
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiquid–liquid extraction for in situ carboxylic acid recovery via continuous membrane-based emulsion separations†
Yian
Chen
a,
Patrick O.
Saboe
 b,
Jacob S.
Kruger‡
a,
Eric C. D.
Tan
c,
Jacob W.
Dempsey
c,
Jeffrey G.
Linger
a,
Violeta
Sànchez i Nogué
a,
Eric M.
Karp‡
a and
Gregg T.
Beckham
*a
b,
Jacob S.
Kruger‡
a,
Eric C. D.
Tan
c,
Jacob W.
Dempsey
c,
Jeffrey G.
Linger
a,
Violeta
Sànchez i Nogué
a,
Eric M.
Karp‡
a and
Gregg T.
Beckham
*a
aRenewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, Golden, CO 80401, USA. E-mail: gregg.beckham@nrel.gov
bStrategic Energy Analysis Center, National Renewable Energy Laboratory, Golden, CO 80401, USA
cCatalytic Carbon Transformation and Scale-up Center, National Renewable Energy Laboratory, Golden, CO 80401, USA
First published on 2nd August 2024
Abstract
The separation and purification of bio-based chemicals from conversion processes often contribute substantially to bioprocessing costs, and for many biochemicals produced through fermentation, solvent-based liquid–liquid extraction (LLE) is a common separations strategy for in situ product recovery (ISPR). Many in situ LLE-based separations for biochemicals are often challenged by emulsion formation between the immiscible aqueous and organic phases. Typically, membrane contactors have been used to overcome emulsion formation, with the aqueous and organic phases coming into contact in the membrane contactor pores, but these unit operations require a large membrane area to compensate for their limited effective interfacial area. In this study, we show that a hydrophobic polytetrafluoroethylene (PTFE) membrane-based emulsion separator (MBES) enables continuous LLE for the recovery of an exemplary fermentation product, butyric acid, with substantially improved throughput relative to a membrane contactor. With a membrane permeate flux of 290 L m−2 h−1, the overall butyric acid flux of the single-stage MBES system was calculated to be 1450 g m−2 h−1, which is substantially higher than the 9 g m−2 h−1 achievable with a membrane contactor. At an equivalent butyric acid throughput, process modeling estimates that MBES-assisted LLE can enable a 55% and 91% reduction in process costs (the sum of CAPEX and OPEX) and greenhouse gas emissions, respectively, compared to a membrane contactor due to a ∼160-fold decrease in the required membrane area. Although membrane fouling from cellular debris led to reduced membrane flux and phase separation efficiency, common fouling mitigation strategies including the incorporation of ultrafiltration and periodic membrane backwashing effectively recovered the membrane performance. Overall, MBES systems can potentially enable continuous LLE processes in bioprocessing separations, including where emulsion formation is a challenge.
Introduction
The conversion of biogenic feedstocks in microbial processes holds substantial promise to displace fossil carbon-derived fuels and chemicals.1 For the production of many bio-based chemicals, downstream separations are often the most expensive and energy-intensive unit operations of an integrated process,2,3 accounting for up to 90% of the total production cost.4 Towards improved separation processes in bioprocessing, in situ product recovery (ISPR) can potentially enhance product biosynthesis rates and titers, and enable continuous fermentation processes.5–9 For many products produced from bioconversion, solvent-based liquid–liquid extraction (LLE) is among the most well studied approaches for ISPR due to its ability to achieve high product recovery yields.4,10–12Traditional in situ LLE is conducted with a solvent overlay in a bioreactor to continuously extract the bioproducts. However, this approach is challenged by emulsion formation between immiscible aqueous and organic phases, which in turn impairs phase separation.13,14 The solvent droplets dispersed in the aqueous phase can be toxic to microorganisms,6 and the water droplets dispersed into the organic phase can increase the energy consumption for the subsequent bioproduct purification and solvent recovery steps. To reduce the potential toxic effects of the solvents on the microbial culture, a cell retention and product clarification step is often added prior to solvent extraction7,15,16 to physically retain the cells in the bioreactor and prevent their direct contact with the organic solvent (Fig. 1A). Continuous membrane-based solvent extraction, a popular separation technology in green chemistry and engineering,17–19 has also been integrated to the ISPR processes. A membrane contactor, in which a hydrophobic porous membrane serves as a physical barrier between the aqueous and organic phases, is typically used downstream of the cell retention unit operation for continuous solvent extraction and to prevent emulsion formation (Fig. 1A).20–23 In previously reported ISPR processes, a membrane contactor enabled continuous carboxylic acid extraction from fermentation broth.7,15,24–27 The acid flux was limited, however, because the aqueous and organic phases were only in contact in the membrane contactor pores,28 and thus, a large membrane area was needed to compensate for the low throughput of butyric acid extraction.
 | ||
| Fig. 1 ISPR process configurations with (A) a membrane contactor unit operation and (B) a membrane-based emulsion separator, both to achieve continuous LLE-based solvent extraction of a target bioproduct. For both process configurations, there is a cell retention unit operation before LLE to ensure that microbial cells and solids do not interact with the membrane systems, and a distillation column is shown downstream of the LLE step for product purification and solvent recovery. The current study examines the use of an MBES system in place of a membrane contactor. Notably, other strategies can be used for the separation of the bioproduct and the organic solvent; distillation is shown here for illustrative purposes. | ||
To increase bioproduct flux in continuous LLE, the use of a membrane-based emulsion separator (MBES)16,29 is a potential alternative approach to a membrane contactor (Fig. 1B). MBES-assisted LLE starts with sufficient mixing of the two phases to promote emulsion formation, shown as a phase mixer in Fig. 1B. Emulsification ensures ample contact between the two phases through increased effective interfacial area, and thus enhances the mass transfer rate of a target product into the extraction solvent. After achieving equilibrium, the MBES, a porous membrane-based system, could be used to continuously separate the emulsion into a product-rich organic solvent and product-lean aqueous phase. Such complete in-line phase separation can be achieved via MBES systems by leveraging surface tension and fine-tuning pressure differential,16 and this approach has been applied successfully in petrochemicals,30–42 pharmaceuticals,43–52 and petroleum refining,53 suggesting that MBES systems could also be promising for bioprocessing applications.
Based on the potential of membrane demulsification systems in bioprocessing applications, we were interested to examine if an MBES unit operation could improve the efficiency and economics of the continuous LLE-based solvent extraction of biochemicals from fermentation broth. To that end, we used a short-chain carboxylic acid, butyric acid, produced via fermentation with Clostridium tyrobutyricum,7 as a case study to evaluate the technical feasibility of an MBES-assisted LLE process (Fig. 1B). Our goal was to optimize the extraction rate, membrane throughput, and phase separation efficiency of the MBES unit operation. The optimized conditions were used to evaluate membrane fouling propensity, and the feasibility of utilizing pre-filtration and membrane backwashing to mitigate fouling. Following process optimization, we compared the economics and greenhouse gas (GHG) emissions of an MBES-assisted LLE process with the membrane contactor-assisted LLE process (Fig. 1) to assess if the MBES systems can potentially enhance the sustainability and economic viability of continuous LLE processes in downstream bioprocessing.
Results
MBES performance for butyric acid recovery and comparison to a membrane contactor
We first aimed to evaluate the performance of MBES-assisted LLE for solvent extraction of butyric acid at different permeate fluxes and phase volume ratios. For these initial experiments, we prepared a solution of butyric acid at 10 g L−1 and pH 5 as a ‘mock broth’, representing a single component model solution to mimic the C. tyrobutyricum fermentation broth (vide infra).7 From our previous work, 10 g L−1 was the target residual butyric acid titer in the bioreactor at pH 5 to prevent microbial product inhibition, while creating a sufficient driving force for partitioning into the organic phase in LLE. For these initial experiments, we used an organic extractant mixture consisting of 70 vol% Cyanex 923 and 30 vol% mineral oil, which exhibits the optimal partition coefficient for butyric acid at pH 5.7,15 Notably, Cyanex 923 is a commercial mixture of phosphine oxides with detailed compositions described previously,15 and is selected in the present study due to its low volatility, commercial availability, miscibility with organic solvents, and relatively low freezing point.15We began by vigorously mixing the aqueous and organic phases in a range of phase volume ratios using a magnetic stir plate to form emulsions (Fig. 2A), with the intention to mimic the in-line phase mixer in Fig. 1B and maximize the contact area between the aqueous and organic phases. The emulsion was then pumped into the MBES unit (SEP-200). We used a hydrophobic polytetrafluoroethylene (PTFE) membrane (OB-2000-S200F) with a pore size of 1 μm and effective membrane area 60 cm2 (Zaiput Flow Technologies) at a range of feed flow rates with different corresponding permeate fluxes for membrane-based demulsification. The MBES unit continuously separated this emulsified feed stream into butyric acid-lean aqueous and butyric acid-rich organic phases (Fig. 2A). The phase separation efficiency (PS) of the MBES system was evaluated by measuring the water content in the feed and permeate streams using Karl Fisher (KF) titration and compared with the conventional overlay LLE as a reference using the same organic solvent composition. PS was defined as:
| PS = 1 − Cw,p/Cw,f | (1) |
 | ||
Fig. 2 (A) Schematic of the experimental setup for continuous membrane demulsification using MBES with an effective total membrane area of 60 cm2 (image created partly with BioRender.com). Mock fermentation broth (10 g L−1 butyric acid, pH 5) and organic solvent (70 vol% Cyanex 923, 30 vol% mineral oil) were vigorously mixed using a magnetic stir plate to form an emulsion for the MBES extraction step, and then continuously separated via MBES into pure aqueous and organic phases. The extraction efficiency of butyric acid into the organic solvent was characterized using HPLC of the aqueous phase before and after the MBES unit, and the phase separation efficiency was measured using Karl Fisher titration with both the organic and aqueous phases after the MBES unit. (B) Images (top) and Karl Fisher titration measurements of water content in the organic solvent phase (values at bottom) after different settling time of an emulsion that is kinetically stable for 1500 min. (C) The impact of the aqueous![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :organic phase volume ratio (ϕ) and permeate flux on MBES phase separation efficiency (PS; eqn (1)). The thermodynamic limits of PS (due to the water partition coefficient) at a ϕ value of 0.5, 1.0, 2.0, and 3.0 are 90.5%, 93.1%, 94.9%, and 95.8%, respectively (eqn (2)). Membrane phase separation efficiency reversibility as indicated by: (D) the phase separation efficiency at different permeate flux (at an exemplary ϕ of 0.5; dark blue curve – direction of increasing permeate flux; light blue curve – direction of decreasing permeate flux) and supplemented by (E) the corresponding photos of the MBES permeate (left) and retentate (right) samples at the tested range of permeate flux. The error bars in (C) and (D) represent the standard deviation from at least 3 replicate measurements. The data shown in (C) and (D) are provided in Tables S2–S5.† :organic phase volume ratio (ϕ) and permeate flux on MBES phase separation efficiency (PS; eqn (1)). The thermodynamic limits of PS (due to the water partition coefficient) at a ϕ value of 0.5, 1.0, 2.0, and 3.0 are 90.5%, 93.1%, 94.9%, and 95.8%, respectively (eqn (2)). Membrane phase separation efficiency reversibility as indicated by: (D) the phase separation efficiency at different permeate flux (at an exemplary ϕ of 0.5; dark blue curve – direction of increasing permeate flux; light blue curve – direction of decreasing permeate flux) and supplemented by (E) the corresponding photos of the MBES permeate (left) and retentate (right) samples at the tested range of permeate flux. The error bars in (C) and (D) represent the standard deviation from at least 3 replicate measurements. The data shown in (C) and (D) are provided in Tables S2–S5.† | ||
Notably, there is a thermodynamic limit of the phase separation efficiency at each aqueous:organic phase volume ratio (ϕ) due to the co-extraction of water. For example, the maximum achievable PS is 93.1% at ϕ of 1.0 and 94.9% at ϕ of 2.0, which is also the thermodynamic limit of PS (PSTL), defined as:
| PSTL = 1 − Cw,min/Cw,f | (2) |
Since membrane demulsification using the MBES system is based on the differences in membrane affinity of the different solvents, membrane surface hydrophilicity is an important indicator of its phase separation efficiency. Membrane surface hydrophilicity can be indicated by membrane surface free energy of hydration (ΔGiw), calculated from the Young-Dupré equation:
| ΔGiw = −γw(1 + cosθw) | (3) |
We also evaluated butyric acid extraction efficiency using the MBES system by measuring the butyric acid concentrations in the aqueous phase before and after the MBES-assisted LLE process using high-performance liquid chromatography (HPLC), and compared that to the previously reported results15 of a membrane contactor-assisted LLE experiment as the reference, again using the same organic solvent composition. The organic solvent partition coefficient (KD) for butyric acid was calculated as:
| KD = CBA,Org,final/CBA,Aq,final | (4) |
| CBA,O,final = (CBA,Aq,initial − CBA,Aq,final) × ϕ | (5) |
The overall MBES butyric acid extraction efficiency (EE) for a specific aqueous:organic phase volume ratio (ϕ) can thus be defined by:
| EE = CBA,Org,final/(CBA,Aq,initial × ϕ) | (6) |
The MBES butyric acid flux (EF) was determined by:
| EF = CBA,Org,final × Jv | (7) |
From the initial phase mixing experiments, only 3 min were required to reach the butyric acid partition equilibrium (with a measured partition coefficient KD = 1; eqn (4)) after a vigorous phase mixing (at a phase volume ratio of 1:1 and a stir rate of 650 rpm). The vigorous phase mixing results in emulsion formation with the aqueous phase present in the form of droplets with an average diameter of 3.5 μm (measured using a Zetasizer) dispersed in the continuous organic phase, which thus allowed droplet-based liquid–liquid microextraction with accelerated partitioning. However, despite the significant increase in the mass transfer rate, the experimentally formed emulsion is kinetically stable and unbreakable via natural separation in overlay LLE. As shown in Fig. 2B, the top organic layer remained as an emulsion with 7.9 wt% water content after 1500 min of settling time. The impractically long time required for natural phase separation is attributed to the presence of a robust oil/water interface, making solvent extraction via a direct overlay infeasible.
To accelerate and achieve complete demulsification, the MBES unit was used to separate the emulsion into a butyric acid-lean aqueous phase and a butyric acid-rich organic phase (Fig. 2A). Due to the high surface hydrophobicity of the chosen membrane, indicated by its ΔGiw (−30.8 mJ m−2; Table S1†) significantly higher than the threshold value of −113.0 mJ m−2,54 the membrane should have higher affinity (i.e., wettability) and preferential transport of the organic phase.55,56 Consequently, it was expected that the organic phase should permeate through the membrane, while the aqueous phase should be retained. Specifically, it required only 10 min for the MBES (at its maximum permeate flux with complete phase separation; Fig. 2C) to continuously separate a 400 mL emulsion with the 60 cm2 membrane used here.
As expected for demulsification, the MBES unit achieved complete phase separation at a lower range of permeate fluxes across phase volume ratios (Fig. 2C). For example, the MBES system enabled complete separations of an emulsion at a ϕ value of 1.0 into a pure aqueous phase as the retentate (water content = 100 wt%; Fig. S1(d)†) and an organic phase as the permeate with water content at the thermodynamic limit for permeate flux values <290.0 L m−2 h−1. At a higher range of permeate flux, membrane phase breakthrough (i.e., intrusion) was observed, as expected (Fig. 2C–E). At these higher permeate fluxes, the aqueous phase wetted the hydrophobic PTFE membrane, resulting in increased membrane surface hydrophilicity (with 40.9% reduced membrane surface water contact angle, and ΔGiw decreased from −30.8 to −92.1 mJ m−2; Table S1†) and greater aqueous phase-membrane affinity, thus leading up to 28.0% aqueous phase intrusion (Fig. 2C). Notably, the increased preferential transport of the aqueous phase also led to membrane rejection of the organic phase, resulting in both permeate and retentate streams composed of mixtures of aqueous and organic phases.
The compromised membrane phase separation efficiency at higher permeate flux values can be 100% reversed by reducing the permeate flux (Fig. 2D). However, the reversal curves (in the direction of decreasing permeate flux) of the permeate and retentate water content do not perfectly overlay with the original ones (in the direction of increasing permeate flux; Fig. S2†), due to the interplay between membrane surface wetting and phase breakthrough. This hysteresis is caused by a more wetted membrane exhibiting lower transport resistance to the aqueous phase, which further increases the potential of aqueous phase breakthrough and surface wetting in a positive feedback loop. It is thus not surprising that a lower permeate flux was needed to achieve the same membrane phase separation efficiency after membrane surface and/or pore wetting.
At ϕ = 1.0, the MBES butyric acid extraction efficiency (EE; eqn (6)) was calculated to be 50%. The butyric acid extraction efficiency was limited by the organic solvent partition coefficient (KD) and aqueous:organic phase volume ratio (ϕ), but can be improved with a counter-current multi-stage membrane system (see ESI† for modeling details). With a membrane permeate flux of 290.0 L m−2 h−1, the overall butyric acid flux (eqn (7)) of the present single-stage MBES system was calculated to be 1450.0 g m−2 h−1, which is substantially higher than 8.9 g m−2 h−1 achievable with a membrane contactor (Table 1). As a result, to have the same butyric acid extraction rate, an MBES would require a ∼160-fold smaller membrane area than a membrane contactor. Notably, this calculated butyric acid flux will change as a function of phase volume ratio. Here, we selected the same ϕ = 1.0 for a direct comparison with the previous membrane contactor-assisted LLE results.15
| Membrane area (m2) | Butyric acid extraction rate (g h−1) | Butyric acid flux (g m−2 h−1) | Membrane area needed (to extract 100 g h−1 butyric acid) (m2) | |
|---|---|---|---|---|
| a Butyric acid extraction rate was measured by using 70 vol% Cyanex 923, 30 vol% mineral oil to extract butyric acid from 10 g L−1 butyric acid aqueous mock solution at pH 5 and a phase volume ratio, ϕ, of 1. b Two Liqui-Cel Extra-Flow 2.5 × 8 membrane contactor units (3 M) were used for membrane contactor LLE. The aqueous and organic phases were continuously circulated through the lumen and shell sides of the membrane contactor, respectively, at the flow rate of 40 mL min−1. c A membrane contactor unit (Liqui-Cel 2.5 × 8, a polypropylene membrane with 1.4 m2 membrane area) can achieve butyric acid extraction efficiency as high as the thermodynamic limit (partition equilibrium) according to a previous study.15 | ||||
| MBES | 0.006 | 8.4 | 1450.0 | 0.07 |
| Membrane contactorb | 1.4 | 12.5c | 8.9 | 11.2 |
Characterization of membrane fouling in the MBES system
The results presented above suggest considerable advantages in using membrane demulsification for butyric acid separations, based on use of a mock broth solution containing only butyric acid at 10 g L−1 in water at pH 5. Importantly, fermentation broth contains impurities including microbial cell debris, thus making membrane fouling a potential challenge for the use of an MBES system, which can lead to both flux decline and compromised permeate quality.57,58 Thus, we were interested in evaluating membrane fouling propensity during its filtration with an emulsified feed stream prepared from the fermentation broth, relative to the emulsified mock broth described above.The emulsified fermentation broth was prepared by mixing 300 mL of filtered C. tyrobutyricum fermentation broth using a rotating microfiltration (MF) ceramic disc (with pore size of 0.2 μm and rotating speed of 750 rpm) and mixing it with 150 mL organic phase. As a comparison, an emulsified mock broth was also prepared by mixing 300 mL of 10 g L−1 butyric acid aqueous solution with 150 mL organic phase. In both systems, we used a stirring rate of 650 rpm for 3 min to achieve ϕ = 2. It was expected that by increasing the aqueous:organic phase volume ratio, we could decrease the volume of organic solvent used in the process and thus improve the efficiency of the separation process.
Membrane fouling propensity is indicated by the normalized membrane permeate flux and flux decline, and the change in water content in the feed, permeate, and retentate streams. The normalized permeate flux (Jov) was calculated by:
| Jov = Jv,t/Jv,o | (8) |
| FD = 1 − Jv,final/Jv,o | (9) |
 | (10) |
After a 1 h membrane filtration experiment with the emulsified fermentation broth (at a feed flowrate 29.5 mL min−1), a 96.1% flux decline (eqn (9)) was observed for the hydrophobic PTFE membrane (Fig. 3A). The reduced membrane permeate flux can be explained by the accumulation of foulants on the membrane surface, which served as a resistance layer (with a calculated Rfouling of 3.0 × 1012 m−1; eqn (10)) and thus decreased the organic solvent permeability through the membrane. Indeed, compared to the pristine membrane, a thick cake layer on the membrane surface was confirmed via scanning electron microscopy (SEM) of the fouled membrane surfaces (Fig. 4). As the C. tyrobutyricum fermentation broth was pre-filtered by a 0.2 μm MF ceramic disc to remove the intact cells before feeding to the MBES unit, only fouling from microbial or organic media components should occur on the membrane surface and/or inside the pores. The extent of organic fouling was also examined by energy-dispersive X-ray spectroscopy (EDS) analysis, which showed the fouled hydrophobic PTFE membrane surface exhibited a 12.5-fold higher carbon-to-fluorine (C/F) ratio relative to the pristine membrane and an oxygen content of 24 wt% (Fig. 4 and Table 2). Notably, even at a 96.1% flux decline, the MBES still exhibited 6.2 times higher butyric acid flux than the membrane contactors.
 | ||
| Fig. 3 Fouling propensities of the hydrophobic PTFE membrane during a 1 h filtration of both emulsified mock broth and emulsified fermentation broth as indicated by (A) the permeate flux profile normalized to initial permeate flux of the pristine membrane (eqn (8)) and (B) the final water content in the permeate and retentate streams measured using Karl Fisher titration. Note: the error bars in (B) represent the standard deviation from at least 3 replicate KF water content measurements. The data shown in this figure are provided in Table S7.† | ||
 | ||
| Fig. 4 EDS (left) and SEM (right) images of the surfaces of (A and B) the pristine hydrophobic PTFE membrane, and the hydrophobic PTFE membranes filtered with (C and D) emulsified mock broth and (E and F) emulsified fermentation broth. | ||
| Membrane | F | C | O | C/F | O/F |
|---|---|---|---|---|---|
| a The membrane filtration was carried out at feed flow rate of 140 mL min−1 for 1 h and membrane phase breakthrough was observed. | |||||
| Pristine hydrophobic PTFE membrane | 79.1% | 20.9% | — | 0.26 | — |
| Hydrophobic PTFE membrane after filtering mock brotha | 50.6% | 42.5% | 2.1% | 0.84 | 0.04 |
| Hydrophobic PTFE membrane after filtering fermentation broth | 14.2% | 46.1% | 23.6% | 3.25 | 1.67 |
Additionally, the foulant layer buildup on the membrane surface not only caused membrane flux decline, but also led to significant aqueous phase breakthrough (Fig. 3B). Unlike the pristine hydrophobic PTFE membrane, which separates almost pure organic phase (3.6 wt% water content; Fig. S1(c)†) as the permeate, the permeate stream from the fouled membrane was composed of 92.0 wt% aqueous and 8.0 wt% organic phases (Fig. 3B). The observed phase breakthrough is attributed to the reduced hydrophobic PTFE membrane surface hydrophobicity (as indicated by the decrease in ΔGiw from −30.8 to −108.4 mJ m−2; Table S1†) with the accumulated fouling layer. Membrane surface wetting thus altered the solvent-membrane surface interactions by significantly increasing the aqueous phase membrane affinity, leading to preferred transport of the aqueous phase.
It may be expected that the observed high fouling propensity is due to the hydrophobic nature of the tested membrane via dispersive forces59 between the membrane surface and organic compounds. Hydrophilic membranes, conversely, could exhibit much lower fouling propensity due to high surface hydrophilicity that could reduce the hydrophobic interactions and thus mitigate surface adsorption or deposition.60–65 Therefore, we also tested the commercial hydrophilic PTFE membrane from Zaiput Flow Technologies (IL-2000-S200F; pore size 1 μm) for demulsification of the emulsified fermentation broth, which should result in the aqueous phase as the permeate while retaining the organic phase. It was observed, as expected, that the hydrophilic PTFE membrane, which has much higher surface hydrophilicity (52.2% reduced water sessile drop contact angle compared to the hydrophobic membrane, Table S1†), exhibited lower fouling propensity as indicated by a 62.5% flux decline over a 1 h filtration experiment, which is significantly less than the 96% flux decrease for the hydrophobic PTFE membrane (Fig. S3A†). The lower fouling propensity of the hydrophilic membrane was consistent with a thinner cake layer observed on the fouled hydrophilic membrane surfaces (with some membrane pores and void fractions exposed) via SEM images (Fig. S4†) and a relatively minor increase in the surface C/F and oxygen-to-fluorine (O/F) ratios compared to the fouled hydrophobic PTFE membrane (Fig. S4 and Table S6†). Moreover, no phase breakthrough was observed for the hydrophilic PTFE membrane, but rather the purity of the permeate stream was maintained even at the end of a 1 h fouling test (Fig. S3B†). The preferred permeation of the aqueous phase throughout the fouling test was due to further increased surface hydrophilicity (and thus membrane affinity to the aqueous phase) after organic fouling (as indicated by the decrease in ΔGiw from −109.1 to −125.6 mJ m−2; Table S1†). Despite this, the hydrophilic membrane is not suitable for separating emulsified fermentation broth (which is a water-in-oil emulsion) due to its significantly lower permeate flux: namely, the initial permeate flux of the hydrophilic PTFE membrane was only 4.0 L m−2 h−1, which is 96% lower than that of the hydrophobic PTFE membrane. Such a low initial permeate flux is likely due to the limited contact area between the dispersive aqueous phase droplets and the membrane surface.
It is also interesting to note that, unlike the hydrophilic membrane, oil wetting (i.e., oil fouling)66 is not a concern for hydrophobic membrane separation of a water-in-oil emulsion. Even though the organic phase attachment does alter the membrane surface morphology, as indicated via the SEM images (by filling in all the pores and void fractions of the membrane; Fig. 4D) and membrane surface chemical composition as indicated by the EDS results (with increased C/F and O/F ratios; Fig. 4 and Table 2), it does not lead to membrane permeate flux decline. Instead, the permeate flux through the hydrophobic PTFE membrane increased by 85.7% after 1 h of membrane filtration with the emulsified mock broth (Fig. 3A), which can be explained by the absorption of organic phase into the membrane matrix (Fig. 4D), leading to membrane solvation (i.e., swelling).67,68 Such membrane solvation phenomenon in organic solvent is widely reported and is consistent to the literature.69–73
Development of fouling mitigation strategies in the MBES system
To address the substantial decreased membrane performance shown in the previous section, fouling mitigation strategies were explored. We hypothesized that proteins (both C. tyrobutyricum-derived extracellular proteins and yeast extract-derived proteins from the media) were the dominant foulants in the emulsified fermentation broth solution, and thus we first evaluated the effectiveness of MF membranes (with pore sizes of 0.2 and 0.1 μm; GNWP04700 and VCTP04700, Millipore Sigma) and ultrafiltration (UF) membranes (with molecular weight cutoff values of 10 and 1 kDa; PLGC04310 and PLAC04310, Millipore Sigma) filtration for protein removal from C. tyrobutyricum fermentation broth. The above MF and UF membrane pre-filtrations of fermentation broth were carried out with a 50 mL dead-end stirred UF cell (Amicon 8050, Millipore Corporation) at 3.4 bar and a stirring rate of 400 rpm, with the intention to mimic a continuous crossflow unit operation in the ISPR process with its retentate stream sent back to the bioreactor to maintain biological productivity. It is noted that throughout the fermentation broth pre-filtration tests, all MF and UF membrane permeate flux remained constant at a permeate volume recovery of up to 80%, suggesting minimum membrane fouling occurred. The effectiveness of protein removal using MF and UF membranes with different pore sizes was indicated by the average solute size (measured by Zetasizer) and the results of protein gel electrophoresis measured for the membrane permeate as compared to the unfiltered C. tyrobutyricum fermentation broth. The MBES performance was then evaluated in a 1 h filtration experiment using the emulsified, pre-filtered fermentation broth in terms of both flux decline and water content in the final permeate and retentate streams. After the filtration test, the fouled membranes were cleaned with a 2 min membrane permeate backwash to evaluate membrane cleaning efficacy.The performance recovery was determined for both permeate flux and phase separation efficiency. Membrane permeate flux recovery (PFR) was defined as:
| PFR = Jv,cleaned/Jv,o | (11) |
| PSR = PScleaned/PS0 | (12) |
With the measured membrane permeate flux recovery, we could calculate the reversible (Rrev) and irreversible (Rirrev) fouling resistances, and their contribution to the overall fouling resistance (Rfouling). It is noted that overall fouling resistance is the sum of reversible and irreversible fouling resistances:
| Rfouling = Rrev + Rirrev | (13) |
 | (14) |
For MF and UF membrane filtration of the C. tyrobutyricum fermentation broth for protein removal, the filtered fermentation broth exhibited a progressively lighter color at higher resolution of filtration, which was consistent with the reduced average solute size as a function of filter cutoff (Fig. 5A). The effectiveness of membrane protein removal was also demonstrated by protein gel electrophoresis, where 100% removal of larger proteins (>14 kDa) was achieved with both 10 kDa and 1 kDa UF membranes (Fig. 5B).
 | ||
| Fig. 5 Pre-filtration of the C. tyrobutyricum fermentation broth using both microfiltration (MF) and ultrafiltration (UF) membranes with different pore size can effectively (A) reduce the average solute size and (B) remove proteins above 14 kDa, thus leading to (C) reduced hydrophobic PTFE membrane fouling propensity and (D) enhanced cleaning efficacy. (Note: membrane fouling propensity was evaluated with 1 h filtration of the emulsified fermentation broth, and membrane cleaning efficacy was assessed with 2 min membrane organic phase backwash with a flow rate of 20 mL min−1.) The error bars in (A) represent the standard deviation from at least 3 replicate measurements and in (C) and (D) represent the systematic error for pressure and flow rate measurements. The data shown in this figure are provided in Table S8.† | ||
MF and UF membrane pre-filtration of the C. tyrobutyricum fermentation broth demonstrated effective protein removal and thus led to significantly reduced MBES membrane fouling propensity. Over a 1 h filtration of emulsified pre-filtered fermentation broth (filtered with membranes of different pore sizes), the hydrophobic PTFE membrane fouling propensity was lowered by 12.5%, 80.0%, and 87.5% (Fig. 5C) using membranes with pore size of 0.1 μm, 10 kDa, and 1 kDa, respectively, relative to 0.2 μm MF pre-filtration. Moreover, phase breakthrough in the permeate was also minimized with membrane protein removal, as indicated by the final permeate water content measurement lowered by up to 63.0%. For membrane performance recovery, a 2 min membrane backwash (where the direction of the permeate flow is reversed to dislodge foulants accumulated on the membrane surface or inside the pores66) was carried out at the end of the 1 h membrane filtration test, leading to 68.6–100.0% and 32.1–100.0% permeate flux and phase separation recovery (Fig. 5D). Especially the 1 kDa UF membrane pre-filtration exhibited complete recovery of both membrane permeate flux and phase separation efficiency indicating the absence of irreversible fouling, while the use of larger pore size membranes led to less effective protein removal and thus 2.6–32.6% irreversible fouling (Table 3).
| Pre-filtration |
R
ma (m−1) |
R
foulingb (m−1) |
R
revc (m−1) |
%Reversible |
R
irrevc (m−1) |
%Irreversible |
|---|---|---|---|---|---|---|
| a Membrane intrinsic resistance, Rm, was determined from the relation Lp = 1/μRm, where Lp is the membrane organic phase permeability, and μ is the organic solvent viscosity (0.0126 Pa s at 20 °C). Membrane permeability (Lp) was determined from the slope of a linear plot of membrane permeate flux as a function of the differences between applied pressure and intrusion pressure (i.e., Lp = Jv/(ΔP − PI), where Jv is membrane permeate flux of the organic phase, ΔP is the applied transmembrane pressure, and PI is the membrane organic phase intrusion pressure).54,74 b At the end of each fouling test, a determination was made of the membrane overall hydraulic resistance (RT) being the sum of the intrinsic membrane resistance (Rm) and fouling resistance (Rfouling).54,75 The fouling resistance (Rfouling) is the sum of reversible and irreversible fouling resistances: Rfouling = Rrev + Rirrev. c Membrane backwash with the permeate organic solvent was conducted by reversing the pump direction at 20 mL min−1 for 2 min. The resistance of the backwashed membrane was then again determined with organic solvent, thereby allowing quantification of the combined intrinsic membrane and irreversible fouling resistances expressed as R′T = Rm + Rirrev. Subsequently, Rrev and Rirrev were determined given the calculated values of RT and R′T.54,75 | ||||||
| 0.2 μm MF | 1.2 × 1011 | 3.0 × 1012 | 2.0 × 1012 | 67.4% | 9.7 × 1011 | 32.6% |
| 0.1 μm MF | 1.2 × 1011 | 6.0 × 1011 | 5.5 × 1011 | 91.7% | 5.0 × 1010 | 8.3% |
| 10 kDa UF | 1.2 × 1011 | 2.3 × 1010 | 2.2 × 1010 | 97.4% | 5.9 × 108 | 2.6% |
| 1 kDa UF | 1.2 × 1011 | 1.1 × 1010 | 1.1 × 1010 | 100.0% | 0.0 | 0.0% |
Process economics and environmental impacts
The use of a single-stage MBES unit operation for ISPR (Fig. 6) was evaluated with techno-economic analysis (TEA) and life cycle assessment (LCA), using a membrane contactor unit operation as a baseline. The ISPR process model and TEA/LCA boundary encompasses a cell retention device, a solvent extraction system, a flash tank, and two-stage distillation columns to continuously extract butyric acid from the C. tyrobutyricum fermentation broth. The process was modeled using Aspen Plus with a process flow diagram and complete mass balance of water, butyric acid, and organic solvent provided in Fig. 6, as well as other model simulation details listed in Fig. S7.† All assumptions and process conditions used are described in the ESI.† We modeled a butyric acid titer in the fermentation broth of 15 g L−1. Other components, such as sugar, acetate, mineral salts, and solids, are not extracted significantly into the organic phase and are of low concentration in the fermentation broth, and these are shown as ‘others’ in process flow diagram (Fig. 6). Based on our bench-scale LLE results, solvent extraction using 70 vol% Cyanex 923 and 30 vol% mineral oil with an aqueous-to-organic volume ratio of 1:1 was assumed to achieve a butyric acid partition coefficient of 1 and water co-extraction of 5.2 wt%. As described above, the MBES system achieved ∼160-fold higher butyric acid flux than the membrane contactor, which affected the process cost but did not change the overall mass balances. After LLE, a flash tank was used to partially recycle the organic solvent at 0.1 atm, followed by two distillation columns for butyric acid purification and ISPR solvent recycling. The operating temperatures of the flash tank and two distillation columns were verified to stay below the decomposition temperature (220 °C) of Cyanex 923. The production rate of butyric acid was 20.6 tonnes per hour (t h−1), which corresponds to a TEA feedstock scale of 2000 t day−1, consistent with previous work.63 The butyric acid purity was 86.7 wt% with an overall recovery of 94.4 wt%. Additionally, the organic solvent recycling efficiency was modeled to be greater than 99.9 wt%, and the unrecovered butyric acid is modeled as being lost to the aqueous waste stream. In this process, organic solvent loss (0.007% of Cyanex 923 and 0.16% of mineral oil) into the aqueous phase76 is consistent with the estimates from our previous work.77 The aqueous waste stream was sent for wastewater treatment to remove organics and salts before recycling back to the process.77
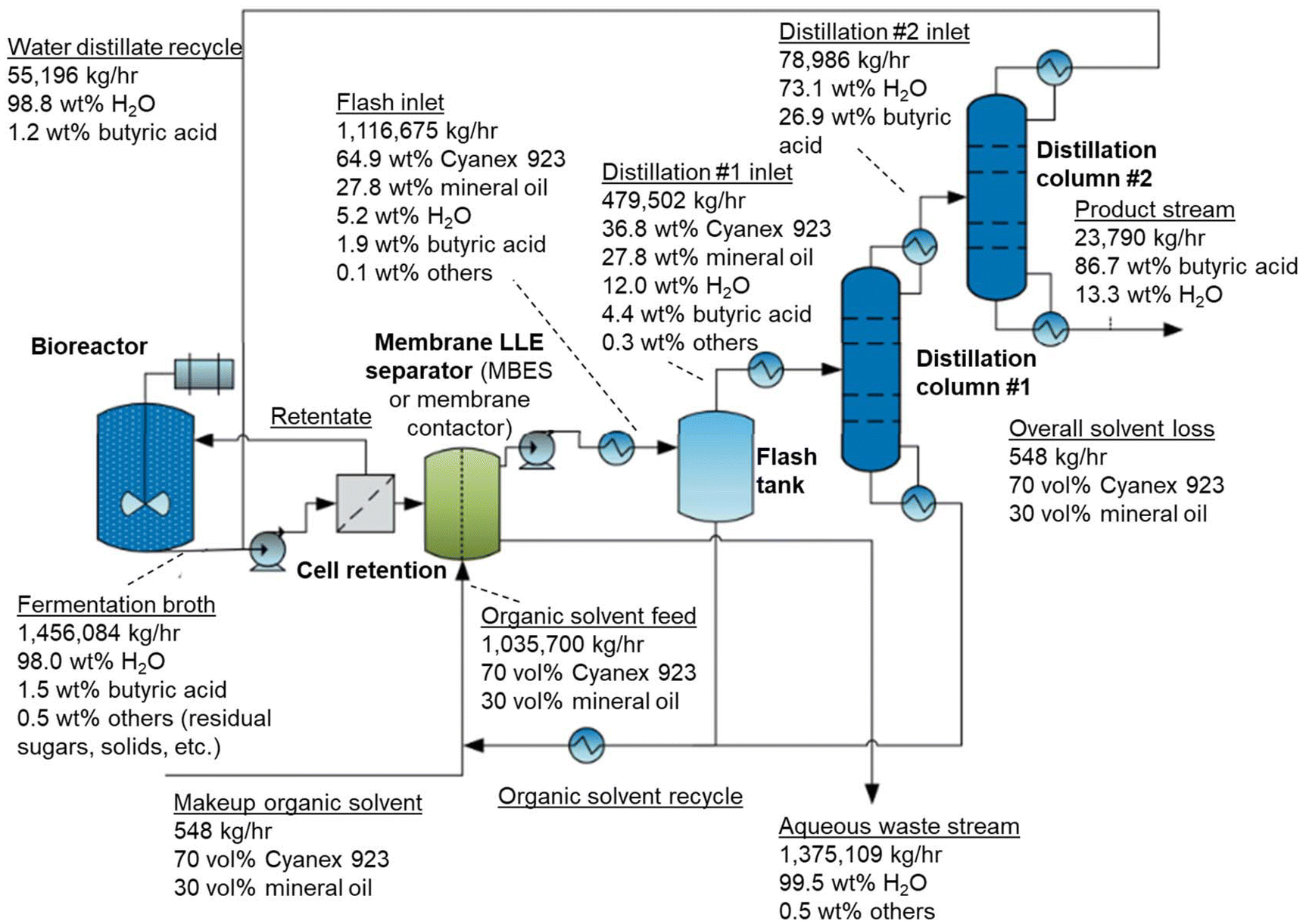 | ||
| Fig. 6 A process flow diagram of the modeled ISPR process for continuous butyric acid extraction from a bioreactor. It is noted that membrane LLE separator can represent both a membrane contactor and an MBES system, only varying the butyric acid flux and thus the membrane area. The flow rates are modeled at the scale of butyric acid production rate of 20.6 t h−1 corresponding to a 2000 t day−1 biorefinery feedstock scale consistent with previous work.63 | ||
The butyric acid separation cost was determined by estimating the process total capital expenses (CAPEX) as the sum of equipment of the major unit operations and the initial material purchase cost for organic solvent and membrane. The major pieces of equipment were assumed to have a lifetime of 30 years. The CAPEX estimates were based on quotes of the commercial products, Aspen scaling model simulations, and empirical correlations from the literature.77 In addition to process CAPEX, we also determined the yearly process operating expenses (OPEX) as a summation of raw material and utility costs for each process. Specifically, the OPEX includes the costs for membrane replacement, solvent makeup, other chemicals (sulfuric acid, ammonium hydroxide, etc.), and energy consumption for the feed pump and distillation columns. Energy costs were estimated by solving the electrical, heating, and cooling demand of the process in Aspen Plus. Labor costs, maintenance costs, insurance, and taxes were not factored into the current OPEX estimate.
The CAPEX and OPEX of both the MBES and membrane contactor-based ISPR processes are shown in Fig. 7A and B and Tables S10 and S11.† For the membrane contactor-based ISPR process, as the membrane contactor needs ∼ a 160-fold larger membrane area compared to the MBES system to achieve the same butyric acid flux, the membrane cost (initial and replacement) becomes the major driver contributing to 61% and 53% of CAPEX and OPEX, respectively (Fig. 7B). While in the MBES-based ISPR process, the membrane cost only contributes to 4% and 3% of CAPEX and OPEX, respectively (Fig. 7A). All other expenditures in this process model are assumed to be the same for both MBES and membrane contactor-based ISPR processes. Consequently, the ISPR process using the MBES unit operation for LLE demonstrated 59.6% lower CAPEX and 51.6% lower OPEX compared to the one using membrane contactors with a $10 per m2 estimated membrane cost (including membrane module) for both MBES and membrane contactor (Fig. 7A and B) consistent with the literature.78 The overall butyric acid ISPR production cost (sum of CAPEX and OPEX) was normalized to the total amount of butyric acid produced. The results showed that the MBES-based ISPR process contributes to a production cost of $0.22 per kg, which is 51.8% lower than the production cost of $0.46 per kg when using membrane contactors. The production cost differences between MBES and membrane contactor-based ISPR processes increase with the estimated cost of membrane, as shown in Fig. 7C.
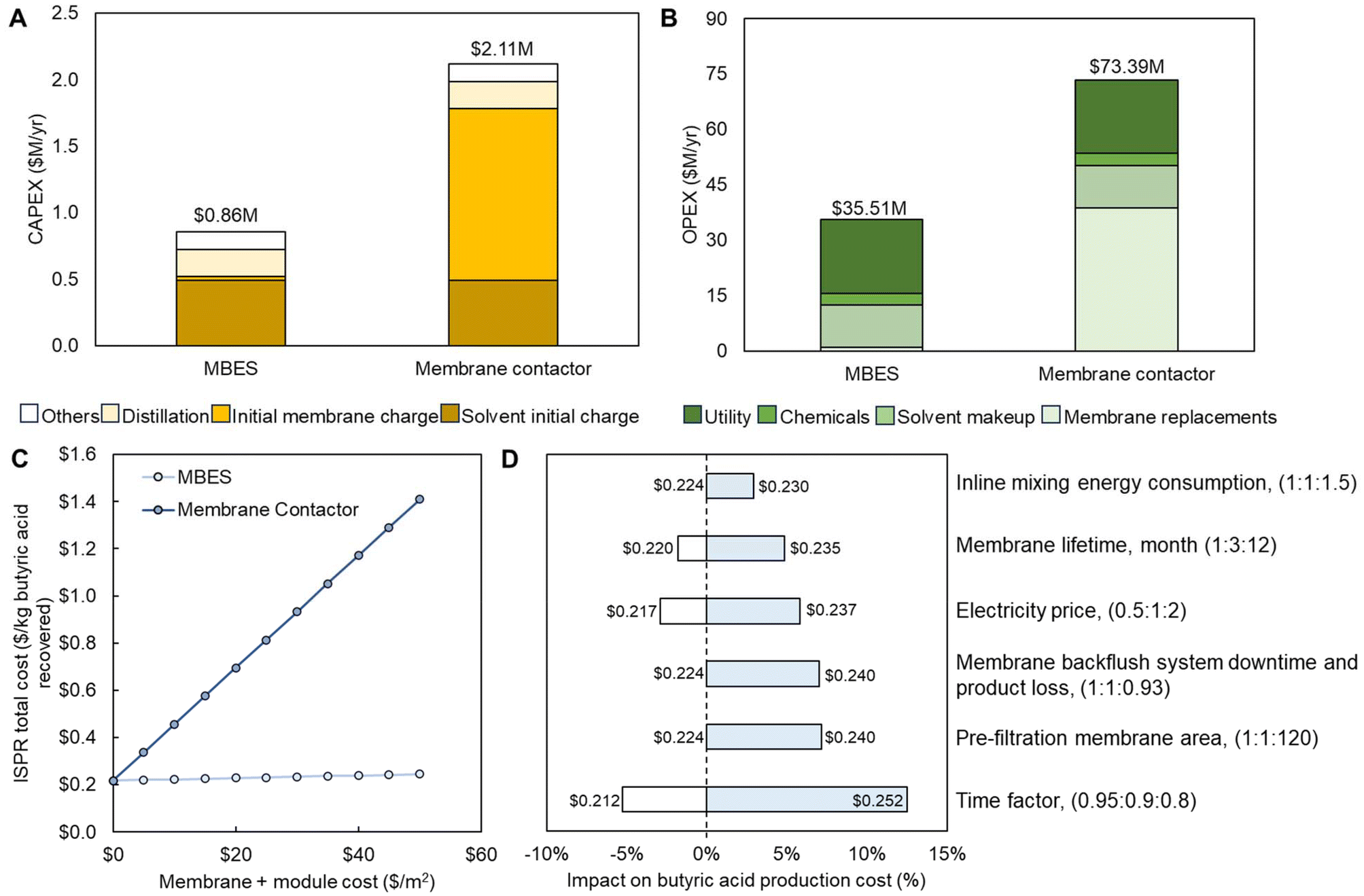 | ||
| Fig. 7 Comparison of TEA estimates for MBES and membrane contactor-based ISPR processes in terms of (A) CAPEX and (B) OPEX, and sensitivity analysis regarding (C) membrane cost and (D) MBES operating conditions as well as other major cost factors. The data shown in this figure are provided in Tables S9–S15.† | ||
Based on our experimental findings, it has become evident that the MBES system is prone to fouling. Therefore, it is necessary to implement a more effective pre-filtration step along with periodic membrane backflush to counteract the issue. However, this may lead to extra membrane area, system downtime, and product loss. To assess the impacts of membrane lifetime, extra membrane area and energy consumption required for pre-filtration, and system downtime and product loss due to membrane backwashing, as well as phase inline mixing and other major cost factors such as electricity price and time factor, we conducted a sensitivity analysis for the MBES-based ISPR process (Fig. 7D). The baseline case assumed an MBES membrane lifetime of 3 months, an electricity price of $0.0682 per kW per h, and a time factor of 0.9. In the sensitivity analysis, we evaluated the impacts of varying membrane lifetime (1–12 months), electricity price ($0.03–0.12 per kW per h), and on-stream time factor (0.80–0.95) on butyric acid production cost using the MBES-based ISPR process. Up to a 50% increase in overall energy consumption was used to simulate the extra energy consumption for inline mixing and pre-filtration. The membrane area needed for the pre-filtration step, ∼350000 m2 for the nth-plant scale of butyric acid production rate of 20.6 t h−1, was estimated based on the simulated process feed flow rate of 24724 L min−1, the membrane permeate flux of 12.6 L m−2 h−1 measured during C. tyrobutyricum fermentation broth clarification using a commercial regenerated cellulose UF membrane with molecular weight cutoff of 1 kDa (at 3.4 bar and a stirring rate of 400 rpm), and an estimated membrane lifetime of 4 months. The extra system downtime and product stream loss leading to a 7.5% butyric acid production cost increase was estimated based on our experimental measurements. It is interesting to know that with the consideration of pre-filtration, inline phase mixing, and periodic membrane backflush (Fig. 7D), the MBES-based ISPR process still demonstrated about 30% lower butyric acid production cost compared to the ISPR with membrane contactor.
In addition to economics, environmental impacts are another major concern of the downstream separation process in the biofuel industry. Notably, the solvent makeup is the major contributor to GHG emissions of the separation process (90% and 99% for MBES and membrane contactor-based ISPR, respectively). Excluding the solvent makeup, the carbon intensity of the MBES-based ISPR process is 90.8% reduced relative to membrane contactor, from 21.7 to 2.0 g CO2e per kg butyric acid (Fig. 8A). The GHG emissions were derived from the energy and material flows and the membrane and organic solvent usage of each process (Table S16†). The life cycle GHG emissions were calculated using a 100-year GHG emission factor in grams of carbon dioxide equivalent (g CO2e). Other than solvent makeup, energy and membrane consumption were the main driving factors. It is noted that the difference in carbon intensity of the MBES and membrane contactor-based processes only lies in their membrane material and consumption in terms of membrane material types and area. The PTFE membrane used for MBES has 16.2 and 13.3 times higher GHG emissions per membrane area during its production process than the regenerated cellulose membrane used for pre-filtration and polypropylene membrane used for the membrane contactor, respectively (Table S16†). Nevertheless, the MBES-based process exhibits 90.8% lower GHG emissions than the membrane contactor-based process due to the 160-fold smaller membrane area needed for MBES (due to its higher flux) than membrane contactor for the same butyric acid production rate. In addition to global warming potential, other impact categories were also considered, and the results are summarized in Fig. 8B and Table S17.† The case with MBES exhibits more favorable impacts than the membrane contactor case in all categories.
 | ||
| Fig. 8 Comparison LCA of MBES and membrane contactor-based ISPR processes in terms of (A) greenhouse gas emissions (excluding solvent makeup) and (B) other environmental impacts. | ||
Discussion
In this work, we aimed to demonstrate the potential of MBES-assisted LLE for enabling continuous LLE processes in bioprocessing separations. It is worth noting that our proposed ISPR process with the integration of continuous membrane-based extraction aligns well with five of the 12 principles of green membrane processes79 in terms of (i) minimizing processing steps compared to the traditional batch processes with crystallization and salting-out approaches, (ii) reducing solvent consumption, (iii) promoting closed-loop systems with the integration of organic solvent and water recycle, (iv) minimizing buffer tanks and auxiliaries, and (v) minimizing footprint by adopting a continuous operating mode.Compared to solvent overlay and membrane contactors, the use of MBES for solvent extraction of bioproducts can lead to increased mass transfer rate, complete in-line phase separation, lower toxicity effects on microorganisms, and higher bioproduct flux. We also developed a process model which estimates that MBES-assisted LLE can result in a 55% and 91% reduction in process costs (the sum of CAPEX and OPEX) and greenhouse gas emissions, respectively, compared to a membrane contactor. Despite the above advantages, using MBES in the downstream bioprocessing is challenged by phase breakthrough and membrane fouling.
The MBES performance evaluation results indicate that whether phase breakthrough of a selected membrane will occur depends on the applied pressure (ΔP), which needs to be between the organic and aqueous phase intrusion pressures to achieve complete phase separation. The intrusion pressure is defined as the maximum pressure that the membrane can withstand without liquid passing through.66,80 Due to the much higher affinity to the organic solvent of the hydrophobic PTFE membrane than the aqueous solvent, it is expected that when the applied pressure is higher than the organic solvent intrusion pressure but lower than that for water (PI,aq), complete phase separation should be achieved with phase separation efficiency equivalent to the thermodynamic limit. With the applied pressure higher than the aqueous phase intrusion pressure, however, phase breakthrough will be observed as the aqueous phase has enough driving force to break the capillary pressure of the organic phase in the membrane pores, leading to both membrane surface and pores partially wetted by the aqueous phase. As in the current system setup, where the transmembrane pressure increases with the feed flow rate (or pump speed), as indicated by a higher membrane permeate flux (Fig. S1†), MBES operation conditions and permeate flux are highly limited by membrane phase intrusion pressures.
In addition to phase breakthrough, membrane fouling is another major challenge when applying MBES-assisted LLE in biorefineries. The high membrane fouling propensity observed for MBES is attributed partly to the high surface hydrophobicity of the used PTFE membrane leading to hydrophobic interactions with the foulant molecules. The low crossflow velocity (i.e., feed flow rate) required to achieve complete in-line phase separation also limits the shear rate on the membrane surface, and thus increases adhesion force between the membrane surface and foulant molecules. Membrane fouling would lead to not only permeate flux decline, but also membrane wetting, and thus a reduced transport resistance (or even preferential transport) of the aqueous phase (Fig. 3B).
The wetted membrane due to both phase breakthrough and membrane fouling can be regenerated by membrane flushing or backflushing using pure organic solvent, but as shown in the process model (Fig. 7D), this will lead to system downtime and additional energy consumption and organic solvent usage. To further reduce the cost of the MBES-assisted LLE process, it is important to prolong membrane service time. The above may be achieved by optimizing operation conditions and permeate flux, developing more effective foulant pre-removal strategies, and designing membranes with larger differences between their affinity for the aqueous and organic phases (for more flexible MBES operations). These membranes should also have low fouling propensity and be suitable for bioprocessing applications.
PTFE membranes, although have various advantages such as high chemical and temperature resistance, excellent cleaning efficacy, long durability, and low hydraulic resistance, are challenged by the adverse environmental effects related to their production and disposal. In fact, a new proposal has been drafted to completely ban the use of PTFE materials under the EU REACH regulation process,81 and thus there is a need to seek sustainable membrane alternatives for continuous membrane-based extraction processes.
Other than membrane development for enhanced performance and sustainability, there are merits to explore the following research directions and thus further improve ISPR process cost efficiency and sustainability: (i) the use of a membrane cascade instead of a single stage MBES unit to achieve increased extraction efficiency with the same solvent volume usage; and (ii) evaluate the potential solvent alternatives for in situ extraction of bio-based carboxylic acids and carefully assess their toxicity, extraction efficiency, recyclability, and availability.
Abbreviations
| CAPEX: | Capital enpenses |
| EDS: | Energy-dispersive X-ray spectroscopy |
| EE | Extraction efficiency |
| GHG: | Greenhouse gas |
| HPLC | High-performance liquid chromatography |
| ISPR: | In situ product recovery |
| KF | Karl Fisher |
| LCA | Life-cycle assessment |
| LLE | Liquid–liquid extraction |
| MBES | Membrane-based emulsion separator |
| MF | Microfiltration |
| M w | Molecular weight |
| OPEX: | Operating expenses |
| P I | Intrusion pressure |
| PS | Phase separation efficiency |
| PTFE | Polytetrafluoroethylene |
| SEM: | Scanning electron microscopy |
| TEA | Technoeconomic analysis |
| UF | Ultrafiltration |
| ΔGiw | Free energy of hydration |
| ΔP | Applied pressure |
| ϕ | Aqueous:organic phase volume ratio |
Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this paper was sponsored by the U.S. Department of Energy (DOE), Energy Efficiency and Renewable Energy Office, Bioenergy Technologies Office (BETO) under the BETO Bioprocessing Separations Consortium and the BETO Biological Upgrading of Sugars project via Contract No. DE-AC36-08GO28308 with the National Renewable Energy Laboratory. The authors acknowledge Neal Hengge, Ashley Berninghaus, and Yannick Bomble for assistance with protein analysis, and surface SEM and EDS from Bobby To and Steven Hayden. This work was authored by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- C. Abels, F. Carstensen and M. Wessling, Membrane processes in biorefinery applications, J. Membr. Sci., 2013, 444, 285–317 CrossRef CAS.
- A. L. Zydney, Continuous downstream processing for high value biological products: a review, Biotechnol. Bioeng., 2016, 113, 465–475 CrossRef CAS PubMed.
- A. L. Zydney, Perspectives on integrated continuous bioprocessing—opportunities and challenges, Curr. Opin. Chem. Eng., 2015, 10, 8–13 CrossRef.
- M. C. Cuellar and A. J. Straathof, Downstream of the bioreactor: advancements in recovering fuels and commodity chemicals, Curr. Opin. Biotechnol., 2020, 62, 189–195 CrossRef CAS PubMed.
- A. Bednarz, A. Jupke, A. C. Spieß and A. Pfennig, Aerated extraction columns for in situ separation of bio–based diamines from cell suspensions, J. Chem. Technol. Biotechnol., 2019, 94, 426–434 CrossRef CAS.
- K. W. Staggs and D. R. Nielsen, Improving n-butanol production in batch and semi-continuous processes through integrated product recovery, Process Biochem., 2015, 50, 1487–1498 CrossRef CAS.
- D. Salvachua, P. O. Saboe, R. S. Nelson, C. Singer, I. McNamara, C. del Cerro, Y.-C. Chou, A. Mohagheghi, D. J. Peterson and S. Haugen, Process intensification for the biological production of the fuel precursor butyric acid from biomass, Cell Rep. Phys. Sci., 2021, 2, 100587 CrossRef CAS.
- F. Carstensen, A. Apel and M. Wessling, In situ product recovery: Submerged membranes vs, external loop membranes, J. Membr. Sci., 2012, 394, 1–36 CrossRef.
- F. Carstensen, T. Klement, J. Büchs, T. Melin and M. Wessling, Continuous production and recovery of itaconic acid in a membrane bioreactor, Bioresour. Technol., 2013, 137, 179–187 CrossRef CAS PubMed.
- S. A. Aktij, A. Zirehpour, A. Mollahosseini, M. J. Taherzadeh, A. Tiraferri and A. Rahimpour, Feasibility of membrane processes for the recovery and purification of bio-based volatile fatty acids: A comprehensive review, J. Ind. Eng. Chem., 2020, 81, 24–40 CrossRef.
- C. S. López-Garzón and A. J. Straathof, Recovery of carboxylic acids produced by fermentation, Biotechnol. Adv., 2014, 32, 873–904 CrossRef PubMed.
- U. A. Salas-Villalobos, R. V. Gómez-Acata, J. Castillo-Reyna and O. Aguilar, In situ product recovery as a strategy for bioprocess integration and depletion of inhibitory products, J. Chem. Technol. Biotechnol., 2021, 96, 2735–2743 CrossRef CAS.
- Z. Berk, Food process engineering and technology, Academic press, 2018 Search PubMed.
- C. Chen, D. Weng, A. Mahmood, S. Chen and J. Wang, Separation mechanism and construction of surfaces with special wettability for oil/water separation, ACS Appl. Mater. Interfaces, 2019, 11, 11006–11027 CrossRef CAS PubMed.
- P. O. Saboe, L. P. Manker, W. E. Michener, D. J. Peterson, D. G. Brandner, S. P. Deutch, M. Kumar, R. M. Cywar, G. T. Beckham and E. M. Karp, In situ recovery of bio-based carboxylic acids, Green Chem., 2018, 20, 1791–1804 RSC.
- X. Ge, Y. Chen, V. Sànchez i Nogué and Y. Li, Volatile Fatty Acid Recovery from Arrested Anaerobic Digestion for the Production of Sustainable Aviation Fuel: A Review, Fermentation, 2023, 9, 821 CrossRef CAS.
- R. Wang, R. He, T. He, M. Elimelech and S. Lin, Performance metrics for nanofiltration-based selective separation for resource extraction and recovery, Nat. Water, 2023, 1, 291–300 CrossRef.
- V. Voros, E. Drioli, C. Fonte and G. Szekely, Process intensification via continuous and simultaneous isolation of antioxidants: an upcycling approach for olive leaf waste, ACS Sustainable Chem. Eng., 2019, 7, 18444–18452 CrossRef CAS.
- B. Adigun, B. P. Thapaliya, H. Luo and S. Dai, Complexation-driven ion-exchange polymer inclusion membranes for separation of cobalt and nickel ions from lithium-ion via proton pumping, RSC Sustainability, 2024, 2, 1859–1867 RSC.
- P. Luis, Fundamental Modeling of Membrane Systems: Membrane and Process Performance, Elsevier, 2018 Search PubMed.
- Z. Trad, J. Akimbomi, C. Vial, C. Larroche, M. J. Taherzadeh and J.-P. Fontaine, Development of a submerged anaerobic membrane bioreactor for concurrent extraction of volatile fatty acids and biohydrogen production, Bioresour. Technol., 2015, 196, 290–300 CrossRef CAS PubMed.
- B. Tao, P. Passanha, P. Kumi, V. Wilson, D. Jones and S. Esteves, Recovery and concentration of thermally hydrolysed waste activated sludge derived volatile fatty acids and nutrients by microfiltration, electrodialysis and struvite precipitation for polyhydroxyalkanoates production, Chem. Eng. J., 2016, 295, 11–19 CrossRef CAS.
- M. Ramos-Suarez, Y. Zhang and V. Outram, Current perspectives on acidogenic fermentation to produce volatile fatty acids from waste, Rev. Environ. Sci. Bio/Technol., 2021, 20, 439–478 CrossRef CAS.
- A. Gössi, F. Burgener, D. Kohler, A. Urso, B. A. Kolvenbach, W. Riedl and B. Schuur, In situ recovery of carboxylic acids from fermentation broths through membrane supported reactive extraction using membrane modules with improved stability, Sep. Purif. Technol., 2020, 241, 116694 CrossRef.
- G. Burgé, F. Chemarin, M. Moussa, C. Saulou-Bérion, F. Allais, H. É. Spinnler and V. Athès, Reactive extraction of bio–based 3−hydroxypropionic acid assisted by hollow–fiber membrane contactor using TOA and Aliquat 336 in n–decanol, J. Chem. Technol. Biotechnol., 2016, 91, 2705–2712 CrossRef.
- S.-T. Yang, H. Huang, A. Tay, W. Qin, L. De Guzman and E. C. San Nicolas, Extractive fermentation for the production of carboxylic acids. in Bioprocessing for value-added products from renewable resources, Elsevier, 2007, pp. 421–446 Search PubMed.
- G. Burgé, M. Moussa, V. Athes-Dutour, C. Saulou-Berion and H.-E. Spinnler, In situ extraction of 3-hydroxypropionic acid assisted by membrane contactor, in 4th International Congress on Green Process Engineering (GPE4), 2014.
- A. Amelio, L. Loise, R. Azhandeh, S. Darvishmanesh, V. Calabró, J. Degrève, P. Luis and B. Van der Bruggen, Purification of biodiesel using a membrane contactor: Liquid–liquid extraction, Fuel Process. Technol., 2016, 142, 352–360 CrossRef CAS.
- A. Adamo, P. L. Heider, N. Weeranoppanant and K. F. Jensen, Membrane-based, liquid–liquid separator with integrated pressure control, Ind. Eng. Chem. Res., 2013, 52, 10802–10808 CrossRef CAS.
- G. Wu, E. Cao, P. Ellis, A. Constantinou, S. Kuhn and A. Gavriilidis, Continuous flow aerobic oxidation of benzyl alcohol on Ru/Al2O3 catalyst in a flat membrane microchannel reactor: An experimental and modelling study, Chem. Eng. Sci., 2019, 201, 386–396 CrossRef CAS.
- M. Waterford, S. Saubern and C. H. Hornung, Evaluation of a continuous-flow photo-bromination using N-bromosuccinimide for use in chemical manufacture, Aust. J. Chem., 2021, 74, 569–573 CrossRef CAS.
- G. Glotz, R. Lebl, D. Dallinger and C. O. Kappe, Integration of bromine and cyanogen bromide generators for the continuous–flow synthesis of cyclic guanidines, Angew. Chem., Int. Ed., 2017, 56, 13786–13789 CrossRef CAS PubMed.
- L. Milani, T. Murray and A. Adamo, Analysis and design of Mass Transfer for Continuous Liquid-liquid extraction with Static Mixers: a practical approach, Zaiput Flow Technologies, 2021 Search PubMed.
- A. C. Wicker, F. A. Leibfarth and T. F. Jamison, Flow-IEG enables programmable thermodynamic properties in sequence-defined unimolecular macromolecules, Polym. Chem., 2017, 8, 5786–5794 RSC.
- P. Szemesi, P. Bana, Z. Szakács and I. Greiner, Safe and Rapid Synthesis and Utilization of 2-Azidopyridine and Related Derivatives via Continuous Flow Diazotization, Curr. Org. Chem., 2022, 26, 2223–2229 CrossRef CAS.
- T. A. Phung Hai, L. J. De Backer, N. D. Cosford and M. D. Burkart, Preparation of mono-and diisocyanates in flow from renewable carboxylic acids, Org. Process Res. Dev., 2020, 24, 2342–2346 CrossRef CAS.
- S. Overmans, G. Ignacz, A. K. Beke, J. Xu, P. E. Saikaly, G. Szekely and K. J. Lauersen, Continuous extraction and concentration of secreted metabolites from engineered microbes using membrane technology, Green Chem., 2022, 24, 5479–5489 RSC.
- Z. Wen, D. Pintossi, M. Nuño and T. Noël, Membrane-based TBADT recovery as a strategy to increase the sustainability of continuous-flow photocatalytic HAT transformations, Nat. Commun., 2022, 13, 6147 CrossRef CAS PubMed.
- É. Lévesque, S. T. Laporte and A. B. Charette, Continuous flow synthesis and purification of aryldiazomethanes through hydrazone fragmentation, Angew. Chem., 2017, 129, 855–859 CrossRef.
- V.-E. H. Kassin, R. Gérardy, T. Toupy, D. Collin, E. Salvadeo, F. Toussaint, K. Van Hecke and J.-C. M. Monbaliu, Expedient preparation of active pharmaceutical ingredient ketamine under sustainable continuous flow conditions, Green Chem., 2019, 21, 2952–2966 RSC.
- G. Wu, E. Cao, S. Kuhn and A. Gavriilidis, A novel approach for measuring gas solubility in liquids using a tube–in–tube membrane contactor, Chem. Eng. Technol., 2017, 40, 2346–2350 CrossRef CAS.
- V.-E. H. Kassin, D. V. Silva-Brenes, T. Bernard, J. Legros and J.-C. M. Monbaliu, A continuous flow generator of organic hypochlorites for the neutralization of chemical warfare agent simulants, Green Chem., 2022, 24, 3167–3179 RSC.
- S. Sacher, I. Castillo, J. Rehrl, P. Sagmeister, R. Lebl, J. Kruisz, S. Celikovic, M. Sipek, J. D. Williams and D. Kirschneck, Automated and continuous synthesis of drug substances, Chem. Eng. Res. Des., 2022, 177, 493–501 CrossRef CAS.
- N. Weeranoppanant and A. Adamo, In-line purification: A key component to facilitate drug synthesis and process development in medicinal chemistry, ACS Med. Chem. Lett., 2019, 11, 9–15 CrossRef PubMed.
- P. Martini, L. Uccelli, A. Duatti, L. Marvelli, J. Esposito and A. Boschi, Highly efficient micro-scale liquid-liquid in-flow extraction of 99mTc from molybdenum, Molecules, 2021, 26, 5699 CrossRef CAS PubMed.
- R. Morodo, R. Gerardy, G. Petit and J.-C. M. Monbaliu, Continuous flow upgrading of glycerol toward oxiranes and active pharmaceutical ingredients thereof, Green Chem., 2019, 21, 4422–4433 RSC.
- D. V. Silva-Brenes, N. Emmanuel, V. L. Mejías, J. Duconge, C. Vlaar, T. Stelzer and J.-C. M. Monbaliu, Out-smarting smart drug modafinil through flow chemistry, Green Chem., 2022, 24, 2094–2103 RSC.
- S. Pollington, 10th International symposium on continuous flow reactor technology for industrial applications, Johnson Matthey Technol. Rev., 2019, 63, 157–165 CrossRef CAS.
- L. Cao, M. Sun, C. Liang, L. Yang, Y. Ma, R. Cheng, Y. Ke, W. Yu and J. Ye, A sequential continuous flow synthesis and purification process of calcium dibutyryladenosine cyclophosphate, Chin. Chem. Lett., 2024, 35, 108758 CrossRef CAS.
- H. P. Gemoets, G. Laudadio, K. Verstraete, V. Hessel and T. Noël, A Modular Flow Design for the meta–Selective C− H Arylation of Anilines, Angew. Chem., 2017, 129, 7267–7271 CrossRef.
- P. Kocienski, Flow Synthesis of Lomustine, Synfacts, 2019, 2019, 0579 CrossRef.
- C. Armstrong, Y. Miyai, A. Formosa, P. Kaushik, L. Rogers and T. D. Roper, Leveraging first-principles and empirical models for disturbance detection in continuous pharmaceutical syntheses, J. Flow Chem., 2023, 1–17 CAS.
- S. Chang, G. Thomas and W. Wang, In-line Partition Coefficient Measurement for Oil Field Tracers Utilizing Microfluidic Mixing Chip. In SPE Middle East Oil and Gas Show and Conference, 2023.
- Y. Chen, J. Zhang and Y. Cohen, Fouling resistant and performance tunable ultrafiltration membranes via surface graft polymerization induced by atmospheric pressure air plasma, Sep. Purif. Technol., 2022, 286, 120490 CrossRef CAS.
- J. Wang and H. Wang, Ultra-hydrophobic and mesoporous silica aerogel membranes for efficient separation of surfactant-stabilized water-in-oil emulsion separation, Sep. Purif. Technol., 2019, 212, 597–604 CrossRef CAS.
- M. Szpakowska and O. B. Nagy, Application of the competitive preferential solvation theory to facilitated ion transport through binary liquid membranes, J. Phys. Chem., 1989, 93, 3851–3854 CrossRef CAS.
- Y. J. Lim, K. Goh, M. Kurihara and R. Wang, Seawater desalination by reverse osmosis: Current development and future challenges in membrane fabrication–A review, J. Membr. Sci., 2021, 119292 CrossRef CAS.
- Y. Chen, Performance Tuning of Ultrafiltration and Reverse Osmosis Membranes Surface Nano-Structured with Tethered Poly (Acrylic Acid) Chains, Doctoral dissertation, UCLA, 2022 Search PubMed.
- C. de Morais Coutinho, M. C. Chiu, R. C. Basso, A. P. B. Ribeiro, L. A. G. Gonçalves and L. A. Viotto, State of art of the application of membrane technology to vegetable oils: A review, Food Res. Int., 2009, 42, 536–550 CrossRef.
- M. Liu, C. Yu, Y. Wu, Z. Lü, S. Yu and C. Gao, In situ modification of polyamide reverse osmosis membrane module for improved fouling resistance, Chem. Eng. Res. Des., 2019, 141, 402–412 CrossRef CAS.
- C. Sun and X. Feng, Enhancing the performance of PVDF membranes by hydrophilic surface modification via amine treatment, Sep. Purif. Technol., 2017, 185, 94–102 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and surface forces, Academic press, 2015 Search PubMed.
- W. J. Feast and H. S. Munro, Polymer surfaces and interfaces, Wiley, 1987 Search PubMed.
- A. Marshall, P. Munro and G. Trägårdh, The effect of protein fouling in microfiltration and ultrafiltration on permeate flux, protein retention and selectivity: a literature review, Desalination, 1993, 91, 65–108 CrossRef CAS.
- A. Fane, C. Fell and A. Suki, The effect of pH and ionic environment on the ultrafiltration of protein solutions with retentive membranes, J. Membr. Sci., 1983, 16, 195–210 CrossRef CAS.
- E. Tummons, Q. Han, H. J. Tanudjaja, C. A. Hejase, J. W. Chew and V. V. Tarabara, Membrane fouling by emulsified oil: A review, Sep. Purif. Technol., 2020, 248, 116919 CrossRef CAS.
- O. W. W. Reif, Microfiltration membranes: characteristics and manufacturing, Sterile Filtr., 2006, 73–103 CAS.
- D. Ren, S. Ren, Y. Lin, J. Xu and X. Wang, Recent developments of organic solvent resistant materials for membrane separations, Chemosphere, 2021, 271, 129425 CrossRef CAS PubMed.
- P. Marchetti, L. Peeva and A. Livingston, The selectivity challenge in organic solvent nanofiltration: membrane and process solutions, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 473–497 CrossRef PubMed.
- L. Nie, C. Y. Chuah, T. H. Bae and J. M. Lee, Graphene–based advanced membrane applications in organic solvent nanofiltration, Adv. Funct. Mater., 2021, 31, 2006949 CrossRef CAS.
- C. M. Sánchez-Arévalo, M. C. Vincent-Vela, M.-J. Luján-Facundo and S. Álvarez-Blanco, Ultrafiltration with organic solvents: A review on achieved results, membrane materials and challenges to face, Process Saf. Environ. Prot., 2023, 177, 118–137 CrossRef.
- L. Xia and J. R. McCutcheon, Understanding the influence of solvents on the intrinsic properties and performance of polyamide thin film composite membranes, Sep. Purif. Technol., 2020, 238, 116398 CrossRef CAS.
- A. Vishnyakov and A. V. Neimark, Molecular simulation study of Nafion membrane solvation in water and methanol, J. Phys. Chem. B, 2000, 104, 4471–4478 CrossRef CAS.
- Y. Chen, S. Kim and Y. Cohen, Tuning the hydraulic permeability and molecular weight cutoff (MWCO) of surface nano-structured ultrafiltration membranes, J. Membr. Sci., 2021, 629, 119180 CrossRef CAS.
- Y. Chen, S. Kim, Y. Kim, J. S. Walker, T. Wolfe, K. Coleman and Y. Cohen, Scale up of polyamide reverse osmosis membranes surface modification with tethered poly (acrylic acid) for fabrication of low fouling spiral-wound elements, Desalination, 2022, 536, 115762 CrossRef CAS.
- W. Li, H. T. Lu, M. S. Doblin, A. Bacic, G. W. Stevens and K. A. Mumford, A solvent loss study for the application of solvent extraction processes in the pharmaceutical industry, Chem. Eng. Sci., 2022, 250, 117400 CrossRef CAS.
- R. E. Davis, N. J. Grundl, L. Tao, M. J. Biddy, E. C. Tan, G. T. Beckham, D. Humbird, D. N. Thompson and M. S. Roni, Process Design and economics for the conversion of lignocellulosic biomass to hydrocarbon fuels and coproducts: 2018 biochemical design case update; biochemical deconstruction and conversion of biomass to fuels and products via integrated biorefinery pathways, National Renewable Energy Lab.(NREL), Golden, CO (United States), 2018 Search PubMed.
- R. W. Baker, Future directions of membrane gas separation technology, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS.
- G. Szekely, The 12 principles of green membrane materials and processes for realizing the United Nations’ sustainable development goals, RSC Sustainability, 2024, 2, 871–880 RSC.
- N. Zhang, X. Yang, Y. Wang, Y. Qi, Y. Zhang, J. Luo, P. Cui and W. Jiang, A review on oil/water emulsion separation membrane material, J. Environ. Chem. Eng., 2022, 107257 CrossRef CAS.
- N. D. Tyrrell, A proposal that would ban manufacture, supply, and use of all fluoropolymers and most fluorinated reagents within the entire EU, Org. Process Res. Dev., 2023, 27, 1422–1426 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc02772h |
| ‡ Current address: Crysalis Biosciences, Louisville, CO 80027. |
| This journal is © The Royal Society of Chemistry 2024 |