
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategies to enable microsized alloy anodes for high-energy and long-life alkali-ion batteries†
Amine
Daali
ab,
Rachid
Amine
c,
Wilkistar
Otieno
b,
Gui-Liang
Xu
 *a and
Khalil
Amine
*a
*a and
Khalil
Amine
*a
aChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA. E-mail: xug@anl.gov; amine@anl.gov
bDepartment of Industrial and Manufacturing Engineering, College of Engineering and Applied Science, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA
cMaterials Science Division, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 16th February 2024
Abstract
Micro-sized anode materials demonstrate greater potential for practical applications than nanomaterials in the aspects of volumetric energy density, coulombic efficiency, fabrication process, and cost. However, the huge volume changes of alloy anodes (up to ∼500%) during repeated charge/discharge has led to a series of challenging issues including pulverization of active material particles and delamination from current collectors, formation of thick and fragile solid-electrolyte interphase (SEI) and depletion of electrolytes, eventually leading to rapid cell degradation. Herein, we review recent progress of rational strategies to enable the use of microsized alloy anodes (Si, P, Sb, Sn, etc.) including electrolyte modulation, binder design and architecture engineering. We also provide perspectives on future directions and remaining challenges of microsized anodes towards practical applications.
Keywords: Volume change; Alloy; Anodes; Microsized; Alkali-ion; Batteries.
 Amine Daali | Amine Daali is currently a PhD candidate at University of Wisconsin-Milwaukee. He is also a co-op student at Argonne National Laboratory. His research focuses on the development of high-capacity electrode materials for room temperature lithium and sodium-ion batteries. |
 Rachid Amine | Rachid Amine is an assistant chemist at Argonne National Laboratory (ANL). He received his PhD in Chemical Engineering at the University of Illinois at Chicago December of 2020 and his Master of Science in Chemical Engineering in December of 2009 from Illinois Institute of Technology. Rachid's research projects included: synthesis and characterization of high energy and high power materials for automotive application; electrochemical evaluation and thermal characterization lithium ion batteries, safety improvement by surface modification of active materials. |
 Wilkistar Otieno | Wilkistar Otieno is an Associate Professor and Chair of Industrial and Manufacturing Engineering at the University of Wisconsin-Milwaukee. Her research interests include data analytics applied to manufacturing, sustainable manufacturing particularly remanufacturing, reliability engineering and overall systems optimization. She has a passion for engineering education and is a principal investigator of an NSF STEM grant and serves as a long-term faculty mentor for the UWM Wisconsin Alliance for Minority Participation, also an NSF grant. |
 Gui-Liang Xu | Dr. Gui-Liang Xu is a chemist in the Electrochemical Energy Storage group under the division of Chemical Sciences and Engineering at Argonne National Laboratory. His research focuses on both fundamental understanding and materials development for advanced lithium-ion batteries and beyond. Dr. Xu earned his Bachelor (2009) and PhD (2014) degree in the department of Chemistry of Xiamen University. Dr. Xu has authors/co-authored over 120 peer-reviewed research articles and has 4 granted U.S. patent and several patent applications. |
 Khalil Amine | Dr. Khalil Amine is Argonne Distinguished Fellow and the leader of the Advanced Battery Technology team at Argonne National Laboratory, where he is responsible for directing the research and development of advanced materials and battery systems for HEV, PHEV, EV, and satellite applications. Among his many awards, Dr. Amine is the 2019 reception of the prestigious Global Energy Prize. He is a six-time recipient of the R&D 100 Award, which is considered as the Oscar of technology and innovation. He is an ECS fellow, and associate editor of the journal of Nano Energy. |
1 Introduction
This smart and digital era is highly energy-consuming and demanding. The depletion of fossil fuels has heightened concerns over energy security, leading to increased research into developing energy storage devices (ESDs).1 These ESDs play a vital role in our daily lives by powering an array of electrical devices, from mobile phones to electric vehicles. Furthermore, they are crucial in addressing the intermittent issues associated with emerging natural resources of energy (tidal, solar, wind, etc.). Over the past few decades, alkali-ion batteries (LIBs, SIBs, …) have garnered significant attention for their potential in energy storage applications. Lithium-ion batteries (LIBs), with their high energy capacity, superior long lifespan, and adaptable design, have emerged as the market leader.2 However, the cost of LIBs remains a concern owing to the limited and uneven distribution of lithium resources on Earth. On the other hand, sodium-ion batteries (SIBs) offer a promising alternative for large-scale energy storage, given the abundance of sodium resources and competitive pricing. As SIBs exhibit electrochemical properties akin to LIBs, it is anticipated that the development of SIBs is expected to leverage the well-mature lithium-ion manufacturing and industry.3,4Modern electronic devices, especially electric vehicles, demand LIBs/SIBs that possess both high energy density and enhanced safety. This has driven the development of electrode materials with substantial capacity and sustained stability.5 Interestingly, when compared to cathode materials, the anode materials in LIBs and SIBs show a higher promise for achieving superior energy densities. This is attributed to their varied nature and impressive theoretical capacity.6
Typically, anodes are categorized into three main types based on their charge storage mechanism: insertion, conversion, and alloying.7
Materials of the insertion variety, like carbon and those based on titanium, support the reversible (de-) insertion of alkali metal ions without harming their crystalline frameworks, resulting in impressive durability and stability during repeated cycles. Graphite, having a capacity of 372 mA h g−1, is the primary choice for LIB anodes due to its consistent performance and superb cycling stability. However, its low discharge plateau (0.2 V vs. Li/Li+) leads to Li dendrite growth during fast charging processes.8
On the other hand, conversion materials, such as phosphide, sulfide, and transition metal oxides, undergo multiple electron reactions with Li/Na. This leads to the formation of metallic particles and alkali metal compounds and, thus, exhibit high specific capacities. However, the challenge of initiating nucleation in mixed phases causes notable voltage hysteresis, substandard reversibility, and a drop in coulombic efficiency (CE), in which the CE is defined as the ratio of the charge specific capacity over the discharge specific capacity. Furthermore, the volume change during the cycling process leads to expedited structural degradation and reduced capacity retention.9
Due to their alloying mechanism with Li/Na, alloy-based anode materials tend to display ∼3 times higher specific capacity than conversion materials, forming stable Li- or Na-rich phase alloys. At room temperature, many metals or metalloids exhibit alloying activity towards Li or Na, including but not limited to Si, Ge, Sn, Sb, Bi, and P.10,11 These alloying materials offer great commercial potential due to their high storage capacities, excellent electronic properties, and appropriate discharge plateaus. Si/C composites with minimal Si content lead the pack as potential anodes for LIBs. However, a loss of capacity is inevitable due to the volume change caused by the high capacity, leading to three major challenges. Firstly, the structural breakdown from expansion disrupts internal electrical connectivity, diminishing the capacity. Next, shattered fragments tend to aggregate and clump together, obstructing ion movement. Lastly, an increase in the charge transfer impedance due to the continuous reactions between active material and electrolytes forming unstable solid-electrolyte interphase (SEI). Additionally, repetitive volume alteration makes electrode films prone to distortion, detachment, and delamination from current collectors. Even though the active material has not collapsed entirely, the detached electrode film ceases to contribute to the system's capacity. This event is a critical factor that hinders the widespread adoption of alloying materials in commercial applications due to their limited lifespan.12
In order to prevent the detrimental effects of lithiation/sodiation-induced volume changes and avoid pulverization, researchers have employed various nano- and micro-scale structures and morphologies to enable reversible (de)lithiation/(de)sodiation in alloying anode materials. These approaches, which utilize both pure alloying elements and composite materials, primarily focus on increasing the surface-to-volume ratio (SVR) of the alloying anode's morphology. Increasing the SVR reduces concentration and stress–strain gradients from lithiation/sodiation significantly by decreasing the required lengths for ion diffusion on the overall lithiation/sodiation behavior. Furthermore, an increase in SVR results in a higher rate of lithiation/sodiation and an increase in the maximum (dis)charging power available in the anode.13–15
One of the most common and well-studied approaches toward increasing the SVR is to create nanostructured morphologies. The introduction of nanowires in 2008 is considered an important milestone, which effectively accommodated volume expansion during the Li–Si alloying process.16 Since then, there's been a surge in the exploration of various nano-dimensional structures, including nanodots,17 nanopores,18 and 2D nanosheets.19 These materials have the potential to mitigate SEI destruction and overconsumption of electrolytes and subsequently improve cyclic stability.19–21 Additionally, due to optimal contact between active material and electrolytes, the high SVR of these materials promotes a better kinetics diffusion and enhances the rate performance.20–22 Yet, it is noteworthy that the initial coulombic efficiency (ICE) is not satisfactory due to an irreversible capacity loss during the first cycle caused by the large contact area between electrolytes and nanostructured materials.23 Moreover, the low tap density of nanostructures might compromise the battery's volumetric energy density.24 It is also noteworthy that well-designed nanomaterials require complicated crafting and intricate production methods, incurring high manufacturing costs.25,26
When comparing micro-sized materials to their nano-sized counterparts, the former exhibits greater potential for practical applications in several aspects, including volumetric energy density, coulombic efficiency, fabrication process, and cost.27–31 The advantages of micro-sized materials include higher tap density, resulting in higher mass loadings and subsequently boosting the volumetric capacity and energy storage. This characteristic is especially beneficial for compact and portable energy storage solutions.32 Additionally, the low specific surface area of micro-sized materials reduces the contact area with electrolytes, leading to a mitigated capacity decay in the first cycle and enhanced ICE.24,27,29,31 Furthermore, the ease of fabrication and resulting cost advantages make micro-sized materials attractive for practical applications.33 Considering these strengths, micro-sized alloy anodes with substantial specific capacity, low reaction potential, and impressive tap density emerge as promising contenders for the large-scale production of high-energy battery systems.
In the realm of material science, it has been observed that the formation of the MxAy alloy phase leads to a significant volume expansion. During the process of sodiation, the high-capacity Pred anode undergoes a volume change of 499%.34 At the same time, the Sn anode experiences a volume expansion of ≈260% and ≈430% during the phase transformation from Sn to Li4.4Sn and Na15Sn4, respectively. Unfortunately, such a notable volume change often results in the pulverization of the active materials, and maintaining an intact SEI layer becomes a significant challenge due to the massive expansion of alloy anodes.23,31,35,36
During the interaction of the alloy anode with the electrolyte, the protective layer (SEI) forms on the anode's surface; when the SEI layer is ruptured due to excessive strain, the electrolyte will further interact with the now unprotected anode surface, intensifying the electrolyte's depletion.36 This freshly developed SEI layer can segregate active components, leading to the disruption of solid electrical connections.31,37 It is crucial to note that these reactions occur in every battery cycle, triggering recurrent breakdowns and persistent SEI layer formation alongside severe electrode pulverization. Consequently, the SEI layer becomes irregular and thick, with extreme consumption of electrolytes and excessive use of cathode charges, leading to decreased reversible capacity. Additionally, the SEI formed on micro-sized alloy particles experiences significant stress, complicating the preservation of its stability. Hence, establishing a premium-quality SEI layer is fundamental for ensuring optimal electrochemical performance.38–40
Structure, composition, and electrolyte engineering are efficient ways to mitigate the huge volume change for micro-size alloy anode materials. Various factors, such as electrolytes, binders, and morphologies, influence the performance of micro-sized alloy anodes. In this comparative review, we will focus on the recent developments in micro-sized alloy anodes, particularly emphasizing the role of electrolyte, binder, structure, and composition in volume expansion mitigation and performance enhancement. The review will provide an in-depth analysis of recent advancements in each segment and highlight the challenges and opportunities for maximizing the performance of high-energy Li and Na-ion batteries.
2 Electrolytes
The electrolyte is an essential and critical component of the battery system.41 The term electrolyte refers to an ion-conducting solution that consists of a solvent and a salt.42 Electrolytes in batteries serve as conductive mediums, enabling the shuttling of ions between the cathode and anode during discharging and charging cycles. This transfer is crucial for the battery's overall function and performance. Due to an energy mismatch between the red/ox potential of the electrolyte and the electrode materials, a spontaneous film or layer is generated on the anode surface. It is called the solid electrolyte interphase (SEI).27,43 In aprotic electrolytes, the formation of an SEI is inevitable for many electrodes, including graphite, Sn, Si, (etc.) Once formed, this layer should protect from undesirable side reactions with the anode and prevent over and continuous consumption of the electrolyte by blocking the electron transfer from the anode to the electrolyte. However, it should allow rapid cation movement to enhance the reaction kinetics.44–46Alloy anodes suffer from repetitive contraction and swelling, rendering the SEI vulnerable. Due to the large volume change, an ideal SEI should have superior mechanical stability, maintaining its integrity without breaking and exposing fresh anode surface to the electrolyte. Thus, electrolyte engineering is an essential approach to designing a flexible and well-suitable SEI for micro-size alloy anode material.
The SEI layer is mainly due to the reduction of both salts and solvents in liquid electrolytes. Electrolyte salts consist of anions and cations. The types of rechargeable batteries determine the cation, such as Li+, Na+, Zn2+, Ca2+, and Al3+. Anions such as sulfonyl imide (bis(fluoro-sulfonyl)imide (FSI−), bis(trifluoro methane sulfonyl)imide (TFSI−)), hexafluorophosphate (PF6−), and others are also commonly used.47–51 The SEI on anodes is formed from inorganic elements like fluorides and oxides, which result from anion breakdown, and organic compounds like fluoroethylene carbonate (FEC).52 The anions play a significant role in determining the various characteristics of the SEI and its reductive kinetics. To build an optimal SEI layer, molecular orbital energy evaluations have been employed to forecast the reductive stability of anions like PF6−, FSI−, and TFSI−. Insights from these evaluations show that PF6− possesses notable reductive stability yet is thermodynamically unsuitable for Li metal anodes. Nonetheless, a robust SEI layer formed by pre-reduced salts counteracts salt decomposition. The distinct properties of various salts highlight the significance of selecting an appropriate salt to optimize the SEI and enhance the overall battery performance.47,53,54
Organic solvents that are frequently utilized include esters and ethers. Up to this point, a significant amount of research has been conducted on ethers, carbonate esters, and phosphates, examining their potential role in designing an optimized SEI for micro-sized alloy anodes. Various aspects of these solvents, including their linear formula, molecular weight, and melting points, have been scrutinized. For instance, carbonate esters can be categorized into two types: cyclic carbonate esters, which include compounds like ethylene carbonate (EC) and propylene carbonate (PC), and linear carbonate esters, such as ethyl methyl carbonate (EMC), diethyl carbonate (DEC), and dimethyl carbonate (DMC).55,56
Carbonate-based electrolytes (CBE) have been preferred in commercial Li-ion batteries due to their improved oxidative stability and higher flash points.50,57 Nonetheless, used in micro-sized alloy anodes presents certain challenges. One such challenge is the formation of a rigid, thick, and subpar SEI, undermining the stability of the anode. This phenomenon is mainly due to the fragile mechanical interactions between the inorganic particles and the polymer matrix. Gao et al.58 highlighted that the SEI produced in CBE showcased a minimal maximum elastic deformation energy. Additionally, Zhang et al.24 observed that SEI layers developed in NaPF6–PC with 5 wt% FEC were not compact, possibly due to the high solubility of certain SEI components, such as sodium oxides and carbonates, in PC-based electrolytes.
In SIBs' electrolytes, employing a mixture of a couple of Na-salts, which are then dissolved in a combination of two or more solvents, is common practice. However, it is worth noting that single solvent formulations are a rarity, with the exception of PC.59 The reason behind this trend lies in the complex and often conflicting demands placed on battery electrolytes, which are challenging to fulfill using a single molecular composition. Therefore, using a combination of solvents and salts has become a primary motivator in developing electrolytes for SIBs. According to recent reports, the EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC combination is the most frequently employed, followed by EC:PC and PC. Concerning the selection of salt, the literature suggests that NaClO4 is the most effective option, followed by NaPF6, NaCF3SO3, and NaTFSI.60,61
:DEC combination is the most frequently employed, followed by EC:PC and PC. Concerning the selection of salt, the literature suggests that NaClO4 is the most effective option, followed by NaPF6, NaCF3SO3, and NaTFSI.60,61
Ethers are an organic compound composed of ether groups, symbolized as R–O–R′, where both R and R′ can be either aryl or alkyl groups. There are two main subcategories within the ether solvent: cyclic ethers include 1,3-dioxolane (DOL), tetrahydrofuran (THF), and 2-methyl tetrahydrofuran (2-MeTHF), and linear ethers include compounds like dimethoxyethane (DME or G1), diglyme (G2), tetraglyme (G4), and ethylene glycol diethyl ether (EGDEE).50,62–65
Electrolytes based on ether exhibit superior reduction stability on the anode side, fostering the development of a thinner SEI compatible with a range of anodes. Within the family of ether solvents, glyme (Gn, CH3O(CH2CH2O)CH3) is a widely used solvent. As the carbon chain length of glyme increases, its viscosity rises, leading to a corresponding decline in ionic conductivity. Early studies of glyme-based electrolytes were conducted by Zhang et al.24 They investigated the use of these electrolytes to stabilize micro-sized alloy anodes without the need for intricate electrode designs. Their findings illuminated the consistent cycling of micro-sized Sn in SIBs employing G1–G4 electrolytes in the presence of 1 M NaPF6. For alloy anodes, diglyme, with its longer carbon chain than DME, provides a balance between oxidation stability and viscosity, positioning it as a prime candidate for full-cell applications. As shown in Fig. 1, Zhang and his team explored the effects of 1 M NaPF6 in diglyme and PC + 5%FEC on the SEI layer's quality and the cycling stability of micro-size Sn anodes in SIBs. Post 100 cycles, the anode in a 1 M NaPF6–diglyme electrolyte showcased superior performance, retaining its structural robustness more effectively than its counterparts. This can be attributed, in large part, to the formation of a highly compact protective film on the surface of the micro-size anode when submerged in 1 M NaPF6–diglyme electrolyte.
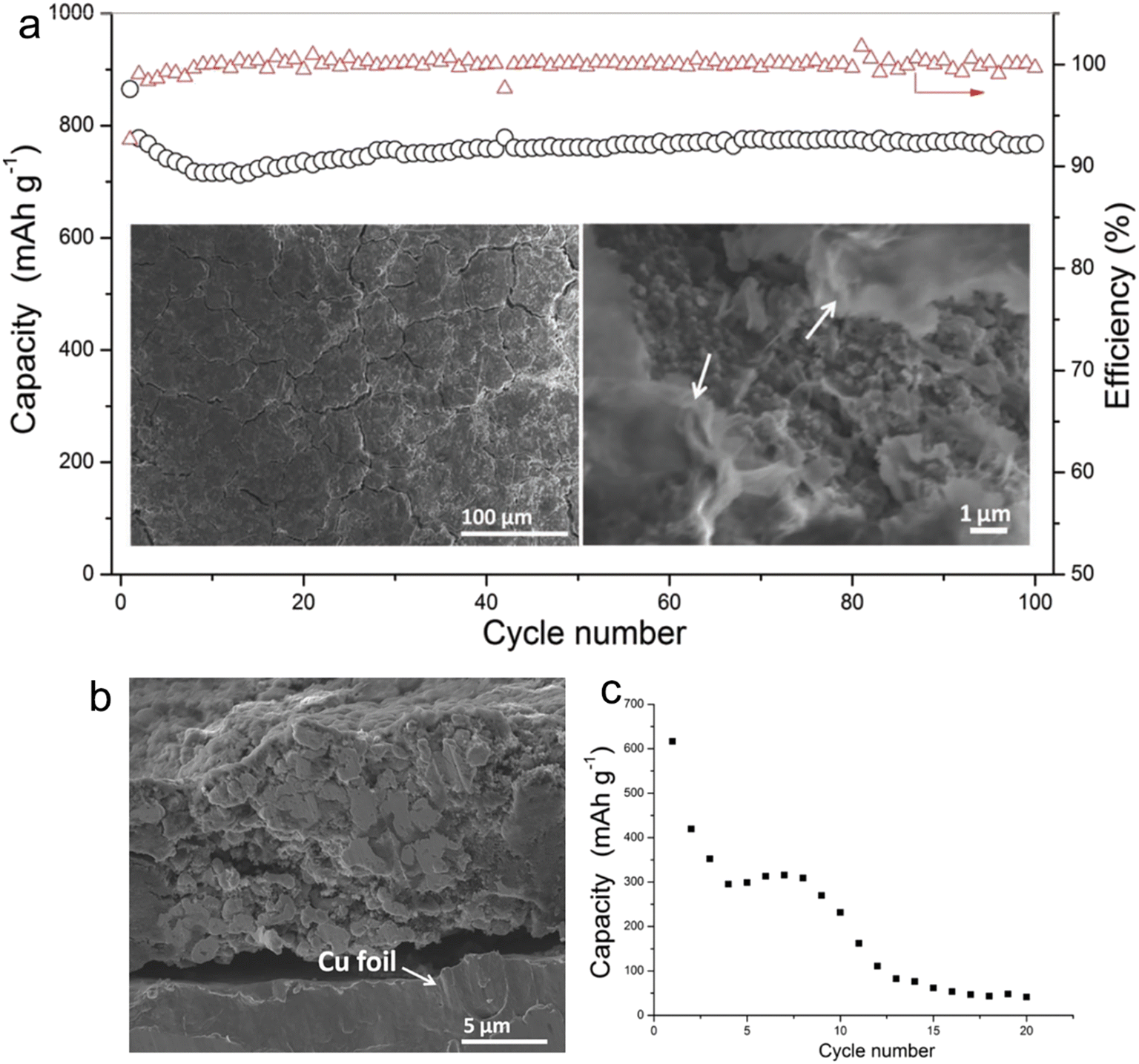 | ||
| Fig. 1 (a) Cycling performance of Sn electrode in 1 M NaPF6/DGME electrolyte at 250 mA g−1 (the morphology of the electrodes after 100 cycles is shown in the insets of (a), and the arrows indicate SEI films, and the red triangles are coulombic efficiency); (b) cross-section image of Sn electrode after cycling in DGME electrolyte; (c) cycling stability of Sn electrode in 1 M NaPF6/PC electrolyte with 5% FEC additives under 100 mA g−1. Reprinted with permission from ref. 24. Copyright 2016, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In a comprehensive study led by Huang et al.,27 the team sought to understand the variations in the microstructure, mechanical attributes, and chemical compositions of SEI layers on micro-size Sn anodes in two distinct electrolytes: 1 M NaBF4–diglyme and NaBF4–EC/DMC. Their investigative tools encompassed cryo-TEM, AFM, XPS, and DFT calculations. As shown in Fig. 2, through cryo-TEM assessments of micro-size anodes post-three cycles, it was deduced that the NaBF4–EC/DMC electrolytes were instrumental in forming substantial SEI layers characterized by polycrystalline domains of Na2CO3 and potential NaF inclusions. On the other hand, SEI layers from NaBF4–diglyme were noticeably thinner and marked by a blend of amorphous Na2CO3/NaF particles interspersed within a polymer-like matrix. This distinctive configuration facilitated enhanced sodium ion movement, boosted its ionic conductivity, and augmented the mechanical resilience of the SEI. Delving deeper with AFM examinations, the researchers discovered that the SEI originating from NaBF4–diglyme showcased superior rigidity (about 355 MPa) and elasticity (roughly 79%), which elucidated the heightened electrochemical efficacy observed in diglyme electrolytes. This investigation successfully revealed the structural and mechanical foundations that render SEI layers in glyme-based electrolytes stable.
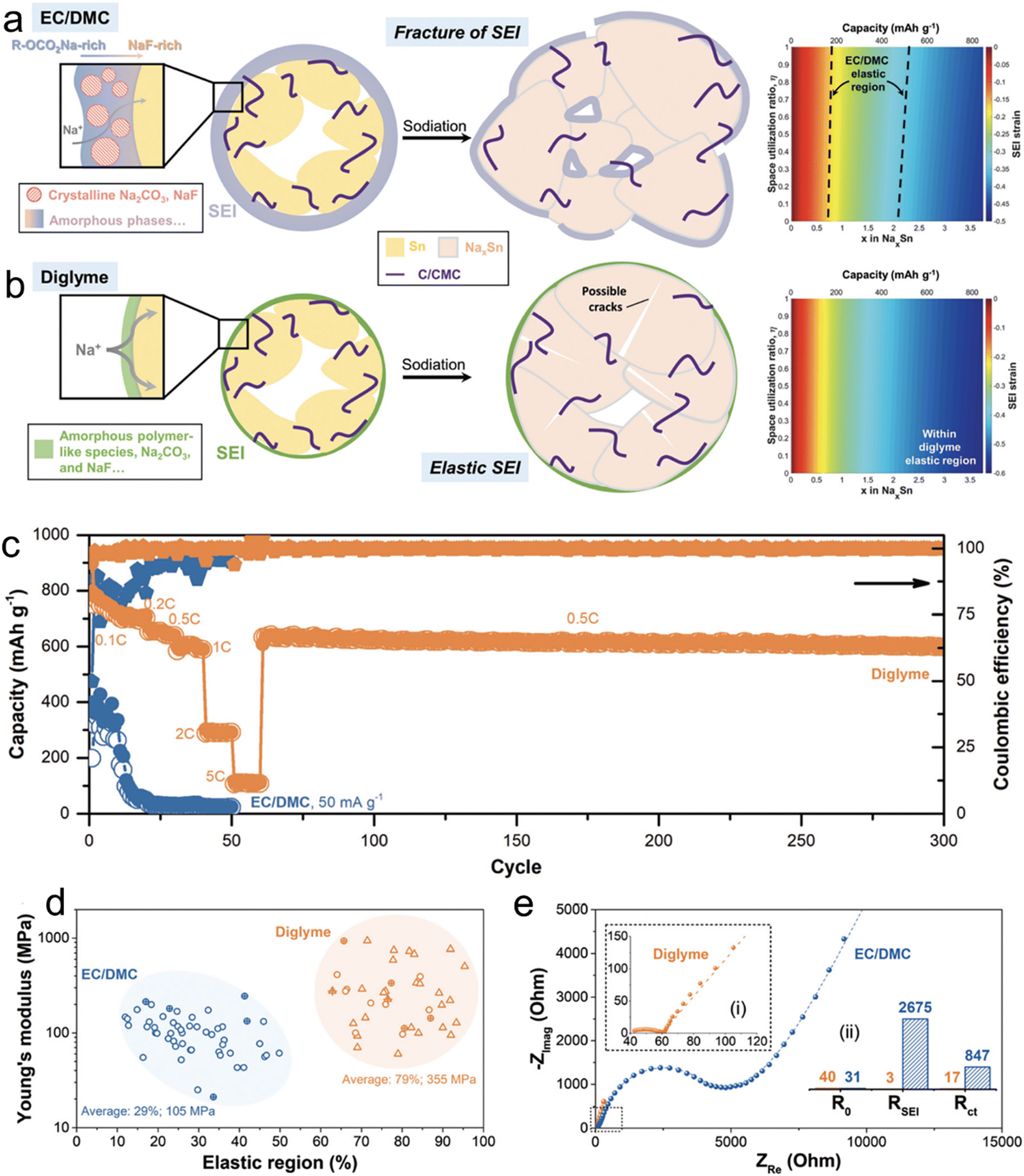 | ||
| Fig. 2 Schematics illustrating the chemical compositions, structures, and mechanical response to the volume expansion of SEIs derived from (a) NaBF4/EC/DMC and (b) NaBF4/diglyme (the strains imposed on the SEI as a function of x in NaxSn and space utilization r ratio in comparison to the relative elastic region of SEIs are also included); (c) rate performance of Sn microparticle electrodes in 1 M NaBF4/diglyme and 1 M NaBF4/EC/DMC electrolytes; (d) distributions of Young's modulus and relative elastic regions of the SEIs with 430 curves for each (the responses with and without fracture/yielding behaviors are plotted by circle and triangle scatters, respectively. The results from electrodeposited Sn without binders and carbons are marked by an additional “+”); (e) three-electrode EIS result of the cycled electrodes with (i) an enlarged Nyquist plot in diglyme and (ii) the fitted impedance as the insets. Reprinted with permission from ref. 27. Copyright 2019, The Royal Society of Chemistry. | ||
The advantages of ether electrolytes can be effectively implemented in the alloy anodes of LIBs. By forming an SEI layer with high interfacial energy (Eint) to the micro-sized Si anode surface, the adhesion of the SEI layer to the anode surface is reduced. This helps accommodate the anode's volume change during the charge and discharge cycles. With its high Eint, LiF is a favorable SEI layer component.66 Ethers are characterized by their low thermodynamic reduction potentials, typically ranging from ≈0.0–0.3 V. This trait amplifies the decomposition of fluorinated salts. Nevertheless, the solubility of LiPF6 in linear ethers, such as DME and diglyme, is not optimal, which has curtailed their prevalent application in LIBs. Cyclic ethers are generally not employed in LIBs due to their pronounced chemical reactivity. Among the possible cyclic ethers, THF and 2-MeTHF are preferable as they contain fewer ether oxygens, which shield them from such reactions.67,68
Chen et al.66 conducted a screening of salts and solvents for microsized alloy anodes in LIBs. The focal point of their research was to discern the reduction potential of the salt and gauge the solvation capabilities of the solvent. In their innovative approach, they formulated a 2 M LiPF6 solution using a combination of THF and 2-MeTHF for different micro-sized anodes (Si, Al, Bi), each exceeding 10 μm in size. Through their experimentation, they discovered that the 2 M LiPF6–THF/2-MeTHF electrolyte facilitated the formation of an SEI layer that was not only thin and homogeneous but also enriched with LiF. This SEI layer, abundant in LiF, exhibited a high Eint when interfacing with the alloy anode. This characteristic acted as a buffer, accommodating the plastic deformation occurring in the lithiated alloy (LixSi) throughout the alloying and dealloying phases. Consequently, micro-sized anodes were able to achieve stable capacities, with initial CEs of higher than 90% (Fig. 3). Additionally, Zhang et al.68 employed 2 M LiPF6 in 2-MeTHF and a blend of 2 M LiPF6 in THF/2-MeTHF as electrolytes. Their aim was to fortify and stabilize the SEI layer on the micro-sized Sn anode for LIBs. The SEI formed on the anode through the 2-MeTHF-based electrolytes was distinguished by its compact and thin structure, which was notably enriched with LiF. This structural characteristic plays a pivotal role in considerably boosting the cycle longevity of the anode.
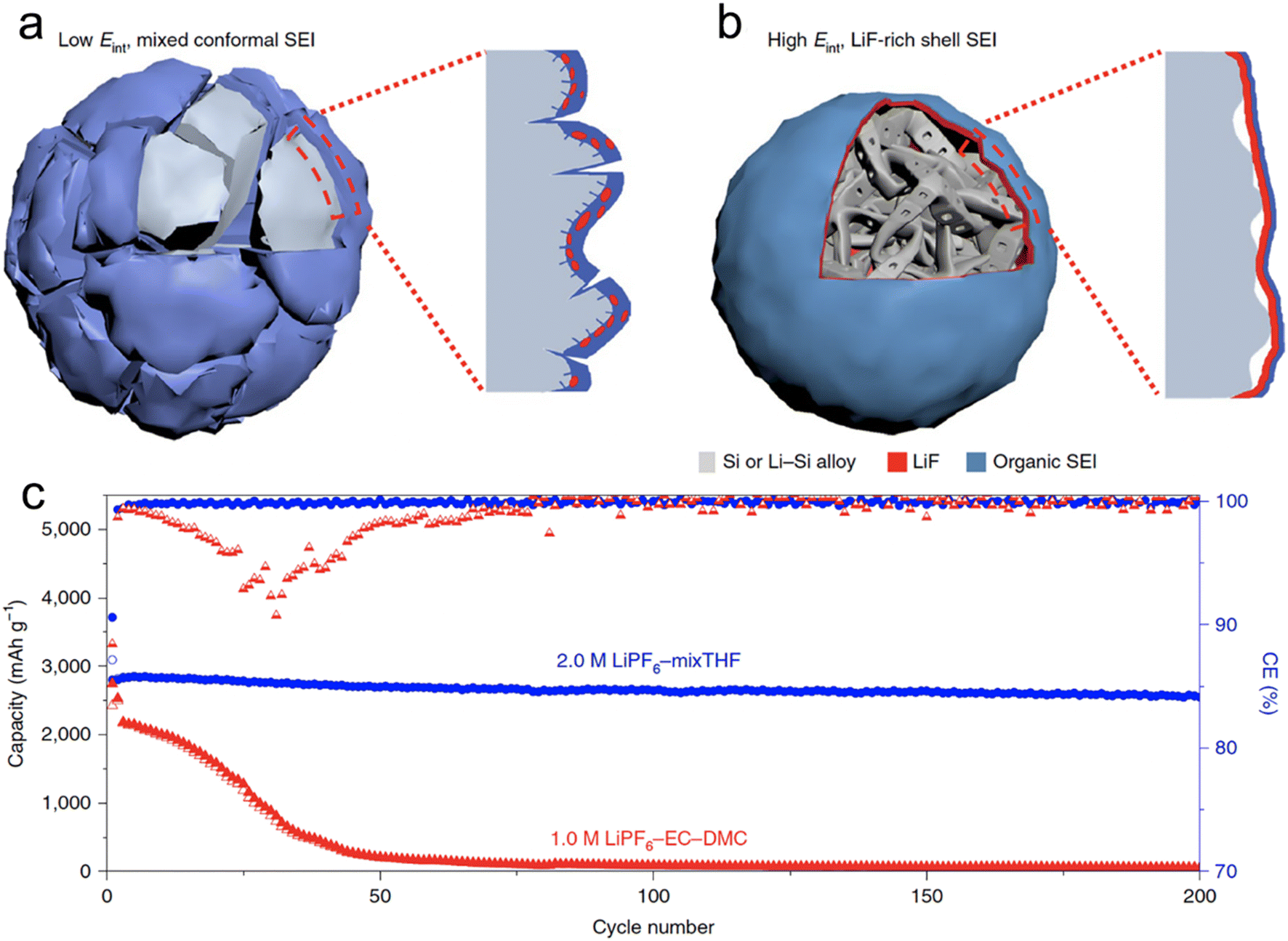 | ||
| Fig. 3 Schematic of the cycled alloy anode with an organic, low Eint and non-uniform (a) and an inorganic, high Eint and uniform (b) Li alloy–SEI interface; (c) cycling stability and CEs of SiMPs cycled in 2.0 M LiPF6–mixTHF and 1.0 M LiPF6–EC–DMC electrolytes at C/5. Reprinted with permission from ref. 66. Copyright 2020, Springer Nature. | ||
Solvents that are phosphorus-based, including phosphates, phosphazenes, and phosphonates, are becoming increasingly popular due to their potential to decrease flammability and enhance anodic stability when used with high-voltage cathodes. Traditional carbonate-based electrolytes are prone to flammability and can result in fire accidents.69,70 Organic phosphates, like trimethyl phosphate and hexamethylphosphoramide, are valued for their non-flammable properties, as phosphorus can serve as a trapping agent for hydrogen radicals.71 However, linear organic phosphates are ineffective at producing a suitable SEI on the anode, leading to inferior battery performance when used with graphite or Si-based anodes, and are, therefore, rarely included unless used in high concentrations.72 Recently, a novel fluorinated cyclic phosphate solvent molecule known as TFEP, has been introduced. This molecule is characterized by its inherent non-flammability and exceptional capability to form SEI on graphite anodes. The SEI, produced by this cyclic phosphate-derived electrolyte, is enriched with polyphosphoesters. These polyphosphoesters are a type of highly flexible elastomer. Alongside other inorganic components, this elastomeric inclusion in the SEI is anticipated to adeptly manage the huge volume change exhibited by Si electrodes.73
In their research, Yang et al.74 have proposed a method for designing a cyclic phosphate (TFEP)/hydrofluoroether (HFE)-based electrolyte that is not flammable. The goal is to enhance the robustness and elasticity of the solid electrolyte interphase (SEI) on Si-based anodes. The design strategy is based on the ability of TFEP to create polymeric components and HFE to increase the association between Li+ and FSI− ions, leading to the formation of additional inorganic components derived from the FSI− anion. This results in a composite SEI that is highly elastic and robust, consisting mainly of LiF, Li2O, LixPOy, sulfur compounds, and polyphosphoesters (Fig. 4). As a result, the micro-sized Si particles can undergo elastic deformation while maintaining their structural integrity during the charge/discharge process.
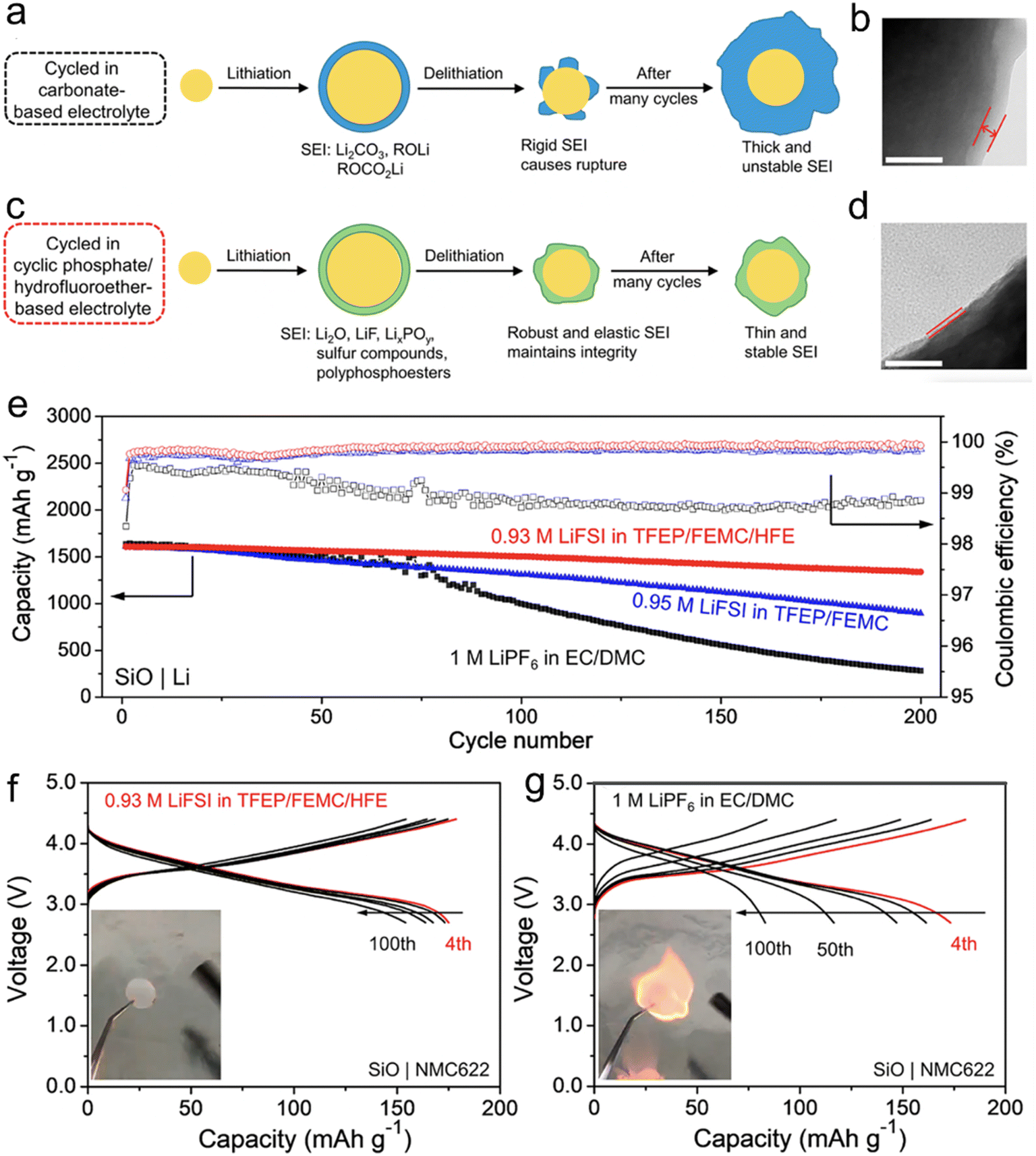 | ||
| Fig. 4 Schematic illustrations of SEIs on an SiO particle derived from (a) 1 M LiPF6 in EC/DMC and (c) 0.93 M LiFSI in TFEP/FEMC/HFE (in the conventional EC based electrolyte, an SEI mainly consisting of Li2CO3, ROCO2Li, and ROLi forms at the first lithiation, which is too rigid to tolerate the volumetric stress during delithiation. On the other hand, the SEI generated by the cyclic phosphate/hydrofluoroether-based electrolyte is mainly composed of Li2O, LiF, LixPOy, sulfur compounds, and polyphosphoesters, which is highly elastic and robust and can sustain the volume changes of SiO upon cycling); TEM images of the SiO electrodes after cycling in (b) 1 M LiPF6 in EC/DMC and (d) 0.93 M LiFSI in TFEP/FEMC/HFE for 100 cycles (the distance between two lines represents the thickness of the SEI, which clearly shows that the SEI formed in 0.93 M LiFSI in TFEP/FEMC/HFE is much thinner than that in 1 M LiPF6 in EC/DMC electrolyte. The white scale bar represents 50 nm); (e) cycling performance of the SiO|Li half-cells using 0.93 M LiFSI in TFEP/FEMC/HFE, 0.95 M LiFSI in TFEP/FEMC, and 1 M LiPF6 in EC/DMC at 0.2C after three formation cycles at 0.05C; selected charge–discharge curves for the full cells at 0.4C with (f) 0.93 M LiFSI in TFEP/FEMC/HFE and (g) 1 M LiPF6 in EC/DMC (the capacity is based on the weight of the NMC622 active material. The insets show the flammability test results). Reprinted with permission from ref. 74. Copyright 2021, American Chemical Society. | ||
Phosphates, despite possessing several benefits, are not compatible with most electrode materials. This is due in part to the low reductive stability of phosphates, which leads to the difficulty of creating a stable SEI on the anode surface, as cited in various sources.50,60 Nevertheless, the use of phosphates as co-solvents or additives can be effective in preventing reductive decomposition and enhancing electrolyte nonflammability. Consequently, organic phosphorus-based solvents are commonly employed as non-flammable co-solvent additives.75 Additionally, a compact and stable SEI layer on micro-sized alloy anodes using phosphate-based electrolytes can be achieved through the concepts of high concentration and localized high concentration.30,50,76
In recent decades, the popularity of highly concentrated electrolytes has grown significantly. As opposed to conventional electrolytes, where the concentration is only 1 M, these highly concentrated electrolytes have a significantly reduced number of free solvent molecules. This leads to forming a unique 3D solution structure and improves stability by minimizing the interplay between unbound solvent molecules and anode materials.76,77 Additionally, increasing the salt concentration of ether-based electrolytes greatly enhances their anodic stability. Moreover, highly concentrated electrolytes promote the creation of a salt-derived SEI layer. This, in turn, beneficially influences the interfacial chemical dynamics of anode surfaces, making them more suitable for high-voltage battery configurations.50
Despite the significant advantages of highly concentrated electrolytes, their practical application is made challenging by their high viscosity, poor wettability, and high cost. Fortunately, there is an alternative in the form of localized high-concentration electrolytes (LHCE), which can enhance the benefits and alleviate the drawbacks of concentrated electrolytes. Typically, some diluents with appropriate quantities are added to the highly concentrated electrolytes. This addition aims to diminish viscosity and enhance ionic conductivity. At the same time, it ensures the retention of the inherent benefits of concentrated electrolytes, like superior oxidative stability and a structured solvation framework. As a practical example, Jia et al.76 introduced a fluorinated bis(2,2,2-trifluoroethyl) ether (BTFE) as a diluent into 1.2 M LiFSI TEP/FEC (TEP/FEC/BTFE: 1.2/0.13/4), resulting in an LHCE for porous micro-sized Si anode (Fig. 5). This electrolyte promoted the creation of locally highly coordinated Li+-TEP solvates and an SEI layer rich in LiF, derived from FSI−. As a direct outcome, when tested in a Si/graphite||NMC333 full cell configuration using the 1.2 M LiFSI-TEP/FEC/BTFE electrolyte, there was a noticeable enhancement in electrochemical performance in comparison to the 1 M LiPF6–EC/EMC–FEC solution.
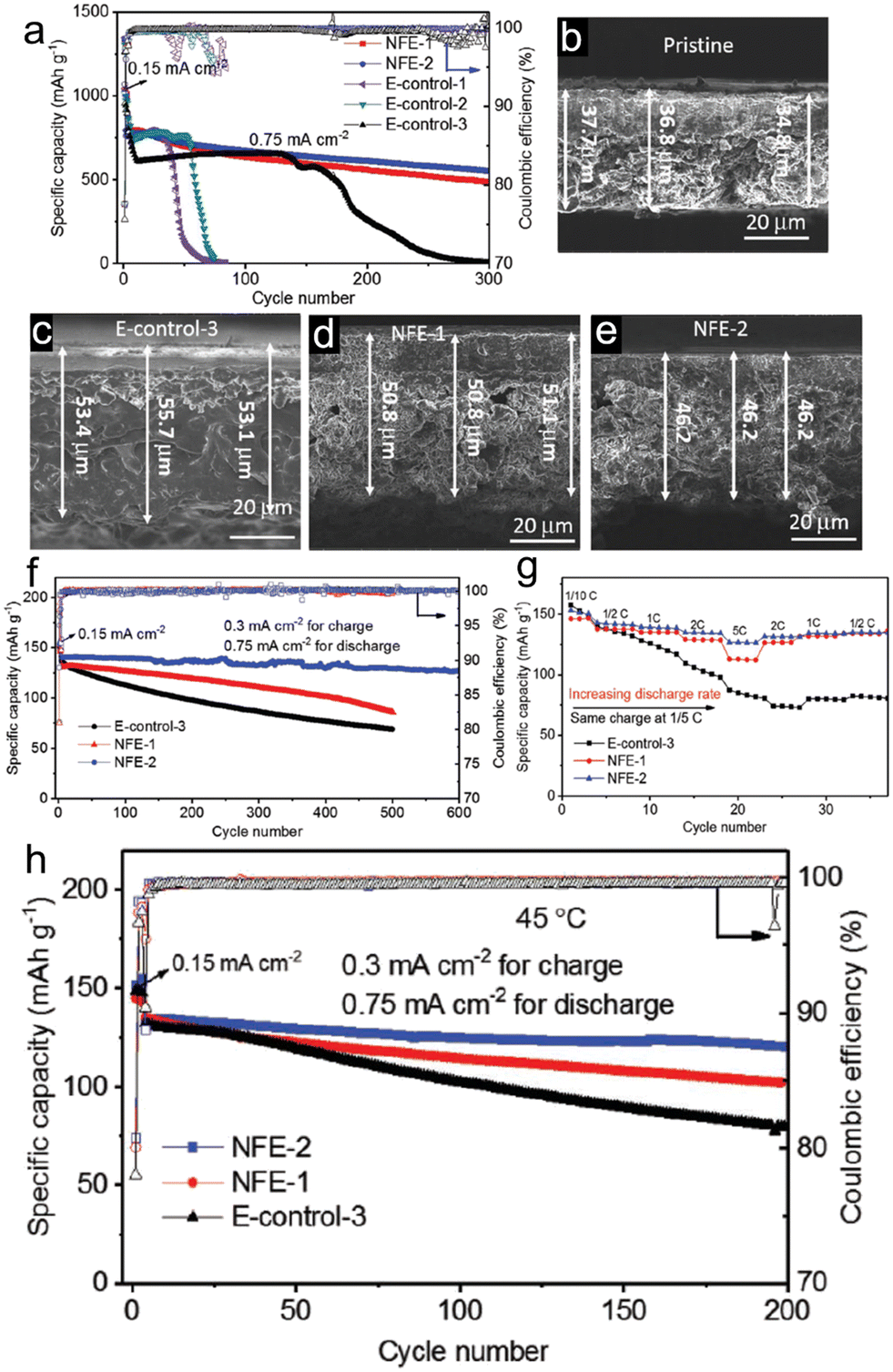 | ||
| Fig. 5 Electrochemical behaviour of Li||Si/Gr half cells in different electrolytes: (a) long-term cycling performance and coulombic efficiency of Li||Si/Gr half cells in different electrolytes; (b) SEM image of a pristine Si/Gr electrode; SEM images of fully lithiated electrodes after 1 cycle in (c) E-control-3, (d) NFE-1, and (e) NFE-2; (f) long-term cycling performance of Si/Gr||NMC333 full cells with different electrolytes at 25 °C; (g) rate capability of the Si/gr||NMC333 cells in different electrolytes at 25 °C; (h) long-term cycling performance of Si/Gr||NMC333 cells cycled at 45 °C in different electrolytes. Reprinted with permission from ref. 76. Copyright 2019, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Film-forming electrolyte additives are also a main constituent of electrolyte engineering. Efforts to tailor and tune the SEI will not be successful without suitable and well-optimized additives. Their role can be summarized in 3 key points: first, stabilizing the anode structure: a stable SEI layer formed by the additives should act as a buffer against the volume changes during cycling, thus maintaining structural integrity. Second, enhancing ionic conductivity: the right SEI composition facilitated by additives is key for efficient ion transport. Finally, suppressing side reactions: additives form a selective SEI layer that prevents undesirable side reactions that lead to capacity fading. There is no clear demarcation between an additive and a solvent/salt, so an arbitrary threshold of 10 vol% is adopted, below which the new components are considered as additives.78 Various additives have been investigated, with the most prevalent ones being salt-type additives, fluorine-containing species, and derivatives of unsaturated carbonates.61,79–83
Additives based on salt compounds, including lithium bis(oxalato)borate (LiBOB), lithium nitrate (LiNO3), sodium carbonate (Na2CO3), and sodium fluoride (NaF), play a crucial role in augmenting the ionic conductivity and thermal stability of batteries. These additives are key in forming a robust SEI and regulating the anode and electrolyte interactions. The strategic application of these salt-type additives leads to enhanced performance and safety of the battery, especially under extreme operational conditions.41,49,84,85 Ma et al.86 employed readily soluble SEI species, Na2CO3 and NaF salts, as additives to saturate the electrolytes. The introduction of NaF salt additive in 1 M NaPF6–PC significantly hindered the dissolution of the SEI and increased NaF concentration within the SEI layer, promoting the formation of a more inorganic and stable SEI layer.
Additives featuring unsaturated C–C bonds in carbonate derivatives are also gaining much attention lately.50,87 A prominent example of such additives is vinyl ethylene carbonate (VC), which has become an essential component in the battery electrolyte industry. The effectiveness of VC lies in its ability to undergo polymerization reactions on the electrode surface, leading to the formation of protective poly(VC) layers. Dahbi et al.88 demonstrated the distinct capabilities of VC additives in a 1 M NaPF6–EC/DEC solution. They found that VC not only contributes to the stability of the SEI but also safeguards the electrolyte against further decomposition, through a unique passivation process. The VC-derived SEI was formed based on a polymerization reaction and consists of a mix of inorganic and organic compounds. This dual composition is key to enhancing battery performance and longevity.
Fluorine-containing (F-containing) species are highly regarded in electrolyte engineering due to their ability to form fluorinated species or polymers, such as LiF, NaF, KF. These fluorinated components are instrumental in building a robust and optimized SEI layer, essential for minimizing potential side reactions in the battery system. Additionally, the intrinsic characteristics of F-containing species, notably their high electron-withdrawing capacity, can significantly enhance the oxidation stability of electrolytes, especially at higher voltages. This property is particularly valuable for maintaining electrolyte integrity under elevated voltage conditions.79,89–91 FEC, in particular, has achieved commercial success as an electrolyte additive. Its effectiveness lies in its ability to reduce undesirable interactions between the electrolytes and electrodes, especially in micro-sized alloy anodes. Moreover, FEC plays a critical role in promoting the formation of a stable, F-rich SEI layer, contributing significantly to the overall performance and longevity of battery systems.68,90,92
Electrolyte additives are crucial for enhancing the performance of micro-size alloy anodes in advanced battery systems. Their ability to stabilize, improve conductivity, and suppress unwanted reactions opens new avenues for high-efficiency, durable batteries. Ongoing research focusing on electrode material compatibility, cost-efficiency, and environmental impact is vital for harnessing the full potential of these energy storage technologies.
For high-capacity ion batteries, a robust chemical reaction accompanies their operation, producing a significant amount of heat that can potentially expedite thermal runaway.93 This presents a safety hazard, given the flammable nature of the liquid electrolyte used. Traditional LIBs face a bottleneck in their operating voltage at 4.5 V due to the decomposition of the electrolyte at higher voltages.94 However, developing high-voltage batteries with wider electrochemical stability windows is crucial for future technological advancements. To address these issues, solid-state ion batteries (SSBs) employing solid-state electrolytes (SSEs) with superior chemical stability, mechanical strength, and flame retardation hold significant promise. SSEs can prevent liquid leakage, mitigate the risk of fire due to high temperatures, and withstand higher temperatures than conventional electrolytes.
Additionally, higher temperatures can enhance SSE conductivity within a certain temperature range instead of posing a danger. Furthermore, it possesses an ultra-high mechanical strength that can effectively inhibit the growth of lithium dendrites. This enhances the safety of the entire battery system.95,96 Implementing solid-state electrolytes (SSE) in the battery design offers several other advantages over traditional liquid-based systems. By eliminating the need for liquid substances and porous separators and replacing them with compact solid electrolytes, the battery design becomes more straightforward and more cost-effective. This shift also results in a reduction in the number of current collectors required for the cell stack. The use of SSBs results in significant increases in both volumetric and gravimetric energy densities. Furthermore, the manufacturing process of SSBs can be streamlined, with conventional procedures such as electrolyte wetting, formation, and aging being reduced or even eliminated, thereby reducing the time and cost associated with production.97
In the course of developing solid electrolytes that exhibit superior electrochemical performance and stability, one encounters numerous challenges. The primary obstacles involve ensuring that the electrolyte possesses sufficient ion-transport capacity, which encompasses both ionic conductivity and ion transference number. Additionally, the electrolyte must exhibit satisfactory chemical/electrochemical stability and interfacial fusion ability. H. S. Tan et al.98 tapped into the interface stabilizing attributes of sulfide solid-state electrolytes to stabilize a non-carbon micro-sized Si anode with an impressively high loading of 99.9 wt% (Fig. 6). Compared to liquid electrolytes, sulfide solid-state electrolytes permit a 2D plane of interfacial contact between themselves and the porous μSi anode, which can be preserved even after the material undergoes large-volume expansion. Furthermore, the absence of carbon in the micro-sized anode curtails any pronounced decomposition of the SSE, leading to a marked surge in coulombic efficiency. The immediate ionic and electronic linkage between Li–Si and micro-size Si paves the way for the Li–Si alloy phase to permeate the entire micro-size Si anode. The study underscored that sulfide SSE counteracts persistent interfacial expansion and the irreversible loss of lithium, culminating in a consistently stable SEI layer upon repeated cycling. Consequently, the carbon-free μSi||SSE||NCM811 full cell demonstrates remarkable reversibility even under severe temperature conditions, ranging from −20 °C to 80 °C, which indicates promising advancements in the field.
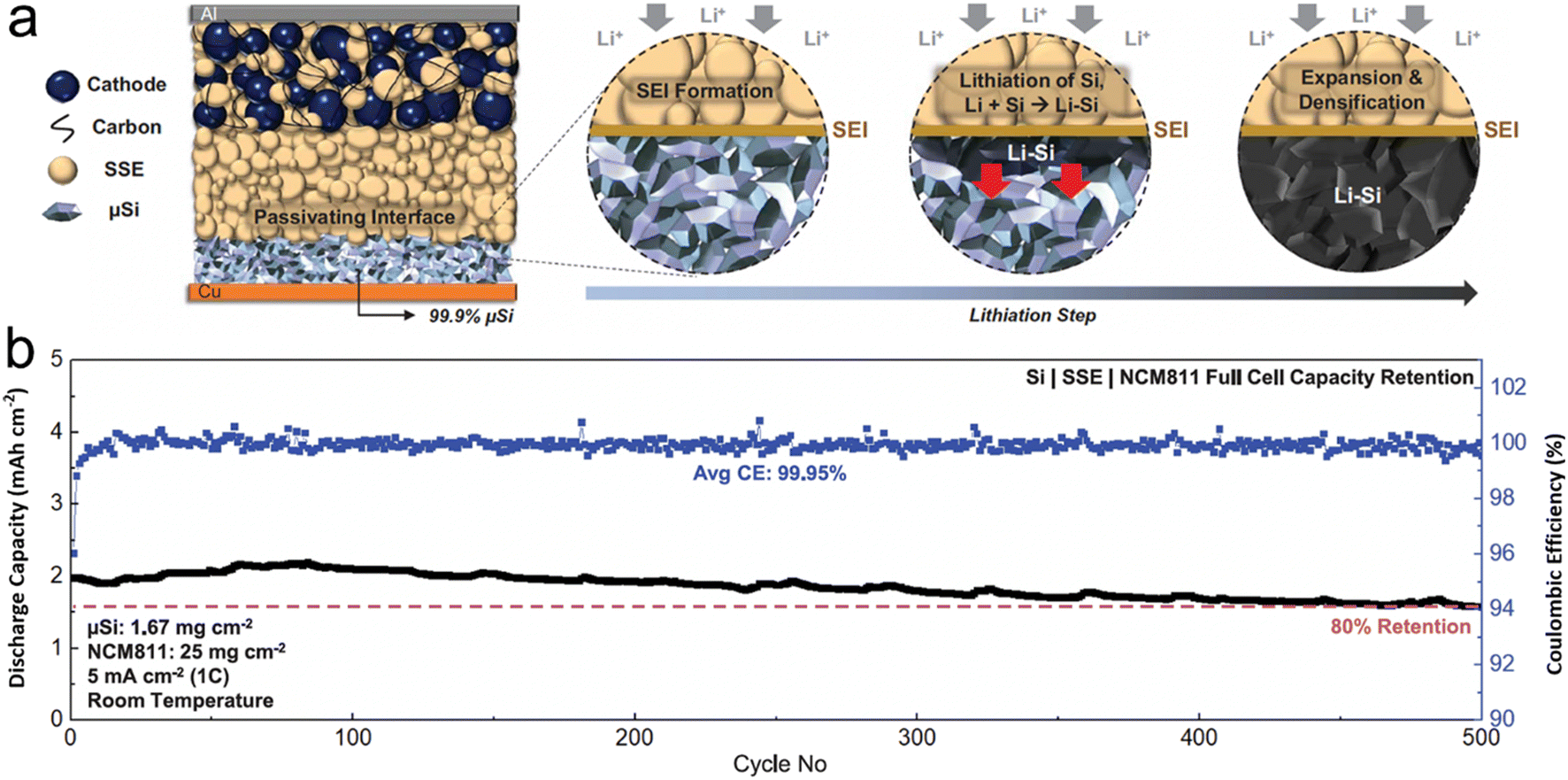 | ||
| Fig. 6 (a) Schematic of 99.9 wt% μSi electrode in an ASSB full cell (during lithiation, a passivating SEI is formed between the μSi and the SSE, followed by lithiation of μSi particles near the interface. The highly reactive Li–Si then reacts with Si particles within its vicinity. The reaction propagates throughout the electrode, forming a densified Li–Si layer); (b) cycle life at room temperature (all cells were tested under similar charge and discharge conditions between 2.0 and 4.3 V. The first cycle voltage profile of each respective cell is plotted in black). Reprinted with permission from ref. 98. Copyright 2021. American Association for the Advancement of Science. | ||
3 Binders
In electrochemistry, the binder stands as a pivotal element within the electrode, acting as the unifying agent for active materials, conductive substances, and current collectors. Its importance cannot be overstated, as it is responsible for facilitating tight electrical connectivity among these components. Additionally, the binder adeptly handles volume expansion, ensures the structural coherence of alloy anode materials, and plays a key role in stabilizing the SEI throughout the cyclical operations.99 According to research, binders can be classified into three categories based on their contact with anode materials: inert adhesion, hydrogen bonding, and covalent cross-linking.Inert adhesion denotes a scenario where no chemical bond forms between the binder and the active materials. Instead, the cohesive integrity of the entire electrode is preserved mechanically, primarily through the van der Waals force. Widely used binders in this context encompass poly(tetrafluoroethylene) (PTFE) and poly(vinylidene difluoride) (PVDF). Research has highlighted that microparticles tend to undergo pulverization due to volume alterations. Consequently, given their subpar mechanical attributes and weak bonding to micro-sized alloy anodes that experience substantial volume expansion, these binders might not be apt choices. In general, inert binders remain passive and do not engage in the SEI formation process.100
Integrating hydrogen bonding in the adhesion process between the binder and alloy anode has proven superior compared to alternative techniques. Given the dominance of hydroxyl groups on alloy anode surfaces, binders like carboxymethyl cellulose (CMC), poly(acrylic acid) (PAA), and sodium alginate (SA), which are abundant in carboxylic acid groups, can innately form hydrogen bonds with the alloy anode surface. This naturally induced bonding mechanism promotes a more robust linkage between the binder and active materials. Simultaneously, it limits the exposure of the active material's surface area to electrolytes, setting the stage for the development of a resilient and optimized SEI.
The covalent bonding between the binder and anode is established through a chemical reaction between the binder's functional groups and the oxide layers present on alloy anodes. This robust bond plays a pivotal role in preserving the electrode's structural integrity, minimizing the degradation of the electrical network throughout the cycling process. The outcome is impressive CE and consistent cycling performance. In contrast to passive binders, those containing functional groups significantly impact the formation of SEI and its spatial distribution and chemical composition. For example, binders like poly(vinyl alcohol) (PVA),101 polyrotaxane–polyacrylic acid (PR–PAA),102 and citric acid (CA)103 have been shown to facilitate the creation of a stable and thin SEI on the surface of alloy anodes.
Binders that exhibit a diverse range of attributes, including pronounced elasticity, dual ionic and electronic conductivity, inherent self-healing mechanisms, and robust adhesive prowess, are highly favored among alloy anode users.
To enhance the self-healing properties of polymeric binders, Wang et al.104 conducted research to develop a binder with a low glass transition temperature (Tg). The authors reported the formation of a self-healing polymer (SHP) with a low Tg value below 0 °C, which was achieved by controlling the reaction time. When the micro-sized Si was expanded, the polymer layer experienced fracturing. Still, the viscous flow of SHP helped in the closure of the cracks through self-healing hydrogen bonding between the urea groups. This highly efficient self-healing property enabled the long-term cyclability of Si micro-particle (SIMP) anodes. In comparison with PVDF SiMP (14%), CMC-SiMP (27%), and Alg-SiMP (47%) after 20 cycles, the SiMP anode with 50 wt% Si delivered 2094 mA h g−1 with a high retention of 80% at a current density of 400 mA g−1 after 90 cycles. The chemical structure and schematic representation of the self-healing working principle, as well as SEM images (Fig. 7).
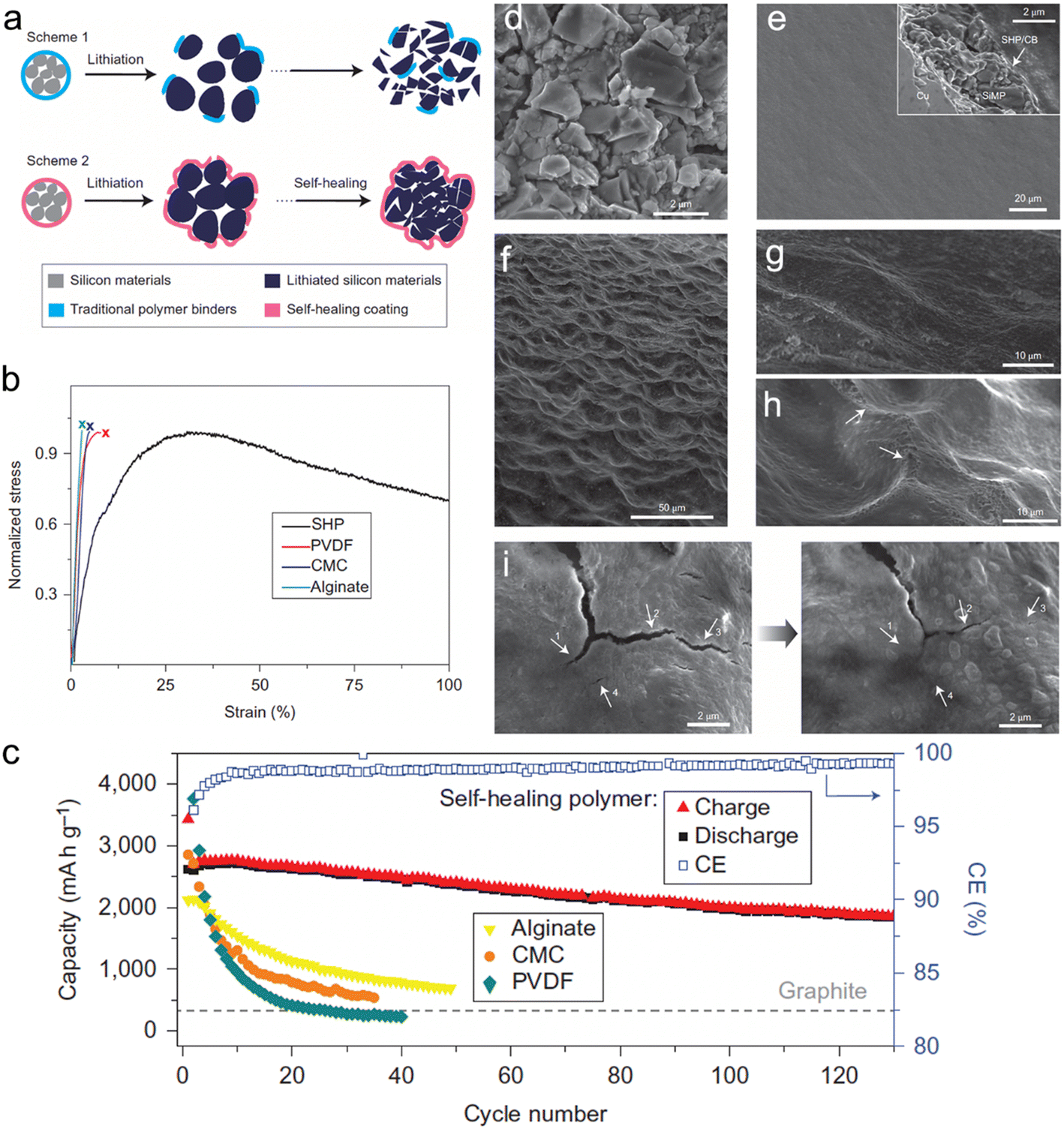 | ||
| Fig. 7 (a) Schematic illustration of the design and behavior of silicon anode with (a) conventional polymer binder and self-healing polymer (SHP) binder; (b) tensile tests of the SHP and other conventional polymer binders; (c) cycling performance of SiMP electrodes with different polymer binders at C/10 with a potential window of 0.01–1 V versus Li/Li+; (d) SEM image of bare SiMPs; (e) surface morphology of the bare SiMPs electrode before cycling. Inset: cross-sectional SEM image of the electrode; (f) low and (g) high magnification SEM images of the bare SiMPs electrode after 20 cycles at a rate of C/10; (h) some scar-like structures (arrows) can be found on the electrode after the cycling process, which appear to be cracks that subsequently healed; (i) left: cracks in the polymer layer in the lithiated state; right: after 5 h the smaller cracks were healed, indicated by the arrows on the images. Reprinted with permission from ref. 104. Copyright 2013. Macmillan. | ||
Choi et al.102 have presented a novel strategy for retaining conductive pathways to whole micro-size Si particles while cycling, accomplished by a highly elastic binder layer that surrounds Si microparticles and prevents the pulverization of Si particles. In order to withstand the mechanical stress caused by Si expansion, the binder must be capable of enduring this continuous stress. They were inspired by the working principle behind moving pulleys, which distribute and equalize localized forces to significantly reduce the tension in the rope. To achieve this principle on a molecular level, they constructed a molecular pulley binder (PR–PAA) by covalently attaching PAA to the ring components of polyrotaxane (PR), as shown in Fig. 8. The stress applied to the polymer is significantly reduced by ring sliding, consistent with the principle of moving pulleys, rendering PR–PAA highly elastic. These unique properties arise from the topological crosslinking between PAA and PR through the ring molecules, which are neither fixed nor dissociated. To examine the mechanical properties of PR–PAA, tensile tests were conducted. During repeated stretch–recovery cycles, the PR–PAA exhibited high elasticity, with a percentage of 390%. Additionally, it displayed less strain hysteresis, indicating its superior performance. The PR–PAA binder was explicitly designed to prevent the disintegration of pulverized Si particles. The effective stabilization of the SEI layer curtailed its recurrent growth, leading to an improved CE and consistent cycling performance at capacities typical of commercial standards. With the employment of the highly elastic PR–PAA binder, the micro-size Si anode showcased a mere 12.1 μm alteration in thickness following ten cycles. In comparison, an anode employing the standard PAA binder underwent a thickness modification of 22.8 μm. This comparison underscores the superior efficacy of high-elasticity binders relative to their traditional PAA counterparts.
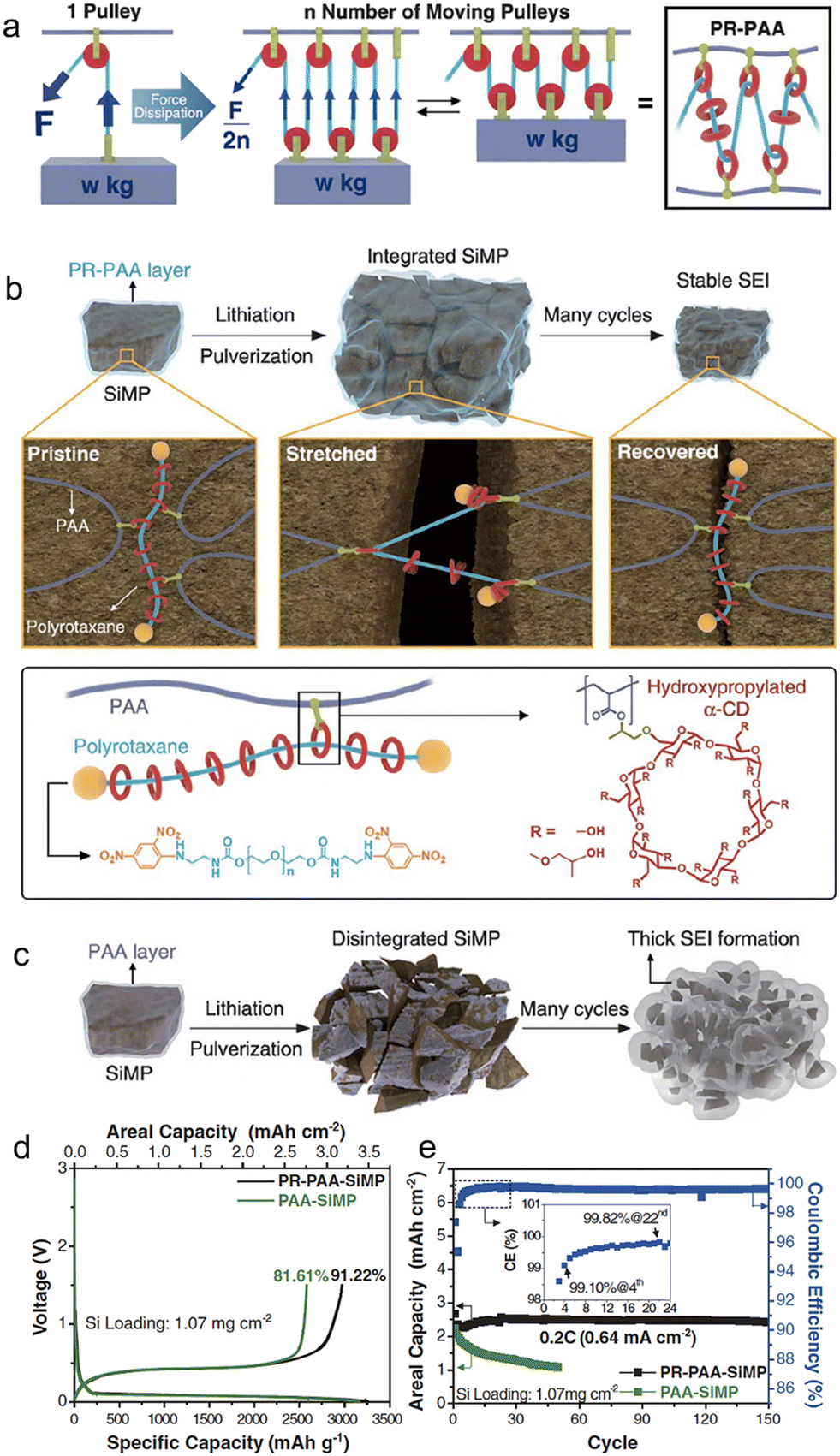 | ||
| Fig. 8 (a) Proposed stress dissipation mechanism of PR–PAA binder for SiMP anodes; (b) graphical representation of the operation of PR–PAA binder to dissipate the stress during repeated volume changes of SiMPs, together with chemical structures of polyrotaxane and PAA; (c) schematic illustration of the pulverization of the PAA-SiMP electrode during cycling and its consequent SEI layer growth; charge/discharge curves at 0.033C (100 mA g−1) (d) and the cycling performance at 0.2C (600 mA g−1) of SiMP electrodes incorporating PR–PAA and PAA as binders (e). Inset is the magnified view of the coulombic efficiencies in the first 24 cycles. Reprinted with permission from ref. 102. Copyright 2017. American Association for the Advancement of Science. | ||
4 Morphology and structure
Architectural engineering stands as a powerful method for meticulously adjusting the physicochemical attributes of electrodes, consequently boosting their electrochemical prowess. Techniques like particle size reduction, interfacial property modification, and lattice chemistry tuning can finetune ionic and electronic conductivities, reaction energy barriers, and the electrochemical responses of electrode materials. The incorporation of intricate structures proves advantageous in modulating stress generation, distribution, and the mechanical characteristics of electrode films, resulting in unmatched structural integrity. Given this context, employing structural methodologies becomes especially pertinent for alloying anodes. These anodes undergo more pronounced volume expansion compared to insertion or conversion materials. There has been a fervent dedication to developing a wide range of material and electrode structures to counteract volume expansion challenges. This dedication has yielded notable improvements in amplifying the electrochemical performance of alloying material in both LIBs and SIBs. Theoretically, volume expansion challenges can be approached in a dual-faceted manner: one, by averting material or electrode fragmentation, and two, by preserving unbroken electrical connections amidst the fragmented sections.To facilitate reversible (de)lithiation/(de)sodiation in alloying anode materials, researchers have implemented various nano- and microscale structures and morphologies.13–15,105,106 These methods, which may involve pure alloying elements or composite materials, typically aim to augment the surface-to-volume ratio (SVR) of the anode's morphology. By amplifying the SVR, the slope of concentration and stress–strain gradients during lithiation can be substantially reduced due to the decreased required lengths and the decreased influence of solid-state lithium/sodium diffusion on the overall process. Additionally, increasing SVR concurrently enhances the rate of lithiation/sodiation and the maximum (dis)charging power available in the anode.
While these procedures are implemented for specific alloying materials, they can be extrapolated to other alloying materials, given their comparable characteristics.
In order to fully utilize the potential capacity of pure alloying anode materials, which tend to crack and break apart during lithiation, innovative methods must be employed to accommodate their large volume changes. One such method is the creation of composite materials, which can significantly decrease overall volume changes, minimize stress gradients, and enhance performance characteristics such as power and capacity. Composite materials provide an array of fabrication options and performance capabilities, as they allow for the selection of multiple elements and materials in a single anode. An active alloying material can be combined with one or more matrix materials in an ideal composite to manage the volume change and prevent plastic deformation and fracturing. This strategy is particularly attractive for alloying anodes, as it provides a broader range of options for fabrication techniques and available properties.107–109 For an ideal matrix material, both electrical and ionic conductivity are necessary to enhance lithiation uniformity and optimize the maximum power capabilities. Additionally, the material must possess a superior yield or fracture strength and low stiffness to accommodate the volume expansion caused by lithiation/sodiation to the fullest extent possible. Various materials and combinations can meet these requirements, and these composites can generally be categorized into two groups: active–inactive and active–active composites. Active–active composites refer to composite materials in which all or the majority of the phases present in the anode participate in (de)lithiation/(de)sodiation during cycling and contribute to the overall capacity of the anode. Unlike active–inactive composites, active–active composites do not suffer the same decreases in gravimetric or volumetric capacity as the volume of inactive phases has been reduced or eliminated. It is noteworthy that all materials undergo (de)lithiation/(de)sodiation during cycling, but ideally, these processes do not happen in the same voltage window.110
In the academic literature, an alternative methodology for fabricating active–active composites has been explored.111–113 This approach involves adding an alloying component, for example, silicon, with graphene, a material that does not undergo notable volume fluctuations during (de)lithiation. Graphene possesses remarkable electrical conductivity and mechanical characteristics that aid in the uniform and regulated performance of the composite.
The process of lithiation of alloying anode elements, such as silicon, has been a focus of research due to its high lithiation capacity. This capacity is estimated at approximately 4200 mA h g−1, making it a promising candidate for developing high-capacity, high-power, and durable batteries. To address the issue of volume changes during (de)lithiation, graphene has been utilized to create a stable SEI between silicon and the electrolyte. This SEI stabilizes the battery's capacity by preventing the continual re-formation of the SEI during the volume changes. Additionally, the use of graphene or other lithiating and conductive agents has shown potential in developing high-performance composite anode designs.114,115
Incorporating both approaches, architectural and material composites, is a logical way for a rational electrode material design. Li et al.116 unveiled a technique to envelop Si microparticles, ranging from 1–3 μm, with meticulously fabricated cages composed of multiple graphene layers (Fig. 9). This graphene enclosure serves as a robust yet flexible shield during intensive galvanostatic cycling. Such a design permits the microparticles to swell and break apart within the confines of the cage, all while ensuring electrical connectivity at both the individual particle and overarching electrode scales. Additionally, the chemically passive nature of the graphene cage fosters a stable SEI. This stability curtails the non-recoverable consumption of Li-ions, leading to a swift augmentation in CE during the initial cycles. Electrochemical data from the half-cell reveals an estimable reversible capacity of 3300 mA h g−1 when subjected to a current density of C/20 (with 1C equating to 4.2 A g−1). This impressive capacity signals that the active materials maintain robust electrical connections and actively participate in both the charge and discharge processes. Notably, this achievement is accomplished devoid of any conductive additives, underscoring the good electrical conductivity intrinsic to the graphene cage. When subjected to rigorous full-cell electrochemical testing, where stable cycling demands are elevated, the graphene-encaged Si microparticles consistently perform, showcasing stable cycling over 100 cycles with a retention of 90% of its capacity.
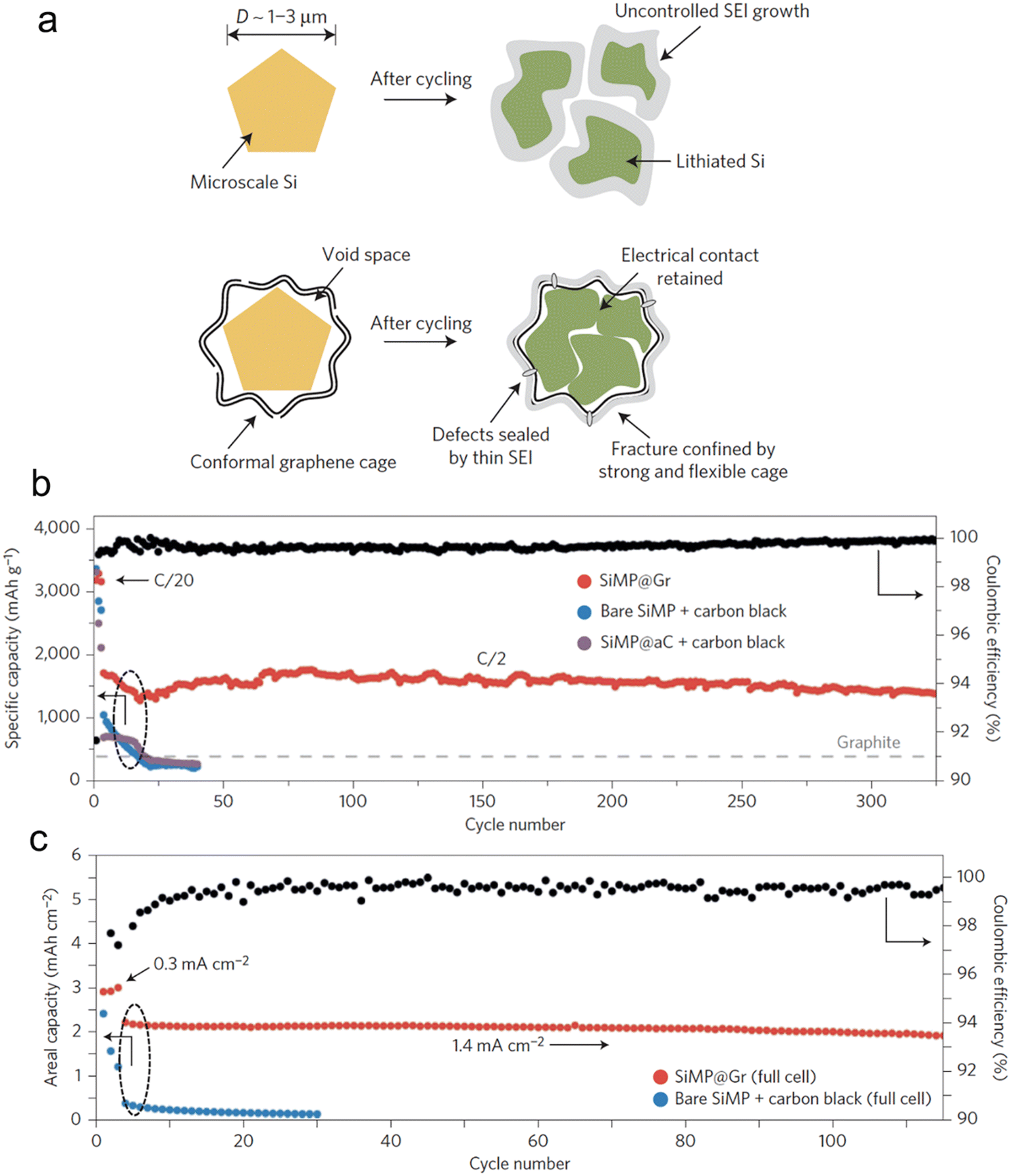 | ||
| Fig. 9 (a) Morphology and structural evolution of Si microparticles without and with graphene cage encapsulation during cycling; (b) cycling performance of bare SiMP with conductive carbon additive, amorphous-carbon-coated SiMP with conductive carbon additive, and SiMP@Gr with no conductive additives (the mass loading was 0.8 mg cm−2); (c) cycling performance of bare SiMP with conductive carbon additive and SiMP@Gr (∼2.0 mg) with no conductive additives when paired with a traditional lithium cobalt oxide cathode. Reprinted with permission from ref. 116. Copyright 2016. Macmillan. | ||
One of the main challenges in designing high-capacity anode material is to combine CE, cycling stability, and volume change. Xu et al.117 prepared nano/micro-structured Si/C composites. Their strategy focused on minimizing the specific surface area while boosting the tap density of Si/C composite. With a judicious selection of particle size, a minimized specific surface area, and a refined structure, they managed to achieve an outstanding initial CE of 91.2%. Further testifying to their design's efficacy, these nano/micro-structured Si/C anodes showcased remarkable cycling stability, retaining a significant 96.5% of their capacity post 100 cycles, all while being subjected to a current density of 0.2 A g−1 (Fig. 10).
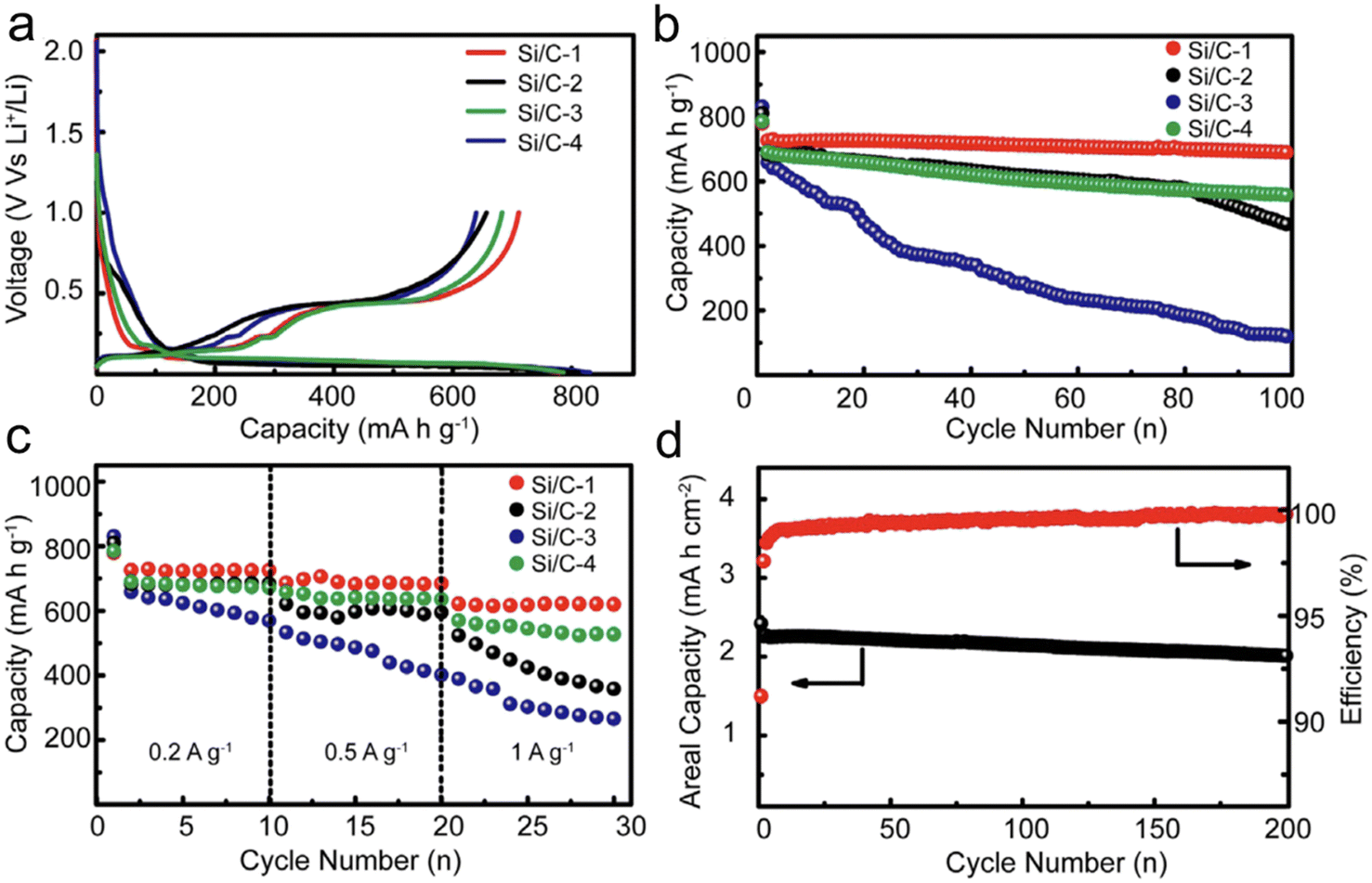 | ||
| Fig. 10 (a) The first charge and discharge profiles; (b) the cycling stability; (c) rate capability of different Si/C anodes; (d) high areal mass loading test and coulombic efficiency of the Si/C-1 anode. Reprinted with permission from ref. 117. Copyright 2016, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Micro-sized 2D layered and 3D porous structural electrodes, such as 3D macro-porous Si,118 mesoporous Si,119 nanoporous Si,120 and layered porous Si@CNT,121 have been widely documented. These configurations, endowed with appropriate void or pore spaces, exhibit enhanced capabilities to manage volume fluctuations. Such designs effectively limit the damage to or growth of the SEI layer. Furthermore, they amplify the electrochemical reaction kinetics during the alloying and dealloying phases, all the while upholding a substantial tap density. Additionally, these structures offer the advantage of reducing the contact surface with the electrolytes, further optimizing performance.
Nanostructured silicon in various architectural designs has demonstrated promising results in battery performance, although its implementation in industrial production poses a significant challenge. This is due to the current limitation in the ratio of silicon in graphite as an anode, which currently stands at less than 5 wt%. Lv et al.122 have presented a scalable approach to the synthesis of a large silicon cage composite, with a size measured in micrometers, which is composed of an ultrathin (<5 nm) mesoporous polypyrrole (PPy) skin and a silicon skeleton using a straightforward wet-chemical technique. They sourced the industrial-grade AlSi alloy as the precursor. The hollow skeleton framework offers generous room to manage the notable volume shifts during cycling. Simultaneously, the conductive polymer acts both as a safeguard and a fast channel for Li+/e− movement. Batteries equipped with these micro-sized silicon cages showcased impressive capacity retention across prolonged cycling at elevated charge/discharge rates and high material loadings. At a 0.2C rate, the battery delivered a specific capacity close to 1660 mA h g−1. Furthermore, its CE hovered around an impressive 99.8% even after 500 cycles at a material loading of 3 mg cm−2. When further charged at 1C, a remarkable capacity of 1149 mA h g−1 was preserved even after 500 cycles despite the high loading of silicon (Fig. 11). This outcome affirms the battery's potential to power large electrical devices, such as electric vehicles, owing to its high energy density.
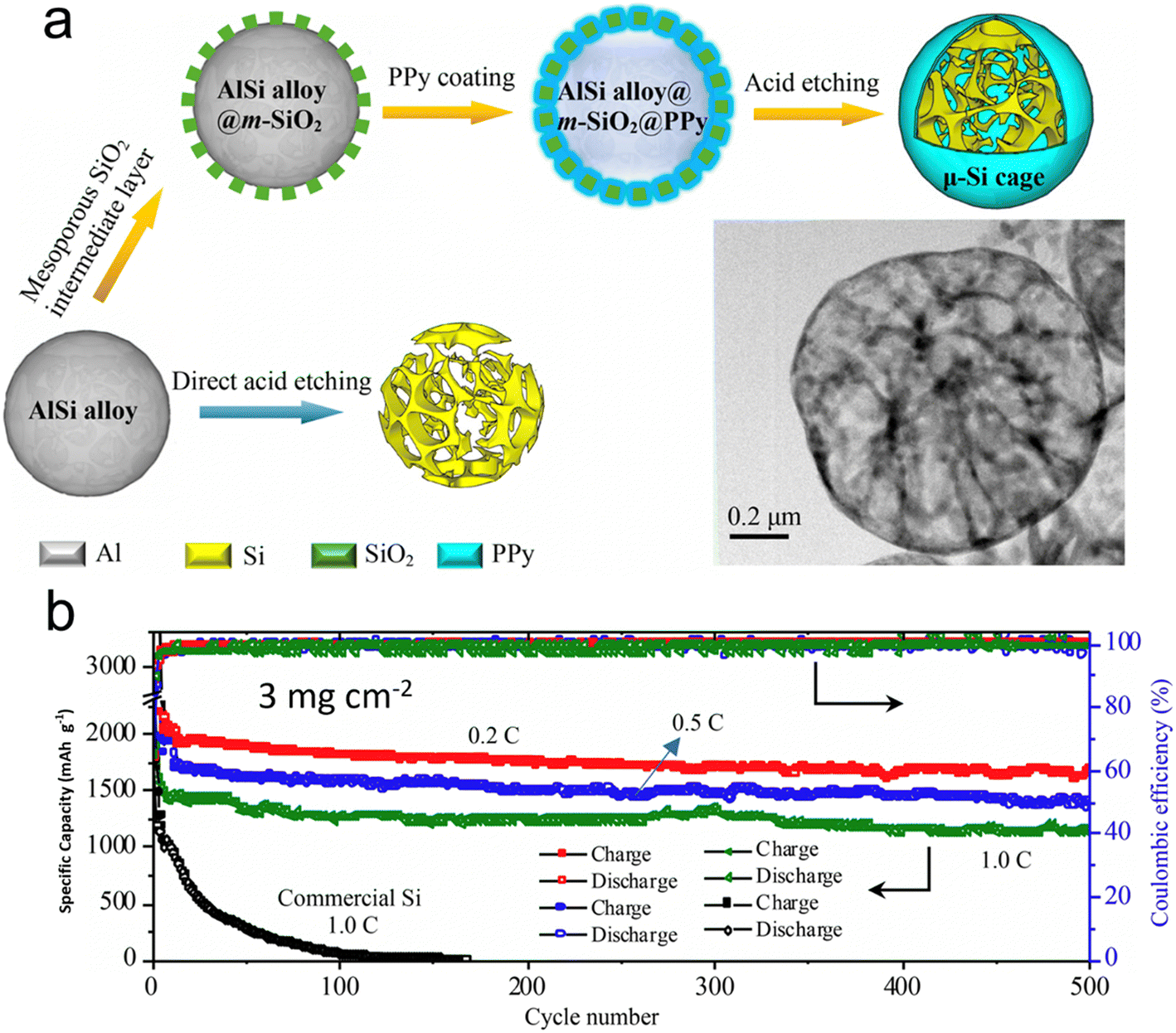 | ||
| Fig. 11 (a) Scalable synthesis of the μ-Si cage using AlSi microspheres as precursor by a chemical method (inset shows the TEM image of the μ-Si cage); (b) cycling performance of the battery with a μ-Si cage as anode with loadings of 3 mg cm−2. Reprinted with permission from ref. 122. Copyright 2019, American Chemical Society. | ||
The transport properties of (Li/Na-ions) within a battery are significantly impacted by morphology and structure. These properties subsequently determine the battery's charge/discharge rates and overall power delivery. One of the essential aspects dictating the charge/discharge rate of LIB/SIB is the combination of the ion diffusion coefficient and diffusion length. Zhao et al.123 have shed light on how, in an amorphous Si/C multilayer electrode, the silicon–carbon (Si–C) junctions play a pivotal role in facilitating the movement of Li-ions. This facilitation occurs in both directions – perpendicular and parallel to the Si–C interfaces, especially after the electrode undergoes cracking. To realize high-performing LIB anodes, Zhao's group introduced a design strategy characterized by a micro-size hierarchical multilayer block (Fig. 12). This approach emphasizes the precise stacking thickness of amorphous Si and carbon. Consequently, the Si/C film anode is formed through the alternating layering of amorphous Si and carbon, with optimal thickness and volume proportion. This layering leads to consistent cracking patterns and the stable formation of the SEI. Impressively, this electrode design reached a significant specific capacity of 2729 mA h g−1. Even after 200 cycles, it retained a capacity that was equivalent to 102.1% of its initial lithiation capacity and 101.8% of its theoretical specific capacity. The strategy hinges on exploiting the stress induced by volume alterations to segment the multilayer Si/C into micro-sized, multilayered hierarchical blocks. Such an approach notably amplifies the electrochemical performance, drawing significant advantages from the Si–C interfaces. While traditionally, the focus has been on confining volume alterations using an inert buffer coating, this research suggests another avenue. It underscores that maintaining impeccable contact between the buffer layer and the active material is crucial. By doing so, it's possible to fully harness the performance of the active material, particularly by amplifying Li-ion diffusion kinetics via these heterointerfaces.
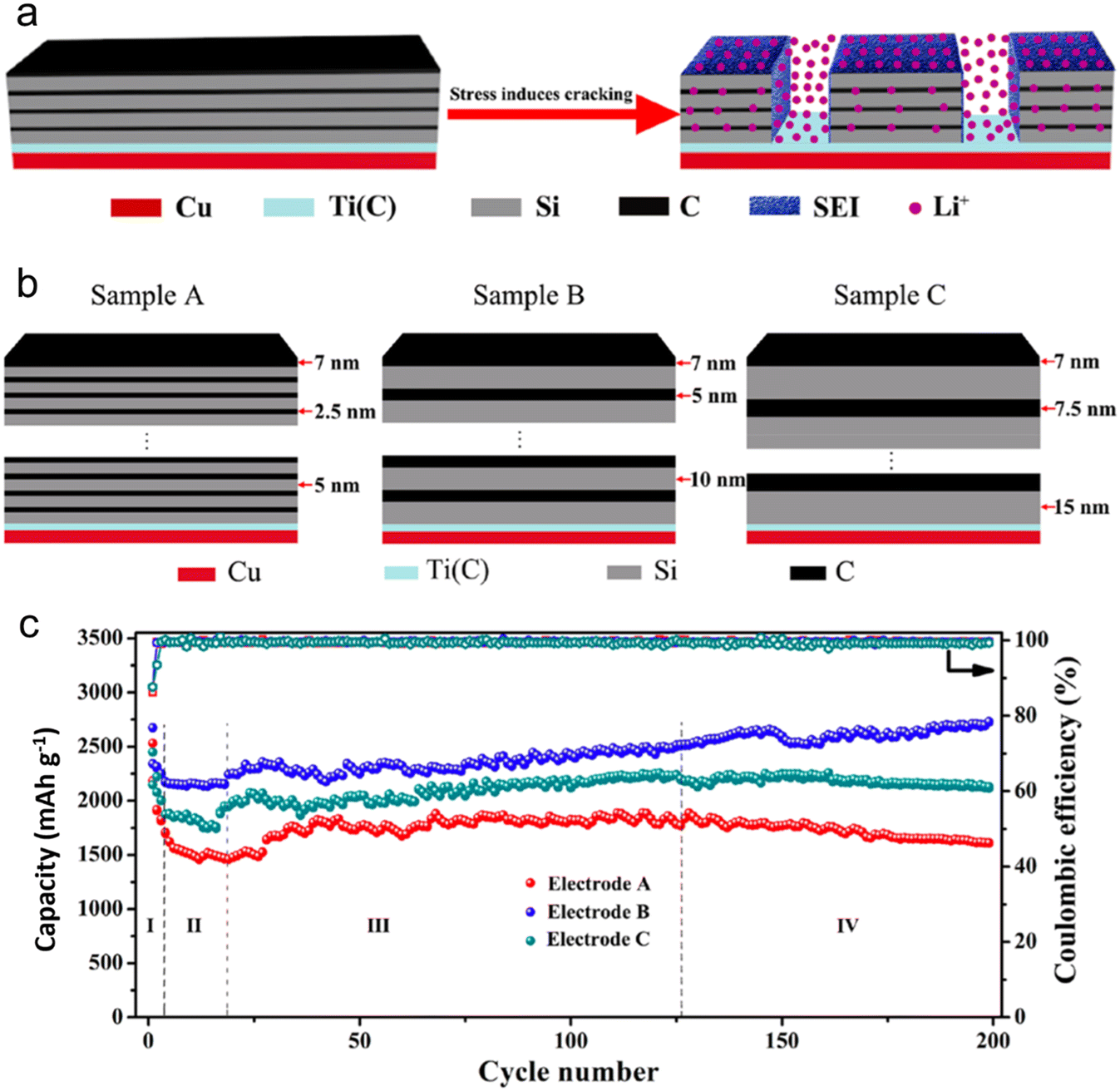 | ||
| Fig. 12 (a) Schematic illustration of Si/C multilayer blocks formed by stress; (b) schematic illustration of Si/C multilayer films structure; (c) cycling performance of electrodes A–C at a current density of 300 mA g−1. Reprinted with permission from ref. 123. Copyright 2019, American Chemical Society. | ||
Employing a structure that incorporates both primary and secondary particles offers insightful benefits,124 particularly in managing the mechanical strain resulting from the significant volume changes of high-capacity alloy anodes during continuous cycling. A recent study led by Liu et al.125 showcased the development of a hierarchical micro/nanostructured Sb-doped Pred/KB-MWCNTs anode tailored for SIBs. Both theoretical analyses and practical experiments harmoniously indicated that this Sb-doped Pred/KB-MWCNTs anode adeptly managed mechanical stresses and effectively hindered unwanted decomposition of the electrolytes. Notably, the Sb-doped Pred/KB-MWCNTs anodes, when placed in traditional carbonate (1 M NaPF6–PC with 2% FEC) and localized high concentration (1.2 M NaFSI–DME/BTFE) electrolytes, exhibited similar initial coulombic efficiency and cycling stability (Fig. 13). This finding underscores the pivotal role played by the anode's structural configuration. Moreover, when juxtaposed with Pred/C30, X-ray photoelectron spectroscopy (XPS) analyses revealed that the Sb-doped Pred/KB-MWCNTs, regardless of the electrolyte type, could foster the formation of a thin, robust, and fluorine-rich SEI layer by controlling the electrolyte decomposition.
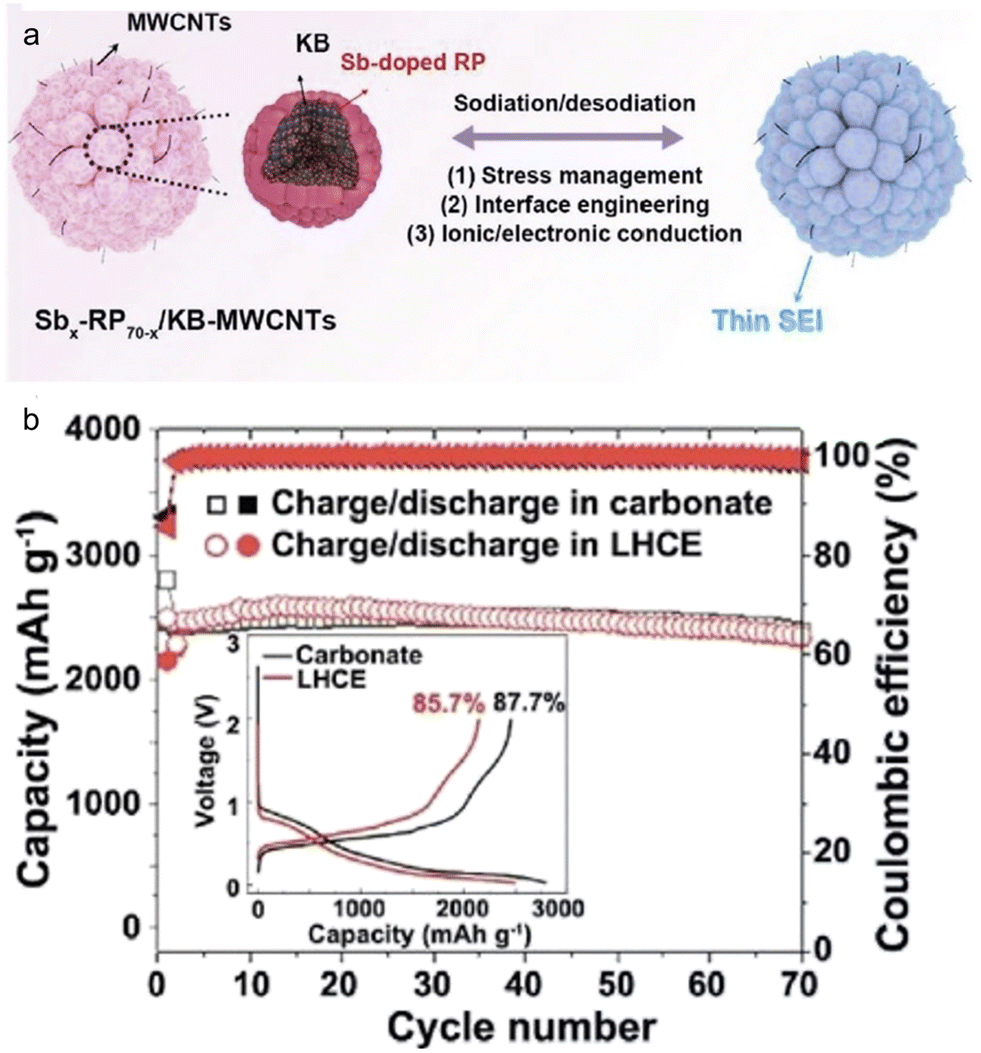 | ||
| Fig. 13 (a) Schematic illustration and (b) cycling performance in carbonate electrolyte and localized highly concentrated ether electrolyte (LHCE) of hierarchical micro/nanostructured Sbx-RP70−x/KB-MWCNTs. Inset: first charge/discharge curve in two electrolytes. Reprinted with permission from ref. 125. Copyright 2021, American Chemical Society. | ||
The use of porous microscale materials, specifically those composed of nano-blocks and nanopores, has garnered significant interest in recent times.126 Their popularity can be attributed to several reasons:
1) Volume accommodation: alloys designed with adjustable pore sizes and porosities can adeptly handle volume changes during the lithiation/sodiation process. This capability minimizes particle-level volume expansion, which in turn restricts electrode swelling and the subsequent detachment of active elements from the current collector.
2) Enhanced kinetics: the porous nature of these materials promotes deeper infiltration of the electrolyte, trims down the distance required for ion/electron transfer, and reduces internal impedance. Collectively, these factors lead to improved kinetic performance of the materials.
3) High tap density and specific BET surface area: these materials exhibit a notable tap density and a reduced specific BET surface area. Such attributes culminate in elevated ICE, mass loading, and energy density. Moreover, their 3D design facilitates electrolyte permeation, endowing the materials with exceptional mass transfer capabilities. This makes them particularly apt for applications even when there's a high mass loading of active components.
4) Increased active sites: the intricate network of interconnected pores within these materials provides additional active sites for reactions, which amplifies their capacity.
In essence, when considering the performance metrics, porous microscale alloys encompassing nanoparticles distinctly outshine their counterparts that are solely nano- or microscale in design, especially in the context of lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs).6 Electrode materials can benefit from the formation of pores, which can improve their electrochemical properties. Nevertheless, the straightforward synthesis of porous structures, particularly in materials like Sn, presents substantial obstacles. This is largely due to Sn's inherent low melting point combined with the inevitable electrochemical agglomeration it undergoes. Song et al.127 tackled this challenge by fabricating 3D continuous microscale Sn with a porous structure from immiscible Al–Sn alloys. Impressively, the resultant microporous Sn anode showcased a high initial capacity, measuring 818 mA h g−1, and a commendable CE of 99% (Fig. 14). Additionally, the structural integrity of the porous Sn was retained post the inaugural cycle, indicating its potential suitability as an anode in LIBs. Nevertheless, the as-dealloyed porous Sn still falls short in terms of ductility and strength, leading to a short cycle life. In other words, the porous Sn structures produced through dealloying are unable to sufficiently buffer the strain during lithiation/delithiation for extended cycle life.
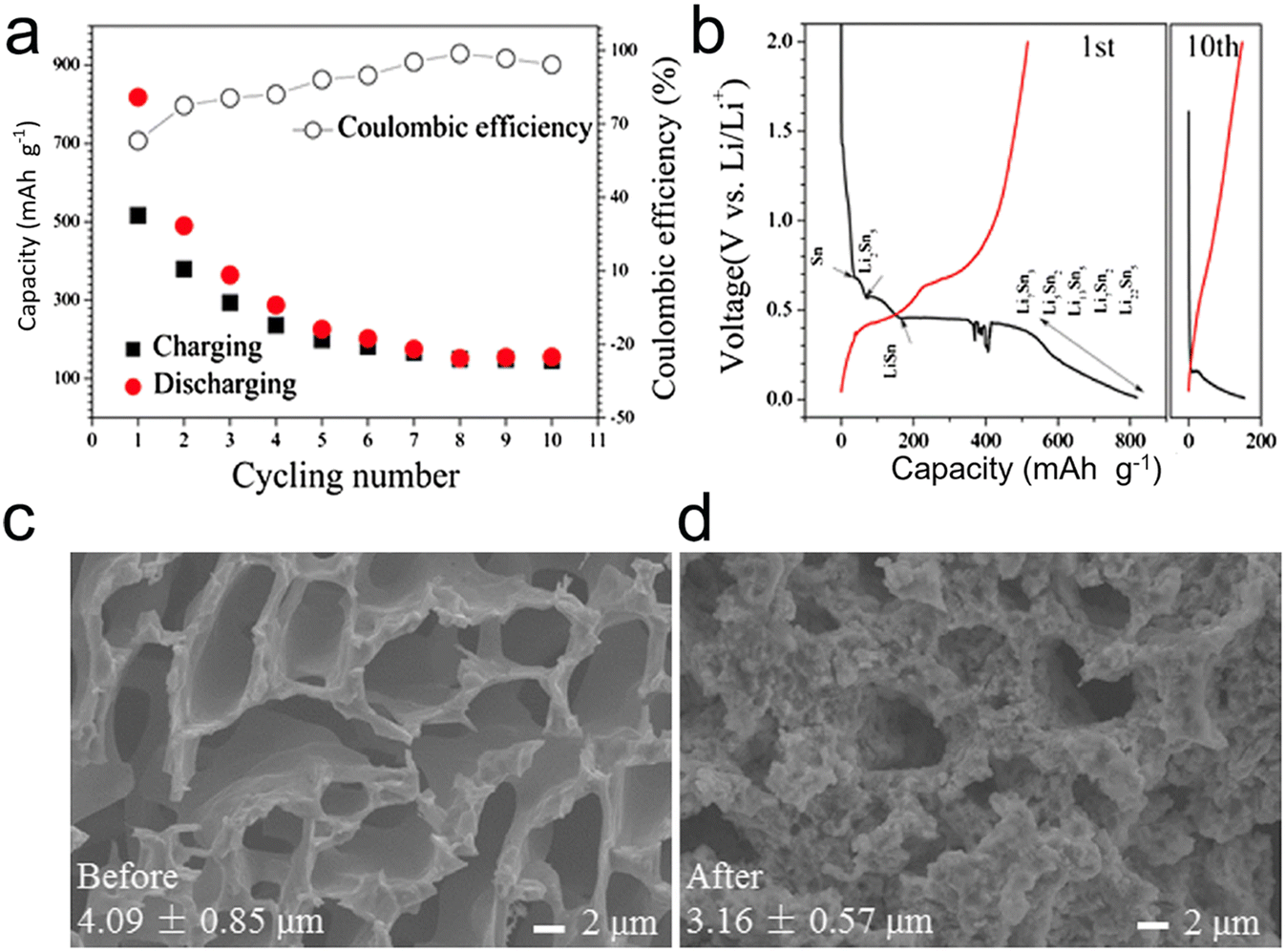 | ||
| Fig. 14 (a) Cycling performance and (b) charge/discharge curve of the porous Sn dealloyed from Al80Sn20; SEM images of the porous Sn structure before (c) and after (d) the lithiation/delithiation process. Reprinted with permission from ref. 127. Copyright 2015, Springer. | ||
5 Conclusion and outlook
In summary, we have reviewed recent progress on the development of micro-sized alloy anodes for alkali-ion batteries through engineering of electrolytes, binder and electrode architecture.Undeniably, electrolyte stands out as a paramount component in battery systems. Its constituents, including anions, cations, solvents, additives, and even the concentration, exert intricate and profoundly impactful effects on the microstructures, composition, and durability, as well as the mechanical and ionic transport characteristics of the SEI layer. To date, a myriad of strategies centered around electrolyte engineering have been put forth to craft a slender yet robust, high-quality SEI. These strategies encompass shifts from carbonate-based solvents to ether derivatives, judicious selection of salts, and modulation of concentration levels, ranging from conventional (1 M) to elevated (≥3 M) or even localized high concentrations and solid-state electrolytes. Despite these advances, the underlying mechanism remains elusive due to the lack of in situ characterization tools to track micro-sized alloy anode's local structure and morphological changes during charge/discharge. For example, in situ TEM, widely used to investigate the volume changes of nano-structured anodes, is not an ideal tool for characterizing micro-sized anodes due to resolution limitations. More importantly, how the larger volume change of micro-sized anode affects the resulting SEI and the consequent charge/discharge kinetics, and reversibility are still open questions.
In electrochemistry, the stabilizing electrode and SEI layer are crucial for the performance of energy storage devices. The stabilizing electrode ensures the stability of the electrode–electrolyte interface by impeding the dissolution of active material during the charge–discharge process. On the other hand, the SEI layer acts as a protective layer that forms on the electrode's surface and mitigates the electrolyte's reactivity with the electrode material. Both the stabilizing electrode and SEI layer are critical components that are fundamental to the overall performance and longevity of energy storage devices. This discussion elucidates the significant role that binders can play in enhancing the electrochemical effectiveness of ion batteries. It is important to note that a tradeoff always exists when selecting a binder and additive, as it can either lead to higher capacities with poor retention or lower capacities with higher retention. As such, discovering new and improved binders and additives is a promising and captivating field for new energy sector researchers.
Additionally, exploring the possible combinations of binders with various electrolytes presents a hot spot for introducing innovative chemistries and augmenting the electrochemical performance of LIB/SIB. Numerous studies have been conducted to investigate multiple polymers, focusing on their chemical bonds, mechanical properties, supramolecular interactions, and electrochemical properties. However, these studies are sporadic and often specific to a particular case, making it challenging to establish a comprehensive design principle for polymeric binders for micro-size alloy anodes. Binder research must delve into in situ analyses to enhance our comprehension of the dynamic physicochemical processes involved in electrode components. By employing such studies, researchers can gain a more nuanced understanding of these processes, much like the approach taken in organic reaction mechanisms. Aiming to establish an in-depth mechanistic understanding of these dynamic processes is therefore imperative.
The quest for effective and economically viable synthesis techniques for electrode materials continues to be at the forefront of contemporary research in battery technology. A plethora of alloying anodes showcased impressive cycling stabilities when tested in half cells. However, a select few have managed to achieve the remarkable coulombic efficiency that is indispensable for their integration into full cells.
An inherent challenge lies in their specific capacity. While these anodes possess a commendable specific capacity, their constrained areal capacity might pose a hindrance, especially when one envisages aligning it with the potentially elevated areal capacities of future cathodes.
The creation of microscale active materials, which combine a high tap density with exceptional performance in lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs), presents its own set of challenges. Beyond the technical aspects of material development, there's a broader perspective to consider. For these alloying-type materials to transition from the laboratory to commercial applications, certain prerequisites need to be met.
The continuous quest for higher energy density, improved safety, and cost-effectiveness drives innovations in LIB technology. In this regard, micro/nanostructured alloy anodes represent a significant leap forward. These anodes offer substantial improvements over conventional graphite anodes, particularly in energy density, cycling stability, and life span, which are crucial for applications ranging from consumer electronics to electric vehicles. Silicon, tin, and their carbon composites exhibit dramatically higher specific capacities than traditional anodes. This translates into batteries that can store more energy without a proportional increase in size or weight, a critical factor for industrial applications where space and weight efficiency are paramount. While the raw material costs for these alloys might be higher, the enhanced energy density means that a smaller amount of active material is required for the same energy output, potentially lowering the overall material cost per kW h of capacity.
The production of micro/nanostructured alloy anodes has become increasingly viable with advancements in battery manufacturing technology. Techniques like electrospinning, chemical vapor deposition, sol–gel processes, and high-energy ball milling enable precise control over the structure of the anode material, leading to better performance and consistency. Importantly, these techniques are scalable and can be integrated into existing battery production lines. This integration is crucial for industrial-scale production, allowing battery manufacturers to adopt these advanced materials without requiring extensive overhauls of current manufacturing facilities. For industrial applications, the longevity and reliability of energy storage systems are as important as their initial costs. These anode materials offer improved cycling stability and are less prone to capacity fade over time than traditional anodes. This longer lifespan of the batteries translates into reduced operational costs as the need for frequent replacements diminishes. Additionally, the enhanced cycle life of these batteries means less downtime and maintenance, which is vital for industrial operations where continuous power is essential. Moreover, the increased energy density of these anodes can lead to smaller and lighter batteries, which is advantageous in terms of material use and logistics, further driving down costs.
Adopting these alloy anodes can also contribute to broader economic and environmental sustainability goals. By improving the efficiency and life span of batteries, these anodes can facilitate a more robust adoption of renewable energy sources in industrial settings, promoting a shift towards greener energy practices. Furthermore, the enhanced battery performance can lead to reductions in waste and a decrease in the environmental footprint of industrial operations, aligning with global efforts toward sustainability.
It's essential to adopt synthesis procedures that are not only efficient but also straightforward. The choice of chemicals used should lean towards those that are cost-effective and environmentally benign. Additionally, the synthesis methods should be capable of producing these materials at a scale and yield that aligns with commercial production demands. In essence, the pathway to commercializing these promising materials entails a balanced amalgamation of performance metrics, sustainable practices, and economic viability.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Argonne National Laboratory is operated for DOE Office of Science by UChicago Argonne, LLC, under contract number DE-AC02-06CH11357. Support from Tien Duong of the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy, Vehicle Technologies Office is gratefully acknowledged.References
- M. Winter, B. Barnett and K. Xu, Before Li ion batteries, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, 30 Years of Lithium-ion batteries, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- H. Zhang, I. Hasa and S. Passerini, Beyond insertion for na-ion batteries: Nanostructured alloying and conversion anode materials, Adv. Energy Mater., 2018, 8, 1702582 CrossRef.
- H. Tan, D. Chen, X. Rui and Y. Yu, Peering into alloy anodes for sodium-ion batteries: Current trends, challenges, and opportunities, Adv. Funct. Mater., 2019, 29, 1808745 CrossRef.
- S. Liang, Y.-J. Cheng, J. Zhu, Y. Xia and P. Müller-Buschbaum, A chronicle review of non-silicon (Sn, Sb, Ge)-based lithium/sodium-ion battery alloying anodes, Small Methods, 2020, 4, 2000218 CrossRef CAS.
- G. Li, S. Guo, B. Xiang, S. Mei, Y. Zheng, X. Zhang, B. Gao, P. K. Chu and K. Huo, Recent advances and perspectives of microsized alloying-type porous anode materials in high-performance Li- and Na-ion batteries, Energy Mater., 2022, 2, 200020 CrossRef CAS.
- V. Aravindan, Y.-S. Lee and S. Madhavi, Research progress on negative electrodes for practical Li-ion batteries: Beyond carbonaceous anodes, Adv. Energy Mater., 2015, 5, 1402225 CrossRef.
- E. J. McShane, A. M. Colclasure, D. E. Brown, Z. M. Konz, K. Smith and B. D. McCloskey, Quantification of inactive lithium and solid–electrolyte interphase species on graphite electrodes after fast charging, ACS Energy Lett., 2020, 5, 2045–2051 CrossRef CAS.
- X. Deng, Z. Chen and Y. Cao, Transition metal oxides based on conversion reaction for sodium-ion battery anodes, Mater. Today Chem., 2018, 9, 114–132 CrossRef CAS.
- S. P. V. Nadimpalli, R. Tripuraneni and V. A. Sethuraman, Real-time stress measurements in germanium thin film electrodes during electrochemical lithiation/delithiation cycling, J. Electrochem. Soc., 2015, 162, A2840 CrossRef CAS.
- Q. Wang, C. Zhao, Y. Lu, Y. Li, Y. Zheng, Y. Qi, X. Rong, L. Jiang, X. Qi, Y. Shao, D. Pan, B. Li, Y.-S. Hu and L. Chen, Advanced nanostructured anode materials for sodium-ion batteries, Small, 2017, 13, 1701835 CrossRef PubMed.
- S. Guo, Y. Feng, L. Wang, Y. Jiang, Y. Yu and X. Hu, Architectural engineering achieves high-performance alloying anodes for lithium and sodium ion batteries, Small, 2021, 17, 2005248 CrossRef CAS PubMed.
- B. L. Ellis, P. Knauth and T. Djenizian, Three-dimensional self-supported metal oxides for advanced energy storage, Adv. Mater., 2014, 26, 3368–3397 CrossRef CAS PubMed.
- K. Gerasopoulos, E. Pomerantseva, M. McCarthy, A. Brown, C. Wang, J. Culver and R. Ghodssi, Hierarchical three-dimensional microbattery electrodes combining bottom-up self-assembly and top-down micromachining, ACS Nano, 2012, 6, 6422–6432 CrossRef CAS PubMed.
- J. W. Long, B. Dunn, D. R. Rolison and H. S. White, Three-dimensional battery architectures, Chem. Rev., 2004, 104, 4463–4492 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, High-performance lithium battery anodes using silicon nanowires, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- Y. Liu, N. Zhang, L. Jiao and J. Chen, Tin nanodots encapsulated in porous nitrogen-doped carbon nanofibers as a free-standing anode for advanced sodium-ion batteries, Adv. Mater., 2015, 27, 6702–6707 CrossRef CAS PubMed.
- Y. An, Y. Tian, L. Ci, S. Xiong, J. Feng and Y. Qian, Micron-sized nanoporous antimony with tunable porosity for high-performance potassium-ion batteries, ACS Nano, 2018, 12, 12932–12940 CrossRef CAS PubMed.
- S. Liu, H. Xu, X. Bian, J. Feng, J. Liu, Y. Yang, C. Yuan, Y. An, R. Fan and L. Ci, Nanoporous red phosphorus on reduced graphene oxide as superior anode for sodium-ion batteries, ACS Nano, 2018, 12, 7380–7387 CrossRef CAS PubMed.
- Y. An, Y. Tian, C. Wei, Y. Tao, B. Xi, S. Xiong, J. Feng and Y. Qian, Dealloying: An effective method for scalable fabrication of 0D, 1D, 2D, 3D materials and its application in energy storage, Nano Today, 2021, 37, 101094 CrossRef CAS.
- Y. An, Y. Tian, C. Wei, H. Jiang, B. Xi, S. Xiong, J. Feng and Y. Qian, Scalable and physical synthesis of 2D silicon from bulk layered alloy for lithium-ion batteries and lithium metal batteries, ACS Nano, 2019, 13, 13690–13701 CrossRef CAS PubMed.
- Y. An, Y. Tian, H. Wei, B. Xi, S. Xiong, J. Feng and Y. Qian, Porosity- and graphitization-controlled fabrication of nanoporous silicon@carbon for lithium storage and its conjugation with mxene for lithium-metal anode, Adv. Funct. Mater., 2020, 30, 1908721 CrossRef CAS.
- J. Huang, X. Lin, H. Tan and B. Zhang, Bismuth microparticles as advanced anodes for potassium-ion battery, Adv. Energy Mater., 2018, 8, 1703496 CrossRef.
- B. Zhang, G. Rousse, D. Foix, R. Dugas, D. A. D. Corte and J.-M. Tarascon, Microsized Sn as advanced anodes in glyme-based electrolyte for Na-ion batteries, Adv. Mater., 2016, 28, 9824–9830 CrossRef CAS PubMed.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Li-ion battery materials: Present and future, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- W. Zhai, Q. Ai, L. Chen, S. Wei, D. Li, L. Zhang, P. Si, J. Feng and L. Ci, Walnut-inspired microsized porous silicon/graphene core–shell composites for high-performance lithium-ion battery anodes, Nano Res., 2017, 10, 4274–4283 CrossRef CAS.
- J. Huang, X. Guo, X. Du, X. Lin, J.-Q. Huang, H. Tan, Y. Zhu and B. Zhang, Nanostructures of solid electrolyte interphases and their consequences for microsized Sn anodes in sodium ion batteries, Energy Environ. Sci., 2019, 12, 1550–1557 RSC.
- J. Wang and Y. Cui, Electrolytes for microsized silicon, Nat. Energy, 2020, 5, 361–362 CrossRef CAS.
- G. Zhu, D. Chao, W. Xu, M. Wu and H. Zhang, Microscale silicon-based anodes: Fundamental understanding and industrial prospects for practical high-energy lithium-ion batteries, ACS Nano, 2021, 15, 15567–15593 CrossRef CAS PubMed.
- X. Du and B. Zhang, Robust solid electrolyte interphases in localized high concentration electrolytes boosting black phosphorus anode for potassium-ion batteries, ACS Nano, 2021, 15, 16851–16860 CrossRef CAS PubMed.
- X. Du, Y. Gao and B. Zhang, Building elastic solid electrolyte interphases for stabilizing microsized antimony anodes in potassium ion batteries, Adv. Funct. Mater., 2021, 31, 2102562 CrossRef CAS.
- R. Jain, A. S. Lakhnot, K. Bhimani, S. Sharma, V. Mahajani, R. A. Panchal, M. Kamble, F. Han, C. Wang and N. Koratkar, Nanostructuring versus microstructuring in battery electrodes, Nat. Rev. Mater., 2022, 7, 736–746 CrossRef CAS.
- Y.-F. Tian, G. Li, D.-X. Xu, Z.-Y. Lu, M.-Y. Yan, J. Wan, J.-Y. Li, Q. Xu, S. Xin, R. Wen and Y.-G. Guo, Micrometer-sized SiMgyOx with stable internal structure evolution for high-performance Li-ion battery anodes, Adv. Mater., 2022, 34, 2200672 CrossRef CAS PubMed.
- Y. Wu, H.-B. Huang, Y. Feng, Z.-S. Wu and Y. Yu, The promise and challenge of phosphorus-based composites as anode materials for potassium-ion batteries, Adv. Mater., 2019, 31, 1901414 CrossRef CAS PubMed.
- H. Zhu, Z. Jia, Y. Chen, N. Weadock, J. Wan, O. Vaaland, X. Han, T. Li and L. Hu, Tin anode for sodium-ion batteries using natural wood fiber as a mechanical buffer and electrolyte reservoir, Nano Lett., 2013, 13, 3093–3100 CrossRef CAS PubMed.
- Y. Gao, R. Yi, Y. C. Li, J. Song, S. Chen, Q. Huang, T. E. Mallouk and D. Wang, General method of manipulating formation, composition, and morphology of solid-electrolyte interphases for stable Li-alloy anodes, J. Am. Chem. Soc., 2017, 139, 17359–17367 CrossRef CAS PubMed.
- Y. Liu, Q. Liu, C. Jian, D. Cui, M. Chen, Z. Li, T. Li, T. Nilges, K. He, Z. Jia and C. Zhou, Red-phosphorus-impregnated carbon nanofibers for sodium-ion batteries and liquefaction of red phosphorus, Nat. Commun., 2020, 11, 2520 CrossRef CAS PubMed.
- H. Ding, J. Wang, J. Zhou, C. Wang and B. Lu, Building electrode skins for ultra-stable potassium metal batteries, Nat. Commun., 2023, 14, 2305 CrossRef CAS PubMed.
- M. Gu, A. M. Rao, J. Zhou and B. Lu, In situ formed uniform and elastic SEI for high-performance batteries, Energy Environ. Sci., 2023, 16, 1166–1175 RSC.
- Y. Zhang, X. Yi, H. Fu, X. Wang, C. Gao, J. Zhou, A. M. Rao and B. Lu, Reticular elastic solid electrolyte interface enabled by an industrial dye for ultrastable potassium-ion batteries, Small Struct., 2024, 5, 2300232 CrossRef CAS.
- K. Xu, Electrolytes and interphases in Li-ion batteries and beyond, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- Handbook of Battery Materials, Wiley-VCH, Weinheim, New York, 1999 Search PubMed.
- P. Peljo and H. H. Girault, Electrochemical potential window of battery electrolytes: The HOMO–LUMO misconception, Energy Environ. Sci., 2018, 11, 2306–2309 RSC.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Sodium metal anodes: Emerging solutions to dendrite growth, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- A. K. Stephan, Completing the picture of the solid electrolyte interphase, Joule, 2019, 3, 1812–1814 CrossRef.
- X. Yi, A. M. Rao, J. Zhou and B. Lu, Trimming the degrees of freedom via a K+ flux rectifier for safe and long-life potassium-ion batteries, Nano-Micro Lett., 2023, 15, 200 CrossRef CAS PubMed.
- J. Wu, M. Ihsan-Ul-Haq, Y. Chen and J.-K. Kim, Understanding solid electrolyte interphases: Advanced characterization techniques and theoretical simulations, Nano Energy, 2021, 89, 106489 CrossRef CAS.
- N. Yao, X. Chen, Z.-H. Fu and Q. Zhang, Applying Classical, Ab initio, and machine-learning molecular dynamics simulations to the liquid electrolyte for rechargeable batteries, Chem. Rev., 2022, 122, 10970–11021 CrossRef CAS PubMed.
- M. Li, R. P. Hicks, Z. Chen, C. Luo, J. Guo, C. Wang and Y. Xu, Electrolytes in organic batteries, Chem. Rev., 2023, 123, 1712–1773 CrossRef CAS PubMed.
- X. Fan and C. Wang, High-voltage liquid electrolytes for Li batteries: Progress and perspectives, Chem. Soc. Rev., 2021, 50, 10486–10566 RSC.
- B. S. Parimalam and B. L. Lucht, Reduction reactions of electrolyte salts for lithium ion batteries: LiPF6, LiBF4, LiDFOB, LiBOB, and LiTFSI, J. Electrochem. Soc., 2018, 165, A251 CrossRef CAS.
- T. Li, X.-Q. Zhang, P. Shi and Q. Zhang, Fluorinated solid-electrolyte interphase in high-voltage lithium metal batteries, Joule, 2019, 3, 2647–2661 CrossRef CAS.
- L. Ma, J. Li, T. Wu, P. Sun, S. Tan, H. Wang, W. Xie, L. Pan, Y. Yamauchi and W. Mai, Re-oxidation reconstruction process of solid electrolyte interphase layer derived from highly active anion for potassium-ion batteries, Nano Energy, 2021, 87, 106150 CrossRef CAS.
- H. Zheng, H. Xiang, F. Jiang, Y. Liu, Y. Sun, X. Liang, Y. Feng and Y. Yu, Lithium difluorophosphate-based dual-salt low concentration electrolytes for lithium metal batteries, Adv. Energy Mater., 2020, 10, 2001440 CrossRef CAS.
- G. G. Eshetu, G. A. Elia, M. Armand, M. Forsyth, S. Komaba, T. Rojo and S. Passerini, Electrolytes and interphases in sodium-based rechargeable batteries: Recent advances and perspectives, Adv. Energy Mater., 2020, 10, 2000093 CrossRef CAS.
- Y. An, H. Fei, G. Zeng, L. Ci, B. Xi, S. Xiong and J. Feng, Commercial expanded graphite as a low–cost, long-cycling life anode for potassium–ion batteries with conventional carbonate electrolyte, J. Power Sources, 2018, 378, 66–72 CrossRef CAS.
- Z. Piao, R. Gao, Y. Liu, G. Zhou and H.-M. Cheng, A review on regulating Li+ solvation structures in carbonate electrolytes for lithium metal batteries, Adv. Mater., 2023, 35, 2206009 CrossRef CAS PubMed.
- Y. Gao, X. Du, Z. Hou, X. Shen, Y.-W. Mai, J.-M. Tarascon and B. Zhang, Unraveling the mechanical origin of stable solid electrolyte interphase, Joule, 2021, 5, 1860–1872 CrossRef CAS.
- A. Meazah Haregewoin, A. Sorsa Wotango and B.-J. Hwang, Electrolyte additives for lithium ion battery electrodes: Progress and perspectives, Energy Environ. Sci., 2016, 9, 1955–1988 RSC.
- Z. Zeng, X. Jiang, R. Li, D. Yuan, X. Ai, H. Yang and Y. Cao, A safer sodium-ion battery based on nonflammable organic phosphate electrolyte, Adv. Sci., 2016, 3, 1600066 CrossRef PubMed.
- Y. Jin, N.-J. H. Kneusels, L. E. Marbella, E. Castillo-Martínez, P. C. M. M. Magusin, R. S. Weatherup, E. Jónsson, T. Liu, S. Paul and C. P. Grey, Understanding fluoroethylene carbonate and vinylene carbonate based electrolytes for Si anodes in lithium ion batteries with NMR spectroscopy, J. Am. Chem. Soc., 2018, 140, 9854–9867 CrossRef CAS PubMed.
- B. Qin, A. Schiele, Z. Jusys, A. Mariani, T. Diemant, X. Liu, T. Brezesinski, R. J. Behm, A. Varzi and S. Passerini, Highly reversible sodiation of tin in glyme electrolytes: The critical role of the solid electrolyte interphase and its formation mechanism, ACS Appl. Mater. Interfaces, 2020, 12, 3697–3708 CrossRef CAS PubMed.
- J. Luis Gómez-Cámer, B. Acebedo, N. Ortiz-Vitoriano, I. Monterrubio, M. Galcerán and T. Rojo, Unravelling the impact of electrolyte nature on Sn4P3/C negative electrodes for Na-ion batteries, J. Mater. Chem. A, 2019, 7, 18434–18441 RSC.
- H. Wang, J. He, J. Liu, S. Qi, M. Wu, J. Wen, Y. Chen, Y. Feng and J. Ma, Electrolytes enriched by crown ethers for lithium metal batteries, Adv. Funct. Mater., 2021, 31, 2002578 CrossRef CAS.
- X. Ma, H. Fu, J. Shen, D. Zhang, J. Zhou, C. Tong, A. M. Rao, J. Zhou, L. Fan and B. Lu, Green ether electrolytes for sustainable high-voltage potassium ion batteries, Angew. Chem., Int. Ed., 2023, 62, e202312973 CrossRef CAS PubMed.
- J. Chen, X. Fan, Q. Li, H. Yang, M. R. Khoshi, Y. Xu, S. Hwang, L. Chen, X. Ji, C. Yang, H. He, C. Wang, E. Garfunkel, D. Su, O. Borodin and C. Wang, Electrolyte design for LiF-rich solid–electrolyte interfaces to enable high-performance microsized alloy anodes for batteries, Nat. Energy, 2020, 5, 386–397 CrossRef CAS.
- Y. Li, F. Wu, Y. Li, M. Liu, X. Feng, Y. Bai and C. Wu, Ether-based electrolytes for sodium ion batteries, Chem. Soc. Rev., 2022, 51, 4484–4536 RSC.
- N. Zhang, C. Sun, Y. Huang, C. Zhu, Z. Wu, L. Lv, X. Zhou, X. Wang, X. Xiao, X. Fan and L. Chen, Tuning electrolyte enables microsized Sn as an advanced anode for Li-ion batteries, J. Mater. Chem. A, 2021, 9, 1812–1821 RSC.
- E. P. Roth and C. J. Orendorff, How electrolytes influence battery safety, Electrochem. Soc. Interface, 2012, 21, 45 CrossRef CAS.
- K. Dokko, N. Tachikawa, K. Yamauchi, M. Tsuchiya, A. Yamazaki, E. Takashima, J.-W. Park, K. Ueno, S. Seki, N. Serizawa and M. Watanabe, Solvate ionic liquid electrolyte for Li–S batteries, J. Electrochem. Soc., 2013, 160, A1304 CrossRef CAS.
- S. Izquierdo-Gonzales, W. Li and B. L. Lucht, Hexamethylphosphoramide as a flame retarding additive for lithium-ion battery electrolytes, J. Power Sources, 2004, 135, 291–296 CrossRef CAS.
- Z. Zeng, B. Wu, L. Xiao, X. Jiang, Y. Chen, X. Ai, H. Yang and Y. Cao, Safer lithium ion batteries based on nonflammable electrolyte, J. Power Sources, 2015, 279, 6–12 CrossRef CAS.
- Q. Zheng, Y. Yamada, R. Shang, S. Ko, Y.-Y. Lee, K. Kim, E. Nakamura and A. Yamada, A cyclic phosphate-based battery electrolyte for high voltage and safe operation, Nat. Energy, 2020, 5, 291–298 CrossRef CAS.
- S. Yang, Y. Zhang, Z. Li, N. Takenaka, Y. Liu, H. Zou, W. Chen, M. Du, X.-J. Hong, R. Shang, E. Nakamura, Y.-P. Cai, Y.-Q. Lan, Q. Zheng, Y. Yamada and A. Yamada, Rational electrolyte design to form inorganic–polymeric interphase on silicon-based anodes, ACS Energy Lett., 2021, 6, 1811–1820 CrossRef CAS.
- G. Zeng, Y. An, S. Xiong and J. Feng, Nonflammable fluorinated carbonate electrolyte with high salt-to-solvent ratios enables stable silicon-based anode for next-generation lithium-ion batteries, ACS Appl. Mater. Interfaces, 2019, 11, 23229–23235 CrossRef CAS PubMed.
- H. Jia, L. Zou, P. Gao, X. Cao, W. Zhao, Y. He, M. H. Engelhard, S. D. Burton, H. Wang, X. Ren, Q. Li, R. Yi, X. Zhang, C. Wang, Z. Xu, X. Li, J.-G. Zhang and W. Xu, High-performance silicon anodes enabled by nonflammable localized high-concentration electrolytes, Adv. Energy Mater., 2019, 9, 1900784 CrossRef.
- L. Wang, B. Zhang, B. Wang, S. Zeng, M. Zhao, X. Sun, Y. Zhai and L. Xu, In-situ nano-crystallization and solvation modulation to promote highly stable anode involving alloy/de-alloy for potassium ion batteries, Angew. Chem., 2021, 133, 15509–15517 CrossRef.
- K. Xu, Nonaqueous liquid electrolytes for lithium-based rechargeable batteries, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- N. M. Johnson, Z. Yang, M. Kim, D.-J. Yoo, Q. Liu and Z. Zhang, Enabling silicon anodes with novel isosorbide-based electrolytes, ACS Energy Lett., 2022, 7, 897–905 CrossRef CAS.
- L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Controlling SEI formation on SnSb-porous carbon nanofibers for improved Na ion storage, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed.
- A. M. Haregewoin, A. S. Wotango and B.-J. Hwang, Electrolyte additives for lithium ion battery electrodes: Progress and perspectives, Energy Environ. Sci., 2016, 9, 1955–1988 RSC.
- Y. Li, Y. An, Y. Tian, H. Fei, S. Xiong, Y. Qian and J. Feng, Stable and safe lithium metal batteries with Ni-rich cathodes enabled by a high efficiency flame retardant additive, J. Electrochem. Soc., 2019, 166, A2736 CrossRef CAS.
- M. Peng, K. Shin, L. Jiang, Y. Jin, K. Zeng, X. Zhou and Y. Tang, Alloy-type anodes for high-performance rechargeable batteries, Angew. Chem., Int. Ed., 2022, 61, e202206770 CrossRef CAS PubMed.
- Q. Sun, Z. Cao, Z. Ma, J. Zhang, W. Wahyudi, G. Liu, H. Cheng, T. Cai, E. Xie, L. Cavallo, Q. Li and J. Ming, Interfacial and interphasial chemistry of electrolyte components to invoke high-performance antimony anodes and non-flammable lithium-ion batteries, Adv. Funct. Mater., 2023, 33, 2210292 CrossRef CAS.
- G. Yan, K. Reeves, D. Foix, Z. Li, C. Cometto, S. Mariyappan, M. Salanne and J.-M. Tarascon, A new electrolyte formulation for securing high temperature cycling and storage performances of Na-ion batteries, Adv. Energy Mater., 2019, 9, 1901431 CrossRef CAS.
- L. A. Ma, A. J. Naylor, L. Nyholm and R. Younesi, Strategies for mitigating dissolution of solid electrolyte interphases in sodium-ion batteries, Angew. Chem., Int. Ed., 2021, 60, 4855–4863 CrossRef CAS PubMed.
- Z. Li, N. Fu and Z. Yang, Particulate modification of lithium-ion battery anode materials and electrolytes, Particuology, 2023, 83, 129–141 CrossRef CAS.
- M. Dahbi, N. Yabuuchi, M. Fukunishi, K. Kubota, K. Chihara, K. Tokiwa, X. Yu, H. Ushiyama, K. Yamashita, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, Black phosphorus as a high-capacity, high-capability negative electrode for sodium-ion batteries: Investigation of the electrode/electrolyte interface, Chem. Mater., 2016, 28, 1625–1635 CrossRef CAS.
- J. Zhang, K. Zhang, J. Yang, V. Wing-hei Lau, G.-H. Lee, M. Park and Y.-M. Kang, Engineering solid electrolyte interphase on red phosphorus for long-term and high-capacity sodium storage, Chem. Mater., 2020, 32, 448–458 CrossRef CAS.
- X. Bian, Y. Dong, D. Zhao, X. Ma, M. Qiu, J. Xu, L. Jiao, F. Cheng and N. Zhang, Microsized antimony as a stable anode in fluoroethylene carbonate containing electrolytes for rechargeable lithium-/sodium-ion batteries, ACS Appl. Mater. Interfaces, 2020, 12, 3554–3562 CrossRef CAS PubMed.
- M. Xu, Y. Li, M. Ihsan-Ul-Haq, N. Mubarak, Z. Liu, J. Wu, Z. Luo and J. K. Kim, NaF-rich solid electrolyte interphase for dendrite-free sodium metal batteries, Energy Storage Mater., 2022, 44, 477–486 CrossRef.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, Better cycling performances of bulk Sb in Na-ion batteries compared to Li-ion systems: An unexpected electrochemical mechanism, J. Am. Chem. Soc., 2012, 134, 20805–20811 CrossRef CAS PubMed.
- X. Feng, J. Sun, M. Ouyang, F. Wang, X. He, L. Lu and H. Peng, Characterization of penetration induced thermal runaway propagation process within a large format lithium ion battery module, J. Power Sources, 2015, 275, 261–273 CrossRef CAS.
- S. Chen, K. Wen, J. Fan, Y. Bando and D. Golberg, Progress and future prospects of high-voltage and high-safety electrolytes in advanced lithium batteries: From liquid to solid electrolytes, J. Mater. Chem. A, 2018, 6, 11631–11663 RSC.
- Z. Shen, W. Zhang, G. Zhu, Y. Huang, Q. Feng and Y. Lu, Design principles of the anode–electrolyte interface for all solid-state lithium metal batteries, Small Methods, 2020, 4, 1900592 CrossRef CAS.
- L.-P. Wang, X.-D. Zhang, T.-S. Wang, Y.-X. Yin, J.-L. Shi, C.-R. Wang and Y.-G. Guo, Ameliorating the interfacial problems of cathode and solid-state electrolytes by interface modification of functional polymers, Adv. Energy Mater., 2018, 8, 1801528 CrossRef.
- D. L. Wood, J. Li and S. J. An, Formation challenges of lithium-ion battery manufacturing, Joule, 2019, 3, 2884–2888 CrossRef.
- D. H. S. Tan, Y.-T. Chen, H. Yang, W. Bao, B. Sreenarayanan, J.-M. Doux, W. Li, B. Lu, S.-Y. Ham, B. Sayahpour, J. Scharf, E. A. Wu, G. Deysher, H. E. Han, H. J. Hah, H. Jeong, J. B. Lee, Z. Chen and Y. S. Meng, Carbon-free high-loading silicon anodes enabled by sulfide solid electrolytes, Science, 2021, 373, 1494–1499 CrossRef CAS PubMed.
- Y.-M. Zhao, F.-S. Yue, S.-C. Li, Y. Zhang, Z.-R. Tian, Q. Xu, S. Xin and Y.-G. Guo, Advances of polymer binders for silicon-based anodes in high energy density lithium-ion batteries, InfoMat, 2021, 3, 460–501 CrossRef CAS.
- L. Han, T. Liu, O. Sheng, Y. Liu, Y. Wang, J. Nai, L. Zhang and X. Tao, Undervalued roles of binder in modulating solid electrolyte interphase formation of silicon-based anode materials, ACS Appl. Mater. Interfaces, 2021, 13, 45139–45148 CrossRef CAS PubMed.
- J. Song, M. Zhou, R. Yi, T. Xu, M. L. Gordin, D. Tang, Z. Yu, M. Regula and D. Wang, Interpenetrated gel polymer binder for high-performance silicon anodes in lithium-ion batteries, Adv. Funct. Mater., 2014, 24, 5904–5910 CrossRef CAS.
- S. Choi, T. Kwon, A. Coskun and J. W. Choi, Highly elastic binders integrating polyrotaxanes for silicon microparticle anodes in lithium ion batteries, Science, 2017, 357, 279–283 CrossRef CAS PubMed.
- Z.-J. Han, N. Yabuuchi, K. Shimomura, M. Murase, H. Yui and S. Komaba, High-capacity Si–graphite composite electrodes with a self-formed porous structure by a partially neutralized polyacrylate for Li-ion batteries, Energy Environ. Sci., 2012, 5, 9014–9020 RSC.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Self-healing chemistry enables the stable operation of silicon microparticle anodes for high-energy lithium-ion batteries, Nat. Chem., 2013, 5, 1042–1048 CrossRef CAS PubMed.
- T. S. Arthur, D. J. Bates, N. Cirigliano, D. C. Johnson, P. Malati, J. M. Mosby, E. Perre, M. T. Rawls, A. L. Prieto and B. Dunn, Three-dimensional electrodes and battery architectures, MRS Bull., 2011, 36, 523–531 CrossRef CAS.
- N. Nitta and G. Yushin, High-capacity anode materials for lithium-ion batteries: Choice of elements and structures for active particles, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS.
- R. Marom, S. Francis Amalraj, N. Leifer, D. Jacob and D. Aurbach, A review of advanced and practical lithium battery materials, J. Mater. Chem., 2011, 21, 9938–9954 RSC.
- B. Liang, Y. Liu and Y. Xu, Silicon-based materials as high capacity anodes for next generation lithium ion batteries, J. Power Sources, 2014, 267, 469–490 CrossRef CAS.
- R. A. Huggins, Lithium alloy negative electrodes, J. Power Sources, 1999, 81–82, 13–19 CrossRef CAS.
- W.-J. Zhang, Lithium insertion/extraction mechanism in alloy anodes for lithium-ion batteries, J. Power Sources, 2011, 196, 877–885 CrossRef CAS.
- M. Ko, S. Chae, S. Jeong, P. Oh and J. Cho, Elastic a-silicon nanoparticle backboned graphene hybrid as a self-compacting anode for high-rate lithium ion batteries, ACS Nano, 2014, 8, 8591–8599 CrossRef CAS PubMed.
- J. K. Lee, K. B. Smith, C. M. Hayner and H. H. Kung, Silicon nanoparticles – graphene paper composites for Li ion battery anodes, Chem. Commun., 2010, 46, 2025–2027 RSC.
- Z. Chen, M. Zhou, Y. Cao, X. Ai, H. Yang and J. Liu, In situ generation of few-layer graphene coatings on SnO2-SiC core-shell nanoparticles for high-performance lithium-ion storage, Adv. Energy Mater., 2012, 2, 95–102 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Mechanisms for lithium insertion in carbonaceous materials, Science, 1995, 270, 590–593 CrossRef CAS.
- R. Mukherjee, A. V. Thomas, A. Krishnamurthy and N. Koratkar, Photothermally reduced graphene as high-power anodes for lithium-ion batteries, ACS Nano, 2012, 6, 7867–7878 CrossRef CAS PubMed.
- Y. Li, K. Yan, H.-W. Lee, Z. Lu, N. Liu and Y. Cui, Growth of conformal graphene cages on micrometre-sized silicon particles as stable battery anodes, Nat. Energy, 2016, 1, 15029 CrossRef CAS.
- Q. Xu, J. Li, Y. Yin, Y. Kong, Y. Guo and L. Wan, Nano/Micro-structured Si/C anodes with high initial coulombic efficiency in Li-ion batteries, Chem. – Asian J., 2016, 11, 1205–1209 CrossRef CAS PubMed.
- B. M. Bang, J.-I. Lee, H. Kim, J. Cho and S. Park, High-performance macroporous bulk silicon anodes synthesized by template-free chemical etching, Adv. Energy Mater., 2012, 2, 878–883 CrossRef CAS.
- X. Li, M. Gu, S. Hu, R. Kennard, P. Yan, X. Chen, C. Wang, M. J. Sailor, J.-G. Zhang and J. Liu, Mesoporous silicon sponge as an anti-pulverization structure for high-performance lithium-ion battery anodes, Nat. Commun., 2014, 5, 4105 CrossRef CAS PubMed.
- Y. An, H. Fei, G. Zeng, L. Ci, S. Xiong, J. Feng and Y. Qian, Green, scalable, and controllable fabrication of nanoporous silicon from commercial alloy precursors for high-energy lithium-ion batteries, ACS Nano, 2018, 12, 4993–5002 CrossRef CAS PubMed.
- Y. Ren, X. Yin, R. Xiao, T. Mu, H. Huo, P. Zuo, Y. Ma, X. Cheng, Y. Gao, G. Yin, Y. Li and C. Du, Layered porous silicon encapsulated in carbon nanotube cage as ultra-stable anode for lithium-ion batteries, Chem. Eng. J., 2022, 431, 133982 CrossRef CAS.
- Y. Lv, M. Shang, X. Chen, P. S. Nezhad and J. Niu, Largely improved battery performance using a microsized silicon skeleton caged by polypyrrole as anode, ACS Nano, 2019, 13, 12032–12041 CrossRef CAS PubMed.
- Y. Zhao, J. Wang, Q. He, J. Shi, Z. Zhang, X. Men, D. Yan and H. Wang, Li-ions transport promoting and highly stable solid–electrolyte interface on Si in multilayer Si/C through thickness control, ACS Nano, 2019, 13, 5602–5610 CrossRef CAS PubMed.
- Y. Feng, Y. Lv, H. Fu, M. Parekh, A. M. Rao, H. Wang, X. Tai, X. Yi, Y. Lin, J. Zhou and B. Lu, Co-activation for enhanced K-ion storage in battery anodes, Natl. Sci. Rev., 2023, 10, nwad118 CrossRef PubMed.
- X. Liu, B. Xiao, A. Daali, X. Zhou, Z. Yu, X. Li, Y. Liu, L. Yin, Z. Yang, C. Zhao, L. Zhu, Y. Ren, L. Cheng, S. Ahmed, Z. Chen, X. Li, G.-L. Xu and K. Amine, Stress- and interface-compatible red phosphorus anode for high-energy and durable sodium-ion batteries, ACS Energy Lett., 2021, 6, 547–556 CrossRef CAS.
- X. Cheng, R. Shao, D. Li, H. Yang, Y. Wu, B. Wang, C. Sun, Y. Jiang, Q. Zhang and Y. Yu, A self-healing volume variation three-dimensional continuous bulk porous bismuth for ultrafast sodium storage, Adv. Funct. Mater., 2021, 31, 2011264 CrossRef CAS.
- T. Song, M. Yan and M. Qian, A dealloying approach to synthesizing micro-sized porous tin (Sn) from immiscible alloy systems for potential lithium-ion battery anode applications, J. Porous Mater., 2015, 22, 713–719 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3im00126a |
| This journal is © Institute of Process Engineering of CAS 2024 |