
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA two-step chromatographic purification method for Ni for its isotopic analysis by MC-ICP-MS
Lingke
Li
a,
Fei
Wu
 *a,
Yongsheng
Liu
a,
Tao
He
a,
Jie
Lin
a,
Wen
Zhang
a,
Rui
Li
a,
Haihong
Chen
a,
Keqing
Zong
a,
Zhen
Zeng
ab and
Zhaochu
Hu
a
*a,
Yongsheng
Liu
a,
Tao
He
a,
Jie
Lin
a,
Wen
Zhang
a,
Rui
Li
a,
Haihong
Chen
a,
Keqing
Zong
a,
Zhen
Zeng
ab and
Zhaochu
Hu
a
aState Key Laboratory of Geological Processes and Mineral Resources, School of Earth Sciences, China University of Geosciences, Wuhan, 430074, China. E-mail: wufei@cug.edu.cn
bState Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science, Chinese Academy of Sciences, Nanjing, 210008, China
First published on 23rd May 2024
Abstract
Stable nickel (Ni) isotopes have shown great potential for investigating planetary accretion processes, mantle–crust magmatism, and paleo-marine evolution. The development of an economical and time-efficient separation method for Ni isotope analysis holds significant value within the realm of Ni isotope research. Previous purification methods for Ni isotope analysis have typically relied on either multi-column procedures or the utilization of significant quantities of organic reagents, such as acetic acid, acetone, and dimethylglyoxime. In this study, a novel two-column chromatographic separation procedure was presented for the determination of Ni isotope compositions in geological materials. A cation resin (AG50W-X8) was utilized in conjunction with diluted HCl and HF to effectively remove a substantial portion of matrix elements. Subsequently, trace amounts (100–200 μL) of Ni-spec resin were used for further purification of Ni. This relatively simple purification scheme for Ni isotope analysis effectively reduces the use of organic reagents and the amount of Ni-spec resin compared to previous methods that relied on multi-column separation and organic reagent extraction. Ni isotopic compositions were analyzed using multi-collector inductively coupled plasma mass spectrometry (MC-ICP-MS). The instrumental mass discrimination of Ni isotopic ratios was corrected for using the double spike (DS) combined with the sample–standard bracketing (SSB) method. The Ni isotope compositions (δ60Ni) of in-house standards (SCP and CUG) were −0.06 ± 0.05‰ (2 SD, n = 104) and 0.36 ± 0.06‰ (2 SD, n = 97), respectively. Furthermore, the measured δ60Ni values of various geological reference materials (BCR-2, BHVO-2, BIR-1, DTS-1, DTS-2b, SDO-1, GSP-2, and W-2a from the USGS; JB-1b from the GSJ; GSR-2, GSR-3, GSR-4, GSR-5, and GSR-10 from the NRCGA) were consistent with previous studies within measurement error. Additionally, the δ60Ni values of AGV-1 from the USGS, as well as those of GSR-10, GSR-12, GSR-15, GSR-17, GSR-18, and GSR-19 from the NRCGA are reported here for the first time. Based on repeated measurements of pure solutions and reference materials, the long-term reproducibility of δ60Ni values was better than ± 0.06‰ (2 SD). Consequently, our study presents a relatively straightforward purification method for Ni for its isotopic analysis using only trace amounts of organic reagents and Ni-spec resin.
1. Introduction
Nickel (Ni) is a first-row transition element and is categorized as a siderophile element. It displays moderately compatible behavior during mantle melting. Nickel has five stable isotopes whose natural abundances vary widely: 58Ni (68.08%), 60Ni (26.22%), 61Ni (1.11%), 62Ni (3.64%), and 64Ni (0.93%).1 Nickel isotopes were initially employed in studies of cosmochemical processes, serving as the daughter isotope of 60Fe and a tracer for nucleosynthetic anomalies.2 Recent investigations have begun to focus on elucidating mass-dependent variations in Ni isotopes in terrestrial samples. Ni isotope compositions are generally reported as δ60Ni values: δ60Ni (‰) = [(60Ni/58Nisample)/(60Ni/58NiSRM 986) − 1] × 1000. Variable Ni isotope fractionation is observed during high-temperature processes, including core–mantle differentiation, mantle partial melting, and magmatic differentiation.3–5 The sub-chondritic Ni isotopic signature of the bulk silicate Earth is suggested to have been established during Earth's late-stage accretion, originating from highly reduced sulfide-rich impactors.6 Additionally, Ni isotopes show potential applications in ore deposit mineralization7 and weathering processes.8,9 Experimental studies and natural observations indicate significant isotope fractionation of Ni during biological processes such as those in methanogens, suggesting the potential application of Ni isotopes in identifying paleo-methane release events and better understanding changes in marine environments through Earth's history.8,10 Nickel isotopes also show promise in environmental sciences.11–14Chemical purification protocols for Ni isotope analysis via MC-ICP-MS have been developed in several studies. Dimethylglyoxime (DMG) serves as a prevalent agent for isolation of Ni from geological materials, owing to its capacity to form a highly selective organic chelate with Ni under alkaline conditions.15 The purification of Ni using Ni-specific resin is also accomplished through the presence of attached DMG functional groups on the resin beads.16,17 During MC-ICP-MS measurements, the quantitative removal of matrix elements is imperative for high-precision Ni isotope analysis. However, due to Ni being a trace element in natural samples, the direct utilization of DMG during chemical purification necessitates a substantial amount of DMG.2,15,18 To attain optimal precision in instrumental measurements, oxidizing agents such as HClO4, H2O2, and aqua regia are also required for complete degradation of residual DMG ligands in the solution or leaching from the resin. This task becomes challenging with excessive DMG quantities.16,19,20
To address these challenges, alternative purification strategies have been explored. These strategies include the utilization of multiple sequential ion exchange chromatography columns, or the application of significant quantities of diverse organic reagents. For instance, in earlier methodologies, more than 50 mL of concentrated (90 to 100% v/v) acetone18 or acetic acid21 was employed to remove matrix elements such as Cu, Mg, K, Na, and Ca. However, the employment of organic reagents may introduce elevated Ni blanks, and the further purification of organic reagents requires a specific operational environment thus incurring relatively high costs. To reduce the utilization of organic reagents, Wu et al.19 devised a separation process employing five columns to isolate Ni from the matrix elements. However, this procedure is relatively time-consuming. Consequently, the development of relatively simple purification protocols that do not rely on large quantities of DMG and organic reagents remains imperative for various terrestrial samples.
Here, our work presents a simplified purification procedure for the purification of nickel in geological materials, achieved through two successive chromatographic steps with minimal utilization of organic reagents. The proposed protocol first employs a cation exchange resin column to effectively remove a substantial portion of matrix elements. Subsequently, a mini-column with a minute quantity of Ni-spec resin (100–200 μL) is utilized to quantitatively separate the remaining matrix elements and obtain high-purity Ni. Rigorous assessments of interferences and matrix effects during instrumental analysis demonstrate the suitability of our purification procedure for high-precision Ni stable isotopic analysis via MC-ICP-MS. To validate the methodology and enhance the existing database of rock standards, a collection of 20 igneous geological reference materials, encompassing both new and well-documented samples with a wide range of Ni content, were also purified and measured for their Ni isotope compositions to contribute to the augmentation of the preexisting database of rock standards.
2. Materials and methods
2.1 Chemical reagents and materials
All operations were conducted in accordance with the standards of a Class 1000 clean room at the State Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences, Wuhan, China. High-purity grades of HNO3, HCl and HF were double distilled using individual sub-boiling distillation systems (Savillex, Eden Prairie, USA). Analytical-grade ammonia (25–28%, w/w) was purchased from Aladdin (Shanghai, China). The citric acid was prepared by dissolving ACS grade anhydrous crystalline particles (≥99.5% (T), Aladdin, Shanghai, China). The guaranteed reagent H2O2 was purchased from LIAN SHI NEW MATERIAL CORP., LTD (LSNM, China). Milli-Q (MQ) water with a resistivity of 18.2 MΩ cm, produced by a Millipore-grade system (Millipore, Bedford, MA, USA), was utilized consistently throughout all procedures.Pure Ni isotope standard solution NIST SRM 986 (National Institute of Standards and Technology) and two in-house standards SCP-Ni (single element standard, lot S120723019; SCP SCIENCE, Spec-pure) and CUG-Ni (single element standard; GSB G 62022-90, Electronic grade) were utilized to evaluate the reliability of instrument measurements. Various geological reference materials (GRMs) purchased from the United States Geological Survey (USGS), National Research Center of Geoanalysis (NRCGA), and Geological Survey of Japan (GSJ) were employed to assess the robustness of our new purification procedure. These GRMs encompassed a variety of rock types, including basalts, andesites, peridotites, sandstone, shales, dolomite, amphibolite, gabbro, granulite, diabase, and granodiorite.
2.2 Sample preparation
The rock sample powders were weighed between 5 and 150 mg to ensure a minimum of 0.3 μg Ni content. Sample powders were firstly digested using a 2 mL mixture of concentrated HF and HNO3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). The 61Ni–62Ni double spike (DS) was added based on the optimized sample/spike ratio of 1:1 (see discussions below). The samples were heated on a hotplate at 120 °C for at least 48 hours and were observed to ensure that all silicate materials were destroyed before being evaporated to dryness. Aqua regia (HCl:HNO3 = 3:1) was then added to ensure thorough digestion and to assist in removing fluoride from the solution. Following approximately 12 hours of reflux, the samples were evaporated to dryness. Subsequently, the samples were treated with 1 mL of 6 mol L−1 HCl at 110 °C for 12 hours at least twice until the solutions became clear without any precipitation. Finally, the solution was dried and dissolved in 1 mL of 1 mol L−1 HCl in preparation for the chromatographic purification of Ni.
:3). The 61Ni–62Ni double spike (DS) was added based on the optimized sample/spike ratio of 1:1 (see discussions below). The samples were heated on a hotplate at 120 °C for at least 48 hours and were observed to ensure that all silicate materials were destroyed before being evaporated to dryness. Aqua regia (HCl:HNO3 = 3:1) was then added to ensure thorough digestion and to assist in removing fluoride from the solution. Following approximately 12 hours of reflux, the samples were evaporated to dryness. Subsequently, the samples were treated with 1 mL of 6 mol L−1 HCl at 110 °C for 12 hours at least twice until the solutions became clear without any precipitation. Finally, the solution was dried and dissolved in 1 mL of 1 mol L−1 HCl in preparation for the chromatographic purification of Ni.
2.3 Chemical purification of Ni
In this study, a two-step ion exchange scheme was developed for the separation of Ni. The detailed procedures are outlined in Table 1. The first step involved the utilization of a cation exchange resin column (AG50W-X8, chloride form, Bio-Rad 200–400 mesh) to remove a substantial portion of matrix elements, as shown in the elution curves (Fig. 1). 2 mL of AG50W-X8 cation resin was loaded into a column with an inner diameter of 6 mm. The loaded cation exchange resins were pre-cleaned with 10 mL of 3.5 mol L−1 HNO3, 10 mL of 6 mol L−1 HCl and 5 mL of MQ water successively. After preconditioning with 5 mL of 1 mol L−1 HCl, 1 mL sample solutions were loaded onto the column. Elution of major matrix elements including Al, Fe, Cr, Na, K, and Ti was done using 8 mL of 0.4 mol L−1 HCl + 0.5 mol L−1 HF mixtures, followed by 13 mL of 1 mol L−1 HCl. Nickel was subsequently collected with 13 mL of 1 mol L−1 HCl, while certain matrix elements (e.g., Ca) remained retained on the cation exchange resin. The collected fractions were then dried down for further purification.| Separation stage | Reagents | Volume/mL | Major collected elements |
|---|---|---|---|
| Column 1: AG50W-X8, 200 to 400 mesh, 2 mL; 9 cm H × 6 mm ID column | |||
| Clean resin | 3.5 mol L−1 HNO3 | 5 + 5 | |
| Clean resin | 6.0 mol L−1 HCl | 5 + 5 | |
| Clean resin | MQ water | 5 | |
| Condition | 1.0 mol L−1 HCl | 2 + 3 | |
| Load sample | 1.0 mol L−1 HCl | 1 | |
| Elute matrix | 0.4 mol L−1 HCl + 0.5 mol L−1 HF | 2 + 6 | Al, Fe, Cr, V, Ti, Sn |
| Elute matrix | 1.0 mol L−1 HCl | 1 + 2 + 10 | Na, K, V, Cr, Cu, Zn, Mg |
| Collect Ni | 1.0 mol L−1 HCl | 1 + 2 + 10 | Ni, Mg, Mn, Co, Zn, Cu |
| Clean resin | 3.5 mol L−1 HNO3 | 6 | Ca |
| Clean resin | MQ water | 5 | |
|
|||
| Column 2: Ni-spec resin, 100 to 200 μL; 7 cm H × 3 mm ID column | |||
| Clean resin | MQ water | 1 | |
| Clean resin | 0.2 mol L−1 ammonium citrate | 1 | |
| Condition | 0.2 mol L−1 ammonium citrate + ammonia (pH = 9–10) | 2 | |
| Load sample | 1 mol L−1 HCl mixed with 1 mol L−1 ammonium citrate + ammonia buffer solution (pH = 9–10) | ∼2 | |
| Elute matrix | 0.2 mol L−1 ammonium citrate + ammonia (pH = 9–10) | 3 | Residue matrices |
| Elute matrix | MQ water | 4 | |
| Collect Ni | 3.5 mol L−1 HNO3 | 2 | Ni |
 | ||
| Fig. 1 Elution curve for Ni purification in the first column with 2 mL AG50W-X8 (200–400 mesh) cation resin. The igneous reference material BHVO-2 was utilized with 2.6 μg of Ni loaded to generate the elution curve. The shaded area denotes the collection Ni elution part. | ||
To further separate Ni from residual matrices, a second purification step was undertaken using a mini-column containing 100 to 200 μL of Ni-spec resins (TrisKem International). The Ni-spec resins were initially washed with MQ water and 0.2 mol L−1 ammonium citrate, and subsequently conditioned using a mixture of ammonium citrate and ammonia with pH of 9–10. The samples were dissolved in 1 mL of 1 mol L−1 HCl, and prior to loading onto the column, the mixture of ammonium citrate and ammonia was added to adjust the pH to 9–10. The matrix was then eluted with 3 mL of mixed ammonium citrate and ammonia solution (pH = 9–10), followed by 4 mL of MQ water, and Ni was collected using 2 mL of 3.5 mol L−1 HNO3. The eluted Ni solution was evaporated to dryness and refluxed with 0.4 mL of a mixture of 15.6 mol L−1 HNO3 and 30% H2O2 (1:1 v/v) to oxidize and remove any potential organic compounds leached from the resin. The solution was evaporated to dryness again and re-dissolved in 2% HNO3 (v/v) for MC-ICP-MS measurements.
2.4 Mass spectrometry
Nickle isotopes were analyzed using MC-ICP-MS (Neptune Plus, Thermo-Fisher Scientific®, Bremen, Germany) at the State Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences, Wuhan. The “Jet” sample cone and “H” skimmer cone made of nickel were employed, with no observed impact on Ni isotope measurements resulting from the use of nickel cones. Medium resolution (MR) mode (m/Δm > 5500) was applied to distinguish molecular interference such as 40Ar18O+. The signals were monitored in static mode using Faraday cups with feedback amplifiers corresponding 1011 Ω resistors. As shown in Table 2, Faraday cups L4, L2, C, H2, H3 and H4 were applied to collect 56Fe, 57Fe, 58Ni, 60Ni, 61Ni and 62Ni, respectively. Sample solutions were introduced into the instrument under either “dry” plasma conditions using an Aridus III desolvator (CETAC Technologies®) or under “wet” plasma conditions using a quartz dual cyclonic-spray chamber. The typical sensitivity of 58Ni was approximately 0.1 V ppb−1 under “dry” plasma conditions with a desolvation system and 0.02 V ppb−1 under “wet” plasma conditions with a quartz dual cyclonic-spray chamber, following optimization of tuning parameters (Table 2).| Instrument settings | |
|---|---|
| RF power, W | 1200 |
| Sampler cone | Jet |
| Skimmer cone | H |
| Cooling gas flow rate, L min−1 | 16 |
| Sample gas flow rate, L min−1 | ∼0.655 |
| Auxiliary gas flow rate, L min−1 | ∼1.25 |
| Nebulizer, mL min−1 | 0.1 |
| Sample introduction systems | Aridus III desolvator (dry plasma) | Quartz dual cyclonic-spray chamber (wet plasma) |
|---|---|---|
| Typical sensitivity | 20–25 V of 58Ni for 200 ng g−1 Ni solution | 4–5 V of 58Ni for 200 ng g−1 Ni solution |
| Resolution mode | Medium resolution | |
| Data acquisition | |
|---|---|
| Mode | Static, multi-collection |
| Integration time, s | 4.194 |
| Number of cycles per block | 40 |
| Number of blocks per analysis | 1 |
| Instrumental mass fractionation correction | Double spike + sample standard bracketing |
| Cup configuration for Ni isotope ratio measurements | ||||||
|---|---|---|---|---|---|---|
| Cup | L4 | L2 | C | H2 | H3 | H4 |
| Amplifier | 1011 Ω | 1011 Ω | 1011 Ω | 1011 Ω | 1011 Ω | 1011 Ω |
| Nuclide | 56Fe | 57Fe | 58Ni | 60Ni | 61Ni | 62Ni |
Standard–sample bracketing (SSB) and double spike (DS) techniques were employed to effectively correct for instrumental mass discrimination. Each measurement for both the sample and standard consisted of 40 cycles with an integration time of 4.194 seconds. To prevent cross-contamination and eliminate memory effects between the standard solution and the sample solution, an extended rinse protocol was employed, including rinsing with 5% HNO3 (v/v) for 90 s and 2% HNO3 (v/v) for 90 s between each analysis. Additionally, to further minimize background interference, isotopic signals were measured in 2% HNO3 (v/v) before each sample analysis and subtracted from the analyzed signal (i.e., the on-peak zero method).
3. Results and discussion
3.1 Optimization of the column separation method for Ni isotope measurement
The use of DMG or Ni-specific resin has been demonstrated as an effective method for the enrichment and purification of nickel.15,20 The formation of the Ni–DMG complex occurs under alkaline conditions (pH = 8–10), leading to strong adsorption onto the ion exchange resin, with subsequent elution of matrix elements. Following this, oxidation reagents such as HNO3 are employed to disrupt the complex, facilitating Ni elution from the resin. However, as Ni is a trace element, fully dissolving a sufficient amount of material in alkaline solutions to attain the necessary amount of Ni for isotope analysis presents challenges. Additionally, substantial quantities of DMG reagent or Ni-specific resin are required to achieve Ni purification in geological samples.16 Implementing pretreatment steps to remove a certain amount of matrix from samples before employing DMG or Ni-specific resin could overcome these shortcomings.In this study, we propose a two-step chromatography strategy for Ni separation from various geological materials for isotope analysis, without the need for large quantities of DMG or other organic reagents (Table 1). In the first ion exchange stage, the cation resin is used with HCl and a small amount of HF to remove a substantial portion of matrix elements. As shown in the elution curve (Fig. 1), after loading samples onto the preconditioned column, Al, Ti, Fe and several other matrix elements were initially eluted using a solution comprising 0.4 mol L−1 HCl + 0.5 mol L−1 HF. These elements form anionic fluorides with low affinity for the cation resin in the presence of fluorion. Conversely, the addition of a trace amount of HF to the HCl solution did not significantly alter the distribution coefficients (Kd) of other elements, including Ni, between the cation resin and the solution.21 Subsequently, 1 mol L−1 HCl was utilized to separate Ni from some major matrix elements such as Na, K, and Ca, as the Kd of these elements differs markedly from that of Ni on the cation resin under these conditions (Fig. 1). Through the utilization of this cation resin procedure, a significant fraction of matrix elements was successfully separated from Ni, leaving residual matrix elements such as Co, Mn, and some Mg, Cu, and Zn (Fig. 2).
 | ||
| Fig. 2 The residual matrix element contents relative to Ni content (i.e., [element]/[Ni] ratios before the purification (horizontal lines), after the first column (hollow circle points) and the whole scheme (hollow triangular points)). Y = 0.001 indicates [element]/[Ni] ratios ≤0.001. The horizontal title element * refers to the corresponding matrix element. | ||
To remove the remaining matrix elements, we utilize Ni-spec resin, adapted from previous studies.16 An ammonium citrate + ammonia mixture solution with a pH of 9–10 is used to separate the matrix elements from Ni. The Ni–DMG complex remains stable and strongly bound to the resin, while other matrix elements exist as metal ions, citrate complexes, or ammonia complexes and are eluted from the resin. Subsequent treatment with 3.5 mol L−1 HNO3 destroys the Ni–DMG complex, allowing Ni to be eluted from the resin. Since a substantial portion of matrix elements has already been removed by the first column, only a trace amount of Ni-spec resin (∼100 μL) is needed in this step.
The elution curves of cations may exhibit drift with varying amounts of elements loaded onto the cation resin columns.22,23 In addition, the efficiency of matrix element removal may fluctuate depending on the ratios of the matrices to nickel, which can vary significantly among different types of samples. To verify the recovery rate and purification efficiency of Ni with our method, the element concentrations of GRM solutions were measured after each column. As shown in Fig. 2, despite variations in Ni and matrix element contents among different types of reference materials, only trace amounts of matrix elements remained in the solution after Ni-spec resin purification. A batch of reference materials was analyzed for their Ni content both before and after the column procedure to calculate the yield of Ni through the two-column separation. The yields exceeded 85% across a range of Ni loading amounts from 0.3 to 15 μg. The blank sample was processed alongside natural samples and measured for Ni content to quantify the total blank. The total blank of our separation scheme was found to be less than 3 ng, which is considered negligible compared to the total amount of Ni loaded onto the columns.
3.2 Double spike optimization
The precision of the double spike correction relies on selecting appropriate double-spike compositions and determining the mixed ratios of samples to double spike. In this study, the 61Ni–62Ni double spike was selected to correct for isotope fractionation similar to previous studies.19,24 Single spikes of 61Ni (99.39%) and 62Ni (99.34%) were purchased from ISOFLEX (USA). In this study, we assessed the optimal combination of double-spike mixtures and appropriate sample/spike ratios based on the methodologies outlined by a previous study.25 The results indicated the optimal double-spike composition was approximately 61Ni/62Ni ≈ 0.8. We employed a 58Ni–60Ni–61Ni–62Ni inversion to minimize the potential for isobaric interferences. The chosen combination of 61Ni–62Ni mixtures exhibited robustness, as evidenced by a flat error surface, indicating accuracy over a broad range of sample–spike mixing ratios (Fig. 3A). Furthermore, the accuracy of the double spike was checked by analyzing spike–sample mixtures with various proportions.26 These tests demonstrated that the double-spike corrected values yielded δ60Ni within typical reproducibility (i.e., 0.06‰, 2 SD) and minimum internal errors across spike proportions ranging from 0.2 to 0.7 (Fig. 3B). | ||
| Fig. 3 (A) Simulation of the influence of isotopic composition of DS and proportion of DS in the DS–sample mix. The vertical axis gives the proportion of 61Ni in the 61Ni–62Ni double spike, and the horizontal axis gives the proportion of double spike in the double spike–sample mixture. (B) δ60Ni values measured for various mixture ratios between the NIST SRM986 solution and the double spike. The double spike consists of 45% 61Ni and 55% 62Ni near the calculated optimum composition as shown in (A). The selected proportion of double spike in the double spike–sample mix in this study is 0.5, which was used in all isotope measurements. Also depicted is the theoretical error curve for the DS with varying proportions in DS–sample mixtures. | ||
3.3 Evaluation of interferences and matrix effects
In this study, we examined the residual levels of matrix elements in variable geological reference materials both in the initial and final purification steps (see Fig. 2). During the two-column procedure, most matrix elements were effectively eliminated, leading to residual matrix levels below the threshold of significance ([X]/[Ni] < 0.1). However, a small amount of residual matrix elements still has the potential to introduce significant spectrum (e.g., polyatomic, double-charged ion and isobaric interferences) and non-spectrum interferences (e.g., matrix effects) in Ni isotope analyses. To ensure the accurate measurement of Ni isotopes, rigorous testing and correction procedures are essential.Isobaric interferences from Fe cannot be eliminated and require correction by monitoring 57Fe. After the two-column purification, the ion beam intensity of 57Fe is substantially lower relative to that of 58Ni, with [Fe]/[Ni] ratios below 0.01 (Fig. 2). A doping test demonstrated that when the ratio of [Fe]/[Ni] is <1, its effects on the measurement of δ60Ni can be well corrected for (Fig. 4). Thus, as described above, isobaric interferences from Fe can be effectively reduced and corrected for.
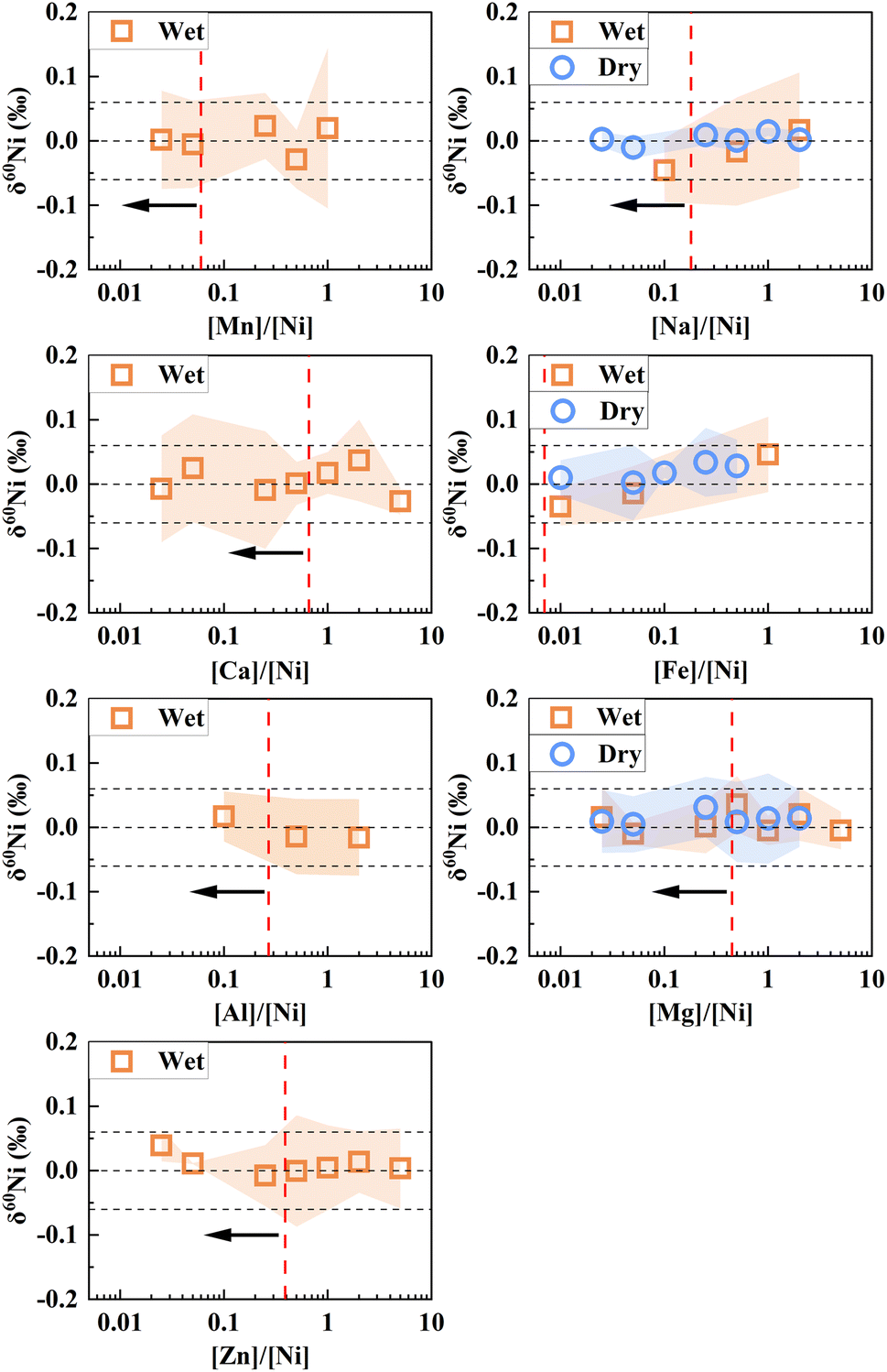 | ||
| Fig. 4 Doping experiments to test the matrix effect of different elements on δ60Ni analyses. The vertical dotted line denotes the upper limit of the residue matrix elements after the two-step purification process for various types of reference materials. | ||
Regarding double-charged ions such as Sn (116Sn++, 120Sn++, 122Sn++, 124Sn++), Cd (116Cd++), Xe (124Xe++) and Te (120Te++, 122Te++, 124Te++),24 they were undetectable due to their relatively low initial contents and effective removal (e.g., [Sn]/[Ni] < 0.01) during purification. Argon, the main component of the carrier gas, can potentially interfere with Ni isotope measurements by generating polyatomic interferences (e.g., 40Ar18O+, 40Ar17OH+). These interferences can be distinguished by MC-ICP-MS in medium-resolution mode, and they can be further avoided by employing the on-peak-zero method. Other matrix elements, such as Mg, Ca, and Ti, can also form polyatomic molecules (e.g., 40Ca18O+, 24Mg36Ar+, 46Ti16O+). However, these matrix elements were sufficiently separated and their relative contents were low ([Mg]/[Ni] < 0.5, [Ca]/[Ni] < 0.6 and [Ti]/[Ni] is below the detection limit) after the two-column purification. The doping test further showed that the influence of Ca and Mg can be negligible when the ratios of [Ca]/[Ni] and [Mg]/[Ni] are less than 5 (Fig. 4). Moreover, some residual elements (e.g., Ti) were present in negligible amounts.
Some matrix elements with a small amount of residue ([X]/[Ni] > 0.1) after the whole procedure were further investigated. Although the mechanism by which residual matrix elements cause isotope measurement drifts is not yet fully understood, they may impact ionization efficiency and transmission of Ni, and interfere with isotopic ratio measurements. To assess the effects of residual matrix elements on Ni isotope analysis, various proportions of Na, Al, Mn, and Zn relative to Ni were also quantitatively added to spiked Ni standard solutions. As shown in Fig. 4, the measurements of δ60Ni exhibit negligible influence within measurement error when the ratios of [Na]/[Ni] and [Al]/[Ni] are <2. Similarly, the effects on δ60Ni are negligible for [Zn]/[Ni] < 5 and [Mn]/[Ni] < 1. In conclusion, the results of these tests demonstrate that following purification using our scheme, the residual matrix elements within the geological materials have no discernible impact on Ni isotope analysis within measurement error.
3.4 Reproducibility and accuracy of measurements
To assess the reproducibility and accuracy of our method, two different approaches were employed: in-house standards (SCP and CUG) not subjected to chemical purification, and rock samples treated with chemical purification (Fig. 5 and Table 3). The Ni isotope ratios (δ60Ni) of standard in-house standards (SCP and CUG) were measured to be −0.06 ± 0.05‰ (2 SD, n = 104) and 0.36 ± 0.06‰ (2 SD, n = 97), respectively. We also analyzed various rock samples including basalts (BCR-2, BHVO-2, BIR-1, GSR-3, GSR-18, JB-1b), andesites (AGV-1, GSR-2), peridotites (DTS-1, DTS-2b, GSR-19), one sandstone (GSR-4), shales (SDO-1, GSR-5), one dolomite (GSR-12), one amphibolite (GSR-15), one gabbro (GSR-10), one granulite (GSR-17), one diabase (W-2a) and one granodiorite (GSP-2). The δ60Ni data of select rock samples (BCR-2, BHVO-2, BIR-1, DTS-1, DTS-2b, GSP-2, GSR-2, GSR-3, GSR-4, GSR-5, JB-1b, SDO-1, W-2a) were consistent with values reported in previous research, within measurement error (Table 3). Other rock samples (AGV-1, GSR-10, GSR-12, GSR-15, GSR-17, GSR-18, GSR-19) are reported here for the first time. The external precisions are ±0.06‰ (2 SD) over a year, indicating that our scheme can be applied to a wide range of samples with different matrix elements and variable Ni content to discern the natural mass-dependent fractionation of Ni isotopes.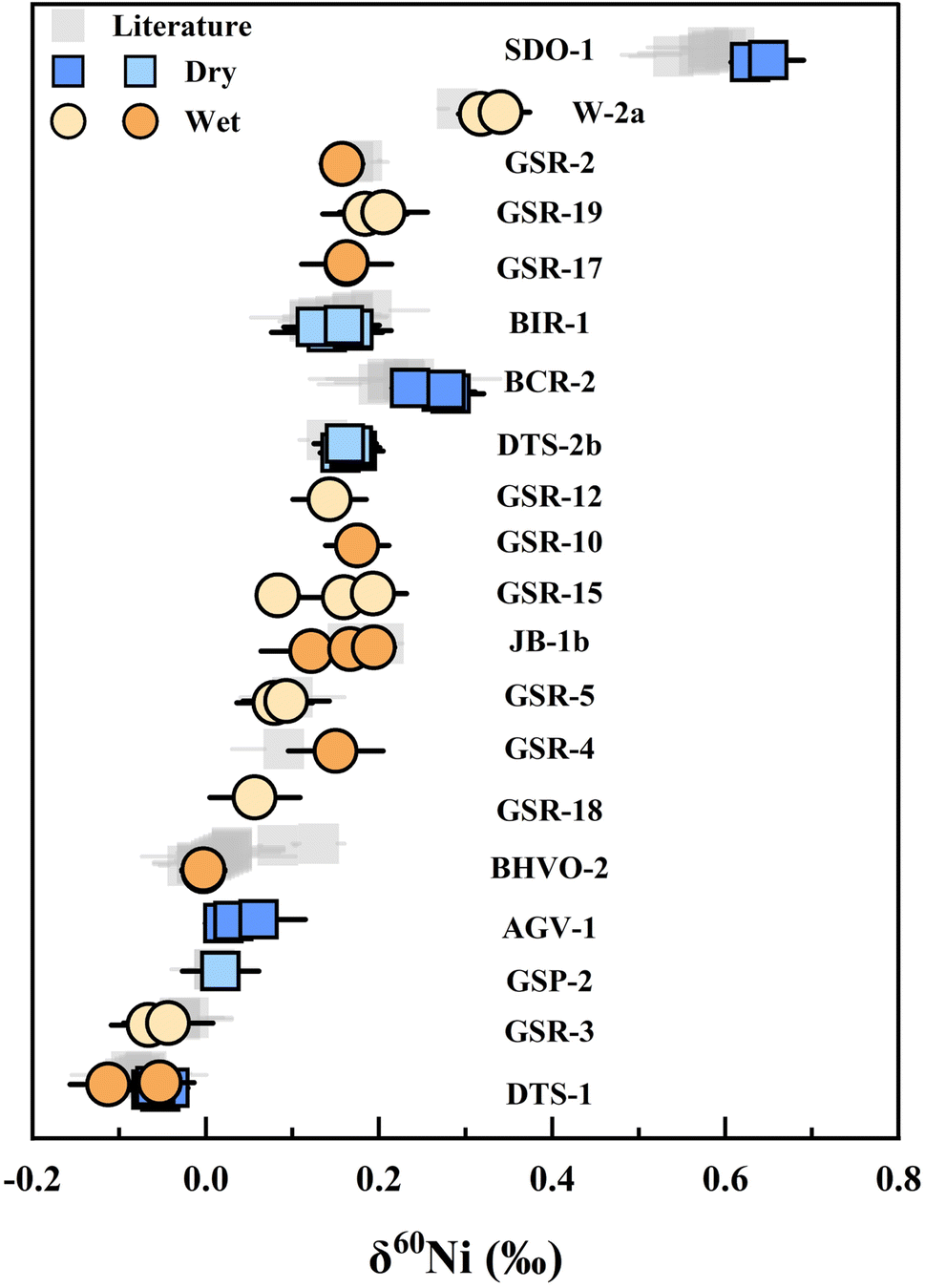 | ||
| Fig. 5 Summary of δ60Ni of the geological reference materials analyzed in this study. The colored square points were measured in the dry plasma, and the colored circle points were measured in the wet plasma. The gray squares represent measurement results from previous studies. The average value of each standard is listed in Table 3. | ||
| Sample name | Rock type | Ni (μg g−1) | n | δ 60Ni (‰) | 2 SD (‰) | References |
|---|---|---|---|---|---|---|
| AGV-1 | Andesite | 16 | 11 | 0.04 | 0.04 | This study |
| BCR-2 | Basalt | 9.5 | 14 | 0.27 | 0.05 | This study |
| 0.20 | 0.07 | Cameron et al.27 | ||||
| 5 | 0.21 | 0.06 | Wu et al.19 | |||
| 3 | 0.23 | 0.04 | Li et al.28 | |||
| 0.24 | 0.10 | Sun et al.29 | ||||
| BHVO-2 | Basalt | 112 | 3 | 0.00 | 0.03 | This study |
| 0.13 | 0.03 | Cameron et al.27 | ||||
| 11 | 0.006 | 0.041 | Gueguen et al.16 | |||
| 4 | 0.01 | 0.02 | Ratié et al.30 | |||
| −0.01 | 0.05 | Estrade et al.31 | ||||
| 5 | 0.083 | 0.019 | Chernonozhkin et al.18 | |||
| 5 | 0.015 | 0.089 | Pasava et al.32 | |||
| 6 | 0.03 | 0.06 | Wu et al.19 | |||
| 0.03 | 0.06 | Sorensen et al.12 | ||||
| 36 | 0.026 | 0.059 | Saunders et al.5 | |||
| 3 | 0.02 | 0.04 | Li et al.28 | |||
| 2 | −0.021 | 0.032 | Beunon et al.33 | |||
| −0.01 | 0.05 | Sun et al.29 | ||||
| 36 | 0.03 | 0.06 | Saunders et al.4 | |||
| BIR-1 | Basalt | 166 | 17 | 0.15 | 0.05 | This study |
| 68 | 0.13 | 0.078 | Gall et al.24 | |||
| 0.17 | 0.02 | Li et al.34 | ||||
| 3 | 0.12 | 0.035 | Gueguen et al.16 | |||
| 2 | 0.191 | 0.066 | Chernonozhkin et al.18 | |||
| 5 | 0.12 | 0.03 | Wang et al.8 | |||
| 2 | 0.169 | 0.032 | Beunon et al.33 | |||
| 20 | 0.15 | 0.06 | Saunders et al.4 | |||
| DTS-1 | Dunite | 2360 | 20 | −0.06 | 0.05 | This study |
| 4 | −0.071 | 0.053 | Gueguen et al.16 | |||
| 2 | −0.069 | 0.006 | Chernonozhkin et al.18 | |||
| 32 | −0.077 | 0.078 | Gall et al.35 | |||
| 8 | −0.082 | 0.009 | Klaver et al.3 | |||
| 3 | −0.093 | 0.032 | Beunon et al.33 | |||
| DTS-2b | Dunite | 3780 | 25 | 0.17 | 0.03 | This study |
| 3 | 0.14 | 0.032 | Beunon et al.33 | |||
| GSP-2 | Granodiorite | 17.1 | 3 | 0.02 | 0.04 | This study |
| 1 | 0.01 | 0.05 | Wu et al.19 | |||
| GSR-10 | Gabbro | 64.5 | 3 | 0.17 | 0.04 | This study |
| GSR-12 | Dolomite | 235 | 6 | 0.14 | 0.04 | This study |
| GSR-15 | Amphibolite | 130 | 9 | 0.15 | 0.10 | This study |
| GSR-17 | Granulite | 44.8 | 6 | 0.16 | 0.04 | This study |
| GSR-18 | Basalt | 69.7 | 6 | 0.06 | 0.05 | This study |
| GSR-19 | Peridotite | 1269 | 6 | 0.19 | 0.05 | This study |
| GSR-2 | Andesite | 16.9 | 3 | 0.16 | 0.01 | This study |
| 3 | 0.18 | 0.02 | Wu et al.19 | |||
| 3 | 0.17 | 0.04 | Li et al.28 | |||
| GSR-3 | Basalt | 139 | 12 | −0.05 | 0.05 | This study |
| 3 | −0.03 | 0.06 | Wu et al.19 | |||
| 2 | −0.03 | 0.06 | Li et al.28 | |||
| GSR-4 | Sandstone | 16.6 | 6 | 0.15 | 0.05 | This study |
| 2 | 0.09 | 0.06 | Li et al.28 | |||
| GSR-5 | Shale | 37 | 13 | 0.09 | 0.05 | This study |
| 2 | 0.10 | 0.06 | Li et al.28 | |||
| JB-1b | Basalt | 144.7 | 9 | 0.16 | 0.07 | This study |
| 1 | 0.205 | 0.017 | Chernonozhkin et al.18 | |||
| SDO-1 | Shale | 101 | 6 | 0.64 | 0.04 | This study |
| 5 | 0.585 | 0.025 | Gueguen et al.16 | |||
| 0.58 | 0.08 | Ventura et al.36 | ||||
| 0.54 | 0.05 | Estrade et al.31 | ||||
| 9 | 0.57 | 0.09 | Wang and Wasylenki37 | |||
| 3 | 0.61 | 0.06 | Wu et al.19 | |||
| 4 | 0.6 | 0.05 | Wang et al.8 | |||
| W-2a | Diabase | 66.1 | 6 | 0.33 | 0.04 | This study |
| 3 | 0.29 | 0.01 | Wu et al.19 |
4. Conclusions
In this study, we developed a straightforward Ni purification procedure for determining Ni isotopes in various samples. A substantial portion of matrix elements was effectively removed using a cation resin in the first column, followed by further separation of Ni using 100–200 μL of Ni-spec resin in the second step. The two-step purification strategy presented in this study simplifies the purification process for different types of geological samples and reduces the need for DMG and organic reagents. The isotopic fractionation during chemical purification and instrument analysis was calibrated using a double-spike combined with sample–standard bracketing approach. The δ60Ni values of various GRMs including basalts (BCR-2, BHVO-2, BIR-1, GSR-3, GSR-18, JB-1b), andesites (AGV-1, GSR-2), peridotites (DTS-1, DTS-2b, GSR-19), one sandstone (GSR-4), shales (SDO-1, GSR-5), one dolomite (GSR-12), one amphibolite (GSR-15), one gabbro (GSR-10), one granulite (GSR-17), one diabase (W-2a) and one granodiorite (GSP-2) were determined using MC-ICP-MS. The δ60Ni values obtained were in agreement with reference values. The long-term precision of the procedure is better than ±0.06‰ (2 SD) for δ60Ni over a year. Thus, this study offers an alternative method to efficiently obtain Ni isotope compositions from various geological and environmental samples, enabling investigation into Ni isotope variations in different geological processes.Author contributions
Lingke Li: investigation, validation, methodology, writing – original draft. Fei Wu: conceptualization, supervision, funding acquisition, methodology, writing – review & editing. Yongsheng Liu: resources, supervision, project administration. Tao He: resources, writing – review & editing. Jie Lin: resources, supervision, writing – review & editing. Wen Zhang: resources. Rui Li: resources. Haihong Chen: resources. Keqing Zong: resources. Zhen Zeng: validation, writing – review & editing. Zhaochu Hu: resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Li-Ping Qin for providing SRM 986 and SCP standard solutions. Major contributions were made by Wen-Gui Liu, Zhi-Fu Liu, Yang Gao, Jia-Wei Li, Zhen-Yi Liu, and Lai-Tong Zhou who provided analytical support in the China University of Geosciences (Wuhan). This work was funded by the National Key R&D Program of China (2023YFF0806200), National Natural Science Foundation of China (42173018), and “CUG Scholar” Scientific Research Funds (Project No. 2022002) from the China University of Geosciences (Wuhan). This manuscript has benefited from reviews by two anonymous reviewers and the editorial handling from Derya Kara Fisher.References
- J. W. Gramlich, L. A. Machlan, I. L. Barnes and P. J. Paulsen, J. Res. Natl. Inst. Stan. Technol., 1989, 94, 347–356 CrossRef CAS PubMed.
- P. Morand and C. J. Allegre, Earth Planet. Sci. Lett., 1983, 63, 167–176 CrossRef CAS.
- M. Klaver, D. A. Ionov, E. Takazawa and T. Elliott, Geochim. Cosmochim. Acta, 2020, 268, 405–421 CrossRef CAS.
- N. J. Saunders, J. Barling, J. Harvey, J. G. Fitton and A. N. Halliday, Geochim. Cosmochim. Acta, 2022, 317, 349–364 CrossRef CAS.
- N. J. Saunders, J. Barling, J. Harvey and A. N. Halliday, Geochim. Cosmochim. Acta, 2020, 285, 129–149 CrossRef CAS.
- S. J. Wang, W. Z. Wang, J. M. Zhu, Z. Q. Wu, J. A. Liu, G. L. Han, F. Z. Teng, S. C. Huang, H. J. Wu, Y. J. Wang, G. L. Wu and W. H. Li, Nat. Commun., 2021, 12, 294 CrossRef PubMed.
- M. Tanimizu and T. Hirata, J. Anal. At. Spectrom., 2006, 21, 1423–1426 RSC.
- S.-J. Wang, R. L. Rudnick, R. M. Gaschnig, H. Wang and L. E. Wasylenki, Nat. Geosci., 2019, 12, 296–300 CrossRef.
- L. J. Spivak-Birndorf, S.-J. Wang, D. L. Bish and L. E. Wasylenki, Chem. Geol., 2018, 476, 316–326 CrossRef CAS.
- C. Chen, J. Wang, T. J. Algeo, J.-M. Zhu, Z. Wang, X. Ma and Y. Cen, Geochim. Cosmochim. Acta, 2023, 349, 81–95 CrossRef CAS.
- B. Gueguen, J. V. Sorensen, S. V. Lalonde, J. Pena, B. M. Toner and O. Rouxel, Chem. Geol., 2018, 481, 38–52 CrossRef CAS.
- J. V. Sorensen, B. Gueguen, B. D. Stewart, J. Peña, O. Rouxel and B. M. Toner, Chem. Geol., 2020, 537, 1–13 CrossRef.
- K. O. Konhauser, E. Pecoits, S. V. Lalonde, D. Papineau, E. G. Nisbet, M. E. Barley, N. T. Arndt, K. Zahnle and B. S. Kamber, Nature, 2009, 458, 750–753 CrossRef CAS PubMed.
- K. O. Konhauser, L. J. Robbins, E. Pecoits, C. Peacock, A. Kappler and S. V. Lalonde, Astrobiology, 2015, 15, 804–815 CrossRef CAS PubMed.
- G. Quitt and F. Oberli, J. Anal. At. Spectrom., 2006, 21, 1249–1255 RSC.
- B. Gueguen, O. Rouxel, E. Ponzevera, A. Bekker and Y. Fouquet, Geostand. Geoanal. Res., 2013, 37, 297–317 CrossRef CAS.
- F. Moynier, J. Blichert-Toft, P. Telouk, J.-M. Luck and F. Albarède, Geochim. Cosmochim. Acta, 2007, 71, 4365–4379 CrossRef CAS.
- S. M. Chernonozhkin, S. Goderis, L. Lobo, P. Claeys and F. Vanhaecke, J. Anal. At. Spectrom., 2015, 30, 1518–1530 RSC.
- G. Wu, J.-M. Zhu, X. Wang, G. Han, D. Tan and S.-J. Wang, J. Anal. At. Spectrom., 2019, 34, 1639–1651 RSC.
- R. C. J. Steele, T. Elliott, C. D. Coath and M. Regelous, Geochem. Cosmochim. Acta, 2011, 75, 7906–7925 CrossRef CAS.
- F. Nelson, R. M. Rush and K. A. Kraus, J. Am. Chem. Soc., 1960, 82, 339–348 CrossRef CAS.
- Y. An, F. Wu, Y. Xiang, X. Nan, X. Yu, J. Yang, H. Yu, L. Xie and F. Huang, Chem. Geol., 2014, 390, 9–21 CrossRef CAS.
- F. Wu, Y. Qi, H. Yu, S. Tian, Z. Hou and F. Huang, Chem. Geol., 2016, 421, 17–25 CrossRef CAS.
- L. Gall, H. Williams, C. Siebert and A. Halliday, J. Anal. At. Spectrom., 2012, 27, 137–145 RSC.
- J. F. Rudge, B. C. Reynolds and B. Bourdon, Chem. Geol., 2009, 265, 420–431 CrossRef CAS.
- F. Liu, Z. Zhang, X. Li and Y. An, Int. J. Mass Spectrom., 2020, 450, 116307 CrossRef CAS.
- V. Cameron, D. Vance, C. Archer and C. H. House, Proc. Natl. Acad. Sci. USA, 2009, 106, 10944–10948 CrossRef CAS PubMed.
- W. Li, J. M. Zhu, D. Tan, G. Han, Z. Zhao and G. Wu, Geostand. Geoanal. Res., 2020, 44, 523–535 CrossRef CAS.
- M. Sun, C. Archer and D. Vance, Geostand. Geoanal. Res., 2021, 45, 643–658 CrossRef CAS.
- G. Ratié, D. Jouvin, J. Garnier, O. Rouxel, S. Miska, E. Guimarães, L. Cruz Vieira, Y. Sivry, I. Zelano, E. Montarges-Pelletier, F. Thil and C. Quantin, Chem. Geol., 2015, 402, 68–76 CrossRef.
- N. Estrade, C. Cloquet, G. Echevarria, T. Sterckeman, T. Deng, Y. Tang and J.-L. Morel, Earth Planet. Sci. Lett., 2015, 423, 24–35 CrossRef CAS.
- J. Pasava, V. Chrastný, K. Loukola-Ruskeeniemi and O. Sebek, Miner. Deposita, 2018, 54, 719–742 CrossRef.
- H. Beunon, S. M. Chernonozhkin, N. Mattielli, S. Goderis, L.-S. Doucet, V. Debaille and F. Vanhaecke, J. Anal. At. Spectrom., 2020, 35, 2213–2223 RSC.
- M. Li, S. E. Grasby, S. J. Wang, X. Zhang, L. E. Wasylenki, Y. Xu, M. Sun, B. Beauchamp, D. Hu and Y. Shen, Nat. Commun., 2021, 12, 1–7 CrossRef PubMed.
- L. Gall, H. M. Williams, A. N. Halliday and A. C. Kerr, Geochim. Cosmochim. Acta, 2017, 199, 196–209 CrossRef CAS.
- G. T. Ventura, L. Gall, C. Siebert, J. Prytulak, P. Szatmari, M. Hürlimann and A. N. Halliday, Appl. Geochem., 2015, 59, 104–117 CrossRef CAS.
- S.-J. Wang and L. E. Wasylenki, Geochim. Cosmochim. Acta, 2017, 206, 137–150 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |