
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous-flow synthesis of cyclic carbonates with polymer-supported imidazolium-based ionic liquid (Im-PSIL) catalysts†‡
Zhibo
Yu
a,
Haruro
Ishitani
*b and
Shu
Kobayashi
 *ab
*ab
aDepartment of Chemistry, School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shu_kobayashi@chem.s.u-tokyo.ac.jp
bGreen & Sustainable Chemistry Social Cooperation Laboratory, Graduate School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: hishitani@chem.s.u-tokyo.ac.jp
First published on 30th October 2024
Abstract
Carbon dioxide (CO2), a major greenhouse gas emitted through human activities, represents a valuable carbon source for chemical production. However, maximizing its efficient utilization requires novel methods for CO2 conversion that leverage the advantages of heterogeneous catalysis in continuous-flow systems. Herein, we report polymer-supported ionic liquids (PSILs) as efficient and recyclable catalysts for the continuous-flow synthesis of cyclic carbonates from epoxides and CO2. We designed imidazolium-based PSIL catalysts specifically for this purpose. These catalysts demonstrated remarkable stability for over 160 h under continuous-flow conditions with gaseous CO2, achieving an average yield of over 90% throughout the reaction. Furthermore, they exhibit broad applicability to 12 different epoxide substrates, yielding moderate to excellent yields. This work suggests an environmentally friendly pathway for the sustainable and scalable production of cyclic carbonates.
Carbon dioxide (CO2), a ubiquitous waste gas emitted in large quantities during industrial processes, agricultural production, and daily human activities, is paradoxically considered the ideal C1 source for producing organic chemicals due to its high availability, renewability, and non-toxic nature.1 In 2022, CO2 emissions were estimated to reach 36.8 billion tonnes, constituting approximately three-quarters of total greenhouse gas emissions.2 This stands in stark contrast to the currently limited capture and storage capabilities. Therefore, capturing and utilizing CO2 from industrial processes, particularly as a chemical feedstock, has recently gained global recognition as a critical strategy. This approach can contribute to both reducing greenhouse gases and transitioning away from a dependence on fossil resources.
Over the past few decades, the conversion of CO2 into various organic chemicals has been extensively studied and optimized. Processes encompass a spectrum, ranging from converting simple molecules like methanol, urea, and formic acid through reductive processes to the production of high-value fine chemicals, including pharmaceuticals.3–7 Among these successful applications, the synthesis of cyclic carbonates from epoxides and CO2 has emerged as one of the most useful and well-studied non-reductive transformations.8,9 This is due to their promising industrial applications as aprotic polar solvents and electrolyte solvents for lithium-ion batteries.10,11 Additionally, cyclic carbonates are identified as benign intermediates for the synthesis of fine chemicals and pharmaceutical products.12
To overcome the low reactivity of CO2 due to its inert nature and the high energy barriers in its interaction with epoxides, numerous catalysts and systems have been investigated and reported. These include well-designed metal complexes,13–16 metal–organic frameworks (MOFs),17,18 metalloporphyrins,19 ionic liquids,20–23 and halogen.24 Among these, ionic liquids have attracted significant interest due to their typically strong affinity for CO2 and the ease of structural modification, which allows for improved catalytic performance.25,26 Additionally, incorporating carboxy- or hydroxy-functional groups onto the cationic moieties facilitates intramolecular hydrogen bonding with epoxides, promoting activation for the desired ring-opening reaction, resulting in enhanced catalytic activity.21,27–29 Furthermore, the straightforward immobilization of ionic liquids onto suitable support materials offers advantages in terms of reusability and cost-effective production. Various modified support materials, such as silica gels,27,30–33 polymers,34–36 and zeolites,37 have been reported to exhibit remarkable catalytic activity, designability, and stability.
Compared to conventional batch methods, continuous-flow methodology has attracted significant interest from researchers in both academia and industry, particularly for the preparation of fine chemicals like pharmaceuticals, agrochemicals, polymers, and functional materials. In fact, continuous-flow methods have long been the standard in the petrochemical industry, proving to be the most efficient method for large-scale production.38–44 While homogeneous micro- and flash-flow chemistry offers exceptional efficiency, especially in reactions involving short-lived active species,45–48 the pursuit of carbon neutrality necessitates the development of heterogeneous catalytic processes for a broader range of chemical transformations under continuous-flow conditions.49 Heterogeneous catalytic flow reactions enable the automatic separation of products from the catalyst bed along with the solvent. Additionally, the high catalyst-to-substrate ratio within the column enhances catalytic activity compared to corresponding batch reactions, leading to increased catalyst turnover number (TON). Precise control over residence time is achievable by adjusting flow rate and column size, preventing undesirable overreactions during the synthesis process.50 Recent research from our group has demonstrated a remarkable acceleration effect, particularly for gas–liquid–solid reactions known as trickle-flow reactions, attributed to the direct contact of gas with the catalyst in the reactor bed.51
Turning to the synthesis of cyclic carbonates from CO2, researchers have explored the transition from batch to continuous-flow methods for synthesizing cyclic carbonates from CO2.52–61 For instance, Xiong et al. employed carbon-tethered dialkyl imidazolium ionic liquids (ILs) for continuous-flow synthesis, achieving successful conversion of epichlorohydrin to the corresponding cyclic carbonate at 140 °C and 1.4 MPa with an average conversion of 82% over 80 h.54 Recent studies by Sans and García-Verdugo,58 Bica-Schröder,59 and Yin60 further demonstrate the versatility of this methodology. However, challenges remain in the continuous-flow synthesis of cyclic carbonates, including the occasional need for high pressure, limited reactivity, and a narrow substrate scope. Additionally, studies comparing the performance of heterogeneous catalysts in batch and continuous-flow systems have often revealed decreased yield and insufficient catalyst durability under continuous-flow conditions.55–59,62,63 These findings suggest that the full potential of trickle-flow reactions has yet to be realized. Despite recent advances, high-productivity continuous-flow systems for cyclic carbonate synthesis still often rely on homogeneous catalysts, highlighting the challenges associated with using heterogeneous catalysts. For example, a benchmark study by Pericàs et al. employed immobilized amine catalysts for the synthesis of cyclic carbonates from glycerol derivatives, but the flow duration was limited to less than 48 h, and the turnover number (TON) was less than 100.63 Simply anchoring active sites on a support appears insufficient for achieving high catalyst stability and activity.64 We believe that directly modifying the polymer backbone is crucial for overcoming these limitations. In gas–liquid flow systems, the performance of heterogeneous catalysts can be significantly influenced by the positioning of polar active sites on the less-polar polymer backbone. To address this challenge, we prepared porous polymers with varying degrees of cross-linking in the presence of a porogen. By fine-tuning the active sites within these porous polymers, we developed catalysts suitable for continuous-flow cyclic carbonate synthesis with high stability (Fig. 1).
 | ||
| Fig. 1 General schematic image of this work. | ||
We initiated the design of a suitable polymer scaffold for gas–liquid–solid reactions and chose a styrene–divinylbenzene (DVB) backbone due to its synthetic scalability, chemical stability under reaction conditions, and affinity for both CO2 and substrate solutions. Methyl isobutylcarbinol (MIBC) was used as a porogen, with its amount set at 90 wt% relative to the total monomer weight.65,66 In this setup, the amount of crosslinking agent (DVB) relative to the other monomers was fixed at 8 wt%. To immobilize imidazolium moieties onto the scaffold, we adopted a post-modification method based on poly(4-vinylbenzyl bromide). Therefore, 4-vinylbenzyl bromide was used as a functionalized comonomer, and styrene served as the basic monomer. As outlined in Scheme 1, the installation of N-substituted imidazole, followed by anion exchange, provided the target butyl-tethered N-substituted imidazolium salt-functionalized porous polystyrene (BRImPS) catalysts. The bromide content in the polymer BBPS was determined by Volhard titration, and its structure was confirmed by infrared spectra and solid-state 13C CP-MAS NMR (see S9 in ESI‡). The structure of BHEImPS-4-890(Cl) was also determined by comparison with that of the parent bromobutyl polystyrene using IR and 13C CP-MAS NMR.
 | ||
| Scheme 1 Synthesis of polystyrene-supported imidazolium ionic liquid catalysts. | ||
Initially, we evaluated the impact of the ratio between the basic monomer and the functionalized comonomer on performance in the cycloaddition reaction using styrene oxide (1a) under batch conditions (Table 1). In this study, we fixed several functionalities: (i) N-methylimidazole was used to construct the imidazolium moiety; (ii) the cross-linking degree, defined by the amount of DVB used, was fixed at 8 wt%; and (iii) the amount of porogen MIBC was set to 90 wt% as a standard, as described previously. In this manner, four BMImPS-X-890(Br) catalysts were prepared with stepwise increasing styrene proportions. Their differences manifested in a trend resembling a volcano shape in yields of the desired styrene carbonate (2). Among them, BMImPS-4-890(Br), where the number 4 indicates the mole-equivalency of styrene to the functionalized comonomer, showed the highest yield (79%).
|
|
|||
|---|---|---|---|
| Entry | X [eq.] | Catalyst | Yieldc [%] |
| a Conditions: 1a, 1.0 mmol, CO2, 1.0 MPa. b 4-Bromobutyl styrene/styrene ratio. c Determined by GC analysis. d The polymer that was prepared from 4-bromobutyl styrene and DVB. e The porogen was not used in the preparation of the catalyst. | |||
| 1 | 0d | BMImPS-0-890(Br) | 58 |
| 2 | 2 | BMImPS-2-890(Br) | 69 |
| 3 | 4 | BMImPS-4-890(Br) | 79 |
| 4 | 8 | BMImPS-8-890(Br) | 74 |
| 5 | 4 | BMImPS-4-800(Br) | 66 |

Following the determination of the optimal monomer-to-comonomer ratio, we investigated the activity of BMImPS-4-800(Br) prepared without porogen. It exhibited a slightly lower yield of 66%. This highlights the importance of the porous structure for efficient substrate access to the catalytically active imidazolium moiety. One of the key advantages of this catalyst is its structural tunability, allowing for tailored catalytic performance for specific reactions. In this study, we extended our investigation beyond the polymer backbone by varying the spacer length between the polymer and the imidazolium group, as well as substituents on the imidazolium ring. A series of candidate catalysts are summarized in Fig. 2. Here, the monomer-to-comonomer ratio, crosslinking degree, and porogen amount were fixed at the optimal values determined previously. The corresponding counter anion is denoted at the end of each catalyst name. All prepared catalysts were evaluated under identical batch conditions in a model reaction, and the results are presented in Table 2.
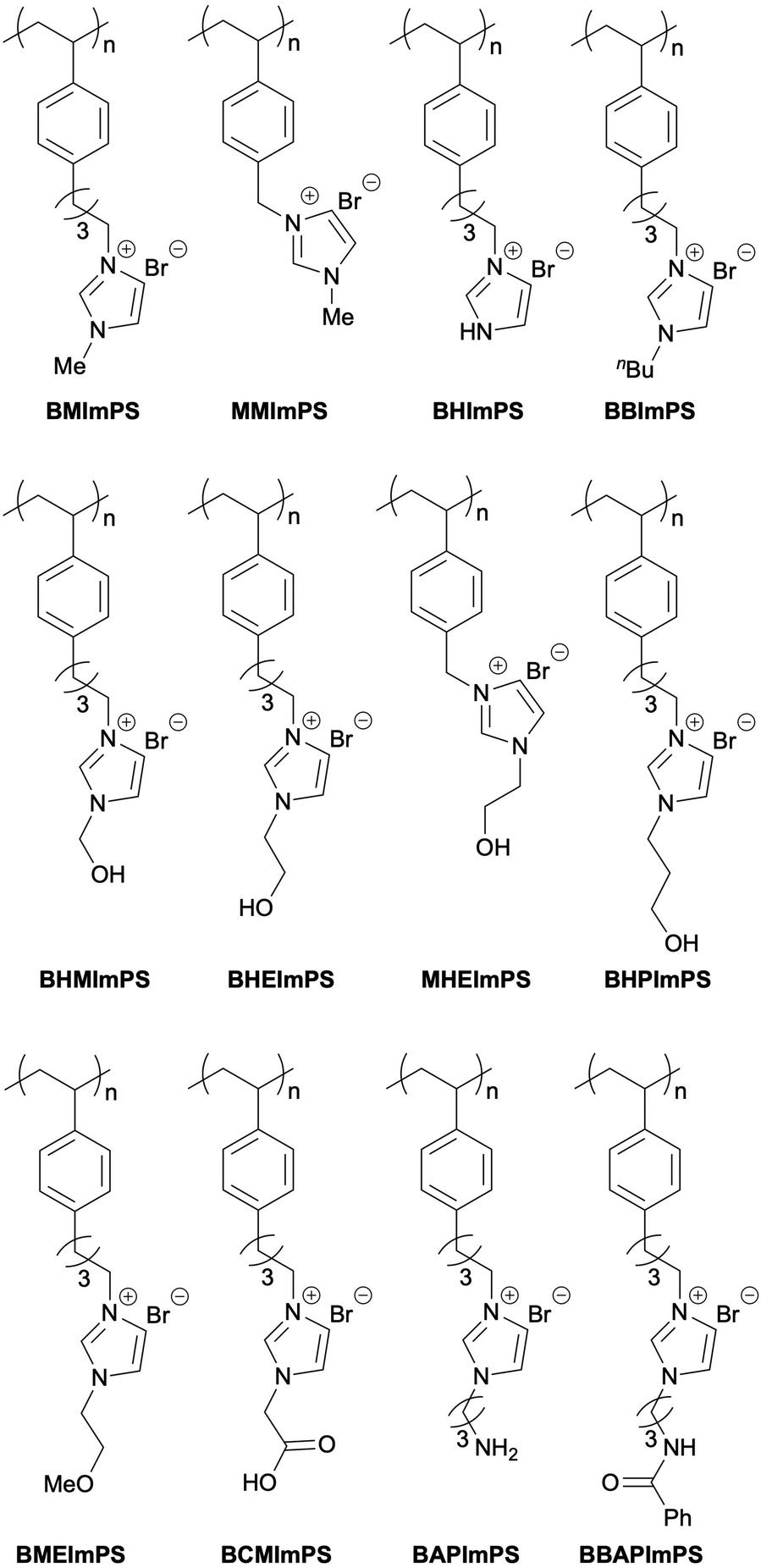 | ||
| Fig. 2 Polystyrene-supported imidazolium ionic liquid catalysts. | ||
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb [%] |
| a Conditions: 1a, 1.0 mmol, CO2, 1.0 MPa. b Determined by GC analysis. | ||
| 1 | BHImPS-4-890(Br) | 31 |
| 2 | BMImPS-4-890(Br) | 79 |
| 3 | MMImPS-4-890(Br) | 56 |
| 4 | BBImPS-4-890(Br) | 75 |
| 5 | BHMImPS-4-890(Br) | 53 |
| 6 | BHEImPS-4-890(Br) | 87 |
| 7 | BHPImPS-4-890(Br) | 70 |
| 8 | MHEImPS-4-890(Br) | 58 |
| 9 | BMEImPS-4-890(Br) | 82 |
| 10 | BCMImPS-4-890(Br) | 31 |
| 11 | BBAPImPS-4-890(Br) | 50 |
| 12 | BAPImPS-4-890(Br) | 55 |
| 13 | BHEImPS-4-890(PF6) | 7 |
| 14 | BHEImPS-4-890(OTf) | 12 |
| 15 | BHEImPS-4-890(I) | 80 |
| 16 | BHEImPS-4-890(Br) | 87 |
| 17 | BHEImPS-4-890(Cl) | 90 |
| 18 | BHEImPS-4-890(OCOCF3) | 31 |
| 19 | BHEImPS-4-890(OH) | 40 |
| 20 | BHEImPS-4-890(OAc) | 57 |

BHImPS-4-890(Br), lacking an imidazolium substituent, displayed low catalytic activity (entry 1). Compared to the standard BMImPS-4-890(Br) (entry 2), shortening the alkyl linker between the polymer backbone and the imidazolium ring from C4 to C1 (MMImPS-4-890(Br)) resulted in decreased yield (entry 3). A similar trend was observed when the imidazolium substituent was changed to a 2-hydroxyethyl (HE) group: BHEImPS-4-890(Br) achieved an 87% yield (entry 6), whereas MHEImPS-4-890(Br) yielded only 58% (entry 8). This difference can be attributed to the influence of the hydrophobic polymer backbone, which may be more pronounced with a shorter alkyl linker.
During the exploration of various functional groups on the imidazolium nitrogen, the hydroxyethyl (HE) group provided the best performance (entry 6). Conversely, hydroxymethyl (HM) and hydroxypropyl (HP) groups significantly decreased the yield (entries 5 and 7). Previous studies have reported a similar positive effect of hydroxyl groups on the cycloaddition of CO2 with epoxides to form cyclic carbonates, proposing hydrogen bonding between epoxides and the –OH group to activate the epoxide ring for subsequent C–O bond cleavage.32,67–71 However, the negligible impact of HM and HP functionalities (entries 5 and 7) and the relatively better performance of methoxyethyl (ME) (entry 9) and also BBImPS-4-890(Br) (entry 4) suggest that hydrogen bond donation is not the critical factor for catalysis. This is further supported by the negative effect of other acidic functionalities like carboxymethyl (CM) and benzoyl aminopropyl (BAP) groups (entries 10 and 11). The aminopropyl (AP) substituent, capable of interacting with CO2, also led to a decrease in yield (entry 12).
Further investigation focused on the anion component using variants of BHEImPS-4-890. The results in Table 2 (entries 13–20) indicate a strong influence of anion nucleophilicity on catalytic activity. The lowest yield observed with the least nucleophilic PF6− anion (entry 13) emphasizes the importance of nucleophilic character. Highly nucleophilic OH− or moderately nucleophilic AcO− resulted in yields of 40% or 57% (entries 19 and 20), possibly due to incompatibility with the hydroxyl group. Halide anions were effective, with yields increasing in the order I− < Br− < Cl− (entries 15–17). This effectiveness suggests that both appropriate nucleophilicity and leaving group ability are crucial for the catalytic cycle. Notably, BHEImPS-4-890(Cl), an improved catalyst, achieved a 90% yield in the model batch reaction (entry 17). These results provide valuable insights into the reaction mechanism, which will be discussed further (vide infra).
With the optimal catalyst in hand, we further applied it in a continuous-flow system. The basic setup of our continuous-flow cycloaddition reaction is illustrated in Scheme 2. A 10 mm (id) × 200 mm (L) stainless steel plug-flow reactor containing 0.23 g of the catalyst BHEImPS-4-890(Cl) was installed in an aluminum block column heater. Under standard conditions consisting of a heater temperature of 140 °C and a back pressure of 0.5 MPa(G), we initiated our continuous-flow investigation by examining the effect of molar flow (F), the amount of substrate provided per unit time, which is a key parameter for flow reactions (Fig. 3. See also S-6 in ESI‡). In the first set of experiments, shown by the three bars on the left end of the line, decreasing the F value by reducing the flow rate from 0.1 mL min−1 to 0.025 mL min−1 resulted in a better yield of 72% at the lowest F value. The same yield was obtained under much higher pressure conditions; however, at this back pressure, conversely, better yields of 84 or 83% were obtained in operations with higher F, using a more concentrated substrate solution. Keeping the F value at 0.30 mmol h−1 and the catalyst amount constant, we employed a larger reactor to extend the residence time. Here, the estimated liquid residence time for the catalyst-packed reactors with inner volumes of 15.7 or 22.4 mL were 280 and 420 min, respectively. The yield of cyclic carbonate was increased to 90%. Trials to lower the column temperature from 140 °C to 110 or 80 °C resulted in lower product yields. To further reduce the environmental impact, we conducted the flow reaction under solvent-free conditions. Under the optimal conditions screened above, the yield of cyclic carbonate could reach 53%, and the STY value remained around 18 mmol h−1 dL. Having established the optimal flow conditions, we proceeded to explore the substrate scope of this system by using various epoxides. As demonstrated in Fig. 4, epoxides bearing mono-substituents readily underwent the reaction, yielding the desired products in good to excellent yields (72–96%), with the exception of 1-hexene oxide. A general trend can be observed: functional groups such as hydroxyl groups, ethers, and halogens were not affected in this reaction, and all resulted in high-yield products. We tested three types of styrene oxide derivatives with different electronic features, and found that only the electron-donating 4-methylstyrene oxide gave a slightly lower yield. Additionally, several di-substituted epoxides were examined as well; however, their low yields indicated their marked inactivity. Similarly, tetrahydrofuran (yielding 2p) exhibited no activity in the reaction,72 possibly due to the higher stability of the five-membered ring ether. We are actively working on expanding the applicability of our system to a wider range of substrates, including internal and gem-disubstituted epoxides.
 | ||
| Scheme 2 Continuous-flow system. | ||
 | ||
| Fig. 3 Optimization of continuous-flow conditions. | ||
 | ||
| Fig. 4 Substrate scope in continuous-flow cyclic carbonate synthesis. Isolated yields except for 2n, 2o, and 2p (determined by 1H-NMR analysis). | ||
The cycloaddition reaction between styrene oxide and CO2 was investigated using BHEImPS-4-890(Cl) as a catalyst under extended operation time (Fig. 5). The estimated amount of catalytically active imidazolium sites in the reactor was 0.2 mmol. A 0.2 M solution of styrene oxide was continuously flowed at a rate of 0.025 mL min−1 (F = 0.3 mmol h−1, SV = 1.5 h−1). The CO2 flow rate was maintained at 5 mL min−1, and the back pressure was kept between 1.0 and 1.1 MPa(G). The reaction mixture was heated to 140 °C using an aluminum column heater and sustained for 168 h. The yield remained within the range of 84% to 97%, 91% yield in average, throughout the operation, demonstrating no significant activity loss. The estimated TON, exceeding 229, and the unchanged yield and turnover frequency (TOF) during the 180 h reaction highlight the high activity and exceptional stability of the catalyst, respectively.
 | ||
| Fig. 5 Extended-time operation for the cycloaddition of 1a and CO2. Conditions: 1a, 0.2 M; back pressure, 1.0–1.1 MPa; heater temp., 140 °C; 15.7 mL stainless reactor was utilized. Symbols: red circle, yield of 2a; green triangle, TON based on the amount of imidazolium moiety in the reactor. | ||
The mechanism of the BHEImPS-4-890(Cl) catalyst-mediated cycloaddition between epoxide and CO2 is proposed based on experimental results, assumptions, and insights from previous reports32,69,70,73–78 and illustrated in Fig. 6. Hydrogen bonding between the epoxide oxygen and the hydroxyl group of the catalyst likely activates the epoxide (intermediate I), facilitating nucleophilic attack by the counter anion and subsequent ring opening. The resulting intermediate III, which possesses a highly nucleophilic alkoxide anion, then reacts with CO2 to form a carbonate anion. Intramolecular nucleophilic attack within this intermediate completes the cyclization, leading to styrene carbonate formation. The effectiveness of imidazolium polymers lacking a hydroxyl group (e.g., BBImPS-4-890(Br) and BMEImPS-4-890(Br), see Table 2, entries 4 and 9) suggests the existence of an alternative catalytic mechanism that doesn't involve hydrogen bonding. This alternative mechanism likely relies on key features like the cationic imidazolium nitrogen and the nucleophilicity/leaving ability of the halides. Intermediate II in Fig. 6 represents a possible non-covalent interaction in this pathway. As mentioned earlier (Table 2), counter anions of the catalysts dramatically influenced their activities. According to the proposed mechanism, halide anions with moderate nucleophilicity and leaving group ability, particularly chloride, exhibit superior catalytic activity. This behavior is consistent with the proposed mechanism. Further investigations into a more detailed reaction mechanism, including the rate-determining step, are ongoing.
 | ||
| Fig. 6 Possible catalytic cycle. | ||
In summary, this work showcases the application of meticulously optimized imidazolium-based ionic liquids immobilized on a polymer scaffold for the continuous-flow synthesis of cyclic carbonates from epoxides and CO2. We prepared and evaluated a library of supported catalysts by varying the monomer/functionalized comonomer ratio, porogen amount, type of catalytically active moiety, and counterion. Among these, the BHEImPS-4-890(Cl) catalyst demonstrated outstanding catalytic performance. Continuous-flow experiments revealed its ability to efficiently produce various mono-substituted cyclic carbonates under optimized reaction conditions. Notably, extended operation for over 160 h maintained a consistent yield of over 90%, highlighting its exceptional stability. Future studies will focus on further enhancing catalytic activity and exploring its potential as a CO2 conversion technology within the framework of Carbon Capture, Utilization, and Storage (CCUS).
Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research from the New Energy and Industrial Technology Development Organization (NEDO) project Grant No. JPNP19004.References
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. MacDowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- International Energy Agency Report, CO2 Emissions in 2022, https://www.iea.org/reports/co2-emissions-in-2022.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC.
- M. Usman, A. Rehman, F. Saleem, A. Abbas, V. C. Eze and A. Harvey, RSC Adv., 2023, 13, 22717–22743 RSC.
- T. Ema, Bull. Chem. Soc. Jpn., 2023, 96, 693–701 CrossRef CAS.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- F.-T. Zangeneh, S. Sahebdelfar and T. T. Ravanchi, J. Nat. Gas Chem., 2011, 20, 219–231 CrossRef CAS.
- G.-C. Chung, H.-J. Kim, S.-H. Jun and M.-H. Kim, Electrochem. Commun., 1999, 1, 493–496 CrossRef CAS.
- Y. Hyun, H.-K. Park, H. S. Park and C.-S. Lee, J. Nanosci. Nanotechnol., 2015, 15, 8951–8960 CrossRef CAS PubMed.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Catal. Lett., 2003, 89, 81–85 CrossRef CAS.
- Y.-M. Shen, W.-L. Duan and M. Shi, Eur. J. Org. Chem., 2004, 3080–3089 CrossRef CAS.
- A. J. Kamphuis, F. Milocco, L. Koiter, P. P. Pescarmona and E. Otten, ChemSusChem, 2019, 12, 3635–3641 CrossRef CAS PubMed.
- Q. Yao, Y. Shi, Y. Wang, X. Zhu, D. Yuan and Y. Yao, Asian J. Org. Chem., 2022, 11, e202200106 CrossRef CAS.
- O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhinimaev and V. P. Fedin, J. Catal., 2013, 298, 179–185 CrossRef CAS.
- Z. Gao, L. Kiang, X. Zhang, P. Xu and J. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 61334–61345 CrossRef CAS PubMed.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304–1309 CrossRef CAS.
- W.-L. Wong, L. Y. K. Lee, K.-P. Ho, Z.-Y. Zhou, T. Fan, Z. Lin and K.-Y. Wong, Appl. Catal., A, 2014, 472, 160–166 CrossRef CAS.
- C. Yue, D. Su, X. Zhang, W. Wu and L. Xiao, Catal. Lett., 2014, 144, 1313–1321 CrossRef CAS.
- S. Yue, P. Wang and X. Hao, Fuel, 2019, 251, 233–241 CrossRef CAS.
- C. Li, F. Liu, T. Zhao, J. Gu, P. Chen and T. Chen, Mol. Catal., 2021, 511, 111756 CrossRef CAS.
- J. A. Kozak, J. Wu, X. Su, F. Simeon, T. A. Hatton and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 18497–18501 CrossRef CAS PubMed.
- C. Cadena, J. L. Anthony, J. K. Shah, T. I. Morrow, J. F. Brennecke and E. J. Maginn, J. Am. Chem. Soc., 2004, 126, 5300–5308 CrossRef CAS PubMed.
- A. S. Martinez, C. Hauzenberger, A. R. Sahoo, Z. Csendes, H. Hoffmann and K. Bica, ACS Sustainable Chem. Eng., 2018, 6, 13131–13139 CrossRef.
- W.-L. Dai, L. Chen, S.-F. Yin, W.-H. Li, Y.-Y. Zhang, S.-L. Luo and C.-T. Au, Catal. Lett., 2010, 137, 74–80 CrossRef CAS.
- S. Wu, B. Wang, Y. Zhang, E. H. M. Elageed, H. Wu and G. Dao, J. Mol. Catal. A: Chem., 2016, 418–419, 1–8 CAS.
- T. Wang, X. Zhu, L. Mao, T. Ren, L. Wang and J. Zhang, J. Mol. Liq., 2019, 296, 111936 CrossRef CAS.
- W.-L. Dai, L. Chen, S.-F. Yin, S.-L. Luo and C.-T. Au, Catal. Lett., 2010, 135, 295–304 CrossRef CAS.
- W. Cheng, X. Chen, J. Sun, J. Wang and S. Zhang, Catal. Today, 2013, 200, 117–124 CrossRef CAS.
- T. Ying, Q. Su, Z. Shi, L. Deng, W. Cheng and W. Hua, Catal. Lett., 2019, 149, 2647–2655 CrossRef CAS.
- A. Siewniak, A. Forajter and K. Szymanska, Catalysts, 2020, 10, 1363 CrossRef CAS.
- L. Han, H.-J. Choi, D.-K. Kim, S.-W. Park, B. Liu and D.-W. Park, J. Mol. Catal. A: Chem., 2011, 338, 58–64 CAS.
- Y. Zhang, E.-S. M. El-Sayed, K. Su, D. Yuan and Z. Han, J. CO2 Util., 2020, 42, 101301 CrossRef CAS.
- W. Zhang, F. Ma, L. Ma, Y. Zhou and J. Wang, ChemSusChem, 2020, 13, 341–350 CrossRef CAS PubMed.
- L. Guo, X. Jin, X. Wang, L. Yin, Y. Wang and Y.-W. Yang, Molecules, 2018, 23, 2710 CrossRef PubMed.
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejuene, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem. – Eur. J., 2014, 20, 15226–15232 CrossRef CAS PubMed.
- I. R. Baxendale, R. D. Braatz, B. K. Hodnett, K. F. Jensen, M. D. Johnson, P. Sharratt, J.-P. Sherlock and A. J. Florence, J. Pharm. Sci., 2015, 104, 781–791 CrossRef CAS PubMed.
- S. Kobayashi, Chem. – Asian J., 2016, 11, 425–436 CrossRef CAS PubMed.
- H. Ishitani, Y. Saito and Y. S. Kobayashi, Top. Organomet. Chem., 2016, 57, 213–248 CrossRef CAS.
- M. Kandasamy, B. Ganesan, M.-Y. Hung and W.-Y. Lin, Eur. J. Org. Chem., 2019, 3183–3189 CrossRef CAS.
- W. J. Yoo, H. Ishitani, Y. Saito, B. Laroche and S. Kobayashi, J. Org. Chem., 2020, 85, 5132–5145 CrossRef CAS PubMed.
- S. B. Ötvös and C. O. Kappe, Green Chem., 2021, 23, 6117–6138 RSC.
- J. Yoshida, Flash Chemistry - Fast Organic Synthesis in Micro Systems, WILEY-VCH, Weinheim, 2008 Search PubMed.
- J. Yoshida, A. Nagaki and T. Yamada, Chem. – Eur. J., 2008, 14, 7450–7459 CrossRef CAS PubMed.
- J. Yoshida, Chem. Rec., 2010, 10, 332–341 CrossRef CAS PubMed.
- J. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- S. Asano, H. Miyamura, M. Matsushita, S. Kudo, S. Kobayashi and J.-I. Hayashi, J. Flow Chem., 2023, 14, 329–335 CrossRef.
- H. Seo, L. V. Nguyen and T. F. Jamison, Adv. Synth. Catal., 2019, 361, 247–264 CrossRef CAS.
- A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 105113 CrossRef CAS.
- Y. Zhang, Z. Tan, B. Liu, D. Mao and C. Xiong, Catal. Commun., 2015, 68, 73–76 CrossRef CAS.
- T. Wang, W. Wang, Y. Lyu, X. Chen, C. Li, Y. Zhang, X. Song and Y. Ding, RSC Adv., 2017, 7, 2836–2841 RSC.
- A. S. Martinez, C. Hauzenberger, A. R. Sahoo, Z. Csendes, H. Hoffmann and K. Bica, ACS Sustainable Chem. Eng., 2018, 6, 13131–13139 CrossRef.
- D. Valverde, P. Porcar, P. Lozano, E. García-Verdugo and S. V. Luis, ACS Sustainable Chem. Eng., 2021, 9, 2309–2318 CrossRef CAS.
- D. Valverde, P. Porcar, M. Zanatta, S. Alcalde, B. Altaba, V. Sans and E. García-Verdugo, Green Chem., 2022, 24, 3300–3308 RSC.
- P. Mikšovsky, E. N. Horn, S. Naghdi, D. Eder, M. Schnürch and K. Bica-Schröder, Org. Process Res. Dev., 2022, 26, 2700–2810 CrossRef PubMed.
- X. Li, J. Sun, M. Xue and J. Yin, J. CO2 Util., 2022, 64, 102168 CrossRef CAS.
- T. Q. Bui, L. J. Konwar, A. Samikannu, D. Nikjoo and J.-P. Mikkola, ACS Sustainable Chem. Eng., 2020, 8, 12582–12869 Search PubMed.
- N. Zanda, L. Zhou, E. Alza, A. W. Kleij and M. À. Pericàs, Green Chem., 2022, 24, 4628–4633 RSC.
- N. Zanda, A. Sobolewska, E. Alza, A. W. Kleij and M. À. Pericàs, ACS Sustainable Chem. Eng., 2021, 9, 4391–4397 CrossRef CAS.
- C. Mizyka, S. Renson, B. Grignard, C. Detrembleur and J.-C. M. Monbaliu, Angew. Chem., Int. Ed., 2024, 63, e202319060 CrossRef PubMed.
- M. Tomoi and W. T. Ford, J. Am. Chem. Soc., 1981, 103, 3828–3832 CrossRef CAS.
- Y.-F. Wang, B.-H. Xu, Y.-R. Du and S.-J. Zhang, Mol. Catal., 2018, 457, 59–66 CrossRef CAS.
- J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- J. Sun, W. Cheng, W. Fan, W. Wang, Z. Meng and S. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS.
- X. Chen, J. Sun, J. Wang and W. Cheng, Tetrahedron Lett., 2012, 53, 2684–2688 CrossRef CAS.
- M. Zhang, B. Chu, G. Li, J. Xiao, H. Zhang, Y. Peng, B. Li, P. Xie, M. Fan and L. Dong, Microporous Mesoporous Mater., 2019, 274, 363–372 CrossRef CAS.
- Y. Liu, J. Li, Z. Zhang, Y. Hou and J. Zhang, Inorg. Chem., 2022, 61, 17438–17447 CrossRef CAS PubMed.
- E. Jie and W. Zhang, CN103058862A, 2012.
- D.-W. Kim, R. Roshan, J. Tharun, A. Cherian and D.-W. Park, Korean J. Chem. Eng., 2013, 30, 1973–1984 CrossRef CAS.
- V. B. Saptal and B. M. Bhanage, Curr. Opin. Green Sustainable Chem., 2017, 3, 1–10 CrossRef.
- A. A. Marciniak, K. J. Lamb, L. P. Ozorio, C. J. A. Mota and M. North, Curr. Opin. Green Sustainable Chem., 2020, 26, 100365 CrossRef.
- M. Liu, F. Wang, L. Shi, L. Liang and J. Sun, RSC Adv., 2015, 5, 14277–14285 RSC.
- S. Denizalti, RSC Adv., 2015, 5, 45454–45458 RSC.
- Z. Guo, Y. Hu, S. Dong, L. Chen, L. Ma, Y. Zhou, L. Wang and J. Wang, Chem. Catal., 2022, 2, 519–530 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Koichi Narasaka on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04499a |
| This journal is © The Royal Society of Chemistry 2024 |