
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of α-methylene-δ-valerolactone and its selective polymerization from a product mixture for concurrent separation and polymer production†
Alexander A.
Khechfe
 a,
Francesca D.
Eckstrom
b,
Eswara Rao
Chokkapu
b,
Lucas A.
Baston
a,
Bowei
Liu
a,
Eugene Y.-X.
Chen
*b and
Yuriy
Román-Leshkov
*a
a,
Francesca D.
Eckstrom
b,
Eswara Rao
Chokkapu
b,
Lucas A.
Baston
a,
Bowei
Liu
a,
Eugene Y.-X.
Chen
*b and
Yuriy
Román-Leshkov
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA. E-mail: yroman@mit.edu
bDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523-1872, USA. E-mail: eugene.chen@colostate.edu
First published on 18th September 2024
Abstract
We report the continuous, gas-phase synthesis of α-methylene-δ-valerolactone (MVL) from δ-valerolactone (DVL) and formaldehyde (FA) over alkaline earth oxide catalysts. MgO, CaO, and BaO supported on silica (∼5 wt%) were active for MVL production (613 K, 0.4 kPa DVL, 1.2 kPa FA, 101 kPa total pressure). CaO and BaO showed 90% and 83% selectivity to MVL at ∼60% DVL conversion, respectively. Decreasing contact times improved MVL selectivity for all three catalysts, achieving near quantitative selectivity at DVL conversions <40% with CaO. Further studies with CaO indicated that increasing the FA partial pressure for a given DVL partial pressure negligibly changed conversion while maintaining high selectivity; however, increasing the reaction temperature generally resulted in lower MVL selectivity. Deactivation and carbon loss were attributed to non-volatile compound formation from series and parallel reactions that consume MVL and DVL and poison the catalyst surface. These side reactions were more pronounced at high temperatures and higher contact times. While slow deactivation poses a challenge, the catalyst could be fully regenerated by calcining at 773 K for 4 h under flowing air. As the product mixture of MVL and DVL is difficult to separate, we developed a selective polymerization strategy to convert either one or both monomers into valuable polymeric materials, thereby achieving efficient separation and concurrent polymer production. Using a model mixture of 30 wt% of MVL in DVL, vinyl-addition polymerization converted MVL to the corresponding vinyl polymer (PMVL)VAP in 98% yield, while DVL was recovered in 96% yield by distillation. Alternatively, ring-opening polymerization of the same mixture resulted in a DVL/MVL copolyester and separatable vinyl homopolymer P(MVL)VAP.
Introduction
Plastics have found use in nearly every aspect of modern life due to their light weight, chemical stability, and tunable mechanical and optical properties.1 As the widespread use of plastics continues to grow, concerns over their environmental impact due to disposal and production have driven the desire for more efficient recycling processes.2–4 State-of-the-art mechanical recycling technologies address only ∼10% of plastic waste production, necessitating new methods to mitigate the growing environmental catastrophe.4Chemical recycling, a method to break a polymer back down to its respective monomer with heat and/or a catalyst for producing virgin-quality polymers repeatedly, has recently attracted attention for recycling polymers that may not be suitable for mechanical processing.5–7 However, the high thermostability of commodity polymers, especially C–C bonded vinyl polymers such as polyolefins and poly(meth)acrylates, makes chemical recycling challenging due to high energy requirements and low selectivity. Common chemical recycling methods for polyethylene and polypropylene include hydrogenolysis,8–11 hydrocracking,12,13 or pyrolysis,14,15 processes that rarely yield the starting monomer in a single step. Several processes have been studied for depolymerizing poly(methyl methacrylate) (PMMA) into methyl methacrylate (MMA) with more success, but these methods require temperatures above 723 K and careful control of PMMA residence time to avoid side reactions that reduce MMA yields.16–18
Efforts to develop biorenewable and recyclable alternatives to PMMA have identified bio-based vinyl lactones as effective monomers for synthesizing chemically recyclable acrylic bioplastics.19–21 Notably, adding a vinyl group to the α-position of bio-based δ-valerolactone (DVL) yields α-methylene-δ-valerolactone (MVL), which can be polymerized via vinyl-addition polymerization (VAP) to form the acrylic (vinyl) polymer (PMVL)VAP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19,20 or ring-opening polymerization (ROP) to form the polyester (PMVL)ROP.19,21,22 (PMVL)ROP can be recycled chemically, with a 96% yield of MVL after heating at 423 K at 0.01 torr for 6 h with a stannous octoate catalyst.19 Further, (PMVL)ROP with a molecular weight of 74.6 kDa has also been demonstrated as a mechanically tough, chemically recyclable thermoplastic with mechanical properties comparable to commodity polyolefins.21 Most recently, (PMVL)VAP has been shown to be quantitatively recyclable, recovering the pure monomer MVL in 99.5% yield from reactive distillation of bulk (PMVL)VAP at 220 °C in 1 h without a catalyst.23
19,20 or ring-opening polymerization (ROP) to form the polyester (PMVL)ROP.19,21,22 (PMVL)ROP can be recycled chemically, with a 96% yield of MVL after heating at 423 K at 0.01 torr for 6 h with a stannous octoate catalyst.19 Further, (PMVL)ROP with a molecular weight of 74.6 kDa has also been demonstrated as a mechanically tough, chemically recyclable thermoplastic with mechanical properties comparable to commodity polyolefins.21 Most recently, (PMVL)VAP has been shown to be quantitatively recyclable, recovering the pure monomer MVL in 99.5% yield from reactive distillation of bulk (PMVL)VAP at 220 °C in 1 h without a catalyst.23
Despite PMVL's promise as a chemically recyclable polymer, the synthesis of MVL has only been demonstrated in batch processes at the lab-scale (∼10–100 g). Typically, this synthesis involves the reaction of DVL with ethyl formate and paraformaldehyde using stoichiometric bases like NaH,19,24 resulting in both high costs and hazardous waste streams. Sustainable and industrial-scale production of PMVL will require the development of a continuous catalytic process for the synthesis of the MVL monomer. Continuous synthesis of the MVL isomer, α-methylene-γ-valerolactone (MGVL), has been reported in the vapor-phase via the aldol condensation of γ-valerolactone (GVL) and formaldehyde (FA) over silica supported alkali and alkaline earth oxide catalysts, with BaO/SiO2 showing >95% selectivity to MGVL at 613 K.25 This reaction was also performed in the liquid-phase over supported Cs oxides, with the highest MGVL production rate observed on 5 wt% Cs oxide supported on a hierarchical beta zeolite (553–583 K, methyl-tetrahydrofuran solvent).26 This chemistry has been further explored in the patent literature.27,28
The reactivity of these oxide catalysts is attributed to acid–base pairs on the metal oxides.29 During the aldol condensation, the carbonyl group of an enolizable aldehyde or ketone binds to the Lewis acidic metal center (e.g. Ba2+ or Cs+), while the neighboring basic lattice oxygen deprotonates the α-hydrogen to form a carbanion. The carbanion can then perform a nucleophilic attack on an adjacent carbonyl to form an aldol product, which can then be dehydrated to form the final aldol condensation product.30 Indeed, a wealth of literature exists on liquid-phase coupling of biomass-derived carbonyls to form biofuels using base catalysts, particularly MgO.31–34
In this work, we studied the aldol condensation of DVL with FA over three supported alkaline earth oxides for the production of MVL (Scheme 1). MgO, CaO, and BaO supported on silica (∼5 wt%) were all active for MVL production (613 K, 0.4 kPa DVL, 1.2 kPa FA, 101 kPa total pressure). CaO and BaO showed 90% and 83% selectivity to MVL at ∼60% DVL conversion, respectively. The results obtained with relatively simple catalysts provide a foundation for further catalyst engineering and mechanistic studies. Our process yielded a product mixture of MVL and unconverted DVL. A model mixture approximating the optimized product composition was subjected to selective polymerization, achieving efficient separation and producing valuable acrylic and polyester materials.
 | ||
| Scheme 1 Aldol condensation reaction pathway for MVL synthesis studied in this work. | ||
Experimental methods
Catalyst synthesis
Supported alkaline earth oxide catalysts were prepared via incipient wetness impregnation. Fumed silica (SiO2, Sigma Aldrich) was used as a support and calcined at 873 K for 6 h (4 K min−1 ramp rate) prior to impregnation. Magnesium acetate tetrahydrate, calcium acetate monohydrate, or barium acetate (all Sigma Aldrich, >99%) were dissolved in enough DI water to saturate the pore volume of a given mass of SiO2 (∼2 gwater gSiO2−1). The metal salt solution was then slowly deposited onto the SiO2, followed by grinding and mixing in a mortar and pestle. The pre-catalyst was then dried for at least 6 h in an oven at 373 K, ground again, and calcined at 773 K (ramp rate 4 K min−1) for 6 h.Catalyst characterization
Weight loadings of Mg, Ca, and Ba on SiO2 were determined by inductively coupled plasma mass spectrometry (ICP-MS) using an Agilent 7900 ICP-MS instrument. Approximately 10 mg of catalyst powder was added to a 15 mL polypropylene centrifuge tube and dissolved in ∼1 mL of 68.0% HNO3 (Veritas Double Distilled, GFS Chemicals Inc.) overnight, followed by dilution in 2 wt% HNO3 and filtration through a 0.2 μm PTFE syringe filter (VWR) to remove the undissolved SiO2 support. Solutions were further diluted to obtain metal concentrations of 100–300 ppb for Mg and Ba. Ca solutions were diluted to a lesser extent (30–60 ppm) to monitor 44Ca (∼2% abundance) due to interference between 40Ca and the Ar plasma. Metal concentrations were determined from calibration curves prepared from standard solutions of 1000 ppm of Mg, Ca, and Ba (all TraceCERT, Sigma-Aldrich), diluted in 2 wt% HNO3.Scanning transmission electron microscopy (STEM) imaging was performed on a probe-corrected Thermo Fisher Scientific Themis Z G3, operated at 200 kV. Images were collected with a 19 mad collection angle and either 70–200 mrad (Z-contrast, HAADF) or 35–200 mrad (diffraction contrast). Transmission electron microscopy (TEM) was performed on an FEI Tecnai F20 electron microscope to determine particle size distributions for each sample. Catalysts were dispersed in ethanol and sonicated until well-dispersed and then dropped onto a copper grid with a carbon film. Measurements were performed using an electron detector at a 200 kV acceleration voltage. Powder X-ray diffraction (PXRD) was used to determine the crystallinity of metal oxide particles. Patterns were collected on a Bruker D8 diffractometer with a Cu Kα radiation source between 2θ = 20–80° and a scan rate of 0.05° s−1.
Carbon dioxide temperature programmed desorption (CO2 TPD) was performed in a Micromeritics Autochem II 2920 unit equipped with a thermal conductivity detector. Approximately 0.15 g of sample was loaded into a quartz U-tube and held in place with plugs of quartz wool on both sides. The sample was pretreated in 50 mL min−1 He at 473 K for 2 h and cooled down to 313 K. The sample was then dosed with 50 mL min−1 of a 1% CO2 in N2 and 30 mL min−1 He mixture for 30 minutes, followed by a 10 minutes purge in 80 mL min−1 of He. This procedure was repeated an additional two times to saturate the surface with CO2. The sample was then heated in a 80 mL min−1 He flow while the temperature was ramped from 313 K to 1073 K at a 10 K min−1 ramp rate while monitoring the reactor effluent. Thermogravimetric analysis (TGA) was performed on a TA Instruments Q500 System. The sample was loaded onto a tared platinum pan and equilibrated at 298 K in 45 mL min−1 of air and 5 mL min−1 N2. After equilibration, the temperature was ramped at 1 K min−1 to 1073 K.
Continuous catalytic MVL synthesis
Aldol condensation between DVL and FA were performed in a gas-phase, fixed-bed reactor. Catalysts were pelletized, crushed, and sieved between 40–60 mesh to reduce the pressure drop across the catalyst bed. Approximately 0.1 g of catalyst was mixed with 0.4 g of silicon carbide (46 mesh) as a diluent and loaded into a ¼′′ 316 stainless steel tube. The catalyst bed was held in place with a plug of quartz wool and topped with ∼1.5 g of borosilicate glass beads to facilitate gas-phase mixing. The bed was heated in a furnace (ATS Systems) and temperature was controlled via a K-type thermocouple touching the base of the catalyst bed connected to a Cole-Parmer Digi-Sense Temperature Controller R/S 68900-11.A mixture of DVL (technical grade, Sigma Aldrich) and formalin (37 wt% FA in water with methanol stabilizer, Sigma Aldrich) were introduced via a Cole-Parmer Masterflex Single-Syringe Infusion Pump with a typical liquid flow rate of ∼0.16 mL h−1. The liquid feed was vaporized at ∼423 K into a stream of N2 (Airgas) with a typical flow rate of 50 mL min−1. Reactor effluents were analyzed by an on-line gas chromatograph (Agilent 8890) equipped with a 30 m HP-5MS-UI column and a flame ionization detector (FID). Product selectivities are calculated as  , where Si is the selectivity of product i,
, where Si is the selectivity of product i,  is the initial molar flow rate of DVL into the reactor, and
is the initial molar flow rate of DVL into the reactor, and  and
and  are the molar flow rates of DVL and product i in the effluent, respectively. Contact times are calculated as moles of active metal loaded in the reactor divided by the molar flow rate of DVL. Reaction rates are normalized by total mass of supported catalyst unless stated otherwise.
are the molar flow rates of DVL and product i in the effluent, respectively. Contact times are calculated as moles of active metal loaded in the reactor divided by the molar flow rate of DVL. Reaction rates are normalized by total mass of supported catalyst unless stated otherwise.
Polymer synthesis
The following reagents were used as received: La[N(SiMe3)2]3 (Sigma-Aldrich) and Al(iBu)3 (Sigma-Aldrich). Butylated hydroxy toluene (BHT) was purchased from TCI and purified via vacuum sublimation at 60 °C. Benzyl alcohol (BnOH) initiator, purchased from TCI, was stirred over CaH2 for 12 h under N2 then vacuum distilled at 100 °C under 50 mTorr. HPLC-grade toluene and hexanes were first sparged extensively with nitrogen during filling 20 L solvent reservoirs and then dried by passage through activated alumina. They were stored under N2 and over molecular sieves. Air and moisture-sensitive reactions were conducted in oven- or flame-dried glassware on a dual manifold N2/vacuum Schlenk line or inside an N2-filled glovebox.DVL was purchased from TCI and MVL was prepared according to the literature procedure.19 They were purified by stirring over CaH2 for 12 h under N2 prior to vacuum distillation at 50 °C under 50 mTorr. The purified monomers were brought into an N2 atmosphere glovebox and filtered through a 0.22 μm filter. They were then mixed to give 30 wt% MVL in DVL and stored at −30 °C in a brown bottle over activated 4 Å molecular sieves. Al(iBu)2BHT was prepared by adding a hexanes solution of Al(iBu)3 dropwise to one equivalent of BHT in hexanes, according to similar literature procedures.35 After letting stir overnight, the solvent was removed in vacuo to reveal a clear viscous liquid. [La(OBn)3]x (oligomeric species) was prepared by mixing La[N(SiMe3)2]3 and BnOH at a 1:3 ratio in toluene. The reaction was allowed to stir for 3 h after which the solvent was removed in vacuo with light heating, a procedure known to also remove the co-product NH(SiMe3)2.36 The resulting solid was washed with and decanted from hexanes and dried at 50 mTorr.
VAP was carried out as follows: in an N2-supplied atmosphere glovebox, 0.333 g of 30 wt% MVL in DVL was added to 6.2 mg of Al(iBu)2BHT in a 10 mL Schlenk flask at −30 °C. After 45 min, the reaction was quenched with a few drops of acidified MeOH. An aliquot of the quenched mixture was taken for analysis with 1H NMR, confirming >99% conversion of MVL to P(MVL)VAP, leaving DVL unreacted. The products were separated via vacuum distillation at 60–70 °C under 40 mTorr, recovering 0.223 g DVL (96% yield). The remaining P(MVL)VAP was dried at 100 °C under 40 mTorr over 12 h to afford 0.113 g of the pure polymer (98% yield).
ROP was carried out as follows: in an N2-supplied atmosphere glovebox, a 0.1 mL aliquot of 0.0071 g mL−1 [La(OBn)3]x toluene solution (0.015 M) was added to a 20 mL glass vial charged with 0.2407 g of 30 wt% MVL in DVL in 0.68 mL toluene (3M monomer) at room temperature (∼23 °C) while stirring vigorously ([M]:[La] = 500:1). After 2.25 h, the reaction was quenched with a few drops of acidified methanol and an additional 10 mL toluene were added. A 0.01 mL aliquot of the quenched mixture was taken for analysis with 1H NMR to determine monomer conversion, revealing 94% conversion of DVL to poly(δ-valerolactone) (PVL) and 98% conversion of MVL to poly(α-methylene-δ-valerolactone) (PMVL), consisting of both ring-opening polymerization (ROP) product P(MVL)ROP and vinyl-addition polymerization (VAP) product P(MVL)VAP in a 29:71 ratio. The mixture was filtered to remove the insoluble minor product PMVLVAP, washed with an additional 50 mL toluene, condensed, and crashed into an excess of cold methanol. The methanol was decanted to reveal a white solid, which was solubilized in CHCl3 and precipitated an additional two times. The purified polymer was dried at ambient temperature for 12 h under 50 mTorr to obtain 0.1346 g ROP copolyester P(MVL-co-VL) (56% yield) with roughly 10% PMVLROP incorporation. The yield of P(MVL-co-VL) was calculated by dividing the isolated yield of the copolyester (0.1346 g) by the initial weight of the monomer mixture (0.2407 g) used for the polymerization. The reported value thus considered unconverted monomer, material lost to P(MVL)VAP formation, and any product loss during the filtration to remove P(MVL)VAP.
Polymer characterization
NMR spectra were collected on a Bruker AV-III 400 MHz spectrometer (400 MHz, 1H; 100 MHz, 13C). Chemical shifts for 1H and 13C spectra were referenced to an internal solvent resonance 7.26 (chloroform) and are reported as parts per million relative to SiMe4. Monomer conversion and copolymer composition were calculated based on relative integrations between monomer and polymer 1H NMR signals.All thermal analyses were performed on polymer samples that were dried at 25 °C for 12 h under 50 mTorr (polyester) or 100 °C for 12 h under 40 mTorr (acrylic polymer). Differential Scanning Calorimetry (DSC) was performed on an Auto Q20, TA Instrument. DSC plots show the melting transition temperature (Tm), crystallization temperature (Tc), and heating of fusion (ΔHf), obtained from a second heating scan. Each cycle was performed at a rate of 10 °C min−1. Decomposition temperatures (Td, defined by the temperature of 5% weight loss) and maximum rate decomposition temperatures (Tmax) of the polymers were measured by thermal gravimetric analysis (TGA) on a Q50 TGA Analyzer, TA Instrument, by heating from ambient temperature to 700 °C at 10 °C min−1 under N2. Values of Tmax were obtained from derivative (wt%/°C) vs. temperature (°C) plots, while Td values were obtained from wt% vs. temperature (°C) plots.
Results and discussion
Catalyst characterization
The presence of alkaline earth metals on the silica supports was confirmed via ICP-MS with their respective weight loadings shown in Table S1.† Reported weight loadings correspond to metal loadings on silica. High-angle annular dark-field scanning transmission electron microscopy (STEM-HAADF, Fig. S1†) and transmission electron microscopy (TEM, Fig. S2–S4†) demonstrate that there is no significant crystalline formation on the surface of the catalysts indicating a high degree of metal oxide dispersion. Greater detail can be found in the ESI.† X-ray diffraction patterns for each of the three catalysts only show a broad peak corresponding to SiO2 at a 2θ value of ∼22°, corroborating the absence of large crystalline domains over these samples (Fig. S5†).CO2 binds to basic lattice oxygens on metal oxides and can thus be used to probe the presence and relative strength of basic sites on these materials.37 CO2 temperature programmed desorption (TPD) was used for this purpose, with curves shown in Fig. S6.† Desorption features at temperatures greater than 600 K in CaO and BaO indicate that these two catalysts have stronger basic sites than MgO, which only shows desorption features below 600 K.
Reactivity studies
Each of the three catalysts in Table S1† was tested for its activity and selectivity toward MVL production from DVL and FA. Introducing 0.4 kPa DVL and 1.2 kPa FA at 613 K into a bed of each catalyst resulted in the formation of MVL and small amounts of other side products, including GVL and α-methyl-δ-valerolactone (methyl-DVL) (representative GC chromatogram shown in Fig. S7†). The formation of the desired MVL isomer was confirmed using proton nuclear magnetic resonance (1H NMR) by the presence of vinyl proton peaks at 5.5 ppm and 6.3 ppm (Fig. S8†). Gas chromatography mass spectrometry (GC-MS) spectra of these products can be found in Fig. S9–S11.†Conversions decreased sharply on all catalysts for approximately the first 2 hours on stream after feed introduction, after which a second regime of slower deactivation began. In this second regime, conversion generally decreased by ∼10% over 6–8 hours under most conditions. Conversion, selectivity, and rate data reported herein were collected in this second regime where deactivation could be neglected over the course of an experiment. Periodic catalyst regenerations in air were performed to restore catalyst reactivity. Under conditions of faster deactivation, typically at temperatures above 613 K, conversion and selectivity data are reported from the first hour of the condition before appreciable deactivation occurred.
Fig. 1 compares the selectivity of each of the three catalysts to MVL across a range of contact times (defined as the moles of active metal divided by the molar flow rate of DVL) and their corresponding conversions. The data in Fig. 1 can be found in tabulated format in Table S2.† BaO generally required lower contact times to obtain a given conversion, as compared to MgO or CaO, indicating the active basic sites on BaO are more active for aldol condensation. MVL selectivity was higher for both CaO and BaO than for MgO, with CaO achieving 90% selectivity to MVL at 60% conversion. This result is consistent with the presence stronger basic sites in CaO and BaO as determined by CO2 TPD experiments. MVL selectivity was only ∼40% for MgO at a similar conversion. For MgO, the dominant side product detected in the vapor effluent was GVL (selectivities of 10–25%), indicating the tendency of MgO to catalyze DVL ring-opening reactions in parallel to aldol condensations. DVL is known to isomerize via ring-opening pathways to GVL over Brønsted acid sites.38 However, ring-opening products from DVL or GVL were not detected in the reactor effluent over our system, indicating that they likely only exist as intermediates between DVL and GVL. We attribute this ring-opening isomerization activity to the presence of hydroxyl groups formed in the presence of water, believed to be active sites for the isomerization of glucose to fructose in water over MgO.39 For BaO and CaO, only small amounts (∼1–2%) of GVL and methyl-DVL were detected, with CaO maintaining slightly higher selectivity to MVL at higher DVL conversion than BaO. Methyl-DVL is hypothesized to form via hydrogenation of MVL by methanol present in the formalin feed.
 | ||
| Fig. 1 Product distributions and DVL conversions as functions of contact time for (a) MgO/SiO2, (b) CaO/SiO2 and (c) BaO/SiO2 (613 K, 0.1 g catalyst, 0.4 kPa DVL, 1.2 kPa FA, 101 kPa, balance N2). | ||
The lower carbon balance at high DVL conversions indicates that high contact times result in DVL and MVL forming non-volatiles that cannot be accurately quantified. Fig. S12† shows TGA curves of spent catalysts (run for 6–8 h on stream) to characterize the deactivation of each catalyst. All catalysts exhibited a small weight loss feature at ∼400 K, hypothesized to correspond to surface-bound species that do not desorb after purging in N2 for ∼30 min prior to catalyst unloading. Larger weight loss features were seen near 570–600 K for CaO and BaO, consistent with heavy organic deposits being formed. Weight-loss features persist to ∼700 K for MgO, indicating the potential formation of more coke-like deposits.
Small GC chromatogram peaks associated with by-products were observed at higher retention times/oven temperatures. The intensities of these peaks did not respond to changes in reactor conditions, indicating slow deposition of organic species on the catalyst, consistent with similar studies on the aldol condensation of GVL and FA.25 Analysis of reactor effluents using GC-MS confirmed the presence of compounds with MS features expected of DVL–DVL or DVL–MVL aldol products (Fig. S13†). We thus conclude that the predominant form of both deactivation and MVL selectivity loss are due to parallel reactions forming DVL–DVL dimers and/or series reactions where DVL reacts with the MVL product to form DVL–MVL dimers, both of which are capable of poisoning the catalyst surface. A summary of the reaction pathways hypothesized for this chemistry is depicted in Scheme 2. While this work seeks to elucidate general reactivity trends for this reaction across these materials, the detailed nature of the active sites and mechanisms for the different observed reactions will be the subject of future studies.
 | ||
| Scheme 2 Reaction scheme showing the hypothesized reaction pathways for this system. The desired reaction converting DVL and FA to MVL is shown in blue alongside pathways that consume both DVL and MVL into vapor-phase side products and non-volatile dimers. | ||
CaO showed the highest MVL selectivity at high DVL conversion. Specifically, at conversions <40% for CaO/SiO2, selectivity to MVL was >95% with no observed side products as detected by GC. As with the other two catalysts, increased contact time decreased selectivity to MVL; however, most of the remaining DVL formed non-volatile products that were not detected in the GC. This provides further evidence that MVL was consumed at higher contact times via series reactions with DVL to form non-volatiles. Based on these data, the process can potentially be operated at conversions <40% with nearly quantitative yields to MVL.
Due to its high selectivity toward MVL at elevated conversions, other process parameters were varied over CaO/SiO2. The effect of temperature on DVL conversion and MVL selectivity is shown in Fig. 2a (shown in tabulated form in Table S3†). At a given contact time, DVL conversion increases monotonically with reaction temperature at the expense of MVL selectivity. Fig. 2a shows that at 653 K, a DVL conversion of 54% resulted in an MVL selectivity of 74%. In contrast, Fig. 1b demonstrates that at 613 K, a similar DVL conversion of 59% led to an MVL selectivity of 86%. This indicates that for a given conversion, MVL selectivity decreases at higher temperatures due to the increased prevalence of side reactions. Therefore, increasing temperature to enhance DVL conversion imposes a greater penalty on MVL selectivity compared to achieving the same conversion through increased contact time in the reactor. Increasing the ratio of FA to DVL to 5:1 negligibly increases the conversion of DVL for a given contact based on Fig. 2b while maintaining >95% selectivity to MVL (tabulated in Table S4†). Decreasing the formaldehyde in the feed to a 1:1 ratio with DVL decreases the DVL conversion by ∼15% with a concomitant loss in MVL selectivity. Thus, MVL selectivities can be optimized over CaO/SiO2 at low to moderate DVL conversions with an excess of FA in the feed.
 | ||
| Fig. 2 Product distribution and DVL conversion over CaO/SiO2 as a function of (a) reaction temperature (contact time of 0.27 h, 0.4 kPa DVL, 1.2 kPa FA, 0.1 g catalyst, 101 kPa) and (b) FA:DVL ratio (613 K, contact time of 0.27 h, 0.4 kPa DVL, 100 mg catalyst, 101 kPa). | ||
Transient curves showing MVL production rate vs. time on stream over CaO/SiO2 are shown in Fig. 3, with dashed lines representing a regeneration in 50 mL min−1 air at 773 K for 4 h. After an initial, faster deactivation period during the first ∼2 h on stream, the second regime of slow deactivation begins and is maintained for several hours. Regeneration in air restores the original reactivity, showing that these catalysts are robust to multiple calcination cycles. This also indicates that minimal chemical or structural changes, such as the formation of inactive chemical surfaces or particle sintering, occur during the course of the reaction.
 | ||
| Fig. 3 MVL production rate (per gcat) as a function of time on stream (613 K, contact time of 0.27 h, 0.4 kPa DVL, 1.2 kPa FA, 101 kPa). Dashed lines represent a regeneration in 50 mL min−1 of air for 4 h. | ||
Polymerization of MVL/DVL mixture
The synthesis of MVL from bio-based DVL using SiO2 supported CaO or BaO described herein produced roughly 30 wt% MVL in DVL as a mixture. A 1H NMR spectrum of the reaction mixture is shown in Fig. S14.† Separation of these two monomers is challenging and energy intensive due to their similar boiling points. Given that MVL carries a highly reactive, conjugated (activated) exocyclic double bond, we hypothesized a selective VAP of MVL over DVL should give useful acrylic polymer P(MVL)VAP and DVL (Scheme 3a). Separation of this polymer/monomer mixture could be done simply by filtration or distillation, which not only promotes energy-efficient separation but also produces useful polymer and monomer products. An alternative hypothesis was that, as both MVL and DVL contain the same six-membered DVL ring, a copolymerization of both monomers together via ROP should yield a high-value copolyester product (Scheme 3b).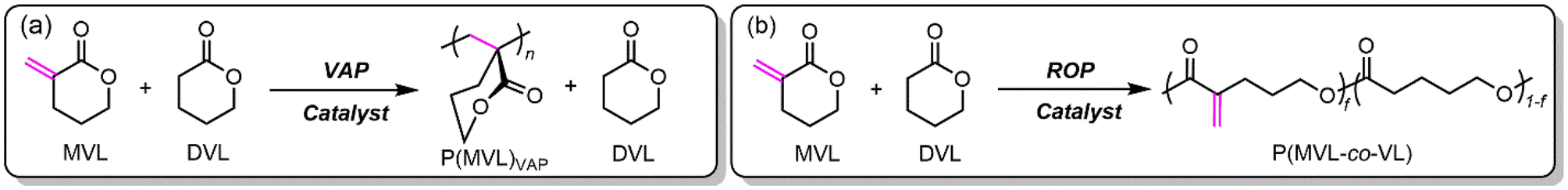 | ||
| Scheme 3 Proposed (co)polymerization strategies to separate the MVL/DVL mixture into (co)polymer and monomer products. (a) Vinyl-addition polymerization pathway to P(MVL)VAP and DVL. (b) Ring-opening polymerization pathway to copolyester P(MVL-co-VL). | ||
The VAP of 30 wt% MVL in DVL (which simulates the product mixture resulted from the MVL synthesis over CaO) was performed without any additional solvent in the presence of a Lewis acid catalyst Al(iBu)2BHT (BHT = butylated hydroxy toluene or 2,6-di-tert-butyl-4-methylphenolate) (1H NMR of catalyst in Fig. S15†). As both an initiator and catalyst, Al(iBu)2BHT was proposed to initiate and catalyze the VAP pathway via a coordination-addition mechanism with the labile alkyl group acting as an initiator (Scheme 4).
 | ||
| Scheme 4 Proposed initiation and propagation mechanism for the VAP pathway to P(MVL)VAP with Al(iBu)2BHT. Nu denotes a nucleophilic (initiating) group on the Al-based catalyst. | ||
The polymerization chemoselectivity was found to be sensitive to reaction temperature. Polymerizations conducted at room temperature (∼23 °C) tended to facilitate ROP of both MVL and DVL, resulting in either crosslinked material or lower recovered yields of DVL. However, when the polymerization of the mixture was conducted at −30 °C, the exclusive selectivity for the VAP of MVL to form P(MVL)VAP was achieved. After 45 min, the polymerization reached >99% conversion of MVL to P(MVL)VAP, leaving DVL unreacted (Fig. S16†). This remarkable selectivity allowed for facile isolation of P(MVL)VAP (98% yield) and recovery of the unreacted DVL (96% yield) via simple vacuum distillation (Fig. S17 and S18†).
The isolated P(MVL)VAP was analyzed by size-exclusion chromatography (SEC) to give a number-average molecular weight of Mn = 25.3 kDa and dispersity of Đ = 1.54 (Fig. S19†). By differential scanning calorimetry (DSC), P(MVL)VAP was shown to exhibit a high glass-transition temperature (Tg) of 201 °C (Fig. S20†). The decomposition temperature (Td, defined by the temperature of 5% weight loss) and maximum rate decomposition temperatures (Tmax) of P(MVL)VAP were obtained by TGA to be 307 °C and 359 °C, respectively (Fig. S21†). The Td value is considerably higher than previously reported Td value (250 °C) for the P(MVL)VAP produced by a free-radical polymerization process.20 These results demonstrated that the direct VAP strategy not only can effectively separate DVL from the MVL/DVL mixture but also produce high-quality acrylic polymer product P(MVL)VAP.
For the ROP of the MVL/DVL mixture, we chose [La(OBn)3]x as the catalyst (which is also an initiator by the OBn ligand), a coordination–insertion polymerization catalyst known to be highly effective towards ROP of lactones and also chemoselective for ROP of bifunctional vinyl lactone monomers.40,41 Catalyst [La(OBn)3]x is often prepared in situ from mixing commercially available La[N(SiMe3)2]3 with three equivalents of benzyl alcohol (BnOH).36,40 To improve the chemoselectivity of MVL towards ROP by minimizing the available VAP pathway, [La(OBn)3]x was isolated from the co-product NH(SiMe3)2 (1H NMR in Fig. S22†). However, bulk polymerizations at room temperature (∼23 °C) were not successful in selecting exclusive ROP. Despite optimization attempts through addition of nonpolar toluene (known to enhance the chemoselectivity for ROP of MVL) and variations in temperature (23 °C, −30 °C, −78 °C), roughly 80% of the MVL in the MVL/DVL mixture consistently polymerized through the VAP pathway, which was hypothesized to be initiated from the beginning of the reaction through a coordination-addition mechanism (Scheme 5c). Nonetheless, the polymerization reaction still reached high conversions of both monomers, 96% and 98% conversion of DVL and MVL, respectively, after 2.25 h at 23 °C in toluene (used to enhance the ROP selectivity, vide supra). Crude reaction products showed a mixture of ring-opening copolymerization product P(MVL-co-VL) (vide infra), composed of poly(δ-valerolactone) (PVL) and P(MVL)ROP, and VAP product P(MVL)VAP, in a ratio of roughly 80:20, based on 1H NMR along with residual monomer (Scheme 5a and Fig. S23†). This distribution was consistent regardless of temperature and solvent addition. Although the alkoxide initiation of the VAP pathway is hypothesized to be less favorable than the carbonyl attack for ring opening of MVL, the VAP pathway is suspected to propagate at a faster rate once initiated, especially in the presence of polar monomer DVL. The observed product distribution is thus likely more reflective of this rate difference rather than competitive rates of initiation.
 | ||
| Scheme 5 (a) General reaction scheme for the separable product mixture of P(MVL-co-VL) and P(MVL)VAP from the polymerization of 30 wt% MVL in DVL with [La(OBn)3]x. (b) Competing initiation pathways: ring-opening of MVL or DVL with [La(OBn)3]x acting as a coordination insertion catalyst (left) and vinyl addition of MVL with [La(OBn)3]x acting as a coordination addition catalyst (right). | ||
Since complete chemoselectivity for the ROP pathway could not be achieved, we attempted post-polymerization separation of the acrylic homopolymer P(MVL)VAP and copolyester P(MVL-co-VL) based on their differing solubility. It was found that the copolyester produced during polymerizations conducted in solution could be solubilized in toluene and filtered to remove P(MVL)VAP, yielding the desired copolyester product, P(MVL-co-VL). This copolyester was determined to consist of a random distribution of P(MVL)ROP units with roughly 10% incorporation based on 1H and 13C NMR (Fig. S24–S26†), an expected result based on the ratio of P(MVL)VAP production and literature reports of similar materials.22 The resulting copolyester P(MVL-co-VL) was analyzed with SEC, which revealed a high molecular weight (Mn = 124 kDa) and a relatively low dispersity (Đ = 1.39) (Fig. S27†).
The purified P(MVL-co-VL) was analyzed with DSC, showing a melting-transition temperature (Tm) of 45 °C with an associated heat of fusion (ΔHf) of 51.5 J per g (Fig. S28†). In comparison, the polyester homopolymer P(MVL)ROP has been reported to exhibit a Tm of 74.4 °C for a sample with Mn = 21.2 kDa.21 Homopolyester PVL with Mn = 55.2 kDa has been reported to have a Tm of 56 °C.40 The lower Tm of P(MVL-co-VL) compared to their parent homopolymers reflects the higher percent composition of PVL and also the likely disruption of PVL crystallization caused by P(MVL)ROP incorporation. The Td and Tmax of P(MVL-co-VL) were obtained by TGA to be 327 °C and 401 °C, respectively (Fig. S29†), consistent with those reported for both polyesters P(MVL)ROP and PVL.21,40 PVL with a high molecular weight (Mn = 99.3 kDa) has been reported to have a Td of 338 °C and a Tmax of 393 °C, values which were strongly dependent on the molecular weight.40 Similar values for the Td (ca. 325 °C) and Tmax (ca. 400 °C) of P(MVL)ROP have also been reported.21 With the Td values of P(MVL)ROP and PVL being similar, it is possible that a higher random incorporation of P(MVL)ROP units could further disrupt the copolymer crystallinity, lowering the Tm, but without reducing the Td. It's worth noting here that the copolyester P(MVL-co-VL) carries pendent double bonds, providing a useful platform for post-polymerization functionalization such as crosslinking or bio-conjugation via click reactions.
Conclusions
In conclusion, we have demonstrated that the acrylic monomer MVL can be continuously produced via the gas-phase aldol condensation of DVL and FA over silica supported alkaline earth oxides. CaO and BaO demonstrated selectivity to MVL >90% at DVL conversions <50%. Selectivity to MVL over CaO generally decreased with increasing conversion and temperature and with lower FA partial pressures. Selectivity loss was attributed to the presence of series reactions that consumed MVL and generated non-volatile lactone dimers at higher conversions. These non-volatiles caused gradual catalyst deactivation over several hours of operation, but full catalyst activity was restored by calcination in air. While separation of DVL and MVL by polarity or boiling point is expected to be difficult, this challenge can be easily overcome by performing polymerization reactions directly in the product mixture. A model product mixture of 30 wt% MVL in DVL was subjected to two different polymerization treatments for efficient, concurrent separation and production of valuable polymers. The VAP of the mixture with Al(iBu)2BHT catalyst at −30 °C produced acrylic polymer P(MVL)VAP with nearly quantitative yields and allowed for 96% recovery of the unreacted DVL. The ROP of the mixture in toluene using [La(OBn)3]x as a catalyst converted nearly all DVL and MVL in the mixture and led to the formation of a DVL/MVL copolymer, polyester P(MVL-co-VL) with some residual and separable P(MVL)VAP. Future work will entail the engineering of catalysts to minimize the formation of lactone dimers, thereby mitigating selectivity loss and catalyst deactivation, as well as studying the effects of minor impurities (such as GVL or methyl-DVL) on the polymerization process.Data availability
The authors confirm that the data supporting this study are available in figures, tables, and schemes within the article body and ESI.†Conflicts of interest
Some of the authors are inventors on patent application 63/675,565, submitted by the Massachusetts Institute of Technology, that covers the MVL synthesis method.Acknowledgements
This work was supported by RePLACE (Redesigning Polymers to Leverage A Circular Economy), funded by the Office of Science of the U.S. Department of Energy via award # DE-SC0022290. ICP-MS measurements were taken at the MIT Center for Environmental Health Sciences, supported by a core center grant P30-ES002109 from the National Institute of Environmental Health Sciences, National Institutes of Health. STEM analysis was carried out in part through the use of MIT.nano's facilities. The authors thank Dr Aubrey Penn for discussion regards microscopic imaging, Dr Reid Gilsdorf for MVL samples that were used for initial GC calibrations, and Dr Ran Zhu for insightful discussions.References
- Ellen McArthur Foundation , The New Plastics Economy: Rethinking the future of plastics & catalysing action https://emf.thirdlight.com/link/cap0qk3wwwk0-l3727v/@/preview/2 (accessed Mar 22, 2022).
- L. Cabernard, S. Pfister, C. Oberschelp and S. Hellweg, Growing Environmental Footprint of Plastics Driven by Coal Combustion, Nat. Sustain., 2021, 5(2), 139–148, DOI:10.1038/s41893-021-00807-2.
- S. B. Borrelle, J. Ringma, K. Lavender Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard and M. A. Hilleary, et al., Predicted Growth in Plastic Waste Exceeds Efforts to Mitigate Plastic Pollution, Science, 2020, 369(6509), 1515–1518, DOI:10.1126/science.aba3656.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, Use, and Fate of All Plastics Ever Made, Sci. Adv., 2017, 3(7), e1700782, DOI:10.1126/sciadv.1700782.
- J. M. Garcia and M. L. Robertson, The Future of Plastics Recycling, Science, 2017, 358(6365), 870–872, DOI:10.1126/science.aaq0324.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, Rational Design of Chemical Catalysis for Plastic Recycling, ACS Catal., 2022, 12(8), 4659–4679, DOI:10.1021/acscatal.2c01286.
- X. Tang and E. Y. X. Chen, Toward Infinitely Recyclable Plastics Derived from Renewable Cyclic Esters, Chem, 2019, 5(2), 284–312, DOI:10.1016/j.chempr.2018.10.011.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, Hydrogenolysis of Polypropylene and Mixed Polyolefin Plastic Waste over Ru/C to Produce Liquid Alkanes, ACS Sustainable Chem. Eng., 2021, 9(35), 11661–11666, DOI:10.1021/acssuschemeng.1c03786.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. Lapointe, S. C. Ammal, A. Heyden and F. A. Perras, et al., Upcycling Single-Use Polyethylene into High-Quality Liquid Products, ACS Cent. Sci., 2019, 5(11), 1795–1803, DOI:10.1021/acscentsci.9b00722.
- L. Chen, Y. Zhu, L. C. Meyer, L. V. Hale, T. T. Le, A. Karkamkar, J. A. Lercher, O. Y. Gutiérrez and J. Szanyi, Effect of Reaction Conditions on the Hydrogenolysis of Polypropylene and Polyethylene into Gas and Liquid Alkanes, React. Chem. Eng., 2022, 7(4), 844–854, 10.1039/d1re00431j.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y. H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Polyethylene Upcycling to Long-Chain Alkylaromatics by Tandem Hydrogenolysis/Aromatization, Science, 2020, 370(6515), 437–441, DOI:10.1126/science.abc5441.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Plastic Waste to Fuels by Hydrocracking at Mild Conditions, Sci. Adv., 2021, 7(17), 8283–8304, DOI:10.1126/sciadv.abf8283.
- J. E. Rorrer, A. M. Ebrahim, Y. Questell-Santiago, J. Zhu, C. Troyano-Valls, A. S. Asundi, A. E. Brenner, S. R. Bare, C. J. Tassone and G. T. Beckham, et al., Role of Bifunctional Ru/Acid Catalysts in the Selective Hydrocracking of Polyethylene and Polypropylene Waste to Liquid Hydrocarbons, ACS Catal., 2022, 12(22), 13969–13979, DOI:10.1021/acscatal.2C03596/.
- S. D. Anuar Sharuddin, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, A Review on Pyrolysis of Plastic Wastes, Energy Convers. Manage., 2016, 115, 308–326, DOI:10.1016/j.enconman.2016.02.037.
- B. Kunwar, H. N. Cheng, S. R. Chandrashekaran and B. K. Sharma, Plastics to Fuel: A Review, Renewable Sustainable Energy Rev., 2016, 54, 421–428, DOI:10.1016/j.rser.2015.10.015.
- D. S. Achilias, Chemical Recycling of Poly(Methyl Methacrylate) by Pyrolysis. Potential Use of the Liquid Fraction as a Raw Material for the Reproduction of the Polymer, Eur. Polym. J., 2007, 43(6), 2564–2575, DOI:10.1016/j.eurpolymj.2007.02.044.
- C. B. Godiya, S. Gabrielli, S. Materazzi, M. S. Pianesi, N. Stefanini and E. Marcantoni, Depolymerization of Waste Poly(Methyl Methacrylate) Scraps and Purification of Depolymerized Products, J. Environ. Manage., 2019, 231, 1012–1020, DOI:10.1016/j.jenvman.2018.10.116.
- W. Kaminsky and J. Franck, Monomer Recovery by Pyrolysis of Poly(Methyl Methacrylate) (PMMA), J. Anal. Appl. Pyrolysis, 1991, 19(C), 311–318, DOI:10.1016/0165-2370(91)80052-A.
- T. Q. Xu, Z. Q. Yu and X. M. Zhang, Recyclable Vinyl-Functionalized Polyesters via Chemoselective Organopolymerization of Bifunctional α-Methylene-δ-Valerolactone, Macromol. Chem. Phys., 2019, 220(12), 1900150, DOI:10.1002/macp.201900150.
- M. Ueda, M. Takahashi, Y. Imai and C. U. Pittman, Synthesis and Homopolymerization Kinetics of α-Methylene-δ-Valerolactone, an Exo-Methylene Cyclic Monomer with a Nonplanar Ring System Spanning the Radical Center, Macromolecules, 1983, 16(8), 1300–1305, DOI:10.1021/ma00242a009.
- J. Li, F. Liu, Y. Liu, Y. Shen and Z. Li, Functionalizable and Chemically Recyclable Thermoplastics from Chemoselective Ring-Opening Polymerization of Bio-Renewable Bifunctional α-Methylene-δ-Valerolactone, Angew. Chem., 2022, 134(32), e202207105, DOI:10.1002/ange.202207105.
- Y. Liu, X. Kou, C. Xu, W. Zhou, H. Zhang, F. Liu, Y. Shen and Z. Li, Chemoselective and Controlled Ring-Opening Copolymerization of Biorenewable α-Methylene-δ-Valerolactone with ε-Caprolactone toward Functional Copolyesters, Polym. Chem., 2023, 14(19), 2326–2332, 10.1039/d3py00188a.
- R. A. Gilsdorf, E. R. Chokkapu, A. Athaley, T. Uekert, R. R. Gowda, A. Singh, J. S. DesVeaux, G. T. Beckham and E. Y. X. Chen, Bio-Based Lactone Acrylic Plastics with Performance and Recyclability Advantages, Cell Rep. Phys. Sci., 2024, 5(5), 101938, DOI:10.1016/J.XCRP.2024.101938.
- M. Ueda, M. Takahashi, Y. Imai and C. U. Pittman, Synthesis and Homopolymerization Kinetics of A-Methylene-6-Valerolactone, an Exo-Methylene Cyclic Monomer with a Nonplanar Ring System Spanning the Radical Center, J. Polym. Sci., Polym. Lett. Ed., 1976, 11(10), 401 Search PubMed.
- L. E. Manzer, Catalytic Synthesis of A-Methylene-y-Valerolactone: A Biomass-Derived Acrylic Monomer, Appl. Catal., A, 2004, 272, 249–256, DOI:10.1016/j.apcata.2004.05.048.
- M. Al-Naji, B. Puértolas, B. Kumru, D. Cruz, M. Bäumel, B. V. K. J. Schmidt, N. V. Tarakina and J. Pérez-Ramírez, Sustainable Continuous Flow Valorization of γ-Valerolactone with Trioxane to α-Methylene-γ-Valerolactone over Basic Beta Zeolites, ChemSusChem, 2019, 12(12), 2628–2636, DOI:10.1002/CSSC.201900418.
- K. W. Hutchenson, K. Kourtakis and L. E. Manzer, Liquid Phase Synthesis of Methylene Lactones Using Novel Catalyst, US7199254B2, 2007.
- K. W. Hutchenson, K. Kourtakis and L. E. Manzer, Supercritical Fluid Phase Synthesis of Methylene Lactones Using Novel Catalyst Field of Invention, US7164032B2, 2007.
- Y. Ono, Solid Base Catalysts for the Synthesis of Fine Chemicals, J. Catal., 2003, 216, 406–415, DOI:10.1016/S0021-9517(02)00120-3.
- V. K. Díez, C. R. Apesteguía and J. I. Di Cosimo, Aldol, Condensation of Citral with Acetone on MgO and Alkali-Promoted MgO Catalysts, J. Catal., 2006, 240(2), 235–244, DOI:10.1016/j.jcat.2006.04.003.
- C. J. Barrett, J. N. Chheda, G. W. Huber and J. A. Dumesic, Single-Reactor Process for Sequential Aldol-Condensation and Hydrogenation of Biomass-Derived Compounds in Water, Appl. Catal., B, 2006, 66(1–2), 111–118, DOI:10.1016/j.apcatb.2006.03.001.
- R. M. West, Z. Y. Liu, M. Peter, C. A. Gärtner and J. A. Dumesic, Carbon–Carbon Bond Formation for Biomass-Derived Furfurals and Ketones by Aldol Condensation in a Biphasic System, J. Mol. Catal. A: Chem., 2008, 296(1–2), 18–27, DOI:10.1016/j.molcata.2008.09.001.
- D. T. Ngo, Q. Tan, B. Wang and D. E. Resasco, Aldol Condensation of Cyclopentanone on Hydrophobized MgO. Promotional Role of Water and Changes in the Rate-Limiting Step upon Organosilane Functionalization, ACS Catal., 2019, 9(4), 2831–2841, DOI:10.1021/acscatal.8b05103.
- D. T. Ngo, T. Sooknoi and D. E. Resasco, Improving Stability of Cyclopentanone Aldol Condensation MgO-Based Catalysts by Surface Hydrophobization with Organosilanes, Appl. Catal., B, 2018, 237, 835–843, DOI:10.1016/j.apcatb.2018.06.044.
- M. D. Healy, M. B. Power and A. R. Barron, Sterically Crowded Aryloxide Compounds of Aluminum, Coord. Chem. Rev., 1994, 130(1–2), 63–135, DOI:10.1016/0010-8545(94)80003-0.
- M. Hong and E. Y. X. Chen, Completely Recyclable Biopolymers with Linear and Cyclic Topologies via Ring-Opening Polymerization of γ-Butyrolactone, Nat. Chem., 2015, 8(1), 42–49, DOI:10.1038/nchem.2391.
- H. Hattori, Heterogeneous Basic Catalysis, Chem. Rev., 1995, 95(3), 537–558, DOI:10.1021/cr00035A005.
- J. Q. Bond, D. Martin Alonso, R. M. West and J. A. Dumesic, γ-Valerolactone Ring-Opening and Decarboxylation over SiO2/Al2O3 in the Presence of Water, Langmuir, 2010, 26(21), 16291–16298, DOI:10.1021/la101424a.
- A. A. Marianou, C. M. Michailof, D. K. Ipsakis, S. A. Karakoulia, K. G. Kalogiannis, H. Yiannoulakis, K. S. Triantafyllidis and A. A. Lappas, Isomerization of Glucose into Fructose over Natural and Synthetic MgO Catalysts, ACS Sustainable Chem. Eng., 2018, 6(12), 16459–16470, DOI:10.1021/acssuschemeng.8B03570.
- X. L. Li, R. W. Clarke, H. Y. An, R. R. Gowda, J. Y. Jiang, T. Q. Xu and E. Y. X. Chen, Dual Recycling of Depolymerization Catalyst and Biodegradable Polyester That Markedly Outperforms Polyolefins, Angew. Chem., Int. Ed., 2023, 62(26), e202303791, DOI:10.1002/ANIE.202303791.
- X. Tang, M. Hong, L. Falivene, L. Caporaso, L. Cavallo and E. Y. X. Chen, The Quest for Converting Biorenewable Bifunctional α-Methylene-γ-Butyrolactone into Degradable and Recyclable Polyester: Controlling Vinyl-Addition/Ring-Opening/Cross-Linking Pathways, J. Am. Chem. Soc., 2016, 138(43), 14326–14337, DOI:10.1021/jacs.6b07974/.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03016h |
| This journal is © The Royal Society of Chemistry 2024 |