
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast and high spatially resolved multielemental mapping of otoliths using IR&UV-femtosecond laser ablation-ICP-TOFMS
Jorge
Pisonero
 *a,
Esteban
Avigliano
*b,
Cristian
Soto-Gancedo
a,
Ana
Méndez-Vicente
c,
Cristina
Méndez-López
a,
Jaime
Orejas
a and
Nerea
Bordel
a
*a,
Esteban
Avigliano
*b,
Cristian
Soto-Gancedo
a,
Ana
Méndez-Vicente
c,
Cristina
Méndez-López
a,
Jaime
Orejas
a and
Nerea
Bordel
a
aGrupo de Espectroscopia, Laseres y Plasmas (GELP), Department of Physics, University of Oviedo, C/ Gonzalo Gutiérrez Quirós S/N, Mieres 33600, Spain. E-mail: pisonerojorge@uniovi.es
bUniversidad de Buenos Aires (UBA)-CONICET, Instituto de Investigaciones en Producción Animal (INPA), Facultad de Ciencias Veterinarias, Av. Chorroarín 280, C1427CWO Buenos Aires, Argentina. E-mail: estebanavigliano@conicet.gov.ar
cScientific and Technological Resources (SCTs), University of Oviedo, Gonzalo Gutierrez Quirós S/N, 33600, Mieres, Spain
First published on 16th January 2024
Abstract
A Femtosecond Laser Ablation-Inductively Coupled Plasma-Time of Flight Mass Spectrometer (fs-LA-ICP-TOFMS) was evaluated for multi-elemental mapping of complex otoliths; in particular, for the analysis of a sample of 8 year-old freshwater fish otolith with irregular growth ring distribution pattern. The fundamental wavelength (λ ∼ 1028 nm) and the 4th harmonic (λ ∼ 257 nm) of a diode pumped Yb:KGW fs-laser (<290 fs) were successfully employed for otolith elemental mapping (map area ∼6000 μm2), at high spatial resolution (spot sizes ≤5 μm) and with negligible thermal effects. Fast, sensitive multi-elemental analysis was achieved combining the use of high laser repetition rates (≥200 Hz), high sample scanning speeds (100 μm s−1), a fast response time ablation cell (<40 ms), and the use of a ICP-TOFMS equipped with a reaction/collision cell (operated in He and H2) to remove plasma gas ion related species and polyatomic interferences. The multi-elemental (from 23Na+ to 238U+) fs-LA-ICP-TOFMS mapping of the Prochilodus lineatus lapillus otolith allowed the identification of the appropriate growth axis based on the chronological chemical record associated with the endogenous and exogenous changes experienced by the fish throughout its life. Moreover, each of the detected and quantified elements provided useful biochemical information about environmental and endogenous factors. For instance, maximum P concentration values were observed during spring-summer, a favorable period for feeding and growth.
Introduction
The chemical composition of biocarbonates has been widely used as an environmental indicator and to describe the life history of both terrestrial and aquatic organisms.1,2 Biocarbonates are calcified structures whose morphology and chemical composition is regulated by endogenous and exogenous factors.3,4 Among the most used are the otoliths of bony fish. These structures are housed in the inner ear of fish, are acellular and are composed of calcium carbonate crystals, precipitated mainly in the form of aragonite immersed in a protein matrix.5 The otoliths grow continuously, forming growth rings (similar to a tree), and they incorporate different elements from the surrounding environment, either through water or diet.5 Because they are metabolically inert, they keep a chronological chemical record of the environments experienced by the fish throughout life and have been used as environmental proxies.6,7 The area of early growth (embryo-larva) is known as the core, while the edge of the structure represents the recent life span.The use of Laser Ablation in combination with Inductively Coupled Plasma Mass Spectrometry (LA-ICP-MS) has allowed the determination of selected number of isotopes with high resolution, through core-to-edge transects.6 Typically, nanosecond pulsed laser ablation units (e.g., Nd:YAG and ArF* excimer) operated in the UV region have been employed for the ablation of otoliths at micrometer scale (e.g., laser spot size ∅ = 10–100 s μm).8,9 Shorter pulsed duration lasers, such as femtosecond lasers (fs-LA), are considered to generate laser-induced aerosols with a broad monomodal particle size distribution in the nanometer range (nano-sized agglomerates in the range of 50 up to 250 nm), resulting in higher ion signals and more stable elemental ratios when analyzed by ICP-MS.10,11 Nevertheless, it is considered that reduced elemental fractionation during the fs laser ablation might still be present.12
Multi-elemental ratios (e.g., Sr/Ca, Mg/Ca, Ba/Ca or Na/Ca) or isotopic (e.g., 87Sr/86Sr) data from otolith analyses can be provided by different LA-ICP-MS systems including quadrupole-based (QMS), sector-field-based (SFMS), or multicollector-based (MC-MS) mass spectrometers. It is also possible to couple a laser ablation system with multiple ICP-MS systems using an aerosol split to achieve simultaneous multi-elemental and isotopic data on a fish otolith surface.13 Quantitative results require the use of external certified reference materials (e.g., NIST 610/612 or MACS3), which are employed for drift corrections and to obtain calibration curves or response factors. Nevertheless, recent studies have also shown other quantitative approaches for the determination of trace elements in otoliths without the requirement of external reference material, making use of online isotope dilution (ID)-LA-ICP-MS. This method was based on two aerosols from an ablated sample and a nebulized isotopically enriched spike solution, which were online mixed using a dual-port spray chamber.14
Otolith analyzes usually assume that the calcium carbonate matrix is deposited homogeneously, forming well-defined layers perpendicular to the axis of growth (approximately concentric). However, it has been documented that the heterogeneity in the elemental distribution within each growth ring is greater than assumed.15 In addition, both environmental and endogenous factors can affect the microstructure of the otolith causing structural discontinuities16 and even the form of crystallization of calcium carbonate,15 which could lead to interpretation errors if the readings are made in regions whose growth does not have a chronological order. This problem might be addressed by evaluating the distribution of elements in two dimensions (mapping the whole otolith). In this sense, in the last two decades different methods based on X-ray fluorescence (XRF) have been explored to perform 2D maps in calcified structures. For instance, nuclear microprobe combined with proton-induced energy emission spectroscopy (micro-PIXE)17 was employed to achieve high spatial resolution (5 μm) and moderate/high limits of detection (e.g., >1–2 μg g−1 for Sr18 and >160 μg g−1 for Ba19) for the detection of masses greater than 27Al. More recently synchrotron X-ray fluorescence microscopy (SXFM)15,20 and wavelength-dispersive X-ray electron probe microanalyzer (EPMA)21 have been used. SXFM provides excellent limits of detection (near ng g−1 for some analytes), but relatively long analytical times, ranging from a few hours to 24 hours in extreme cases (depending on the instrument configuration it can exceed 8 h for surfaces >4 mm2).22 Furthermore, this technique require large-scale installations and it has the limitation of mainly detecting masses greater than 40Ca. On the other hand, EPMA achieves high spatial resolution (<3 μm),23 but it provides relatively poor detection limits (>10 μg g−1), not allowing the detection of some key analytes frequently present in otoliths (e.g., Li, Mn, Ba, Se).24 LA-ICP-MS based on the use of sequential mass spectrometers might also allow multiple scans to cover the entire sample surface; however, depending on the size of the spot, the sample scanning speed, the required resolution, and the total integration times, it could take relatively long times. For instance, mapping an area of about 1500 × 4000 μm (6 mm2) using a spot size of 5 μm and a scanning speed of 5 μm s−1 (e.g., low speed for multi-elemental analysis (>20 isotopes)), might take more than 66 h (more than 2 days and a half), resulting in highly expensive and impractical. Nevertheless, the recent combination of advanced laser ablation units (including fast laser repetition rate, high sample scanning speeds and ultra-fast wash-out ablation cells), with ICP-TOFMS systems (including collision/reaction cells and fast detectors that provide high mass spectra acquisition rates), has tremendously enhanced the analytical potential of LA-ICP-MS for fast multi-elemental mapping.25,26 Therefore, in this study, a IR&UV-fs-LA-ICP-TOFMS system is evaluated for fast and sensitive quantitative multi-elemental mapping of complex otoliths. In particular, a sample of 8 year-old Prochilodus lineatus lapillus otolith from the Bermejo River Basin (Argentina), which is subject to regular periods of floods and droughts (summer–winter), was consider as a model of a complex otolith with irregular growth ring distribution pattern.
Experimental
Instrumentation
A NWRFemto UltraCompact laser ablation system from Elemental Scientific Inc (ESL) was used in these experiments. This instrument consists of a diode pumped Yb:KGW fs-laser (<290 fs, Pharos, Light Conversion) that can be operated at the two different wavelengths (λ ∼ 1028 nm (fundamental), and at λ ∼ 257 nm (4th harmonic)). It provides repetition rates from 1 Hz up to 900 Hz, fluence at the sample surface up to 20 J cm−2, and nominal spot sizes from 2 μm to 65 μm of diameter (∅). A fast wash-out ablation cell (TwoVol 2) combined with a capillary inlet system provided washout times shorter than 40 ms. It should be remarked that ultra-fast wash-out ablation cells with response times even shorter than 1 ms are nowadays commercially available. Helium gas (99.999% minimum purity) from AirLiquide was used as cell carrier gas at a constant flow rate of 350 mL min−1. The laser-induced aerosol was transported from the inner cup of the ablation cell to the ICP-TOFMS through a flexible peek tube (<1 m length and 1 mm i. d).The detection was carried out by a Vitesse ICP-TOFMS (Nu Instruments, UK). Operation conditions in the ICP-MS (e.g., ion optics, gas flows) are optimized on a daily basis. In particular, the ion optics are adjusted to obtain the maximum sensitivity monitoring 44Ca+, 89Y+, 139La+, 232Th+, and 238U+ signals produced by the ablation of the standard reference material SRM NIST 610 in raster mode at 100 Hz, 10 μm spot size, 10 μm s−1 and at a fluence of 3 J cm−2. Additionally, the makeup gas flow is adjusted to keep the ratios 238U+/232Th+ below 120%. High purity Ar (99.999% minimum purity) from AirLiquide was employed as plasma and make up gas. Ar was coaxially mixed with the He carrier gas before entering the ICP. Moreover, the mass spectrometer is equipped with a collision/reaction cell operated with a flow rate of He (0.016 L min−1) and H2 (0.006 L min−1) to remove polyatomic interferences, and to remove Ar+ related-ions, protecting the detector from this major plasma element. In this sense, it was possible to detect some polyatomic-interfered key-isotopes in otolith analysis, including 31P, or 80Se. The TOFMS was operated at 30 kHz. Moreover, every two spectra, isotope ion signal are integrated, and background subtracted (considering the ion signals at both sides of the isotope-peak region). This process is repeated 40 times to achieve a full discrete spectrum from Na+ to U+ every 2.13 ms. Additionally, blanking voltage (200 V) based on the use of Bradbury–Nielsen style timed ion gates were applied to remove selected isotopes from major elements before reaching the detector. ICP-TOFMS operating conditions are listed in Table 1.
| Applied power | 1300 W |
| Coolant Ar flow rate | 13 L min−1 |
| Auxiliary Ar flow rate | 2 L min−1 |
| Nebulizer Ar flow rate | 1080 mL min−1 |
| Sample depth | 17.2 mm |
| Entry focus lens | −120 V |
| Collision cell gas | He at 0.016 L min−1 and H2 at 0.006 L min−1 |
| Pushout & pullout HV voltages | 1630 V |
| Reflectron HV | 2117 V |
| Detector HV | 2492 V |
| Acquired mass to charge range | 20–260 m/z |
Sample description
A sample of 8 year-old Prochilodus lineatus lapillus otolith from the Bermejo system (La Plata Basin, Argentina), which is deposited in the COLV (ID-3987) otolith collection (INPA-CONICET-UBA, Argentina), was analyzed. This sample is a good analysis model because the Bermejo system is subject to regular periods of floods and droughts (summer–winter), which severely modifies the environments and physicochemical parameters of the water (e.g., salinity, temperature). These seasonal variations affect otolith growth, resulting in structures with an irregular growth ring distribution pattern. Furthermore, these seasonal variations produce alterations in the incorporation of elements to the otolith. Particularly for this species, including the bermejo river, a direct relationship in Sr (expressed as Sr/Ca ratio) between otolith, water and conductivity was reported, positioning this element in otolith as a good environmental proxy.27The otolith was embedded in epoxy resin (Cristal Tack, Argentina) and then cut a section at core level 800 μm thick using a low-speed saw (Buehler Isomet, China). Otolith slice was mounted on a glass slide using epoxy resin and manually polished with 800–2000 μm grid wet sandpapers. The otolith section was sonicated for 10 min submerged in ultrapure water (18.2 MΩ cm−1) to remove particles generated during polishing.
Reference materials and data treatment software
NIST 610, NIST 612 (trace elements in glass) and MACS3 (trace elements in synthetic calcium carbonate) were employed as reference materials. These samples were analyzed at the beginning and at the end of the otolith mapping studies (bracketing method) to be used for drifts corrections and to calculate relative sensitive factors that allowed the conversion of qualitative data into quantitative multi-elemental maps (Ca was used as an internal standard).Iolite software (v4.8.7)28 was employed for data reduction to obtain multi-elemental maps and transects along different directions in the otolith; and for calculations of LODs and concentration recoveries.
Results and discussion
IR-fs versus UV-fs for otolith analysis
The fs-LA-ICP-TOFMS experimental conditions for fast multi-elemental otolith mapping were optimized considering a compromise among spatial resolution, sensitivity and time of analysis. In particular, laser ablation conditions were selected to achieve the smallest detectable spot sizes in the otolith for each of the two available wavelengths (λ ∼ 1028 nm and λ ∼ 257 nm, respectively) and to apply the same laser spot overlapping percentage while scanning the sample at a constant speed (100 μm s−1). When employing the fundamental wavelength of the Yb:KGW fs-laser (IR wavelength), the smallest circular spot size that provided a proper ablation of otoliths was ∅5 μm, using a fluence of about 7.4 J cm−2. For this spot size the fs laser was operated at 200 Hz to achieve a sample displacement of 0.5 μm per laser shot (90% overlapping requiring 10 laser shots per spot in a total time of 50 ms). However, using the 4th harmonic of the Yb:KGW fs-laser (UV wavelength), the smallest circular spot size that provided a proper ablation of otoliths was ∅4 μm, using a fluence of about 6.6 J cm−2. In that case, the fs laser was operated at 250 Hz to achieve a sample displacement of 0.4 μm per laser shot (90% overlapping requiring 10 laser shots per spot in a total time of 40 ms, which is approximately the response time of the TwoVol2 ablation cell combined with the capillary inlet system).The TOFMS acquisition method was adapted for the different matrices employed in this study to avoid the saturation of the detector. When analyzing NIST 610 and NIST 612 reference materials, blanking was employed to remove 23Na+, 28Si+ and 40Ca+ ion signals considering the high concentration of these elements in these glass samples. In this case, mass spectra were collected from 23.2 to 260 m/z, and a blanking region was applied between 27.8 and 40.2 m/z (also removing 31P+ ion signal). During the analysis of MACS3 reference material and the otolith (both CaCO3 matrices), mass spectra were collected from 20 to 260 m/z, and a blanking region was applied between 33.8 and 42.8 m/z to remove 40Ca+ ion signals. It should be highlighted that the minimum mass blanking window increase, from 5 m/z width to 15 m/z width, at increasing masses because blanking is based on the application of fast time ion gates (≥50 ns). Moreover, blanking process is calibrated to completely remove the signals of the blanked masses while not affecting the sensitivity of the nearby masses.
The otolith multi-elemental mapping analysis covered a region of 1540 μm (height) × 4000 μm (length). This area was divided into two different mapping regions: the upper region (height × length = 768 × 4000 μm2) was ablated using the UV-fs laser (192 parallel raster lines) and it took about 2 h 10 min; while the lower region (height × length = 770 × 4000 μm2) was ablated using the IR-fs laser (154 parallel raster lines) and it took about 1 h 30 min. Therefore, the total multielemental map of the otolith took about 3 h 40 min, which was up to 20 times faster than using typical operating conditions employed in multielemental (≥20 isotopes) LA-ICP-QMS analysis and even faster than the SXFM.20 As an example, Fig. 1 shows the quantitative spatial distribution of P within the otolith (calculated using MACS3 as external reference material), which demonstrates the analytical potential of this method. It is noticed that using both wavelengths at the selected operating conditions it is possible to resolve the growing rings from the otolith core and to determine the heterogenous distribution of this minor element (concentrations lower than 80 μg g−1). Scale based on non-linear ECDF (Empirical Cumulative Distribution Function) that maximizes contrast of the image.29Fig. 1 inlet shows in higher detail the spatial distribution of P in the otolith (zoom of the region highlighted with a dashed rectangle). Mass spectra could be exported every 2.13 ms; however, maps were built up considering an average mass spectra per spot in the sample in order to minimize the amount of data providing comprehensive information. Therefore, considering 10 laser shots per spot, approx. 1520 spectra (# spectra measured in 40 ms) were average per 4 μm spot when using UV-fs-LA, and approx. 1920 (# spectra measured in 50 ms) spectra were average per 5 μm spot when using the IR-fs-laser. Furthermore, the use of both femtosecond laser wavelengths (IR and UV) provided analogous P concentrations, highlighting the potential use of both of them for the analysis of otoliths.
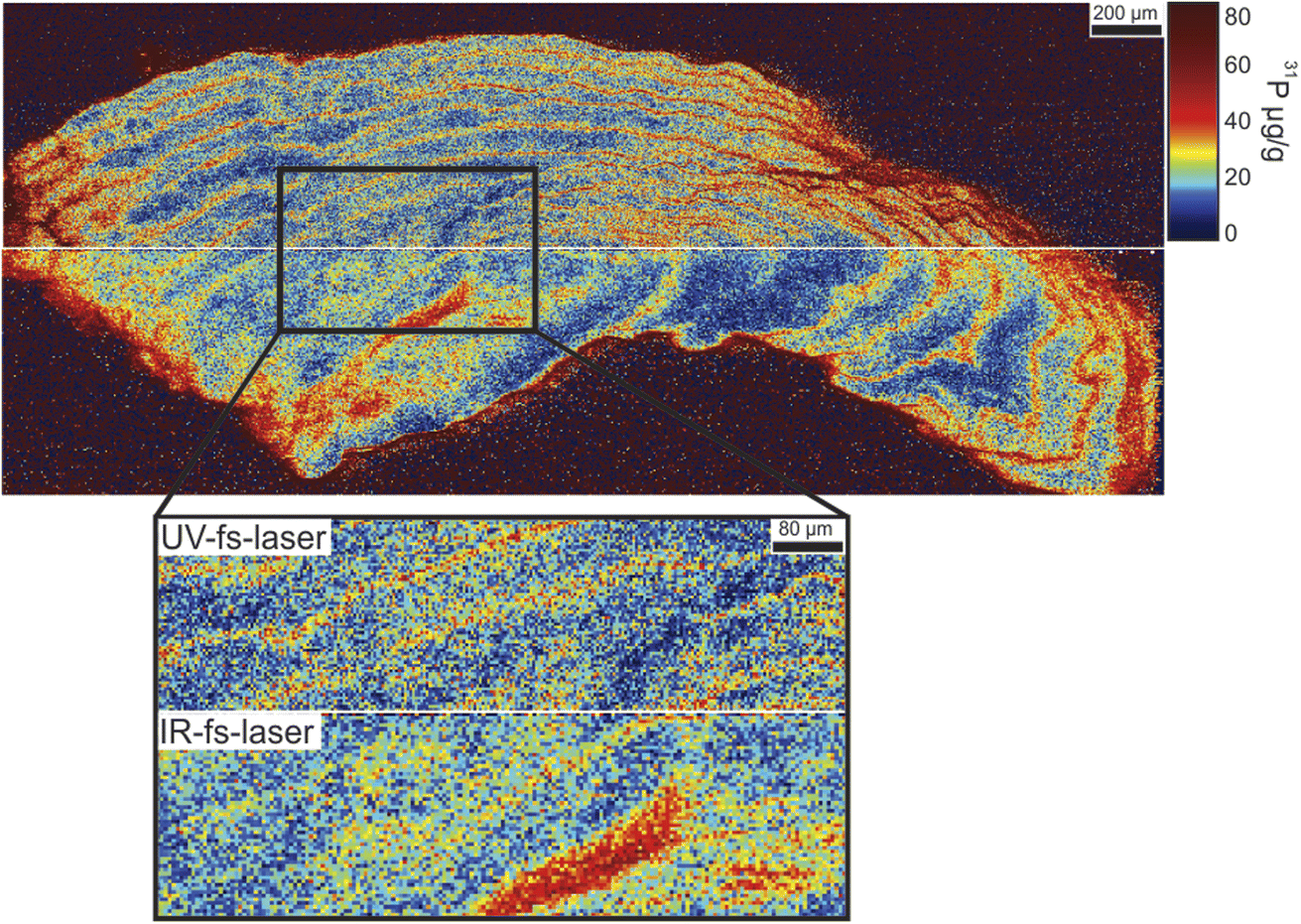 | ||
| Fig. 1 Quantitative spatial distribution of P within the otolith. Upper map was obtained using UV-fs-LA; while the lower map was obtained using IR-fs-LA. Figure inlet shows in higher detail the spatial distribution of P in the otolith (zoom of the region highlighted with a dashed rectangle). Scale based on non-linear ECDF (Empirical Cumulative Distribution Function). | ||
Otolith elemental distribution
The usefulness of otolith features as indicators depends, at least in part, on knowledge about the dynamics of incorporation into the otolith. Various elements present in otoliths can be used as environmental (e.g., temperature and salinity) or physiological (e.g., hatching, reproduction, growth) indicators. Trace element incorporation into the otolith is governed by exogenous and endogenous factors. Among all the different isotopes that were measured from Na to U, 23Na, 24Mg, 55Mn, 80Se, 88Sr, and 138Ba are shown in detail as they show spatial heterogeneity, and their concentrations are consistently above the LOD. Fig. 2 shows an image of the otolith morphology, and the full quantitative spatial distributions of the other analytes within the otolith, merging the analysis carried out using both laser wavelengths (scale based on non-linear ECDF, except for the trace analytes 55Mn and 80Se where a linear scale was employed for clarity purposes). Qualitative data (analyte ion signals measured in cps) were converted into concentration values (μg g−1) using 44Ca as internal standard (38.8 wt%),30 and using MACS3 as external reference material, except for 80Se were NIST 610 was used as external reference material (Se concentration is not certified in MACS3). Moreover, Table 2 lists the LODs estimated for the selected isotopes using Iolite software. Calculations were performed on MACS3 reference material and using Longerich et al. criteria based on background noise.31 In particular, minimum and maximum values of calculated LODs from the n = 8 analysis of MACS3 reference material (4 before and 4 after otolith analysis) are listed. It is observed that UV-fs-LA provided slightly improved LODs versus IR-fs-LA. Furthermore, it is remarked that the applied methodology based on fs-LA-ICP-TOFMS showed lower detection limits than those reported for other methods such as micro-PIXE (>160 μg g−1 for Ba19) and EPMA (>10 μg g−1 for Mn, Ba and Se24), which is optimal for the detection of elements present at relatively low concentrations. | ||
| Fig. 2 Quantitative spatial distribution of elements in a cross section of a Pochilodus lineatus lapillus otolith determined by UV&IR-fs-LA-ICPTOFMS (upper and lower maps were merged). V: ventral; D: dorsal; I: internal; E: external face. Scale based on non-linear ECDF (Empirical Cumulative Distribution Function); except for Mn and Se trace analytes, whose scale is linear. Scale concentration units: μg g−1. | ||
| Analyte | Limit of detection (μg g−1) using UV-fs-LA-ICP-TOFMS | Limit of detection (μg g−1) using IR-fs-LA-ICP-TOFMS |
|---|---|---|
| 23Na | 15–20 | 15–45 |
| 24Mg | 1.5–3.5 | 5–9 |
| 31P | 3–7 | 3–4 |
| 55Mn | 0.55–0.65 | 0.55–0.7 |
| 80Se** | 0.4–1.4 | 1.5–3 |
| 88Sr | 0.5–4 | 1.5–14 |
| 138Ba | 0.1–0.4 | 0.3–0.5 |
Analyte concentrations and the recoveries were also calculated for NIST 610 and for NIST 612, using MACS3 as calibrating material (non-matrix matched calibration). Tables 3 and 4 list the certified analyte concentrations of MACS3, and the calculated analyte concentrations and recoveries in NIST 610 and NIST 612 obtained using UV- and IR-fs-LA, respectively. Data related to analytes that were not detected during the analysis of the otoliths (highlighted with “*”) were included in the Tables to expand the mass spectra range of the study. It is noticed that most recoveries showed improved values when using ultraviolet radiation. Additionally, it is observed that worse values were obtained for the quantification of NIST 612 (more transparent glass), except for analytes such as Pb and Th, whose concentration was very similar in NIST 612 and in MACS3. The worst recovery case was found for the quantification of Fe in NIST 612, probably due to the fact that Fe is highly concentrated in MACS3 and also heterogeneously distributed.
| Analyte | Certified concentration MACS3 (μg g−1) | Calculated – nominal concentrations (μg g−1) and recoveries from NIST 610 | Calculated – nominal concentrations (μg g−1) and recoveries (%) from NIST 612 |
|---|---|---|---|
| 55Mn | 534 ± 24 | 387–444 | 28–39 |
| 87% | 72% | ||
| 56Fe | 9987 ± 1728 | 205–458 | 14–51 |
| 45% | 28% | ||
| 59Co* | 54 ± 4 | 406–410 | 26–35 |
| 99% | 73% | ||
| 63Cu* | 113 ± 4 | 393–441 | 37–38 |
| 89% | 98% | ||
| 88Sr | 6675 ± 245 | 593–515 | 82–78 |
| 115% | 105% | ||
| 138Ba | 58 ± 3 | 480–452 | 37–39 |
| 106% | 94% | ||
| 208Pb* | 60 ± 3 | 247–426 | 30–39 |
| 58% | 79% | ||
| 232Th* | 54 ± 2 | 568–457 | 38–38 |
| 124% | 101% |
| Analyte | Certified concentration MACS3 (μg g−1) | Calculated – nominal concentrations (μg g−1) and recoveries (%) from NIST 610 | Calculated – nominal concentrations (μg g−1) and recoveries (%) from NIST 612 |
|---|---|---|---|
| 55Mn | 534 ± 24 | 346–444 | 22–39 |
| 78% | 57% | ||
| 56Fe | 9987 ± 1728 | 153–458 | 1–51 |
| 34% | 2% | ||
| 59Co* | 54 ± 4 | 385–410 | 20–35 |
| 94% | 56% | ||
| 63Cu* | 113 ± 4 | 362–441 | 27–38 |
| 82% | 71% | ||
| 88Sr | 6675 ± 245 | 513–515 | 58–78 |
| 99% | 74% | ||
| 138Ba | 58 ± 3 | 441–452 | 28–39 |
| 98% | 73% | ||
| 208Pb* | 60 ± 3 | 292–426 | 20–39 |
| 69% | 52% | ||
| 232Th* | 54 ± 2 | 688–457 | 41–38 |
| 150% | 108% |
Fig. 2 provided elemental-dependent information obtained from the high-spatially resolved multi-elemental maps. It is observed that these analytes are heterogeneously distributed, both in the transversal axis to the direction of growth (temporal variation) and within the same period or growth ring. Specifically, 23Na showed a heterogeneous distribution within the growth rings, with the quantity being greater in the external edge and internal area of the otolith. 24Mg showed the highest levels around the core (<36.5 μg g−1). Moreover, it showed discontinuous rings (approximately at the first annual mark) and discrete areas (e.g., external-ventral area) with relatively high values (<75.9 μg g−1). 55Mn distribution pattern similar to Mg. It showed a temporal distribution with relatively low heterogeneity, except for the core where high values (<1.15 μg g−1) were recorded with a distribution parallel to the dorsal edge. The rest of the structure showed discontinuous bands with relatively high concentration (<0.73 μg g−1). 80Se showed a weak annual periodicity. Two discontinuous rings (age 7 and 8 years) of relatively high concentration (5.1–5.6 μg g−1) were observed. A cyclic temporal variation was observed for 88Sr, with higher concentrations (<6850 μg g−1) at the inner and external area of the otolith. Annual periodicity was also found for 138Ba, with heterogeneous distribution within the growth marks.
Concentration variations from three core-to-edge profiles (transects) were extracted as a representation of three classic scenarios of elemental analysis in otoliths. For instance, Fig. 3 shows the concentration variations for 88Sr and 31P along these three core-to-edge transects. The three core-to-edge transects showed very different profiles for Sr: transect 1, which was carried out in the portion where the growth rings showed a homogeneous structuring, revealed a dominant peak (5408 μg g−1) in the core area (age < 1 year) and eight oscillations with annual periodicity starting from the first year; transect 2 also showed an annual periodicity, but with a different pattern, especially in the core where a double top is evident. These peaks are also of lesser magnitude in relation to the first profile (5489 μg g−1); in transect 3 the first annual mark is lost, and 7 oscillations are observed. A more pronounced double top is observed in the core, with highest values (<6854 μg g−1) than the other transects. In relation to P, only transect 3 registered the maximum concentration values due to the high heterogeneity in the core.
 | ||
| Fig. 3 Core-to-edge Sr and P profiles for transects performed in three different sections of the otolith. The red arrow on the otolith indicates the trajectory of the core-to-edge analysis and the white dots the position of the annual growth rings. The position of the rings (age) is also represented with gray vertical lines, which can be followed between the different analytical trajectories. The colored boxes within the first year of life indicate the same period represented in the different trajectories. | ||
Moreover, Fig. 4 shows the concentration variations for 23Na, 24Mg, and 138Ba, along these three core-to-edge transects. Sodium was the element with the highest concentration (808–2790 μg g−1) and showed a temporally heterogeneous distribution with periodic oscillations mainly in the ventral-external region. Like Mg, the core was the area with the highest concentration. In relation to Ba distribution, within the first year of life, a plateau was observed in the three transects, with the distribution being greater in the third (transect 1: <11.7 μg g; transect 2: <11.0 μg g−1, transect 3: <23.8 μg g−1). Transect 3 also shows an area of high concentration of 138Ba (∼63.0 μg g−1) in the first annual rings and restricted to the ventral-internal area of the otolith.
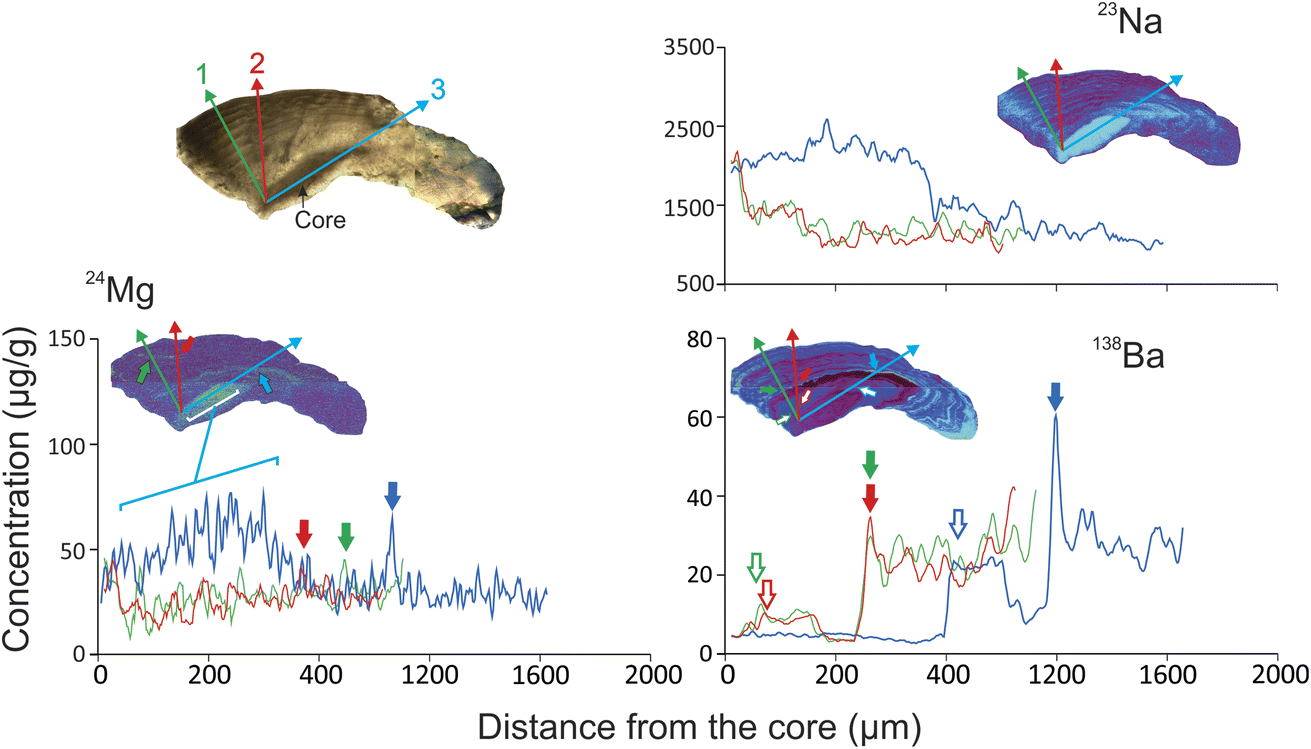 | ||
| Fig. 4 Core-to-edge elemental profiles for transects performed in three different sections of the otolith. The arrows indicate the same period represented in the different trajectories. | ||
Otolith chemistry as a bioindicator
The elements that make up metalloproteins in the hemolymph of the inner ear (such as 31P) are physiologically regulated and reflect endogenous processes (e.g., reproduction, growth, metamorphosis) rather than environmental (e.g., physicochemistry of water).32 Only one study reported a pilot analysis of the seasonal distribution of P in the otolith, suggesting that the incorporation of this element would be linked to the deposition of proteins in the biocarbonate matrix and would be favored by periods of rapid growth.33 Our results support this hypothesis because maximum P values were observed during spring-summer, a favorable period for feeding and growth.Other elements such as Mg, Mn, Sr, Ba are mainly associated with the salt-fraction of the endolymph and would be the best option to resolve unknowns associated with habitat use.32 Reports on Se in otoliths are scarce, with one study suggesting that this could be a natural marker in environments with heterogeneous distribution of this element.34 In the present study, two marks with relatively high Se concentration were reported, so it is worth exploring the use of this element as an environmental proxy.
Conclusions
IR- and UV-fs-LA-ICPTOFMS has demonstrated a great potential for fast, high-spatially resolved, multi-elemental analysis of otoliths, allowing the visualization of the growing rings from the otolith core and the determination of heterogenous distribution of major and minor elements. IR- and UV-fs-LA-ICPTOFMS can be up to 20 times faster than using typical operating conditions employed in multi-elemental LA-ICP-QMS analysis and even faster than SXFM. Both methods also provided lower detection limits than those reported for other micro-PIXE (e.g., Ba) and EPMA (e.g., Mn, Ba and Se) resulting in a good analytical option for elements that have a relatively low concentration. Despite the advantages of reduced analysis times, high lateral resolution and low LODs, access to fs-LA-ICPTOFMS system is relatively limited. Furthermore, it was found that the use of UV-fs-LA, at the selected operating conditions, provided slightly improved results in terms of limits of detection and recoveries calculated using non-matrix matched calibrations.Elements such as Sr, Ba and P revealed a heterogeneous growth pattern with overlapping and even discontinuous growth rings. In addition to the heterogeneity in the microstructure, a heterogeneous distribution of elements within the same growth ring was observed. For example, Sr, Ba, and P showed higher values in the inner portion of the otolith, while discrete or discontinuous areas with relatively high values of Se, Mg, and Mn were observed. This fact highlights the usefulness of mapping to decide which area should be analyzed and avoid errors in interpretation.
The heterogeneous distribution of growth rings and elements (even within continuous growth rings) suggests differential crystallization mechanisms. Many species exhibit vaterite incrustations in addition to aragonite. These areas usually present rather disorganized growth that can be difficult (or impossible) to identify using optical equipment and divergent element concentrations with respect to aragonite.35 In addition to the form of crystallization, the structural heterogeneity may be due to cessation of calcium carbonate deposition that affects the “rhythmic growth”,16 which could be due to different factors such as environmental stress (very cold winters, temperature changes, flow) or reproduction, among others.36
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from project MCI-21-PID2020-113951GB-I00, funded by the Spanish Ministerio de Ciencia e Innovación, and PIBAA 2872021010 supported by CONICET. Acknowledgments are also given to Lukas Schlatt and Phil Shaw from Nu Instruments, and to David Douglas, Jay Wilkins and Bence Paul from ESI, for all their support and assistance in the instrumental and software set-up and maintenance. Cristina Méndez-López also acknowledges the support of the “Severo Ochoa Grant” (BP20-059) from the Principality of Asturias, Spain.References
- O. E. Tzadik, J. S. Curtis, J. E. Granneman, B. N. Kurth, T. J. Pusack, A. A. Wallace, D. J. Hollander, E. B. Peebles and C. D. Stallings, Chemical archives in fishes beyond otoliths: A review on the use of other body parts as chronological recorders of microchemical constituents for expanding interpretations of environmental, ecological, and life-history changes. Limnol Oceanogr Methods, Limnol. Oceanogr.: Methods, 2017, 15, 238–263 CrossRef
.
- S. Barakat, M. Le Corre, M. Willmes, J. Cohen, M. Vuillien, E. Desclaux and K. Britton, Laser ablation strontium isotopes and spatial assignment show seasonal mobility in red deer (Cervus elaphus) at Lazaret Cave, France (MIS 6), Front. Ecol. Evol., 2023, 11, 988837 CrossRef
- A. M. Sturrock, E. Hunter, J. A. Milton, R. C. Johnson, C. P. Waring and C. N. Trueman, Quantifying physiological influences on otolith microchemistry, Methods Ecol. Evol., 2015, 6, 806–816 CrossRef
- A. Annabi, K. Said and B. Reichenbacher, Inter-population differences in otolith morphology are genetically encoded in the killifish Aphanius fasciatus (Cyprinodontiformes), Sci. Mar., 2013, 77, 269–279 CrossRef
- O. R. B. Thomas and S. E. Swearer, Rev. Fish. Sci. Aquac., 2019, 27, 458–489 CrossRef
- E. Avigliano, Optimizing the methodological design in fish stock delineation from otolith chemistry: review of spatio-temporal analysis scales, Rev. Fish. Sci. Aquac., 2022, 30(3), 330–345 CrossRef
- R. J. Brown and K. P. Severin, Otolith chemistry analyses indicate that water Sr:Ca is the primary factor influencing otolith Sr:Ca for freshwater and diadromous fish but not for marine fish, Can. J. Fish. Aquat. Sci., 2009, 66, 1790–1808 CrossRef CAS
- E. Avigliano, A. Ibañez, N. Fabré, R. C. Fortunato, A. Méndez, J. Pisonero and A. V. Volpedo, White mullet Mugil curema population structure from Mexico and Brazil revealed by otolith chemistry, J. Fish Biol., 2020, 97(4), 1187–1200 CrossRef CAS PubMed
- E. Avigliano, J. Pisonero, A. Dománico, N. Silva, S. Sánchez and A. V. Volpedo, Spatial segregation and connectivity in young and adult stages of Megaleporinus obtusidens inferred from otolith elemental signatures: implications for managment, Fish. Res., 2018, 204, 239–244 CrossRef
- V. Moná, J. Pisonero, M. Holá, V. Kanický and D. Günther, Quantitative analysis of Fe-based samples using ultraviolet nanosecond and femtosecond laser ablation-ICP-MS, J. Anal. At. Spectrom., 2006, 21, 1194–1201 RSC
- J. Koch, M. Wälle, J. Pisonero and D. Günther, Performance characteristics of ultra-violet femtosecond laser ablation inductively coupled plasma mass spectrometry at ∼265 and ∼200 nm, J. Anal. At. Spectrom., 2006, 21, 932–940 RSC
- R. Le Harzic, N. Huot, E. Audouard, C. Jonin, P. Laporte, S. Valette, A. Fraczkiewicz and R. Fortunier, Comparison of heat-affected zones due to nanosecond and femtosecond laser pulses using transmission electronic microscopy, Appl. Phys. Lett., 2002, 80, 3886 CrossRef CAS
- T. Prohaska, J. Irrgeher and A. Zitek, Simultaneous multi-element and isotope ratio imaging of fish otoliths by laser ablation split stream ICP-MS/MC ICP-MS, J. Anal. At. Spectrom., 2016, 31, 1612–1621 RSC
- K. Yanagisawa, M. Matsueda, M. Furukawa, H. Ishiniwa, T. Wada, T. Hirata and Y. Takagai,
Analyst Quantitative imaging of trace elements in solid samples by online isotope dilution laser ablation, -inductively coupled plasma-mass spectrometry, 2023, 148, 4291–4299 CAS
- K. E. Limburg and M. Elfman, Insights from two-dimensional mapping of otolith chemistry, J. Fish Biol., 2017, 90, 480–491 CrossRef CAS PubMed
-
J. Panfili, J. Tomas and B. Morales-Nin Otolith, Microstructure in Tropical Fish, In Tropical fish otoliths: information for assessment, management and ecology, Springer Netherlands, Dordrecht, 2009, pp. 212–248 Search PubMed
- M. Elfman, K. E. Limburg, P. Kristiansson, H. Svedäng, L. Westin, H. Wickström, K. Malmqvist and J. Pallon, Complex life histories of fishes revealed through natural information storage devices: case studies of diadromous events as recorded by otoliths, Nucl. Instrum. Methods Phys. Res., Sect. B, 2000, 161–163, 877–881 CrossRef CAS
- J. A. Babaluk, J. L. Campbell, C. L. Evans, N. M. Halden, S. R. Mejia, Z. Nejedly, J. D. Reist and W. J. Teesdale, Micro-PIXE analysis of strontium in Arctic char, Salvelinus alpinus, otoliths from Quttinirpaaq National Park, Nunavut, Canada, Nucl. Instrum. Methods Phys. Res., Sect. B, 2002, 189, 190–195 CrossRef CAS
- P. Kuisma-Kursula Accuracy, Precision and Detection Limits of SEM-WDS, SEM-EDS and PIXE in the Multi-Elemental Analysis of Medieval Glass, X-Ray Spectrom., 2000, 29, 111–118 CrossRef
- K. E. Limburg, R. Huang and D. H. Bilderback, Fish otolith trace element maps: new approaches with synchrotron microbeam x-ray fluorescence, X-Ray Spectrom., 2007, 36, 336–342 CrossRef CAS
- Y. Hu, T. Jiang, H. Liu, X. Chen and J. Yang, Otolith Microchemistry Reveals Life History and Habitat Use of Coilia nasus from the Dayang River of China, Fishes, 2022, 7, 306 CrossRef
- I. Del Real, L. Smieska, J. F. H. Thompson, C. Martinez, J. Thomas and D. Layton-Matthews, Using multiple micro-analytical techniques for evaluating quantitative synchrotron-XRF elemental mapping of hydrothermal pyrite, J. Anal. At. Spectrom., 2019, 34, 1724–1738 RSC
- A. McFadden, B. Wade, C. Izzo, B. M. Gillanders, C. E. Lenehan and A. Pring, Quantitative electron microprobe mapping of otoliths suggests elemental incorporation is affected by organic matrices: implications for the interpretation of otolith chemistry, Mar. Freshwater Res., 2015, 67, 889–898 CrossRef
- V. G. Batanova, A. V Sobolev and V. Magnin, Trace element analysis by EPMA in geosciences: detection limit, precision and accuracy, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 304, 012001 Search PubMed
- S. Strekopytov, K. Billimoria and H. Goenaga-Infante, A systematic study of high resolution multielemental quantitative bioimaging of animal tissue using LA-ICP-TOFMS, J. Anal. At. Spectrom., 2023, 38, 704–715 RSC
- T. Van Helden, S. Braeuer, T. Van Acker, O. Leroux, D. Van Der Straeten and F. Vanhaecke, High-speed mapping of Hg and Se in biological tissue via laser ablation-inductively coupled plasma-mass spectrometry, J. Anal. At. Spectrom., 2022, 37, 1455–1461 RSC
- E. Avigliano, J. Pisonero, J. Bouchez, M. Pouilly, A. Domanico, S. Sánchez, C. Clavijo, P. Scarabotti, J. Facetti, J. Caffetti, F. Del Rosso, S. Llamazares Vegh and A. Volpedo, Otolith Sr/Ca ratio complements Sr isotopes to reveal fish migration in large basins with heterogeneous geochemical landscapes, Environ. Biol. Fishes, 2021, 104, 277–292 CrossRef
- C. Paton, J. Hellstrom, B. Paul, J. Woodhead and J. Hergt Iolite, Freeware for the visualisation and processing of mass spectrometric data, J. Anal. At. Spectrom., 2011, 26, 2508 RSC
- M. Rittner and W. Müller, 2D mapping of LA-ICPMS trace element distributions using R, Comput. Geosci., 2012, 42, 152–161 CrossRef CAS
- J. Yoshinaga, A. Nakama, M. Morita and J. S. Edmonds, Fish otolith reference material for quality assurance of chemical analyses, Mar. Chem., 2000, 69, 91–97 CrossRef CAS
- H. P. Longerich, S. E. Jackson and D. Günther Inter-laboratory note, Laser ablation inductively coupled plasma mass spectrometric transient signal data acquisition and analyte concentration calculation, J. Anal. At. Spectrom., 1996, 11, 899–904 RSC
- O. R. B. Thomas, K. Ganio, B. R. Roberts and S. E. Swearer, Trace element-protein interactions in endolymph from the
inner ear of fish: implications for environmental reconstructions using fish otolith chemistry, Metallomics, 2017, 9, 239–249 CrossRef CAS PubMed
- K. Hüssy, K. E. Limburg, H. De Pontual, O. R. B. Thomas, K. Cook, Y. Heimbrand, M. Blass and A. M. Sturrock, Element patterns in otoliths: the role of biomineralization, Rev. Fish. Sci. Aquac., 2020, 1–33 Search PubMed
- K. E. Limburg, A. Lochet, D. Driscoll, D. S. Dale and R. Huang, Selenium detected in fish otoliths: A novel tracer for a polluted lake?, Environ. Biol. Fishes, 2010, 89, 433–440 CrossRef
- B. M. Pracheil, B. C. Chakoumakos, M. Feygenson, G. W. Whitledge, R. P. Koenigs and R. M. Bruch, Sturgeon and paddlefish (Acipenseridae) sagittal otoliths are composed of the calcium carbonate polymorphs vaterite and calcite, J. Fish Biol., 2017, 90, 549–558 CrossRef CAS PubMed
-
J. Panfili, H. Pontual, H. Troadec and P. J. Wright, Manual of Fish Sclerochronology, Ifremet-IRD, Brest, 2002 Search PubMed
| This journal is © The Royal Society of Chemistry 2024 |