
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-equilibrium plasma co-upcycling of waste plastics and CO2 for carbon-negative oleochemicals†
Harish
Radhakrishnan‡
 a,
Samirah
Gnangbe‡
a,
Alif
Duereh
a,
Sultan Ul
Iffat Uday
a,
Lusi
A
a,
Haiyang
Hu
b,
Hui
Hu
b,
Mark Mba
Wright
*a and
Xianglan
Bai
*ac
a,
Samirah
Gnangbe‡
a,
Alif
Duereh
a,
Sultan Ul
Iffat Uday
a,
Lusi
A
a,
Haiyang
Hu
b,
Hui
Hu
b,
Mark Mba
Wright
*a and
Xianglan
Bai
*ac
aDepartment of Mechanical Engineering, Iowa State University, Ames, IA 50011, USA. E-mail: bxl9801@iastate.edu
bDepartment of Aerospace Engineering, Iowa State University, Ames, IA 50011, USA
cDepartment of Chemical & Biological Engineering, Iowa State University, Ames, IA 50011, USA
First published on 25th July 2024
Abstract
Mechanical recycling and chemical upcycling by thermochemical reactions have been the major approaches for recycling end-of-life plastics. Herein, we report an electrified approach to upcycle waste plastics into carbon-negative commodity chemicals using greenhouse gas CO2 as the oxidant and additional carbon source. In this non-equilibrium plasma process, waste polyolefins were oxidatively depolymerized by plasma-activated CO2 to produce oleochemicals and hydrocarbon chemicals in a single-step process at high reaction rates. In addition, a mixture of CO2 and a small amount of O2 was employed as plasma gases to selectively produce fatty alcohols from polyolefins. Based on this atmospheric pressure, non-solvent, and non-catalyst process, up to 97.6% of fatty alcohols could be produced within minutes. In this article, the co-conversion approach was demonstrated using common polyolefins and real-world mixed waste plastics to obtain comparable results. The techno-economic analysis estimates the internal rate of return to be 42.2% and 43.5% for the plasma-based conversion of waste plastics, depending on the plasma gas composition. Lifecycle assessment indicates the global warming potential is between −3.33 and −3.07 kg CO2e per kg of plastic.
1. Introduction
The utilization of plastic materials has grown over the years because of their lightweight, low-cost, durability, and processability attributes.1,2 Plastics can be shaped into various products for several applications, such as packaging, automotive, electronics, and medical applications. An alarming consequence of the rise in production and usage is the amount of plastic waste generated and its inefficient management leading to plastic pollution.Global plastic production reached 390.7 million metric tons (MMT) in 2021, which corresponds to a significant increase of 44% within ten years.3 Single-use plastics, mainly composed of polyolefins, constitute over 50% of the plastic production.4 United States (US) generated the largest quantity of plastics, accounting for 18% of global production, after China.3 The EPA reported that 4.5 MMT out of 32.4 MMT of plastic generated in the US in 2018 were recycled, ∼15% were incinerated, and the remaining waste was disposed of into landfills.5 Polyolefins, such as polyethylene (PE) and polypropylene (PP), have a meager recycling rate and are discarded in landfills or oceans.4 Waste plastic litter is piling up in rivers, oceans, and soil, posing adverse effects on the environment and human health.4
Recycling, incineration, and landfilling are the most common methods to manage waste plastics. However, they are found to be ineffective in managing plastic waste. Landfilling occupies valuable land, emits greenhouse gases, including CH4 and CO2, and pollutes the aquifer.6 The conversion of waste to energy through incineration releases CO2 and toxic chemicals into the atmosphere. Additionally, mechanical recycling has several limitations, including challenges in handling contaminated and mixed waste streams and the decrease in the mechanical properties of the materials at each cycle, resulting in downcycling.6
Research efforts have been undertaken to explore technologies to increase the value of plastic waste. Plastics are made from fossil fuels and contain hydrocarbon resources that can be recovered and converted to valuable chemicals at their end-of-life. Thermochemical conversion has been considered a promising technology to upcycle plastic waste and reduce waste disposal in landfills and natural habitats.1 Pyrolysis and gasification are the most industrialized chemical recycling technologies.1,6 However, these endothermic reactions require high energy input to depolymerize and decompose plastics.6 Plastic waste gasification occurs at 750–900 °C in the presence of steam or oxygen to yield a gaseous product known as syngas. The latter requires intensive cleaning to remove tar and contaminants and additional upgrading processes to produce marketable commodities.7 Pyrolysis of polyolefins is performed under oxygen-starved conditions at 500–700 °C (or lower than 500 °C when pressures and extended conversion times are allowed) to produce wax and oil, a complex long-chain hydrocarbon mixture.8 Pyrolysis products can be regarded as low-value, which requires further upgrading, such as thermal catalytic or hydroformylation, to generate transportation fuel or chemical commodities.1
Other sustainable techniques, which can convert polyolefins into value-added products at low energy requirements, have gained greater interest. Recent studies identified non-equilibrium plasma processes as a viable technology for producing light hydrocarbons and hydrogen from plastic waste at low temperatures and atmospheric pressure operations.9 Plasma, often referred to as the “fourth state of matter,” is an ionized gas composed of free electrons and heavy species such as atoms, ions, and molecules.10 Plasma is created by applying electrical discharge to a gas and is generally classified as either thermal or non-thermal, depending on the energy level. Thermal plasma operates at high temperature, 6000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 K, and the electrons and heavy particles are in thermal equilibrium.11 On the contrary, non-thermal plasma (NTP) performs at non-equilibrium conditions, where the gas temperature can remain as low as room temperature while the electron temperature can range from 10000–100000 K.10
000 K, and the electrons and heavy particles are in thermal equilibrium.11 On the contrary, non-thermal plasma (NTP) performs at non-equilibrium conditions, where the gas temperature can remain as low as room temperature while the electron temperature can range from 10000–100000 K.10
The energy levels of plasma species are high and can break any hydrocarbon chains without an external heating source. Thermal plasma is a well-established technology in metallurgical processing, hazardous waste treatment, and solid waste treatment.12 In recent decades, thermal plasma for waste plastic treatment has been intensively examined, leading to the construction of pilot plants.11,13 The high energy density and high temperature of thermal plasma result in low thermal efficiency and product selectivity, and it is also associated with high capital and operational costs when executed on a large scale.11,14 On the other hand, non-thermal plasma processes offer greater energy efficiency and selectivity in plasma chemical reactions. Under non-thermal plasma discharge, the collision between high-temperature electrons and cold gas molecules leads to the generation of radicals, ions, activated atoms and molecules. Energetic electrons and active particles generated by plasma discharge can break a wide range of chemical bonds without needing to establish thermodynamic equilibrium. Since the reactions occur under non-equilibrium conditions, thermodynamically unfavorable reactions can proceed with plasma at unconventionally low temperatures.10,15–17 This distinctive property makes this process chemically selective and advantageous for synthesizing fuels and chemicals from plastic waste. Several experimental studies have reported using non-thermal plasma to decompose PE into hydrogen and light hydrocarbons.9,11,18–20 For instance, Yao et al.19 used non-thermal plasma-activated H2 to deconstruct high-density PE to lower alkanes (C1–C3). Diaz-Silvarrey et al.21 applied nitrogen plasma to crack PE pyrolysis vapor containing long-chain hydrocarbons into ethylene-rich gases. Rather than plastics, previous studies also applied non-thermal plasma to CO2 and single molecules (H2, H2O, or CH4) to produce synthetic fuel or other higher-value platform chemicals.10,22 While non-thermal plasma has been recognized as an efficient pathway to upcycle biomass and various waste streams to in-demand products with excellent potential for scale-up,9,10 the technical and economic feasibility of non-thermal plasma for plastic waste conversion has not yet been verified.
In this article, we present a novel process to co-upcycle plastic wastes and waste CO2 to commodity chemicals using a non-equilibrium plasma approach developed and demonstrated at a laboratory scale. Based on the experimental results in this work, we evaluated the technical feasibility and economic viability of producing oleochemicals from waste resources. Additionally, we performed a life cycle assessment (LCA) to investigate the global warming potential (GWP) of this technology.
Oleochemicals, such as fatty alcohols and fatty acids, are employed in various industries, such as cosmetics, pharmaceuticals, lubricants, plastic, and surfactants. The global market was valued at $22.66 billion in 2022 and is forecasted to grow at a compound annual growth rate (CAGR) of 7.1% from 2023 to 2030.23 Oleochemicals not only have a substantial market size but also are valuable chemical commodities. These chemicals have many industrial applications. Fatty alcohol mixtures find direct applications such as emollients, emulsifiers, and thickeners in cosmetics.24 Long-chain fatty alcohols as a mixture form (C12+) are widely used as intermediates in the production of surfactants and detergents, while C5–C12 fatty alcohols find applications as plasticizers in polymer production and solvents in coatings and resin formulations.24–26 Fatty acids can be used as cosmetic and food additives and have wide applications in textile treatment and in producing surfactants, soaps, and detergents.24,25 Further, fatty acids are also used as intermediates in the production of esters, amines, and amides.25 In 2022, the market prices of fatty alcohol and fatty acid were $2.5–3 per kg and $1.6–1.9 per kg, respectively,27,28 much higher than hydrocarbons of the same chain length. The cost of oleochemicals can be associated with the feedstock used and the production process. Conventionally, fatty alcohol is manufactured from petrochemicals or palm kernel oil. Fossil-derived fatty alcohol is produced via the Ziegler or oxo process, which involves multi-step processes such as the alkylation of ethylene using a hydrogenated catalyst and hydroformylation of long-chain olefins utilizing syngas.29 Such methods are energy-intensive, and the life cycle greenhouse gas (GHG) emission for 1 kg of fatty alcohol is estimated to be 2.97 kg CO2e.29,30 Meanwhile, palm kernel oil-derived fatty alcohol was found to have a high GHG emission of 5.27 kg CO2e due to the land use change for palm oil plantations.29 Hence, producing oleochemicals from less energy-intensive pathways and abundant feedstock, like plastic waste, would be beneficial in minimizing GHG emissions. In their recent study, Li et al.31 produced fatty alcohols from polyolefin waste through a multi-step process involving pyrolysis, hydroformylation, and hydrogenation. The GHG of converting 1 kg of plastic waste using this process was calculated at 1.6 kg CO2e, lower than the conventionally produced fatty alcohols.
In our recent study, a single-step process was developed to produce oleochemicals and hydrocarbons from waste plastics and plasma-activated CO2. The reaction system was also tailored to selectively produce fatty alcohols by introducing a small amount of O2. An isotopic study was performed to confirm the role of polyolefin as a sink for chemically capturing CO2. The applicability of the selective co-conversion by plasma was also demonstrated using common polyolefins and real-world mixed polyolefins. Technoeconomic analysis (TEA) and LCA results suggest this non-thermal plasma, catalyst and solvent-free approach can produce carbon-negative chemicals and fuels using wastes at low costs. The concept of green upcycling of waste plastics and CO2 for carbon-negative commodity chemicals is illustrated in Scheme 1.
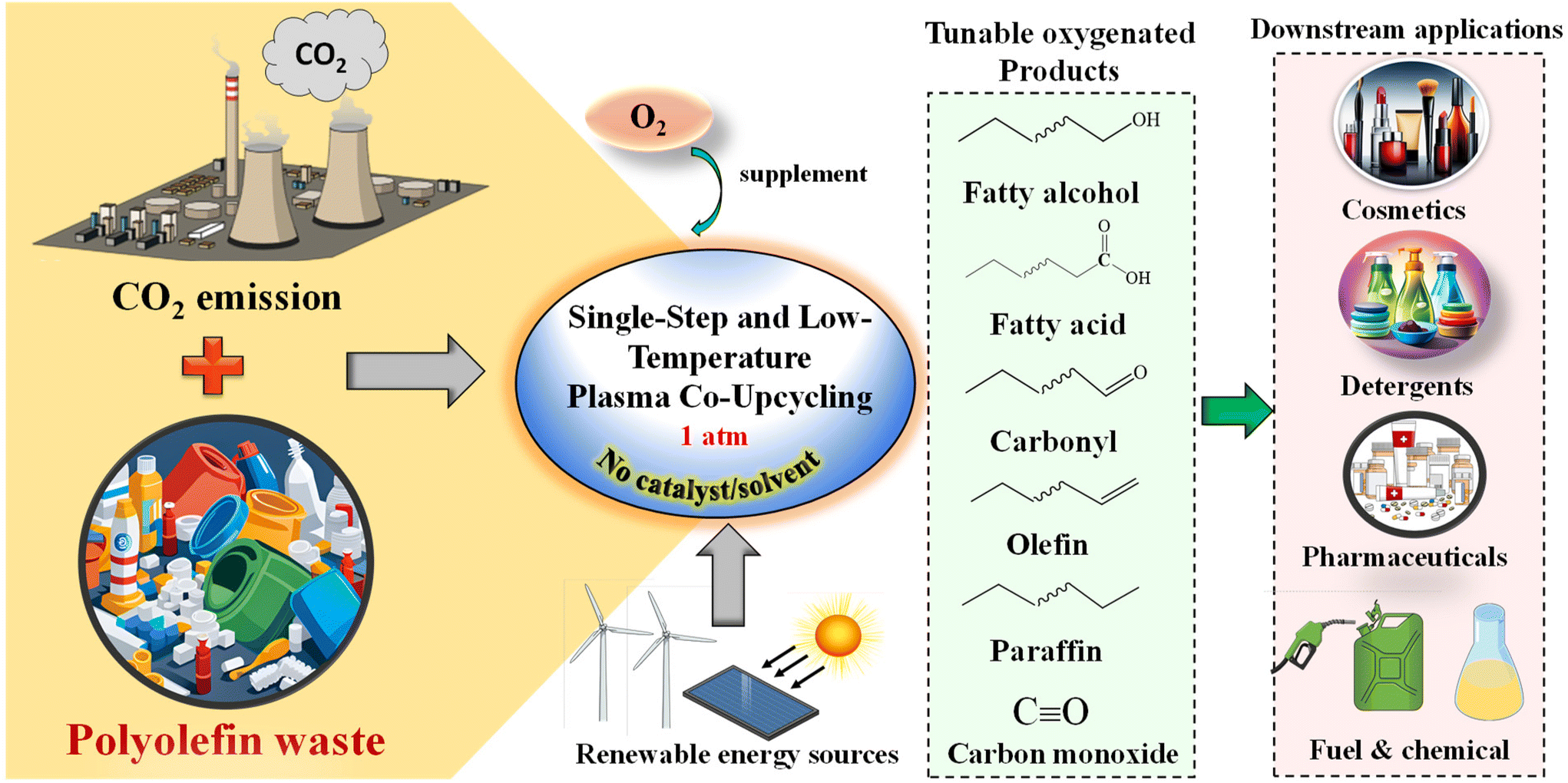 | ||
| Scheme 1 Concept of plasma co-upcycling waste plastics and CO2. CO2 serves as an oxidant and carbon source to depolymerize polyolefins into platform chemicals, whereas plastics enhance CO2 conversion. Fatty alcohols are selectively produced by adding O2. | ||
2. Results and discussion
2.1. PE conversion using CO2 plasma
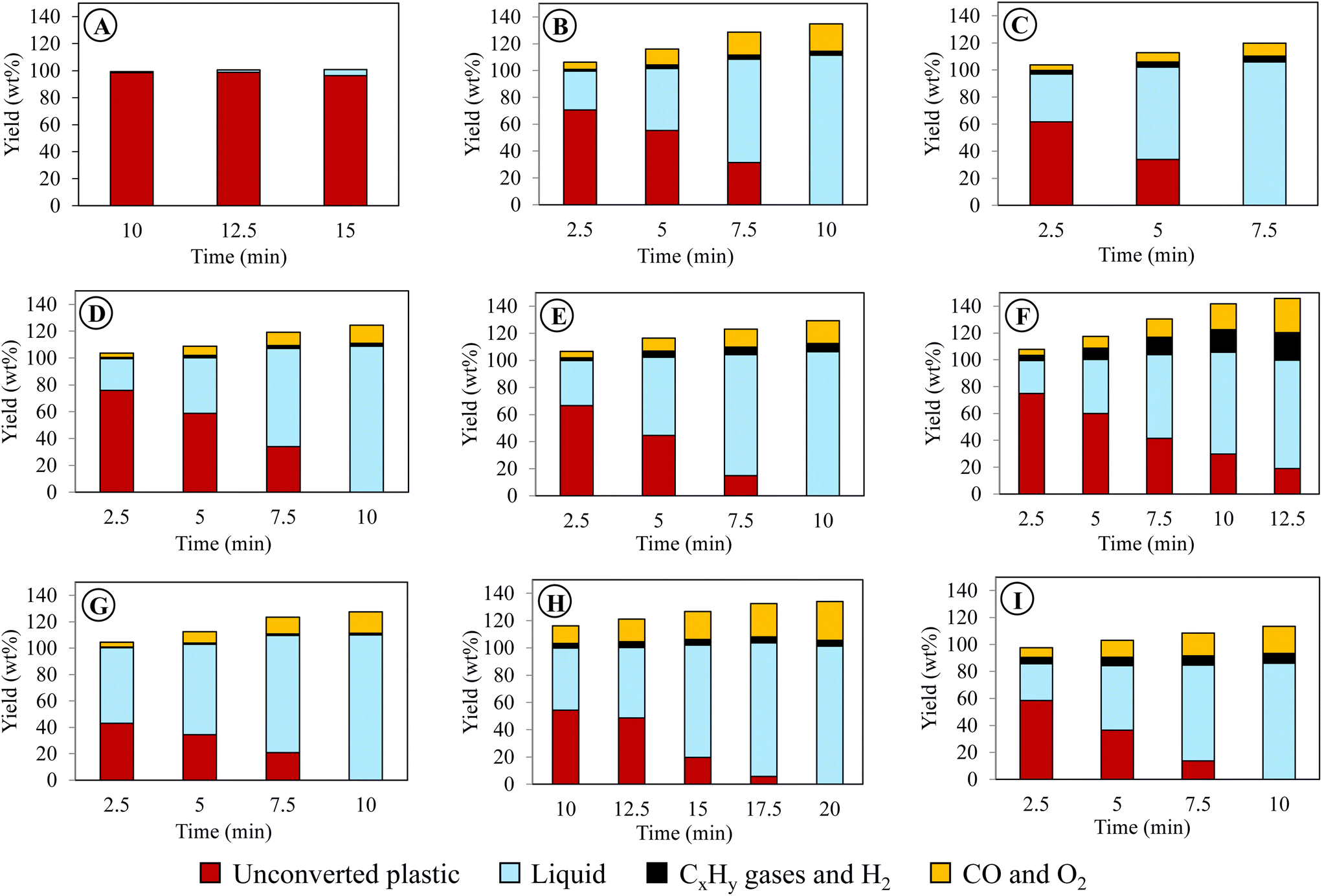 | ||
| Fig. 1 Time-dependent product distribution calculated per initial PE mass for different plasma conditions. Refer to Table 1 for the alphabetical nomenclatures in circles and their corresponding reaction conditions. | ||
| Condition | Feed | Gas | T i | t R | V | f | Time |
|---|---|---|---|---|---|---|---|
| (°C) | (s) | (kV) | (kHz) | (min) | |||
| In the above, Ti is the initial temperature before plasma discharge, CO2/O2 concentration is represented in vol%, tR is the vapor-phase gas residence time, V is the voltage, f is the frequency, and “Time” is the reaction completion time. This nomenclature applies for the entire article including ESI.† | |||||||
| A | PE | CO2 | 350 | 13 | 12.5 | 8 | 15 |
| B | PE | CO2 | 350 | 13 | 15 | 8 | 10 |
| C | PE | CO2 | 350 | 13 | 17.5 | 8 | 7.5 |
| D | PE | CO2 | 350 | 13 | 15 | 7.5 | 10 |
| E | PE | CO2 | 350 | 13 | 15 | 8.5 | 10 |
| F | PE | CO2 | 350 | 10 | 15 | 8 | 12.5 |
| G | PE | CO2 | 350 | 20 | 15 | 8 | 10 |
| H | PE | CO2 | 300 | 13 | 15 | 8 | 20 |
| I | PE | CO2 | 400 | 13 | 15 | 8 | 10 |
| J | PE | CO2/8% O2 | 350 | 13 | 15 | 8 | 10 |
| K | PE | CO2/8% O2 | 350 | 20 | 15 | 8 | 7.5 |
| L | PE | CO2/14% O2 | 350 | 13 | 15 | 8 | 7.5 |
| M | LDPE | CO2 | 325 | 20 | 15 | 8 | 10 |
| N | PP | CO2 | 325 | 20 | 15 | 8 | 10 |
| O | LDPE | CO2/8% O2 | 350 | 13 | 15 | 8 | 10 |
| P | PP | CO2/8% O2 | 350 | 13 | 15 | 8 | 10 |
| Q | PC-PE | CO2 | 325 | 20 | 15 | 8 | 10 |
| R | PC-PE | CO2/8% O2 | 350 | 13 | 15 | 8 | 10 |
The gas selectivity for different reaction conditions is provided in Fig. 2. CO was the primary gas product in all conditions, with its selectivity ranging from 50% to 90.3%. The highest and lowest CO selectivity was observed with the longest gas residence time (case G) and the highest initial temperature (case I), respectively. Although dissociation of 1 mol of CO2 theoretically produces 1 mol of CO and 0.5 mol of O2, O2 content in the gas stream was nearly negligible. Thus, CO2-derived O was preferentially used to oxidize the liquid molecules.
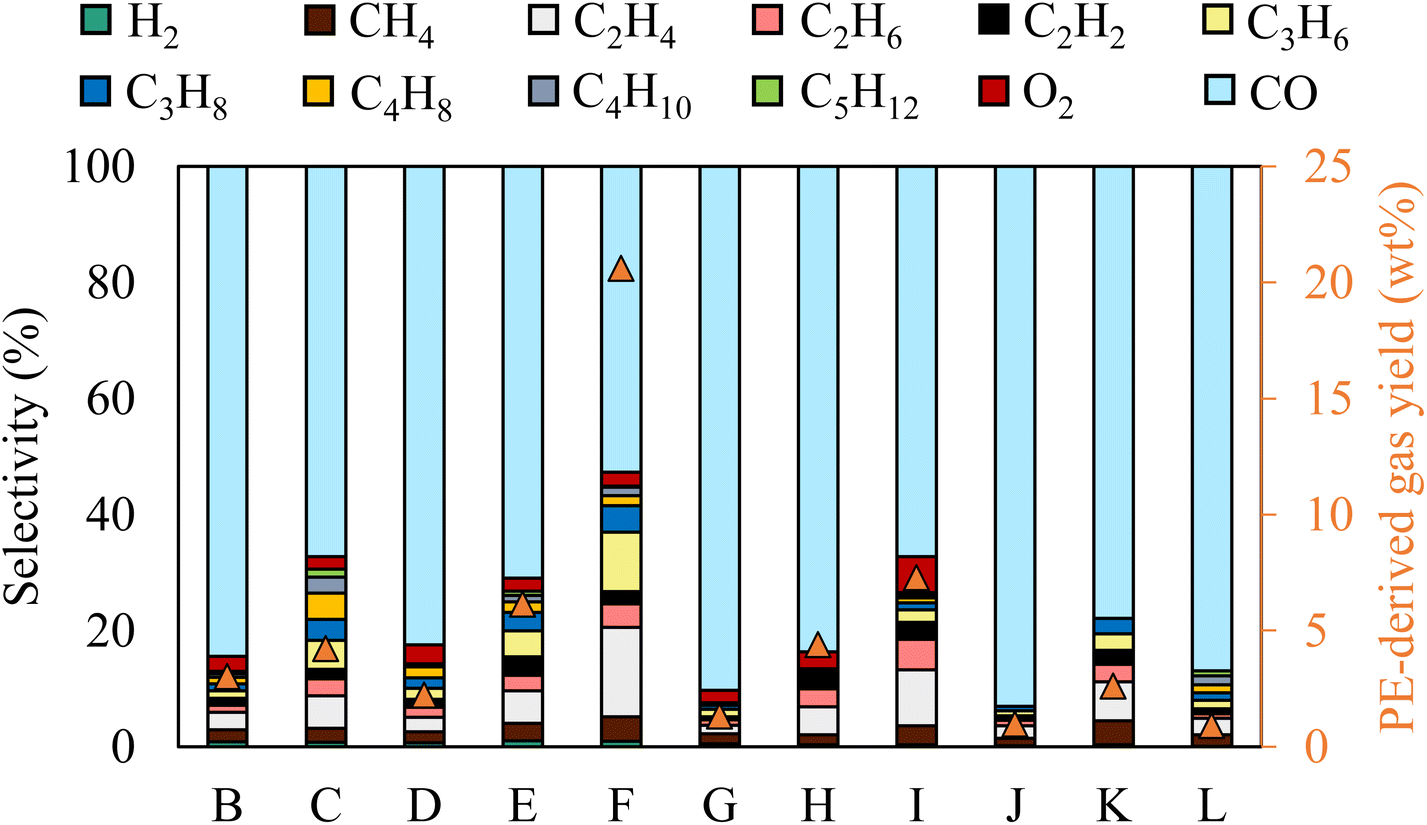 | ||
| Fig. 2 Gas product selectivity and PE-derived gas yield (H2 and CxHy) for different reaction conditions. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, δ = 180–210 ppm), R–CHCH2 bonds (δ = 135–140 ppm), methyl (–CH3, δ = 10–20 ppm) and methylene (–CH2, δ = 110–15 ppm), supporting the GC/MS analysis results. As shown in Table 2, the cases with higher voltage, higher frequency, longer gas resistance time, and higher initial temperature resulted in liquids with narrower molecular weight distributions of shorter-carbon chain-length compounds, attributing to the higher degrees of PE depolymerization and β–β carbon scissions. However, the oxygen content of the liquids did not follow the exact trend as their carbon chain-length distributions. Liquid oxygen content was 6.2% in case B, with the highest liquid yield. However, increasing the voltage caused a decrease in oxygen content to 4.7% in case C. In comparison, increasing frequency at case E had no significant impact on the oxygen content. The liquid with the highest oxygen content of 7.8% was obtained in case G with tR of 20 s, compared to 6.2% in case B with tR of 13 s and 3.9% in case F with tR of 10 s. Thus, the longer gas residence time was beneficial in enhancing oxidative depolymerization of PE. The initial temperature also had a significant impact on PE oxidation. While the liquid oxygen content was 6.2% in case B with Ti of 350 °C and 5.3% in case H with 300 °C, it was only 1.7% in case I with 400 °C despite this case having a higher average reactor temperature during plasma discharge than the other two cases.
O, δ = 180–210 ppm), R–CHCH2 bonds (δ = 135–140 ppm), methyl (–CH3, δ = 10–20 ppm) and methylene (–CH2, δ = 110–15 ppm), supporting the GC/MS analysis results. As shown in Table 2, the cases with higher voltage, higher frequency, longer gas resistance time, and higher initial temperature resulted in liquids with narrower molecular weight distributions of shorter-carbon chain-length compounds, attributing to the higher degrees of PE depolymerization and β–β carbon scissions. However, the oxygen content of the liquids did not follow the exact trend as their carbon chain-length distributions. Liquid oxygen content was 6.2% in case B, with the highest liquid yield. However, increasing the voltage caused a decrease in oxygen content to 4.7% in case C. In comparison, increasing frequency at case E had no significant impact on the oxygen content. The liquid with the highest oxygen content of 7.8% was obtained in case G with tR of 20 s, compared to 6.2% in case B with tR of 13 s and 3.9% in case F with tR of 10 s. Thus, the longer gas residence time was beneficial in enhancing oxidative depolymerization of PE. The initial temperature also had a significant impact on PE oxidation. While the liquid oxygen content was 6.2% in case B with Ti of 350 °C and 5.3% in case H with 300 °C, it was only 1.7% in case I with 400 °C despite this case having a higher average reactor temperature during plasma discharge than the other two cases.
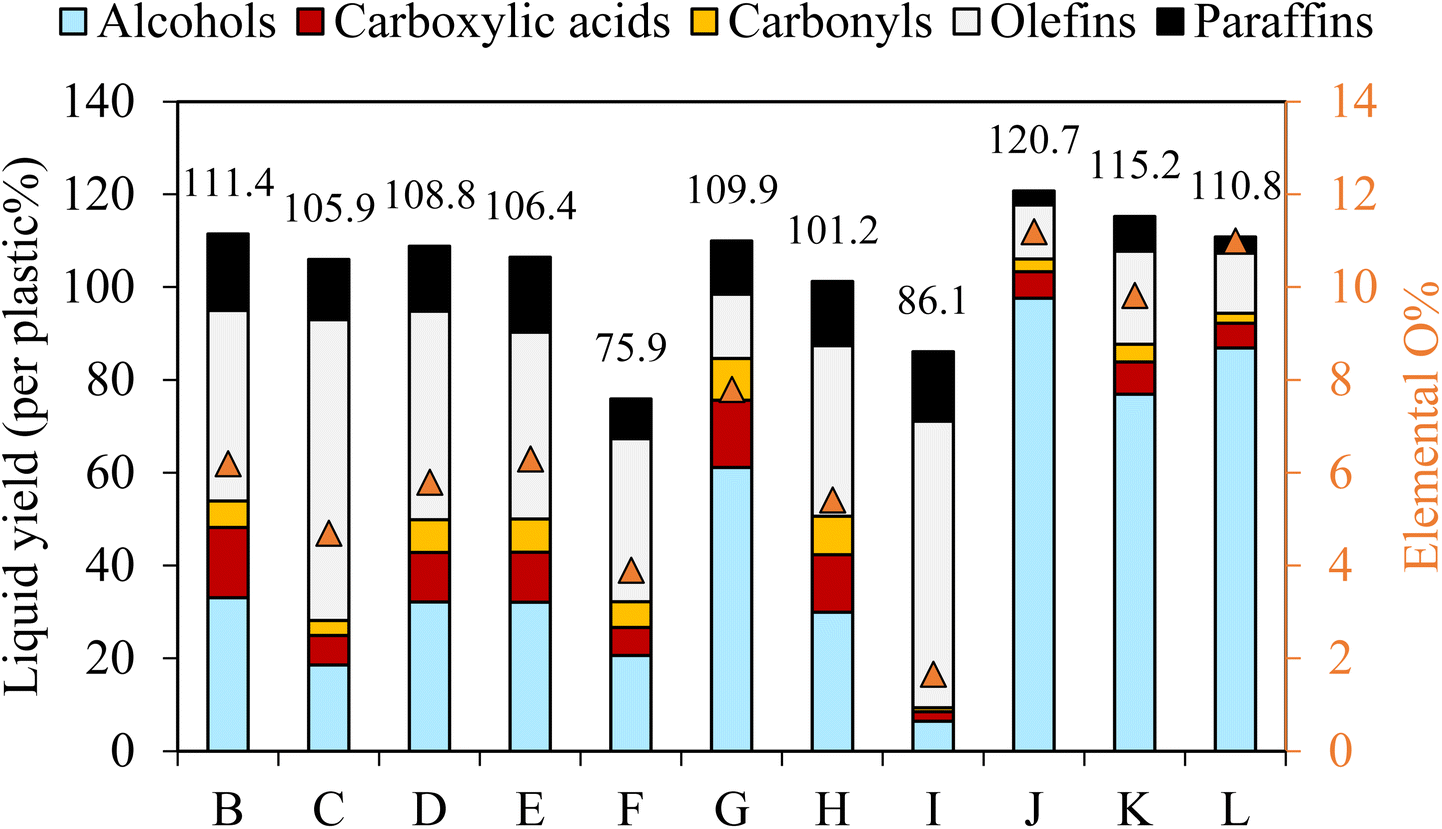 | ||
| Fig. 3 Liquid product compositions and oxygen content for PE conversion using different reaction conditions. | ||
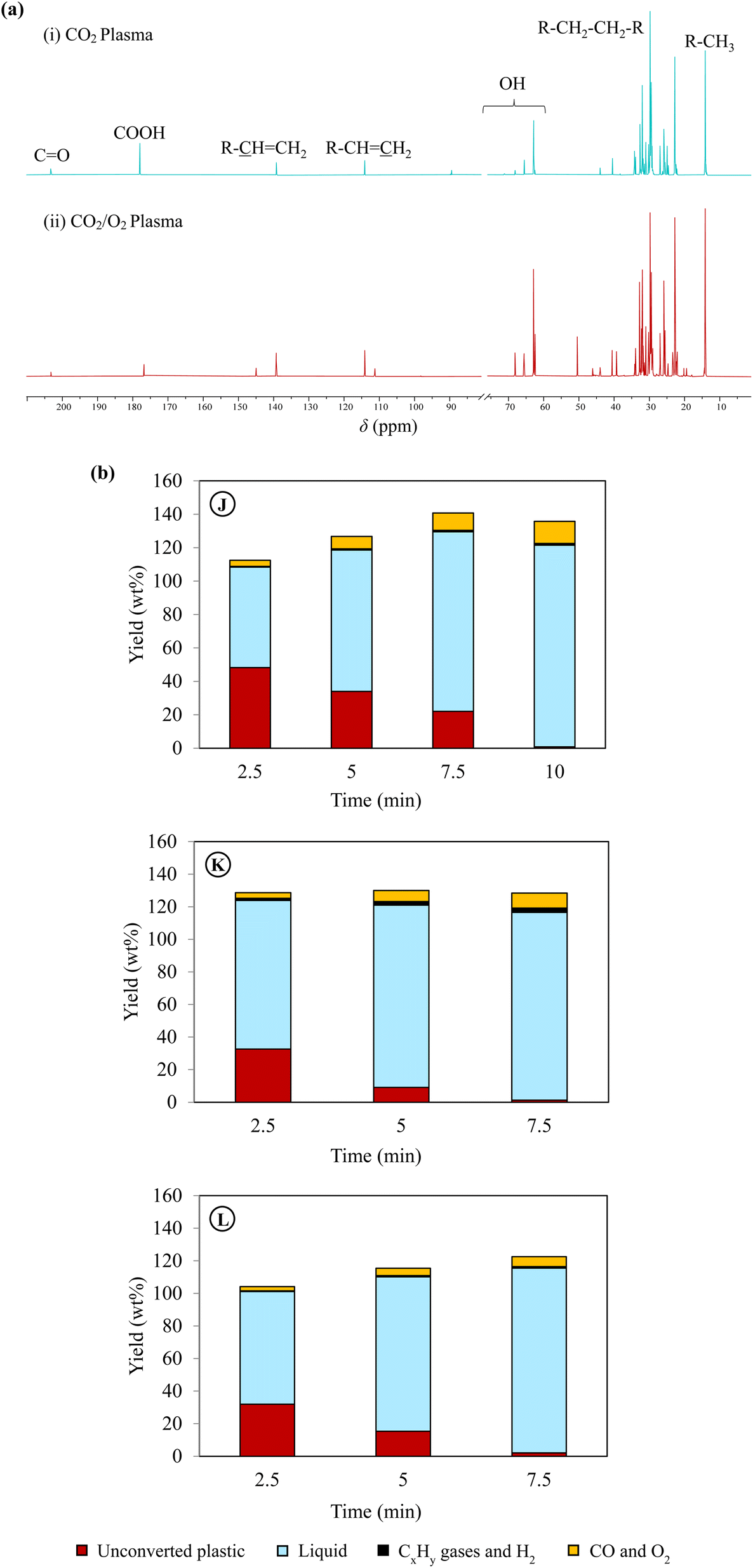 | ||
| Fig. 4 Effect of oxygen addition on plasma co-upcycling of plastics and CO2. (a) 13C NMR spectra of liquid products obtained using (i) CO2 plasma (case G), and (ii) CO2/O2 plasma (case J). The chloroform-d (CDCl3) peak between chemical shifts 75 and 80 was hidden to improve the visibility of desired peaks, and (b) time-dependent product distribution during PE conversion using CO2/O2 plasma with different oxygen concentrations and gas residence times. Product characterizations can be found in Table 2 and Fig. 2, 3 for cases J, K and L. | ||
| Condition | Liquid yield (wt%) | Product carbon number distribution (wt%) | Elemental analysis (wt%) | |||||
|---|---|---|---|---|---|---|---|---|
| C5–C12 | C13–C20 | C21–C28 | C28+ | C (%) | H (%) | O (%) | ||
| Products from case A were not analyzed due to low conversion. | ||||||||
| B | 111.4 | 28.2 | 23.4 | 22.6 | 37.2 | 80.9 | 12.9 | 6.2 |
| C | 105.9 | 39.6 | 33.3 | 20.1 | 12.9 | 82.2 | 13.1 | 4.7 |
| D | 108.8 | 22.4 | 21.9 | 21.2 | 43.3 | 81.2 | 13 | 5.8 |
| E | 106.4 | 39.1 | 25.2 | 27.1 | 15.0 | 80.6 | 13.1 | 6.3 |
| F | 80.6 | 15.6 | 13.1 | 12.0 | 39.9 | 82.8 | 13.3 | 3.9 |
| G | 109.9 | 50.1 | 24.4 | 19.2 | 16.2 | 79.6 | 12.6 | 7.8 |
| H | 101.2 | 29.8 | 24.2 | 10.5 | 36.7 | 81.5 | 13.1 | 5.4 |
| I | 86.1 | 18.5 | 16.8 | 43.3 | 7.5 | 84.9 | 13.5 | 1.7 |
| J | 120.7 | 64.4 | 37.1 | 13.2 | 6.0 | 76.7 | 12.1 | 11.2 |
| K | 115.3 | 61.0 | 41.2 | 12.9 | 0.0 | 78.1 | 12.1 | 9.8 |
| L | 110.8 | 56.7 | 29.2 | 16.5 | 8.4 | 76.8 | 12.2 | 11.0 |
| M | 105.8 | 31.5 | 53.8 | 15.2 | 5.2 | 78.8 | 12.5 | 7.5 |
| N | 98.1 | 36.5 | 40.8 | 17.0 | 3.8 | 78.7 | 12.5 | 7.6 |
| O | 113.5 | 76.7 | 22.5 | 6.5 | 7.8 | 76.8 | 12.2 | 11.0 |
| P | 109.5 | 66.8 | 30.5 | 7.6 | 4.7 | 77.2 | 12.3 | 10.5 |
| Q | 104.4 | 61.1 | 23.3 | 11.1 | 8.9 | 80.5 | 12.4 | 7.1 |
| R | 111.1 | 60.1 | 32.7 | 15.6 | 2.6 | 77.0 | 12.1 | 10.9 |
Notably, the oxygen content in liquids was not contributed by water as the amounts of water produced in this work were negligible (<0.7%, Table S3†). The oxygen content of the liquids well corresponded to the abundance of oleochemicals products. Case B resulted in 33% fatty alcohols, 15.2% fatty acids, and 5.7% carbonyls, totaling 53.9% (or 48.4% per liquid) oxygenated compounds. The rest included 41.1% of olefins and 16.5% of paraffins. Increasing voltage in case C reduced the oxygenated products, whereas it increased the hydrocarbons. The frequency effect was less noticeable in case E than the voltage effect. The highest yield of total oxygenated products was obtained in case G with the longest gas residence time, producing 61.1% of fatty alcohols, 14.5% of fatty acids, and 9.0% of carbonyls with a total 84.6% of oxygenated products (or 77.0% per liquid) (Fig. 3). The rest included 13.1% olefins and 11.4% paraffin. As described earlier, the sum yield of H2 and hydrocarbon gases was the lowest in this case because more PE elements are converted to oleochemicals. As predicted from its oxygen content, the liquid produced at case I with the highest initial temperature of 400 °C was mainly hydrocarbons (61.7% olefins and 15% paraffin). The total oxygenated compounds were only 9.3% in this case. In comparison, case H, with the lowest initial temperature of 300 °C, could still produce 50.6% oxygenated compounds, although it took longer to convert PE.
As shown above, the plasma-activated CO2 can effectively depolymerize and oxidize PE to produce oleochemicals, olefins, and paraffin in high product yields. Among the variables, gas residence time and initial reactor temperature had the most pronounced effects on oleochemical selectivity. Case G, with the longest gas residence time, delivered the liquid with the highest oleochemical yields and narrower molecular distribution. As described earlier, the gaseous product with the highest CO selectivity was also observed in this case.
2.2. PE conversion by CO2/O2 plasma
The chemical selectivity of PE conversion by CO2 plasma was further tailored by entraining small amounts of O2 to CO2 at the reactor inlet. Three CO2/O2 plasma conditions were studied by varying gas residence time and O2 concentration (cases J, K and L). The plasma power and the reactor gas temperature for the respective cases are included in Table S1† to compare with the CO2 plasma-base conversion. Employing CO2/O2 plasma reduced plasma power compared to using CO2 plasma, from 98 W in case B (0% O2 and 13 s) to 84 W in case J (8 vol% O2 and 13 s), and 75 W in case K (14 vol% O2 and 13 s), and from 100 W in case G (0% O2 and 20 s) to 77 W in case L (8 vol% O2 and 20 s). On the other hand, it increased the average reactor gas temperature from 339 °C to 344 °C and 370 °C, and from 353 °C to 367 °C in the corresponding cases. The time-resolved product distributions of the CO2/O2 plasma-based conversion reported in Fig. 4(b) show that compared to CO2 plasma, CO2/O2 plasma increased the PE conversion rate and promoted higher liquid yields. The liquid yields per PE mass were 120.7% after 10 min for case J, 113.4% after 7.5 min for case K, and 115.3% after 7.5 min for case L, respectively.The total gas yields per PE mass were 14.2%, 7.0%, and 11.9% for cases of J, K, and L, as also shown in Fig. 4(b). The corresponding gas selectivity and the yields of PE-derived gases are included in Fig. 2 to compare with the CO2 plasma-based cases. The sum yields of H2 and hydrocarbon gases were 1.0%, 0.9%, and 2.6% per PE mass for the three cases, suggesting CO2/O2 plasma can more effectively convert PE into liquid products than CO2 plasma. CO was also the dominant gas with CO2/O2 plasma with the highest selectivity of 93%, as observed in case J.
PE bond cleavages were more intensive with CO2/O2 plasma than CO2 plasma, leading to liquid products with narrower molecular weight distributions of shorter-carbon chain-length compounds (the results included in Table 2). CO2/O2 plasma also resulted in liquids with a higher degree of oxidation. The liquid oxygen content was 11.2% in case J, followed by 11.0% in case K and 9.8% in case J.
The liquid compositions of the three CO2/O2 plasma cases are also included in Fig. 3 to compare with the CO2 plasma-based liquids. The advantages of O2 as a supplementary gas were not only limited to increasing the reaction rate of PE, producing higher yields of liquids with a higher degree of oxidation, and reducing molecular weights of the liquid products. Most importantly, employing CO2/O2 plasma caused a dramatic shift in product selectivity toward fatty alcohols. Case J was the optimal condition, producing the maximum fatty alcohol yield of 97.6% (or 80.9% per liquid). The fatty alcohols produced at this condition consisted of 49.7% of C5–C12 alcohols, 44.1% of C13–C28 alcohols, and 6.2% of C28+ alcohols. Among them, 12.7% were di-alcohols, and the rest were mono-alcohols. While products with other functional groups were reduced significantly, hydrocarbons in the liquid were only C6–C10 paraffin and olefins. The shift in the product functional groups by CO2/O2 plasma was validated in the 13C NMR result of the liquid (Fig. 4(a(ii))). In the NMR spectrum, OH, methyl, and methylene are the dominant peaks in the liquid produced using CO2/O2 plasma, suggesting a prevalence of mono-alcohol structures. The peaks of other oxygen-containing functional groups are minor due to their low concentrations. The 13C NMR results-based quantification of the functional group selectivity (Table S4†), calculated using a method detailed in Section B of the ESI text,† agrees with the GC/MS-based results above, confirming that fatty alcohols were selectively produced by CO2/O2 plasma. Compared to case J, the alcohol yield was 86.9% (or 78.4% per liquid) in case K and 76.9% (66.7% per liquid) in case L. While selective production of fatty alcohols was confirmed in all CO2/O2 plasma cases, olefins and paraffin increased at longer residence time or higher O2 concentrations. The oxygenated products were probably further deoxygenated under such conditions. Noteworthy, fatty alcohols in such high selectivity were not observed when PE was converted using air plasma. Although a high degree of oxygenation was also observed with air plasma, it only produced 51% fatty alcohols (Fig. S4†). Employing air plasma produced a much more complex mixture containing fatty alcohols, fatty acids, carbonyl, acetoxy esters, and ether functional group compounds.
2.3. Effect of PE on CO2 conversion
In this plasma-based system, CO2 was also converted by forming CO and oxidizing PE-derived hydrocarbons to produce oleochemicals. CO2 conversion in the plasma reactor with and without PE for the different reaction conditions is compared in Fig. 5(a) to evaluate the effect of PE presence on CO2 conversion. CO2 conversion ranged from 2.9% to 4.9% when PE was absent, which is similar to CO2 conversion using DBD plasma reactors reported in literature36 (Fig. 5(b)). CO2 conversion was always higher when PE was added (Fig. 5(a)), indicating that interactions between PE and CO2 under plasma discharge can synergistically enhance CO2 conversion. CO2 conversion in case B was 6.3% with PE compared to 3.6% without PE. However, CO2 conversion did not further increase by increasing the voltage in case C or frequency in case E, lowering the extent of synergistic increase in CO2 conversion by PE. On the other hand, increasing the gas retention time could further improve synergy. The highest CO2 conversion of 7.5% was achieved in case G with PE, compared to 4.9% without PE. It also showed that the excessively high initial temperature reduces synergy. CO2 conversions with and without PE were 4.0% and 3.5% for case I with the highest initial temperature of 400 °C. Overall, the synergistic increases of CO2 conversion due to PE were positively correlated to the oxidation degree of the liquids. The most significant increase of CO2 due to PE was observed in case G, which had the longest gas residence time, and delivered the highest yield of oleochemicals. CO2/O2 plasma slightly reduced CO2 conversion compared to CO2 plasma. The CO2 conversion was 5.6% in case J and increased to 7.1% in case L with a longer gas residence time. On the other hand, CO2 conversion was similar in cases J and K for different O2 concentrations.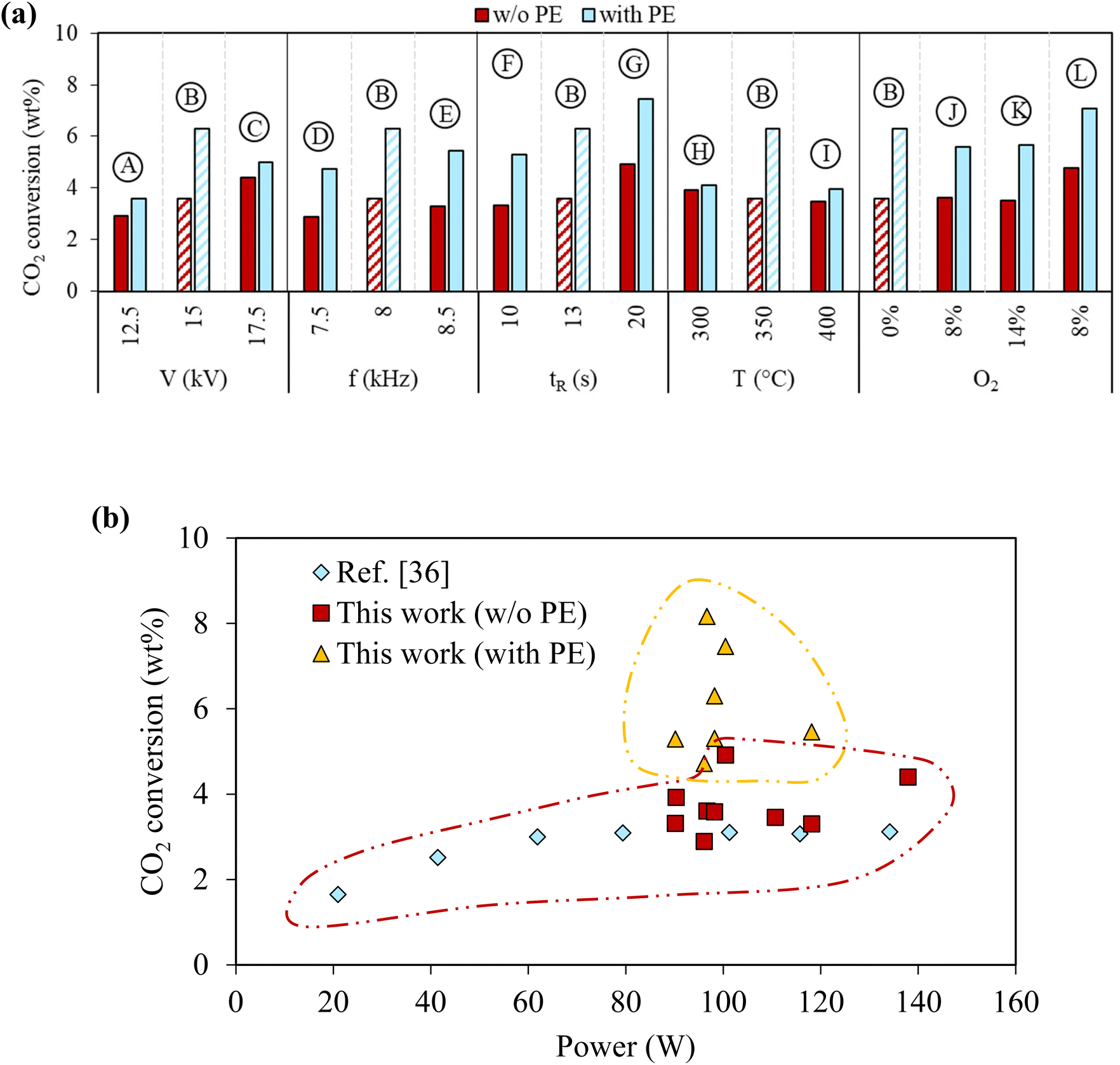 | ||
| Fig. 5 Effect of PE on CO2 conversion. (a) Cumulative CO2 conversion after complete PE conversion under various reaction conditions, and (b) comparison of CO2 conversion from this work with ref. 36 values. | ||
2.4. Mass balance of co-conversion system
PE, CO2, and O2 are all reactants in this plasma-based approach. The mass balances of the most promising conversions were reported: case G for CO2 plasma and case J for CO2/O2 plasma. Mass closures of 96.9% and 98.6% were achieved when all reactants and quantified products were counted (Table S5†). In the case of CO2 plasma, 75.9 g of PE and 24.1 g of CO2 are converted to produce 46.4 g of fatty alcohols, 11 g of fatty acids, 10.5 g of olefins and 8.7 g of paraffin, assuming 100 g of converted feedstock PE and CO2. Additionally, 12.1 g of CO and other minor gas products are produced. With CO2/O2 plasma, 72.6 g of PE, 17.6 g of CO2, and 9.8 g of O2 are converted to yield 70.9 g of fatty alcohols, 4.2 g of fatty acids, 8.5 g of olefins, 2.2 g of paraffin, and 9.6 g of CO. Sankey diagrams of the mass closures can be found in Fig. S6.†2.5. Isotopic study-assisted reaction mechanisms
The plasma-based approach can effectively co-convert PE and CO2 to produce commodity and platform chemicals in which CO2 acts as the robust oxidant and cracking agent to depolymerize PE. In contrast, PE serves as a sink to insert CO2 into its hydrocarbon chain. The formation of oxygenated hydrocarbons as the primary products and synergistic increase of CO2 conversion together indicate strong interactions between PE and CO2 under plasma discharge. When plasma discharge occurs to CO2, electron collision impact generates radicals, ions, and other metastable particles of CO2, CO, O, O2, C, and many more.36 However, the CO2-derived particles can recombine, reducing the overall CO2 conversion.37,38 When PE is exposed to CO2 plasma discharge, energetic electrons and active species of CO2 plasma can attack the PE polymer chain to cleave its C–C and C–H bonds.21,39 Not only does PE depolymerize, but the reactive CO2 plasma species could further bond with the PE-derived hydrocarbons and hydrogen radicals.40 This way, PE can act as a scavenger to inhibit the reverse reactions among CO2 plasma species. The synergistic increase of CO2 conversion by PE was pronounced with the longer gas residence time, implying that the chemical quenching effect was promoted by extending CO2 and PE interactions under plasma discharge. In this work, we also found that doubling PE mass with the same CO2 flow rate could increase CO2 conversion from 6.3% to 8.2% for CO2 plasma for case B, which can be explained by the increased scavenger (i.e., plastic) concentration inside the reactor.Although plasma discharge forms a complex mixture and catalysts are absent in this study, the products displayed a high selectivity. The extensive optimization performed in this study suggests that multiple reactions involving different plasma species can be funneled down to the same type of products when the optimal reaction parameters are chosen. The interactions between CO2 and PE were investigated by converting isotopic 13CO2 and eicosane as a model compound of PE. The CO2-originated C atoms in the compounds were tracked by analyzing the liquid product using GC/MS and comparing the mass-to-charge ratios (m/z) of the compounds resulting from the isotopic test with that of the corresponding standard compounds. Despite 13CO2 being more challenging to dissociate than regular CO2,4113C carbons were successfully identified in the conversion products (see Fig. S7–S11, Tables S6, S7, and the ESI text, Section C† for a discussion of isotopic study).
The reaction mechanisms of PE and CO2 co-conversion are proposed (Fig. 6). Under plasma discharge, electrons and metastable CO2-derived plasma species (e.g., CO2, CO, O, O2, C, C2, and other CxOy) in the gas phase could attract PE to cleave its C–H and C–C bonds, forming the radicals of hydrogen and hydrocarbons (H, CxHy) with reduced chain lengths. The subsequent β-scission of the hydrocarbon radicals leads to alkenes and hydrogen radicals. Alternatively, the hydrocarbon radicals could also be saturated to form alkanes and alkenes by eqn (1) and (2), respectively. From the isotope results, CO2-derived C atoms (up to 3 atoms) presented themselves in alkene products at their chain ends (Fig. S7†). Accordingly, the possible reaction pathway for CO2-originated C atom to enter the alkene products can be a two-step process: (i) formation of CxHy radicals from CO2-originated C and PE-derived H and (ii) further coupling reaction with hydrocarbon radicals (eqn (3)). Carbon coke formation was minor in this work because C further interacted with H or O.
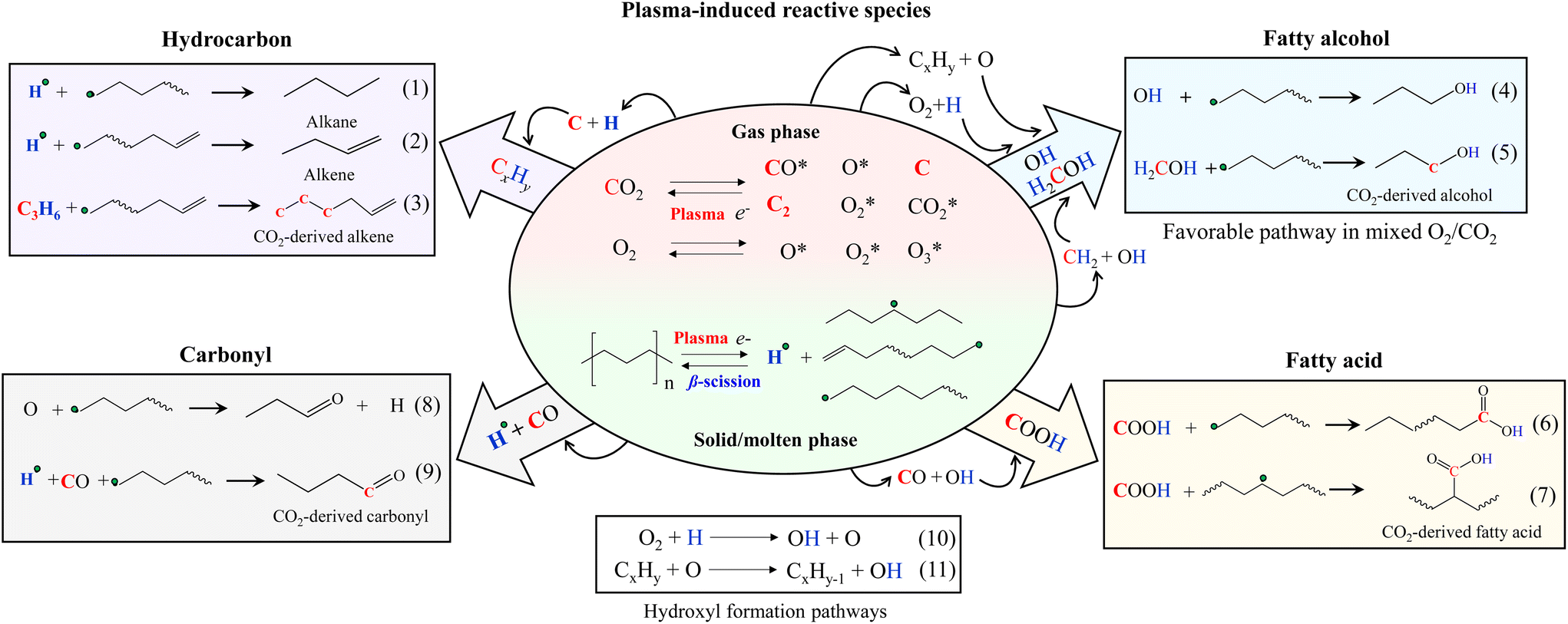 | ||
| Fig. 6 Proposed reaction pathways of plasma-based co-upcycling of polyolefin and CO2 with CO2 or CO2/O2 plasma. | ||
PE-derived H and CO2-derived O could form OH, which can further combine hydrocarbon radicals to produce fatty alcohols (eqn (4)). The CO2-originated C atom linked to the OH in alcohols was also detected in the isotope results (Fig. S8†), suggesting the alcohol products can also form via with H2COH intermediates containing CO2-originated carbon40,42,43 (eqn (5)). Although OH can combine with H to form water, this undesired reaction was not significant because the water content in the liquid products was negligible.
CO and OH can react with hydrocarbon radicals to form fatty acids (eqn (6) and (7)).40,44 The metastable CO2 and H could directly react with the hydrocarbon radicals to form the acids. However, this route has a much higher energy barrier than the previous route.44,45 Carbonyl products are generated from O bonding with the hydrogen-abstracted hydrocarbon radicals (eqn (8)) or carbonylation reactions of CO with hydrocarbon radicals (eqn (9)). Our isotope results confirmed the CO2-originated C atoms in carboxylic (COOH) (Fig. S9, S10 and Table S6†) and carbonyl (CO) groups (Fig. S11 and Table S7†), which were in accordance with the reaction pathways shown in eqn (6) and (9), respectively.
Adding O2 to CO2 can affect the reaction rate, producing shorter carbon-chain molecules and shifting the product selectivity. Compared to CO2, O2 is easier to form plasma discharge to generate electrons and active species of O, O2, and O3.37,38,46 Therefore, introducing a small amount of O2 to CO2 can increase the concentrations of energetic electrons and oxidative species in the system. CO2/O2 plasma resulted in the liquid with a lower carbon number distribution than CO2 plasma, which can be explained by the stronger collision impact of CO2/O2 plasma that cleaves increased numbers of C–C and C–H bonds. Increased C–H bond scissions will also increase hydrogen radicals. Accordingly, the higher O and H concentrations in CO2/O2 plasma discharge (eqn (10) and (11)) compared to CO2 plasma discharge will significantly increase OH formation,46 promoting the alcohol-forming reactions (eqn (4) and (5)) and reducing hydrocarbon products. Fatty acids decreased considerably, possibly due to the reduced availability of the reactive CO intermediate species that suppress the reactions shown in eqn (6) and (7). Notably, CO2 conversion was lower when PE was converted by CO2/O2 plasma than CO2 plasma, suggesting the increased O in the system partially consumes CO2-derived CO species to form CO2. This increased combination reaction due to the added O2 will reduce the PE-induced chemical quenching effect of CO2 compared to PE conversion by CO2 plasma. This statement was also supported in Fig. 5(a) result, as the extent of synergistic increase in CO2 conversion was lower for CO2/O2 plasma. However, fewer CO species and higher OH concentration, paired with the scarcity of unbonded O species, combined with higher reactivity of OH-derived species,47,48 resulted in the selective production of fatty alcohols under CO2/O2 plasma. If excess oxygen is supplied, the overwhelming presence of O species could introduce additional oxidative reactions, as found with air plasma. The enhanced collision impact by higher O2 concentration may also promote secondary cracking reactions of the oxygenated hydrocarbons to increase hydrocarbons. Adding an optimal amount of O2 to CO2 during PE conversion could balance the reactions above, effectively controlling the product selectivity toward fatty alcohols without catalysts.
It should be emphasized that besides plasma gas composition, other reaction conditions also greatly impact reaction selectivity during the plasma-based co-conversion. Although the non-thermal plasma reactions do not need thermodynamic equilibrium, the reactor gas temperatures strongly affect the reaction rate and oleochemical formation. There was an optimal temperature range that could promote the oxidation of PE. Higher degrees of oxidation were usually observed with an intermediate reactor temperature range around 350 °C. Temperatures higher than the optimal temperature increased the PE conversion rate, but it preferentially produced hydrocarbons than oleochemicals. For example, case I (Ti of 400 °C) had the highest average reactor temperature but produced mainly olefins and paraffins with the least oxygenated products. On the other hand, slower PE conversion rates and less oxidized products were observed with the reactor gas temperatures lower than the optimal temperature range. This observation suggests that product selectivity is determined by both chemical reactions and physical phenomena of feedstock and products. The plasma co-conversion of PE by activated CO2 or CO2 and O2 involves multi-phase changes and reactions because the solid plastic changes to the molten/liquid phase and vapor/gas phase. A higher initial temperature or higher electricity voltage can promote strong plasma intensity. Activated CO2 under such conditions can more easily attack PE for chain scissions, producing hydrocarbon molecules with shorter chain lengths. Higher initial temperatures and stronger plasma also raise overall reactor gas temperatures. At higher reactor temperatures, the produced hydrocarbons with smaller molecular weights would have sufficient volatility to evaporate from the molten/liquid phase. Once entrained in the gas stream, the hydrocarbon vapors and CO2-derived species could quickly exit the reactor without sufficiently interacting with each other. This will increase hydrocarbons in the products and reduce oleochemical formation. Higher temperatures can also cause secondary cracking of oleochemicals to hydrocarbons. The reduced interaction between CO2 and hydrocarbons would also hinder the chemical scavenger effect of PE to lower CO2 conversion. On the other hand, lower reactor temperatures correspond to lower plasma intensity, which can reduce CO2 conversion and, therefore, hinder PE depolymerization by activated CO2. Under optimal reactor temperatures, activated CO2 can depolymerize PE but the hydrocarbon products are unlikely to evaporate immediately at the reactor temperature conditions due to low volatilities. Thus, the hydrocarbons in their liquid phase could continue to react with CO2 plasma species inside the reactor to produce oleochemicals. Increasing gas resistance time also significantly improved the oxygenated chemicals. The slower gas flow at the reactor inlet hinders the evaporation rate of the hydrocarbons and extends their residence time inside the reactor. This allows hydrocarbons to interact with CO2-derived species more fully before products are swept away. While CO2 or CO2/O2 plasma must depolymerize the polymer chain before the hydrocarbons are oxidatively functionalized, careful tuning of the reaction conditions and gas composition can balance polymer depolymerization, hydrocarbon volatilization, and the type of oxidation reactions to produce oleochemicals selectively. The chemical reactions and physical phenomena of plastic polymers and CO2 under plasma discharge involving mass transfer in multi-phases warrant further investigation in future studies.
2.6. Conversion of common polyolefins and post-consumer mixed waste PE
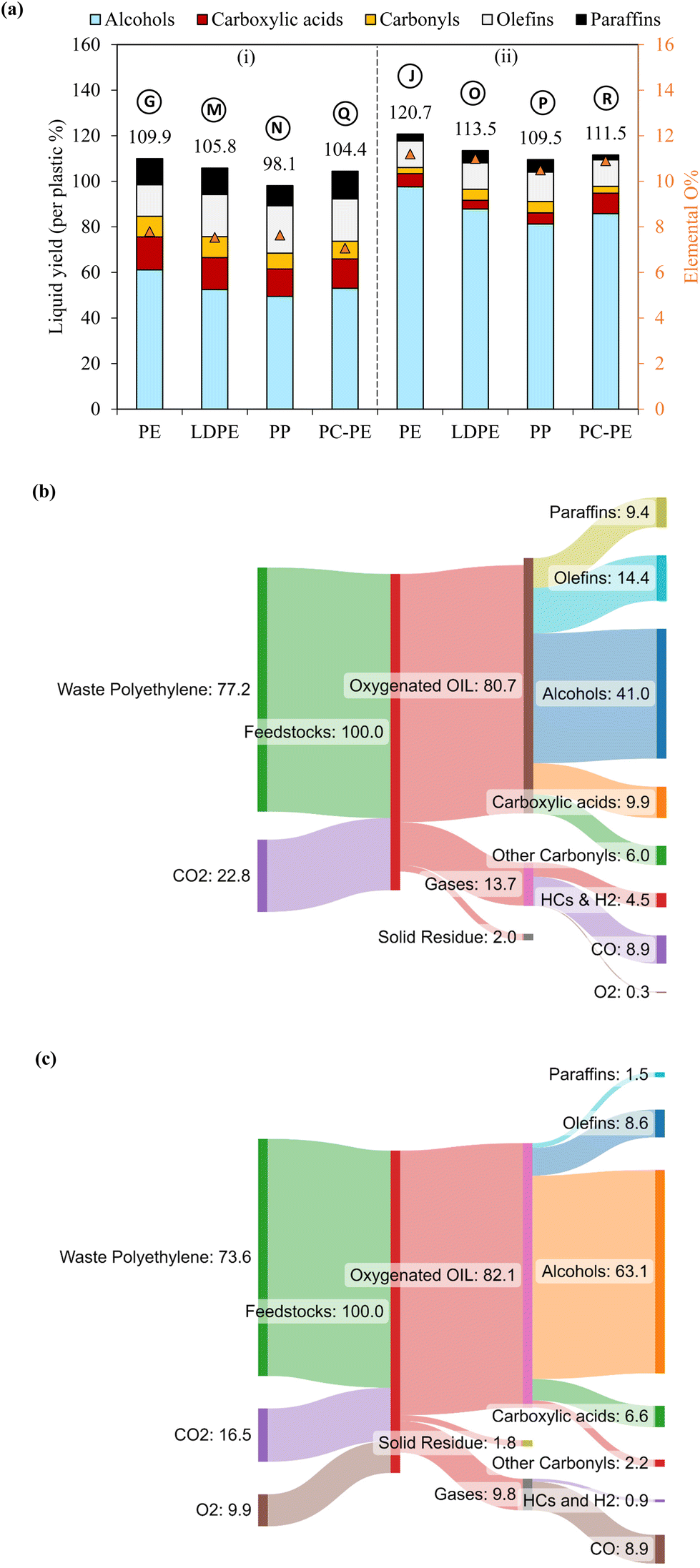 | ||
| Fig. 7 Plasma-based upcycling of common polyolefins and post-consumer mixed PE. (a) Liquid product composition from conversion of PE, LDPE, PP and PC-PE by (i) CO2 and (ii) CO2/O2 plasma, (b) overall mass balance from PC-PE conversion by CO2 plasma, and (c) overall mass balance from PC-PE conversion by CO2/O2 plasma. | ||
Sankey diagrams of the PC-PE conversion systems using CO2 plasma and CO2/O2 plasma are shown in Fig. 7(b), with 96.8% and 93.7% of mass closures (given in Table S7†). In the case of CO2 plasma, 77.2 g of PC-PE and 22.8 g of CO2 are converted to produce 41 g of fatty alcohols, 9.9 g of fatty acids, 6 g of carbonyls, 14.4 g of olefins and 9.4 g of paraffin, assuming 100 g of converted feedstock PC-PE and CO2. Additionally, it produces 8.9 g of CO and 4.5 g of H2 and hydrocarbon gases. When CO2/O2 plasma is employed, 73.6 g of PC-PE, 16.5 g of CO2, and 9.9 g of O2 are converted to produce 63.1 g of fatty alcohols, 6.6 g of fatty acids, 8.6 g of olefins, and 8.9 g of CO gas. These results show the presented approach can be applied to waste polyolefin plastics.
2.7. TEA and LCA
The presented approach can convert waste plastics and CO2 mixtures into highly desired oleochemicals and syngas-like products in a single step without catalysts or other costly co-agents. Notably, current chemical recycling technologies such as pyrolysis of polyolefins only produce low-quality hydrocarbon fuels requiring catalytic upgrading to chemicals.31,49,50 The economic and environmental impact of commercializing the plasma-based conversion of waste plastics and CO2 is evaluated based on the experimental results of PC-PE conversion, specifically, case Q (CO2 plasma) and case R (CO2/O2 plasma). Fig. 8(a) shows a simplified block diagram for a commercial-scale plasma-based waste plastic to fuels and chemicals facility. The plasma decomposition of PC-PE yields light gases and liquid products. The liquid products contain olefins, paraffin, fatty alcohols, fatty acids, and carbonyls, and the gases consist of CO, H2, and C1–C5 hydrocarbons. Multiple fractional distillation and distillation columns were utilized to separate the compounds based on their boiling points, and the gases were combusted to generate power and heat to meet the facility's energy requirements. The material and energy flows of the 200 metric tons per day (MTPD) facility are presented in Fig. S18 and S19.† A 200 MTPD PE waste plasma facility produces 98–142 TPD of fatty alcohols, 24–19 TPD of fatty acids, 37–22 TPD of olefins, 24–4 TPD of paraffin, 15–7 TPD of carbonyls for case Q and case R respectively. The annual processing capacity of the plasma-based waste plastic conversion plant represents 0.29% of the total PE waste produced in the United States in 2019.51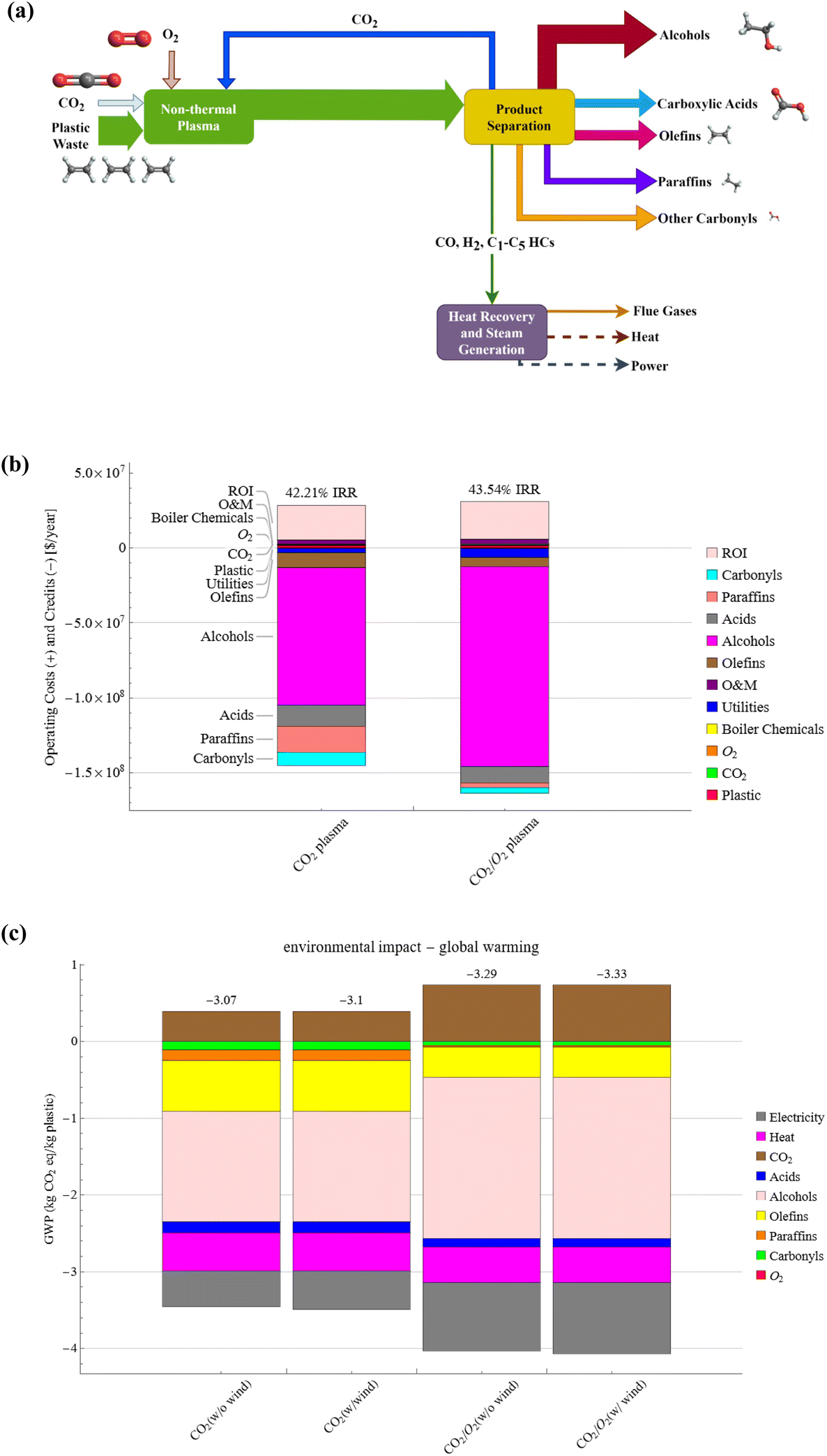 | ||
| Fig. 8 TEA and LCA for the plasma-based conversion process. (a) Simplified block flow diagram for plasma-based plastic waste to fuels and chemicals, (b) operating costs and internal rates of return for a commercial-scale plasma-based waste plastic to fuels and chemicals facility, and (c) global warming potential (GWP) for a commercial-scale plasma-based waste plastic to fuels and chemicals facility using on-site power production or wind electricity. | ||
The facility processes waste plastic collected for $25 per metric ton (MT), including transportation and pretreatment, which is consistent with previous TEA studies on waste PE thermochemical conversion.8,52 The PE waste considered in this study cannot be mechanically recycled and is routinely landfilled. Material recovery facilities in the US collect raw waste with a tipping fee of up to $50 per MT, representing a significant revenue source.53,54 The plastic fraction can be separated and pretreated to remove impurities that impact the conversion performance and quality of the plasma process, which increases the value of the plastic waste to $25 per MT. According to the US Billion Ton report of 2023, the price per MT of CO2 ranges from $30–$200 depending on the source and the purity of the CO2 stream.55 The study assumed a standard cost of $35 per MT of CO2, consistent with prior studies on CO2 utilization processes.56,57 Sensitivity analysis is conducted to evaluate the importance of the plastic and CO2 stream price in the plant's economics. The plasma-based conversion of waste plastic and CO2 generates multiple chemicals, and the on-site combustion of the gases produces surplus power and heat that are sold to the grid. The average market price of the products was obtained from online databases and documented in Table S10.†27,28,58–60 The average price of fatty alcohol and fatty acids is $2.82 per kg and $1.79 per kg, respectively, and the average price of the other chemicals ranges from $0.83 to $2.19 per kg.
The estimated total installed equipment cost (TIC) breakdown for a 200 MT per day plastic waste facility is presented in Fig. S20.† The TIC for PC-PE/CO2 plasma and PC-PE/CO2/O2 plasma is $52.7 million and $56.9 million, respectively. At a Lang factor of 5, the capital cost ranges from $188–202 million. The plasma reactor and heat recovery and steam generation (HRSG) systems are the most expensive pieces of equipment, representing over 85% of TIC in both cases.
The internal rate of return (IRR) and net present value (NPV) were determined from the discounted cash flow analysis to assess the economic performance of the facilities. The financial parameters used in the TEA are presented in Table S9.† The cash flow result is shown in Fig. 8(b). The IRR and NPV are 42.2% and 739.6 $MM, respectively, for the CO2 plasma-based facility, and 43.5% and 854.2 $MM for the CO2/O2 plasma-based facility. The latter has a higher IRR because of the high alcohol yield, generating considerable revenue.
The minimum selling price (MSP) is not a primary economic indicator in this study, but it was calculated to provide a comparison of the chemical products of this process with current petroleum processes. The MSP of the product with the highest yield in each scenario was estimated, and the sale of the co-products contributed to the revenue in estimating the MSP. The MSP of the primary product was calculated from the discounted cash flow, assuming an internal rate of return of 10% over a 20-year lifespan of the plant. In both cases, fatty alcohols were selected as the primary product. The CO2 plasma-based approach has an MSP of $-0.59 per kg, while the CO2/O2 plasma-based approach has an MSP of $0.14 per kg for fatty alcohols. The MSP calculated for both cases is significantly lower than the market price of fatty alcohols ($2.82 per kg). The negative MSP for the CO2 plasma-based facility indicates that the other chemical products (acids, paraffin, olefins and carbonyls) generate sufficient revenue for the NPV to remain positive. Fig. S21† displays the contributions of the co-product revenues and operating expenses on the MSP of fatty alcohol for each scenario.
Sensitivity analysis was carried out to assess the effect of different process variables on the net present value in each scenario. The profitability of the plant heavily depends on the product's sales, which are solely determined by market dynamics. Throughout a plant's lifespan, economic conditions can vary. Sensitivity was determined in a ±20% variation in the baseline cost of capital investment, feedstock prices, plasma energy, and product prices, as shown in Fig. S22.† The sensitivity results indicate that the capital cost and fatty alcohol price are the most significant determinants of the NPV. The plastic price, CO2 price, and plasma energy consumption have a limited impact on the NPV.
This study utilized LCA to evaluate the environmental impact of producing chemicals from the non-thermal plasma deconstruction of plastic waste. As mentioned above, electricity and heat are generated from the combustion of gases and heavy compounds to cover the plant's energy requirement, and the surplus is injected into the grid, which accounts for displacement credits. In the analysis, we consider additional scenarios where the facility energy requirements are supplied by wind energy, and the total on-site energy generation displaces grid power. Furthermore, we assumed all the products substitute similar products from fossil-based processes, resulting in credits earned. The life cycle inventory is available in Table S11.†
LCA results are presented in Fig. 8(c) and show that the process has net negative global warming potentials (GWP) of −3.07 and −3.29 kg CO2e per kg of plastic for PC-PE/CO2 plasma and PC-PE/CO2/O2 plasma, respectively. The GWP of the scenarios with wind energy is slightly lower. The negative emissions indicate that the process results in an overall reduction of emissions in the carbon economy. This occurs because of the displacement of sourced fossil chemicals and fuels. The main positive contributor that increases GWP in the process is the carbon emissions produced from the on-site combustion of flue gases in the power generation unit. However, the product credits from chemicals, fuels, excess heat, and power exceed the emissions. Sensitivity analysis indicates that fatty alcohol, olefins, process heat, and electricity impact GWP most, and the results are shown in Fig. S23.† Carbonyls and paraffin have a relatively low impact on the GWP due to their low yields. Table S12† shows the results for all TRACI environmental impact categories, including eutrophication, ecotoxicity, and others.
3. Conclusion
The study effectively demonstrates the potential of non-thermal plasma-activated CO2/O2 technology for upcycling plastic waste into oleochemicals. During their plasma-based process, CO2 and plastic conversions were synergistically enhanced. Using O2 as a supplementary gas was a highly effective and simple approach to tailor chemical selectivity toward fatty alcohols and increase product yield.The 200 MTPD waste plastic facility produces 32524 MT and 47286 MT per year of fatty alcohol for the CO2 and CO2/O2 plasma-based facility, respectively. This represents ∼2.4% of the global market demand for fatty alcohol in 2019.61 TEA results indicate a promising IRR for the proposed process, underlining its economic viability as an alternative to conventional recycling and waste management methods. The analysis further suggests a negative GWP due to the avoided emissions of the various products. This GWP compares favorably to existing waste management practices, emphasizing the environmental benefits of this innovative approach. Our evaluation across different scenarios, varying in feedstock compositions, reaffirmed the robustness and flexibility of the plasma-based technology. The sensitivity analysis further highlighted the significant impact of the fatty alcohol yield, which not only impacts the process economics but also the GHG emissions. The viability of this technology highly depends on the yield of the products, particularly fatty alcohol. Therefore, enhancing the process selectivity of alcohols will significantly increase the economic and environmental benefits of plasma-based technology.
The research opens avenues for future exploration, notably in optimizing process conditions, scaling up the technology for industrial application, and exploring the integration of renewable energy sources to reduce the GWP further. Additionally, investigating the feasibility of other value-added products from plastic waste using non-thermal plasma could expand the economic and environmental benefits of this promising technology.
Future work should also focus on detailed environmental impact assessments, exploring partnerships with waste management and recycling industries, and conducting pilot studies to validate the TEA presented. This would provide a comprehensive understanding of the technology's practical implications and facilitate the global transition towards more sustainable waste management practices.
4. Experimental
4.1. Materials
High-density polyethylene (PE or HDPE; Mw ∼ 60000 Da) and polypropylene (PP; Mw ∼ 200000 Da) were purchased from Yangli Tech Company (China), and low-density (LDPE; Mw ∼ 80000 Da) were procured from DOW Chemicals. The colored and mixed post-consumer polyethylene (PC-PE; ∼75000 Da) was collected from material recovery facilities. All HPLC-grade solvents (dichloromethane, toluene, pyridine, and tetrahydrofuran) were purchased from Fisher Scientific. High-purity GC carrier gases were purchased from Airgas. The silylation agent (N,O-bis(trimethylsilyl)trifluoroacetamide with trimethylchlorosilane) for identification of carboxylic acid and alcohols compounds, and NMR relaxation agent (chromium(III) acetylacetonate) were supplied by Sigma Aldrich. High-purity standard gases (CO, CO2, H2, O2) and light hydrocarbon gases were purchased from Praxair. Isotopic 13CO2 was supplied by Cambridge Isotope Laboratories, Inc. The standard chemicals of alkanes (C6–C40), alkenes (C5–C23), dienes (C6–C14), alcohols (C6–C30), carboxylic acids (C6–C24) and carbonyl (C6–C18) compounds used for the GCMS calibration were purchased from Fischer Scientific, Thermo Scientific, TCI America, and Sigma Aldrich. A detailed list of the standard chemicals and their sources is provided in Section A of the ESI text.†
4.2. Plasma-based plastics conversion process
Fig. S1† shows a schematic diagram of the plasma reactor and product recovery system. The experiments were conducted in a co-axial dielectric barrier discharge (DBD) reactor made of quartz. A tungsten rod was inserted as a high-voltage electrode. The outer surface of the reactor was covered by a copper sheet, which acted as a grounded electrode, and was insulated. The high-voltage electrode was connected to a high-voltage AC power supply (Nanjing Suman Company, CTP-2000 K), with a maximum peak-to-peak sinusoidal voltage of 30 kV and a center frequency of 10 kHz. Electric voltage and current were measured using a high-voltage probe (i.e., P6015A from Tektronix) and a high-response current probe (Pearson Electronics, Inc., Pearson 2877) equipped with an oscilloscope (Tektronix MDO3102 mixed domain). About 0.15 g of plastics was placed inside the plasma reactor. Reaction gases entered the reactor at the inlet. The volume flow rates were controlled by volumetric flowmeters, calibrated prior to the experiments by a high-accuracy universal flow controller (Agilent, model ADM G6691) with an accuracy of ±0.2 mL min−1. The gas flow rate was between 32.5 and 65 mL min−1. When external heating was required, the reactor was heated by a coil heater.Initially, the reactor was heated externally until the internal reactor gas temperature reached the preset temperatures. Later, the heater was turned off before the plasma generator was turned on. The reaction was carried out under atmospheric pressure, and reaction time was calculated from when the plasma generator was turned on. The vapors and gases leaving the reactor at the other end were passed through a two-stage condenser cooled with methanol–dry ice mixtures to collect liquids and non-condensable gases were sent to an online micro-GC for gas analysis. During the reaction, the reactor outlet gas flow was continuously measured downstream of the condenser using the high-accuracy universal gas flowmeter. The reactor gas temperature was measured, and the recorded current and voltage were used to determine plasma power. The masses of the solid residues and liquids were determined by weighting the reactor and the condenser before and after the conversion using an analytical balance with an accuracy of 0.0001 g (Veritas, M124AS).
For converting eicosane using isotopic 13CO2 plasma, the experiment was carried out using a DBD plasma reactor. The reactor had similar dimensions to the above-mentioned plasma reactor, except it had inlet and exit valves. Plasma discharge was generated in the reactor with closed values containing about 0.15 g of eicosane and 13CO2 gas. After conversion, the products were collected from the reactor by dissolving in a mixture of toluene and pyridine solvents (2.5/1.5, v/v). Eicosane was also converted in the same reactor using regular CO2 as plasma gas to collect products, aiding product identification during the isotopic tests.
Definitions for product yield, selectivity, and energy efficiency
Liquid or solid yields based on the initial plastic mass are calculated as shown below: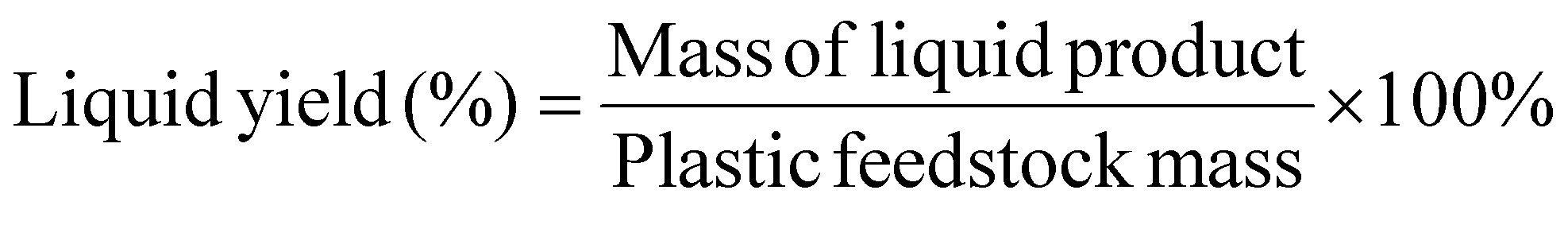
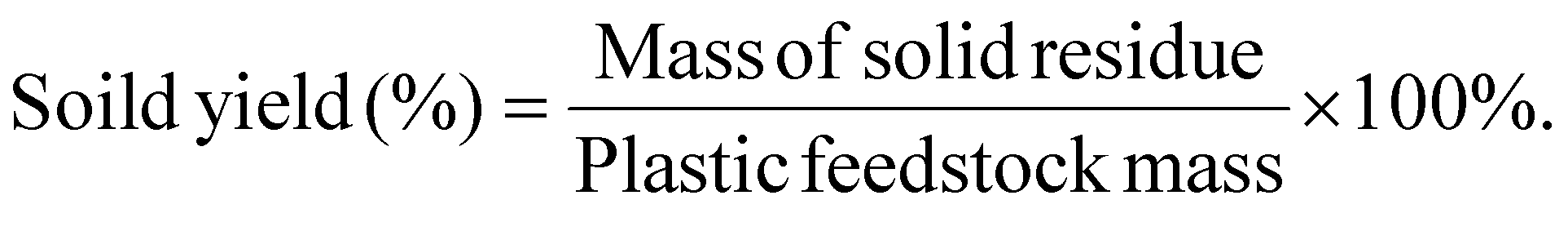
The mass yield of an individual gas compound per plastics are calculated as:
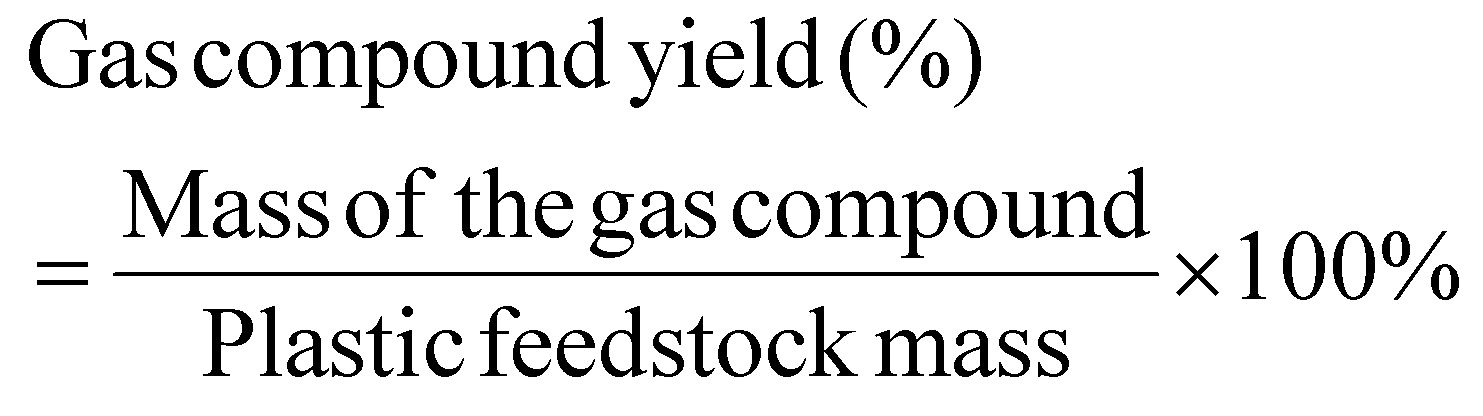
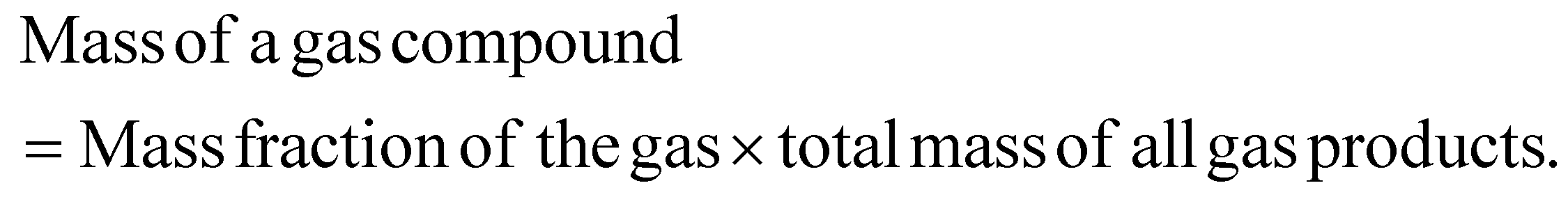
The mass selectivity of individual gas compound among the total gas product was calculated as:
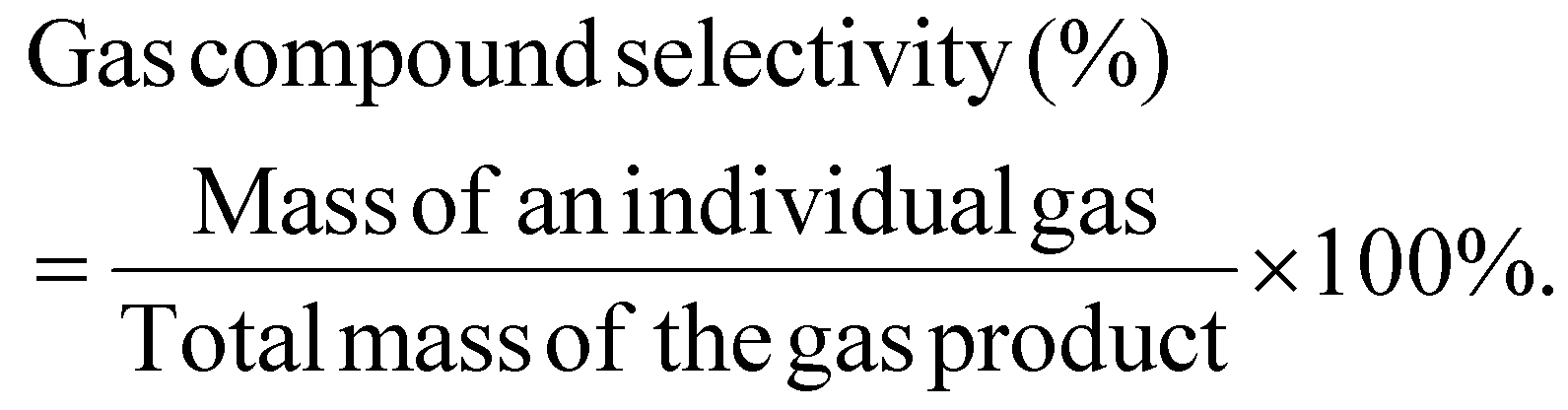
The yield of an individual liquid compound per initial plastic mass is calculated as:
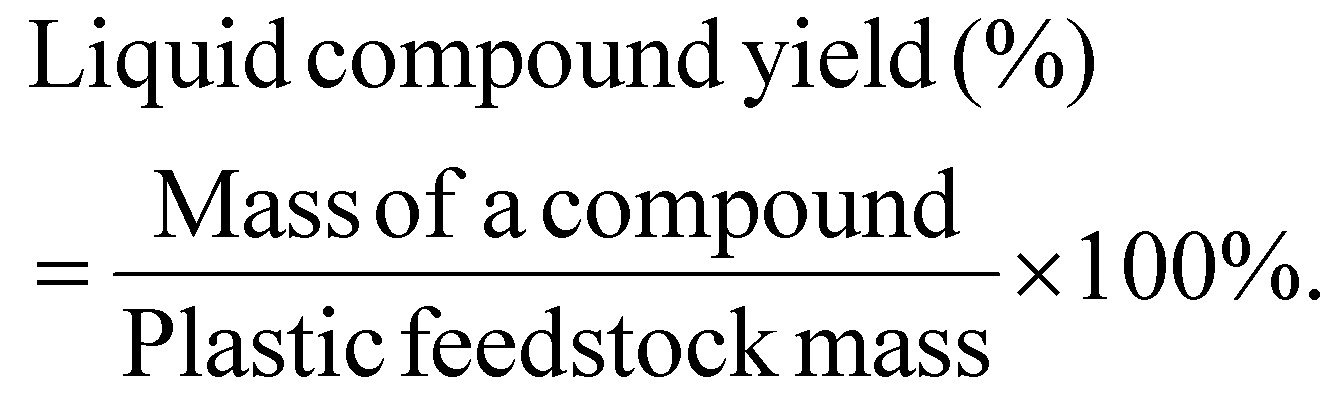
The mass selectivity of a compound with a functional group in the liquid is calculated as:
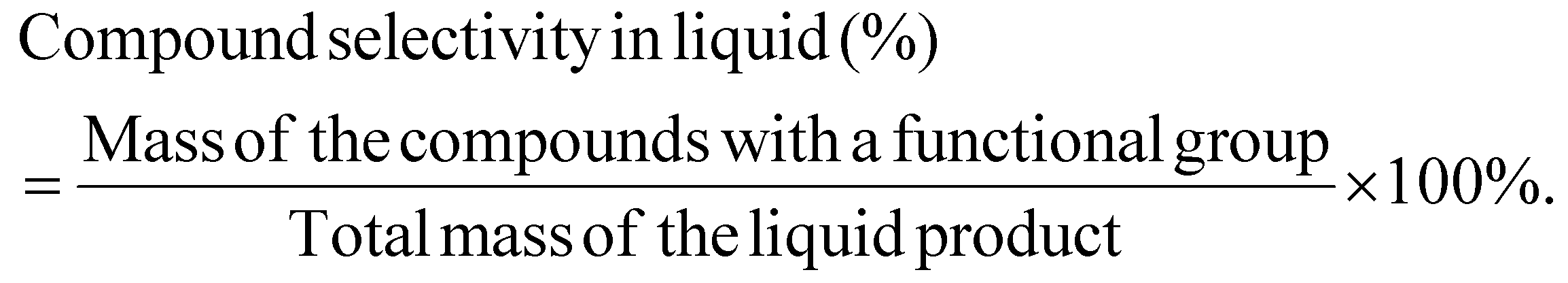
CO2 conversion is calculated as:
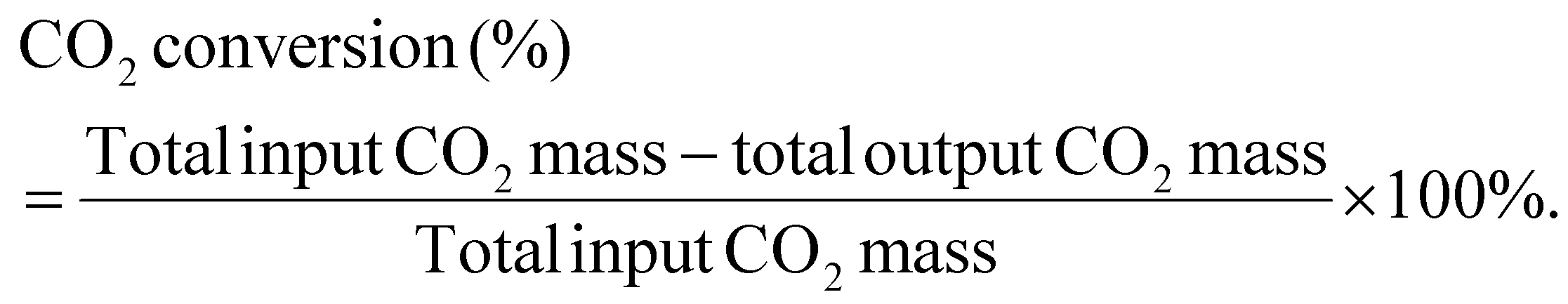
For mass balance of the conversion system including all reactants, the calculations are given below:
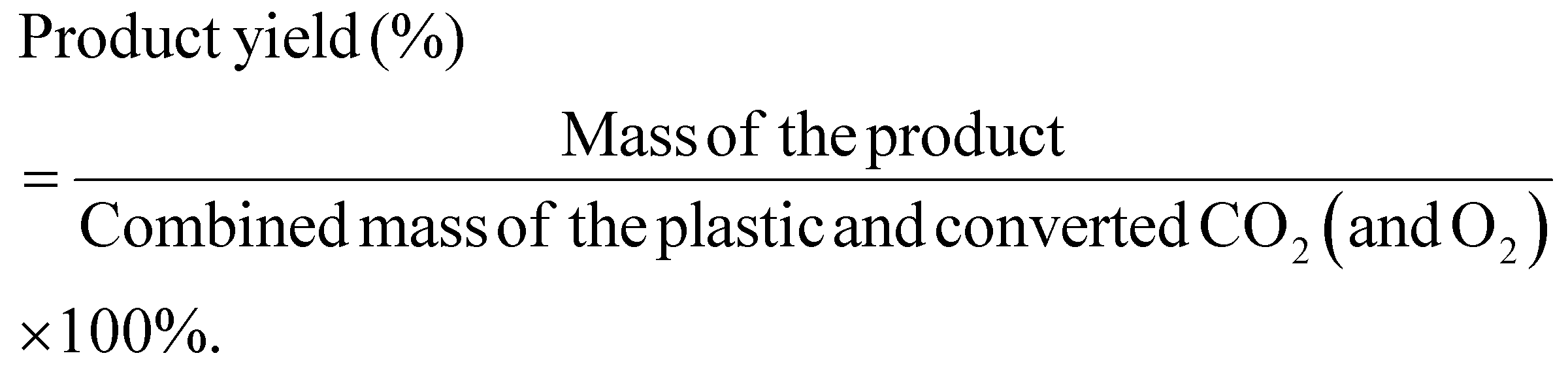
4.3. Characterization methods
:1. The temperature of the FID detector was set at 375 °C. Agilent MassHunter software was used to process the GC chromatograms and measure peak areas. The compounds in the liquid products were identified using a combination of tools, including the NIST MS spectral and mass ion database. High-purity standards of alkane, alkene, alcohol, diol, carboxylic acid, and aldehyde were injected into the GC to aid MS identification. Five different concentrations of the alkane standards were injected to calibrate the Polyarc-FID for liquid product quantification. Since the Polyarc-FID calibration is based on carbon response, the calibration factor from a particular carbon number of alkane can be used for any compound containing the same number of carbons. The resultant calibration curves had regression coefficients higher than 0.99. Unless specified otherwise, the mass selectivity and yields of individual liquid compounds or a specific functional group in the liquid product are from GC analysis.
Individual products up to C28 carbon number could be quantified using the Polyarc-FID due to their good peak separations in the MS chromatograms. In the >C28 compound region, co-elution of different class compound peaks was noticed in some liquid samples for higher molecular weight products. In these limited cases, the functional group selectivity of compounds up to C28 was considered for the entire liquid product.
4.4. TEA
TEA was conducted to evaluate the commercialization potential of a plasma-based plastic waste to chemicals and fuel facility. The facility capacity was modeled as a 200 MT per day unit capable of processing plastic waste streams from multiple material recovery facilities.53 The TEA includes a detailed process design described in Fig. S15.† Rigorous material and energy balance calculations were conducted using the US DOE-funded BioSTEAM chemical process modeling package.65 Equipment costs and financial assumptions are based on the NREL report by Humbird et al.66 Material and utility costs are based on historical market price data. NPV and IRR were calculated based on a 20-year project lifetime. The process generates multiple products, so the primary comparison metric is the IRR instead of the MSP. However, the MSP of the product with the highest yield in the process was calculated. More information on the TEA methodology is provided in Section D of the ESI text.†4.5. LCA
LCA was developed to estimate the GWP and other environmental impact factors of the process. The LCA was performed in OpenLCA 1.11.0.67 The Ecoinvent version 3.7 database and the Tool for Reduction and Assessment of Chemicals and Other Environmental Impacts (TRACI) were used to calculate the ecotoxicity, acidification, eutrophication, global warming, ozone depletion, photochemical oxidation, and carcinogenic impacts of the process. The impact allocation methodology is the system expansion or displacement method. The functional unit is a kg of waste plastic. The system boundary is described in Fig. S17,† and consists of a cradle-to-factory gate recycling analysis. Plastic is received as a material with no environmental burden. The TEA and LCA analyses include sensitivity analysis of key technical, economic, and environmental impact parameters. More information on the lifecycle assessment methodology is provided in Section D of the ESI text.†Data availability
All data needed to evaluate this work are available in the main manuscript and/or the ESI.†Conflicts of interest
X. B., L. A, and H. R. have filed US Patent Application 18/451,482 with the support of the Iowa State University Research Foundation. All other authors declare no competing interests.Acknowledgements
This research is supported by the United States Department of Energy's Office of Energy Efficiency and Renewable Energy under contract no. DE-EE0009943. The authors greatly acknowledge Drs. Keith Vorst and Victor Sanfins Cecon for providing waste plastic material used in this work, and the Bioeconomy Institute of Iowa State University for providing analytical instrument support.References
- H. Li, H. A. Aguirre-Villegas and R. D. Allen, Expanding plastics recycling technologies: chemical aspects, technology status and challenges, Green Chem., 2022, 24, 8899–9002 RSC.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- Statista, Plastic Production Worldwide, 2021, available at: https://www.statista.com/statistics/282732/global-production-of-plastics-since-1950/, accessed October 20, 2023.
- Y. Chen, A. K. Awasthi, F. Wei, Q. Tan and J. Li, Single-use plastics: Production, usage, disposal, and adverse impacts, Sci. Total Environ., 2021, 752, 141772 CrossRef CAS PubMed.
- U.S. Environmental Protection Agency, Plastics: Material-Specific Data, available at: https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/plastics-material-specific-data, accessed October 20, 2023.
- X. Zhao, M. Korey and K. Li, Plastic waste upcycling toward a circular economy, Chem. Eng. J., 2022, 428, 131928 CrossRef CAS.
- P. J. Woolcock and R. C. Brown, A review of cleaning technologies for biomass-derived syngas, Biomass Bioenergy, 2013, 52, 54–84 CrossRef CAS.
- O. Olafasakin, J. Ma, V. Zavala, R. C. Brown, G. W. Huber and M. Mba-Wright, Comparative Techno-economic Analysis and Life Cycle Assessment of Producing High-Value Chemicals and Fuels from Waste Plastic via Conventional Pyrolysis and Thermal Oxo-degradation, Energy Fuels, 2023, 37, 15832–15842 CrossRef CAS.
- H. M. Nguyen and M. L. Carreon, Non-thermal Plasma-Assisted Deconstruction of High-Density Polyethylene to Hydrogen and Light Hydrocarbons over Hollow ZSM-5 Microspheres, ACS Sustainable Chem. Eng., 2022, 10, 9480–9491 CrossRef CAS.
- A. Bogaerts and E. C. Neyts, Plasma Technology: An Emerging Technology for Energy Storage, ACS Energy Lett., 2018, 3, 1013–1027 CrossRef CAS.
- B. Tabu, K. Akers and P. Yu, Non-thermal atmospheric plasma reactors for hydrogen production from low-density polyethylene, Int. J. Hydrogen Energy, 2022, 47, 39743–39757 CrossRef CAS.
- C. Ducharme, N. J. Themelis and M. J. Castaldi, Technical and Economic Analysis of Plasma-assisted Waste-to-Energy processes, 2010 Search PubMed.
- T. Maczka, M. Wnukowski, E. Sliwka and L. Niedzwiecki, Pilot Installation for the Thermal Plasma Treatment of Plastic Wastes, 2013 Search PubMed.
- S. Kwon and S. k. Im, Feasibility of non-thermal plasma gasification for a waste-to-energy power plant, Energy Convers. Manage., 2022, 251, 114978 CrossRef.
- A. Lusi, H. Hu and X. Bai, Producing high yield of levoglucosan by pyrolyzing nonthermal plasma-pretreated cellulose, Green Chem., 2020, 22, 2036–2048 RSC.
- A. Lusi, H. Radhakrishnan, H. Hu and X. Bai, Plasma electrolysis of cellulose in polar aprotic solvents for production of levoglucosenone, Green Chem., 2020, 22, 7871–7883 RSC.
- A. Lusi, H. Radhakrishnan, H. Hu and X. Bai, One-pot production of oxygenated monomers and selectively oxidized lignin from biomass based on plasma electrolysis, Green Chem., 2021, 23, 9109–9125 RSC.
- I. Aminu, M. A. Nahil and P. T. Williams, Hydrogen from Waste Plastics by Two-Stage Pyrolysis/Low-Temperature Plasma Catalytic Processing, Energy Fuels, 2020, 34, 11679–11689 CrossRef CAS.
- L. Yao, J. King, D. Wu, S. S. C. Chuang and Z. Peng, Non-thermal plasma-assisted hydrogenolysis of polyethylene to light hydrocarbons, Catal. Commun., 2021, 150, 106274 CrossRef CAS.
- I. Aminu, M. A. Nahil and P. T. Williams, Hydrogen Production by Pyrolysis–Nonthermal Plasma/Catalytic Reforming of Waste Plastic over Different Catalyst Support Materials, Energy Fuels, 2022, 36, 3788–3801 CrossRef CAS.
- L. S. Diaz-Silvarrey, K. Zhang and A. N. Phan, Monomer recovery through advanced pyrolysis of waste high density polyethylene (HDPE), Green Chem., 2018, 20, 1813–1823 RSC.
- A. George, B. Shen and M. Craven, A Review of Non-Thermal Plasma Technology: A novel solution for CO2 conversion and utilization, Renewable Sustainable Energy Rev., 2021, 135, 109702 CrossRef CAS.
- Grand View Research, Oleochemicals Market Size, available at: https://www.grandviewresearch.com/industry-analysis/oleochemicals-industry, accessed November 10, 2023.
- P. Munkajohnpong, C. Kesornpun, S. Buttranon, J. Jaroensuk, N. Weeranoppanant and P. Chaiyen, Fatty alcohol production: an opportunity of bioprocess, Biofuels, Bioprod. Biorefin., 2020, 14, 986–1009 CrossRef CAS.
- H. A. Wittcoff, B. G. Reuben and J. S. Plotkin, Industrial Organic Chemicals, John Wiley & Sons, 2012 Search PubMed.
- J. Salimon, N. Salih and E. Yousif, Industrial development and applications of plant oils and their biobased oleochemicals, Arabian J. Chem., 2012, 5, 135–145 CrossRef CAS.
- ChemAnalyst, Fatty Alcohol Price Trend and Forecast, available at: https://www.chemanalyst.com/Pricing-data/fatty-alcohol-1084, accessed November 10, 2023.
- ChemAnalyst, Fatty Acid Price Trend and Forecast, available at: https://www.chemanalyst.com/Pricing-data/fatty-acid-1257, accessed November 10, 2023.
- J. Shah, E. Arslan, J. Cirucci, J. O'Brien and D. Moss, Comparison of Oleo- vs Petro-Sourcing of Fatty Alcohols via Cradle-to-Gate Life Cycle Assessment, J. Surfactants Deterg., 2016, 19, 1333–1351 CrossRef CAS PubMed.
- C. Liang, U. R. Gracida-Alvarez, T. R. Hawkins and J. B. Dunn, Lifecycle Assessment of Biochemicals with Clear Near-Term Market Potential, ACS Sustainable Chem. Eng., 2023, 11, 2773–2783 CrossRef CAS.
- H. Li, J. Wu and Z. Jiang, Hydroformylation of pyrolysis oils to aldehydes and alcohols from polyolefin waste, Science, 2023, 381, 660–666 CrossRef CAS PubMed.
- Y. Zhang, Z. Fu, W. Wang, G. Ji, M. Zhao and A. Li, Kinetics, Product Evolution, and Mechanism for the Pyrolysis of Typical Plastic Waste, ACS Sustainable Chem. Eng., 2022, 10, 91–103 CrossRef CAS.
- A. Aboulkas, K. El Harfi and A. El Bouadili, Thermal degradation behaviors of polyethylene and polypropylene. Part I: Pyrolysis kinetics and mechanisms, Energy Convers. Manage., 2010, 51, 1363–1369 CrossRef CAS.
- C. Abdy, Y. Zhang, J. Wang, Y. Yang, I. Artamendi and B. Allen, Pyrolysis of polyolefin plastic waste and potential applications in asphalt road construction: A technical review, Resour., Conserv. Recycl., 2022, 180, 106213 CrossRef CAS.
- D. Choi, S. Jung, S. Lee, K. A. Lin, Y. Park, H. Kim, Y. Tsang and E. Kwon, Leveraging carbon dioxide to control the H2/CO ratio in catalytic pyrolysis of fishing net waste, Renewable Sustainable Energy Rev., 2021, 138, 110559 CrossRef CAS.
- R. Aerts, W. Somers and A. Bogaerts, Carbon Dioxide Splitting in a Dielectric Barrier Discharge Plasma: A Combined Experimental and Computational Study, ChemSusChem, 2015, 4, 702–716 CrossRef PubMed.
- K. Zhang and A. P. Harvey, CO2 decomposition to CO in the presence of up to 50% O2 using a non-thermal plasma at atmospheric temperature and pressure, Chem. Eng. J., 2021, 405, 126625 CrossRef CAS.
- C. Fromentin, T. Silva, T. Dias, A. Morillo-Candas, O. Biondo, O. Guaitella and V. Guerra, Study of vibrational kinetics of CO2 and CO in CO2–O2 plasmas under non-equilibrium conditions, Plasma Sources Sci. Technol., 2023, 32, 024001 CrossRef.
- H. Kang, Y.-N. Kim, H. Song, H. Lee, K.-T. Kim, Y.-H. Song, D. H. Lee and O. L. Li, Feasibility test of a concurrent process for CO2 reduction and plastic upcycling based on CO2 plasma jet, J. CO2 Util., 2021, 52, 101701 CrossRef CAS.
- L. M. Martini, G. Dilecce, G. Guella, A. Maranzana, G. Tonachini and P. Tosi, Oxidation of CH4 by CO2 in a dielectric barrier discharge, Chem. Phys. Lett., 2014, 593, 55–60 CrossRef CAS.
- M. Zeng, Z. Kan, Z. Wang and M. Shen, Carbon isotope effects in the artificial photosynthesis reactions catalyzed by nanostructured Co/CoO, Chem. Phys. Lett., 2020, 754, 137731 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- L. Wang, Y. Yi, H. Guo and X. Tu, Atmospheric Pressure and Room Temperature Synthesis of Methanol through Plasma-Catalytic Hydrogenation of CO2, ACS Catal., 2018, 8, 90–100 CrossRef CAS.
- H.-G. Yu, J. T. Muckerman and T. J. Sears, A theoretical study of the potential energy surface for the reaction OH + CO → H + CO2, Chem. Phys. Lett., 2001, 349, 547–554 CrossRef CAS.
- J.-g. Wang, C.-j. Liu, Y.-p. Zhang and B. Eliasson, A DFT study of synthesis of acetic acid from methane and carbon dioxide, Chem. Phys. Lett., 2003, 368, 313–318 CrossRef CAS.
- S. Li, J. Sun, Y. Gorbanev, K. van't Veer, B. Loenders, Y. Yi, T. Kenis, Q. Chen and A. Bogaerts, Plasma-Assisted Dry Reforming of CH4: How Small Amounts of O2 Addition Can Drastically Enhance the Oxygenate Production—Experiments and Insights from Plasma Chemical Kinetics Modeling, ACS Sustainable Chem. Eng., 2023, 11, 15373–15384 CrossRef CAS.
- W. Wang, R. Snoeckx, X. Zhang, M. S. Cha and A. Bogaerts, Modeling Plasma-based CO2 and CH4 Conversion in Mixtures with N2, O2, and H2O: The Bigger Plasma Chemistry Picture, J. Phys. Chem. C, 2018, 122, 8704–8723 CrossRef CAS.
- J. Slaets, M. Aghaei, S. Ceulemans, S. Van Alphen and A. Bogaerts, CO2 and CH4 conversion in “real” gas mixtures in a gliding arc plasmatron: how do N2 and O2 affect the performance?, Green Chem., 2020, 22, 1366–1377 RSC.
- T. Thi Bui, L. Pazdur, S. Janssens, V. Vande Velde, P. Billen and S. Tavernier, Production of Long Chain Alcohols from Waste Polyethylene Pyrolysis Oil Using a One-Pot Hydroboration–Oxidation Process, ACS Sustainable Chem. Eng., 2023, 11, 11729–11736 CrossRef.
- Z. Xu, N. E. Munyaneza, Q. Zhnag, M. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. Liu, Chemical upcycling of polyethylene, polypropylene, and mixtures to high-value surfactants, Science, 2023, 381, 666–671 CrossRef CAS PubMed.
- A. Milbrandt, K. Coney, A. Badgett and G. T. Beckham, Quantification and evaluation of plastic waste in the United States, Resour., Conserv. Recycl., 2022, 182, 106363 CrossRef.
- U. R. Gracida-Alvarez, O. Winjobi, J. C. Sacramento-Rivero and D. R. Shonnard, System Analyses of High-Value Chemicals and Fuels from a Waste High-Density Polyethylene Refinery. Part 1: Conceptual Design and Techno-Economic Assessment, ACS Sustainable Chem. Eng., 2019, 7, 18254–18266 CrossRef CAS.
- O. Olafasakin, J. Ma, S. L. Bradshaw, H. A. Aguirre-Villegas, C. Benson, G. W. Huber, V. M. Zavala and M. Mba-Wright, Techno-Economic and life cycle assessment of standalone Single-Stream material recovery facilities in the United States, Waste Manage., 2023, 166, 368–376 CrossRef CAS PubMed.
- Environmental Research and Educational Foundation, Analysis of MSW Landfill Tipping Fees, available at: https://www.erefdn.org, accessed March 23, 2024.
- A. Badgett, G. Cooney, J. Hoffmann and A. Milbrandt, CO2 Emissions from Stationary Sources, 2023 Billion-Ton Report, M. H. Langholtz (Lead), Oak Ridge National Laboratory, 2024, ch. 7.3 Search PubMed.
- T. N. Do, C. You and J. Kim, A CO2 utilization framework for liquid fuels and chemical production: techno-economic and environmental analysis, Energy Environ. Sci., 2022, 15, 169–184 RSC.
- J. Kim, T. A. Johnson, J. E. Miller, E. B. Stechel and C. T. Maravelias, Fuel production from CO2 using solar-thermal energy: system level analysis, Energy Environ. Sci., 2022, 15, 169–184 RSC.
- ChemAnalyst, Paraffin Wax Price Trend and Forecast, available at: https://www.chemanalyst.com/Pricing-data/paraffin-wax-1205, accessed November 10, 2023.
- ChemAnalyst, Linear Alpha Olefin Price Trend and Forecast, available at: https://www.chemanalyst.com/Pricing-data/linear-alpha-olefin-1103, accessed November 10, 2023.
- ChemAnalyst, Paraformaldehyde Price Trend and Forecast, available at: https://www.chemanalyst.com/Pricing-data/paraformaldehyde-pfa-1195, accessed November 10, 2023.
- J. Hambalko, P. Gajdoš and J. M. Nicaud, et al., Production of Long Chain Fatty Alcohols Found in Bumblebee Pheromones by Yarrowia lipolytica, Front. Bioeng. Biotechnol., 2020, 8, 593419 CrossRef PubMed.
- R. Wang, Y. Luo, H. Jia, J. R. Ferrell and H. Ben, Development of quantitative 13C NMR characterization and simulation of C, H, and O content for pyrolysis oils based on 13C NMR analysis, RSC Adv., 2020, 10, 25918–25928 RSC.
- R. Partington, J. Clarkson, J. Paterson, K. Sullivan and J. Wilson, Quantitative carbon distribution analysis of hydrocarbons, alcohols, and carboxylic acids in a Fischer-Tropsch product from a Co/TiO2 catalyst during gas phase pilot plant operation, J. Anal. Sci. Technol., 2020, 11, 42 CrossRef CAS.
- R. J. Speight, 1H and 13C solution- and solid-state NMR investigation into wax products from the Fischer–Tropsch process, Solid State Nucl. Magn. Reson., 2011, 39, 58–64 CrossRef CAS PubMed.
- Y. Cortes-Peña, D. Kumar, V. Singh and J. S. Guest, BioSTEAM: A Fast and Flexible Platform for the Design, Simulation, and Techno-Economic Analysis of Biorefineries under Uncertainty, ACS Sustainable Chem. Eng., 2020, 8, 3302–3310 CrossRef.
- D. Humbird, R. Davis and L. Tao, Process Design and Economics for Biochemical Conversion of Lignocellulosic Biomass to Ethanol: Dilute-Acid Pretreatment and Enzymatic Hydrolysis of Corn Stover, NREL/TP-5100-47764, 1013269, 2011.
- openLCA Modeling Suite, GreenDelta GmbH, Berlin, Germany, 2023, available at: https://www.openlca.org/openlca/, accessed November 30, 2023.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc02340d |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |