
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStraightforward solid-phase modification of TiO2 with propylphosphonic acid via manual grinding and shaker mixing: enhancing modification degree by thermal control while improving atom economy†
Kaimin
Zhang
 ab,
Jinxin
Wang‡
*b,
Nick
Gys
bcde,
Elien
Derveaux
f,
Nahal
Ghanemnia
f,
Wouter
Marchal
f,
Peter
Adriaensens
f and
Vera
Meynen
*bd
ab,
Jinxin
Wang‡
*b,
Nick
Gys
bcde,
Elien
Derveaux
f,
Nahal
Ghanemnia
f,
Wouter
Marchal
f,
Peter
Adriaensens
f and
Vera
Meynen
*bd
aSchool of Resources and Safety Engineering, Wuhan Institute of Technology, Wuhan 430074, China
bLaboratory for Adsorption and Catalysis (LADCA), Department of Chemistry, University of Antwerp, Universiteitsplein 1, 2610, Wilrijk, Belgium. E-mail: angjinxin@yeah.net; vera.meynen@uantwerpen.be
cCenter for Membrane Separations, Adsorption, Catalysis, and Spectroscopy KU Leuven, University of Leuven, Celestijnenlaan 200F, 3001, Leuven, Belgium
dFlemish Institute for Technological research, VITO NV, Boeretang 200, 2400 Mol, Belgium
eMaterials and Chemistry, Electrochemical and Surface Engineering, Vrije Universiteit Brussel, Pleinlaan 2, 1050, Brussels, Belgium
fNMR Group, Applied and Circular Chemistry, Hasselt University, Campus Diepenbeek – Gebouw D, 3590 Diepenbeek, Belgium
First published on 25th September 2024
Abstract
Grafting organophosphonic acids (PAs) on metal oxides has shown to be a flexible technology to tune the surface properties of metal oxides for various applications. The solvents applied in the commonly used synthesis method have associated impeding effect on tailoring the resulting modification degree. In this work, an alternative solid-phase manual grinding method is proposed that (i) is straightforward, (ii) can achieve controllable and higher modification degree, and (iii) excludes the use of solvent during the synthesis. Specifically, propylphosphonic acid (3PA) was grafted onto titania by manual grinding, and different modification degrees were obtained by varying the duration of the post-synthetic thermal treatment. Importantly, the solid-phase method can achieve a modification degree that is 25.0% higher than the maximal modification degree reached by the liquid-phase method, while its atom utilization efficiency is 4.8 times (toluene-based) or 7.5 times (water-based) that of the liquid-phase method.
Introduction
Surface modification with organic molecules is a flexible method to adjust the surface properties of metal oxides (MOx).1,2 Organophosphonic acids (PAs) are of particular interest, as the formed M–O–P bonds possess a higher hydrolytic and thermal stability than the bonds resulting from other organic modifiers, such as organosilanes, thiols, and carboxylic acids.1,3–6 PAs-modified metal oxides thus have been used in various applications, ranging from adsorption,7,8 separation,9,10 catalysis11,12 to biosensors.13The P–OH groups and phosphoryl oxygen atoms (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the PAs can interact and bond (grafting) with the reactive sites (i.e., hydroxyl groups and Lewis acid sites) on metal oxides supports (via different routes, see Scheme 1).1,2,14 However, in a typical PAs surface modification method, solvents (water or organic solvents) are applied that can also create (competitive) interactions with these hydroxyl groups and Lewis acid sites on supports as well as with the applied solute (type of phosphonic acid), which can limit the resulting modification degree.14–17 Nevertheless, in some applications, a higher modification degree is desired, as it can lead to a better performance, e.g., in CO2 adsorption,18 Pd recovery,19etc. Hence, to achieve a higher modification degree, the concentration of PAs or the reaction temperature needs to be elevated.15 However, this results in increasing costs, particularly when increasing their concentration, as PAs are expensive reagents (e.g., propylphosphonic acid used in this study costs ∼ 38 € per g). Importantly, an increase in concentration or temperature is also restricted to the solubility of the PAs in the solvent and the boiling point of the solvent. Moreover, solvent itself, such as organic solvents, might pose environmental harm or health risks.20 Furthermore, depending on the type of PAs and pH, at high concentration (e.g., 0.1 M) of PAs and/or high reaction temperature (e.g., ≥90 °C) in aqueous systems, metal oxides can be partially dissolved promoting the formation of layered titanium phosphonate phase(s) by a dissolution-precipitation side reaction at the surface,14,15,21 which might influence the performance of the modified materials.
O) of the PAs can interact and bond (grafting) with the reactive sites (i.e., hydroxyl groups and Lewis acid sites) on metal oxides supports (via different routes, see Scheme 1).1,2,14 However, in a typical PAs surface modification method, solvents (water or organic solvents) are applied that can also create (competitive) interactions with these hydroxyl groups and Lewis acid sites on supports as well as with the applied solute (type of phosphonic acid), which can limit the resulting modification degree.14–17 Nevertheless, in some applications, a higher modification degree is desired, as it can lead to a better performance, e.g., in CO2 adsorption,18 Pd recovery,19etc. Hence, to achieve a higher modification degree, the concentration of PAs or the reaction temperature needs to be elevated.15 However, this results in increasing costs, particularly when increasing their concentration, as PAs are expensive reagents (e.g., propylphosphonic acid used in this study costs ∼ 38 € per g). Importantly, an increase in concentration or temperature is also restricted to the solubility of the PAs in the solvent and the boiling point of the solvent. Moreover, solvent itself, such as organic solvents, might pose environmental harm or health risks.20 Furthermore, depending on the type of PAs and pH, at high concentration (e.g., 0.1 M) of PAs and/or high reaction temperature (e.g., ≥90 °C) in aqueous systems, metal oxides can be partially dissolved promoting the formation of layered titanium phosphonate phase(s) by a dissolution-precipitation side reaction at the surface,14,15,21 which might influence the performance of the modified materials.
 | ||
| Scheme 1 Examples of organophosphonic acids (PAs) surface modification process, adapted from ref. 1, with the permission from the John Wiley and Sons. | ||
To mitigate the negative impact of solvents on grafting, some researchers have attempted to reduce their usage during the synthesis process.22–24 Hanson et al. introduced the T-BAG method (tethering by aggregation and growth), improving the grafting of octadecylphosphonic acid (ODPA) onto the Si (SiO2/Si) that was less affinity to PAs.22 The ODPA solutions, with the support inside, evaporated slowly (approximately 3 h, relative humidity < 16%) until the support could not be submerged anymore. Next, a heating at 140 °C (2 days) was carried out to enhance the bonding between the PAs and the Si (SiO2/Si). This method demonstrated the formation of a ODPA monolayer on Si (SiO2/Si) support. However, the grafting was sensitive to moisture (interface water concentration), making the method plagued by inhomogeneity due to poorly controlled humidity levels.23
Researchers have also attempted to completely omit the use of solvents during the synthesis process,25–29 but new problems rise and the use of uncommon equipment is also necessary and/or more complex synthesis procedures are required. For instance, Fischer et al. and Betke et al. validated the feasibility of grafting phenylphosphonic acid (PhPA) onto TiO2via reactive milling using a high-energy planetary ball milling setup. They investigated the influence of milling parameters, such as the materials of the grind bowl, the grinding sphere, rotation speed etc., on the resulting crystal phase of TiO2 and modification degrees.27,28 However, the crystal phase of TiO2 changed from anatase to rutile after the modification due to the high-energy vibrational impact of the process.27–29 This can be a drawback for applications, e.g., anatase TiO2 outperforms than rutile in photocatalytic degradation of volatile organic compounds.30–32 Moreover, Gupta et al. used special vacuum vapour deposition to achieve the deposition of monolayers of n-octylphosphonic acid (C8PA) onto the surface of AlOx, but no specific modification degree was provided.25
In this study, a straightforward manual grinding method is proposed to provide a controllable and higher modification degree of PAs on metal oxides. It not only avoids the impeding effect of a solvent, but also preserves the crystal phase of the support. Moreover, it increases the greenness of the modification method as the use of solvent is avoided during the modification process and a higher atom efficiency can be achieved, while using the same or lower PA/TiO2 ratio. Specifically, propylphosphonic acid (3PA) was grafted onto a commercially available TiO2 support, P25, by the proposed manual grinding method, followed by a thermal treatment (of different durations) to achieve different modification degrees. The material properties obtained after each synthesis step, including manual grinding, washing, and thermal treatment, were elucidated by (in situ) diffuse reflectance infrared Fourier transform (DRIFT), thermogravimetric analyses (TGA) coupled with mass spectrometry (MS), solid-state 31P-MAS (magic angle spinning) NMR, X-ray powder diffraction (XRD) and scanning electron microscope (FEG-ESEM) measurements. Therefore, the role of every step during the solid-phase modification is thoroughly revealed. Additionally, the reproducibility of the proposed method was validated by characterizing samples prepared from different batches. The obtained 3PA-modified TiO2 is compared with those prepared by conventional liquid-phase water- or toluene-based methods, demonstrating that the proposed method is not only straightforward but also more atom economic. To proof that the method can also be used for other types of PAs, another organophosphonic acid, phenylphosphonic acid (PhPA), was grafted onto TiO2.
Experimental section
Materials
Propylphosphonic acid (3PA) and phenylphosphonic acid (PhPA), see Scheme 2, were purchased from Cayman and Sigma, respectively. Commercial TiO2 P25 (Sigma-Aldrich) was used as the metal oxide support in this work. Toluene, ethanol (99.9%+, absolute) and acetone (99.5%+, for analysis) were purchased from Fischer Chemical and Acros, respectively. All chemicals were used without pre-treatment. | ||
| Scheme 2 Chemical structures of 3PA (a) and PhPA (b). | ||
Surface modification
In the case of the solid-phase grafting method, 1 g of TiO2 and 0.065 g of 3PA were ground by hand in a mortar for 20 min, transferred to an open flask and heated at 90 °C in oil bath for 4 h, 24 h, 48 h, 72 h, and 96 h. The reference sample was only ground but not heated. After grinding and heating, the samples were washed with 3 × 30 mL ethanol and 3 × 30 mL water, followed by a drying procedure overnight in an oven at 60 °C (ambient conditions). The obtained samples were denoted as 3PA-G-P25 (without heating), 3PA-G-P25-4 h, 3PA-G-P25-24 h, 3PA-G-P25-48 h, 3PA-G-P25-72 h, and 3PA-G-P25-96 h, after 4 h, 24 h, 48 h, 72 h and 96 h of heating, respectively.To make a comparison with the solid-phase method, a liquid-phase method was also applied. During the liquid-phase synthesis, the same amounts of TiO2 and 3PA, as used in the solid-phase method, were put into an open flask, and 20 mL of solvent (water or toluene, both are good solvents for 3PA) was then added, resulting in a solution of 0.025 M 3PA. The solution was stirred at 90 °C for X (4, 24, 48, 72, 96) h and an additional 20 minutes, under reflux conditions. The reaction time X of the liquid-phase method was prolonged by 20 min since the solid-phase method has 20 min of manual grinding prior to the heating process. When the reaction was finished, the sample modified in water was washed with 3 × 30 mL water and dried overnight in an oven at 60 °C. For the samples prepared in toluene, additional washing steps were applied to remove weakly interacting 3PA. Hence, the samples were additionally washed with 3 × 30 mL toluene and 3 × 30 mL acetone before the washing steps with water, followed by drying overnight in an oven at 60 °C. To obtain the highest modification degree in water and toluene, also experimentally optimized 3PA concentrations of 0.150 M (water) and 0.100 M (toluene) were applied.14,16 In both cases, 3PA remained completely soluble. For convenience of description, samples prepared by the liquid-phase reaction were denoted as 3PA-P25-W (in water) and 3PA-P25-T (in toluene).
To further look into the solid-phase method, a sample was prepared through simple mixing with a shaker rather than via manual grinding to compare with the sample prepared by manual grinding but without thermal treatment (3PA-G-P25). No extra thermal treatment was applied for this shaker-mixed sample (3PA-M-P25) either. The preparation is as follows: 3PA and P25 were mixed in a digital shaker (VWR, advanced digital shaker) at a speed of 300 rpm for 20 minutes. The reaction time, as well as the amounts of 3PA (0.065 g) and P25 (1.0 g) applied were kept identical to those used for the manually ground sample, 3PA-G-P25. Subsequently, the mixed sample was treated with the same washing and drying procedures as those used for the preparation of 3PA-G-P25.
In addition, to create a blank sample for comparison with washed 3PA-M-P25 and 3PA-G-P25, some unmodified P25 support underwent the same washing and drying procedure but without the addition of 3PA.
To extend the solid-phase method, phenylphosphonic acid (PhPA) was selected as the modifier. The grafting of PhPA on P25 was achieved by both solid- and liquid-phase (water or toluene as the solvent) methods. The amount of P25 was 1.0 g for these two methods, the amount of PhPA was 0.0790 g (75% less than the required amounts in liquid-phase method) and 0.3162 g (i.e., 0.1 M) for the solid- and liquid-phase methods, respectively. The reaction temperature was kept at 90 °C for both methods, while the reaction time was 48 h and 4 h for the solid- and liquid-phase methods, respectively. The washing steps were kept the same as those applied during the 3PA grafting. The modified samples, obtained via the solid- and liquid-phase methods, were denoted as PhPA-G-P25, PhPA-P25-W (water-based) and PhPA-P25-T (toluene-based).
Before characterization, the samples obtained after washing and drying were stored in an Ar atmosphere in a UV-resistant case, preventing photo- and moist mediated decomposition of the grafted 3PA.33 The characterization of unwashed samples was performed immediately after the synthesis to minimize potential changes over time.
Characterizations
Thermogravimetric analyses (TGA) and differential thermogravimetric analysis (DTG) were recorded on a Mettler Toledo TGA-DSC 3+. The measurements were carried out under a continuous flow of O2 (30–600 °C, 80 mL min−1) or Ar (30–800 °C, 50 mL min−1) with a heating rate of 10 °C min−1. For the measurements in Ar atmosphere, a flushing of 30 min at 30 °C was added prior to the temperature ramping, to remove traces of oxygen. The modification degree, denoted as #/nm2 (i.e., groups per nm2), was calculated from the weight loss of burned hydrocarbon groups in O2 by the following formula (1), since the phosphorus group remains at the surface after heating.34 The temperature range for the calculation is 240–420 °C or 270–420 °C for 3PA modification, depending on the samples analyzed. For PhPA-modified P25, the temperature range for calculating the modification degree is 300–480 °C. The calculation of the modification degrees excludes the weight of the titania phosphonate formed by the dissolution-precipitation process (weight losses at temperature range of 420–475 °C for 3 PA14 or 480–550 °C for PhPA15). | (1) |
In which wt%(R) is the weight percentage loss of the organic group, MM(R) is the molar mass of the propyl (43 g mol−1) or phenyl (77 g mol−1) group, SBET is the specific surface area of P25 (50 m2 g−1), and NA (6.02 × 10−23 mol−1) is Avogadro's constant. The experimental error is around 0.1 #/nm2 based on five repeated measurements of different batches of samples.15
The atom utilization efficiency (AUE) is calculated as follows:
 | (2) |
In which mod. deg. is the modification degree of the sample (#/nm2), m(PA) and MW are the amount (g) and the molar weight (124.08 g mol−1 for 3PA, 158.09 g mol−1 for PhPA) of the phosphonic acid used, respectively; SBET is the specific surface area of P25 (50 m2 g−1), and NA (6.02 × 10−23 mol−1) is Avogadro's constant. The experimental errors are calculated as standard deviation errors based on three different batches of samples.
Thermogravimetric analysis coupled with mass spectrometry (TGA-MS) measurements were performed using a Mettler Toledo TGA-DSC 3+ coupled with a Hiden HPR20 Mass spectrometer. Prior to the Ar measurement, the samples were purged with an Ar flow of 50 mL min−1 for 30 min at room temperature to ensure a completely inert atmosphere. The inert atmosphere was confirmed by the oxygen spectra (m/z = 32) in the MS, with a response lower than 1 × 10−10. Then, the temperature was ramped up to 800 °C with a heating rate of 10 °C min−1 under an Ar flow of 50 mL min−1. For the measurements under an O2 atmosphere, the samples were heated from RT to 600 °C with a heating rate of 10 °C min−1 under an O2 flow of 80 mL min−1. The data was recorded and analyzed with the EGA software package. The m/z range of 2–150 was collected on the MS with the Secondary Electron Multiplier (SEM) detector for measurements conducted in both Ar and O2 atmospheres.
The (in situ) diffuse reflectance infrared Fourier transform (DRIFT) measurements were carried out on a Nicolet 6700 Fourier Transform IR spectrometer (Thermo Scientific), equipped with an electromagnetic source in the mid-infrared region (4000–400 cm−1) and a DTGS detector. The applied DRIFT accessory for the measurements at different temperatures was a Praying Mantis High Temperature Reaction Chamber (Harrick, USA). The resolution was 4 cm−1, and 100 scans were accumulated for each spectrum. The sample holder contained a 2 wt% diluted sample in KBr, and the sample was then measured at room temperature (30 °C) after 30 min under an Ar flow of 50 mL min−1 to reduce the amount of molecular adsorbed water and CO2. All the spectra were normalized with respect to the Ti–O–Ti peak between 1000–800 cm−1 (except for the spectrum of pure 3PA that was normalized towards its highest peak at 945 cm−1).
The in situ DRIFT spectra of undiluted samples at different temperatures (30 °C to 600 °C) were obtained against a background of KBr, collected at 500 °C under an Ar flow of 50 mL min−1, using a 30 min constant heating at each temperature. The spectra of samples were collected when the sample was cooled back to room temperature to exclude thermal effects.35,36
Solid-state 31 P-MAS (magic angle spinning) NMR spectra were obtained at room temperature using an Agilent VNMRS DirectDrive 400 MHz spectrometer (9.4 T wide bore magnet) equipped with a T3HX 3.2 mm VT probe dedicated for small sample volumes and high decoupling powers. Magic angle spinning was carried out at 15 kHz using ceramic zirconia rotors of 3.2 mm in diameter (22 μL rotors). The phosphorus chemical shift scale was calibrated to monopotassium phosphate (KH2PO4) at 3.9 ppm. Other acquisition parameters applied were: a spectral width of 60 kHz, a 90° pulse length of 3.4 μs, an acquisition time of 25 ms, a recycle delay time of 20 s and between 3000–5000 accumulations. High power proton dipolar decoupling during the acquisition time was set at 80 kHz.
X-ray powder diffraction (XRD) spectra were obtained by a D8 advance Eco diffractometer (Bruker) equipped with Cu-Kα radiation (λ = 1.5406 Å). The measurement parameters included a scanning range of 5–80° 2θ and a scanning rate of 0.04°/4 s. The software used for Rietveld analysis was x'pert highscore plus from Panalytical.
The field emission gun – environmental scanning electron microscope (FEG-ESEM) was used to characterize the samples’ morphology before and after modification. The applied measurement parameters comprise 20 kV of accelerating voltage, 10 mm of measurement distance, 30° take-off angle and 10−4 Pa chamber pressure. The samples were sputter-coated with gold before the measurement. Limited magnification (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×) was used to allow a sufficiently broad overview of the particles that are representative for the batch of materials.
000×) was used to allow a sufficiently broad overview of the particles that are representative for the batch of materials.
Results and discussion
No thermal treatment, prior to washing and drying
In order to study the role of manual grinding in 3PA modification on P25, 3PA-G-P25 (manual grinding with a pestle in a mortar), 3PA-M-P25 (mixing 3PA and P25 by a shaker instead of manual grinding, providing much less energy and no mechanical forces on the system), native P25, and pure 3PA were analyzed with DRIFT at room temperature (RT) under an Ar flow (Fig. 1). These samples were measured prior to further processing by washing or thermal treatment. The spectra of 3PA-G-P25, 3PA-M-P25, and native P25 were measured undiluted (Fig. 1a, b and e) or as a 2 wt% dilution in KBr (Fig. S1†), to provide qualitative or semi-quantitative analysis, respectively. The spectra of the undiluted samples display more prominent signals compared to the diluted samples but cannot be quantified as signal intensity depends on the depth of penetration of the reflected IR signal, which is absorption dependent. The DRIFT measurement of pure 3PA was performed using a 2 wt% diluted sample in KBr (Fig. 1c and d). All the spectra in Fig. 1 were normalized, the unnormalized spectra can be found in Fig. S2.† All the measurements were performed immediately after the samples’ synthesis to avoid potential changes over time. | ||
| Fig. 1 Normalized DRIFT spectra of diluted and undiluted samples were measured under an Ar flow of 50 mL min−1 at RT. The spectra of undiluted 3PA-G-P25, 3PA-M-P25, and native P25, before washing and without thermal treatment, are presented in (a), and enlarged spectra of the 1300–1080 cm−1 and 3800–3500 cm−1 region are shown in (b) and (e), respectively. The spectrum of diluted 3PA (2 wt% in KBr) is shown in (c) of which a zoom-in of the 1300–1100 cm−1 region is shown in (d). | ||
In contrast to native P25, 3PA-M-P25 and 3PA-G-P25 both show the characteristic peaks of C–H stretching and deformation in alkyl groups between 3000–2800 cm−1 and 1500–1300 cm−1, respectively,37–40 a band at 1280 cm−1 might be related to the P–O bond,38,40 and a broad band at 2302 cm−1, characteristic for P–OH stretching vibrations (Fig. 1a).38,40 The band at 1248 cm−1 (Enlarged in Fig. 1b) and the subtle shoulder at 1108 cm−1 were exclusively observed for 3PA-G-P25. The band at 1248 cm−1 might be assigned to phosphoryl vibrations (PO, a minor signal at 1257 cm−1 for pure 3PA in Fig. 1d)40 even though DFT calculations proposed a shift of the bond of free PO (1260 cm−1) to much lower wavenumbers on clean surfaces.41 The small feature at 1108 cm−1 might be related to PO in a bidentate bonding to anatase (001)41 or PO3 stretching.42 Although reproducible, the band at 1108 cm−1 is very small, therefore, the band at 1248 cm−1 was used to highlight the differences between 3PA-G-P25 and 3PA-M-P25. These mentioned signals are not observed for native P25, while they can be found in the spectra of 3PA (Fig. 1c). The presence versus absence of a peak at 1248 cm−1 for 3PA-G-P25 and 3PA-M-P25 can be attributed to the manual grinding process, which is the primary distinction between the synthesis methods of these two samples.
Additionally, differences in the stretching vibrations of OH groups between 3750–3600 cm−143,44 are also visible when comparing 3PA-G-P25 and 3PA-M-P25. Fig. 1e shows the zoomed spectra to display the differences more clearly. 3PA-M-P25 and P25 reveal a broad absorption band characterized by a superposition of multiple (partly) overlapping signals at 3720 cm−1, 3690 cm−1, 3670 cm−1, 3655 cm−1 and 3630 cm−1 (see Table S1† for specific assignments). In contrast to 3PA-M-P25 and P25, a similar absorption band pattern, composed of multiple signals, was absent for 3PA-G-P25, i.e., these OH group signals are barely visible or featureless for 3PA-G-P25. Therefore, homogeneous mixing during manual grinding of P25 and 3PA seems to promote the adsorption and grafting of 3PA groups through condensation reactions or hydrogen bonding. Furthermore, differences in water signal (1620 cm−1 or 1630 cm−1) can also be found in 3PA-G-P25, 3PA-M-P25, and native P25, again suggesting a promotion of the interaction between 3PA and P25 during the manual grinding process. See text S1† for detailed analysis.
In order to further correlate the observed changes with grafting and to confirm the effect of manual grinding on the modification, in situ DRIFT measurements were performed at elevated temperatures under an Ar flow (Fig. 2) for the same samples. All the spectra in Fig. 2 were normalized, the unnormalized spectra can be found in Fig. S3.†
 | ||
| Fig. 2 Normalized in situ DRIFT spectra in function of temperature of undiluted 3PA-G-P25, 3PA-M-P25, and native P25, before washing and without thermal treatment, measured under an Ar flow of 50 mL min−1 from RT to 600 °C. (a–c) Spectra between 3800–3500 cm−1 and 1700–1550 cm−1, (d–f) spectra between 3000–2800 cm−1 and 1280–1200 cm−1. | ||
Fig. 2a–c display the hydroxyl stretching region between 3800–3500 cm−1 and water deformation mode between 1630–1620 cm−1. The hydroxyl signals decrease in intensity upon increasing temperature, and different evolutions were observed for 3PA-G-P25 and 3PA-M-P25. The signals of hydroxyl groups between 3750–3600 cm−1 were barely visible between 150–200 °C for 3PA-G-P25 (Fig. 2a). For 3PA-M-P25 (Fig. 2b), the intensity of OH groups largely decreased from RT to 250 °C, and became almost invisible between 350–400 °C; which is higher than the temperature at which the signals disappeared for 3PA-G-P25. However, although the intensity of OH groups gradually decreased for native P25, these signals remain pronouncedly present even up to 600 °C (Fig. 2c). Apparently, the presence of 3PA molecules affects the interaction of hydroxyl groups, also when 3PA and P25 are only mixed via the shaker (3PA-M-P25), indicating that 3PA molecules could condense/interact with hydroxyl groups on TiO2 while heating in situ. The differences in the OH-region in function of heat between 3PA-M-P25 and 3PA-G-P25 again suggest that the manual grinding process already induced the interaction or condensation between 3PA and TiO2. Furthermore, the desorption temperature of surface water (1620–1630 cm−1) was lower for 3PA-G-P25 and 3PA-M-P25 (200–250 °C) than for native P25 (250–300 °C), suggesting their weaker interactions with surface water. Fig. 2d–f show the band at 1248 cm−1 and the C–H stretching in alkyl groups between 3000–2800 cm−1 as a function of temperature for 3PA-G-P25, 3PA-M-P25, and native P25. The peak at 1248 cm−1 became more pronounced or became visible when increasing the temperature from RT to 250 °C, for 3PA-G-P25 and 3PA-M-P25, respectively. As previously discussed, the peak at 1248 cm−1 is not present for pure P25 and might be originally situated at 1257 cm−1 for pure 3PA (Fig. 1), therefore, its presence could be closely correlated with the grafting or H-bond interaction of 3PA with the surface (covalently bonded via condensation or coordination of PO to Lewis acid sites or H-bond interaction with surface OH). As the 1248 cm−1 peak is already clearly present after manual grinding (at RT, 3PA-G-P25) while not present in the RT spectrum of 3PA-M-P25, but only appears with temperature, it is suggested that the grafting of 3PA on P25 does not occur by simple shaker mixing but only forms upon heating the material in situ. This points again to a difference between manual grinding and shaker mixing with respect to the interaction of 3PA with the TiO2 surface. Other changes in 3000–2800 cm−1 and 1248 cm−1 can be found in text S2.†
To further confirm the effect of manual grinding on the modification, to understand the difference with shaker mixing and to provide insights in the occurring reactions for 3PA-G-P25 and 3PA-M-P25, TG/DTG-MS analyses were performed from RT to 800 °C under an Ar flow for these samples (Fig. 3). Fig. 3a and b summarizes the TG/DTG results of 3PA-G-P25, 3PA-M-P25, and native P25, and their corresponding DTG-MS results are displayed in Fig. 3c–e.
 | ||
| Fig. 3 DTG (a) and TG (b) results of 3PA-G-P25, 3PA-M-P25, and native P25, obtained before washing and without thermal treatment. (c–e) The corresponding DTG-MS results of 3PA-G-P25 (c), 3PA-M-P25 (d), and native P25 (e). The solid and dash-dot lines represent the MS and DTG results, respectively. TG/DTG-MS were measured under an Ar flow of 50 mL min−1. | ||
Different weight losses and maxima were observed in the (D)TG curves of 3PA-G-P25, 3PA-M-P25, and native P25 (Fig. 3a and b). In the region of 30–330 °C, 3PA-G-P25, 3PA-M-P25, and native P25 show divergent weight losses which are all attributed to the loss of water, according to their water fragment (m/z = 18) in MS (Fig. 3c–e). The water signals of these three samples originate from molecular surface water desorption and (partial) condensation of TiO2 surface OH groups (Fig. 2a–c). For 3PA-G-P25 and 3PA-M-P25, the condensation between Ti–OH and P–OH (3PA) could also occur in the same temperature region, by which water would be released. When closely looking at the DTG maxima between 30–330 °C, both 3PA-G-P25 (Fig. 3c) and native P25 (Fig. 3e) presented two water signals in this temperature region, while these signals are less resolved for 3PA-G-P25, which coincides with the lower water signal at 1630 cm−1 and the invisible OH groups between 3750–3600 cm−1 in the DRIFT of 3PA-G-P25 (Fig. 1a and e). In contrast, three pronounced DTG maxima were observed for 3PA-M-P25, including the additional DTG maximum between 270–330 °C, which can be related to the large decrease in intensity of OH groups between RT – 250 °C in Fig. 2b. Due to the absence of this signal in native P25, this weight loss can be attributed to the condensation of the 3PA with the titania surface, also explaining the higher decomposition temperature of this grafted 3PA (Fig. 3d) compared to pure 3PA (Fig. S4†). The temperature range for the condensation was lower in in situ DRIFT (between RT and 250 °C based on the OH signals between 3750–3600 cm−1) compared to that in TG/DTG. This can be caused by the 30 min stabilization at each temperature applied in in situ DRIFT, which is not present in TGA. Thus, the condensation reaction between 3PA and the surface might be affected by the duration of the heating. Moreover, as suggested by in situ DRIFT, this condensation reaction of the 3PA with the surface OH-groups on P25 already occurred for 3PA-G-P25 during the manual grinding process, explaining the absence of the weight loss between 270–330 °C in 3PA-G-P25 (Fig. 3c). This absence of the water loss in this temperature range also coincides with the observations in 3PA liquid-phase modification of P25 in our previous work.17 Other (D)TG results and discussion can be found in Table S2 and text S3.†
To gain further insights into the differences between manual grinding and shaker mixing, solid-state 31P-MAS NMR measurements were performed to study the phosphorous environment in more detail. Fig. 4 shows the 31P-MAS spectra of pure 3PA, 3PA-M-P25, and 3PA-G-P25, and different resonance signals are clearly observed for 3PA-M-P25 and 3PA-G-P25. Both pure 3PA and 3PA-M-P25 exhibit a main sharp resonance signal at 37 ppm, indicating that the phosphorus environment remained unchanged after mixing 3PA with P25, and so confirming that no modification occurred during the shaker mixing process. However, the spectrum of 3PA-G-P25 does not show this signal of pure 3PA anymore and is characterized by three upfield signals around 33 ppm, 28 ppm (broad), and 8 ppm (besides small peaks around 25 and 18 ppm), indicating that 3PA molecules are attached to the P25 surface.15 The broad upfield shifted peak around 28 ppm was assigned to a multitude of slightly different chemical environments around phosphorus nuclei that are covalently grafted to the titania via P–O–Ti condensation bonds (non-uniform binding) in grafting P25 with 3PA via a liquid-phase process.15 In all, it is evidenced that the grafting of 3PA to P25 is occurring in the manual grinding process, while not taking place during the shaker mixing. This finding is in accordance with DRIFT and TG/DTG-MS results. Other detailed assignments of these NMR peaks can be found in text S4.
 | ||
| Fig. 4 Solid-state 31P-MAS NMR spectra of pure 3PA, 3PA-M-P25 and 3PA-G-P25 obtained before washing and without thermal treatment. | ||
In addition, the preparation of manually ground samples showed good reproducibility compared to shaker mixed samples, as proven by the TG/DTG and 31P-MAS measurements of different independent batches of samples (Fig. S5†). See ESI† for detailed analysis.
No thermal treatment, after washing and drying
As the 31P-MAS NMR also indicated the presence of weakly interacting 3PA next to grafted 3PA (text S4†), a washing procedure was performed on 3PA-G-P25 to remove them and unreacted 3PA, if any. Moreover, 3PA-M-P25 was washed using the same washing procedure as a reference blank sample. These two samples, before and after washing, were characterized by in situ DRIFT (Fig. S6†) and 31P-MAS NMR spectra (Fig. S7†). The results confirm that more weakly bonded 3PA on both 3PA-M-P25 and 3PA-G-P25 were removed after washing step. More detailed discussion can be found in ESI.†The washed samples 3PA-M-P25 and 3PA-G-P25 were further measured by TG/DTG-MS under an oxygen flow to determine the number of grafted organic groups, as shown in Fig. 5. Native P25 was also treated with the same washing procedure as a blank sample. Fig. 5a displays the TG/DTG results, and the corresponding DTG-MS profiles are presented in Fig. 5b–d. Fig. 5a shows that 3PA-G-P25, 3PA-M-P25, and native P25 displayed different DTG maxima between 270 °C and 420 °C. 3PA-G-P25 and 3PA-M-P25 show two additional DTG maxima around 325 °C and 360 °C (they are less resolved for 3PA-M-P25), while native P25 only exhibits a DTG maximum at 320 °C. Given that (i) native P25 shows a sharp but tiny DTG maximum resulting from the release of only CO2 (m/z = 44) in this temperature range (Fig. 5d), (ii) while the DTG maxima of 3PA-G-P25 and 3PA-M-P25 in the same temperature range are caused by simultaneous release of water (m/z = 18) and CO2 (m/z = 44) (Fig. 5b–c), the weight loss between 270–420 °C of 3PA-G-P25 and 3PA-M-P25 can be assigned to the burning of the propyl groups of 3PA. Based on this weight loss in this region, the modification degree (the number of grafted 3PA groups) of 3PA-G-P25 and 3PA-M-P25 was determined, i.e., 1.3 #/nm2 and 0.9 #/nm2, respectively (see Table S3† for modification degrees calculated by different methods). The higher modification degree for 3PA-G-P25 than 3PA-M-P25 confirms the positive effect of manual grinding on modification. The presence of grafted organic groups on 3PA-M-P25 probably results from the washing, which can create a short liquid-phase grafting condition, and/or the following overnight drying at 60 °C, as the possibility of grafting during the mixing is small (Fig. 1–4) and less controllable (Fig. S5†). Other detailed TG/DTG results and discussion of 3PA-G-P25 and 3PA-M-P25 can be found in Table S4 and text S5.†
 | ||
| Fig. 5 (a) TG and DTG results for 3PA-G-P25, 3PA-M-P25, and native P25. These samples were obtained after washing and overnight drying at 60 °C. The solid and dash-dot lines represent the TG and DTG results, respectively. The experimental error on the modification degree is 0.1 #/nm2, based on five repeated TG/DTG measurements on different batches of samples. (b–d) The corresponding DTG-MS results of 3PA-G-P25 (b), 3PA-M-P25 (c), and native P25 (d). The solid and dash-dot lines represent the MS and DTG results, respectively. TG/DTG-MS were measured under an O2 flow of 80 mL min−1. | ||
XRD measurements were performed on 3PA-G-P25 (after washing and overnight drying at 60 °C) and native P25 to evaluate if any changes in the crystal phases of P25 occurred during the manual grinding process. The XRD patterns in Fig. 6 showed that 3PA-G-P25 consists of the same phase mixture as the P25 reference, as indicated by asterisk symbols (*) for anatase (JCPDS No. 21-1272) and diamond symbols (◆) for rutile (JCDPS 21-1276).45,46 As the corresponding relative peak intensities remain unaffected, it could be inferred that the crystal phases were not changed by the manual grinding process. Moreover, a quantitative Rietveld phase analysis (the table inserted in Fig. 6) showed that the proportions of anatase and rutile were similar for 3PA-G-P25 and native P25, although changes in the amorphous phase cannot be excluded. This confirms that the manual grinding process did not cause a change in the crystal phases and phase composition. Moreover, the SEM results indicate no obvious changes in morphology or particle size after the grinding process (Fig. S8†).
 | ||
| Fig. 6 XRD patterns of native P25 and 3PA-G-P25. ◆ is for anatase, * is for rutile. The inserted table is the phase composition of 3PA-G-P25 and native P25 based on a Rietveld analysis. 3PA-G-P25 was obtained after washing and drying at 60 °C overnight. | ||
The impact of an additional thermal treatment
In the case of manual grinding at RT, 3PA molecules at least partially interact with the TiO2 surface through covalent bonding, while 31P-MAS NMR evidenced the presence of also more weakly surface interacting 3PA (Fig. 4 and text S4†). Moreover, as in situ DRIFT (Fig. 2a, b, d and e) and TG/DTG (Fig. 3) results demonstrated that a thermal treatment can enhance the bonding between 3PA and TiO2 and the duration of the thermal treatment can also influence the bonding. Here, an extra thermal treatment with different durations was introduced after the manual grinding step to strengthen the bonding and increase the relative amount of strongly bonded 3PA.In this study, 90 °C was selected as the thermal treatment temperature based on both the in situ DRIFT results (condensation occurs between RT – 250 °C) and the reaction temperature (50 °C, 90 °C, 130 °C) used in our previous work on liquid-phase modification.15,16,47 Different thermal durations from 0 h to 96 h were investigated, and modification degrees are summarized in Table 1. It is clear that the modification degree rises from 1.3 #/nm2 (0 h, i.e., without thermal treatment), to 1.8 #/nm2 (4 h) and to 2.5 #/nm2 (96 h), caused by an enhanced amount of strongly grafted 3PA with increasing thermal treatment, which remains after washing. Corresponding TG/DTG results and more detailed discussions can be found in Fig. S9.†
| Thermal durations (h) | 0 | 4 | 24 | 48 | 96 |
| Modification degree (#/nm2) | 1.3 | 1.8 | 1.9 | 2.3 | 2.5 |
In addition, to confirm the effect of thermal treatment on the grafting and type of bonds formed, the sample prepared with a 4-hour thermal treatment was selected as an example to compare with the one prepared without the thermal treatment. Both were measured by solid-state 31P-NMR prior to washing (Fig. S10†). The results confirm the positive effect of the thermal treatment on enhancing the relatively amount of strongly bonded 3PA molecules, i.e., the modification degree. More detailed discussion can be found in ESI.† Additionally, the SEM and XRD results show that the thermal effect did not cause changes in the samples morphology, particle size (Fig. S11†), or crystal composition (Fig. S12†), respectively.
Solid-phase grafting vs. liquid-phase grafting
To further understand the proposed solid-phase grafting method, the 3PA-G-P25-4 h sample was compared with samples prepared by a common liquid-phase method under the same reaction conditions, including the same reaction temperature (90 °C), time (4 h), and 3PA/TiO2 amounts (0.065 g/1.0 g). The samples synthesized by the liquid-phase method were obtained by grafting TiO2 in a 3PA solution under reflux using water or toluene as the solvent. The TG/DTG results are shown in Fig. S13† and the modification degrees are summarized in Table 2.| Samples | 3PA-G-P25-4 h | 3PA-P25-W-4 h | 3PA-P25-T-4 h |
| Modification degree (#/nm2) | 1.8 | 1.4 | 1.8 |
Table 2 shows that 3PA-G-P25-4 h reveals a similar modification degree as 3PA-P25-T-4 h (1.8 #/nm2), which is higher than this of 3PA-P25-W-4 h (1.4 #/nm2). The lower modification degree in water compared to toluene was previously explained in literature and correlated to the existence of competitive interactions between water and 3PA for adsorption at the TiO2 surface, while less competition between toluene and 3PA accounts for the higher modification degree of 3PA-P25-T-4 h.14,16 Therefore, the similar higher modification degree of 3PA-G-P25-4 h might be explained by the absence of a competitive solvent during modification, excluding competitive solvent-solute interactions (except for moisture from the ambient atmosphere and adsorbed surface water on the TiO2). Additionally, 3PA-G-P25-4 h and 3PA-P25-T-4 h also show more similarities in surface properties as compared to 3PA-P25-W-4 h, e.g., surface OH groups, which was confirmed by TGA (Fig. S13†) and DRIFT (Fig. S14†). Moreover, their similarities in phosphorus environment was also validated by solid-state 31P-MAS NMR measurements (Fig. S15†). More detailed discussion can be found in ESI.†
Furthermore, the impact of the reaction time at 90 °C (4 h, 24 h, 48 h, 72 h, and 96 h) on the modification degree was explored, studying the kinetics for the solid-phase vs. liquid-phase methods (Fig. S16†). With increasing reaction time from 4 h to 96 h, the modification degree gradually increased for 3PA-G-P25, while it remained unchanged for samples prepared by the liquid-phase water- or toluene-based method, within the experimental error of 0.1 #/nm2.
As the reaction time does not allow to enhance the modification degree in the liquid-phase method, the amount of 3PA used in water and toluene was increased to respectively six and four times the amount used in the solid-phase method. The detailed results are summarized in Fig. 7 and TG/DTG results are displayed in Fig. S17.†
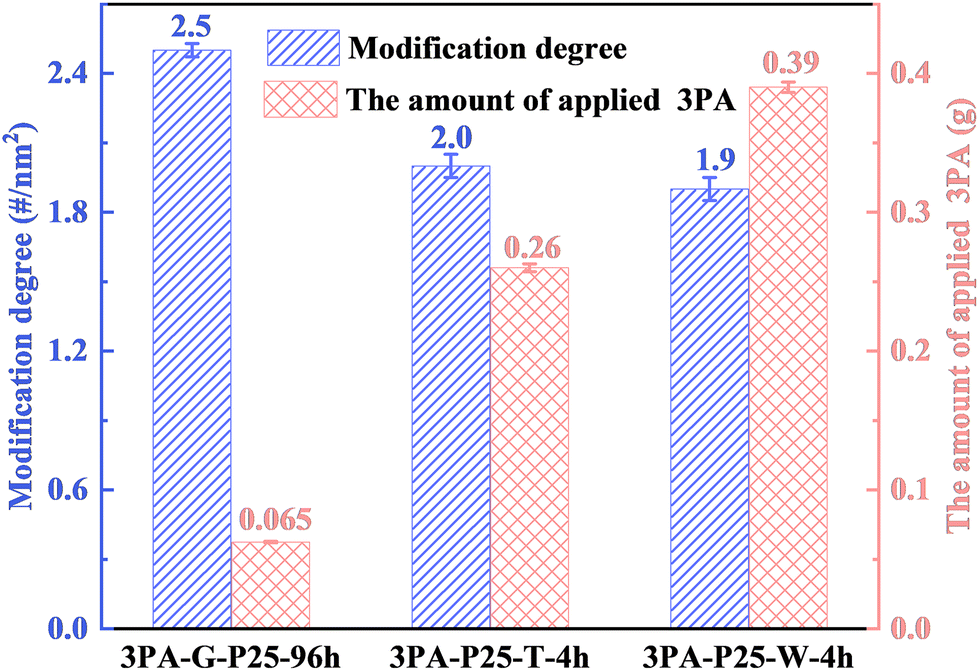 | ||
| Fig. 7 The modification degrees and the amount of 3PA used in samples prepared by the solid- and liquid-phase (water- or toluene-based) methods at 90 °C using the same amount of TiO2 (1.0 g). The error bars were calculated as standard deviation errors based on three repeated experiments. | ||
In Fig. 7, the modification degrees reached by the liquid-phase water- or toluene-based method can be regarded as the maximal modification degrees that can be reached at 90 °C by the liquid-phase method, according to our previous research.14 However, the maximal modification degrees obtained by the liquid-phase method in water or toluene (1.9 #/nm2 or 2.0 #/nm2) are still lower than the modification degree achieved by the here applied solid-phase method (2.5 #/nm2, obtained after a 96-hours thermal treatment). Therefore, higher surface coverage with 3PA, i.e., the reactivity of 3PA towards TiO2, seems to be suppressed in the liquid-phase method, irrespective of the type of solvent used (water or toluene). This higher modification degree obtained from the solid-phase method is desired in some applications, such as CO2 adsorption,18 Pd recovery,19etc. Moreover, apart from differences in surface coverage, the formation of undesired side product, titanium phosphonate, was formed in liquid-phase water-based sample, (3PA-P25-W-4 h, 1.9 #/nm2), while was limited for the sample prepared by the solid-phase method (3PA-G-P25-96 h, 2.5 #/nm2, Fig. S17 and S18†). This phenomenon again highlights merits of the proposed solid-phase method. See ESI† for detailed analysis.
Furthermore, Fig. 7 indicates that the solid-phase method achieves a 25.0% higher modification degree than the maximal modification degree reached by the liquid-phase method even with an 83.3% (in water) or 75.0% (in toluene) lower amount of 3PA in the mixture. Therefore, the here applied solid-phase method shows a pronouncedly improved atom economy, the quantitative discussion of the atom utilization efficiency can be found later.
In-depth assessment of greenness of the solid-phase method
To further evaluate the greenness of the solid-phase method, the atom utilization efficiency of the samples used in Fig. 7, comprising the new solid-state grinding method in comparison to the state-of-the-art liquid-phase methods, was calculated. Moreover, the modification processes to synthesize these samples were comprehensively analyzed and compared (Table 3 and Fig. 8). | ||
| Fig. 8 The human toxicity (a and b) and ecotoxicity (c and d) of the organic solvents used in the solid-phase method and toluene-based liquid-phase method. (a) and (b) The rat's median lethal dose (LD50) and concentration (LC50) values of different solvents, respectively; (c) and (d) the daphnia magna's and minnow's LC50 values of different solvents, respectively. The data is obtained from the safety data sheet (Fisher Chemical, 2023) of these organic solvents. | ||
| Samples | Atom utilization efficiency (%) | Amount of solvents used (mL) | Thermal treatment time (h) | |||||
|---|---|---|---|---|---|---|---|---|
| During the modification process | During the washing steps | |||||||
| Toluene | water | Toluene | Acetone | Ethanol | Water | |||
| 3PA-G-P25-96 h | 39.6 ± 0.5 | ✗ | ✗ | ✗ | ✗ | 90 | 90 | 96 |
| 3PA-P25-T-4 h | 8.3 ± 0.2 | 20 | ✗ | 90 | 90 | ✗ | 90 | 4 |
| 3PA-P25-W-4 h | 5.3 ± 0.1 | ✗ | 20 | ✗ | ✗ | ✗ | 90 | 4 |
Table 3 summarizes the atom utilization efficiency, the required amount of solvents and reaction/thermal treatment time in synthesizing these samples (in Fig. 7) by the solid- and liquid-phase methods. The atom uitlization efficiency of the solid-phase method is much higher than that of the liquid-phase method (toluene- or water-based), which is almost 4.8 times (toluene-based) or 7.5 (water-based) times as efficient. To further confirm the higher atom utilization efficiency of the solid-phase method, another organophsophonic acid, phenylphosphonic acid (PhPA), was grafted onto P25 (Fig. S19 and Table S3†). The results indicate that the atom utilization efficiency of the solid-phase method (28.2%) is again much higher than that of the liquid-phase toluene- (5.0%) or water-based (6.6%) methods, being approximately 5.6 times (toluene-based) or 4.3 (water-based) times as efficient, respectively. These results confirm the improved atom efficiency of the solid-phase method also when using a different PA with another functional group (phenyl). The better atom economy can also indirectly lower the impact of the synthesis of the PAs, as less PAs is needed to achieve the same modification degree. This means that lower amounts of reactants, such as phosphonates and concentrated HCl (35%–37% in water)/HBr, are needed, and less waste, such as excess (HCl/HBr), is produced.48 For some PAs, the improved atom economy might also provide financial benefits for the modification, although this depends on the type of PA: e.g. 3PA costs ∼38 € per g vs. PhPA ∼0.39 € per g.
Additionally, as compared to the liquid-phase toluene- and water-based methods, the solid-phase method also omits the use of solvent during the modification process. Therefore, the solid-phase method reduces the mass of solvent waste besides the amounts of PAs required (based on its high atom utilization efficiency). Table 3 also shows that the solid-phase method requires longer thermal treatment (96 h) than the liquid-phase methods (4 h) during the modification process. However, the reaction temperature is only 90 °C, which does not require much energy and can be achieved by using waste heat, such as steam or electrical heating. Therefore, the longer reaction time is not necessarily an issue. While the use of solvents during the modification process in the liquid-phase methods is a problem that can only be addressed by avoiding them.
Although washing remains necessary after the modification, the solid-phase method uses less amounts of organic solvent (90 mL of ethanol) as compared to the toluene-based liquid-phase method (90 mL toluene and 90 mL of acetone) during the washing steps. Moreover, it avoids the use of toxic organic solvents, using ethanol instead of toluene and acetone. More detailed discussion about the greenness or toxicity of the applied solvents will be provided later (Fig. 8). To sum up, this proposed method exhibits greater atom utilization efficiency and/or is greener than the liquid-phase methods (toluene- or water-based).
Fig. 8 further compares the human toxicity and ecotoxicity, indicated by median lethal dose (LD50) or concentration (LC50), of the organic solvents used during the washing and/or synthesis steps for the proposed solid-phase method (ethanol) and conventional toluene-based liquid-phase method (toluene and acetone). Fig. 8a–d display that toluene has the lowest LD50 and LC50 values among these three organic solvents, indicating its highest human toxicity and ecotoxicity. Ethanol has the highest LD50 and LC50 values, demonstrating the greener nature among the three organic solvents. Acetone has a medium human toxicity and ecotoxicity in this case. Similarly, the Chem21 solvent selection guide49 and GlaxoSmithKline's(GSK) solvent selection guide50 also reported that ethanol is a recommended solvent, while toluene is highlighted as a problematic solvent and acetone is intermediate between them, based on the combined metrics of safety, health and environment. Therefore, it is clear that the solid-phase method is greener.
Further improvement could however still be achieved by optimizing the solid-state method to a water-based washing method. A critical note, although the solid-phase method is greener and more atom efficient, it has one drawback with respect to sustainability, as it requires longer heating during the post-treatment. Its impact depends on the duration of the thermal treatment and the energy source used. If electrical furnaces would be used, the energy mix used will determine its footprint, which is expected lower when a renewable energy source such as solar or wind is applied.
Conclusions
In this work, a straightforward and controllable solid-phase manual grinding method (i.e. low energy) is proposed to graft propylphosphonic acid on a commercially available TiO2 (P25) support. It (i) avoids the suppressive role of the solvent on achieving a high modification degree; and (ii) preserves the native crystal phase of the support as compared to the reactive milling method. Furthermore, it allows to decrease environmental harm or health risk while increase atom economy compared to liquid-phase methods.In contrast to shaker mixing, the manual grinding process promotes the grafting interaction between 3PA and P25. Moreover, a post-thermal treatment at 90 °C, explored as a function of duration, allows to further increase the modification degree. For the same reaction temperature and 3PA/TiO2 amounts, the solid-phase method shows an increase in modification degree with increasing duration of the thermal treatment, while little to no changes were found for samples obtained by the liquid-phase method.
Importantly, the green solid-phase method allows to achieve a modification degree that is 25.0% higher than the maximal modification degree obtained by the liquid-phase methods. It is worth to note that it does so even with an 83.3% or 75.0% reduction in 3PA usage, while at the same time excluding the use (and purification) of solvents. Therefore, this method clearly demonstrates a prominent atom economy advantage (7.5 or 4.8 times atom economy of the liquid-phase methods). Moreover, the atom economy merit was also confirmed for PhPA-modified TiO2. Therefore, the atom economy improved by the solid-phase method is regardless of the type of PAs.
Additionally, this work provides some insights into the reaction engineering principles necessary for scale-up, i.e., providing appropriate energy. This means that mechanical grinding can be used, when the energy generated/provided in the system is sufficiently controlled. However, further exploration is needed to identify how this control should be implemented (e.g., grinding time, grinding rate, grinding system and materials, possible use of cooling) to ensure effective scale-up.
Author contributions
Kaimin Zhang: conceptualization, methodology, formal analysis, validation, investigation, writing-original draft; Jinxin Wang: conceptualization, methodology, validation, investigation, writing – review & editing; Nick Gys: validation, investigation, methodology, writing-review & editing; Elien Derveaux (31P-MAS NMR): validation, investigation, writing-review & editing; Nahal Ghanemnia (31P-MAS NMR): validation, investigation, writing-review & editing; Wouter Marchal (31P-MAS NMR): supervision, conceptualization, resources, writing-review & editing. Peter Adriaensens (31P-MAS NMR): supervision, conceptualization, resources, writing-review & editing. Vera Meynen: supervision, conceptualization, methodology, resources, writing-review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the EASICHEM project funded by the Flemish Strategic Basic Research Program of the Catalisti cluster and Flanders Innovation & Entrepreneurship (HBC.2018.0484). J. Wang (201806060123) acknowledges the supporting from the China Scholarship Council (CSC). N. Gys, N. Ghanemnia and E. Derveaux acknowledge EOS project PHOSPORE funded by FWO–FNRS with grant number G0H0522N (EOS ID: 40007504). This work is also supported by the Research Foundation Flanders (FWO) and Hasselt University via the Hercules project (AUHL/15/2−GOH3816N). The XRD equipment was funded by an UAntwerp BOF equipment project. We thank Wouter Van Hoey for the Rietveld data process, and thank Prof. Christophe Vande Velde for the discussion about the results of Rietveld analysis.References
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS PubMed.
- P. J. Hotchkiss, S. C. Jones, S. A. Paniagua, A. Sharma, B. Kippelen, N. R. Armstrong and S. R. Marder, Acc. Chem. Res., 2012, 45, 337–346 CrossRef CAS PubMed.
- G. Guerrero, J. G. Alauzun, M. Granier, D. Laurencin and P. H. Mutin, Dalton Trans., 2013, 42, 12569–12585 RSC.
- M. Cichomski, K. Kośla, J. Grobelny, W. Kozłowski and W. Szmaja, Appl. Surf. Sci., 2013, 273, 570–577 CrossRef CAS.
- B. M. Silverman, K. A. Wieghaus and J. Schwartz, Langmuir, 2005, 21, 225–228 CrossRef CAS PubMed.
- I. L. Liakos, R. C. Newman, E. McAlpine and M. R. Alexander, Surf. Interface Anal., 2004, 36, 347–354 CrossRef CAS.
- J. Zhang, S. Deo, M. J. Janik and J. Will Medlin, J. Am. Chem. Soc., 2020, 142, 5184–5193 CrossRef CAS PubMed.
- C. Theile-Rasche, M. Wiesing, S. Schwiderek, M. Noeske and G. Grundmeier, Appl. Surf. Sci., 2020, 513, 145701 CrossRef CAS.
- G. Mustafa, K. Wyns, S. Janssens, A. Buekenhoudt and V. Meynen, Sep. Purif. Technol., 2018, 193, 29–37 CrossRef CAS.
- G. Mustafa, K. Wyns, P. Vandezande, A. Buekenhoudt and V. Meynen, J. Membr. Sci., 2014, 470, 369–377 CrossRef CAS.
- L. D. Ellis, J. Ballesteros-Soberanas, D. K. Schwartz and J. W. Medlin, Appl. Catal., A, 2019, 571, 102–106 CrossRef CAS.
- A. H. Jenkins and J. W. Medlin, Acc. Chem. Res., 2021, 54, 4080–4090 CrossRef CAS PubMed.
- M.-J. Bañuls, R. Puchades and Á. Maquieira, Anal. Chim. Acta, 2013, 777, 1–16 CrossRef PubMed.
- K. Zhang, J. Wang and V. Meynen, Surf. Interfaces, 2024, 44, 103697 CrossRef CAS.
- A. Roevens, J. G. Van Dijck, M. Tassi, J. D'Haen, R. Carleer, P. Adriaensens, F. Blockhuys and V. Meynen, Mater. Chem. Phys., 2016, 184, 324–334 CrossRef CAS.
- A. Roevens, J. G. Van Dijck, D. Geldof, F. Blockhuys, B. Prelot, J. Zajac and V. Meynen, Appl. Surf. Sci., 2017, 416, 716–724 CrossRef CAS.
- X. Chen, E. Luais, N. Darwish, S. Ciampi, P. Thordarson and J. J. Gooding, Langmuir, 2012, 28, 9487–9495 CrossRef CAS PubMed.
- C. C. Aquino, G. Richner, M. C. Kimling, D. Chen, G. Puxty, P. H. M. Feron and R. A. Caruso, J. Phys. Chem. C, 2013, 117, 9747–9757 CrossRef CAS.
- N. Gys, B. Pawlak, L. L. Lufungula, K. Marcoen, K. Wyns, K. Baert, T. A. Atia, J. Spooren, P. Adriaensens, F. Blockhuys, T. Hauffman, V. Meynen, S. Mullens and B. Michielsen, RSC Adv., 2022, 12, 36046–36062 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- R. An, L. C. Quiñones, N. Gys, E. Derveaux, K. Baert, T. Hauffman, P. Adriaensens, F. Blockhuys and V. Meynen, Appl. Surf. Sci., 2023, 639, 158179 CrossRef CAS.
- E. L. Hanson, J. Schwartz, B. Nickel, N. Koch and M. F. Danisman, J. Am. Chem. Soc., 2003, 125, 16074–16080 CrossRef CAS PubMed.
- A. Vega, P. Thissen and Y. J. Chabal, Langmuir, 2012, 28, 8046–8051 CrossRef CAS PubMed.
- A. Bulusu, S. A. Paniagua, B. A. MacLeod, A. K. Sigdel, J. J. Berry, D. C. Olson, S. R. Marder and S. Graham, Langmuir, 2013, 29, 3935–3942 CrossRef CAS PubMed.
- S. Gupta and H. Gleskova, Org. Electron., 2013, 14, 354–361 CrossRef CAS.
- G. Ilia, V. Simulescu, R. Gheonea, E. Crasmareanu and I. Hulka, J. Iran. Chem. Soc., 2021, 18, 1815–1823 CrossRef CAS.
- A. Betke and G. Kickelbick, New J. Chem., 2014, 38, 1264–1270 RSC.
- A. Fischer, C. Ney and G. Kickelbick, Eur. J. Inorg. Chem., 2013, 5701–5707 CrossRef CAS.
- A. Betke and G. Kickelbick, Inorganics, 2014, 2, 410–423 CrossRef CAS.
- B. Tryba, P. Rychtowski, A. Markowska-Szczupak and J. Przepiórski, Catalysts, 2020, 10, 1–26 CrossRef.
- A. Chakraborty, Samriti, O. Ruzimuradov, R. K. Gupta, J. Cho and J. Prakash, Environ. Res., 2022, 212, 113550 CrossRef CAS PubMed.
- K. Zhang, J. Wang, R. Ninakanti and S. W. Verbruggen, Chem. Eng. J., 2023, 474, 145188 CrossRef CAS.
- N. Gys, B. Pawlak, K. Marcoen, G. Reekmans, L. F. Velasco, R. An, K. Wyns, K. Baert, K. Zhang, L. L. Lufungula, A. Piras, L. Siemons, B. Michielsen, S. Van Doorslaer, F. Blockhuys, T. Hauffman, P. Adriaensens, S. Mullens and V. Meynen, Chempluschem, 2023, n/a, e202200441 CrossRef PubMed.
- J. McElwee, R. Helmy and A. Y. Fadeev, J. Colloid Interface Sci., 2005, 285, 551–556 CrossRef CAS PubMed.
- Y. Yang, Q. Xia, M. Feng and P. Zhang, Am. Mineral., 2010, 95, 1439–1443 CrossRef CAS.
- A. Mahdavi-Shakib, J. M. Arce-Ramos, R. N. Austin, T. J. Schwartz, L. C. Grabow and B. G. Frederick, J. Phys. Chem. C, 2019, 123, 24533–24548 CrossRef CAS.
- S. Pawsey, M. McCormick, S. De Paul, R. Graf, Y. S. Lee, L. Reven and H. W. Spiess, J. Am. Chem. Soc., 2003, 125, 4174–4184 CrossRef CAS PubMed.
- G. Socrates, Infrared and Raman characteristic group frequencies. Tables and charts, 2001 Search PubMed.
- B. Guo, X. Lin, P. Liu, Y. Zeng and H. Fan, Mater. Lett., 2019, 236, 85–88 CrossRef CAS.
- A. H. Carrieri and J. O. Jensen, Phosphorus, Sulfur Silicon Relat. Elem., 1992, 66, 1–11 CrossRef.
- D. Geldof, M. Tassi, R. Carleer, P. Adriaensens, A. Roevens, V. Meynen and F. Blockhuys, Surf. Sci., 2017, 655, 31–38 CrossRef CAS.
- W. Gao, L. Dickinson, C. Grozinger, F. G. Morin and L. Reven, Langmuir, 1996, 12, 6429–6435 CrossRef CAS.
- C. Morterra, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 1617–1637 RSC.
- M. Takeuchi, G. Martra, S. Coluccia and M. Anpo, J. Near Infrared Spectrosc., 2009, 17, 373–384 CrossRef CAS.
- K. Li, Z. Huang, X. Zeng, B. Huang, S. Gao and J. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 11577–11586 CrossRef CAS PubMed.
- Y. R. Lv, C. J. Liu, R. K. He, X. Li and Y. H. Xu, Mater. Res. Bull., 2019, 117, 35–40 CrossRef CAS.
- J. G. Van Dijck, H. Lenaerts, L. Siemons, F. Blockhuys and V. Meynen, Surf. Interfaces, 2020, 21, 100710 CrossRef CAS.
- C. M. Sevrain, M. Berchel, H. Couthon and P.-A. Jaffrès, Beilstein J. Org. Chem., 2017, 13, 2186–2213 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03330b |
| ‡ School of Electrical and Electronic Engineering, Hubei University of Technology, Wuhan 430068, China (current address). |
| This journal is © The Royal Society of Chemistry 2024 |