45,47 The latter, despite being the most reliable mass bias correction approach, is labour-intensive and time-consuming. As a result, we aimed for a more straightforward approach that allows for higher sample throughput and faster processing, while still displaying fit-for-purpose performance at the low Cd concentrations encountered.
For MC-ICP-MS isotopic analysis at low concentration, an aerosol desolvation unit such as an Aridus II (Teledyne Cetac Technologies, USA) is typically used to introduce Cd into the ICP. The dry plasma conditions thus obtained offer a signal enhancement and therefore, theoretically also an improved isotope ratio precision (cf. counting statistics). In addition, the use of such aerosol desolvation unit also reduces the level of oxide formation, which is advantageous for minimising the signal intensities of potentially present MoO+ ions that can overlap with the signals of Cd+ isotopes, especially when relatively high Mo/Cd post-isolation ratios persist in the purified Cd fractions.26 However, notwithstanding these advantages, Pallavicini and colleagues reported that this introduction system suffers from memory effects and poor signal stability, which can potentially jeopardise the precision of the Cd isotope ratio measurements.26 While the standard sample introduction system (wet plasma) yields more stable Cd+ signal intensities and therefore a better precision compared to that obtained when using an Aridus II introduction system, the corresponding application range is limited to relatively high Cd concentrations, due to the low analyte introduction efficiency. As a result, the minimum Cd concentration that was measured with the standard sample introduction system was reported to be 100 ng mL−1.48,49
Recently, Faraday cup amplifiers equipped with a 1013 Ω resistor became commercially available and were demonstrated advantageous for high-precision MC-ICP-MS isotopic analysis of several elements at low concentration levels.50 To the best of the authors' knowledge, the combined use of a standard introduction system and Faraday cup amplifiers equipped with a 1013 Ω resistor for high-precision Cd isotopic analysis at low concentration levels has not been described yet in the literature.
The aim of this work was to develop a method for accurate and precise Cd isotopic analysis at low Cd concentration levels using MC-ICP-MS equipped with the standard sample introduction system (wet plasma conditions) and Faraday cup amplifiers with a 1013 Ω resistor. Once developed, the method was used to measure Cd isotope ratios in Adamussium colbecki bivalve and Trematomus bernacchii fish samples collected in the 1990s and 2020s in Terra Nova Bay – Antarctica, with the aim of obtaining information on the natural/anthropogenic origin of Cd and on potential differences in its biogeochemical cycle in this environment over the years.
2 Experimental
2.1. Chemical and reagents
Ultrapure water (18.2 MΩ cm at 25 °C) was obtained from a Milli-Q purification system (Millipore, France). Pro analysis grade 12 M HCl and 14 M HNO3 were supplied by Fisher Chemicals (UK) and further subjected to a sub-boiling distillation in a Savillex DST purification system (Savillex, USA). Suprapure grade hydrogen peroxide 9.8 M H2O2, used for sample digestion in addition to HNO3, was purchased from Sigma Aldrich (Belgium).Standard solutions of Cd (NIST SRM 3108) and Ag, used for external and internal mass bias correction were purchased from the National Institute of Standards and Technology (NIST, USA) and ChemLab (Belgium), respectively.
The certified reference materials (CRMs)31,48,51,52 Montana Soil (NIST SRM 2711a) and Lobster hepatopancreas (NRC TORT-3), previously characterized for their Cd isotopic composition were used for method validation and were purchased from the National Institute of Standards and Technology (NIST, USA) and the National Research Council of Canada (NRC), respectively. An “in-house” Cd standard solution from ChemLab, Belgium (Batch number 31.4372108) was characterized for its Cd isotopic composition and was measured after every 6 samples in each sequence for quality control purposes.
2.2. Antarctic samples
Two marine species, the bivalve mollusc Adamussium colbecki and the fish Trematomus bernacchii, were selected and collected at Terra Nova Bay (Ross Sea—Northern Victoria Land), during the XI, XIII and XXXVII Italian Antarctic Expeditions and stored at −80 °C until analysis. The organisms were dissected, separating the soft tissue from the shell for the bivalve and collecting liver, spleen and gonads from the fish. All samples were weighed and subsequently lyophilized. Organisms were pooled and further stored in the Antarctic Environmental Specimen Bank.532.3. Sample digestion
Three replicates of 250 mg of lyophilized and homogenized (agate mortar) sample, from pooled individuals, were subjected to acid digestion with 2 mL of 14 M HNO3 and 0.5 mL of 9.8 M H2O2, using a MARS-5 microwave digestion system (CEM, USA). Following the mineralization, samples were diluted to 10 mL with ultrapure water. The selected certified reference materials (NIST SRM 2711a, NRC TORT-3) were digested following the same procedure as the samples. Finally, digests of samples and reference materials were evaporated to dryness in 15 mL Teflon beakers heated to 110 °C on a ceramic-top hot plate and the residues thus obtained were redissolved in 3 mL of 2 M HCl for subsequent chromatographic Cd isolation.2.4. Chromatographic isolation of Cd
All sample purification steps were conducted in a class-10 clean lab (PicoTrace, Germany) at Ghent University. All Savillex™ PFA beakers used in this work were previously kept soaked in 7 M HNO3 for 2 × 24 h and subsequently in 6 M HCl at 110 °C for 2 × 24 h. The chromatographic isolation of Cd was carried out using 2 mL of AG®MP-1M anion exchange resin from Bio-rad, Belgium (100–200 μm dry mesh size, chloride anionic form). The resin was pre-cleaned in bulk 3 times using 7 M HNO3 and 3 times using ultrapure water, then loaded into 2 mL polypropylene columns (Eichrom, France). The columns were connected to a polycarbonate vacuum box (24 positions, Triskem, France) attached to a chemically resistant diaphragm pump purchased from Fisher Scientific (UK). The elution flow rate under vacuum conditions was 0.07 mL s−1.The isolation protocol selected for separating Cd from the matrix elements was originally proposed by Cloquet et al.,39 and modifications were suggested by Li et al.48 The protocol is summarised in Table 1.
| Step | Volume (mL) | HCl conc (M) | Element(s) eluted |
|---|---|---|---|
| Washing and conditioning | 20 | 2 | — |
| Sample load | 2 | 2 | — |
| Elution matrix 1 | 10 | 2 | Mg, Ca, Fe, Ni, Ga, Ge, Zr, Ag, Mo, In, Pd etc. |
| Elution matrix 2 | 20 | 0.3 | Pb, Mo |
| Elution matrix 3 | 20 | 0.06 | Zn, Sn |
| Elution matrix 4 | 10 | 0.012 | Residual Sn |
| Cd elution | 20 | 0.0012 | Cd |
The amount of Cd loaded into the column ranged from 20 to 100 ng. The collected pure Cd fractions were evaporated to dryness at 110 °C and then redissolved in 2 mL of 2% HNO3 for subsequent elemental and isotopic analysis.
2.5. Instrumentation
| a O2 mode was used to avoid interference from potentially present isobaric MO+ ions. | |
|---|---|
| Plasma parameters | |
| RF power | 1550 W |
| Plasma gas flow rate | 15 L min−1 |
| Auxiliary gas flow rate | 0.90 L min−1 |
| Nebulizer gas flow rate | 1.12 L min−1 |
| Sample uptake rate | 350 μL min−1 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Mass/charge ratios monitored (Q1→Q2) | |
| No gas (on mass) | 90→90Zr+, 95→95Mo+, 105→105Pd+, 107→107Ag+, 111→111Cd+, 115→115In+, 208→208Pb+, 103→103Rh+ (IS) |
| NH3/He (mass shift) | 23Na→57Na(NH3)2+, 40Ca→57, Ca(NH3)+, 56Fe→90Fe(NH3)2+, 63Cu→97Cu(NH3)2+, 64Zn→98Zn(NH3)2+, 74Ge→90Ge(NH2)+, 103→103Rh+ (IS) |
| O2 (on mass)a | 103→103Rh+ (IS), 120→120Sn+ |
|
|
| Gas flow rates | |
| He | Collision gas, 1 mL min−1 |
| NH3/He (10%/90%) | Reaction gas, 3 mL min−1 |
| O2 (30%) | 0.45 mL min−1 |
|
|
| Acquisition parameters | |
| Q2 peak pattern | 1 point per peak |
| Integration time | 1 s |
| Replicates | 10 |
| Sweeps/replicate | 100 |
| Neptune XT settings | |
| Sample uptake rate | 100 μL min−1 |
| Plasma gas flow rate | 15 L min−1 |
| Auxiliary gas flow rate | 0.8 L min−1 |
| Nebulizer gas flow rate | 1.015–1.050 L min−1 |
| RF power | 1200 W |
| Sampling cone | Ni; Jet-type; 1.1 mm Ø orifice |
| Skimmer cone | Ni; X-type; 0.8 mm Ø orifice |
| Resolution mode | Low |
|
|
| Data acquisition parameters | |
| Scan type | Static, multi-collection |
| Number of blocks | 1 |
| Number of cycles/block | 50 |
| Integration time | 4.194 s |
| Number of integrations | 1 |
| Idle time | 3.000 s |
| Cup configuration | |||||||
|---|---|---|---|---|---|---|---|
| L4 | L3 | L2 | L1 | C | H1 | H2 | H4 |
| 1011 Ω | 1011 Ω | 1011 Ω | 1013 Ω | 1013 Ω | 1013 Ω | 1013 Ω | 1011 Ω |
| 107Ag | 108Cd | 109Ag | 110Cd | 111Cd | 112Cd | 114Cd | 117Sn |
| 108Pd | 112Sn | 114Sn | |||||
Cd isotope ratios were reported in delta notation, as per mil deviation (‰), relative to the NIST SRM 3108 standard:
 | (1) |
Instrumental mass bias was corrected for via a double correction approach based on the use of Ag as an internal standard using the protocol described by Baxter, combined with external correction using a standard measured in a SSB sequence.54
A blank solution of 2% HNO3 was introduced in-between every sample/standard measurement to rinse the sample introduction system and control for possible memory effects. The signal intensity for the blanks was typically lower than 0.2 fA (0.002 V) for 110Cd, 111Cd 112Cd and 114Cd and lower than 10 fA (0.001 V) for 117Sn, which corresponds to approximately 0.86 fA at a mass-to-charge ratio of 114. For 10 ng mL−1 of Cd, the signal intensity for 110Cd ranged between 12–15 fA (0.12–0.15 V) and that for 114Cd between 29–32 fA (0.29–0.32 V). Blank correction was applied when the instrumental 117Sn background was ≥10 fA. For each sample, three digests were subjected to chromatographic isolation and the purified Cd fraction was subsequently measured with MC-ICP-MS for its isotopic composition and unless stated otherwise, the mean δ value of the three measurements was reported throughout this work ±2SD (n = 3).
3 Results and discussion
3.1. Chromatographic isolation of Cd from matrix elements
As mentioned previously, the single-column Cd purification procedure (Table 1), originally proposed by Cloquet et al.39 and modified by Li et al.48 was employed. As this protocol requires large volumes of HCl (up to 80 mL),39 the time required for completing the isolation would be excessive using gravity-driven elution, reaching 5–6 hours for the entire procedure.26 Therefore, the selected isolation procedure was accelerated through the use of a vacuum box (Fig. S1†), permitting halving the isolation time compared to that with standard gravity-driven elution to approximately 2.5–3 hours. In order to evaluate the efficiency of the vacuum-assisted isolation protocol for separating Cd from the matrix elements, two relevant reference materials, previously characterized for their Cd isotope ratios, Lobster hepatopancreas (NRC TORT-3)55 and Montana soil (NIST SRM 2711a),31,36,37,45,48,51,56 were digested in the same way as the samples. Then, an amount of sample digest corresponding to 20 ng of Cd was loaded into the column and elution fractions of 1 mL each were collected individually and subsequently measured for elemental concentrations using ICP-MS/MS (see Table 2). The elution profiles obtained for both reference materials are presented in Fig. 1.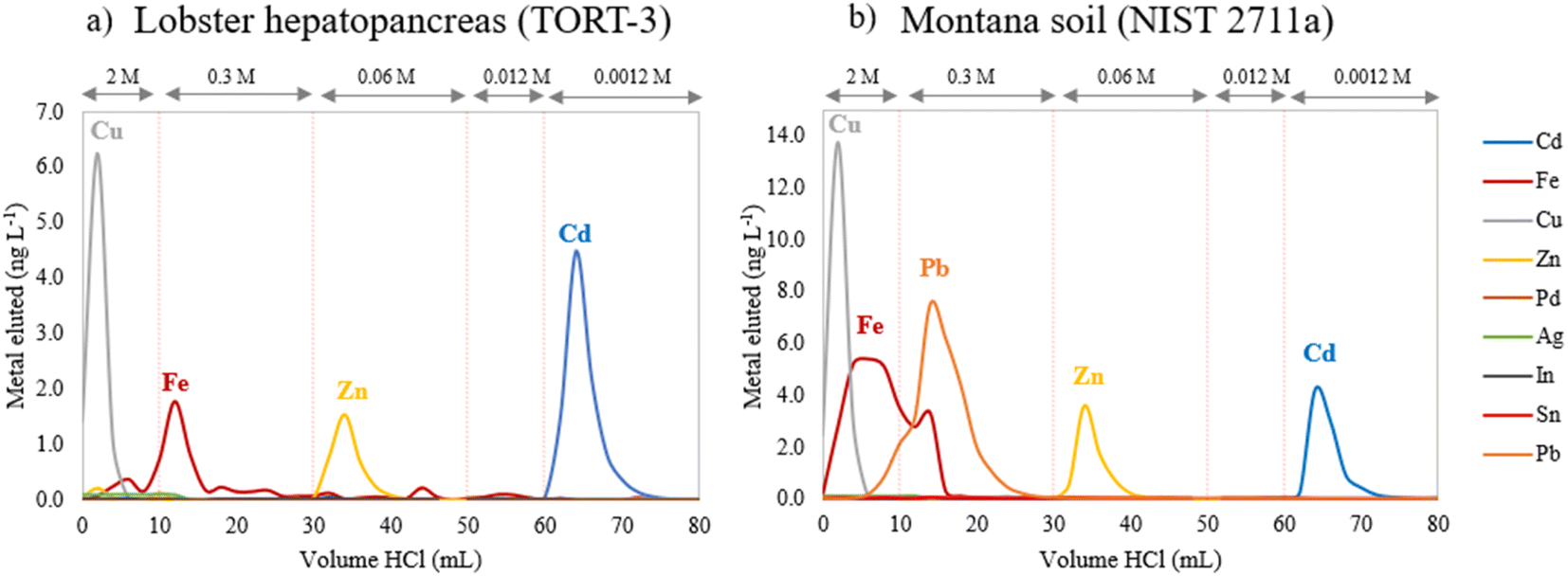 | ||
| Fig. 1 Elution curves for (a) NRC TORT-3 Lobster hepatopancreas and (b) NIST SRM 2711a Montana soil reference materials (the concentrations of Fe, Cu and Zn are divided by 10 for a better visualization). | ||
The elution profile obtained under vacuum conditions is identical to that obtained using gravity-driven elution48 and allowed successful isolation of Cd from the matrix elements using 20 mL of 0.0012 M HCl as reported in other studies.4,26,31,42,43,48,49 Cd recoveries of 97 ± 6% (n = 3) and 92 ± 6% (n = 5) were obtained for NRC TORT-3 and NIST SRM 2711a, respectively, showing (near) quantitative recovery of Cd using this method. Moreover, matrix elements (E) such as Cu, Fe and Zn, present at an E/Cd pre-isolation ratio of 8.75, 3.22, 2.51 for NRC TORT-3 and of 2.06, 329 and 5.2 for NIST SRM 2711a were successfully separated from the Cd fraction, with an E/Cd post-isolation ratio ranging between 0.02 and 0.2. Critical elements such as Sn, Pd, Mo, Ag and In already displayed very low E/Cd pre-isolation ratios for these reference materials, ranging from 0.001 to 0.02, which is also representative for the concentration of these elements in our target samples. The concentrations of the latter elements in the collected Cd fraction were lower than the corresponding detection limits and therefore their levels were considered to be negligible with respect to that of Cd.31 In addition, Zn was eluted with 20 mL of 0.06 M HCl with a total recovery (>99%), meaning that this fraction can be kept for Zn isotopic analysis if required, as was proposed by Pallavicini et al.26
3.2. Accuracy of Cd isotope ratio measurements
The accuracy of Cd MC-ICP-MS measurements can be affected by several parameters such as (i) high procedural blank contribution, (ii) instrumental mass discrimination, (iii) the presence of residual isobaric/polyatomic ions causing spectral interference following chemical purification and (iv) matrix effects. As the measurement of Cd isotope ratios was carried out at lower concentrations than usually reported in the literature, the effect of these parameters on the accuracy of the Cd isotope ratio data was revisited and observations were discussed in comparison to previously published results.To confirm this, the effect of the blank was further evaluated by processing 20 ng of Cd NIST SRM 3108 solution through the column and comparing the δ114/110Cd value obtained for this processed standard to that for an unprocessed solution. The recovery obtained for the processed NIST solution was 88 ± 3% for 3 replicates and was thus not entirely quantitative. However, the δ114/110Cd value of −0.03 ± 0.07‰ (2SD, n = 6) obtained for the processed solution measured at 10 ng mL−1 matches the δ114/110Cd value of the unprocessed solution measured at the same concentration, 0.01 ± 0.09‰ (2SD, n = 9) within the experimental uncertainty. Hence, it was concluded that the contribution of the blank was negligible (<0.15%) and the incomplete (ca. 90%) recovery does not have an impact on the accuracy of the isotope ratio measurements performed at low Cd concentration.
3.2.2.1 Impact of sample – (bracketing) standard concentration mismatch. As the extent of instrumental mass bias is affected by the elemental concentration, samples and the bracketing standard must be matched in concentration to the largest extent possible for accurate mass bias correction. The acceptable mismatch range was evaluated by fixing the concentration of the NIST SRM 3108 bracketing standard at 10 ng mL−1 and measuring the same solution with a concentration mismatch varying from −25 to +15%. Results obtained under those conditions are reported in Fig. 2.
 | ||
| Fig. 2 Effect of concentration mismatch between sample and bracketing standard bracketing used for external correction for the mass bias on the accuracy of the δ114/110Cd measurement result. | ||
As can be seen in Fig. 2, the accuracy seems unaffected when the mismatch between the samples and standard is within ±10%. Beyond this limit, the δ114/110Cd value deviates from the correct value by up to ±0.2‰ in the range investigated. Based on these results, samples and standard concentrations were always matched within ±10% in all further work.
3.2.2.2 Impact of internal standard concentration. Ag was used by several authors for the correction of the mass bias affecting the Cd isotope ratios and different Ag/Cd ratios were used; Shiel et al.,5e.g., used a ratio of 1/2 and Pallavicini et al. of 1/4.26 In this part of the work, we have evaluated the impact of different Ag/Cd ratios on the accuracy of the Cd isotope ratio results corrected for mass bias using the Baxter + SSB approach. For this purpose, the NIST SRM 3018 concentration was fixed at 10 ng mL−1 and the solution was spiked with Ag to final concentrations ranging from 2.5 to 15 ng mL−1, corresponding to an Ag/Cd ratio ranging from 1/4 to 1/0.7. The corrected δ114/110CdNIST SRM 3018 values obtained at each Ag concentration are reported in Fig. 3.
 | ||
| Fig. 3 δ 114/110Cd measurement results for 10 ng mL−1 Cd NIST SRM 3108 solutions spiked with different concentrations of the internal standard Ag. Error bars correspond to 2SD (n = 10). | ||
As illustrated by the data in Fig. 3, the concentration of the internal standard did not have a significant impact on the accuracy of the corrected Cd isotope ratio within the range tested. The same observation was also reported for mass bias correction of Hg isotope ratios using Tl as an internal standard in a combined Baxter + SSB approach.57 As for the rest of the study, the Ag concentration was systematically fixed at 5 ng mL−1, corresponding to an Ag/Cd ratio of 1/2 as this showed to be a good compromise in terms of sensitivity (0.4 V for 107Ag and 109Ag) and internal precision (2SE of 0.04‰ for the 109Ag/107Ag ratio).
The effect of potential residual amounts of the elements mentioned above on the accuracy of Cd isotope ratio measurements was assessed by doping NIST SRM 3108 standard solution with increasing amounts of Zn, Zr, Mo and Ge. Despite being one of the major isobaric interferences affecting measurement of the Cd isotopes, Sn was excluded from this test as doping with increasing concentrations of Sn resulted in higher instrumental Sn backgrounds due to memory effects, affecting the signals of 114Cd and 112Cd, especially when low Cd concentrations were measured. In addition, elemental analysis of the actual samples showed that Sn was initially not present in our samples (Table S1†) and therefore it was not considered a critical element that could potentially jeopardise the accuracy of our results. Other elements with isobaric nuclides were completely separated from Cd during the chromatographic isolation (see paragraph 2.5). The effect of the selected interfering elements (E) present at different E/Cd ratios on the accuracy of the δ114/110Cd measurement result is presented in Fig. 4 when using either external correction (SSB) only or the combined Baxter + SSB correction approach.
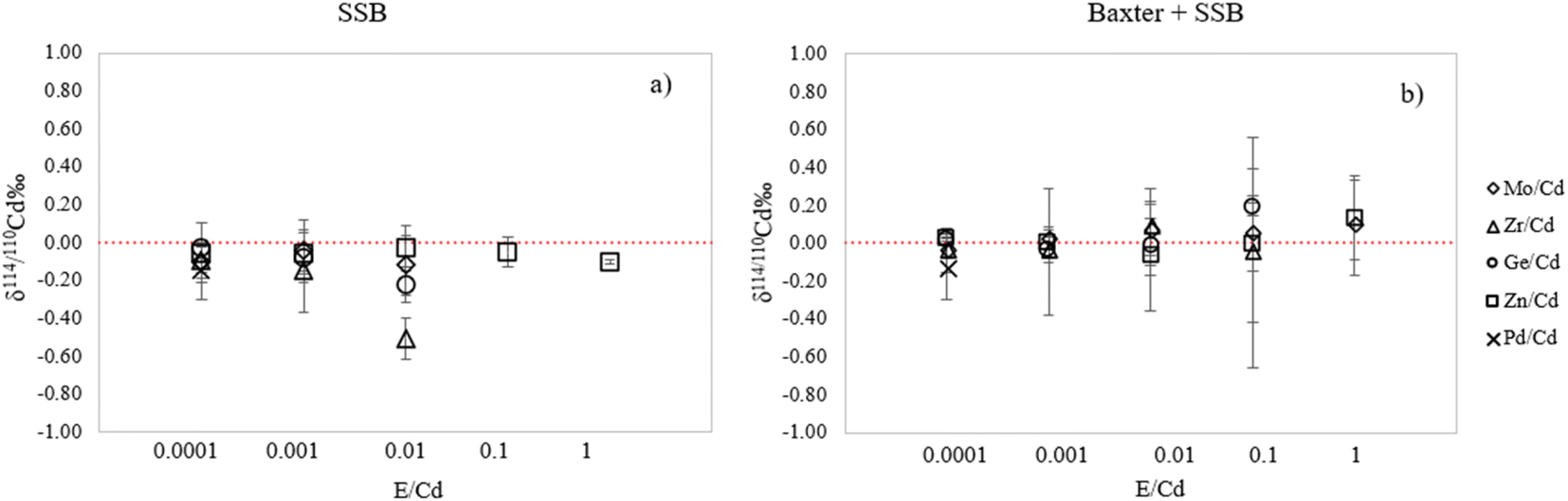 | ||
| Fig. 4 Effect of the presence of different elements on the δ114/110Cd measurement result for 10 ng mL−1 NIST SRM 3108 as a function of the element/Cd ratio, using (a) external and (b) internal + external correction of the mass bias. Error bars correspond to 2SD (n = 3). | ||
As can be observed in Fig. 4, when the Baxter + SSB correction approach is employed, the accuracy is not significantly affected by polyatomic interferences due to Zr, Ge, Mo and Zn when these matrix elements (E) occur at E/Cd ratios < 0.1 for Zr and Ge and <1 for Mo and Zn. This result is in accordance with the literature, where a Zn/Cd ratio < 1 was also reported to be acceptable by Cloquet et al.39 When only external correction is used, the deviation of the measured δ114/110Cd value is already present at Zr/Cd and Ge/Cd ratios ≤ 0.01, thus demonstrating the advantage of using Ag as an internal standard. On the other hand, the presence of trace amounts of isobaric interferences such as Pd can obviously not be corrected for using an internal standard. A deviation of about 0.1‰ was observed when the Pd/Cd ratio is 0.0001 and about 1.35‰ when the Pd/Cd ratio is 0.001, using both the SSB and Baxter + SSB correction approaches. Therefore, total removal of Pd needs to be achieved upon chromatographic isolation to ensure accurate Cd isotope ratio results.
In order to evaluate the effect of such potential contamination, the NIST SRM 3108 standard solution was doped with increasing amounts of a multi-element solution containing equal concentrations of Na, K, Ca, Fe, Cu and Mg (referred to as “Matrix”) with the matrix element/Cd ratio varying from 0.0001 to 1 (Fig. 5).
 | ||
| Fig. 5 Effect of the presence of a matrix (combination of Na, K, Ca, Fe, Cu and Mg) on the δ114/110Cd measurement result for 10 ng mL−1 NIST SRM 3108 as a function of the element/Cd ratio using (a) external correction and (b) internal + external correction. Error bars correspond to 2SD (n = 3). | ||
As shown in Fig. 5a, when doping the sample solution with increasing amounts of the multi-element matrix solution, the delta values obtained using external correction only deviate from the expected value, in accordance with the data reported by Shiel et al.54 However, when correcting with the Baxter + SSB approach (Fig. 5b), the Ag internal standard compensates for the matrix effect caused by these elements and yields correct results at all matrix/Cd ratios tested. This observation is consistent with the results of Wombacher et al.,29 who observed that the Ag normalization procedure exhibits lower susceptibility to matrix effects compared to the simple SSB approach.29
3.3. Precision and reproducibility of Cd isotope ratio measurements
 | ||
| Fig. 6 Impact of Cd concentration on the internal and external precision for (a) δ114/110Cd and (b) δ112/110Cd at 10, 5 and 2.5 ng mL−1. Solid red lines represent the mean values of the ten replicates and the dashed lines ±2SD (n = 10). Error bars correspond to 2SE for single runs. | ||
For a block of 50 measurement cycles, internal precision (2SE) values of 0.09, 0.11 and 0.14‰ for δ114/110Cd and of 0.08, 0.12 and 0.17‰ for δ112/110Cd were obtained at Cd concentrations of 10, 5 and 2.5 ng mL−1, respectively. This shows that by reducing the Cd concentration and, therefore, the Cd signal intensity 4-fold, the internal precision was slightly degraded only when using the 1013 Ω resistors. Pallavicini reported an internal precision of up to 0.04‰ for δ114/110Cd at 200 ng mL−1 with the standard introduction system and up to 0.20‰ at 10 ng mL−1 when using dry plasma conditions.26 Our data show that good internal precision can be obtained even at a Cd concentration as low as 2.5 ng mL−1.
Moreover, the external precision for δ114/110Cd for 10 measurements showed to be similar when Cd is measured at 2.5 and 5 ng mL−1 with 2SD values of 0.11 and 0.15‰, respectively. However, this figure was improved to 0.05‰ when the measurement was conducted at 10 ng mL−1 of Cd. The external precision obtained is similar to values reported in the literature, where a δ114/110Cd external precision of 0.06‰ (ref. 26) and 0.08‰ (ref. 31) was reported at 10 ng mL−1 and 25 ng mL−1, respectively, using the Aridus introduction system. On the other hand, an external precision of 0.09‰ was reported at 100 ng mL−1 Cd concentration levels using the standard introduction system and 1011 Ω resistors.48
 | ||
| Fig. 7 Long-term reproducibility of the δ114/110Cd measurement value for NIST SRM 3108 at 10 ng mL−1 over five months. The grey area represents ±2SD (n = 170). | ||
The long-term precision measured for δ114/110Cd was 0.09‰ (2SD, n = 170) and fits the precision reported in the literature (which generally falls within the range of 0.05–0.10‰) well.25–27,31,34,37,38,48,51,59 It is important to emphasize that most authors report the use of a desolvating sample introduction system and the double-spike method for mass bias correction. In this study, it was possible to achieve a similar precision at lower concentrations and using wet plasma conditions, while employing the combined Baxter + SSB approach using Ag as internal standard for mass bias correction.
3.4. Method validation
In order to validate our proposed method, δ114/110Cd was measured for two digests and several chromatographic isolations of Montana soil (NIST SRM 2711a) and Lobster hepatopancreas (NRC TORT-3) reference materials and the results were compared with the previously published data (Table 4).| Instrument | Introduction system | Concentration (ng mL−1) | δ 114/110Cd (‰) ± 2SD | Sensitivity (V ppm−1) | Mass bias correction | Reference |
|---|---|---|---|---|---|---|
| a NR: Not Reported. b The absolute amount of Cd needed for a single measurement is 10 ng. | ||||||
| NRC TORT-3 | ||||||
| Neptune plus | Aridus II | 10 | 0.07 ± 0.04 n = 1 | NR | SSB | Scott et al.55 2019 |
| Neptune plus | Wet plasma | 10b | 0.00 ± 0.10 n = 6 | 25–28 (112Cd) | Baxter + SSB | This study |
|
||||||
| NIST SRM 2711a | ||||||
| Neptune plus | Aridus II | 50–100 | 0.55 ± 0.05 n = 8 | 103 (112Cd) | DS | Liu et al.51 2019 |
| Neptune plus | Aridus II | 5–25 | 0.53 ± 0.04 n = 26 | 640 (112Cd) | DS | Tan et al.31 2020 |
| Nu plasma II | Aridus II | 100 | 0.62 ± 0.03 | NR | DS | Peng et al.36 2021 |
| Neptune plus | Aridus II | 40 | 0.55 ± 0.05 n = 10 | 110 (114Cd) | DS | Guo et al.37 2022 |
| Neptune plus | Aridus II | 100 | 0.55 ± 0.05 | 130 (114Cd) | DS | Dong et al.44 2022 |
| Neptune plus | Wet plasma | 100 | 0.57 ± 0.07 n = 5 | 11 (112Cd) | DS | Li et al.48 2018 |
| Neptune plus | Wet plasma | 10b | 0.54 ± 0.12 n = 8 | 25–28 (112Cd) | Baxter + SSB | This study |
|
||||||
| In-house Cd solution | ||||||
| Neptune plus | Wet plasma | 10b | −0.01 ± 0.11 n = 25 | 25–28 (112Cd) | Baxter + SSB | This study |
The δ114/110Cd values obtained in this study are in good agreement with the data reported in the literature, which were measured at higher concentrations using the standard introduction system (wet plasma) or at similar/higher concentrations using an aerosol desolvation unit (dry plasma).
3.5. Application to Antarctic samples
The tissues and shells of the mollusc bivalve Adamussium colbecki (Antarctic scallop) and spleen, liver and gonads of the fish Trematomus bernacchii (emerald rockcod) collected during the 1996–1998 and 2021–2022 sampling campaigns at Terra Nova Bay in Antarctica were analysed for their Cd concentration and Cd isotopic composition. Each organism was digested in 3-fold and each digest was subjected to Cd chromatographic purification (30–100 ng of Cd load, depending on the Cd concentration in the sample) and followed by isotope ratio measurement in duplicate at 10 ng per mL Cd concentration. The isolation procedure successfully achieved the separation of Cd from all matrix elements and the trace elements with isobaric nuclides. The critical element-to-cadmium (element/Cd) ratios in the Cd fraction were drastically reduced, from 18 to 0.11 for Zn/Cd, from 0.04 to 0.00 for Mo/Cd, from 0.13 to 0.00 for Ge/Cd, from 0.2 to 0.00 for Pd/Cd, from 65 to 0.00 for Ag/Cd and from 0.09 to 0.00 for Sn/Cd (Table S1†). The Cd recovery for all the samples ranged from 80 to 99%. The average δ114/110Cd, δ112/110Cd and δ111/110Cd values for three digests of each organism collected in the 1990s and 2020s are presented in Table 5.| (Mean ± 2SD, n = 3 digests) | N | [Cd] μg g−1 | δ 114/110Cd‰ | δ 112/110Cd‰ | δ 111/110Cd‰ |
|---|---|---|---|---|---|
| a N: number of organisms in a pool. | |||||
| Adamassium colbecki (bivalve) | |||||
| Shell 1990s | 10 | 0.9 ± 0.1 | −0.12 ± 0.08 | −0.07 ± 0.12 | −0.04 ± 0.12 |
| Tissue 1990s | 10 | 29.5 ± 1.6 | −0.18 ± 0.09 | −0.10 ± 0.05 | −0.03 ± 0.06 |
| Tissue 2022 | 10 | 14.6 ± 0.2 | −0.24 ± 0.12 | −0.11 ± 0.04 | −0.04 ± 0.09 |
|
|||||
| Trematomus bernacchi (fish) | |||||
| Gonads 1990s | 20 | 1.1 ± 0.0 | −0.09 ± 0.09 | −0.00 ± 0.05 | −0.05 ± 0.15 |
| Spleen 1990s | 20 | 19.4 ± 0.6 | −0.14 ± 0.08 | −0.08 ± 0.06 | −0.02 ± 0.00 |
| Liver 1990s | 20 | 17.7 ± 0.5 | −0.18 ± 0.12 | −0.12 ± 0.08 | −0.06 ± 0.06 |
| Liver 2022 | 11 | 5.0 ± 0.9 | −0.12 ± 0.03 | −0.04 ± 0.15 | −0.06 ± 0.07 |
For both species, a light Cd isotopic composition was obtained, characterized by a negative δ114/110Cd value, indicating that these species show a preferential uptake of the lighter Cd isotopes.28 Indeed, these δ114/110Cd values are significantly lighter compared to the δ114/110Cd value of +0.4‰ reported by Xue et al. for Southern Ocean water at a depth of 150 meters, representative of the depth at which these species are present.34,60 This is in accordance with the findings of Shiel et al. who report that δ114/110Cd values for oyster and bivalve species were significantly lighter than the value of the seawater, suggesting Cd fractionation upon biological uptake.28
For the Adamussium colbecki bivalve, the tissue exhibits insignificant variations in δ114/110Cd values only (determined by a two-sided t-test, p > 0.05) for the organisms collected during the 1990s and the 2020s. Although no clear difference was observed in the Cd isotopic composition, the pooled tissue samples collected from the 1990s show higher Cd concentration (2-fold) in comparison with those collected in the 2020s (Fig. 8).
 | ||
| Fig. 8 Diagrams of (a) δ114/110Cd and (b) δ112/110Cd versus Cd concentrations in bivalve Adamussium colbecki and the organs of the fish Trematomus bernacchii collected at Terra Nova Bay during the 1990s and in 2022. Error bars correspond to the SD (n = 3). | ||
Similarly, no statistical difference was observed between the δ114/110Cd values for the shell and the bivalve tissue. Thus, it can be suggested that the high Cd content in the tissues of the bivalve collected in Terra Nova Bay during the 1990s in comparison with those collected during the 2020s is probably related to the same source, as no clear difference was observed in δ114/110Cd within the measurement precision. This is in accordance with the observations of Shiel et al.,17 who did not report any significant correlation between Cd concentration and isotopic composition for bivalves collected from smelting-polluted areas during the 1980s and 2004–2005.
Like bivalves, fish can also accumulate harmful heavy metals in their tissues.61 While the liver and kidneys are primary organs for accumulation, other tissues such as muscle, intestinal mucosa, spleen, brain, pancreas and gonads also play a significant role.62,63 As shown in Table 5, for the organs of the Trematomus bernacchii fish collected during the 1990s, higher Cd contents were found in the liver and spleen in comparison with the gonads. Moreover, they exhibited slightly lighter δ-values (−0.14 ± 0.08‰ and −0.18 ± 0.12‰, 2SD, n = 3, respectively) in comparison to the gonads (−0.09 ± 0.08‰, 2SD, n = 3), although the differences were not significant (two-sided t-test, p > 0.05). Furthermore, as indicated in Table 5, liver samples collected during the 1990s showed a significantly higher Cd content in comparison with those collected during the 2020s and their isotopic composition showed to be slightly lighter although the differences were not significant (two-sided t-test, p > 0.05) (Fig. 8). This means that, similarly to the bivalve, the Cd content in the liver of the fish collected during the 1990s and 2022s is most likely related to the same source, as no clear difference was observed in δ114/110Cd at the instrumental precision obtained.
The insignificant difference in the δ114/110Cd values between the different organs of the fish collected in the 1990s suggests that there is no clear Cd isotope fractionation during the Cd distribution over the different organs15 Moreover, no significant difference in the δ114/110Cd values between the bivalve and the fish was observed. This could be hypothetically explained by the fact that the bivalve in question is one of the most consumed items by the fish T. bernacchii,64 suggesting no significant Cd fractionation along the trophic chain. Finally, δ114/110Cd values for fish and bivalves collected at Terra Nova Bay in the 1990s and 2020s ranged between −0.24 and −0.09‰ and did not show a significant difference, which suggests a stable Cd biogeochemical cycle over the 30 years timeframe. As for the source of Cd, the δ114/110Cd results reported in the literature for bivalves collected from Cd-polluted areas are characterised by significantly negative values, down to −1.2‰.28 Since our values are isotopically heavier compared to the Cd-polluted areas, we can suggest that the source of Cd in these samples is probably linked to natural processes. This aligns with the findings of Bargagli and Rota findings, who explain that Cd acting as a substitute for Zn, is actively absorbed by primary producers and transferred to consumers.13 However, more Cd isotope ratio data for various sample types from the Antarctic continent still need to be gathered in order to further unravel the origin of the Cd present in marine samples.
4 Conclusions
This study aimed at the development, characterization, validation and use of a method for precise and accurate Cd isotopic analysis at low concentration levels using MC-ICP-MS equipped with a standard sample introduction system (wet plasma conditions) and with Faraday cups connected to amplifiers with a 1013 Ω resistor. A long-term precision of 0.09‰ (2SD) was achieved for δ114/110Cd at a Cd concentration of 10 ng mL−1 using the Baxter + SSB mass bias correction approach with Ag as an internal standard. This value is similar to those documented in the literature, for which Cd was measured with Faraday cup amplifiers with a 1011 Ω resistor at higher concentrations using the standard introduction system (0.09‰ at 100 ng mL−1 of Cd)48 or similar concentrations using an Aridus aerosol desolvation unit (0.06‰26 at 10 ng mL−1). The method developed was validated by characterizing NIST SRM 2711a and NRC TORT-3 reference materials for their δ114/110Cd values, which were in agreement with the data reported in the literature. Finally, the method was applied to the Antarctic marine organisms Adamussium colbecki and Trematomus bernacchi collected during the 1990s and 2020s to explore the biogeochemical cycle of Cd throughout the years and to potentially identify the element's natural/anthropogenic sources. The Cd isotopic composition in these organisms showed a preferential uptake of light Cd isotopes, characterised by a negative δ114/110Cd value which ranged between −0.24 and −0.09‰, in accordance with earlier literature reporting Cd fractionation upon biological uptake by marine organisms. No significant difference in the δ114/110Cd value was observed between organs of the same species or between the same species collected in the 1990s and 2020s. This allowed to conclude that (i) there is no clear Cd isotope fractionation during the distribution of this element over the different organs and that (ii) the source of the Cd is most likely the same in the 1990s and 2020s and that the element is probably present due to a natural process, suggesting a stable Cd biogeochemical cycle in the area of Terra Nova Bay throughout the 30 years timeframe.Data availability
The authors believe that all relevant data are made available in the manuscript and the corresponding ESI.† Should data be missing, these will be made available by the authors upon simple request.Author contributions
Maria Alessia Vecchio: investigation; writing – original draft; formal analysis. Lana Abou-Zeid: investigation; writing – review & editing. Marco Grotti: conceptualization; writing – review & editing; supervision; funding acquisition. Frank Vanhaecke: conceptualization; writing – review & editing; supervision; funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the Italian National Program of Research in Antarctica (projects PNRA18_00229 and PNRA18_00216-B2). Frank Vanhaecke acknowledges FWO-Vlaanderen for providing the funding for the acquisition of MC-ICP-MS instrumentation (ZW15-02 – G0H6216N).References
- K. Hans Wedepohl, Geochim. Cosmochim. Acta, 1995, 59, 1217–1232 CrossRef.
- J. R. Larison, G. E. Likens, J. W. Fitzpatrick and J. G. Crock, Nature, 2000, 406, 181–183 CrossRef CAS PubMed.
- C. Cloquet, J. Carignan, G. Libourel, T. Sterckeman and E. Perdrix, Environ. Sci. Technol., 2006, 40, 2525–2530 CrossRef CAS PubMed.
- B. Gao, Y. Liu, K. Sun, X. Liang, P. Peng, G. Sheng and J. Fu, Anal. Chim. Acta, 2008, 612, 114–120 CrossRef CAS PubMed.
- A. Shiel, D. Weis and K. Orians, Sci. Total Environ., 2010, 408, 2357–2368 CrossRef CAS PubMed.
- J. O. Nriagu and J. M. Pacyna, Nature, 1988, 333, 134–139 CrossRef CAS PubMed.
- F. Vagnoni, S. Illuminati, A. Annibaldi, F. Memmola, G. Giglione, A. M. Falgiani, F. Girolametti, M. Fanelli, G. Scarponi and C. Truzzi, Atmosphere, 2021, 12, 1030 CrossRef CAS.
- N. M. Mahowald, A. R. Baker, G. Bergametti, N. Brooks, R. A. Duce, T. D. Jickells, N. Kubilay, J. M. Prospero and I. Tegen, Global Biogeochem. Cycles, 2005, 19, 4 Search PubMed.
- D. D. Deheyn, P. Gendreau, R. J. Baldwin and M. I. Latz, Mar. Environ. Res., 2005, 60, 1–33 CrossRef CAS PubMed.
- M. Grotti, F. Soggia, C. Ianni and R. Frache, Antarct. Sci., 2005, 17, 289–300 CrossRef.
- D. J. H. Phillips, Environ. Pollut., 1977, 13, 281–317 CAS.
- R. Bargagli, L. Nelli, S. Ancora and S. Focardi, Polar Biol., 1996, 16, 513–520 CrossRef.
- R. Bargagli and E. Rota, Environ. Sci.: Adv., 2024, 3, 543–560 Search PubMed.
- M. Mauri, M. Nigro and F. Regoli, Mar. Ecol. Prog. Ser., 1990, 67, 27–33 CrossRef CAS.
- S. D. Riva, M. L. Abelmoschi, E. Magi and F. Soggia, Chemosphere, 2004, 56, 59–69 CrossRef PubMed.
- F. Vanhaecke and P. Degryse, Isotopic Analysis - Fundamentals and Applications using ICP-MS, Wiley-VCH, 2012 Search PubMed.
- A. E. Shiel, D. Weis, D. Cossa and K. J. Orians, Geochim. Cosmochim. Acta, 2013, 121, 155–167 CrossRef CAS.
- M. Salmanzadeh, A. Hartland, C. H. Stirling, M. R. Balks, L. A. Schipper, C. Joshi and E. George, Environ. Sci. Technol., 2017, 51, 7369–7377 CrossRef CAS PubMed.
- K. J. R. Rosman and J. R. De Laeter, Int. J. Mass Spectrom. Ion Phys., 1975, 16, 385–394 CrossRef CAS.
- K. J. R. Rosman and J. R. De Laeter, Nature, 1976, 261, 216–218 CrossRef CAS.
- K. J. R. Rosman, J. R. De Laeter and M. P. Gorton, Earth Planet. Sci. Lett., 1980, 48, 166–170 CrossRef CAS.
- F. Lacan, R. Francois, Y. Ji and R. Sherrell, Geochim. Cosmochim. Acta, 2006, 70, 5118 CrossRef.
- A.-D. Schmitt, S. J. G. Galer and W. Abouchami, J. Anal. At. Spectrom., 2009, 24, 1079–1088 RSC.
- S. Ripperger, M. Rehkämper, D. Porcelli and A. N. Halliday, Earth Planet. Sci. Lett., 2007, 261, 670–684 CrossRef CAS.
- M. Gault-Ringold and C. H. Stirling, J. Anal. At. Spectrom., 2012, 27, 449 RSC.
- N. Pallavicini, E. Engström, D. C. Baxter, B. Öhlander, J. Ingri and I. Rodushkin, J. Anal. At. Spectrom., 2014, 29, 1570–1584 RSC.
- E. Martinková, V. Chrastný, M. Francová, A. Šípková, J. Čuřík, O. Myška and L. Mižič, J. Hazard. Mater., 2016, 302, 114–119 CrossRef PubMed.
- A. E. Shiel, D. Weis and K. J. Orians, Geochim. Cosmochim. Acta, 2012, 76, 175–190 CrossRef CAS.
- F. Wombacher, M. Rehkämper, K. Mezger and C. Münker, Geochim. Cosmochim. Acta, 2003, 67, 4639–4654 CrossRef CAS.
- D. J. Weiss, M. Rehkämper, R. Schoenberg, M. McLaughlin, J. Kirby, P. G. C. Campbell, T. Arnold, J. Chapman, K. Peel and S. Gioia, Environ. Sci. Technol., 2008, 42, 655–664 CrossRef CAS PubMed.
- D. Tan, J.-M. Zhu, X. Wang, G. Han, Z. Lu and W. Xu, J. Anal. At. Spectrom., 2020, 35, 713–727 RSC.
- D. C. Baxter, I. Rodushkin, E. Engström and D. Malinovsky, J. Anal. At. Spectrom., 2006, 21, 427–430 RSC.
- S.-C. Yang, D.-C. Lee and T.-Y. Ho, Geochim. Cosmochim. Acta, 2012, 98, 66–77 CrossRef CAS.
- Z. Xue, M. Rehkämper, M. Schönbächler, P. J. Statham and B. J. Coles, Anal. Bioanal. Chem., 2012, 402, 883–893 CrossRef CAS PubMed.
- L. Bridgestock, M. Rehkämper, T. van de Flierdt, K. Murphy, R. Khondoker, A. R. Baker, R. Chance, S. Strekopytov, E. Humphreys-Williams and E. P. Achterberg, Geophys. Res. Lett., 2017, 44, 2932–2940 CrossRef CAS.
- H. Peng, D. He, R. Guo, X. Liu, N. S. Belshaw, H. Zheng, S. Hu and Z. Zhu, J. Anal. At. Spectrom., 2021, 36, 390–398 RSC.
- C. Guo, T. Li, G. Li, T. Chen, L. Li, L. Zhao and J. Ji, J. Anal. At. Spectrom., 2022, 37, 2470–2479 RSC.
- Q.-H. Zhong, J. Li, L. Yin, N.-P. Shen, S. Yan, Z.-Y. Wang and C.-H. Zhu, J. Anal. At. Spectrom., 2023, 38, 939–949 RSC.
- C. Cloquet, O. Rouxel, J. Carignan and G. Libourel, Geostand. Geoanal. Res., 2005, 29, 95–106 CrossRef CAS.
- S. Ripperger and M. Rehkämper, Geochim. Cosmochim. Acta, 2007, 71, 631–642 CrossRef CAS.
- T. M. Conway, A. D. Rosenberg, J. F. Adkins and S. G. John, Anal. Chim. Acta, 2013, 793, 44–52 CrossRef CAS PubMed.
- R. Wei, Q. Guo, H. Wen, J. Yang, M. Peters, C. Zhu, J. Ma, G. Zhu, H. Zhang, L. Tian, C. Wang and Y. Wan, Anal. Methods, 2015, 7, 2479–2487 RSC.
- L. Yang, S. Tong, L. Zhou, Z. Hu, Z. Mester and J. Meija, J. Anal. At. Spectrom., 2018, 33, 1849–1861 RSC.
- C. X. Qiang Dong, At. Spectrosc., 2022, 43, 145–153 Search PubMed.
- Z. Zhang, T. Li, B. Li, Y. Lin, G. Li and T. Chen, J. Anal. At. Spectrom., 2024, 39, 1142–1151 RSC.
- M. Grotti, F. Soggia, C. Lagomarsino, S. D. Riva, W. Goessler and K. A. Francesconi, Antarct. Sci., 2008, 20, 39–52 CrossRef.
- H. Chang, J.-M. Zhu, X. Wang and T. Gao, J. Anal. At. Spectrom., 2023, 38, 950–962 RSC.
- D. Li, M.-L. Li, W.-R. Liu, Z.-Z. Qin and S.-A. Liu, Geostand. Geoanal. Res., 2018, 42, 593–605 CrossRef CAS.
- Y. Zhang, H. Wen, C. Zhu, H. Fan and C. Cloquet, Chem. Geol., 2018, 481, 110–118 CrossRef CAS.
- T. Breton, N. S. Lloyd, A. Trinquier, C. Bouman and J. B. Schwieters, Procedia Earth Planet. Sci., 2015, 13, 240–243 CrossRef CAS.
- M.-S. Liu, Q. Zhang, Y. Zhang, Z. Zhang, F. Huang and H.-M. Yu, Geostand. Geoanal. Res., 2020, 44, 169–182 CrossRef CAS.
- J. Borovička, L. Ackerman and J. Rejšek, Talanta, 2021, 221, 121389 CrossRef PubMed.
- F. Soggia, M. Abelmoschi, S. Riva, R. Pellegrini and R. Frache, Int. J. Environ. Anal. Chem., 2001, 79, 367–378 CrossRef CAS.
- A. E. Shiel, J. Barling, K. J. Orians and D. Weis, Anal. Chim. Acta, 2009, 633, 29–37 CrossRef CAS PubMed.
- S. R. Scott, K. E. Smith, C. Dahman, P. R. Gorski, S. V. Adams and M. M. Shafer, Sci. Total Environ., 2019, 688, 600–608 CrossRef CAS PubMed.
- Y. Yin and Y. Liang, At. Spectrosc., 2022, 43, 145–153 CrossRef.
- A. Rua-Ibarz, E. Bolea-Fernandez and F. Vanhaecke, Anal. Bioanal. Chem., 2016, 408, 417–429 CrossRef CAS PubMed.
- W. Abouchami, S. J. G. Galer, T. J. Horner, M. Rehkämper, F. Wombacher, Z. Xue, M. Lambelet, M. Gault-Ringold, C. H. Stirling, M. Schönbächler, A. E. Shiel, D. Weis and P. F. Holdship, Geostand. Geoanal. Res., 2013, 37, 5–17 CrossRef CAS.
- T. J. Horner, M. Schönbächler, M. Rehkämper, S. G. Nielsen, H. Williams, A. N. Halliday, Z. Xue and J. R. Hein, Geochem., Geophys., Geosyst., 2010, 11, 1525–2027 CrossRef.
- O. Dell'Acqua, T. Brey, M. Vacchi and M. Chiantore, Polar Biol., 2017, 40, 1557–1568 CrossRef.
- M. Chevreuil, A.-M. Carru, A. Chesterikoff, P. Boët, E. Tales and J. Allardi, Sci. Total Environ., 1995, 162, 31–42 CrossRef CAS PubMed.
- I. Kar and A. K. Patra, Biol. Trace Elem. Res., 2021, 199, 3846–3868 CrossRef CAS PubMed.
- S. G. George, K. Todd and J. Wright, Comp. Biochem. Physiol., Part C: Pharmacol., Toxicol. Endocrinol., 1996, 113, 109–115 CAS.
- M. La Mesa, M. Dalú and M. Vacchi, Polar Biol., 2004, 27, 721–728 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00214h |
| This journal is © The Royal Society of Chemistry 2024 |