
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical recovery of Pt/C electrocatalyst: optimization of the potential range on the leaching process and application to an aged MEA†
François
Guillet
a,
Marian
Chatenet
 a,
Alex
Paul
b,
Lenka
Svecova
*a and
Laetitia
Dubau
*a
a,
Alex
Paul
b,
Lenka
Svecova
*a and
Laetitia
Dubau
*a
aUniv. Grenoble Alpes, Univ. Savoie Mont Blanc, CNRS, Grenoble INP, LEPMI, 38000 Grenoble, France. E-mail: laetitia.dubau@grenoble-inp.fr
bSYMBIO, 5 rue Simone Veil, 69200 Vénissieux, France
First published on 3rd October 2023
Abstract
Carbon-supported platinum nanoparticles (Pt/C) are widely used electrocatalysts in proton exchange membrane fuel cell and electrolyzer applications and represent a substantial part of the capital expenditure of these devices. Platinum being a critical raw material, its recovery is critical for the deployment of these technologies. In this contribution, the first step of a recycling protocol, i.e. the leaching of Pt/C, is studied. To avoid the use of concentrated acids and oxidants, the focus of the present study is on the design of an efficient electrochemical protocol. In particular, the values of the upper and lower potential limits have an impact on Pt dissolution efficiency. The upper potential limit should avoid (or at least limit) Pt particles' detachment from the carbon support and the lower potential limit should take into account the competition between the platinum dissolution and the unwanted platinum redeposition. The evolution of the particle morphology and dissolution rate were monitored by coupling a statistical analysis of TEM images and ICP-MS concentration measurements. The cycling potential window was first optimized for a model commercial Pt/C catalyst in a low-chloride concentration electrolyte, leading to a full Pt leaching efficiency (99%). A similar protocol was transferred to more technological objects: MEA aged under realistic conditions. The MEAs were electrochemically treated without any prior GDL separation and the efficiency of the process was demonstrated.
Keywords: MEA recycling; Platinum electrodissolution; Platinum recovery.
1 Introduction
Hydrogen (H2) is one of the most promising energy carriers.1,2 Although the vast majority of H2 is currently produced from fossil fuel by methane reforming, it would be more advantageous from an environmental point of view if it was produced by water electrolysis, using renewable resources of energy such as sun or wind.3,4 Currently, proton exchange membrane fuel cells (PEMFC) are the most used technology to convert the chemical energy of hydrogen into electrical energy, in particular for the mobility sector.5 The concomitant hydrogen oxidation at the anode side and oxygen reduction at the cathode side are considered as environmentally friendly, as the final products are only water, heat and electricity. The core of a PEMFC device is the membrane electrode assembly (MEA), composed of the anodic and cathodic catalytic layers, each of them being sandwiched between the polymeric proton exchange membrane (PEM) and a carbon-based gas diffusion layer (GDL). In state-of-the-art PEMFCs, the catalytic layers are composed of platinum (or platinum-based) nanoparticles supported on a high surface area carbon and proton exchange membrane ionomer. It is essential that the platinum (alloyed) electrocatalyst is organized in nanoparticles of well-controlled size and shape, to optimize the use of this rare and expensive metal,6 that is now on the list of critical raw materials of the European Union7 and represents almost half of the price of a PEMFC.8 Needless to add that recycling platinum is mandatory, both for the deployment of the hydrogen sector and the PEMFC global environmental impact.Platinum is a noble metal, resistant to corrosion and dissolution in acidic conditions, these properties explaining why it is the state-of-the-art base metal in PEMFCs; this however complicates its recycling. Different approaches have been investigated to recover platinum from aged PEMFC catalysts by hydrometallurgy.6 In all processes, the first step, which is related to its leaching, remains the same. After this step, platinum can be further recovered in the form of either metallic platinum or a platinum salt, depending on the targeted application. One solution consists of the incineration of the MEA, followed by extraction of Pt from the remaining ashes by leaching in aqua regia.9 Although this process is quite simple to implement, it cannot be considered ecological, owing to the membrane thermal destruction. The combustion of a perfluorosulfonated membrane indeed produces HF, which is highly toxic, and SO2, which is an acidic gas. The process also generates substantial amounts of CO2, which is unwanted. Furthermore, in such a recycling scenario, no valorization of the polymer of the membrane could be further considered. Greener strategies based on a closed loop recycling process have been proposed in the literature. They consist in redesigning new MEA catalysts from platinum salt originating from the MEA recovery process, without membrane combustion.10,11
Regarding leaching, two conditions are required for the dissolution of noble metals, namely the presence of an oxidant and a complexing ligand. A mix of hot HCl/HNO3 (aqua regia), which combines acidic and oxidizing properties, constitutes a well-known leaching agent for platinum, palladium and gold.12 Nitric acid oxidizes the Pt and chlorides (from HCl) react with the oxidized Pt species, resulting in an anionic PtCl62− complex. However, this solution is not very environmentally friendly and presents some safety issues too (high temperature, gas evolution, etc.). The presence of nitrates in the effluents should also be avoided, which might necessitate another post-leaching treatment. Several different approaches have been attempted in the literature to find an alternative to aqua regia:13 organic solvents with SOCl2 as complexing agent,14 highly concentrated solutions of AlCl3·6H2O and Al(NO3)3·9H2O,15 or even the replacement of HNO3 by another and stronger oxidant such as H2O2. The mix of HCl/H2O2 was used as a leaching agent for a complete recycling process of the MEA.16 Hodnik et al. present another interesting approach to dissolve Pt by alternating oxidative and reducing atmospheres: gaseous O3 and CO were used, chloride ions being added as a complexing agent.17 O3 allowed formation of platinum oxides (PtOx) and CO to reduce them. This study highlights that the amount of leached platinum is more important by alternating oxidation and reduction steps rather than the simple oxidation of platinum by O3. In the same vein, electrodissolution and electroleaching are other approaches to address the first step of the recycling protocol. Their advantage is that neither oxidant nor reducing agent is needed, these being replaced by application of a proper potential protocol to induce the metals' oxidation and facilitate their further dissolution. The electrochemical dissolution of platinum has been largely studied from the standpoint of PEMFC catalyst durability.18–20 All this knowledge represents a very useful base to explore the electrochemical leaching of Pt or Pt-based catalysts.
Platinum electrodissolution in acidic medium results from a complex mechanism, depending on the applied potential as well as the initial oxidation state of the Pt surface. Xing et al. report different reactions for dissolution of platinum, depending on the oxidation state of platinum (II or IV).21 The electrodissolution under oxidizing potentials (1) occurs simultaneously with formation of PtOx(1′), while electrodissolution under reducing potentials occurs simultaneously with the reduction of PtOx. The proposed mechanism can be thus simplified and described as follows:
| Pt → Pt2+ + 2e− | (1) |
| Pt + xH2O → PtOx + 2xH+ + 2xe− | (1′) |
| PtOx + 2xH+ + 2xe− → Pt + xH2O | (2) |
| PtOx + 2xH+ + 2xe− → Pt2x+ + xH2O | (2′) |
Significant advances in the understanding of Pt electrochemical dissolution have been made after coupling of electrochemical measurements with a direct inductively coupled plasma-mass spectroscopy (ICP-MS) analysis. First, in situ experiments were conducted on a polycrystalline platinum in a 0.1 M HClO4 electrolyte and shows that above 1.1 V vs. reversible hydrogen electrode (RHE), the Pt dissolution and its surface passivation are in competition.22 In contrast, below 1 V vs. RHE during the cathodic polarization, a competition occurs between the dissolution of PtOx and Pt redeposition. Consequently, when platinum dissolution is carried out by an alternation of oxidation and reduction potentials, the value of the lower potential limit appears to impact more the PtOx dissolution process than the upper potential limit. Similar conclusions were drawn with other electrolytes (H2SO4 and NaOH), both for platinum and gold.23 In addition, the amount of dissolved platinum increases significantly when chloride ions are added as complexing agent, even at a very low concentration of 1 μM. In this case, the amount of platinum leached remains higher during the cathodic polarization than during the anodic one (without the addition of chloride), but these two values are closer in the presence of chloride. The cathodic dissolution is 3 times higher than the anodic one in the presence of chloride, and 10 times higher without chloride.24,25
Compared to chemical leaching, the recycling of platinum based on electrodissolution is more environmentally friendly, as it requires lower concentration acidic solution, lower chloride concentration, no additional oxidizing agents and might be carried out at ambient temperature, which represents substantial benefits in terms of environmental impacts. However, another challenge is the control of potential with a reference electrode: industrial electrochemical processes are generally monitored by the value of the current or by the (2-electrode cell) voltage. Moreover, as mentioned by Sharma et al., aged platinum nanoparticles might also have lost their electric connection within the catalytic layer due to the harsh operating conditions, preventing their further electrodissolution.26 This is the case for the platinum particles present in the membrane after ageing (the so-called platinum-band, originating from the corrosion of cathodic Pt nanoparticles leading to Pt ionic species which are chemically reduced in the membrane by H2 cross-over27); these can be reached by the leaching agents only if the membrane is dissolved.
Rare studies dealing with electrochemical leaching of PEMFC platinum electrocatalysts for recycling purposes have been identified in the literature.28–31 Whereas all these processes are carried out at room temperature and use HCl as electrolyte, the comparison between these different studies remains complicated, because impacting parameters such as the nature of the support of platinum nanoparticles, the mass-transport properties, the electrochemical operating conditions and especially the chloride concentration are different. Latsuzbaia and Sharma confirm that chloride concentration is the main driver for the platinum dissolution rate.29,30 The chloride concentrations as well as the efficiency of platinum recovery obtained in the cited articles are reported in Table 1. In all these studies the ratios of leached platinum are high, between 70% and 100% (Table 1), but the comparison is difficult because of the different initial support of platinum (commercial catalyst or real MEA, Table 1) and the different operating conditions. Interestingly, these studies have in common the same type of electrochemical protocol: linear cycling between a higher and a lower potential. These methods are inspired by accelerated stress tests (AST), which are electrochemical protocols specifically designed to study the stability of PEMFC catalysts. Basically, a triangle or square (steps) protocol is applied by cycling between an upper and a lower potential at a defined scan rate.32 The applied values of the upper and the lower potential limit are reported in Table 1. Each of these three parameters (upper potential, lower potential and scan rate) has an impact on the morphological changes and the dissolution of the catalyst. In general, the dissolution of platinum increases with the number of cycles and the increase of temperature. Under ideal conditions, upper and lower potential limits are set up to be the highest and the lowest to achieve an improved dissolution of Pt. However, in real conditions, there are some limitations. In particular, the corrosion of the carbon support (usually carbon black) occurs thermodynamically at 0.207 V vs. RHE, but this reaction is kinetically slow.19 In practice, carbon oxidation starts above 0.8 V vs. RHE (and is catalyzed by platinum) but it becomes significant above 1.2 vs. RHE and can lead to a detachment of platinum; this effect must be avoided, as it may decrease the recycling efficiency, since the electrolyte is not “strong” enough to further chemically dissolve the detached platinum particles.33,34 This issue is overlooked in the literature and it is important to have in mind that the upper potential limit must be chosen to avoid or at least to minimize the particle detachment phenomenon.31 In addition, there are only few studies dealing with the impact of the reduction potential, which is often fixed around 0.4 V vs. RHE.30,31 This lower potential enables complete reduction of the PtOx formed during steps at high potential and to regenerate a metallic platinum surface for the next step of oxidation (PtOx passivates the surface, hence slows down dissolution). However, such a low potential limit could be non-optimal, considering the competition between the PtOx reduction and platinum redeposition mentioned previously, it being admitted that platinum electrodeposition reduces the leaching efficiency. For example, Sharma et al. report that with a reduction potential of 0.4 V vs. RHE, the ratio of leached platinum increases from 16% with stagnant volume to 80% with an agitated electrolyte, a result they attribute to a higher extent of platinum redeposition in the stagnant electrolyte conditions.30
| Electrolyte | Lower potential (V vs. RHE) | Higher potential (V vs. RHE) | Nature of Pt catalyst | Platinum dissolution (%) | Ref. | |
|---|---|---|---|---|---|---|
| Shiroishi, 2012 | 1 M HCl | 0.3 | 1.4 | Fresh MEA | 83 | 28 |
| Latsuzbaia, 2015 | 0.1 M HCl | 0.5 | 1.1 | Commercial catalyst Hispec 9100 | 100 | 29 |
| Sharma, 2019 | 1 M HCl | 0.4 | 1.6 | Fresh MEA | 95 | 30 |
| Montiel, 2023 | 0.1 M HCl | 0.4 | 1.6 | Aged MEA | 70 | 31 |
This selected literature review shows not only the interest in electrochemical procedures to leach Pt but also the uncertainty concerning the “ideal” parameters to render this methodology viable in practice. Hence, the present study aims at optimizing the electrochemical dissolution protocol of platinum electrocatalysts. The novelty of the present approach is to propose a rational potential window allowing an efficient dissolution of Pt. The choice of the upper potential limit is driven by the limitation of Pt particle detachment induced by carbon support corrosion and the lower potential limit by the limitation of Pt redeposition. Two different lower potential limits will be compared: a value of 0.40 V vs. RHE (which is quite classical in the literature) and a value of 0.85 V vs. RHE to limit platinum redeposition, such higher potential being less favorable to fast/complete reduction of PtOx. The upper potential limit was fixed at 1.2 V vs. RHE. The quantitative evolution of Pt dissolution will be monitored by transmission electron microscopy (TEM) imaging of the catalyst at different stages of the protocol, associated to the determination of Pt concentration in the electrolyte by ICP-MS. The process will be first optimized on well-defined commercial catalysts (Pt/C 20% provided by Tanaka Kikinzoku Kogyo K.K.), with an average size of the Pt particles close to 2.5 nm, and transferred to a technological object, typically an aged MEA, as a proof of concept. It has to be noted that for all the studies for platinum electrodissolution, the electrochemistry on platinum is carried out after the separation of the electrodes from the MEA or with catalyst deposited on a carbon sheet.28–31 In the present work the electrochemistry for catalyst dissolution is performed directly on the MEA.
2 Results and discussion
2.1 Electrochemical dissolution of Pt – optimizing the lower potential limit
Preliminary experiments were carried out using HUPD loss as a descriptor of Pt dissolution with the model Pt/C catalyst to confirm the role of chloride in platinum electrodissolution (see section S2 and S3 in the ESI†). However, it has to be noted that HUPD loss is not directly linked to the Pt dissolution and could induce some experimental bias in the interpretation of the results. HUPD reflects the electrochemical surface area and depends on several parameters such as the particle size and morphology, the poisoning of the Pt surface (e.g. by chloride ions),35 and the occurrence of particle detachment.31 Moreover, there is no way to estimate the extent of platinum redeposition. For all these reasons, the study is focused on the follow-up of the evolution of particle size and morphology by TEM imaging coupled to the monitoring of dissolved Pt concentration by ICP-MS. The population of particles is classified into two groups, isolated and aggregated particles, as explained in section 4.6.1. In the absence of chloride ions, the morphological changes of platinum nanoparticles upon cycling are driven by two main mechanisms: Ostwald ripening, due to dissolution of the smallest particles at the expense of larger ones, and crystallite migration, leading to aggregated and possibly coalesced particles.36 These mechanisms can explain the aggregation of particles for cycling from 1.2 to 0.4 V vs. RHE without chloride ions (see Fig. S4†). Interestingly, it has been shown in the literature that the amount of leached platinum is at least 100 times higher in the presence of chloride ions (NaCl 0.1 M) than in their absence.24 In this chloride concentration, the only plausible mechanism explaining the change in particle morphology is related to significant Pt dissolution combined to its redeposition.24,37 In this work it is assumed that the formation of new aggregated particles is related to mechanisms of platinum deposition. On the opposite, a constant, or even decreasing, number of aggregated particles is linked to a lack of deposition.Different parameters are defined to quantify the number, rn,agg, and the part of the surface of aggregated particles on the total area of platinum, rA,agg, as detailed in section 4.6.1.
2.2 Reduction potential at 0.4 V vs. RHE
Fig. 1 shows some representative TEM images and the morphological evolution of the Pt/C catalyst submitted to an alternation of oxidation (1.2 V vs. RHE) and a reduction potential (0.4 V vs. RHE). This potential range has been chosen according to the literature, where a lower potential limit of 0.4 V vs. RHE is traditionally chosen as it favors the reduction of the PtOx layer generated during steps at high potential. Both agglomerated (squares) and isolated (ellipses) particles can be distinguished in the TEM snapshots, both for the pristine and for the aged samples. Visually, it might appear that the Pt dissolution is not a very pronounced phenomenon as the particle size of Pt particles does not change very much after 1000 cycles, compared to the pristine material. It seems also that the density of isolated particles (number of isolated particles per area of carbon support) decreases simultaneously with an increase of aggregated ones (the phenomenon not being very pronounced). Since TEM is a very local analysis, it is difficult to go further without a statistical analysis of several TEM images. To distinguish between the decrease of Pt particle size and their aggregation, all obtained parameters related to both isolated and agglomerated particles have thus been extracted (see the experimental section 4.6.1). Fig. 2 presents the distribution of the diameters for the isolated particles (Fig. 2a) and the distribution of area for aggregated particles (Fig. 2b) as well as the characteristic ratios for both populations, the average area Aiso, rn,iso and rA,iso for isolated particles (Fig. 2c), and the average area Aagg, rn,agg and rA,agg for aggregated particles (Fig. 2d). For both populations, i.e. the aggregated and isolated particles, a continuous decrease of size and area occurs upon cycling, proving the occurrence of platinum dissolution. | ||
| Fig. 1 Representative TEM images of platinum nanoparticles from a Pt/C 20% TKK at 20 μg cm−2 loading at different stages of cycling between 1.2 and 0.4 V vs. RHE in 1 M H2SO4 + 0.01 M NaCl. The ellipses highlight some examples of isolated particles and the rectangles show some examples of aggregated particles. | ||
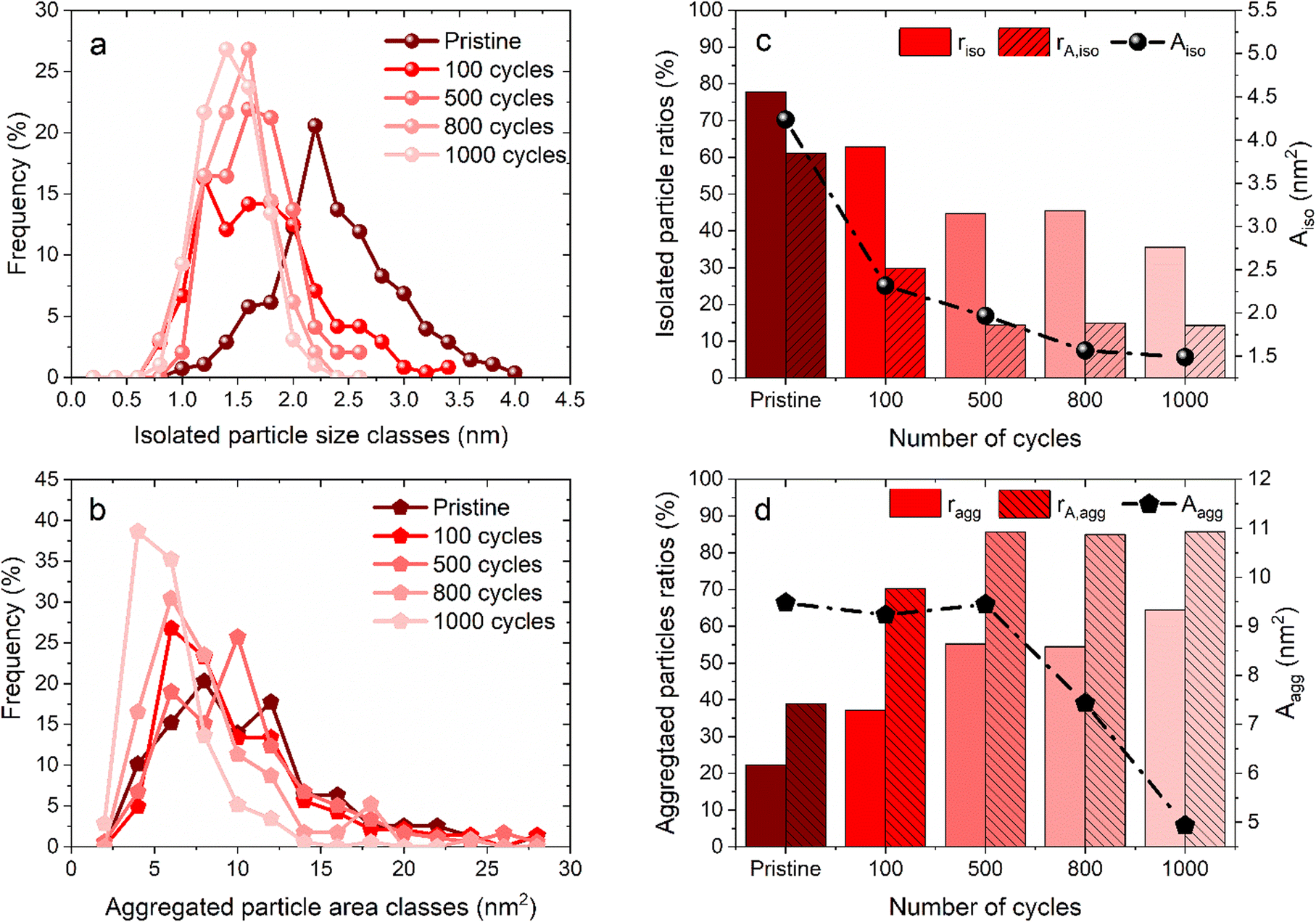 | ||
| Fig. 2 Distribution of particle (a) diameter for isolated particles and (b) area for aggregated particles; parameters and average area for (c) isolated particles rn,iso and rA,iso and (d) aggregated particles rn,agg and rA,agg are measured from pristine to 1000 cycles. These parameters are evaluated at different stages of the cycling protocol between 0.4 and 1.2 V vs. RHE in 1 M H2SO4 + 0.01 M NaCl. | ||
The majority of pristine isolated particles are characterized by a diameter of 2.25 nm, while the aggregated ones exhibit an area of 7 to 12 nm2. After 1000 cycles between 1.2 and 0.4 V vs. RHE, the average size of isolated particles drops to 1.25, while the surface of agglomerated particles decreases to 5 nm2. However, the evolution seems to be faster for isolated than for aggregated particles. The decrease of isolated particle diameter is pronounced at the beginning of the protocol, from initial state to 100 cycles, and tends to slow down after (Fig. 2a). This decrease is confirmed by the evolution of projected area Aiso, from 4.24 nm2 at the beginning to 2.32 nm2 after 100 cycles. Even if this parameter continues to decrease, the change becomes less significant. On the other hand, the distribution of area for aggregated particles stagnates for the first cycles of the protocol. Moreover, the particles are slightly larger in size after 500 cycles than after 100 cycles: Aagg moves from 9.23 to 9.45 nm2. An opposite trend is reported beyond 500 cycles: the area of aggregated particles starts to decrease, illustrating that aggregated particles are decreasing in size following their dissolution. However, the ratio of aggregated particles to the total number of particles, rn,agg, increases all along the protocol. This might be explained by a simultaneous dissolution of Pt (both isolated and aggregated particles) and a redeposition of dissolved platinum. The proportion of area of aggregated particles to the total area considering the area of both isolated and aggregated particles increases from 40% (pristine particles) to 85% (after 500 cycles) and remains stable up to 1000 cycles. Meanwhile, the population of aggregated particles continues to increase, rn,agg passing from 22% (pristine) to 65% for 1000 cycles (Fig. 2d). In conclusion, the process is sequential. From pristine to 500 cycles, the platinum from isolated particles is leached preferentially and the occurrence of platinum redeposition induces a higher extent of particle aggregation. Beyond 500 cycles, the mechanism is different, since the aggregated particles are dissolving too at the same rate as the isolated particles (signed by a constant rA,agg at 85%, meaning the majority of the platinum are aggregated particles). These observations can be related to Oswald ripening mechanism leading to the preferential dissolution of the smallest particles at the expense of the larger ones. The platinum ionic species are then preferentially reduced on bigger particles, according to the Gibbs–Thomson effect.36 The same effect is observed in AST, i.e. the disappearance of small particles and the bigger particles' growth after ageing of the MEA platinum catalysts.18,38 The main difference between the literature on PEMFC durability and in the present work is related to the presence of chloride, inducing a significant platinum dissolution compared to AST. The area of aggregated particles grows due to the recovery of platinum from isolated particles by dissolution and redeposition mechanism but at the same time their electrodissolution also occurs, thus partially inhibiting their growth. We posit that until 500 cycles, the fast dissolution of isolated particles provides a large input of platinum ionic species to the electrolyte, a part of which is then being redeposited on aggregated particles, which temporarily protects them from dissolution. After 500 cycles, the amount of platinum provided from the leached isolated particles is lower, because there are not enough isolated particles to be dissolved. Thus, the redeposition of platinum becomes less significant and aggregated particles are no longer protected and they begin to shrink; that is why rA,agg remains stable. This hypothesis is consistent with the evolution of the remaining platinum quantity at the electrode for 1.2–0.4 V vs. RHE cycling plotted in Fig. 4d (red curve), showing that the majority of the platinum is leached between the pristine state and 500 cycles.
2.3 Reduction potential at 0.85 V vs. RHE and comparison with 0.4 V vs. RHE
The previous section demonstrates that (unwanted) Pt redeposition is substantial when the lower potential limit is 0.4 V vs. RHE, which decreases the efficiency of the platinum leaching. The lower potential limit has consequently been increased from 0.4 to 0.85 V vs. RHE. A higher reduction potential leads to a slower electrodissolution rate.24 However, the standard potential of Pt/PtCl62− is 0.74 V vs. RHE.39 Platinum electrodeposition from chloride complex is usually performed at a potential lower than 0.5 V vs. RHE.39,40 Hence a reduction potential of 0.85 V is expected to nearly inhibit the platinum redeposition. Fig. 3 displays representative TEM images of the Pt/C catalyst at different stages of this new protocol. | ||
| Fig. 3 Representative TEM images of platinum particles from a Pt/C 20% TKK at 20 μg cm−2 loading at different stages of cycling between 1.2 and 0.85 V vs. RHE in 1 M H2SO4 + 0.01 M NaCl. | ||
Unlike for the experiments with a lower potential at 0.4 V vs. RHE, the TEM images for the 0.85 V vs. RHE reduction potential experiments reveal a continuous decrease of both the size and the density of the particles, with no sign of any Pt redeposition. Moreover, after 500 cycles no aggregated particles can be identified. Fig. 4 displays the distribution of diameters for isolated particles (Fig. 4a) and of area for aggregated ones (Fig. 4b). For comparison, the ratio of the number of aggregated particles to the total number of particles rn,agg (Fig. 4c) and the evolution of platinum content remaining on the electrode are shown for the two different reduction potentials (0.4 and 0.85 V vs. RHE, Fig. 4d). First of all, unlike what happened at 0.4 V vs. RHE, the number of aggregated particles decreases rapidly up to their complete disappearance after 300 cycles. Moreover, there is no formation of aggregated particles, confirming the absence of platinum redeposition. The reduction of their area is also fast, as testified by the value of 5% for rn,agg. However, it should be highlighted that the determination of the distribution of aggregated particle area was carried out with only 13 particles after 300 cycles (Table S2†), since almost none remained visible at that stage, a clear sign of their disappearance. The decrease in size of isolated particles is faster for the first cycles (Fig. 4a) and particularly from 0 to 20 cycles. Finally, the comparison of the remaining platinum on the electrode (as defined in section 4.6.2) for cycling between 1.2 and 0.85 V vs. RHE reveals a faster platinum dissolution than for cycling between 1.2 and 0.4 V vs. RHE.
 | ||
| Fig. 4 Distribution of particle size and area for (a) isolated particles and (b) aggregated particles. These parameters are evaluated at different stages of the cycling protocol between 0.85 and 1.2 V vs. RHE in 1 M H2SO4 + 0.01 M NaCl; comparison between the two reduction potentials 0.4 and 0.85 V vs. RHE for (c) the ratio of the number of aggregated particles to the total number of particles and (d) the evolution of platinum content remaining on the electrode. | ||
The amount of platinum remaining on the working electrode decreases more rapidly up to 500 cycles when 0.85 V vs. RHE is the reduction potential, 43% for 100 cycles and 14% for 500 cycles (against 83% and 22% for 100 and 500 cycles, respectively, at 0.4 V vs. RHE). This result originates from the substantial minimization of platinum redeposition, even though the electrodissolution rate is more important for the 0.4–1.2 V vs. RHE protocol. However, beyond 500 cycles and up to 700–800 cycles, the values of the platinum content remaining on the electrode become closer for the two protocols. Interestingly, this occurs simultaneously with the decrease in area reported for aggregated particles for the 1.2–0.4 V vs. RHE protocol. This point supports the hypothesis that aggregated particles are protected from dissolution by redeposition of platinum when the reduction potential is 0.4 V vs. RHE. Beyond the fact that increasing the lower potential limit to 0.85 V vs. RHE is beneficial for the efficiency of platinum dissolution at the earlier stages of the process, it has to be noted that the dissolution is almost complete as less than 1% Pt remains on the electrode surface after 3500 cycles (against 3.5% for the 0.4–1.2 V vs. RHE protocol). This agrees with Sharma et al.'s findings for cycling between 0.4 and 1.6 V vs. RHE: electrodeposition of platinum increases with platinum concentration if the electrolyte is immobile.30 In the present case, with no agitation and a concentration of 1 mM H2PtCl6, there is still net platinum dissolution with reduction potential at 0.85 V vs. RHE but not at 0.4 V vs. RHE (see section S6†).
At that stage, it is important to obtain some insights into the nature of the platinum lost (detached Pt metallic particles or Pt ions) having in mind that the Pt dissolution in the form of platinum ionic species is highly wanted for the efficiency of the recycling process. Further dissolution of Pt particles detached from the carbon support would indeed require additional steps prior to any recovery process (such as solvent extraction, for example). Based on the obtained results, the particle detachment from the carbon support cannot be completely excluded, even for the 0.85–1.2 V vs. RHE protocol and is the focus of the next part.
2.4 Quantification of particle detachment for cycles 1.2–0.85 V
The evolution of the amount of platinum remaining on the electrode (Fig. 4d) proves that any lower potential limit (0.4 or 0.85 V vs. RHE) yields a large loss of platinum with respect to the initial platinum loaded on the electrode. However, this loss might also be attributed, at least partially, to the detachment of Pt particles induced by the corrosion of the carbon support, and not only to dissolution. ICP-MS measurements are sensitive to both platinum ions and Pt nanoparticles, provided that they are present in the liquid phase further nebulized. Since the electrolyte is agitated, the platinum particles provided from the electrode (section 2.6.2) could be collected during the sampling. To test whether this is the case (and biases the results), two different models of estimation of Pt dissolution efficiency based on the TEM image analysis are proposed in order to compare with the one estimated by ICP-MS measurements. The evolution of concentration of platinum in the electrolyte is measured in cycles by ICP-MS and is normalized with respect to the initial Pt loading on the electrode; this gives the remaining amount of platinum Ptrem defined in eqn (11). For the two proposed models, two different ways to determine the remaining platinum are defined. The evolution of the Pt dissolution efficiency as a function of number of oxidation/reduction cycles determined by the three alternatives (the ICP-MS measurement and the two models) is compared in Fig. 5. This approach is applied to the experimental results obtained for the 1.2–0.85 V vs. RHE protocol, which is much more efficient for platinum dissolution than the 1.2–0.4 V vs. RHE one.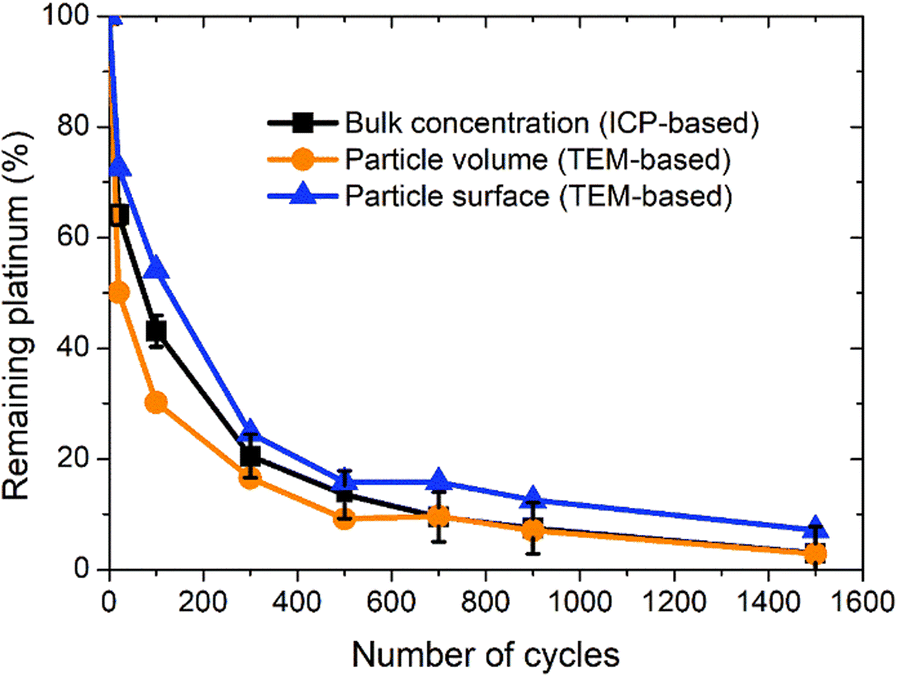 | ||
| Fig. 5 Comparison of evolution of remaining platinum rate on the electrode as a function of the number of cycles estimated with three different methods: (i) from ICP measurements (black), (ii) from averaged particle volume (orange) and (iii) from averaged surface of particles (deep blue). | ||
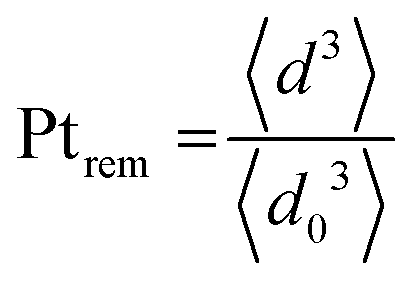 | (3) |
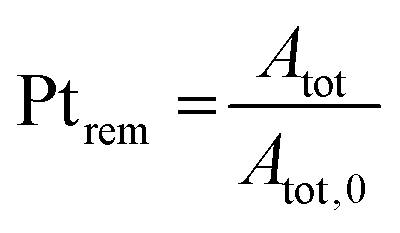 | (4) |
The three curves follow the same trend: the remaining Pt% significantly decreases after the first cycles (up to 300 cycles approx.); then, the Pt dissolution slows down (without stopping). The experimental points obtained by ICP-MS analysis are located between the curves derived using the two TEM-based models. From 0 to 300 cycles the particle volume model seems to overestimate the dissolved quantity compared to the ICP-MS determination (that is believed to represent the real Pt concentration). This phenomenon might come from the fact that the aggregated particles are not taken into account in this model. At the earliest stages of the protocol, the fraction of aggregated particles is more pronounced, which could explain the non-negligible discrepancy between the two curves at the beginning of the test. In contrast, after 300 cycles there are almost no aggregated particles (Fig. 4c) and the value derived from the particle volume model is very close to the ICP-MS one. After 700 cycles, the values obtained by the particles' volume-based model and the ICP-MS measurements can be considered similar. This model seems to be more adapted to the second part of the protocol, after 500 cycles when the number of aggregated particles start to be negligible. In contrast, the particle surface model gives a higher value of the remaining platinum, underestimating the Pt dissolution compared to the ICP-MS based Pt concentration. This trend lasts all along the test up to 700 cycles. Although the differences between the three models are noticeable from 0 to 300 cycles, the values remain consistent after 300 cycles, especially for the particle volume-based model. These two models are based on dissolution of platinum only and do not consider any particle detachment. Their excellent correlation with the evolution of Pt concentration in the electrolyte enables us to conclude that there is no significant nanoparticle detachment occurring during this 0.85–1.2 V vs. RHE protocol.
Moreover, since the gap between the real Pt concentration and the one obtained indirectly via the TEM-based surface particle model is only 5%, the platinum losses are mostly driven by a dissolution mechanism. This result confirms that the experimental conditions are properly chosen to guarantee a very efficient Pt dissolution process in view of its recycling. Other future studies could be led with another value of reduction potential between 0.4 and 0.85 but also different durations of different steps of oxidation and reduction could potentially as well lead to optimized dissolution rates.
2.5 Transferring the protocol on a real aged MEA
The optimized 1.2–0.85 V vs. RHE cycling protocol was applied to technological objects, such as MEA samples aged under real conditions with a long-term test (LTT, see section 4.4), in order to validate the industrial feasibility of Pt electrodissolution. This leaching process occurs directly in the MEA, without separation of the GDL or redeposition of particles in another electrode and thus avoids preliminary steps for MEA recycling. The fresh state of the catalytic layers was provided, for comparison of the size and morphology of Pt/C nanoparticles between fresh and aged MEA. However, the composition or loading of this fresh MEA cannot be communicated here for confidentiality reasons (the anode catalyst was Pt/C whereas the cathode catalyst was a bimetallic PtCo/C catalyst). Fig. 6 compares the Pt nanoparticles before and after the LTT. Post LTT, the Pt nanoparticles exhibit features in line with the classical degradation mechanisms reported in the literature for ageing of PEMFC catalysts: compared to the reference state-of-the-art catalyst, the particles are rounder, larger (size ranging from 5 to 10 nm) and more uniform in size,18,36 a sign of Ostwald ripening.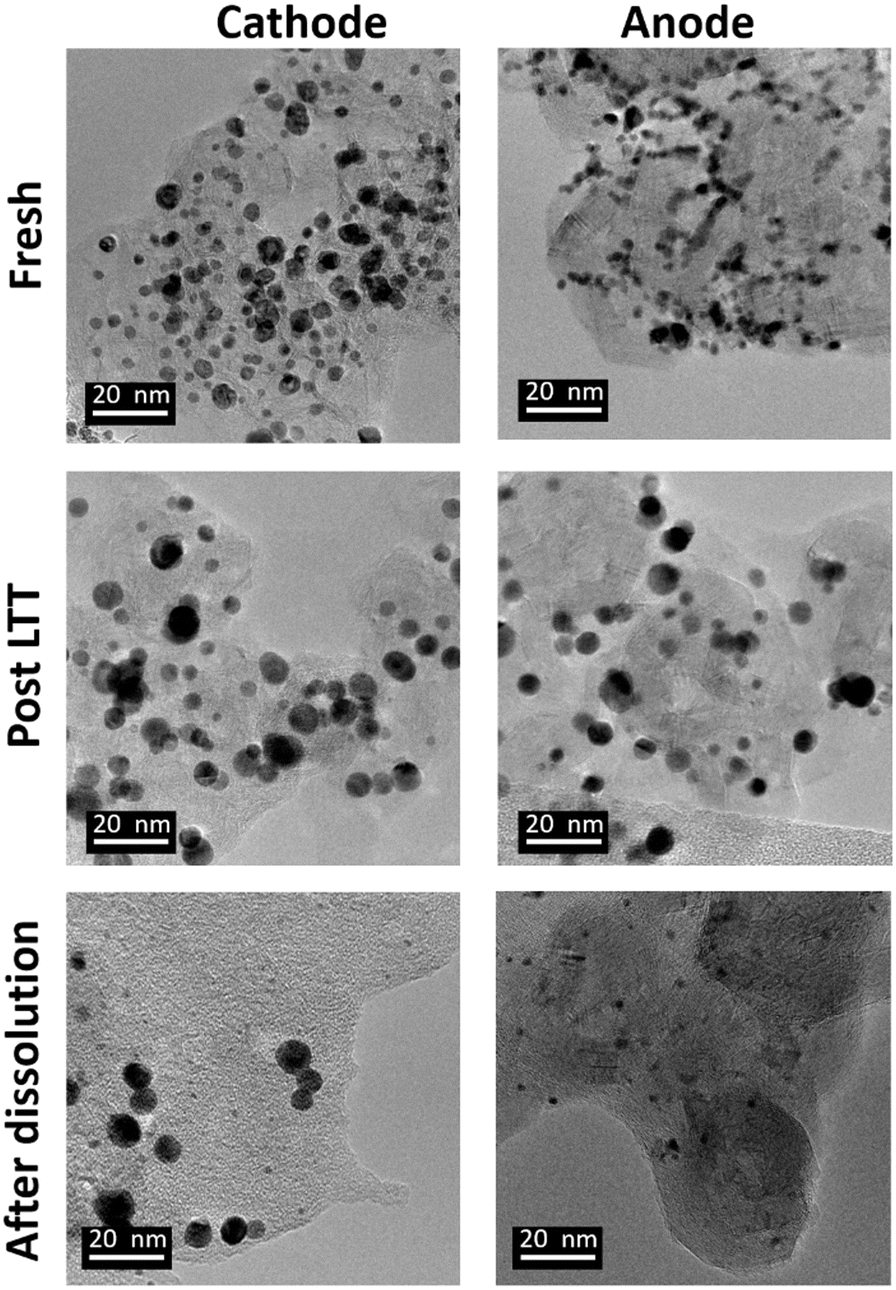 | ||
| Fig. 6 Representative TEM images of platinum particles for fresh MEA, post LTT MEA and for MEA after Pt electrochemical dissolution protocol consisting in 4000 cycles between 1.2 and 0.85 V vs. RHE in 1 M H2SO4 + 0.01 M NaCl. | ||
Since after 3500 cycles between 1.2 and 0.85 V vs. RHE, less than 1% platinum was remaining on the model Pt/C electrode, it was decided to submit the post LTT MEA to 4000 cycles under similar conditions (as it was posited that larger particle size would be harder to dissolve). The TEM images after the dissolution protocol reveal that Pt particles can be separated into two groups for both electrodes. First, very small particles are highly represented, which results from substantial dissolution of aged platinum particles. If the number of cycles was further increased, one could expect that these small particles would be completely leached. However, a second population of particles can be observed; these seem to keep their initial post LTT size and shape, as if they could not electrochemically react. The most likely explanation for this phenomenon is that the electrical connection was lost during the LTT and no electrochemical reaction was further possible for this type of particles. Another explanation could be that the liquid electrolyte did not reach the particles, maybe because they are covered by carbon and/or ionomer. In any case, these (electronically disconnected or “inaccessible”) particles will not be leached even with a larger number of cycles, meaning that reaching 100% Pt leaching is unlikely with this process for spent MEA. Despite these unreactive particles and in view of a leaching process directly in the MEA, after 4000 cycles, an acceptable dissolution yield, yPt, of 85% is obtained. It is slightly lower than the 99% achieved with the model Pt/C catalyst but it can be increased by a higher number of cycles with the end of dissolution of small particles.
3 Conclusions
In the present work, the influence of the reduction potential on the efficiency of platinum electrodissolution with a low-chloride concentration is investigated in view of the recycling of Pt/C electrocatalysts or spent PEMFC MEAs. While the literature states that a reduction potential of 0.4 V vs. RHE leads to higher dissolution rate than a reduction potential of 0.85 V vs. RHE, the impact of such a low lower potential limit on the unwanted Pt particles' detachment mechanism has not been deeply investigated; this is the focus of the present work. The results obtained on a model commercial Pt/C catalyst show that when the reduction potential is set at 0.4 V vs. RHE, Pt redeposition is significant, overall leading to a net Pt dissolution rate that is smaller than when the reduction potential is 0.85 V vs. RHE: adopting a “high” reduction potential of 0.85 V vs. RHE prevents (or at least slows down) Pt redeposition, which is preferable for an efficient Pt leaching process; this effect is especially favorable when the leached platinum concentration increases in the electrolyte, which shall be the case in a scaled industrial process. The amount of platinum remaining on the electrode, monitored by ICP-MS after digestion in aqua regia, is in agreement with the values obtained with two TEM-based dissolution models; this proves that there is no, or very limited, particle detachment for this electrochemical leaching protocol (detached particles – not in electric contact with the catalyst powder – would not be dissolved by the present electrochemical leaching procedure).As a proof of concept, a similar electrochemical protocol was tested directly on a spent MEA that had been previously aged under realistic conditions by our industrial partner. In that case, it cannot be excluded that some Pt particles had lost their electrical contact with the carbon support during the MEA aging procedure, thus rendering complex the electrochemical leaching of the corresponding fraction of platinum. Whatever this bias, a platinum dissolution efficiency of 85% was measured, which is very promising for such a simple and rather green electrochemical leaching process. This efficiency could be further improved by a longer duration of the protocol.
4 Material and methods
The experiments of electrochemical dissolution were conducted with two different types of carbon-supported Pt (Pt/C) electrocatalysts. The first one is a commercial platinum catalyst purchased from Tanaka Kikinzoku Kogyo K.K. (TEC10V20E 20.0% Pt, called TKK), which is used as a model catalyst (section 4.3). The second one is from the catalytic layers of a post LTT MEA (section 4.4) provided by an industrial partner.4.1 Electrochemical protocol
The electrochemical dissolution consists of cycling between a high and a low potential limit, using an Autolab PGSTAT12 potentiostat. A cycle is composed of two steps at high and low potential, during which the potential is maintained for 5 s. The upper potential limit (which is the first step of each experiment) is fixed at 1.20 V vs. RHE. The effect of the lower potential limit is the key variable in this work. Two values are studied: 0.40 and 0.85 V vs. RHE. All potentials are measured via a homemade reversible hydrogen electrode (RHE) and reported versus RHE.4.2 Electrochemical cell
The electrochemical experiments are performed in a three-electrode H-cell, described in the ESI† (Fig. S1), equipped with a P4 glass frit (pores sizes between 10 and 16 μm) as a separator between the working and the counter electrodes. The separator is used to avoid the presence of dissolved platinum ions in the counter cell, which could be further electrodeposited at the counter electrode. Prior to all experiments, the cell is immersed in a mixture of H2SO4/H2O2 for one night and carefully rinsed with boiling water. The volume of the working cell is approx. 50 mL and the volume of the counter cell around 20 mL. For the experiments, almost 30 mL of electrolyte are added in the working cell and 20 mL in the counter cell. Prior to electrochemical measurements, oxygen dissolved in the electrolyte of the two compartments is purged with a flow of argon for at least 20 min and an argon atmosphere is maintained during all experiments.4.3 Model Pt/C catalyst
The working electrode is an ink containing platinum/carbon particles and Nafion® ionomer deposited on a glassy-carbon tip of 5 mm diameter. The composition of the ink is 5 mg of TKK Pt/C 20%, 2 μL of Nafion®, 523 μL of isopropanol and 1.8 mL of pure water. The targeted loading of platinum is 20 μg cm−2 so 10 μL of ink are deposited on the glassy-carbon tips. The counter electrode is a glassy-carbon plate of 5 cm2 area. Fig. S2† presents a typical voltamperogram of this TKK Pt/C 20% catalyst between 0 and 1.5 V vs. RHE. The platinum oxidation begins close to 1.1 V vs. RHE and the peak of the reduction of platinum oxide starts at 0.9 V vs. RHE, with a minimum at 0.75 V vs. RHE, which is consistent with studies carried out with the same catalyst.23,244.4 Pt/C from the catalytic layers of aged MEA
The feasibility of platinum electrochemical dissolution from the catalytic layers of an aged MEA supplied by SYMBIO, an industrial partner, is then verified. The MEA were aged through a long-term test (LTT) representing fuel cell lifetime according to a light commercial vehicle duty cycle. The MEAs were directly used for Pt recovery and anodic and cathodic catalytic layers were simultaneously submitted to a similar electrochemical protocol in liquid electrolyte. The Pt/C catalyst coated layer of aged MEA was not electrically conductive enough, thus doing electrochemistry on the MEA was not possible without keeping a GDL to ensure a good electrical conductivity. However, on aged MEAs, the GDL can be partially detached from the catalyst layer. To improve the contact between the catalytic layers and the GDL, the MEA was pressed under a weight of 500 g in an oven at 110 °C for 4 hours. After this treatment, the MEA was left at room temperature until complete cooling. To counter the hydrophobicity of the GDL, the MEA was first soaked in 96% ethanol for 10 s, then in pure water for 30 min, then immersed in the electrolyte. The MEA was connected with a carbon clip.The area of this MEA is substantially larger than the glassy carbon tip used under model conditions. According to the higher value of the current, the size of the counter electrode was adjusted: a 25 cm2 carbon sheet was used in that case.
4.5 Electrolyte
All electrolytes are prepared by dilution of sulfuric acid (1 M Suprapur®, 96%). When necessary, chlorides are added via a 1 M NaCl solution (Honeywell, Reagent Grade >99%). Milli-Q water (Millipore, 18.2 MΩ cm, total organic compounds <3 ppb) is used for all dilutions.4.6 Physicochemical characterization
This section deals with the characterization of the catalyst morphology and platinum concentration.The images are analyzed with ImageJ software to determine the size of Pt particles and to plot the particle size distribution. Two types of particle populations are distinguished: well-defined isolated particles and aggregated ones (Fig. 1). In this work, the isolated particles are considered as spherical particles. The aggregated particles exhibit different, quite irregular shapes, and it was not possible to determine their “diameter” accurately. At different stages of the electrochemical protocol, two parameters are determined: the diameter of isolated particles and the projected area for aggregated particles. The diameters of isolated particles are measured using the function “straight”, whereas the enclosed area of aggregated particles is measured by the function “freehand selection” of the software ImageJ. Statistical analysis of different TEM images (between 6 and 10) representing more than 300 particles for each experiment (see Tables S1 and S2†) leads to a frequency distribution of diameter with a range of 0.5 nm for isolated particles and a frequency distribution of area with a range of 2 nm2 for aggregated particles. Since the number of analyzed particles can be different, all the distributions are expressed in frequency, meaning that the number of particles (isolated or aggregated) for all classes is divided by the total number of particles in the distribution.
Some parameters are needed to monitor the evolution of particle morphologies and their numbers. Since the distribution is not Gaussian or compliant with other probability distributions, the chosen parameters reduce inevitably the information of total distribution. For each cycle, the isolated and aggregated particles are compared with respect to their number and to their projected area, respectively. For a given number of cycles, the amount of isolated and aggregated particles is called niso and nagg, respectively. The ratio of isolated particles, rn,iso, and the ratio of aggregated particles, rn,agg, to the total number of particles are defined by eqn (3) and (4), respectively:
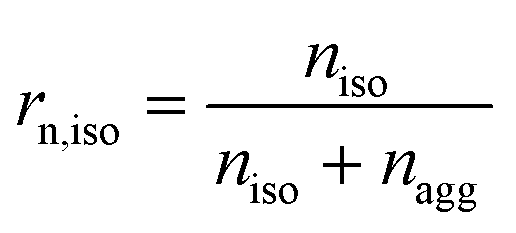 | (5) |
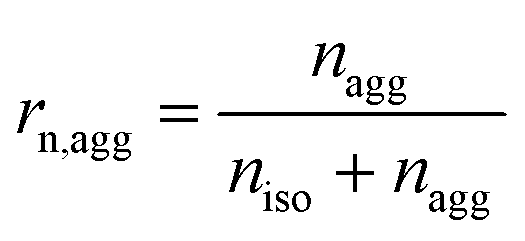 | (6) |
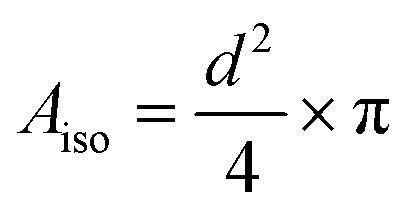 | (7) |
| Atot = rn,iso × Aiso + rn,agg × Aagg | (8) |
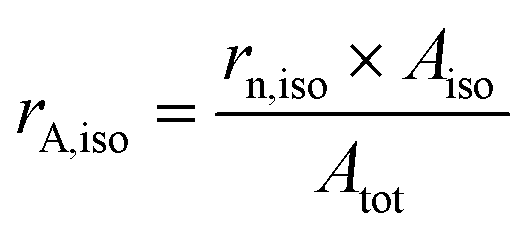 | (9) |
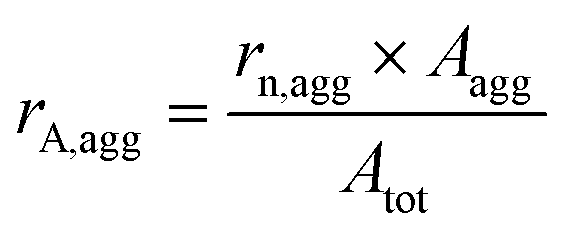 | (10) |
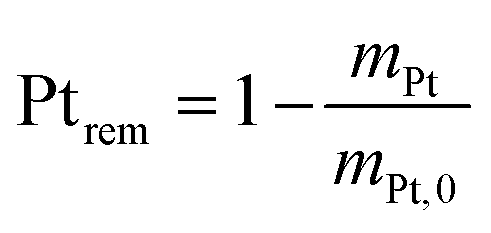 | (11) |
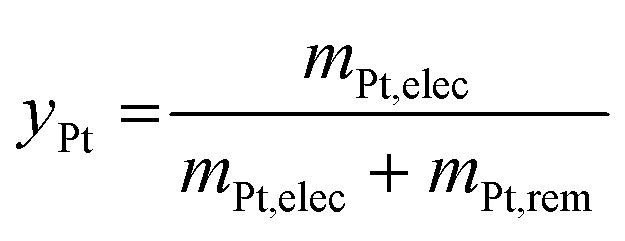 | (12) |
Author contributions
FG: conceptualization, investigation, data curation, formal analysis, writing – original draft, MC: methodology, writing – review and editing, AP: validation, LS: conceptualisation, methodology, writing – review and editing, funding acquisition, supervision, LD: conceptualization, methodology, writing – review and editing, funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed within the framework of the European Union via European Regional Development Fund (ERDF) through the Operation RA0027886 – STACK NEXT GEN project. The authors acknowledge the financial support of ANR CARNOT Energie du Futur (EF) through the HERMOS project. The authors thank V. Martin for ICP-MS measurements.References
- K. T. Møller, T. R. Jensen, E. Akiba and H. Li, Hydrogen - A sustainable energy carrier, Prog. Nat. Sci.: Mater. Int., 2017, 27, 34–40 CrossRef.
- M. Ball and M. Weeda, The hydrogen economy – Vision or reality?, Int. J. Hydrogen Energy, 2015, 40, 7903–7919 CrossRef CAS.
- C. Acar and I. Dincer, Impact assessment and efficiency evaluation of hydrogen production methods, Int. J. Energy Res., 2015, 39, 1757–1768 CrossRef CAS.
- E. Cetinkaya, I. Dincer and G. F. Naterer, Life cycle assessment of various hydrogen production methods, Int. J. Hydrogen Energy, 2011, 37, 2071–2080 CrossRef.
- J. Shin, W.-S. Hwang and H. Choi, Can hydrogen fuel vehicles be a sustainable alternative on vehicle market?: Comparison of electric and hydrogen fuel cell vehicles, Technol. Forecast. Soc. Change, 2019, 143, 239–248 CrossRef.
- V. W. W. Yam, Behind platinum's sparkle, Nat. Chem., 2010, 2, 790 CrossRef CAS.
- Communication from the commission to the European economic and social committee and the committee of the regions, Brussels, 3.9.2020 COM(2020) 474 final, https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52020DC0474&from=EN, (accessed Mar. 10, 2023).
- B. James, J. Moton and W. Colella, Mass production cost estimation of direct H2 PEM fuel cell systems for transportation applications: 2014 update, 2014 Search PubMed.
- J. Zhao, X. He, J. Tian, C. Wan and C. Jiang, Reclaim/recycle of Pt/C catalysts for PEMFC, Energy Convers. Manage., 2007, 48, 450–453 CrossRef CAS.
- L. Duclos, R. Chattot, L. Dubau, P.-X. Thivel, G. Mandil, V. Laforest, M. Bolloli, R. Vincent and L. Svecova, Closing the loop: Life cycle assessment and optimization of a PEMFC platinum-based catalyst recycling process, Green Chem., 2020, 22, 1919–1933 RSC.
- M. Chourashiya, R. Sharma, S. Gyergyek and S. M. Andersen, Gram-size Pt/C catalyst synthesized using Pt compound directly recovered from an end-of-life PEM fuel cell stack, Mater. Chem. Phys., 2022, 276, 125439 CrossRef CAS.
- A. G. Chmielewski, T. S. Urbański and W. Migdał, Separation technologies for metals recovery from industrial wastes, Hydrometallurgy, 1997, 45, 333–344 CrossRef CAS.
- M. Gökelma, A. Birich, S. Stopic and B. Friedrich, A review on alternative gold recovery Re-agents to cyanide, J. Mater. Sci. Chem. Eng., 2016, 04, 8–17 Search PubMed.
- W. Lin, R.-W. Zhang, S.-S. Jang, C.-P. Wong and J.-I. Hong, ‘Organic Aqua Regia’—Powerful liquids for dissolving noble metals, Angew. Chem., Int. Ed., 2010, 49, 7929–7932 CrossRef CAS PubMed.
- F. Forte, S. Riaño and K. Binnemans, Dissolution of noble metals in highly concentrated acidic salt solutions, Chem. Commun., 2020, 56, 8230–8232 RSC.
- L. Duclos, M. Lupsea, G. Mandil, L. Svecova, P.-X. Thivel and V. Laforest, Environmental assessment of proton exchange membrane fuel cell platinum catalyst recycling, J. Cleaner Prod., 2017, 142, 2618–2628 CrossRef CAS.
- N. Hodnik, et al., Platinum recycling going green via induced surface potential alteration enabling fast and efficient dissolution, Nat. Commun., 2016, 7, 13164 CrossRef CAS.
- L. Dubau, M. Lopez-Haro, L. Castanheira, J. Durst, M. Chatenet, P. Bayle-Guillemaud, L. Guétaz, N. Caqué, E. Rossinot and F. Maillard, Probing the structure, the composition and the ORR activity of Pt3Co/C nanocrystallites during a 3422h PEMFC ageing test, Appl. Catal., B, 2013, 142–143, 801–808 CrossRef CAS.
- L. Castanheira, L. Dubau, M. Mermoux, G. Berthomé, N. Caqué, E. Rossinot, M. Chatenet and F. Maillard, Carbon corrosion in proton-exchange membrane fuel cells: From model experiments to real-life operation in membrane electrode assemblies, ACS Catal., 2014, 4, 2258–2267 CrossRef CAS.
- L. Castanheira, W. O. Silva, F. H. B. Lima, A. Crisci, L. Dubau and F. Maillard, Carbon corrosion in proton-exchange membrane fuel cells: Effect of the carbon structure, the degradation protocol, and the gas atmosphere, ACS Catal., 2015, 5, 2184–2194 CrossRef CAS.
- L. Xing, M. A. Hossain, M. Tian, D. Beauchemin, K. T. Adjemian and G. Jerkiewicz, Platinum electro-dissolution in acidic media upon potential cycling, Electrocatalysis, 2014, 5, 96–112 CrossRef CAS.
- A. A. Topalov, S. Cherevko, A. R. Zeradjanin, J. C. Meier, I. Katsounaros and K. J. J. Mayrhofer, Towards a comprehensive understanding of platinum dissolution in acidic media, Chem. Sci., 2014, 5, 631–638 RSC.
- S. Cherevko, A. R. Zeradjanin, G. P. Keeley and K. J. J. Mayrhofer, A comparative study on gold and platinum dissolution in acidic and alkaline media, J. Electrochem. Soc., 2014, 161, H822–H830 CrossRef.
- S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Dissolution of platinum in presence of chloride traces, Electrochim. Acta, 2015, 179, 24–31 CrossRef CAS.
- A. Pavlišič, P. Jovanovič, V. S. Šelih, M. Šala, N. Hodnik, S. Hočevar and M. Gaberšček, The influence of chloride impurities on Pt/C fuel cell catalyst corrosion, Chem. Commun., 2014, 50, 3732–3734 RSC.
- R. Sharma, S. J. Andreasen, J. Chamier and S. M. Andersen, Pt/C electrocatalyst synthesis from recycling of the spent PEMFC membrane electrode assembly: A closed loop circular economy, J. Electrochem. Soc., 2019, 166, F963 CrossRef CAS.
- T. Hyeon, S. Han, Y.-E. Sung, K.-W. Park and Y.-W. Kim, High-performance direct methanol fuel cell electrodes using solid-phase-synthesized carbon nanocoils, Angew. Chem., 2003, 115, 4488–4492 CrossRef.
- H. Shiroishi, S. Hayashi, M. Yonekawa, R. Shoji, I. Kato and M. Kunimatsu, Dissolution rate of noble metals for electrochemical recycle in polymer electrolyte fuel cells, Electrochemistry, 2012, 80, 898–903 CrossRef CAS.
- R. Latsuzbaia, E. Negro and G. J. M. Koper, Environmentally friendly carbon-preserving recovery of noble metals from supported fuel cell catalysts, ChemSusChem, 2015, 8, 1926–1934 CrossRef CAS.
- R. Sharma, K. Rode Nielsen, P. Brilner Lund, S. Bredmose Simonsen, L. Grahl-Madsen and S. Ma Andersen, Sustainable platinum recycling through electrochemical dissolution of platinum nanoparticles from fuel cell electrodes, ChemElectroChem, 2019, 6(17), 4471–4482 CrossRef CAS.
- M. A. Montiel, R. Granados-Fernández, S. Díaz-Abad, C. Sáez, C. M. Fernández-Marchante, M. A. Rodrigo and J. Lobato, Towards a circular economy for Pt catalysts. Case study: Pt recovery from electrodes for hydrogen production, Appl. Catal., B, 2023, 327, 122414 CrossRef CAS.
- S. Zhang, X. Yuan, H. Wang, W. Mérida, H. Zhu, J. Shen, S. Wu and J. Zhang, A review of accelerated stress tests of MEA durability in PEM fuel cells, Int. J. Hydrogen Energy, 2009, 34, 388–404 CrossRef CAS.
- S. J. Ashton and M. Arenz, A DEMS study on the electrochemical oxidation of a high surface area carbon black, Electrochem. Commun., 2011, 13, 1473–1475 CAS.
- S. J. Ashton and M. Arenz, Comparative DEMS study on the electrochemical oxidation of carbon blacks, J. Power Sources, 2012, 217, 392–399 CrossRef CAS.
- N. Job, M. Chatenet, S. Berthon-Fabry, S. Hermans and F. Maillard, Efficient Pt/carbon electrocatalysts for proton exchange membrane fuel cells: Avoid chloride-based Pt salts!, J. Power Sources, 2013, 240, 294–305 CrossRef CAS.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Instability of supported platinum nanoparticles in low-temperature fuel cells, Top. Catal., 2007, 46, 285–305 CrossRef CAS.
- A. P. Yadav, A. Nishikata and T. Tsuru, Effect of halogen ions on platinum dissolution under potential cycling in 0.5M H2SO4 solution, Electrochim. Acta, 2007, 52, 7444–7452 CrossRef CAS.
- N. Hodnik, M. Zorko, B. Jozinović, M. Bele, G. Dražič, S. Hočevar and M. Gaberšček, Severe accelerated degradation of PEMFC platinum catalyst: A thin film IL-SEM study, Electrochem. Commun., 2013, 30, 75–78 CrossRef CAS.
- C. Rao and D. Trivedi, Chemical and electrochemical depositions of platinum group metals and their applications, Coord. Chem. Rev., 2004, 249, 613–631 CrossRef.
- M. Inaba, M. Ando, A. Hatanaka, A. Nomoto, K. Matsuzawa, A. Tasaka, T. Kinumoto, Y. Iriyama and Z. Ogumi, Controlled growth and shape formation of platinum nanoparticles and their electrochemical properties, Electrochim. Acta, 2006, 52, 1632–1638 CrossRef CAS.
- L. Dubau, L. Castanheira, G. Berthomé and F. Maillard, An identical-location transmission electron microscopy study on the degradation of Pt/C nanoparticles under oxidizing, reducing and neutral atmosphere, Electrochim. Acta, 2013, 110, 273–281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3im00085k |
| This journal is © Institute of Process Engineering of CAS 2024 |