
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iodine-mediated, chalcogen–chalcogen bond formation in water: green synthesis of carbamo(dithioperoxo)thioates, carbamo(selenothioperoxo)thioates, carbono(dithioperoxo)thioates, and carbono(selenothioperoxo)thioates†
Appanapalli N. V.
Satyanarayana
,
Paramita
Pattanayak
 ,
Pranav Pramod
Nayar
and
Tanmay
Chatterjee
*
,
Pranav Pramod
Nayar
and
Tanmay
Chatterjee
*
Department of Chemistry, Birla Institute of Technology and Science, Pilani, Hyderabad Campus, Jawahar Nagar, Hyderabad – 500078, India. E-mail: tanmay@hyderabad.bits-pilani.ac.in
First published on 11th December 2024
Abstract
Herein, we disclose an iodine-mediated, three-component versatile synthetic strategy for the green synthesis of a wide variety of biologically active carbamo(dithioperoxo)thioates including a couple of anti-filarial and anti-tumour agents, along with their new classes of promising analogues, such as carbamo(selenothioperoxo)thioates, carbono(dithioperoxo)thioates, and carbono(selenothioperoxo)thioates. This strategy employs only readily available and inexpensive reactants or reagents, such as iodine, diorganyl disulfides or diselenides, amines, and carbon disulfide (CS2) or potassium xanthate to access potential classes of molecules by enabling the formation of chalcogen–chalcogen bonds (S–S and S–Se) in an aqueous environment at ambient temperature under aerobic conditions. Notably, two synthetically challenging yet promising classes of molecules, namely carbamo(selenothioperoxo)thioates and carbono(selenothioperoxo)thioates, have been successfully synthesized in a sustainable manner in water at room temperature through the formation of the challenging S–Se bond for the first time. The reaction proceeded through the in situ formation of the corresponding chalcogenyl iodides, followed by nucleophilic substitution at the chalcogen center by the in situ generated dithiocarbamates or xanthate, as the major path, to afford the desired product along with the formation of non-hazardous by-products or waste, i.e., NaI and H2O or KI.
Green foundation1. The green advances of this work as compared to those of previously developed ones are (a) a metal-free, versatile, and scalable protocol using only commercially available and inexpensive reactants or reagents and (b) use of water as a green solvent and generation of non-hazardous waste, i.e., NaI and water or KI.2. This work demonstrates a significant reduction in waste generation, as indicated by the E-factor. The E-factor of our protocol for the synthesis of propyl pyrrolidine-1-carbo(dithioperoxo)thioate was calculated to be 16.96 g waste per g product formation, where water contributed the majority of the waste. However, the E-factor of recent literature for the synthesis of the same molecule was found to be 122.13 g waste per g product formation. 3. This work could be made greener by eliminating the formation of any by-products and thus further decreasing the E-factor. |
Introduction
Dithiocarbamates have received tremendous attention from synthetic organic, medicinal, and materials chemists because of their widespread application in biochemistry, medicinal chemistry, agriculture, and organic synthesis.1 Among the various classes of dithiocarbamates, carbamo(dithioperoxo)thioates are the most valuable class of molecules, exhibiting a wide variety of biological activities such as anti-cancer,2 anti-tumor,3 anti-filarial, etc. (Fig. 1).4 For example, disulfiram or Antabuse (A, Fig. 1), an acetaldehyde dehydrogenase (ALDH) inhibitor, is a well-known drug for the treatment of alcoholism (aversion therapy). It is also a well-known inhibitor of the E3 ubiquitin ligase enzyme and BCA2 (breast cancer associated protein 2).5 Moreover, 2-hydroxyethyl diethylcarbamo(dithioperoxo)thioate2a (B) is also found to inhibit the expression of BCA2 in human breast cancer cell lines owing to its anti-tumor activity and p-tolyl piperidine-1-carbo(dithioperoxo) thioate2b (C), and 4-methoxyphenyl 4-benzylpiperidine-1-carbo(dithioperoxo)thioate (D) exhibits excellent anti-filarial activity (Fig. 1).4 | ||
| Fig. 1 Examples of biologically active carbamo(dithioperoxo)thioates. | ||
Consequently, several synthetic methods have been developed so far for the synthesis of carbamo(dithioperoxo)thioates. Earlier methods involved nucleophilic substitution (SN2 type) at dialkyl disulfides by sodium dithiocarbamates or at the dimer of dithiocarbamates by alkyl thiols for the synthesis of only alkylsulfenyl N,N-dialkyldithiocarbamates (Scheme 1A).2b,6–8 These methods suffer from several serious limitations, such as a very limited substrate scope, poor yields of products, the requirement of pre-synthesized sodium dithiocarbamates or the dimer of dithiocarbamates, the use of toxic/hazardous chlorine gas and CCl4 solvent, and/or high temperature. Later, several multicomponent synthetic strategies were developed for the synthesis of carbamo(dithioperoxo)thioates. Westwell et al. developed a one-pot, two-step protocol for the synthesis of only carbamo(dithioperoxo)thioates using 2 equiv. of CBr4 (Scheme 1B).2a This method suffered from some limitations, such as a limited substrate scope for accessing carbamo(dithioperoxo)thioates using only alkyl mercaptans and secondary amines, the requirement of excess CBr4 (2 equiv.), leading to the formation of organic halides as by-products, hazardous solvent (CH2Cl2), and harsh reaction conditions, such as the requirement of anhydrous CH2Cl2 and low temperature.
 | ||
| Scheme 1 Previous synthetic strategies to access carbamo(dithioperoxo)thioates and this work. | ||
Subsequently, the Misra group reported N-chlorosuccinimide (NCS) mediated, one-pot, two-step synthesis of only aryl carbamo(dithioperoxo)thioates from arylthiols, CS2, and secondary amines, which suffered from a limited substrate scope, poor atom-economy and stoichiometric generation of succinimide waste (Scheme 1C).4a Saha and co-workers also developed a similar strategy for synthesizing only aryl carbamo(dithioperoxo)thioates using N-(arylthio)phthalimides as the source of electrophilic sulfur, CS2, and secondary amines in water (Scheme 1C).9 Recently, Duan and Tang et al. developed a hydrazine-mediated, indirect electrochemical synthesis of carbamo(dithioperoxo)thioates (Scheme 1D).10 Despite the broad substrate scope, this strategy also suffers from some limitations, such as the requirement of hydrazine as a mediator, Et4NBF4 as the electrolyte, Na2CO3 as a base, electricity, and MeCN as the toxic solvent. Moreover, all these methods are limited to the synthesis of only carbamo(dithioperoxo)thioates via S–S bond formation alone. Since carbamo(dithioperoxo)thioates are a highly valuable class of molecules in medicinal chemistry, there is an urgent need for the development of a green and versatile synthetic strategy for accessing carbamo(dithioperoxo)thioate and other new classes of its potential analogues. As a part of our continued interest in developing iodine-catalyzed/mediated sustainable synthetic strategies for chalcogenative C–H annulation11 or functionalization reactions,12 herein we report an iodine-mediated versatile and sustainable synthetic strategy for the synthesis of novel carbamo(dithioperoxo)thioates and some of the new classes of its potential analogues, such as carbamo(selenothioperoxo)thioates, carbono(dithioperoxo)thioates, and carbono(selenothioperoxo)thioates, via S–S or challenging S–Se bond formation in water at room temperature (Scheme 1E).
Results and discussion
We commenced our investigation by treating dipropyl disulfide 1a with CS2 and piperidine 2a in the presence of iodine (0.5 equiv.) and NaOH (1 equiv.) in water at room temperature, and to our delight, the desired product, propyl piperidine-1-carbo(dithioperoxo)thioate 3aa, was formed in 86% yield in just 30 min (entry 1, Table 1). The change of solvent from water to ethanol, DCM, MeCN, and DMSO negatively impacted the reaction outcome (entries 2–5 vs. 1, Table 1). Both increasing the loading of iodine to 1 equiv. and decreasing the same to 0.2 equiv., i.e., use of a catalytic (20 mol%) amount of iodine, lower the yield of 3aa to 47% and 18%, respectively, which revealed that the use of 0.5 equiv. of iodine is required for the effective synthesis of 3aa (entries 6 and 7 vs. 1, Table 1). Use of 0.5 equiv. of 1a also negatively impacted the reaction outcome, leading to the formation of 50% 3aa (entry 8, Table 1). When 1 equiv. of iodine was used in the presence of 0.5 equiv. of 1a, the yield of 3aa further went down to 30% (entry 9, Table 1). Blank experiments revealed the important roles of NaOH and iodine in the efficient synthesis of 3aa (entries 10 and 11 vs. 1, Table 1).|
|
||
|---|---|---|
| Entry | Variation in conditions from the scheme | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol, 1 equiv.), CS2 (1.5 equiv.), 2a (1 equiv.), I2 (0.5 equiv.), NaOH (1 equiv.), H2O (0.6 mL), rt, and 30 min. b Yield of 3aa was determined by the 1H NMR of the crude reaction mixture using 1,3,5-trimethoxybenzene as the internal standard. | ||
| 1 | None | 86 |
| 2 | EtOH (0.6 mL) was used instead of DMSO | 46 |
| 3 | DCM (0.6 mL) was used instead of DMSO | 52 |
| 4 | MeCN (0.6 mL) was used instead of DMSO | 62 |
| 5 | DMSO (0.6 mL) was used instead of DMSO | 55 |
| 6 | 1 equiv. of I2 was used | 47 |
| 7 | 0.2 equiv. (20 mol%) of I2 was used | 18 |
| 8 | 0.5 of 1a was used | 50 |
| 9 | 1 equiv. of I2 and 0.5 of 1a were used | 30 |
| 10 | Blank experiment without using NaOH | 60 |
| 11 | Blank experiment without using I2 | 0 |

Next, we explored the scope of the synthesis of carbamo(dithioperoxo)thioates first by iodine-mediated, three-component coupling between disulfides, CS2, and amines in water at room temperature, and the results are summarized in Table 2. All kinds of aliphatic disulfides, i.e., cyclic and acyclic, including primary, secondary, and tertiary dialkyl disulfides, smoothly reacted with CS2 and piperidine to furnish the desired products: propyl piperidine-1-carbo(dithioperoxo)thioate 3aa (86%), cyclohexyl piperidine-1-carbo(dithioperoxo)thioate 3ba (66%), and tert-butyl piperidine-1-carbo(dithioperoxo)thioate 3ca (48%) in moderate to good yields. When 2-mercaptoethanol was reacted with CS2 and piperidine under the optimized conditions, the desired product, 2-hydroxyethyl piperidine-1-carbo(dithioperoxo)thioate 3da, was formed, which possesses interesting biological activities.2a Diphenyl disulfide and various diaryl disulfides bearing an electron-donating (Me and OMe) or electron-withdrawing (Br, Cl, F, and CF3) substituent participated in the three-component reaction in water at room temperature without any difficulties to afford a library of aryl piperidine-1-carbo(dithioperoxo)thioates (3ea–3ka) in moderate to excellent yields (65–90%), irrespective of the electronic nature and the position of the substituent. Notably, an anti-filarial agent, p-tolyl piperidine-1-carbo(dithioperoxo)thioate 3fa, was synthesized, in excellent yield (90%) under standard conditions. A heteroaryl disulfide, 1,2-di(thiophene-2-yl)disulfane, also participated in the three-component coupling reaction in water at room temperature to furnish the desired thiophene-2-yl piperidine-1-carbo(dithioperoxo)thioate 3la in 60% yield. Subsequently, the scope of amines was explored, and various cyclic and acyclic amines were tested. Like piperidine, another cyclic amine, i.e., morpholine, also participated in the reaction with CS2 and dipropyl disulfide 1a to synthesize propyl morpholine-4-carbo(dithioperoxo)thioate 3ab in 70% yield. A five-membered cyclic amine, pyrrolidine, also reacted smoothly with both dialkyl disulfides and diaryl disulfides to afford the desired products (3ac and 3fc) in good yields (82–88%). Acyclic amine (diethyl amine) reacted with various dialkyl disulfides and diphenyl disulfide to produce the corresponding diethylcarbamo(dithioperoxo)thioates (3md, 3dd, and 3ed), including a bioactive molecule, BCA2 inhibitor and anti-tumor agent (3dd). Notably, primary amines such as benzylamine and 4-methoxy benzylamine also reacted with CS2 and 1a to afford the desired products 3ae and 3af in 94% and 71% yields, respectively.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:
Next, we explored the scope of the synthesis of a new class of potential analogues of carbamo(dithioperoxo)thioates, i.e., carbamo(selenothioperoxo)thioates, by a three-component coupling between diselenides, CS2, and amines through the formation of challenging S–Se bonds in water at room temperature. Aliphatic diselenides, such as dimethyl diselenide and dibenzyl diselenide, smoothly reacted with CS2, and various cyclic/acyclic amines, such as piperidine, morpholine, 1-methylpiperazine, pyrrolidine, and diethyl amine, to furnish the desired new class of molecules: SSe-methyl piperidine-1-carbo(selenothioperoxo)thioate 3na, SSe-benzyl piperidine-1-carbo(selenothioperoxo)thioate 3oa, SSe-methyl morpholine-4-carbo(selenothioperoxo)thioate 3nb, SSe-methyl diethylcarbamo(selenothioperoxo)thioate 3nd, SSe-methyl 4-methylpiperazine-1-carbo(selenothioperoxo)thioate 3ng, and SSe-benzyl diethylcarbamo(selenothioperoxo)thioate 3od in yields of up to 67%. Unfortunately, the S–Se bond forming reactions with diaryl diselenides and primary amines were unsuccessful.
The environmental factor or E-factor is a unique green chemistry metric to measure the greenness of a protocol quantitatively based on the waste generated.11 To determine the greenness of our developed protocol as compared to recent literature,10 we evaluated the E-factors of both protocols for the synthesis of propyl pyrrolidine-1-carbo(dithioperoxo)thioate 3ac (see Tables S1 and S2 in the ESI†). Notably, the E-factor of our protocol was calculated to be 16.96 g waste per g product formation, considering water as the majority of the waste. However, considering water as not a waste, the E-factor was found to be only 1.57 g waste per g product formation. On the other hand, the E-factor of recent literature for the synthesis of the same molecule, propyl pyrrolidine-1-carbo(dithioperoxo)thioate 3ac, was found to be 122.13 g waste per g product formation, which decreases to 118.94 g waste per g product formation considering water as not a waste. Even without considering the solvent, i.e., acetonitrile (MeCN), as waste, the E-factor of the recent literature was found to be 4.47 g waste per g product formation, which is around three times higher than that of our protocol. Hence, the E-factor of our protocol is superior to that of the recent literature, and most importantly, the solvent used in our protocol is water. Hence, our method could be considered as greener as compared to the previous literature.10
After synthesizing a series of carbamo(dithioperoxo)thioates and carbamo(selenothioperoxo)thioates, we became interested in employing our developed protocol for synthesizing two more new classes of molecules, i.e., carbono(dithioperoxo)thioates and carbono(selenothioperoxo)thioates, using xanthate and diorganyl dichalcogenides (Table 3). When dialkyl disulfides, such as dimethyl disulfide, dipropyl disulfide, and dicyclohexyl disulfide, were treated with xanthate, in particular, potassium O-ethyl carbonodithioate (potassium ethylxanthate), under the standard conditions, desired carbono(dithioperoxo)thioates, i.e., O-ethyl SS-methyl carbono(dithioperoxo)thioate 4a, O-ethyl SS-propyl carbono(dithioperoxo)thioate 4b, and SS-cyclohexyl O-ethyl carbono(dithioperoxo)thioate 4c, were formed in moderate to good yields (53–75%). Diaryl disulfides, such as diphenyl disulfide and di-p-tolyl disulfide, also participated in the reaction to afford the desired carbono(dithioperoxo)thioates 4d and 4e in 72% and 65% yields, respectively.

Notably, when dimethyl diselenide was treated with potassium ethylxanthate, the desired new class of molecules, O-ethyl SSe-methyl carbono(selenothioperoxo)thioates, was formed. Unfortunately, the reactions with diaryl disulfides or diaryl diselenide were unsuccessful. For all the reactions mentioned above, non-hazardous and environmentally benign by-products, such as NaI and water or KI, were formed.
To demonstrate the practicality of the developed protocol, a gram-scale reaction was conducted using 1f (1.23 g, 5 mmol), CS2 (1.5 equiv.), and pyrrolidine 2c (1 equiv.), and to our delight, 3fc was formed in 82% yield (1.1 g), which revealed that the developed protocol could efficiently be scaled up to the gram scale without any compromise in the reaction outcome (Scheme 2).
 | ||
| Scheme 2 Gram-scale synthesis of 3fc. | ||
To gain insights into the reaction mechanism, several radical quenching and control experiments were conducted, which are summarized in Scheme 3. In the presence of various radical quenchers (3 equiv.), such as TEMPO, BHT, and ethene-1,1-diyldibenzene, 3aa was formed in 80%, 86%, and 78% yields, respectively, from 1a under standard conditions (Scheme 3A), which revealed that the reaction does not follow a radical pathway. Next, we performed a series of control experiments, which are presented in Scheme 3B. When piperidine 2a was treated with CS2 in the presence of NaOH (1 equiv.), sodium piperidine-1-carbodithioate 5 was formed quantitatively [Scheme 3B, (i)]; however, in the presence of iodine (1 equiv.), the same reaction furnished piperidine-1-carbothermic dithioperoxyanhydride 6 in 99% yield [Scheme 3B, (ii)], which revealed that iodine can oxidize 5 to 6. Iodine could also oxidize potassium xanthate to its dimer 7 almost quantitatively in water at room temperature [Scheme 3B, (iii)]. When 1a was treated with sodium piperidine-1-carbodithioate 6 in the presence of iodine (0.5 equiv.) in water, the desired product 3aa was formed in 76% yield [Scheme 3B, (iv)], which revealed that 6 is one of the intermediates of the reaction. However, in the absence of iodine, 3aa was not formed at all from 1a and 6, which again supported the crucial role of iodine in the reaction [Scheme 3B, (iv)]. When 1a was treated with 6 and 7 in water, 3aa6 and 4b were formed in 33% and 10% yields, respectively [Scheme 3B, (v) and (vi)]. Interestingly, while in the presence of NaI (1 equiv.), the formation of 3aa from 1a and 6 decreased to 14% from 33% [Scheme 3B, (vii)], in the presence of KI (1 equiv.), the formation of 4b from 1a and 7 enhanced significantly to 88% from 10% [Scheme 3B, (viii)]. We speculate that the iodine-mediated conversion of 5 to 6 is irreversible, while the iodine-mediated conversion of potassium xanthate to 7 is a reversible reaction.
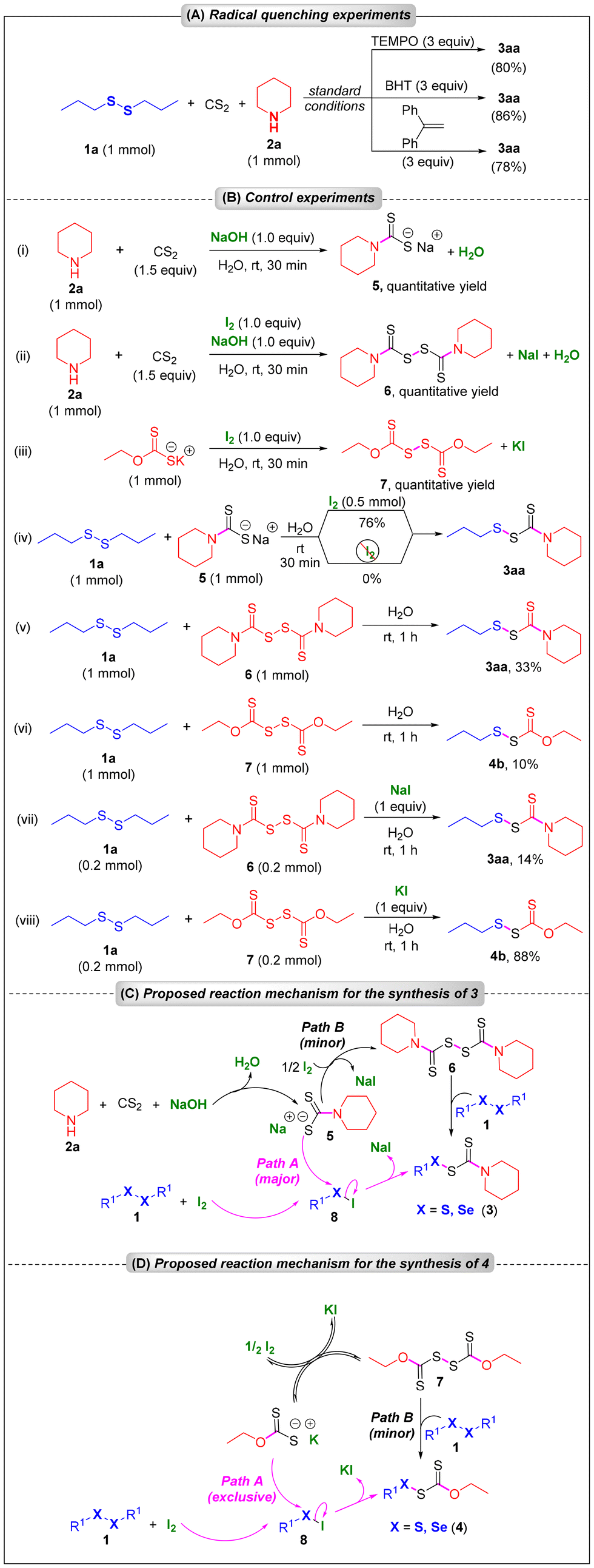 | ||
| Scheme 3 (A) Radical quenching experiments, (B) control experiments, and (C) proposed reaction mechanism. | ||
Based on the experimental results and the literature precedence,6,11 we proposed plausible reaction mechanisms for the synthesis of 3 and 4, which are presented in Schemes 3C and D, respectively. For the synthesis of 3, the amine spontaneously reacted with CS2 in the presence of NaOH to generate the crucial intermediate, sodium piperidine-1-carbodithioate 5, in situ (Scheme 3C). On the other hand, diorganyl disulfides or diselenides also reacted spontaneously with iodine to furnish the corresponding chalcogenyl halides 8.11 A direct substitution at the chalcogen center of 8 by the in situ generated sodium piperidine-1-carbodithioate 5 furnished the desired products (3) via chalcogen–chalcogen (S–S or S–Se) bond formation (part A) along with the formation of sodium iodide and water as by-products (Scheme 3C). However, we cannot fully rule out another pathway (path B) where sodium piperidine-1-carbodithioate 5 could get oxidized by iodine irreversibly to form 6, which then reacted with 1 to afford the final product (3). However, based on the experimental procedures (see the ESI†) and some controlled experiments [Scheme 3B, (v) and (vi)], we believe that path A is predominantly operative for this reaction as compared to path B.
For the synthesis of 4, we proposed that at first the corresponding chalcogenyl halide 8 was formed from the diorganyl disulfides or diselenides and iodine, which then immediately underwent a nucleophilic substitution at the chalcogen centre by potassium xanthate to furnish the desired product 4 (path A). However, during the reaction, potassium xanthate could get oxidized by iodine reversibly to form 7, which then could react with 1 to form 4 (path B). However, based on the experimental results [Scheme 3B, (vi) and (viii)], we speculate that path A is exclusively operative for the synthesis of 4.
Conclusions
In conclusion, we have developed a versatile yet sustainable synthetic strategy for the general synthesis of a biologically active class of molecules, i.e., carbamo(dithioperoxo)thioates, and their new classes of highly potential analogues, i.e., carbamo(selenothioperoxo)thioates, carbono(dithioperoxo)thioates, and carbono(selenothioperoxo)thioates, by using readily available and inexpensive reactants or reagents such as iodine, diorganyl disulfides or diselenides, amines, and carbon disulfide (CS2) or potassium xanthate in water at room temperature via the formation of chalcogen–chalcogen (S–S and S–Se) bonds. This protocol offers several advantages and green advancements over previously developed ones, such as (a) a versatile, sustainable, and scalable protocol, (b) use of only commercially available and inexpensive reactants or reagents, (c) use of water as a green solvent and generation of non-hazardous waste, i.e., NaI and water or KI, with a superior E-factor, (d) a broad substrate scope in accessing a wide variety of biologically active carbamo(dithioperoxo)thioates including a couple of anti-filarial and anti-tumour agents in good yields, and (e) access to new classes of novel analogues of carbamo(dithioperoxo)thioates, i.e., carbamo(selenothioperoxo)thioates, carbono(dithioperoxo)thioates, and carbono(selenothioperoxo)thioates, via the formation of challenging S–S and S–Se bonds in water. To the best of our knowledge, this is the first report of both S–S and S–Se bond-forming reactions in water, and we believe that this strategy will be utilized for the synthesis of other new and novel classes of organochalcogens.Author contributions
A. N. V. Satyanarayana carried out most of the experiments, including optimization of the reaction conditions, synthesis of products, their synthetic diversifications, and mechanistic studies. P. Pattanayak and P. P. Nayar also synthesized some carbamo(dithioperoxo)thioates. T. Chatterjee conceived and supervised the work and wrote and edited the manuscript. All the authors have given their final approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. Chatterjee gratefully acknowledges the financial support from the Science and Engineering Research Board (SERB), Govt. of India (file no. CRG/2023/003045), and the Council of Scientific and Industrial Research (CSIR), Govt. of India [file no. 02(0390)/21/EMR-II]. The NMR facility at the Central Analytical Laboratory (CAL) of BITS Pilani, Hyderabad campus, and the HRMS facility, funded by DST-FIST (grant no: SR/FST/CS-I/2020/158), at BITS Pilani, Hyderabad campus, are acknowledged. A. N. V. Satyanarayana thanks CSIR [File No. 09/1026(17969)/2024- EMR-I] for the senior research fellowship (SRF Direct).References
- For a couple of recent review articles on the importance of dithiocarbamates, see: (a) L. Kaul, R. Suss, A. Zannettino and K. Richter, The revival of dithiocarbamates: from pesticides to innovative medical treatments, iScience, 2021, 24, 102092 CrossRef CAS PubMed; (b) J. W. F. Oliveira, H. A. O. Rocha, W. M. T. Q. Medeiros and M. S. Silva, Application of Dithiocarbamates as Potential New Antitrypanosomatids-Drugs: Approach Chemistry, Functional and Biological, Molecules, 2019, 24, 2806 CrossRef PubMed.
- (a) V. Cilibrasi, K. Tsang, M. Morelli, F. Solfa, H. L. Wiggins, A. T. Jones and A. D. Westwell, Synthesis of substituted carbamo(dithioperoxo)thioates as potential BCA2-inhibitory anticancer agents, Tetrahedron Lett., 2015, 56, 2583–2585 CrossRef CAS; (b) G. Brahemi, F. R. Kona, A. Fiasella, D. Buac, J. Soukupov, A. Brancale, A. M. Burger and A. D. Westwell, Exploring the Structural Requirements for Inhibition of the Ubiquitin E3 Ligase Breast Cancer Associated Protein 2 (BCA2) as a Treatment for Breast Cancer, J. Med. Chem., 2010, 53, 2757–2765 CrossRef CAS PubMed; (c) B. Cvek, Nonprofit drugs as the salvation of the world's healthcare systems: the case of Antabuse (disulfiram), Drug Discovery Today, 2012, 17, 409–412 CrossRef PubMed.
- (a) S. H. Lee, Disulfide and Multisulfide Antitumor Agents and Their Modes of Action, Arch. Pharmacal Res., 2009, 32, 299–315 CrossRef CAS PubMed; (b) L. Ronconi, C. Marzano, P. Zanello, M. Corsini, G. Miolo, C. Macca, A. Trevisan and D. Fregona, Gold(III) Dithiocarbamate Derivatives for the Treatment of Cancer: Solution Chemistry, DNABinding, and Hemolytic Properties, J. Med. Chem., 2006, 49, 1648–1657 CrossRef CAS PubMed.
- (a) A. Gucchait, N. Joardar, P. K. Parida, P. Roy, N. Mukherjee, A. Dutta, R. Yesuvadian, S. P. SinhaBabu, K. Jana and A. K. Misra, Development of novel anti-filarial agents using carbamo(dithioperoxo)thioate derivatives, Eur. J. Med. Chem., 2018, 143, 598 CrossRef CAS PubMed; (b) A. Gucchait, N. Joardar, P. K. Parida, P. Roy, N. Mukherjee, A. Dutta, R. Yesuvadian, S. P. SinhaBabu, K. Jana and A. K. Misra, Development of novel anti-filarial agents using carbamo(dithioperoxo)thioate derivatives, Eur. J. Med. Chem., 2018, 143, 598e610 CrossRef PubMed.
- A. Burger, Y. Amemiya, R. Kitching and A. K. Seth, Novel RING E3 Ubiquitin Ligases in Breast Cancer, Neoplasia, 2006, 8, 689–695 CrossRef CAS PubMed.
- K. Peng, H. Zhu, X. Liu, H. Peng, J. Chen and Z. Dong, Chemoselective C–S/S–S Formation between Diaryl Disulfides and Tetraalkylthiuram Disulfides, Eur. J. Org. Chem., 2019, 7629–7634 CrossRef CAS.
- W. F. Gilmore and R. N. Clark, New Alkylsulfenyl N,N-Dialkyldithiocarbamates, J. Chem. Eng. Data, 1969, 14, 119–120 Search PubMed.
- J. G. Sheppard, K. R. Frazier, P. Saralkar, M. F. Hossain, W. J. Geldenhuys and T. E. Long, Disulfiram-based disulfides as narrow-spectrum antibacterial agents, Bioorg. Med. Chem. Lett., 2018, 28, 1298–1302 CrossRef CAS PubMed.
- S. Dutta and A. Saha, Benign One-Pot Synthesis of Carbamo(dithioperoxo)thioate Compounds in Water Medium Using N-(Arylthio) phthalimides as the Electrophilic Sulfur Source, ChemistrySelect, 2018, 3, 11895–11897 CrossRef CAS.
- Q. Wang, C.-H. Xu, Y.-C. Wang, Y.-M. Pan, W.-G. Duan and H.-T. Tang, Electrochemical synthesis of carbamo (dithioperoxo)thioates through the dehydrogenation coupling of thiols and amines and the insertion of CS2, Green Chem., 2022, 24, 7362 RSC.
- (a) N. Mukherjee and T. Chatterjee, Iodine-Catalyzed Methylthiolative Annulation of 2-Alkynyl Biaryls with DMSO: A Metal-Free Approach to 9-Sulfenylphenanthrenes, J. Org. Chem., 2021, 86, 7881–7890 CrossRef CAS PubMed; (b) N. Mukherjee and T. Chatterjee, Iodine-catalyzed, highly atom-economic synthesis of 9-sulfenylphenanthrenes and polycyclic heteroaromatics in water, Green Chem., 2021, 23, 10006–10013 RSC; (c) N. Mukherjee, A. N. V. Satyanarayana, P. Singh, M. Dixit and T. Chatterjee, Recyclable iodine-catalyzed radical selenylative annulation of 2-alkynyl biaryls with diselenides in water: a green approach to selanyl polycyclic aromatic hydrocarbons and polycyclic heteroaromatics, Green Chem., 2022, 24, 7029–7038 RSC.
- (a) A. N. V. Satyanarayana, N. Mukherjee and T. Chatterjee, 100% atom-economical and highly regio- and stereoselective iodosulfenylation of alkynes: a reagentless and sustainable approach to access (E)-β-iodoalkenyl sulfides and (Z)-tamoxifen, Green Chem., 2022, 25, 779–788 RSC; (b) N. Mukherjee and T. Chatterjee, Recyclable iodine-catalyzed oxidative C–H chalcogenation of 1,1-diarylethenes in water: green synthesis of trisubstituted vinyl sulfides and selenides, Green Chem., 2023, 25, 8798–8807 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data of the synthesized molecules, and 1H, 13C, 19F and 77Se-NMR spectra of the synthesized compounds. See DOI: https://doi.org/10.1039/d4gc04699d |
| This journal is © The Royal Society of Chemistry 2025 |