
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainability in a can: upcycling aluminium scrap in the waste-minimized electrochemical synthesis of 2-oxazoline†
Simone
Trastulli Colangeli
,
Francesco
Ferlin
 * and
Luigi
Vaccaro
*
* and
Luigi
Vaccaro
*
Laboratory of Green S.O.C. – Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123 – Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: https://greensoc.chm.unipg.it
First published on 20th June 2024
Abstract
The use and consumption of electrodes is a crucial aspect regarding the overall efficiency and sustainability of electrochemical reactions. When metal electrodes are used, the cost of the material combined with the processing costs assumes relevance in terms of economics and sustainability. Herein we report a case study that aims to define an electrochemical synthetic protocol using electrodes prepared from recovered aluminium scrap. We approached the problem by evaluating and comparing the different carbon footprints associated with the use of different electrode materials from primary sources and secondary (recycled) sources. We optimized the use of electrodes made from secondary aluminium to develop a simple, oxidant-free protocol for the representative synthesis of 2-oxazolines from amino alcohols and aldehydes using generally elusive concentrated conditions and a recoverable reaction media. A further evaluation of the developed process using green metrics allowed us to quantify the waste distribution of our procedure in comparison with the literature processes as well as the progress in terms of sustainability and intrinsic reaction efficiency.
Introduction
To limit global warming, carbon neutrality is becoming an increasingly urgent challenge.1 The decarbonization of the chemical industry is of particular strategic importance as it is responsible for 15% of global industrial greenhouse gas (GHG) emissions.2 In this context, electrochemical production based on the use of electricity from renewable sources is becoming a promising alternative to reduce the carbon footprint of chemicals.3 While research on the use of electrolytic processes for the production of green hydrogen and CO2 reduction is expanding, electrochemical industrial processes are so far mainly limited to the chlor-alkali and aluminium sectors.4To succeed in the challenging application of electrochemistry in industrial chemical synthesis, researchers are focusing their efforts on optimizing key process parameters such as energy efficiency, product selectivity, productivity per time unit, and electrocatalyst efficiency.5
The consumption of electrodes represents a key aspect influencing the efficiency and sustainability of electrosynthesis.6 For example, in the case of the Hall-Héroult process that uses non-metallic electrodes for aluminium smelting, about 400 kg of carbon anode are consumed per ton of aluminium produced.7 When electrodes are based on metals, their cost and preparation are even more critical, and it is not easy to generalize information on the electrode consumption. For example, in the electrocoagulation processes employed in wastewater treatment, the aluminium consumption rate is approximately 2 kg per kg of contaminant removed.8
While research on finding durable and stable electrodes is extensive, more emphasis should be placed on using electrodes made from waste materials.9 This can be even more crucial considering the availability of metallic materials compared to polymers.10
Aluminium, while being widely used in electrochemistry, is currently included in the list of critical raw materials and is classified as “primary” or “secondary”.
Primary aluminium is derived directly from bauxite, while secondary aluminium is derived from the recycling of primary aluminium.11 The production of secondary aluminium is a more sustainable access route to this metal as its energy cost is only 5% of that of the primary production, and in addition, it includes the advantage of reducing the waste and the consumption of bauxite.12 In fact, recycling and upcycling are fundamental methodologies in a circular economy strategy that allows more sustainable access to the desired materials while making the process more ready to be integrated into modern production principles.13
In this contribution, we report our study aimed at the definition of novel electrochemical synthetic protocols employing electrodes prepared from recovered scrap aluminium. In coherence with our circular-economy approach to the definition of effective synthetic methodologies,14 we have prepared electrodes from aluminium can waste. We have therefore optimized their use to develop a simple, oxidant-free protocol for the representative synthesis of 2-oxazolines starting from amino alcohols and aldehydes. To confirm the validity of our approach, we compared the different carbon footprints associated with the use of different electrodes. Finally, we decided to prove the practical utility of our methodology we have also reported a larger-scale synthesis and the preparation of chiral PyOX ligand.
We have decided to test our aluminium can waste-derived electrodes in the synthesis of 2-oxazoline as this process is of general representative interest and this class of compounds find application in several different areas.15
Generally, 2-oxazolines are prepared by (A) reactions between an amino alcohol and a nitrile or carboxylic acid using various catalysts in combination with heat or MW irradiation;16 (B) the coupling between an amino alcohol and an aldehyde using a chemical oxidant, i.e. molecular iodine in combination of heat and bases, hydrobromide perbromide (PHPB), diacetoxyiodobenzene (PIDA) or N-bromosuccinimide (NBS).17
Electrochemistry offers an intriguing opportunity to replace chemical oxidants with electrons. Different well established electrochemical protocols based on the oxidative cyclization of β-amino arylketones or olefinic amides (Fig. 1).18 Anyway a direct electrochemical approach between an amino alcohol and an aldehyde is not reported.
 | ||
| Fig. 1 Known approaches to the electrosynthesis of 2-oxazolines. | ||
Results and discussion
We began our investigation by employing 4-fluorobenzaldehyde 1a and ethanolamine 2 as model substrates to optimize the best reaction conditions. We found that product 3a was obtained in 88% isolated yield (Table 1, entry 1) using LiBr as supporting electrolyte, graphite anode and aluminium cathode in an undivided cell at 50 mA, after the passage of 7 F mol−1 of charge (Faraday efficiency equal to 27%). The reaction medium influences the efficiency of the process and the best results were achieved in a mixture of methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile (1:1) (entry 1) while changing the solvent to methanol, acetonitrile, ethanol, or water conversions decreased (Table 1, entries 2–5).
:acetonitrile (1:1) (entry 1) while changing the solvent to methanol, acetonitrile, ethanol, or water conversions decreased (Table 1, entries 2–5).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Supporting electr. | Conversionb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2 (0.5 mmol), supporting electrolyte (0.4 M), solvent (5 mL, 0.1 M), graphite anode and aluminium cathode, 50 mA of current for 7 F mol−1 of charge based on 1a (about 110 minutes) at 25 °C. b GLC conversion has been determined using samples of pure compound as reference standards; the remaining materials are 1a and 2, isolated yield in parenthesis. c 2 equivalents of 2 were used. d 4 equivalents of 2 were used. e Without current. | |||
| 1 | CH3CN:MeOH 1:1 |
LiBr | 93 (88) |
| 2 | MeOH | LiBr | 61 |
| 3 | CH3CN | LiBr | 0 |
| 4 | EtOH | LiBr | 31 |
| 5 | H2O | LiBr | 5 |
| 6c | CH3CN:MeOH 1:1 |
LiBr | 84 |
| 7d | CH3CN:MeOH 1:1 |
LiBr | 62 |
| 8 | CH3CN:MeOH 1:1 |
KBr | 56 |
| 9 | CH3CN:MeOH 1:1 |
nBu4NPF6 | 0 |
| 10 | CH3CN:MeOH 1:1 |
NaI | 5 |
| 11 | CH3CN:MeOH 1:1 |
KPF6 | 0 |
| 12e | CH3CN:MeOH 1:1 |
LiBr | 0 |

It is noteworthy that in our conditions, the use of stoichiometric amounts of 1a and 2 is functional for achieving best results. Increasing the amount of 2, leads to poorer conversions (Table 1, entries 6 and 7). Without using bromide salts as electrolytes, the reaction does not proceed satisfactorily, suggesting that electrolyte participates in the mechanism as a redox mediator (Table 1, entries 8–11). Finally, by performing the process with no current, product 3a was not observed at all (Table 1, entry 12).
Investigation continued to compare different materials for the electrodes. In Table 2 are shown the results that confirm how important is the use of graphite anode. This behaviour indicates that probably, among the materials tested, graphite has the least overpotential towards bromide oxidation. On the other hand, different metallic cathodes were selected, taking into consideration the carbon footprint associated with the different materials. Carbon footprint, measured in kg CO2 equivalent per kg of product, is an intuitive parameter that measures the quantity of CO2 emitted in the entire production cycle of the material of interest.19
|
|
||
|---|---|---|
| Entry | Anode material | Conversion (%)b |
|
a Reaction conditions: 1a (0.5 mmol), 2 (0.5 mmol), LiBr (0.4 M) as supporting electrolyte, CH3CN:CH3OH (1:1, 5 mL) as solvent, aluminium cathode, 50 mA of current for 7 F mol−1 of charge based on 1a (about 110 minutes) at 25 °C.
b GLC conversion has been determined using samples of pure compound as reference standards; the remaining materials are 1a and 2, isolated yield in parenthesis.
|
||
| 1 | Graphite | 93 |
| 2 | Stainless steel | 0 |
| 3 | Copper | 0 |
| 4 | Zinc | 0 |

From the data reported in Table 3, we can see that the efficiency of the reaction remains almost constant while varying the cathode materials. Based on this evidence, we could focus on cathode materials with the lowest environmental impact and lowest carbon footprint. Aluminium is on the list of critical raw materials, but it is also highly recyclable.13
|
|
|||
|---|---|---|---|
| Entry | Cathode material | C.F. (kg CO2 eq.)c | Conversion (%)b |
|
a Reaction conditions: 1a (0.5 mmol), 2 (0.5 mmol), LiBr (0.4 M) as supporting electrolyte, CH3CN:CH3OH (1:1, 5 mL) as solvent, graphite anode, 50 mA of current for 7 F mol−1 of charge based on 1a (about 110 minutes) at 25 °C, cathode material as described in the Table 3.
b GLC conversion has been determined using samples of pure compound as reference standards; the remaining materials are 1a and 2.
c Carbon footprint associated with the production of different pure metals22 and stainless steel.23
|
|||
| 1 | Silver | 196 | 91 |
| 2 | Primary aluminium | 8.2 | 93 |
| 3 | Titanium | 8.1 | 95 |
| 4 | Nickel | 6.5 | 86 |
| 5 | Zinc | 3.1 | 96 |
| 6 | Copper | 2.8 | 87 |
| 7 | Stainless steel | 1.8 | 92 |
| 8 | Iron | 1.5 | 92 |
| 9 | Upcycled aluminium | 0.5 | 93 |
Based on the 2021 Climate Action report,20 post-consumer aluminium scrap has an associated carbon footprint of about 0.5 kg CO2 per kg of aluminium that is related to its collection, transport, and smelting.20 Additionally, it should be mentioned that recovered post-consumer aluminium scrap leads to a lower purity product, often unattractive for most markets that instead prefer to use higher purity materials.21
For all these reasons, we have decided to focus on using aluminium from recovered cans as cathode material. Recovered cans were used without any further manipulation, as the cathode gave an excellent 93% conversion to 3a and confirmed the effective use of this highly interesting source of aluminium from post-consumer scrap (Table 3, entry 9).
At this stage, we also investigated the possibility of recovering the acetonitrile/methanol mixture used as reaction medium. Due to their similarity, their direct evaporation allowed us to recover 80% of the original 1:1 ACN:MeOH mixture (as confirmed by 1H-NMR). Encouraged by these results, we next decided to demonstrate the synthetic utility and tolerability of our protocol, exploring the scope of various substrates (Scheme 1).
 | ||
| Scheme 1 Scope of the electrochemical synthesis of oxazoline. aReaction conditions: 1a (1 mmol), 2 (1 mmol), LiBr (0.4 M) as supporting electrolyte, CH3CN:CH3OH (1:1, 10 mL) as solvent, graphite anode and aluminum cathode, 100 mA of current for 7 F mol−1 of charge based on 1a (about 110 minutes). All reported yields are isolated yields. b8 F mol−1 of charge passed (about 130 minutes). c2 equivalents of ethanolamine (2) and 12 F mol−1 of charge passed (about 190 minutes). d(S)-2-Amino-3-methylbutan-1-ol (2a) was used instead of ethanolamine (2). | ||
In general, substrates featuring electron-withdrawing substituents led to high yields with a faraday efficiency equal to. By using halogen-substituted benzaldehydes in the para (2a, 2b), meta (2c, 2d) or ortho (2e) positions, products 3 were obtained satisfactory yields. Also, halogen poly-substituted rings (2f, 2g) show similar good results, making the procedure quite interesting for the post-functionalization of substrates by cross-coupling reactions. Other electron-withdrawing groups, such as cyano (2h), nitro (2i, 2j, 2k) or pyridyl (2l), lead to high yields of the corresponding products 3 as well. However, nitro-substituted benzaldehydes required a longer reaction time (8 F mol−1, ca. 130 minutes).
Electron-donor substituents showed lower efficiency. ortho-Tolualdehyde (2m) and ortho-trifluoromethylbenzaldehyde (2s) required longer reaction time, probably due to the steric hindrance effect. meta-substitution with methoxy group (2n) drastically decreases the yield in 3n. However, the propoxy group in para position (2o) led to a good, isolated yield. Concerning the product of terephthaldehyde (2r), it was not possible to steer the reaction towards the formation of the mono-oxazoline product using less amount of ethanolamine. We assume that the higher reactivity of the mono-oxazoline product compared to terephthaldehyde is due to electronic effects, making the procedure ineffective for the formation of aldehydes-substituted oxazolines.
It worthy to notice that with our procedure, we successfully synthesized a chiral ligand of the PyOX type (4), that has been used in various nickel, iridium, copper or palladium-catalyzed reactions.24 Product 4 could be isolated in 84% yield and 95% ee, therefore preserving the enantiopurity of the starting material. Substrates 2t and 2u, let to unsuccessful results presumably due to steric and electronic effects (Scheme 1). In addition, a gram-scale test was also performed showing that scalability of our synthetic protocol is possible (Scheme 2).
 | ||
| Scheme 2 Large scale electrochemical synthesis of 3a. | ||
Interestingly, under such slightly larger scale conditions, solvent distillation was expectedly more effective with 87% of the initial mass recovered.
Finally, to quantify the advances in terms of chemical and environmental efficiency, we calculated different green metrics for our electrochemical approach (E-factor, Reaction Mass Efficiency (RME), and Material Recovery Parameter (MRP)), and for some of the common procedures reported in the literature25 (Scheme 3). We compared seven different protocols that varied in nature of the process (intramolecular or intermolecular), mechanism (oxidative or reductive), solvent and additives. From the data reported Scheme 3 and ESI,† we can confirm the advances in terms of sustainability of our protocol.
 | ||
| Scheme 3 Sustainability assessment and comparison. | ||
The E-factor distribution analysis (see ESI† for complete details of the percentages) shows that, in the gram-scale procedure, recovering the reaction mixture and avoiding the use of a chemical oxidant led to an E-factor as low as 18.7, value that derives only from the mass of non-recovered solvent, with a minor contribution of the excess of LiBr. On the contrary, most of the compared literature processes require tedious and wasteful work-up procedures to remove oxidant, reductant or their byproducts and obviously result strongly in larger total E-factor values (range 84.4–465.7).
Reaction mass efficiency (RME) for our protocol is 5.1% (3.7% for the one-mmol scale), while other procedures never exceed an RME value of 1.2%, with most of them falling in the range of 0.2–0.6%.
To conclude this study, we have decided to have more insights into the reaction mechanism, and therefore, some control experiments were conducted (Scheme 4). First, the reaction was carried out without electrolysis in the presence of 4 equivalents of molecular bromine, but the reaction did not proceed at all. Nevertheless, using 4 equivalents of Br2 in combination with 1 equivalent of lithium methoxide led to the formation of 3a in 52% yield after 2 hours, demonstrating the key role of the base.
 | ||
| Scheme 4 Control experiment for the electrochemical synthesis of 3a. | ||
No product formation was observed when the reaction was performed in the presence of 1 equivalent of TEMPO, therefore suggesting a radical-type reaction pathway.
Performing the reaction without electrochemical conditions and using molecular bromine/lithium methoxide but in the presence of 1 equivalent of TEMPO, the reaction proceeds anyway without significant variation in conversion or yield. Therefore, this allowed to conclude that the traditional chemical conditions an anionic reaction pathway is operative.
By performing the electrochemical reaction in presence of 1 equivalent of BHT, product 3a was not observed. These results together with the fact that electrolysis does not lead to product formation in the absence of bromide salts, suggest that the reaction procced via halogen radicals rather than through a simple hydrogen atom transfer (HAT) pathway.
Furthermore, a cyclic voltammetry analysis was conducted (Fig. 2). As it can be seen from these measurements, the oxidation peak doesn't change in the case of lithium bromide alone (upper chart) or in the presence of reagents 1a and 2 (bottom chart). The oxidation peak occurs around 0.95 V vs. Ag/AgNO3 reference electrode and no appreciable catalytic current is observed upon the addition of substrates. This experimental observation leads to the conclusion that when graphite is used as anode, only bromide undergoes oxidation, suggesting its ability to mediate the oxidation of substrate. Based on the control experiments and on the data reported in literature, the plausible mechanism is described in Scheme 5. First, benzaldehyde undergoes a nucleophilic attack by ethanolamine, giving the imine condensation product I by eliminating a molecule of water.
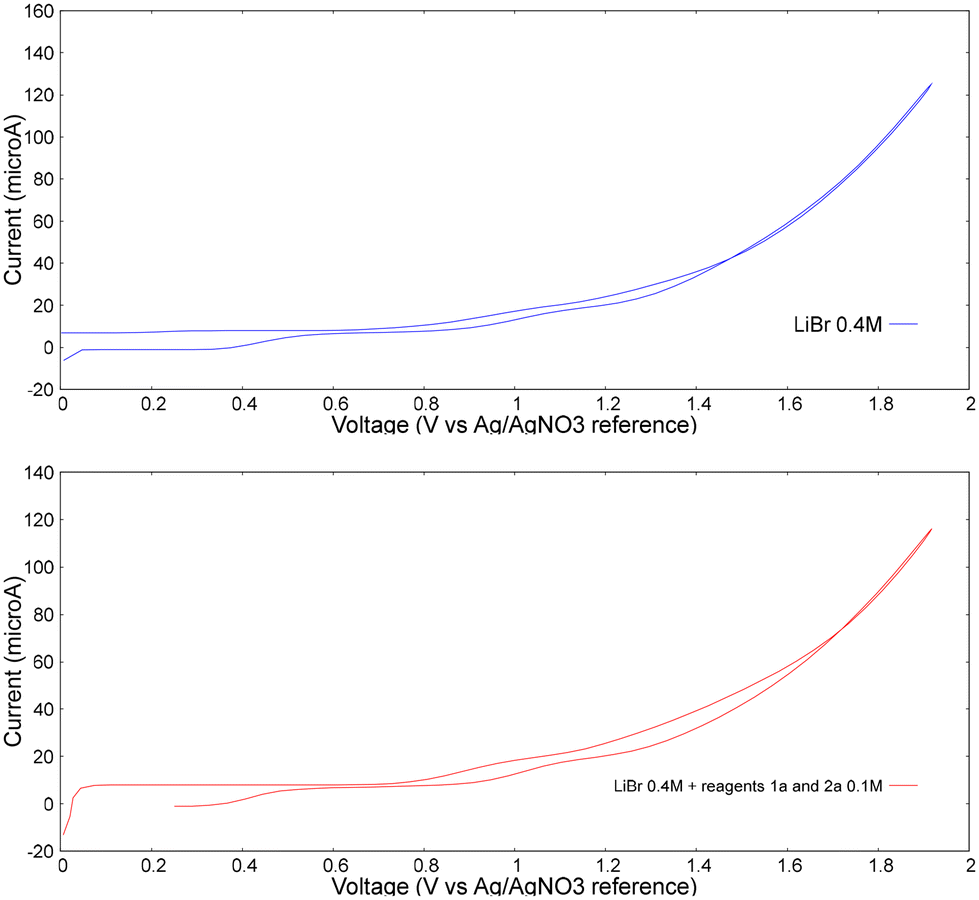 | ||
| Fig. 2 Cyclic voltammetry experiments: upper chart: LiBr 0.4 M, MeCN:MeOH 1:1, sweep 200 mV s−1; lower chart: LiBr 0.4 M, 1a 0.1 M, 2 0.1 M, MeCN:MeOH 1:1, sweep 200 mV s−1. | ||
 | ||
| Scheme 5 Plausible reaction mechanism. | ||
Species I tautomerizes to oxazolidine II that is oxidized to III with an electron loss by halogen radical induced process. The radical–radical coupling between III and radical bromine furnish IV, which, after subsequent base-mediated elimination, gives final 2-oxazoline product 3. From the substrate scope we can highlight that: (1) benzaldehydes with electron-withdrawing groups give better yields; (2) benzaldehydes with ortho substituents give slower reactions. These considerations suggest that the most probable rate-limiting step is the formation of the imine I.
Conclusions
In conclusion, we have developed an electrochemical approach to synthesize 2-oxazoline 3a–s and 4 with a broad range of substitutions, starting from inexpensive and commercially available starting materials, also on a gram-scale. The reaction was carried out using cheap electrode materials, in particular, using cathode deriving from aluminium scrap. The utilization of electrodes made by post-consumer aluminium cans has also allowed to minimize the carbon footprint for making effective electrodes. Control experiments were performed to elucidate the reaction mechanism. Finally, a sustainability assessment using green metrics allowed to quantify the advances in terms of sustainability and the inherent environmental efficiency and potential of the electrochemical approach reported.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript. S. T. C.: investigation, methodology, data analysis, manuscript preparation; F. F. and L. V.: conceptualization, project administration, manuscript review/editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. We acknowledge Università degli Studi di Perugia and MUR for support within the project Vitality. MUR is also thanked for PRIN-PNRR 2022 project “P2022XKWH7 – CircularWaste”.References
- (a) L. De La Peña, R. Guo, X. Cao, X. Ni and W. Zhang, Resour., Conserv. Recycl., 2022, 177, 105957 CrossRef; (b) A. Elshkaki and L. Shen, Energies, 2022, 15, 4967 CrossRef.
- M. H. Barecka and J. W. Ager, Energy Adv., 2023, 2, 268–279 RSC.
- (a) D. S. Mallapragada, Y. Dvorkin, M. A. Modestino, D. V. Esposito, W. A. Smith, B.-M. Hodge, M. P. Harold, V. M. Donnelly, A. Nuz, C. Bloomquist, K. Baker, L. C. Grabow, Y. Yan, N. N. Rajput, R. L. Hartman, E. J. Biddinger, E. S. Aydil and A. D. Taylor, Joule, 2023, 7, 23–41 CrossRef CAS; (b) R. Xia, S. Overa and F. Jiao, JACS Au, 2022, 2, 1054–1070 CrossRef CAS; (c) Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10–11 CrossRef.
- (a) M. Ostadi, K. G. Paso, S. Rodriguez-Fabia, L. E. Øi, F. Manenti and M. Hillestad, Energies, 2020, 13, 4859 CrossRef CAS; (b) M.-Y. Lee, K. T. Park, W. Lee, H. Lim, Y. Kwon and S. Kang, Crit. Rev. Environ. Sci. Technol., 2019, 50, 769–815 CrossRef; (c) G. G. Botte, Electrochem. Soc. Interface, 2014, 23, 49–55 CrossRef.
- (a) T. Noël, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858–2869 CrossRef PubMed; (b) N. Tanbouza, T. Ollevier and K. Lam, iScience, 2020, 23, 101720 CrossRef CAS PubMed.
- M. Moradi, Y. Vasseghian, A. Khataee, M. Kobya, H. Arabzade and E.-N. Dragoi, J. Ind. Eng. Chem., 2020, 87, 18–39 CrossRef CAS.
- (a) S. K. Padamata, K. Singh, G. M. Haarberg and G. Saevarsdottir, J. Electrochem. Soc., 2023, 170, 073501 CrossRef; (b) A. T. Tabereaux and R. D. Peterson, Treatise Process Metall., 2014, 839–917 CAS.
- (a) A. Arabameri, M. R. Alavi Moghaddam, A. R. Azadmehr and M. Payami Shabestar, Chem. Eng. Process., 2022, 174, 108869 CrossRef CAS; (b) M. A. Sadik, Adv. Chem. Eng. Sci., 2019, 09, 109–126 CrossRef CAS; (c) D. T. Moussa, M. H. El-Naas, M. Nasser and M. J. Al-Marri, J. Environ. Manage., 2017, 186, 24–41 CrossRef PubMed.
- (a) N. Idusuyi, O. O. Ajide, R. Abu, O. A. Okewole and O. O. Ibiyemi, Mater. Today: Proc., 2022, 56, 1712–1716 CAS; (b) M. Moradi, Y. Vasseghian, A. Khataee, M. Kobya, H. Arabzade and E.-N. Dragoi, J. Ind. Eng., 2020, 87, 18–39 CAS.
- S. Du, S. Zhang, J. Wang, Z. Lv, Z. Xu, C. Liu, J. Liu and B. Liu, J. Cleaner Prod., 2024, 141176 CrossRef CAS.
- G. Wallace, Fundamentals of Aluminium Metallurgy, 2011, pp. 70–82 Search PubMed.
- https://rmis.jrc.ec.europa.eu/uploads/CRM_2020_Factsheets_critical_Final.pdf .
- (a) J. Cho, B. Kim, T. Kwon, K. Lee and S.-I. Choi, Green Chem., 2023, 25, 8444–8458 RSC; (b) Z. Yu, Y. J. Lim, T. Williams and S. Nutt, Green Chem., 2023, 25, 7058–7061 RSC; (c) Y. Wang, K. Liu, F. Liu, C. Liu, R. Shi and Y. Chen, Green Chem., 2023, 25, 5872–5877 RSC.
- (a) F. Ferlin, F. Valentini, D. Sciosci, M. Calamante, E. Petricci and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 12196–12204 CrossRef CAS; (b) F. Valentini, F. Ferlin, S. Lilli, A. Marrocchi, L. Ping, Y. Gu and L. Vaccaro, Green Chem., 2021, 23, 5887–5895 RSC; (c) N. Salameh, F. Ferlin, F. Valentini, I. Anastasiou and L. Vaccaro, ACS Sustainable Chem. Eng., 2022, 10, 3766–3776 CrossRef CAS; (d) F. Campana, F. Valentini, A. Marrocchi and L. Vaccaro, Biofuel Res. J., 2023, 10, 1989–1998 CrossRef CAS; (e) F. Ferlin, F. Valentini, A. Marrocchi and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 9604–9624 CrossRef CAS.
- (a) V. Facchinetti, C. R. B. Gomes and M. V. N. de Souza, Tetrahedron, 2021, 102, 132544 CrossRef CAS; (b) R. Connon, B. Roche, B. V. Rokade and P. J. Guiry, Chem. Rev., 2021, 121, 6373–6521 CrossRef CAS; (c) D. Lu, J. Cui, S. Yang and Y. Gong, ACS Catal., 2021, 11, 4288–4293 CrossRef CAS; (d) L. Ju, Q. Lin, N. J. LiBretto, C. L. Wagner, C. T. Hu, J. T. Miller and T. Diao, J. Am. Chem. Soc., 2021, 143, 14458–14463 CrossRef CAS PubMed; (e) S. R. Shinde, S. N. Inamdar, M. Shinde, C. Pawar, B. Kushwaha, V. A. Obakachi, A. Kajee, R. Chauhan and R. Karpoormath, J. Mol. Struct., 2023, 1273, 134243 CrossRef CAS; (f) X. Yu, Y. Zhang, Y. Liu, Y. Li and Q. Wang, J. Agric. Food Chem., 2019, 67, 4224–4231 CrossRef CAS PubMed; (g) M. Zhou, W. Jiang, J. Xie, W. Zhang, Z. Ji, J. Zou, Z. Cong, X. Xiao, J. Gu and R. Liu, ChemMedChem, 2020, 16, 309–315 CrossRef PubMed; (h) M. Zhou, Y. Qian, J. Xie, W. Zhang, W. Jiang, X. Xiao, S. Chen, C. Dai, Z. Cong, Z. Ji, N. Shao, L. Liu, Y. Wu and R. Liu, Angew. Chem., Int. Ed., 2020, 59, 6412–6419 CrossRef CAS PubMed; (i) W. Jiang, M. Zhou, Z. Cong, J. Xie, W. Zhang, S. Chen, J. Zou, Z. Ji, N. Shao, X. Chen, M. Li and R. Liu, Angew. Chem., 2022, 134, e202200778 CrossRef.
- (a) K. A. Jadhav, S. D. Bhosle, S. V. Itage, R. S. Bhosale, G. Eppa and J. S. Yadav, Tetrahedron Lett., 2022, 106, 154048 CrossRef CAS; (b) M. Trose, F. Lazreg, M. Lesieur and C. S. Cazin, J. Org. Chem., 2015, 80, 9910–9914 CrossRef CAS PubMed; (c) V. Mirkhani, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork and M. Mahdavi, Monatsh. Chem., 2009, 140, 1489–1494 CrossRef CAS; (d) A. Cwik, Z. Hell, A. Hegedüs, Z. Finta and Z. Horváth, Tetrahedron Lett., 2002, 43, 3985–3987 CrossRef CAS; (e) L. Wang, B. Guo, H.-X. Li, Q. Li, H.-Y. Li and J.-P. Lang, Dalton Trans., 2013, 42, 15570 RSC.
- (a) M. Ishihara and H. Togo, Tetrahedron, 2007, 63, 1474–1480 CrossRef CAS; (b) H. Fujioka, K. Murai, Y. Ohba, A. Hiramatsu and Y. Kita, Tetrahedron Lett., 2005, 46, 2197–2199 CrossRef CAS; (c) S. Sayama, Synlett, 2006, 1479–1484 CrossRef CAS; (d) N. Karade, G. Tiwari and S. Gampawar, Synlett, 2007, 1921–1924 CrossRef CAS; (e) W. Wei, R. Li, N. Huber, G. Kizilsavas, C. T. Ferguson, K. Landfester and K. A. Zhang, ChemCatChem, 2021, 13, 3410–3413 CrossRef CAS.
- (a) H. Wang, J. Zhang, J. Tan, L. Xin, Y. Li, S. Zhang and K. Xu, Org. Lett., 2018, 20, 2505–2508 CrossRef CAS PubMed; (b) F. Lu, J. Xu, H. Li, K. Wang, D. Ouyang, L. Sun, M. Huang, J. Jiang, J. Hu, H. Alhumade, L. Lu and A. Lei, Green Chem., 2021, 23, 7982–7986 RSC; (c) S. Mallick, M. Baidya, K. Mahanty, D. Maiti and S. De Sarkar, Adv. Synth. Catal., 2020, 362, 1046–1052 CrossRef CAS; (d) Y. He, Y. Yin, C. Liu, X.-F. Wu and Z. Yin, New J. Chem., 2022, 46, 663–667 RSC.
- L. J. Müller, A. Kätelhön, S. Bringezu, S. McCoy, S. Suh, R. Edwards, V. Sick, S. Kaiser, R. Cuéllar-Franca, A. El Khamlichi, J. H. Lee, N. von der Assen and A. Bardow, Energy Environ. Sci., 2020, 13, 2979–2992 RSC.
- https://www.climateaction.org/news/carbon-footprint-of-recycled-aluminium .
- (a) X. Lu, Z. Zhang, T. Hiraki, O. Takeda, H. Zhu, K. Matsubae and T. Nagasaka, Nature, 2022, 606, 511–515 CrossRef CAS; (b) S. Kosai, K. Matsui, K. Matsubae, E. Yamasue and T. Nagasaka, Resour., Conserv. Recycl., 2021, 166, 105256 CrossRef CAS; (c) J. M. Cullen and J. M. Allwood, Environ. Sci. Technol., 2013, 47, 3057–3064 CrossRef CAS PubMed; (d) D. Raabe, D. Ponge, P. J. Uggowitzer, M. Roscher, M. Paolantonio, C. Liu, H. Antrekowitsch, E. Kozeschnik, D. Seidmann, B. Gault, F. De Geuser, A. Deschamps, C. Hutchinson, C. Liu, Z. Li, P. Prangnell, J. Robson, P. Shanthraj, S. Vakili, C. Sinclair, L. Bourgeois and S. Pogatscher, Prog. Mater. Sci., 2022, 128, 100947 CrossRef CAS.
- (a) P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef PubMed; (b) J. Torrubia, A. Valero and A. Valero, Resour., Conserv. Recycl., 2023, 199, 107281 CrossRef CAS.
- https://worldsteel.org/steel-topics/sustainability/sustainability-indicators-2023-report/ .
- (a) P. Zhou and T. Xu, ChemComm, 2020, 56, 8194–8197 RSC; (b) K. S. Yoo, C. P. Park, C. H. Yoon, S. Sakaguchi, J. O'Neil and K. W. Jung, Org. Lett., 2007, 9, 3933–3935 CrossRef CAS PubMed; (c) W. L. Lyon and D. W. MacMillan, J. Am. Chem. Soc., 2023, 145, 7736–7742 CrossRef CAS PubMed; (d) L. Zhao, X. Lu and W. Xu, J. Org. Chem., 2005, 70, 4059–4063 CrossRef CAS PubMed; (e) A. J. Hallett, T. M. O'Brien, E. Carter, B. M. Kariuki, D. M. Murphy and B. D. Ward, Inorg. Chim. Acta, 2016, 441, 86–94 CrossRef CAS.
- Literature process for E-factor comparison in order of appearance in Scheme 3: (a) F. Lu, J. Xu, H. Li, K. Wang, D. Ouyang, L. Sun, M. Huang, J. Jiang, J. Hu, H. Alhumade, L. Lu and A. Lei, Green Chem., 2021, 23, 7982 RSC; (b) C. Uma Maheswari, G. Satish Kumar and M. Venkateshwar, RSC Adv., 2014, 4, 39897–39900 RSC; (c) M. Ishihara and H. Togo, Tetrahedron, 2007, 63, 1474–1480 CrossRef CAS; (d) A. Alhalib, S. Kamouka and W. J. Moran, Org. Lett., 2015, 17, 1453 CrossRef CAS; (e) F. Glorius and K. Schwekendiek, Synthesis, 2006, 2996 CrossRef; (f) W. Wei, R. Li, N. Huber, G. Kizilsavas, C. T. J. Ferguson, K. Landfester and K. A. I. Zhang, ChemCatChem, 2021, 13, 3410 CrossRef CAS; (g) N. N. Karade, G. B. Tiwari and S. V. Gampawar, Synlett, 2007, 1921 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, full characterization of the synthetized compounds and copies of 1H, 13C and 19F NMR spectra. See DOI: https://doi.org/10.1039/d4gc02564d |
| This journal is © The Royal Society of Chemistry 2024 |