
Unraveling the secrets of harnessing a surfactant-modified strategy in organosolv pretreatment of lignocellulosic biomass for efficient fermentable sugar production†
Guojie
Song‡
a,
Hui
Zhang‡
a,
Meysam
Madadi
a,
Zhixiangpeng
Chen
a,
Hao
Wang
a,
Ao
Xia
 b,
Abdolreza
Samimi
c,
Chihe
Sun
*a,
Xianzhi
Meng
d,
Arthur J.
Ragauskas
de and
Fubao
Sun
a
b,
Abdolreza
Samimi
c,
Chihe
Sun
*a,
Xianzhi
Meng
d,
Arthur J.
Ragauskas
de and
Fubao
Sun
a
aKey Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi 214122, China. E-mail: chihesun@jiangnan.edu.cn
bKey Laboratory of Low-grade Energy Utilization Technologies and Systems, Chongqing University, Ministry of Education, Chongqing 400044, China
cDepartment of Chemical Engineering, University of Sistan and Baluchestan, Zahedan, Iran
dDepartment of Chemical & Biomolecular Engineering, University of Tennessee, Knoxville, TN 37996, USA
eJoint Institute of Biological Sciences, Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 23rd August 2024
Abstract
Alkaline-catalyzed organosolv pretreatment of lignocellulosic biomass affords excellent delignification, yielding a holocellulose-rich substrate for fermentable sugar production. However, complete lignin removal is impractical, and residual lignin also exacerbates negative effects on subsequent enzymatic hydrolysis. Herein, a novel strategy of developing surfactant-assisted organosolv pretreatment (Triton-X 100, AEO 9 and Tween 80) for in situ lignin modification was proposed to overcome this issue. The results indicated that the pretreated substrates showed considerable enzymatic hydrolyzability with a 15.7%–38.3% higher sugar yield compared to the control group without surfactants. Surfactants could graft on both residual and dissolved lignin through etherification, forming α-etherified lignin, though without significantly changing the component distribution and substrate-related properties. Quantum chemical calculations provided theoretical evidence of strong H-bonding and pronounced interaction energy between lignin and surfactants (maximally at −48.4 kcal mol−1). In particular, this surfactant modification decreased the aliphatic –OH and phenolic –OH contents of residual lignin by 16.0%–22.4% and 13.8%–28.8%, respectively. The reduction of –OH groups mitigated non-productive adsorption between lignin and cellulases via H-bonding interaction, which exhibited a significant correlation with the increased enzymatic hydrolyzability (>−0.9). Overall, this study offers valuable insight into the fundamental understanding of the mechanism involved in lignin modification during surfactant-assisted pretreatment and lignin–enzyme interaction during enzymatic hydrolysis. The new findings underscore the potential application of surfactants in organosolv pretreatment to achieve a feasible approach for developing an efficient enzyme-mediated lignocellulosic sugar platform.
1. Introduction
The transition towards a sustainable, bio-based economy is crucial due to dwindling fossil fuel reserves and the escalating impact of greenhouse gas emissions.1 Lignocellulosic biomass (LCB), a renewable resource, has immense potential for biofuel and biochemical production. Sugar platform biorefineries, which convert LCB into fermentable sugars, are considered a pivotal step in numerous biochemical production processes.2 However, the inherent recalcitrance of LCB, particularly its intricate lignin network, poses significant challenges for conversion into fermentable sugars. Thus, an efficient pretreatment method targeting lignin is considered a pioneering step in enzyme-mediated lignocellulosic sugar platform biorefineries to overcome this recalcitrance.3Among the various pretreatment strategies, organosolv pretreatment is notable for effectively disrupting lignin–carbohydrate complexes, enhancing cellulose and hemicellulose accessibility for subsequent sugar production.4 As a high-boiling organic solvent, glycerol is low-cost, non-toxic, and user-friendly under atmospheric conditions.5 In particular, glycerol organosolv (GO) pretreatment with alkaline catalysts afforded efficient delignification while preserving cellulose and hemicellulose with limited inhibitory compounds, making it an ideal substrate for fermentable sugar production.6 However, like other pretreatment methods, GO pretreatment still faces some common problems: (1) the formation of stable C–C bonds caused by lignin repolymerization reactions; (2) the non-productive interaction between lignin and cellulases; and (3) the balance of easily hydrolysable substrates and high quality lignin.7 Overall, the residual lignin in the pretreated substrates still undergoes structural alterations and significantly hampers substrate hydrolyzability.8 Advanced pretreatments targeting residual lignin are desperately warranted to moderate the adverse effects on enzymatic hydrolysis.
Recently, in situ lignin modification during pretreatment has emerged as a promising approach to mitigate the negative effects of lignin on sugar production, offering advantages such as no additional processing steps, no sacrifice of holocellulosic sugars and no excess wastewater discharge.7,9 To name a few, Chu et al. reported that adding 5% of 2-naphthol-7-sulfonate, syringic acid, or mannitol in dilute acid pretreatment enhanced the enzymatic hydrolyzability of poplar by 24%–71%.1 Sheng et al. also found that nucleophilic derivatives like 3-hydroxylic-2-naphthoic acid in dilute acid pretreatment increased carboxylic acid groups and negative charge in lignin, reducing its inhibition of fermentable sugar production.10 These additives interact with lignin to form Cα-etherified lignin, which mitigates lignin recondensation and decreases the non-productive adsorption onto enzymes.11 Evidently, the above-mentioned additives primarily target aqueous phase solvents used in hydrothermal and dilute acid pretreatments prone to lignin recondensation. So far, there are few studies on exogenous additive-assisted organosolv pretreatment with a more complex lignin modification. More importantly, naphthol and its derivatives are petroleum-based, expensive, and potentially toxic, limiting the overall sustainability and cost-effectiveness.12 Therefore, it is crucial to explore more suitable additives to enhance the efficiency of organosolv pretreatment for lignocellulosic sugar platform biorefineries.
Surfactants, especially non-ionic surfactants, such as polysorbate (Tween), fatty alcohol polyoxyethylene ether (AEO) and polyethylene glycol octyl phenyl ether (Triton-X) series, have been widely employed as low-cost, less toxic and eco-friendly exogenous additives to stabilize enzymes and mitigate lignin–enzyme interactions during enzymatic hydrolysis.13,14 However, their application in upstream pretreatment processes for in situ lignin modification is limited, with only a few studies reporting positive effects on component deconstruction.14–16 Fan et al. reported that the addition of Tween 80 in FeCl3-catalyzed aqueous-phase pretreatment increased delignification from bagasse by nearly 50%.16 Even so, some studies have reported the opposite result that surfactants had no obvious promotional effects on removing the LCB components.17 In our recent studies, the authors found that adding PEG 6000 to glycerol pretreatment could not improve the fractionation of lignocellulose, but significantly enhanced the enzymatic hydrolysis of the pretreated substrates.18 Structurally, the –OH tails of PEG or other surfactants could react with the phenolic –OH group of the isolated lignin under acidic conditions, forming etherified lignin like the above-mentioned nucleophilic derivatives.19 It was reported that the phenolic –OH group of lignin significantly affects the non-productive adsorption among lignin–enzymes.7 This suggested that the structural modification of residual lignin induced by surfactants, rather than just the deconstruction of components, may be the fundamental reason for improving the substrate hydrolyzability. Furthermore, the coupled effects of different surfactants and organic solvents may also lead to the diversity of lignin structures, which could positively or negatively affect the non-productive adsorption of lignin and enzymes through hydrogen bonding, hydrophobicity, and electrostatic interactions.20 However, the changes of lignin properties during organosolv pretreatment in the presence of different surfactants are virtually a mystery, and the mechanism of how the alterations in the structural/physicochemical properties of modified lignin dominate the substrate hydrolyzability is not fully understood.
This work aims to shed light on the potential of various non-ionic surfactants (i.e., Triton-X 100, AEO 9, and Tween 80) for in situ lignin structural modification during the organosolv pretreatment and elucidates the mechanisms underlying the relationship between the altered lignin properties and enhanced substrate's enzymatic hydrolysis. First, biomass component fractionation and substrate hydrolyzability after the surfactant-assisted GO pretreatment were analyzed to evaluate the availability of surfactants in LCB pretreatment. Subsequently, to fully understand the structural alteration due to such modification behavior, the changes in the physicochemical properties of both residual and dissolved lignin (i.e., chemical functional groups, key linkage bonds, and surface properties) were evaluated with and without surfactant treatment. Molecular dynamics simulations and density functional theory (DFT) calculations were also employed to reveal the interactions between surfactants and lignin. Furthermore, the relationship between the structural changes of lignin and the non-productive adsorption of enzymes was elucidated by quantitative analysis to assess the key structure-related parameters affecting hydrolysis performance. Overall, this work will propose underlying mechanisms to explain how in situ lignin modification during the surfactant-assisted pretreatment contributes to the favorable enhancement of enzymatic hydrolysis.
2. Materials and methods
2.1. Materials and reagents
Sugarcane bagasse was obtained from Guangxi Province, China. It was oven-dried at 60 °C overnight and passed through a 40-mesh sieve. The contents of cellulose, hemicelluloses, lignin, and ash were 42.7%, 24.1%, 23.8%, and 4.3%, respectively. Cellulase Cellic® CTec2 (filter paper activity: 120 FPU g−1) and endo-xylanase (protein content: 150 mg g−1) were provided by Novozymes Investment Co. Ltd (Beijing, China) and Qingdao Vland Biotech Inc. (Qingdao, China). Industrial-grade glycerol with 99.5% purity was obtained from Wuxi Chemical Factory, China. All other reagents were purchased from Sinopharm Chemical Reagent Factory Ltd, China.2.2. Surfactant-assisted glycerol pretreatment
In a typical process, 10 g of dried sugarcane bagasse and 100 g of glycerol along with NaOH (45 mg g−1, based on raw substrates) were mixed in a three-necked round-bottom flask equipped with an electric heating jacket.21 The GO pretreatment (control group) was performed at 195 °C with stirring at 180 rpm for 45 min based on our previously published work.18 As for surfactant-assisted GO pretreatment, three non-ionic surfactants, including Triton-X 100, AEO 9, and Tween 80 (40 mg g−1, based on the raw substrates), were individually added to the pretreatment system; other operations were the same as for the above GO pretreatment. After the pretreatment, 150 g of tap water was added to the flask with vigorous stirring when the temperature was reduced to 100 °C. Subsequently, the slurry was filtered through a G1 filter to obtain an undissolved solid substrate and a liquid phase. The solid fraction was washed twice with 200 mL of tap water, and then oven-dried at 60 °C for further analyses.2.3. Enzymatic hydrolysis
The pretreated substrates under 2% (w/v) solid loading were mixed with citrate buffer (0.05 M, pH 4.8) and Cellic CTec 2 (6 FPU g−1 dry substrate) in an Erlenmeyer flask with an effective working volume of 25 mL. Enzymatic hydrolysis was performed in an incubator shaker at 50 °C for 72 h at 180 rpm. After hydrolysis, 0.4 mL of samples were withdrawn and heated at 100 °C for 5 min to deactivate the enzymes. The samples were centrifuged at 8000 rpm. The supernatants containing glucose/xylose and enzyme protein were diluted, in which the glucose/xylose titer was detected by high-performance liquid chromatography (HPLC, Chomaster CM5110, Hitachi, Japan) with a refractive index detector and a packed column (Bio-Rad Aminex HPX-87H, 9 μm, 7.8 × 300 mm). The mobile phase was 5 mM dilute H2SO4 with a flow rate of 0.6 mL min−1 under isocratic elution, and the column temperature was set at 60 °C.22 The glucose yield was calculated using eqn (1) as follows. | (1) |
The enzyme protein in the supernatants was estimated using a UV-Vis spectrometer with an absorbance maximum at 595 nm using the Bradford protocol, as reported by Lai et al.23 The value of free enzyme concentration was calculated as a percentage of the total enzyme protein concentration.
To evaluate the non-productive adsorption between enzymes and lignin, the pretreated substrates were incubated with bovine serum albumin (BSA) before enzymolysis. In detail, 25 mg g−1 (dry substrate) of BSA and 2% (w/v) pretreated substrates were mixed with citrate buffer, and the mixtures were incubated at 50 °C for 24 h at 180 rpm.11 Then, 6 FPU g−1 enzyme was added to induce the enzymolysis reaction. The differences between the glucose yields with and without BSA treatment revealed a non-productive enzyme adsorption onto lignin.4
2.4. Compositional and characterization analyses
The lignocellulosic components were determined by two-step H2SO4 processes according to the National Renewable Energy Laboratory method.24 The component analysis was performed using eqn (2)–(4), respectively. | (2) |
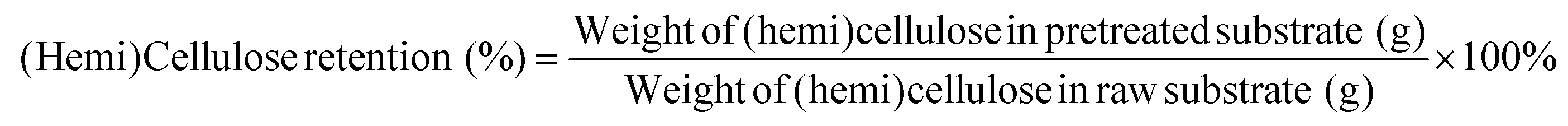 | (3) |
 | (4) |
Cellulose accessibility was assessed by the direct red dye (DR28) staining protocol.11 In detail, the experiment was performed in 50 mL flasks with 1% (w/v) of the pretreated substrates. The concentrations of the dye were varied at 0.05–4.0 g L−1 and incubated at 50 °C for 24 h at 180 rpm. After this, the mixtures were centrifuged for 5 min at 8000 rpm, and the dye concentration in the supernatant was measured at 498 nm using a UV-Vis spectrometer. The quantity of adsorbed dye was determined by calculating the differences between the initial dye concentration and that in the supernatant. The maximum amount of dye adsorbed onto the pretreated substrates was estimated using the Langmuir adsorption isotherm.
The porosity of the pretreated substrates was measured using a Micromeritics analyzer (Autosorb IQ3, Quantachrome).25 The contents of different organic elements (C, H, N, and S) were detected using an elemental analyzer (Elementar Unicube, Germany), and the O content was calculated by subtracting the content of the other elements from 100%. Additionally, X-ray diffraction (XRD) analysis was used to analyze the cellulose crystallinity index (CrI) of the pretreated substrates, as reported elsewhere.5 The CrI was calculated based on eqn (5).
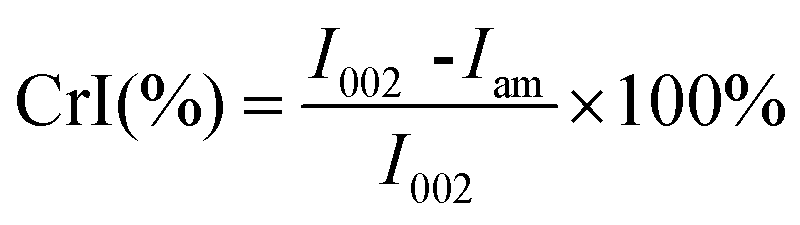 | (5) |
2.5. Lignin isolation and characterization
The dissolved lignin (DL) in the pretreatment liquid was extracted using an acid-precipitation method. The pretreatment liquid was first mixed with a 10-fold acidic water solution (with the pH of the HCl solution adjusted to 2.0), and the mixture was centrifuged (8000 rpm for 10 min) to obtain lignin precipitates. The resulted lignin was water-washed, followed by freeze-drying. The lignin samples are labelled as GO–DL, GO + Triton-X 100-DL, GO + AEO 9-DL, and GO + Tween 80-DL.Residual lignin (i.e., cellulolytic enzyme lignin) was obtained from the pretreated substrates by double enzymatic hydrolysis and 1,4-dioxane extraction methods.17 Initially, 10% (w/v) pretreated substrates with an excess enzyme loading (CTec2: 60 FPU g−1, endo-xylanase: 2.4 mg g−1 of substrate) were hydrolyzed in a 72 h reaction. Thereafter, the slurry was centrifuged, and the solid residues were hydrolyzed again with a fresh enzyme. The resulting residues were collected and rinsed with deionized water to eliminate any remaining sugars and enzyme proteins. Finally, the residues were freeze-dried and extracted with dioxane–water (96% v/v) at a solid-to-liquid ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. The extraction process was conducted twice for 24 h in a dark environment. The mixtures were centrifuged and the supernatant was concentrated using a rotary evaporator. The concentrated liquid was mixed with a 10-fold acidic water solution, followed by centrifugation, washing and freeze-drying. The lignin samples are designated as GO, GO + Triton-X 100, GO + AEO 9, and GO + Tween 80.
:20. The extraction process was conducted twice for 24 h in a dark environment. The mixtures were centrifuged and the supernatant was concentrated using a rotary evaporator. The concentrated liquid was mixed with a 10-fold acidic water solution, followed by centrifugation, washing and freeze-drying. The lignin samples are designated as GO, GO + Triton-X 100, GO + AEO 9, and GO + Tween 80.
2D-HSQC NMR spectroscopy was carried out using a Bruker AVIII 400 MHz (Bruker, Germany) spectrometer to analyze the lignin structure.26 Specifically, 60–80 mg of lignin was dissolved in DMSO-d6 (0.5 mL). The F2 (1H) and F1 (13C) spectral widths were 5000 and 20000 Hz, respectively. The data points for the 1H dimension were 1024 with 1.5 s of recycling delay and 64 scans, whereas the data points for the 13C dimension were 256. The obtained 2D spectra and integrals were processed based on the previous reports.27 The contents of hydroxyl and carboxylic groups were detected by 31P NMR measurements.28 Fourier transform infrared (FTIR) spectroscopy was employed to analyze the changes in the functional groups using a Nicolet Nexus 670 instrument (Thermo Fisher, USA) within the wavenumber range of 800–4000 cm−1. The thermogravimetric analysis of different isolated lignin samples was performed using a TG209 instrument (Netzsch, Germany), and approximately 3 mg of each sample was heated from 30 °C to 850 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere.29 The weight-average molecular weight (Mw) and number-average molecular weight (Mn) of lignin were determined using gel permeation chromatography (GPC). Additionally, lignin's hydrophobicity was estimated by a water contact angle measurement using an automatic video contact angle detector (JY-82, Chengde Dingsheng, China). A Zetasizer Nano ZS90 (Malvern, UK) was used evaluate the zeta potential of lignin.30
2.6. Simulation calculations
A lignin model, named veratrylglycerol-b-guaiacyl ether (VG), was employed to investigate its interaction with glycerol and surfactants. Quantum chemical calculations were conducted using the DFT method within the Gaussian 09 software package.28,31 Geometry optimization and energy calculations of all the molecules were performed at the B3LYP and M06-2X levels of theory and the 6-311++G (d,p) basis set with the DFT-D3 dispersion correction.32 The weak interactions were computed using the Multiwfn 3.6 program, and these interactions were visualized using the reduced density gradient (RDG), the distribution of electrostatic potential (ESP), and extreme points at the van der Waals surface.33,34 The interaction energy was calculated using eqn (6).| Eint = Etotal − E1 − E2 − E3 | (6) |
3. Results and discussion
3.1. Effects of surfactants on glycerol organosolv pretreatment performance
 | ||
| Fig. 1 Substrate hydrolyzability from surfactant-assisted glycerol organosolv pretreatment. (a) Glucose yield and (b) xylose yield. | ||
| Pretreated substrates | SR (%) | Chemical composition (%) | CR (%) | HR (%) | DL (%) | |||
|---|---|---|---|---|---|---|---|---|
| C (%) | H (%) | L (%) | Ash (%) | |||||
| SR, solid recovery; C, cellulose; H, hemicellulose; L, lignin; CR, cellulose retention; HR, hemicellulose retention; and DL, delignification. | ||||||||
| GO | 69.1 ± 1.2 | 57.5 ± 0.7 | 29.7 ± 0.2 | 9.1 ± 0.4 | 2.6 ± 0.2 | 93.1 ± 1.1 | 81.8 ± 0.6 | 73.6 ± 1.2 |
| Triton-X 100-assisted GO | 68.8 ± 1.1 | 58.0 ± 0.7 | 29.5 ± 0.3 | 8.6 ± 0.7 | 2.8 ± 0.3 | 93.6 ± 1.1 | 82.5 ± 0.8 | 75.1 ± 1.9 |
| AEO 9-assisted GO | 69.9 ± 0.8 | 57.4 ± 0.8 | 30.0 ± 0.5 | 9.6 ± 0.3 | 2.3 ± 0.2 | 93.5 ± 0.8 | 82.4 ± 1.5 | 72.0 ± 0.9 |
| Tween 80-assisted GO | 70.2 ± 1.3 | 56.6 ± 0.5 | 29.9 ± 0.3 | 9.4 ± 0.4 | 2.5 ± 0.1 | 92.7 ± 0.7 | 83.2 ± 0.7 | 72.5 ± 1.0 |
Even so, some researchers obtained opposite results that the assistance of PEG and Tween series in the pretreatment was effective for promoting the removal of lignin and hemicellulose components.37,38 To name a few, 20% (w/w, based on the raw substrates) of PEG 8000 and Tween 80 in the ethylenediamine pretreatment of corn stover maximally increased the delignification by 32% as compared to the control group without adding surfactants.37 Such differences may be attributed to the used surfactant dosages. In this study, only 4% (w/w) of surfactants was employed, which was far less than the above reported values. The authors previously found that once the usage of the surfactant exceeded the 8% (w/w) threshold, it indeed promoted lignin removal from LCB.39 It was speculated that excessive surfactants had a significant effect on surface tension and solvent transfer due to the changes of hydrophilicity/hydrophobicity in the pretreatment system, thus enhancing the component solubility. Notably, there is no denying that all non-ionic surfactants used in pretreatment indeed enhanced the enzymatic hydrolysis, though without changing the component distribution. In other words, the surfactant in pretreatment facilitated substrate hydrolyzability was not rely solely on the physically aided component removal/dissolution, but the chemically modified substrates may play a dominant role. Such alteration may alleviate the non-productive adsorption between lignin and cellulases, facilitating subsequent hydrolysis.
Considering the various structures of surfactants, the coupled effects of different surfactants may also lead to diverse changes in the pretreated substrate's structure, which could positively or negatively affect the enzymatic hydrolysis performance. Moreover, it is crucial to verify the relationship between the altered substrate properties and the enhanced enzymatic hydrolysis of the substrates. Accordingly, the full characterization of the pretreated substrates was performed in the subsequent section to reveal the promotion mechanism of surfactant-assisted GO pretreatment on substrate hydrolyzability.
3.2. Changes in the physicochemical properties of the pretreated substrates
To elucidate the reason why surfactants in the GO pretreatment can facilitate the hydrolysis performance of the pretreated substrates, various potential factors, such as cellulose crystallinity, accessibility, biomass porosity, and specific surface area, were analyzed, as shown in Table 2. The crystallinity index of the GO-pretreated substrates was calculated to be 52.3%, while that of the surfactant-assisted GO pretreated substrates remained virtually unchanged (51.5–52.8%). This was in line with lignocellulosic component analysis results. Chen et al. also reported that adding Tween 80 in the FeCl3-catalyzed pretreatment of eucalyptus waste fibers did not cause a dramatic change in the CrI value of the pretreated substrates.40 As expected, the correlation between the CrI value and glucose yield was indistinct (r = −0.32, p > 0.05, see Fig. S1†), implying that the cellulose crystallinity may not play a significant role in facilitating the hydrolysis performance of the surfactant-assisted GO-pretreated substrates.| Pretreated substrates | CrI (%) | Specific surface area (m2 g−1) | Pore volume (cm3 mg−1) | Γ max/DR28 (mg g−1) |
|---|---|---|---|---|
| GO | 52.3 | 3.1 ± 0.4 | 6.2 ± 0.9 | 560.5 ± 40.3 |
| Triton-X 100-assisted GO | 52.8 | 3.5 ± 0.7 | 5.5 ± 1.0 | 520.3 ± 50.5 |
| AEO 9-assisted GO | 52.6 | 2.7 ± 0.6 | 5.6 ± 0.8 | 580.3 ± 35.5 |
| Tween 80-assisted GO | 51.5 | 2.9 ± 0.5 | 6.3 ± 0.6 | 601.6 ± 30.8 |
Moreover, the specific surface area and pore volume of all samples were estimated to be around 3.0 m2 g−1 and 6.0 cm3 mg−1, respectively, with no significant differences in their porosity properties. This observation was corroborated by the extremely low correlation coefficients between the specific surface area/pore volume and the glucose yield (r = −0.19 or −0.24, p > 0.05, see Fig. S1†). The changes of cellulose accessibility (based on dye adsorption analysis) exhibited certain differences, while its correlation with the glucose yield was still inapparent (r = 0.36, p > 0.05, see Fig. S1†). Thus, the contribution of these factors to improving the substrate hydrolyzability was also very low, regardless of surfactant addition during the GO pretreatment.
Regarding the organic elements in all pretreated substrates (Table 3), the main elements were C, O and H, accounting for around 44%, 49% and 6%, respectively. Predictably, the elemental compositions of the surfactant-assisted GO substrates were very close to those of the GO substrates, with only minor differences of around 0.5%. This indicated that the non-ionic surfactants such as Triton X-100, AEO-9, and Tween 80 had a limited impact on the component and element distribution of the GO substrates (Table 1). The alterations in the chemical compositions of different surfactants were also not reflected in the elemental analysis results. Thus, exploring other chemical characterization processes is necessary to better understand the effects of the surfactant-assisted pretreatment on the GO substrates.
| Pretreated substrates | C (%) | H (%) | Oa (%) | N (%) | S (%) |
|---|---|---|---|---|---|
| a O content was calculated by subtracting the content of the other elements from 100%. | |||||
| GO | 44.19 ± 0.10 | 6.21 ± 0.05 | 49.25 ± 0.16 | 0.12 ± 0.01 | 0.24 ± 0.01 |
| Triton-X 100-assisted GO | 44.74 ± 0.02 | 6.25 ± 0.01 | 48.74 ± 0.02 | 0.13 ± 0.01 | 0.14 ± 0.01 |
| AEO 9-assisted GO | 43.82 ± 0.05 | 6.58 ± 0.07 | 49.38 ± 0.09 | 0.09 ± 0.01 | 0.13 ± 0.03 |
| Tween 80-assisted GO | 44.33 ± 0.01 | 6.30 ± 0.01 | 49.10 ± 0.13 | 0.11 ± 0.01 | 0.17 ± 0.02 |
The discussion thus far suggests that introducing various surfactants in the GO pretreatment did not significantly alter the properties of the pretreated substrates. However, beyond the substrate properties, lignin has been identified as a predominant factor influencing biomass hydrolysis.7 Lignin typically impedes hydrolysis through two mechanisms: (1) physical obstruction that restricts enzyme accessibility and (2) non-productive adsorption binding with enzymes by chemical interactions.41 As the pretreatment severity remained comparable with/without surfactant addition and the lignin contents of the pretreated substrates were nearly identical, the physical barriers imposed by lignin on enzymatic hydrolysis may be negligible in this study. In this case, the authors hypothesized that the high substrate hydrolyzability stemmed from the surfactant-induced modifications to the lignin structure during the GO pretreatment.
3.3. Structural modification of residual lignin in the pretreated substrates
2D-HSQC NMR was employed to characterize the monolignol units and inter-unit linkages of lignin. As depicted in Fig. 2, the NMR spectra were divided into the aliphatic region (δC/δH 50–90/2.5–6.0 ppm) and the aromatic region (δC/δH 100–135/6.0–8.0). In the aliphatic region, the prominent signals corresponded to the methoxy (OCH3) groups, aryl ether linkages (A, β-O-4), and phenylcoumaran (C, β-5), while in the aromatic region, signals associated with S, G, H, and pcoumaric acid (PCA) units were observed for the lignin samples. The results suggested that the addition of surfactants during the GO pretreatment had negligible impact on the distribution of lignin subunits.17 Furthermore, the signal intensity at δC/δH 71.8/4.87 is connected to the α-position (Aα) of β-O-4 linkages. A previous study reported that the β-O-4 linkage content in CEL (i.e., lignin extracted from raw sugarcane bagasse) was estimated to be 59.3/100 Ar.12 In contrast, the content of β-O-4 linkages (20/100 Ar) in all detected lignin samples was significantly lower, indicating the cleavage of ether linkages within the lignin structure during the pretreatment. | ||
| Fig. 2 2D HSQC spectra of the residual lignin isolated from different pretreated substrates. (a) GO lignin; (b) GO + Triton-X 100 lignin; (c) GO + AEO 9 lignin; and (d) GO + Tween 80 lignin. | ||
Interestingly, additional new signals were detected in all lignin spectra, marked in green (δC/δH 70.4–73.5/3.33–3.58 ppm and 62.5/3.40 ppm). This phenomenon primarily arose from the grafting of glycerol moieties onto the α-position of β-O-4 linkages in lignin through etherification/esterification reactions.42 Such protective modification of the lignin structure by glycerol aided in preventing further repolymerization. Surfactant moieties have been reported to exhibit intensified signals at δC/δH 68.0–75.0/2.5–4.3,29 suggesting that the surfactants were also grafted onto lignin, as observed in the GO + surfactant lignin spectra (Fig. 2). However, no distinct difference in the β-O-4 linkage contents was observed between the GO lignin and GO + surfactant lignin. Surfactant-assisted GO pretreatment had little effect on the distribution of inter-unit linkages.
Polyethylene glycol (PEG) is commonly used to promote the pretreatment efficiency of LCB. Cai et al. demonstrated that PEG 8000 could be grafted onto the α-position of β-O-4 in lignin during the ethylenediamine pretreatment of corn stover, thus alleviating lignin recondensation and preserving a higher content of β-O-4 linkages as compared with the lignin isolated without surfactants.37 In another study, new intense signals attributed to the α-PEG-β-O-4 units were detected at δC/δH 60.80/3.74 ppm for PEG 600-modified lignin from steam-exploded hardwood.43 Although these studies have stated that PEG series preserved the β-O-4 linkages, the present investigation indicated that adding Triton-X 100, AEO 9, and Tween 80 in the GO pretreatment had minimal influence on the content of lignin β-O-4 linkages. PEG possesses two terminal –OH tails that have been reported to act as carbonium ion scavengers by reacting at the α-position of the lignin side chain, forming α-PEG-β-O-4. Besides, PEG-based surfactants contain distinct hydrophilic heads and hydrophobic tails, as well as large spatial site resistance. The linear structure and high hydrophilicity may facilitate its interaction with the α-position of β-O-4 in lignin through hydrogen bonding or van der Waals force. Such effects may be insignificant in other non-ionic surfactants (such as Triton-X 100, AEO 9, and Tween 80 in this study). The inherent surfactant properties and the pretreatment methods may also contribute to these differences.
A quantitative analysis of the aliphatic –OH, phenolic –OH, and carboxyl groups based on 31P NMR is shown in Fig. 3a and Fig. S2.† The 31P NMR data unequivocally demonstrated the successful grafting of surfactants onto lignin by revealing changes in both the aliphatic and phenolic –OH groups after structural modification. The GO lignin exhibited the highest content of aliphatic –OH groups as compared with CEL (6.69 vs. 4.23 mmol g−1 (ref. 12)). This confirmed that the glycerol grafting induced –OH tails on the benzylic carbocation structure at the α-site of lignin, coinciding with the 2D-HSQC analysis. However, a notable decrease in the aliphatic –OH content was observed in all GO + surfactant lignin samples. Specifically, the aliphatic –OH content of the GO + Triton-X, GO + AEO 9, and GO + Tween 80 samples was 5.19, 5.62, and 5.31 mmol g−1, respectively, corresponding to 16.0%–22.4% reduction as compared with the GO lignin. The OH groups of the surfactants presumably reacted with the aliphatic –OH and/or phenolic –OH groups within the lignin structure via etherification, as depicted in Fig. 3b. Despite the decrease in the aliphatic –OH content present in the GO + surfactant lignin, it remained higher than those obtained from the conventional pretreatments, such as dilute acid/alkaline, ionic liquid, and organosolv pretreatments using monomeric alcohols (i.e., methanol and ethanol). In the latter cases, the aliphatic –OH content was typically decreased due to the dehydration and oxidation reactions.44,45
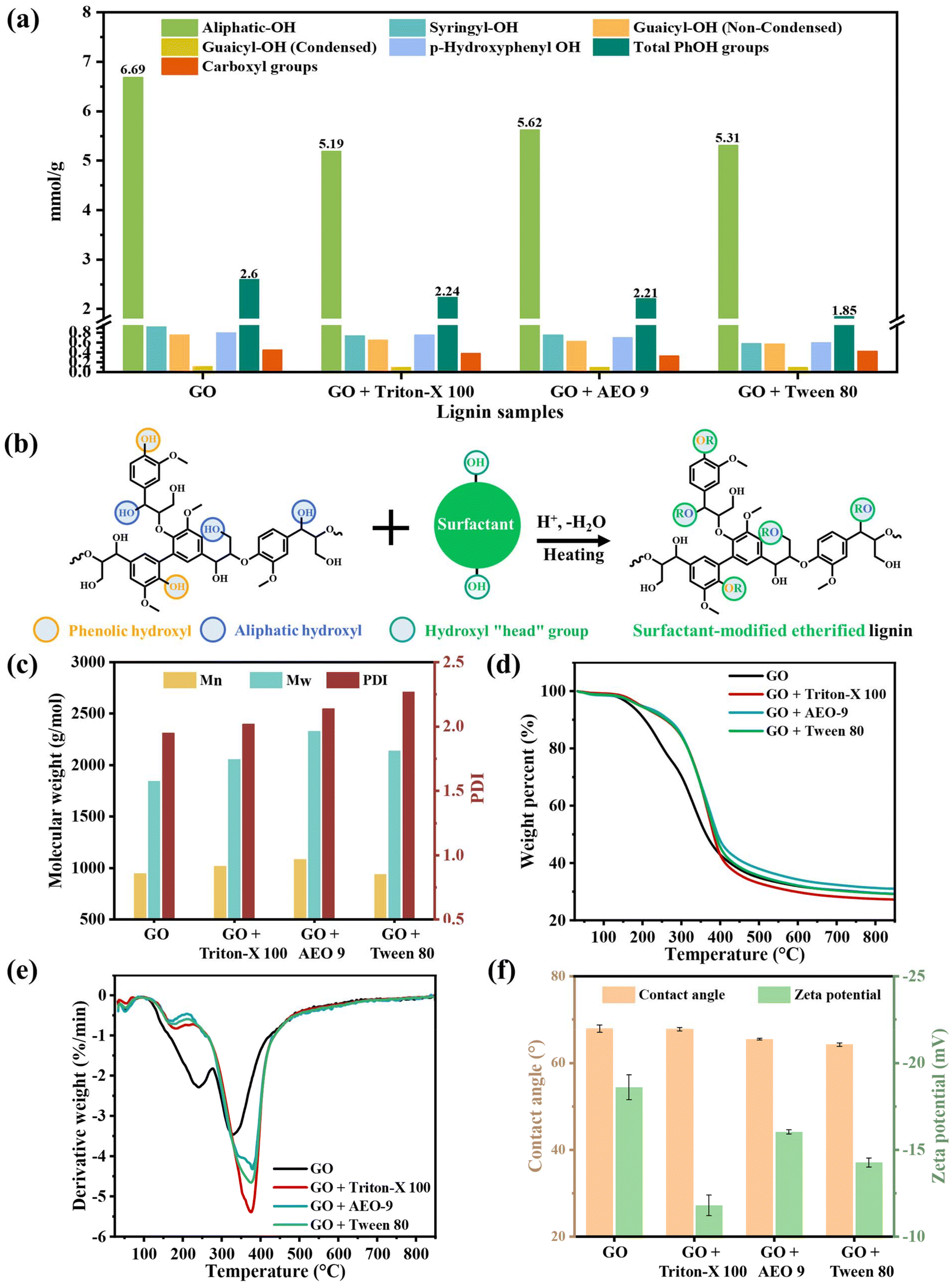 | ||
| Fig. 3 (a) Quantification of functional groups (mmol g−1) in the residual lignin based on 31P NMR; (b) proposed routes of surfactant modification on the lignin structure via etherification; (c) molecular weight distribution; (d) TG and (e) DTG curves of different lignin samples; and (f) contact angle measurement and zeta potential analysis. | ||
The GO + surfactant lignin (1.85–2.24 mmol g−1) possessed lower total phenolic –OH content as compared with the GO lignin (2.6 mmol g−1). Among these, the GO + Tween 80 lignin with the lowest phenolic –OH content exhibited more significant structural modification caused by etherification grafting. The phenolic –OH groups are primarily responsible for the non-productive adsorption of cellulases, which interact with lignin through hydrogen bonding interactions.7 Thus, the reduction in the phenolic –OH groups of lignin during the surfactant-assisted GO pretreatment may play an important role in facilitating substrate hydrolyzability. It is worth noting that although other naphthols and their derivatives can inhibit lignin recondensation and reduce the inhibition of subsequent enzymatic hydrolysis, this effect often increases the phenolic hydroxyl content of lignin, which, to a certain extent, enhances the hydrogen bonding interactions with cellulases. In contrast, this study found that the hydroxyl groups of surfactants undergo etherification reactions with the hydroxyl groups of lignin, precisely reducing the content of aliphatic and phenolic –OH groups in lignin that can interact with cellulases. This finding opens up new avenues for the in situ modification of the lignin structure to achieve efficient enzymatic hydrolysis of lignocellulosic biomass. Also, the GO lignin contained 0.45 mmol g−1 of the carboxylic group, which was 4.4%–26.7% higher than that of the GO + surfactant lignin (0.33–0.43 mmol g−1). Incorporating surfactants in the GO pretreatment mitigated the oxidation of phenolic –OH and/or aliphatic –OH groups into carboxyl groups.46
The FTIR analysis further confirmed the successful grafting of the surfactants onto the lignin structure (Fig. S3†). The characteristic hydroxyl (–OH) peak in the GO + surfactant lignin had a broader profile at around 3345 cm−1 compared to the GO lignin, indicating the formation of extensive intramolecular H-bonding between the surfactant and lignin molecules.19 Moreover, the absorption peak at 2873 cm−1, attributed to the methylene and methyl groups, was more pronounced in the surfactant-modified lignin than in the GO lignin.43 The emergence of new characteristic peaks of the surfactants, such as the C–H shear vibration at 1352 cm−1 and C–OH vibrations at 949 cm−1, also provided strong evidence for surfactant modification.47 Notably, these surfactant-derived signals persisted even after thorough extraction and washing of the GO + surfactant lignin, further substantiating the covalent nature of grafting. Interestingly, the small peak at 1150 cm−1 derived from the GO lignin was not discernible in the GO + surfactant lignin. This can be attributed to the strong C–O–C shear vibration peak of the grafted PEG chains, which overshadowed the C–H shear vibration peak of lignin at 1150 cm−1.48 Collectively, these findings demonstrate the occurrence of a chemical etherification reaction between the hydroxyl groups of lignin and the terminal hydroxyl groups of the surfactants, resulting in the formation of a novel covalent bond.
GPC analysis revealed the alteration of residual lignin molecular weight after surfactant grafting, as depicted in Fig. 3c. The Mw and Mn of the GO lignin were 1845 and 947 g mol−1, respectively, whereas the GO + surfactant lignin exhibited a higher molecular weight, with Mw ranging from 2058 to 2329 g mol−1 and Mn varying from 942 to 1085 of g mol−1. This suggested that the grafting of long-chain surfactants would increase the molecular weight of residual lignin. Additionally, it was observed that the polydispersity index (PDI) of the GO lignin (1.95) was lower than that of the GO + surfactant lignin (2.02–2.27). The presence of surfactants resulted in a broader molecular weight distribution of lignin.
Besides, the thermal stability of lignin is closely related to its structure. Fig. 3d and e present the thermogravimetric (TG) and derivative thermogravimetric (DTG) curves of various lignin samples. Expectedly, surfactant modification significantly influenced their pyrolysis characteristics, with GO + surfactant lignin exhibiting enhanced thermal stability. In conjunction with 31P NMR, FTIR and GPC measurements, it can be deduced that the surfactants facilitated the etherification of lignin during the GO pretreatment process. The pyrolysis of GO lignin proceeded slowly over a broad temperature range, while the pyrolysis of GO + surfactant lignin occurred more rapidly. At temperatures above 300 °C, the C–C and C–O bonds in the molecular chains were easily cleaved into small molecules.43 The surfactant moiety formed chain-initiating groups under thermal activation, laying the foundation for subsequent chain-breaking reactions. When further increasing the temperature, the extensive bond cleavage occurred within the molecular chains. For the TGA-derived maximum weight loss temperature (Tdmax) in Fig. 3e, the GO lignin had the lowest Tdmax (≈329.3 °C), whereas that of GO + surfactant lignin increased from 375.6 °C to 379.8 °C. The high thermal stability of surfactant-modified lignin enhances its potential for value-added applications, such as bio-based polymers and carbon materials.29
Except structural modification reflected by chemical bonds and functional groups, such effects can affect the hydrophobicity and surface charge of lignin. They are also essential factors in alleviating non-productive adsorption between lignin and cellulases.49 As shown in Fig. 3f and Fig. S4,† the contact angle of the GO lignin was 67.89°, while those of the GO + surfactant lignin samples ranged from 64.22° to 67.78°. There was only around a 5% difference in contact angles, indicating that the surfactants did not obviously change the hydrophilicity of lignin. Generally, the phenolic –OH group exhibits hydrophobic behavior, and the aliphatic –OH group demonstrates hydrophilicity. Based on this, the considerably reduced phenolic –OH contents in the GO + surfactant lignin samples should correspond to a relatively higher hydrophilicity. However, the Triton-X 100, AEO 9, and Tween 80 surfactants contain some hydrophobic benzene rings and methylene structures. With the reduction of aliphatic –OH, the increase in hydrophilicity was largely offset. Overall, the residual lignin derived from the pretreated substrates had similar contact angles, and the hydrophobic interaction was not the main factor affecting the non-productive adsorption of lignin and cellulases.
Zeta potential is another crucial factor governing the electrostatic interaction (i.e., repulsion or attraction) between lignin and cellulases during the non-productive adsorption.50 The GO lignin had the highest zeta potential of −18.6 mV. In contrast, the zeta potential of GO + surfactant lignin was measured in the range of −11.8 to −16.0 mV (Fig. 3f) with an absolute value reduction of 14.0%–36.6%. This result indicated that surfactant modification could shift the surface charge of lignin, which was tightly related to the carboxyl group content. A higher carboxyl group content in lignin can endow lignin with more negative charges.51 Commonly, the electrostatic interaction between lignin and cellulases highly depends on the pH of the hydrolysis environment. Both lignin and cellulases possess a negative charge under a representative condition with pH 4.8.52 Previously studies have pointed out that when lignin carried a more negative charge, the resulting strong electrostatic repulsion between lignin and cellulases could improve enzymatic hydrolysis.20 However, notably, all the GO + surfactant lignin displayed a reduced negative charge, which made it difficult to produce an elevated electrostatic repulsion among lignin and cellulases. Huang et al. also provided insights that lignin after adsorption with Tween 80 decreased the zeta potential (−28.2 mV) as compared to the control group without adsorption (−34.4 mV).49 They speculated that the electrostatic interaction between lignin after Tween 80 adsorption and cellulases did not dominate the non-productive adsorption.
In summary, the non-productive adsorption between lignin and cellulases is closely related to the hydrolysis performance of the pretreated substrates, and it is mainly influenced by three interactions, including hydrogen bonding, hydrophobic interaction, and electrostatic interaction. In this study, the surfactant-induced lignin modification through etherification remarkably decreased the phenolic –OH group of lignin, while barely changing its hydrophilicity and zeta potential. The reduction of hydrogen bonding between the phenolic –OH group and cellulases seems to be the prevailing factor governing the eventual result of non-productive adsorption, thereby enhancing the substrate hydrolyzability.
3.4. Structural modification of dissolved lignin in the pretreatment liquor
To better elucidate the structural modification in the presence of surfactants during GO pretreatment, the dissolved lignin was also characterized, as shown in Table 4. Both GO–DL and GO + surfactant–DL displayed relatively higher aliphatic –OH contents (5.70–6.88 mmol g−1) as compared with the residual lignin (Fig. 3a). The dissolved lignin was more likely to react with glycerol than the residual lignin. Moreover, the aliphatic –OH content of GO–DL was 6.88 mmol g−1, whereas that of GO + surfactant–DL decreased to 5.70–6.69 mmol g−1. Similarly, the total phenolic –OH content of GO + surfactant–DL (2.00–2.29 mmol g−1) was also lower than that of GO–DL (2.65 mmol g−1). These observations collectively supported the hypothesis that the surfactants chemically grafted onto lignin through etherification, leading to a reduction in both aliphatic and phenolic –OH groups. Interestingly, such a modification effect was also demonstrated in the appearance color of dissolved lignin. As shown in Fig. S5,† the surfactant-modified GO lignin presented a light brown color, while the GO lignin showed black color. The color change of lignin caused by surfactant modification is of great significance to broaden its application in specific fields, such as UV-blocking lignin nanoparticles for sunscreen products.| Lignin sample | Aliphatic –OH | S–OH | G–OH | H–OH | Total phenolic –OH | Carboxylic group | |
|---|---|---|---|---|---|---|---|
| NCa | Cb | ||||||
| a Non-condensed unit. b Condensed unit. | |||||||
| GO-DL | 6.88 | 0.91 | 0.68 | 0.15 | 0.91 | 2.65 | 0.63 |
| GO + Triton-X 100-DL | 5.70 | 0.85 | 0.60 | 0.08 | 0.76 | 2.29 | 0.50 |
| GO + AEO 9-DL | 6.69 | 0.80 | 0.64 | 0.10 | 0.65 | 2.19 | 0.37 |
| GO + Tween 80-DL | 5.79 | 0.74 | 0.53 | 0.08 | 0.65 | 2.00 | 0.55 |
Table 5 summarizes the molecular weight distribution of the dissolved lignin. Compare with the residual lignin (Mw: 1845–2329 g mol−1 and Mn: 942–1085 g mol−1), the dissolved lignin had a relatively low molecular weight (Mw: 1331–1794 g mol−1 and Mn: 712–944 g mol−1), suggesting its preferential depolymerization into smaller fragments during the pretreatment process. However, GO + surfactant–DL exhibited a higher molecular weight and PDI than GO–DL. This result aligned with the expectation that the grafting of surfactants onto the lignin structure would increase the molecular weight. For the physical properties of dissolved lignin (i.e., hydrophobicity and zeta potential), no statistically significant difference in the contact angle was observed between GO + surfactant–DL and GO–DL. Although the decrease in phenolic –OH content corresponded to the increased hydrophilicity, the hydrophobic moieties within the surfactant itself could potentially counteract this effect, as previously discussed. The reduced carboxylic group content also resulted in a decreased zeta potential in GO + surfactant–DL.
| Lignin sample | M w (g mol−1) | M n (g mol−1) | PDI (Mw/Mn) | Contact angle (°) | Zeta potential (mV) |
|---|---|---|---|---|---|
| GO-DL | 1331 | 712 | 1.87 | 65.12 ± 0.66 | −14.33 ± 0.57 |
| GO + Triton-X 100-DL | 1604 | 823 | 1.95 | 67.62 ± 0.87 | −12.67 ± 0.60 |
| GO + AEO 9-DL | 1794 | 944 | 1.90 | 66.31 ± 0.51 | −12.03 ± 0.25 |
| GO + Tween 80-DL | 1691 | 855 | 1.98 | 64.81 ± 0.02 | −10.83 ± 0.41 |
Overall, structural modification occurs in both dissolved lignin from the pretreatment liquor and residual lignin from the pretreated substrates. With the depolymerization of the lignin supramolecular structure and its dissolution into the solvent phase during the pretreatment, the modification behavior of dissolved lignin is more significant. Moreover, lignin modification is a dual in situ grafting reaction by glycerol and different surfactants, involving the multisite etherification of –OH groups in the aromatic skeleton and side chain of lignin. Considering the different structural variations from the widely used PEG and the selected surfactants in this study (Triton-X 100, AEO 9, and Tween 80), modification for promoting the Cα reactive-site end-capping of lignin and suppressing its condensation depends on the physicochemical properties of the surfactants.37 However, the extensive etherification of phenolic –OH groups by these surfactants is still a common occurrence, which can substantially mitigate the adverse hydrogen bonding interactions between lignin and cellulases during enzymatic hydrolysis. This presents a pioneering concept for forging an efficient lignocellulose-based sugar platform based on surfactant-assisted organosolv pretreatment.
3.5. Molecular interactions of surfactants with lignin by DFT simulation
To better assess the interactions generated by surfactants with lignin, this study used DFT calculations to investigate the weak interactions between glycerol–lignin and surfactant systems. During the DFT simulation, VG was selected as a lignin model compound. The energy-optimized molecular structures of glycerol, VG, and the targeted surfactants have been molded using the Gasteiger method and are shown in Fig. 4a. The atomic energy densities on the molecular surfaces of different combined chemical structures annotating their respective positive, neutral, and negative electrostatic potential points, have been marked in red, white, and blue, respectively (Fig. 4b). When the structure is subjected to any atomic force fields, the positional electrostatic energy is found to be regulated by the position of O atoms and the number of H-bonds formed with each O atom. Based on the frequency of O–H interaction, a prominent negative electrostatic potential surface was observed for the glycerol–VG-surfactant system compared with the glycerol–VG system. Significantly, the introduced Tween 80 endowed the system with more negative electrostatic potential, which may be attributed to the large number of O atoms in Tween 80 that easily form hydrogen bonds with lignin and glycerol, thus facilitating the lignin modification. | ||
| Fig. 4 (a) Molecular structures of glycerol, Triton-X 100, AEO 9, Tween 80 and VG (lignin model); (b) geometries and electrostatic potential surfaces of different compounds; (c) reduced density gradient analysis of glycerol–VG and glycerol–VG surfactant; and (d) portion of hydrogen bonding interaction. | ||
To further investigate the effects arising between the surfactants and the glycerol–lignin systems, RDG analysis was used to examine the weak interactions. As shown in Fig. 4c, the blue (λ2 < 0, negative values), green (λ2 ≈ 0), and red regions (λ2 > 0, positive values) present the H-bonding interactions, van der Waals interactions, and strong repulsion effects, respectively.34 It clearly shows four low gradient peaks in the blue region (negative values) for both the glycerol–VG system and the glycerol–VG-surfactant system, indicating strong hydrogen bonds present in the above systems. Compared with glycerol–VG, the interaction spikes of surfactants with glycerol–VG are more intensive, indicating a higher strength of H-bonding interactions between the surfactants and glycerol–VG system. Moreover, the intermolecular interaction energies of different systems were also calculated. The interaction energy of the glycerol–VG system was −32.5 kcal mol−1, whereas the presence of different surfactants led to a substantial decrease ranging from −38.1 to 48.4 kcal mol−1, suggesting that surfactants could effectively promote the reaction with glycerol–lignin complexes. Commonly, glycerol, with its higher number of –OH groups, has more H-bonding interactions with lignin and modifies lignin.6 Incorporating surfactants into the system further synergistically intensifies the weak interaction with lignin due to the presence of extra –OH groups in surfactants, as depicted in the 2D-HSQC and 31P NMR results. As Tween-80 has a long straight ‘alkyl chain’, it becomes more exposed to the energy force fields in a solvent system than other surfactants.
The hydrogen bond number is a dependable indicator that represents the interaction of glycerol–VG with different surfactants. More specifically, the formation of H-bonds can be categorized into four positions: O1, O2, O3, and O4.53 Among these, O1 and O2 refer to oxygen atoms participating in the β-O-4 linkages and the two benzene rings connected by ether bonds in VG, respectively.54 O3 denotes the phenolic –OH group, while O4 includes the aliphatic –OH groups (i.e., O4aH, α-OH, O4bH, and γ-OH) of VG.55 Apparently, considering the maximal number of H-bonds concerning the interaction strengths of both the internal and auxiliary O atoms, the glycerol–VG-Tween 80 system was vigorous than all others, possessing 18%, 26%, 11%, 23%, and 22% of O1, O2, O3, O4a, and O4b, respectively. This analysis represents a more evenly distributed H-bond formation with the O atoms compared to that with glycerol–VG. Overall, adding surfactants to the glycerol–lignin systems could effectively promote the electrophilic reaction and enhance the weak interaction with lignin.
3.6. Quantitative analysis between surfactant modification and substrate hydrolyzability
The above characterization validated that the lignin structure was modified during surfactant-assisted GO pretreatment. To further evaluate the “structure–function” relationship between surfactant modification and substrate hydrolyzability, a quantitative analysis was conducted based on various hydrolysis-related parameters, including the pore structure, surface properties, cellulose accessibility, and functional groups.Fig. 5a shows the free enzyme content in the supernatant after 72 h enzymatic hydrolysis. For the GO-pretreated substrates, the free enzyme content was 27.8%, which was much lower than those of the surfactant-assisted GO-pretreated substrates (38.9%–46.9%). Generally, the higher free enzyme suggests that the cellulases can quickly act on the substrates by adsorption/desorption.9 Since adding surfactants did not significantly change the component distribution and the physical pore size of the pretreated substrates (see section 3.1), the physical barrier of cellulase accessibility to substrates could be neglected. However, notably, the chemical structure of residual lignin, which tends to dominate the non-productive adsorption of cellulases, was obviously changed via etherification grafting of the surfactants. Therefore, such structural modification led to a greater free enzyme concentration in the supernatant.
 | ||
| Fig. 5 (a) Free enzyme content in the supernatant after 72 h enzymatic hydrolysis; (b) evaluation of non-productive adsorption by BSA incubation; and (c) quantitative correlation analysis of the factors affecting enzymatic hydrolysis after surfactant-assisted pretreatment. | ||
To identify the inhibition degree of enzymatic hydrolysis by the non-productive adsorption of the residual lignin, the pretreated substrates were incubated with BSA (without cellulase addition). The differences in 72 h glucose yields before and after incubation with/without BSA were considered as the contribution of non-productive adsorption. Apparently, the contribution of the GO-pretreated substrates to the non-productive adsorption was calculated as 37.3%, whereas that of the surfactant-assisted GO-pretreated substrates showed a sharp reduction in the range of 4.9%–8.3% (Fig. 5b). The low non-productive adsorption effect was also due to the surfactant-induced lignin modification, which decreased the hydrogen bonding interaction between the phenolic –OH groups of lignin and cellulases. Altogether, the results involving the free enzyme content, non-productive adsorption, and glucose yield (as indicators to analyze substrate hydrolyzability) were used to develop the quantitative correlation analysis.
As shown in Fig. 5c, the glucose yield exhibited a strong positive correlation with the free enzyme content (0.98), while it had an opposite negative correlation with the non-productive adsorption (−0.99). Typically, lignin can bind into cellulases through non-productive adsorption, thereby reducing the free enzyme availability.7 For the substrate properties, the correlation coefficients of the specific surface area, pore volume, cellulose accessibility, and crystalline nature with glucose yield (−0.24–0.33), free enzyme content (−0.07–0.46), and non-productive adsorption (−0.18–0.39) were extremely low. Thus, the contributions of these parameters to improving substrate hydrolyzability were limited during the surfactant-assisted GO pretreatment. Differently, there was a high negative correlation between the lignin structure and substrate hydrolyzability. The correlation coefficients of the aliphatic –OH group content with glucose yield and free enzyme content were −0.92 and −0.87, respectively, and those of the total phenolic –OH content reached up to −0.93 and −0.98, respectively. The strong correlation between the phenolic/aliphatic –OH content and non-productive adsorption (0.87/0.96) also underscored the importance of surfactant modification in promoting substrate hydrolyzability by decreasing the total phenolic/aliphatic –OH content. On the other hand, the PDI of lignin showed a good correlation with the glucose yield (0.85) and free enzyme content (0.92), and this was due to the changes in the molecular weight distribution of lignin through surfactant grafting. Considering that both electrostatic repulsion and hydrophobic interaction between lignin and cellulases were weakened by surfactant-induced lignin modification, the zeta potential of lignin had a moderate negative relevance to non-productive adsorption (−0.77), while that of the contact angle was only 0.60.
Combined with characterization and quantitative correlation analysis, the secrets of harnessing a surfactant-assisted organosolv pretreatment strategy for efficient enzymatic hydrolysis are attributed to the dual solvent/surfactant modification of the residual lignin structure in the pretreated substrates, rather than the component fractionation of LCB. Compared with the conventional organosolv pretreatment, the introduction of surfactants plays an important role in reducing H-bonding interaction between lignin and cellulases through decreasing the phenolic –OH content of lignin. This positive effect in promoting enzymatic hydrolysis is superior to mitigating hydrophobic and electrostatic interactions through altering lignin surface properties. The structural and physical properties of surfactants may be closely related to the structural modification of lignin. Thus, customizing surfactants with structure-oriented modifications to facilitate substrate hydrolyzability is a potential research focus.
4. Conclusions
This study investigated the incorporation of different non-ionic surfactants (i.e., Triton-X 100, AEO 9, and Tween 80) in the organosolv pretreatment for in situ lignin modification to suppress the adverse effects of residual lignin on enzymatic hydrolysis. Adding these non-ionic surfactants significantly enhanced the substrate hydrolyzability, though without changing the main component distribution. Notably, they could chemically react with the hydroxyl groups of lignin through etherification. Based on the molecular dynamic simulations, the surfactant–lignin mixture had strong H-bonding interaction and van der Waals force. Furthermore, such grafting reaction altered the surface properties and functional groups of lignin, endowing the etherified lignin with fewer hydroxyl groups and reduced negative charge. The relationships between the structure and function indicated that the etherified lignin exhibited lower non-productive adsorption onto enzymes; the reduced hydroxyl groups (especially phenolic –OH) played a predominant role in mitigating the H-bonding interaction between lignin and cellulases, affording satisfactory fermentable sugar production. Overall, the advanced surfactant-assisted pretreatment holds great promise as an efficient process for enzyme-mediated lignocellulosic biorefineries. Surfactant types are closely related to the pretreatment effects and lignin modification. The mechanism by which etherification grafting modifies the lignin structure to enhance substrate hydrolyzability still requires further investigation in future studies to assess its applicability across different surfactants.Data availability
All data generated or analyzed during this study are included in this published article and its ESI†. Additional data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the State Key R&D Program (2022YFC2104601), the National Natural Science Foundation of China (U23A20135 and 22278189), the International Cooperation and Exchange of the National Natural Science Foundation of China (22361142705), and the National First-class Discipline Program of Light Industry Technology and Engineering (QGJC20230203). The authors also thank the 111 Project (111-2-06) and Iran National Science Foundation (INSF, 4021538).References
- Q. Chu, W. Tong, S. Wu, Y. Jin, J. Hu and K. Song, Green Chem., 2021, 23, 4074–4086 RSC.
- M. Madadi, G. Song, V. K. Gupta, M. Aghbashloh, C. Sun, F. Sun and M. Tabatabaei, Green Chem., 2023, 25, 7141–7156 RSC.
- X. Yuan, G. Shen, S. Chen, W. Shen, X. Chen, S. Liu and M. Jin, Green Chem., 2023, 25, 6759–6773 RSC.
- S. C. Rabelo, P. Y. S. Nakasu, E. Scopel, M. F. Araújo, L. H. Cardoso and A. C. da Costa, Bioresour. Technol., 2023, 369, 128331 CrossRef PubMed.
- F. F. Sun, L. Wang, J. Hong, J. Ren, F. Du, J. Hu, Z. Zhang and B. Zhou, Bioresour. Technol., 2015, 187, 354–361 CrossRef.
- C. Sun, H. Ren, F. Sun, Y. Hu, Q. Liu, G. Song, A. Abdulkhani and P. Loke Show, Bioresour. Technol., 2022, 344, 126264 CrossRef.
- C. Huang, X. Jiang, X. Shen, J. Hu, W. Tang, X. Wu, A. Ragauskas, H. Jameel, X. Meng and Q. Yong, Renewable Sustainable Energy Rev., 2022, 154, 111822 CrossRef.
- M. Karimi, B. Jenkins and P. Stroeve, Renewable Sustainable Energy Rev., 2014, 40, 400–421 CrossRef.
- X. Li and Y. Zheng, Biotechnol. Adv., 2017, 35, 466–489 CrossRef CAS PubMed.
- Y. Sheng, M. Liu, C. Xia, J. Song, S. Ge, L. Cai, S. S. Lam and C. Sonne, Chem. Eng. J., 2021, 420, 130258 CrossRef CAS.
- M. Madadi, M. M. A. Bakr, A. Abdulkhani, Zahoor, M. A. Asadollahi, C. Sun, F. Sun and A. E.-F. Abomohra, Energy Convers. Manage., 2022, 251, 114956 CrossRef.
- G. Song, M. Madadi, X. Meng, C. Sun, M. Aghbashlo, F. Sun, A. J. Ragauskas, M. Tabatabaei and A. Ashori, Chem. Eng. J., 2024, 481, 148713 CrossRef.
- T. Zheng, J. Jiang and J. Yao, Fuel Process. Technol., 2021, 213, 106660 CrossRef.
- S. Sánchez Muñoz, T. Rocha Balbino, E. Mier Alba, F. Gonçalves Barbosa, F. Tonet de Pier, A. Lazuroz Moura de Almeida, A. Helena Balan Zilla, F. Antonio Fernandes Antunes, R. Terán Hilares, N. Balagurusamy, J. César dos Santos and S. Silvério da Silva, Bioresour. Technol., 2022, 345, 126477 CrossRef.
- Q. Ma, W. Zhou, X. Du, H. Huang and Z. Gong, Bioresour. Technol., 2023, 382, 129218 CrossRef CAS.
- M. Fan, C. Zhao, X. Huang, H. Zhang and J. Xie, Ind. Crops Prod., 2021, 172, 114006 CrossRef CAS.
- C. Gong, N. Bryant, X. Meng, S. Bhagia, Y. Pu, D. Xin, C. B. Koch, C. Felby, L. Garbrecht Thygesen, A. Ragauskas and S. Tjalfe Thomsen, Green Chem., 2021, 23, 1050–1061 RSC.
- G. Song, M. Madadi, C. Sun, L. Shao, M. Tu, A. Abdulkhani, Q. Zhou, X. Lu, J. Hu and F. Sun, Bioresour. Technol., 2023, 383, 129178 CrossRef CAS.
- Q. Li, D. Cao, M. T. Naik, Y. Pu, X. Sun, P. Luan, A. J. Ragauskas, T. Ji, Y. Zhao, F. Chen, Y. Zheng and H. Zhu, ACS Sustainable Chem. Eng., 2022, 10, 8704–8714 CrossRef CAS.
- M. Li, B. Jiang, W. Wu, S. Wu, Y. Yang, J. Song, M. Ahmad and Y. Jin, Int. J. Biol. Macromol., 2022, 195, 274–286 CrossRef CAS PubMed.
- H. Chen, C. Sun, Y. Hu, C. Xia, F. Sun and Z. Zhang, J. Environ. Chem. Eng., 2023, 11, 109531 CrossRef CAS.
- F. F. Sun, X. Zhao, J. Hong, Y. Tang, L. Wang, H. Sun, X. Li and J. Hu, Biotechnol. Biofuels, 2016, 9, 59 CrossRef PubMed.
- C. Lai, M. Tu, Q. Yong and S. Yu, RSC Adv., 2015, 5, 97966–97974 RSC.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Natl. Renewable. Energy Lab., 2008, TP-510-42618 Search PubMed.
- M. Madadi, M. M. A. Bakr, G. Song, C. Sun, F. Sun, Z. Hao, Zahoor and A. Abomohra, J. Cleaner Prod., 2022, 366, 132817 CrossRef CAS.
- M. Madadi, Zahoor, G. Song, K. Karimi, D. Zhu, M. Elsayed, F. Sun and A. Abomohra, Bioresour. Technol., 2022, 359, 127503 CrossRef CAS.
- Y. Xu, S.-C. Sun, C. Zhang, C.-Y. Ma, J.-L. Wen and T.-Q. Yuan, Chem. Eng. J., 2023, 462, 142213 CrossRef.
- M. Madadi, M. Elsayed, G. Song, R. Kumar, M. Mahmoud-Aly, B. Basak, B.-H. Jeon and F. Sun, Chem. Eng. J., 2023, 465, 142881 CrossRef.
- T. T. Nge, Y. Tobimatsu, S. Takahashi, E. Takata, M. Yamamura, Y. Miyagawa, T. Ikeda, T. Umezawa and T. Yamada, ACS Sustainable Chem. Eng., 2018, 6, 7841–7848 CrossRef.
- W. Ying, J. Yang and J. Zhang, Ind. Crops Prod., 2022, 182, 114863 CrossRef.
- Z. Wang, Y. Sun, Y. Mao, F. Zhang, L. Zheng, D. Fu, Y. Shen, J. Hu, H. Dong, J. Xu and X. Wu, Energy Storage Mater., 2020, 30, 228–237 CrossRef.
- J. Řezáč and P. Hobza, J. Chem. Theory Comput., 2012, 8, 141–151 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
- D. Fan, X. Xie, C. Li, X. Liu, J. Zhong, X. Ouyang, Q. Liu and X. Qiu, J. Agric. Food Chem., 2021, 69, 10838–10847 CrossRef PubMed.
- J.-Q. Cui, Y.-Q. Li, S. Ntakirutimana, Z.-H. Liu, B.-Z. Li and Y.-J. Yuan, Bioresour. Technol., 2023, 382, 129174 CrossRef PubMed.
- B. Qi, X. Chen and Y. Wan, Bioresour. Technol., 2010, 101, 4875–4883 CrossRef CAS.
- D. Cai, J. Wen, Y. Wu, C. Su, H. Bi, Y. Wang, Y. Jiang, P. Qin, T. Tan and C. Zhang, Bioresour. Technol., 2023, 394, 130231 CrossRef.
- X. Cheng, Y. Luo, Y. Gao, S. Li, C. Xu, S. Tang, Y. Yang, Z. Zhang, H. Jiang, H. Xu, S. Shi and Q. Yan, Front. Bioeng. Biotechnol., 2022, 10, 918126 CrossRef PubMed.
- G. Song, C. Sun, M. Madadi, S. Dou, J. Yan, H. Huan, M. Aghbashlo, M. Tabatabaei, F. Sun and A. Ashori, Bioresour. Technol., 2024, 395, 130358 CrossRef PubMed.
- L. Chen and S. Fu, J. Agric. Food Chem., 2013, 61, 3293–3300 CrossRef.
- X. Zhao, X. Meng, A. J. Ragauskas, C. Lai, Z. Ling, C. Huang and Q. Yong, Biotechnol. Adv., 2022, 54, 107830 CrossRef PubMed.
- M. Hassanpour, M. Abbasabadi, L. Moghaddam, F. F. Sun, L. Gebbie, V. S. J. Te'o, I. M. O'Hara and Z. Zhang, Bioresour. Technol., 2020, 318, 124059 CrossRef PubMed.
- Z. Ju, I. Ziegler-Devin, L. Chrusciel, B. Lu, Z. Wang, H. Zhang and N. Brosse, Ind. Crops Prod., 2023, 206, 117614 CrossRef.
- Z. Pan, Y. Li, B. Wang, F. Sun, F. Xu and X. Zhang, Bioresour. Technol., 2022, 343, 126122 CrossRef PubMed.
- J.-L. Wen, T.-Q. Yuan, S.-L. Sun, F. Xu and R.-C. Sun, Green Chem., 2014, 16, 181–190 RSC.
- X. Cheng, L. Zhang, F. Zhang, P. Li, L. Ji, K. Wang and J. Jiang, Bioresour. Technol., 2023, 386, 129527 CrossRef PubMed.
- Z. Ju, N. Brosse, S. Hoppe, Z. Wang, I. Ziegler-Devin, H. Zhang and B. Shu, Int. J. Biol. Macromol., 2024, 262, 129997 CrossRef PubMed.
- Y. Feng, J. Lan, P. Ma, X. Dong, J. Qu and H. He, Wood Sci. Technol., 2017, 51, 135–150 CrossRef.
- C. Huang, X. Zhao, Y. Zheng, W. Lin, C. Lai, Q. Yong, A. J. Ragauskas and X. Meng, Bioresour. Technol., 2022, 360, 127524 CrossRef.
- C. Huang, W. Lin, Y. Zheng, X. Zhao, A. Ragauskas and X. Meng, Green Chem., 2022, 24, 5263–5279 RSC.
- C.-Y. Ma, L.-H. Xu, Q. Sun, X.-J. Shen, J.-L. Wen and T.-Q. Yuan, Chem. Eng. J., 2022, 450, 138315 CrossRef.
- M. Madadi, G. Song, F. Sun, C. Sun, C. Xia, E. Zhang, K. Karimi and M. Tu, Environ. Res., 2022, 215, 114291 CrossRef PubMed.
- W. Wang, Y. Xu, B. Zhu, H. Ge, S. Wang, B. Li and H. Xu, Bioresour. Technol., 2023, 385, 129401 CrossRef PubMed.
- M. Madadi, M. Elsayed, F. Sun, J. Wang, K. Karimi, G. Song, M. Tabatabaei and M. Aghbashlo, Bioresour. Technol., 2023, 371, 128591 CrossRef.
- W. Wang, B. Zhu, Y. Xu, B. Li and H. Xu, Bioresour. Technol., 2022, 363, 127887 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc02775b |
| ‡ These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |