
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWaterborne polymers and coatings from bio-based butenolides†
Andries
Jensma
 a,
Niels
Elders
*b,
Keimpe J.
van den Berg
b and
Ben L.
Feringa
*a
a,
Niels
Elders
*b,
Keimpe J.
van den Berg
b and
Ben L.
Feringa
*a
aStratingh Institute for Chemistry, Advanced Research Center Chemical Building Blocks Consortium (ARC CBBC), University of Groningen, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bDepartment Resin Technology, Akzo Nobel Car Refinishes BV, 2171 AJ Sassenheim, The Netherlands. E-mail: niels.elders@akzonobel.com
First published on 20th August 2024
Abstract
In the quest for sustainable paints and coatings, bio-based resources for the polymeric binder constituents are key. Recently, we introduced poly-butenolides as bio-based acrylate replacement for solventborne and 100% solids (UV-curing) coatings. Here, we report the first step towards aqueous poly-butenolide dispersions, enabling the use of this novel binder technology platform in waterborne coatings.
Facing the challenge to lower its environmental impact, the paints and coatings industry has made significant progress over the last decades.1 For example, by increasing the efficiencies of products & processes (i.e., reducing waste/energy requirements) and by improving the coating durability at lower film-thicknesses. In addition, the content of harmful components has been reduced in the formulations (e.g. lowering/eliminating solvents and replacing poisonous pigments & additives).
The sustainability of paints & coatings can be increased further by switching from petrochemical to bio-based feedstocks, ultimately enabling a negative CO2 emission for the polymeric binder constituent.2 Although the first binders for paints were completely derived from bio-based feedstocks (e.g. linseed oil, tree resins and shellac), the petrochemical industry enabled the development of synthetic resin technologies with superior performance (e.g. polyester, polyacrylic, polyurethane, and epoxy-based resins). Developing coatings using bio-based resources, one strategy is to pursue the same commodity monomers as derived from fossil supply.3,4 This route currently facilitates the production of identical paints with a targeted bio-based content. Unfortunately, additional process steps accompanied with transforming biomass to drop-in materials increases the price, making the commercial viability of bio-based paints challenging when oil prices are low. Alternatively, the unique (heteroatom) functionalities of bio-based materials may provide polymers with inherently different architectures possibly leading to performance benefits which could accelerate the transition to bio-based coatings.4,5
Taking advantage of these opportunities, our group recently used furfural (1) (obtained from cheap xylan-rich lignocellulosic biomass6) in the synthesis of butenolide co-polymers (5)7 which represents a novel polymeric backbone for the coatings industry, potentially replacing acrylates (Fig. 1).
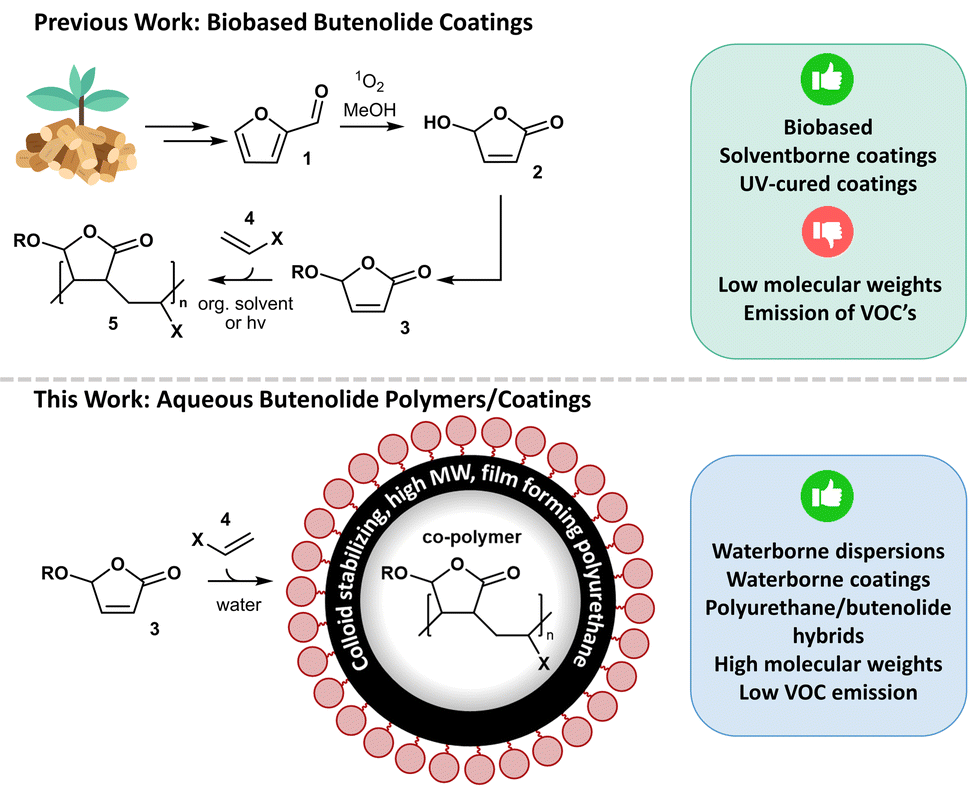 | ||
| Fig. 1 Previous work: biobased coating binder technology based on poly-butenolides using photo-oxidation, condensation, and co-polymerization.6,10–12 This work: waterborne polyurethane/poly-butenolide hybrid dispersions. | ||
Furfural is first converted to hydroxy butenolide (2) using an environmentally benign photo-chemical reaction with singlet oxygen.8 Hydroxy butenolide is subsequently converted using a condensation reaction with (bio-based) alcohols, allowing the synthesis of a wide variety of alkoxy butenolide monomers (3, R = alkyl).9 Subsequent co-polymerization with vinyl monomers (vinyl esters, ethers and lactams) was performed using free radical polymerization conditions.7,10
In recent studies we: (i) improved the efficiency of hydroxy butenolide (2) synthesis by optimising the photo-flow chemistry conditions,11 (ii) demonstrated a fully bio-based carbon resin technology,12 (iii) increased the monomer scope to acyloxy- (R = (CO)R′), carbonoxy- (R = (CO)OR′), and carbamoxy butenolides (R = (CO)NR′R′′) which significantly increased the rate of polymerization,13 and (iv) elucidated that the co-polymerization process occurs in an alternating fashion, using reaction kinetics experiments and DFT calculations.13
Various examples of solventborne (low molecular weight) and 100% solids/UV-curing (monomeric) bio-based butenolide coatings have been developed by our group in recent studies.14 However, to extend the scope of coating segments which can benefit from our novel binder technology, applying water as mobile phase is key. Waterborne coatings are more benign for the painter and the environment because they limit the emission of volatile organic compounds (VOC's) to the atmosphere and have a reduced flammability. In addition, higher molecular weight polymers can be targeted in waterborne dispersions, which further reduces the hazard profile of the binder component compared to its monomers/oligomers. In this paper we will present, to the best of our knowledge, the first synthetic method to obtain aqueous butenolide based co-polymers and coatings. Initially we focused on conventional emulsion polymerization, but unstable dispersions were observed. Switching to a different system, using hybrid dispersions with high molecular weight (MW) polyurethane (PU) as colloid stabilizing moiety,15 changed the outcome of the experiments to stable dispersions.
The PU/poly-butenolide hybrid dispersions were synthesized at a 60/40 weight ratio according to the procedure depicted in Fig. 2. First, a carboxylic acid and isocyanate functional PU pre-polymer (9) was produced by reacting dimethylolpropionic acid (6, DMPA) and poly(tetramethylene ether) glycol (Mn 1000 g mol−1) (7, PTMEG) with isophorone diisocyanate (8, IPDI). For the latter two constituents, bio-based supply is available on industrial scale (from bio-butanediol/tetrahydrofuran16 and bio-acetone,17 respectively). The obtained low molecular weight PU (9) was diluted to 80% non-volatiles by the addition of 20 wt% dipropylene glycol dimethyl ether (DPGDME). The resulting viscous solution was further diluted with butenolide-monomer (3) + co-monomer (4) (at equimolar ratio) at a 9/monomers weight ratio of 60/40. Emulsification was accomplished by neutralization of the pendant carboxylic acid groups using triethyl amine (TEA) followed by transfer of the viscous polymer solution to a reactor containing water while stirring. The molecular weight of the PU was increased by coupling of the remaining isocyanate groups using ethylene diamine (EDA) followed by radical polymerization of 3 + co-monomer by the addition of a tBuOOH/FeII/sodium ascorbate redox couple (two subsequent additions, 30 min interval). The resulting products are expected to form spherical particles of 30–100 nm consisting of high MW polyurethane (10) and high MW butenolide co-polymer (5), based on previous acrylate based PU dispersions.15 (Part of) the PU will be positioned at the particle surface to enable the required electrostatic stabilisation. The interior of the particle is either phase separated to e.g. a core/shell morphology or phase mixed forming an interpenetrated network.15 Finally, the DPGDME solvent may be present inside the dispersed particle but can also partially have migrated to the aqueous continuous phase.
 | ||
| Fig. 2 Methodology for producing aqueous polyurethane/poly-butenolide hybrid dispersions. | ||
As control experiments, two dispersions were made applying the procedure described above, using isobutyl methacrylate (iBMA) and methyl isobutylketone (MiBK) representing a polymethacrylate analogue and a polyurethane dispersion (PUD) variant containing a hydrophobic solvent. Next, 15 unique PU/poly-butenolide hybrid dispersions were synthesized differing in the butenolide and co-monomer constituent. Three alkoxy- (3a–c) and two acyloxy butenolides (3d–e) were evaluated all in combination with a vinyl ester (vinyl neononanoate, VeoVa 9), vinyl ether (butyl vinylether, BVE), and vinyl lactam (N-vinyl pyrrolidone, NVP). For clarity reasons, Table 1 only displays a summary of the results. For complete and detailed analysis, we would like to refer to the ESI (S36†). Reference experiment 1 (using iBMA) demonstrated a reaction exotherm of +8 °C in 5 min during the first radical initiation phase while no exotherm is observed in the second. This indicates that the methacrylate homo-polymerization using this process is (nearly) fully completed in the first radical polymerization stage. A volume-weighted mean particle diameter (d43) of 50 nm was obtained for this dispersion and a pH of 7.7, which is a typical value of these type of dispersions (pH 7–8). The measured solid content (SC) corresponds very well with the expected value (27.4%) considering water, DPGDME and TEA are the only volatile constituents in the final product (i.e. all volatile iBMA is converted to non-volatile poly-iBMA) (Table 1).
| Monomer | Co-monomera | Exothermb (°C) | Solid contentc (%) | pHc |
d
43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (nm) c (nm) |
Storage stabilityd | |
|---|---|---|---|---|---|---|---|
| 1st shot | 2nd shot | ||||||
| a iBMA = isobutyl methacrylate, MiBK = methyl isobutylketone, VeoVa 9 = vinyl neononanoate, BE = butyl vinylether, and NVP = N-vinylpyrrolidone. b Reaction exotherm measured 30 min after radical initiator redox couple is added, unless noted differently. c Measured after 1 day storage at room temperature. d Storage stability is further discussed in ESI.† | |||||||
| iBMA (ref. 1)b | — | +7.9 (5 min) | — | 27.3 | 7.7 | 50 | Good |
| MiBK (ref. 2)b | — | — | — | 16.7 | 8.1 | 44 | Good |

|
VeoVa 9 | +2.3 | +0.8 | 26.3 | 6.5 | 77 | Destabilized (1 week) |
| BVE | +3.9 | +0.5 | 25.7 | 6.6 | 138 | Destabilized (1 month) | |
| NVP | — | +0.4 | 25.1 | 6.4 | 260 | Destabilized (1 week) | |
|
VeoVa 9 | +1.0 | +1.4 | 27.6 | 7.5 | 99 | Good |
| BVE | +3.6 | +0.8 | 27.0 | 7.7 | 86 | Good | |
| NVP | +5.6 (20 min) | — | 27.8 | 7.3 | 30 | Good | |

|
VeoVa 9 | +0.9 | +0.9 (25 min) | 27.2 | 7.5 | 64 | Good |
| BVE | +1.4 | +1.6 | 26.8 | 7.5 | 65 | Good | |
| NVP | +4.4 (20 min) | — | 27.1 | 7.5 | 37 | Good | |
|
VeoVa 9 | +2.9 (15 min) | +0.4 (10 min) | — | — | — | Destabilized (1 day) |
| BVE | +6.3 (5 min) | — | 26.3 | 6.5 | 137 | Destabilized (1 month) | |
| NVP | — | +0.4 (15 min) | — | — | — | Destabilized (1 day) | |

|
VeoVa 9 | +2.1 | +0.5 (7 min) | 27.0 | 7.2 | 75 | Good |
| BVE | +5.3 (11 min) | — | 26.8 | 7.2 | 62 | Good | |
| NVP | +7.1 (4 min) | — | 27.3 | 7.1 | 40 | Good | |
For reference experiment 2 (using MiBK), a similar particle size and pH was obtained. Again, the measured SC perfectly corresponds to the expected value because volatile MiBK will not polymerize. The match in predicted and measured solid content for the two reference experiments showed that these measurements provide a rough estimate of monomer conversion.‡
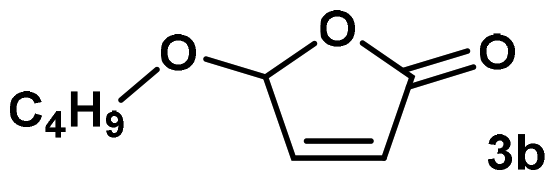
Applying the synthetic procedure using methoxy butenolide (3a) unfortunately resulted in poor results. Although the dispersions could be synthesized, the measured solid content for all three co-monomers were 1.1–2.3% lower than expected, indicating incomplete monomer conversions (90–80% conversion, respectively). The measured pH's were lower and the particle size using BVE and NVP significantly larger, compared to the reference experiments. Consequently, the PU hybrid dispersions using 3a/VeoVa 9 and 3a/NVP proved unstable during RT storage leading to complete phase separation within one week. The combination 3a/BVE did withstand one month RT storage, however, the particle size grew from 138 to 164 nm, indicating that also this hybrid dispersion suffered from some particle instability.
Much to our delights, replacing methoxy butenolide for n-butoxy- (3b) and n-hexyloxy butenolide (3c) completely changed the experimental outcome advantageous (Table 1). Stable dispersions which easily tolerated one month RT storage were obtained with solid contents, pH's and particle sizes much closer to those of reference experiment 1.§ Based on the measured reaction exotherms it can be concluded that the polymerization rate is increasing in the order VeoVa 9 < BVE < NVP which is in line with our reported findings on co-polymerization rates in solution.13 Clearly, the alkoxy chain-length has an important influence on monomer conversion and dispersion stability. Based on previous investigations this cannot be explained by differences in reactivity and is most likely caused by the hydrophobic alkyl chains protecting the acetal moiety at the (poly-)butenolide backbone from hydrolysis.
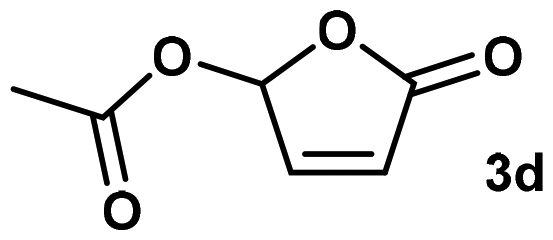
With these promising results in hand, we investigated acyloxy butenolides (3d–e), in which similar trends were found. Acetoxy butenolide (3d) gave rather poor results comparable to methoxy butenolide (3a). The initial properties of combinations 3d/VeoVa 9 and 3d/NVP could not even be measured because they did not resist overnight storage while the dispersion particles using combination 3d/BVE grew from 137 to 286 nm over one month. However, a longer acyloxy chain, with butenolide 3e, again resulted in perfectly stable hybrid dispersions over one month RT storage. Radical polymerization of combination 3e/NVP resulted in a peak reaction exotherm of +7 °C in only 4 min, which confirmed the previously observed faster reaction rate of acyloxy butenolides compared to their alkoxy derivatives.13
Encouraged by the promising results with the aforementioned liquid, relatively hydrophobic, butenolide monomers we opted to remove the solvent (DPGDME) and increase the butenolide co-polymer content in the hybrid dispersions (Table 2). The combination 3b/NVP was selected for this investigation, applying the same experimental procedure and simply removing the solvent, without correcting for the increased solid content (31.5 wt% expected at full monomer conversion). The hybrid dispersions were made at PU/butenolide co-polymer weight ratios of 60/40, 50/50, and 40/60 (see Table 2). Comparing the 60/40 co-polymer ratio result from Table 2 with the 3b/NVP result from entry 1 (with DPGDME) it can be concluded that eliminating the solvent resulted in a slightly lower slope of the reaction exotherm. In addition, a very small exotherm could be measured during the second initiation stage indicating that the rate of polymerization is slightly reduced and there was a minor amount of unreacted monomer left after the first 30 min of radical polymerization. Naturally, by increasing the 3b + NVP content, and thus the bio-based content, in the hybrid dispersion from 40 → 50 → 60 wt%, an increased reaction exotherm slope was observed but also an increased exotherm in the second initiation phase. The solvent-free hybrid dispersions all had similar solid contents and pH, and as expected, the particle size was growing at increased poly-butenolide content since the hydrophilic PU (surfactant) concentration is reduced.
| PU/(3b + NVP) wt ratio | Exotherma (°C) | 1 day storage | 1 month storage | |||||
|---|---|---|---|---|---|---|---|---|
| 1st shot | 2nd shot | Solid content (%) | pH | d 43 (nm) | Solid content (%) | pH | d 43 (nm) | |
| a Reaction exotherm measured 30 min after radical initiator redox couple is added, unless noted differently. | ||||||||
| 60/40 (with DPGDME) | +5.6 (20 min) | — | 27.6 | 7.3 | 30 | 27.6 | 7.1 | 37 |
| 60/40 | +5.3 | +0.1 (3 min) | 31.1 | 7.1 | 42 | 31.4 | 6.8 | 40 |
| 50/50 | +6.3 | +0.6 (5 min) | 31.4 | 7.0 | 41 | 31.4 | 6.8 | 48 |
| 40/60 | +6.9 | +2.4 (7 min) | 31.3 | 7.0 | 67 | 31.5 | 6.6 | 69 |
To gain insight into the morphology of our particles, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) measurements were performed. Exposing the dispersion particles to phosphotungstic acid (PTA), followed by drying and analysis revealed PTA-staining predominantly occured at the shell of the hybrid dispersion particles suggesting a core/shell particle morphology (Fig. 3, top row).
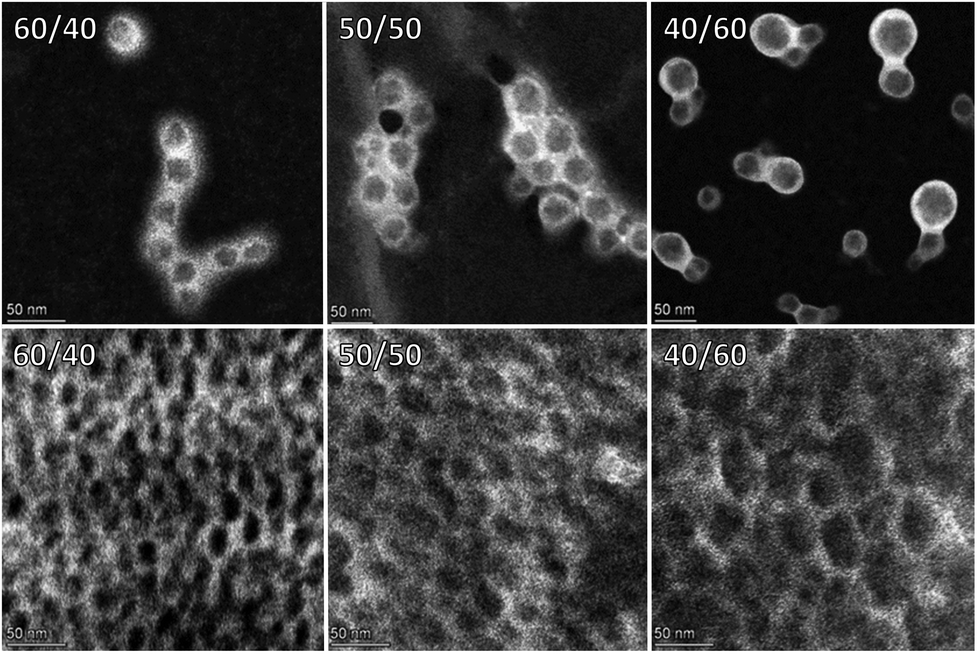 | ||
| Fig. 3 HAADF-STEM imaging of PTA stained solvent-free hybrid dispersion particles (top) and coating films (bottom) where the weight ratios PU/(3b/BE) are respectively 60/40, 50/50, and 40/60. | ||
As expected, molecular weight distribution (MWD) analysis of the dispersions revealed significantly higher Mw's (>150000 g mol−1)§ compared to our previously synthesized poly-butenolides in solvent,7,12–14 due to longer chains of butenolide co-polymer are synthesized but also due to present PU polymer. The hybrid dispersions show most often a bimodal distribution, which probably originates from two polymeric backbones dispersed into one particle.
Having established the key features of our butenolide dispersions, we proceeded to the formation of waterborne coatings. First, the dispersion was applied uniformly to a glass plate, followed by air-drying to form a clear hard coating (Fig. 4). Preliminary evaluation of the produced dispersions from Tables 1 & 2 as coatings demonstrated that the physical properties are promising for further developments. Clear, defect free films were obtained with water/solvent resistances, and hardnesses often exceeding the values of films obtained from reference experiment 1 with iBMA.§ Besides the poly-butenolide, also the polyurethane design can be altered in many ways enabling the development of tailor-made properties for many coating applications using our waterborne hybrid dispersion technology. Finally, cross section imaging of the corresponding coatings with HAADF-STEM after PTA exposure revealed the presence of unstained domains in a stained continues matrix (Fig. 3, bottom row) supporting the assumed core/shell morphology of the original hybrid particles.
 | ||
| Fig. 4 Coating of PU/(3b + NVP) dispersion with a ratio of 40/60. (A) Application of dispersion on glass plate. (B) In front of black/white paper after application but before drying. (C) Clear, hard coating in front of window after drying. | ||
Since the unstained domains are increasing in size with increasing 3b + NVP content, these domains are probably rich in poly-butenolide. Increasing the 3b + NVP content in the coatings led to an increase in Knoop hardness, while reducing the water resistance indicating that these coatings can be altered. The PU/poly-butenolide hybrids can potentially find applications where hard coatings are required, such as for the automotive industry or for protection of wooden floors or furniture, similarly to the acrylate counterpart.15
Conclusions
We have succeeded to transform our previously reported bio-based poly-butenolide binder technology to aqueous hybrid dispersions using high molecular weight polyurethane as colloid stabilizing moiety. Alkoxy- and acyloxy butenolides can be used in combination with a vinyl-ester, ether, or lactam in co-polymer dispersion systems. The hybrid dispersions can be made solvent free in a wide range of PU/poly-butenolide weight ratio. These findings enable the future design of high MW poly-butenolide resin dispersions for waterborne bio-based coating applications, with low emission of VOCs. As toxic isocyanates are used for the synthesis of the polyurethane pre-polymer, incorporation of non-isocyanate based polyurethanes18 is a next step to develop. Furthermore, studying the butenolide monomer scope, noting the excellent properties of the dispersions with hydrophobic butenolides, will be part of our further studies, as well as investigations on possible coating performance benefits accompanied with this novel polymeric backbone for coatings. Finally, with the knowledge gained from this study, other conventional and more cost-effective dispersion techniques will be investigated.Author contributions
B. L. F. and K. J. v. d. B. conceptualized the research project. A. J. and N. E. performed the research and prepared the manuscript with input from B. L. F. and K. J. v. d. B. All authors reviewed the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the Advanced Research Center for Chemical Building Blocks, ARC CBBC, which is co-founded and co-financed by the Netherlands Organization for Scientific Research (NWO, contract 736.000.000) and the Netherlands Ministry of Economic Affairs and Climate. We thank Lizet Brugman, Connie Hermans, and René Freeke (AkzoNobel) for GPC analysis and coating evaluation. We thank Brenda Rossenaar (Nouryon) for (S)TEM imaging. We thank J. L. Sneep and J. Hekelaar (University of Groningen) for assistance with HRMS. Johannes G. H. Hermens (AkzoNobel), Jelmer T. Meijer, and Mathieu L. Lepage (University of Groningen) are gratefully acknowledged for their input during scientific discussions.References
- C. Challener, CoatingsTech, 2018, 15, 4 Search PubMed; M. F. Cunningham, J. D. Campbell, Z. Fu, J. Bohling, J. G. Leroux, W. Mabeee and T. Robert, Green Chem., 2019, 21, 4919 Search PubMed.
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Nature, 2022, 612, 272 Search PubMed; C. C. N. de Oliveira, M. Z. Zotin, P. R. R. Rochedo and A. Szklo, Biofuels, Bioprod. Biorefin., 2021, 15, 430 Search PubMed.
- D. R. Dodds and R. A. Gross, Science, 2007, 318, 1250 Search PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950 Search PubMed.
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y.-X. Chen, Nat. Rev. Mater., 2022, 7, 83 Search PubMed.
- A. Jaswal, P. P. Singh and T. Mondal, Green Chem., 2022, 24, 510 Search PubMed; A. P. Dunlop, 1951, US2536732A; K. J. Zeitsch, 2004, US6743928B1; L. Liu, H. M. Chang, H. Jameel and S. Park, Bioresour. Technol., 2018, 252, 165 Search PubMed.
- J. G. H. Hermens, T. Freese, K. J. van den Berg, R. van Gemert and B. L. Feringa, Sci. Adv., 2020, 6, eabe0026 Search PubMed.
- G. O. Schenck, Ann. Chem., 1953, 584, 156 Search PubMed; F. W. Machado-Araujo and J. Gore, Tetrahedron Lett., 1981, 22, 1969 Search PubMed; B. L. Feringa, Recl. Trav. Chim. Pays-Bas, 1987, 106, 469 Search PubMed.
- H.-D. Scharf and J. Janus, Chem. Ber., 1978, 111, 2741 Search PubMed; B. L. Feringa and B. de Lange, Tetrahedron, 1988, 44, 7213 Search PubMed; B. L. Feringa and J. C. de Jong, J. Org. Chem., 1988, 53, 1125 Search PubMed.
- V. V. Poskonin, D. N. Yakovlev, V. A. Kovardakov and L. A. Badovskaya, Russ. J. Org. Chem., 1999, 35, 721 Search PubMed.
- J. G. H. Hermens, M. L. Lepage, A. Kloekhorst, E. Keller, R. Bloem, M. Meijer and B. L. Feringa, React. Chem. Eng., 2022, 7, 2280 Search PubMed; M. D. Edwards, M. T. Pratley, C. M. Gordon, R. I. Teixeira, H. Ali, I. Mahmood, R. Lester, A. Love, J. G. H. Hermens, T. Freese, B. L. Feringa, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2024, 28, 1917 Search PubMed.
- J. G. H. Hermens, T. Freese, G. Alachouzos, M. L. Lepage, K. J. van den Berg, N. Elders and B. L. Feringa, Green Chem., 2022, 24, 9772 Search PubMed.
- M. L. Lepage, G. Alachouzos, J. G. H. Hermens, N. Elders, K. J. van den Berg and B. L. Feringa, J. Am. Chem. Soc., 2023, 145, 17211 Search PubMed.
- B. L. Feringa, J. G. H. Hermens, K. J. van den Berg and R. van Gemert, 2021, WO2021/084066A1; B. L. Feringa, J. G. H. Hermens, K. J. van den Berg and R. van Gemert, 2021, WO2021/259819A1; M. L. P. Lepage, B. L. Feringa, J. G. H. Hermens, K. J. van den Berg and N. Elders, 2024, WO2024/126828A1; M. L. P. Lepage, B. L. Feringa, J. G. H. Hermens, K. J. van den Berg and N. Elders, 2024, WO2024/126829A1; M. L. P. Lepage, B. L. Feringa, J. G. H. Hermens, K. J. van den Berg and N. Elders, 2024, WO2024/126832A1.
- H. L. Manock, Pigm. Resin Technol., 2000, 29, 143 Search PubMed; J. Kozakiewicz, Polimery, 2016, 61, 81 Search PubMed; S. Mehravar, N. Ballard, R. Tomovska and J. M. Asua, Ind. Eng. Chem. Res., 2019, 58, 20902 Search PubMed.
- Y. Zhu, J. Yang, F. Mei, X. Li and C. Zhao, Green Chem., 2022, 24, 6450 Search PubMed.
- A. Delavarde, G. Savin, P. Derkenne, M. Boursier, R. Morales-Cerrada, B. Nottelet, J. Pinaud and S. Caillol, Prog. Polym. Sci., 2024, 151, 101805 Search PubMed.
- O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431 Search PubMed; E. Pichon, D. De Smet, P. Rouster, K. Freulings, A. Pich and K. V. Bernaerts, Mater. Today Chem., 2023, 34, 101822 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Extended discussions, experimental procedures, monomer and dispersion synthesis, characterisation, and coating properties. See DOI: https://doi.org/10.1039/d4gc03466j |
| ‡ Only for the volatile monomers used (3a–d, VeoVa 9, BVE, NVP). |
| § Full data available in ESI.† |
| This journal is © The Royal Society of Chemistry 2024 |