
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature hydrogen production via electro-dehydrogenation of amines into nitriles: advancements in liquid organic hydrogen carriers†
Nihal
Guenani
,
Jose
Solera-Rojas
 ,
David
Carvajal
,
Carmen
Mejuto
,
Andrés
Mollar-Cuni
,
Antonio
Guerrero
,
Francisco
Fabregat-Santiago
,
José A.
Mata
* and
Elena
Mas-Marzá
*
,
David
Carvajal
,
Carmen
Mejuto
,
Andrés
Mollar-Cuni
,
Antonio
Guerrero
,
Francisco
Fabregat-Santiago
,
José A.
Mata
* and
Elena
Mas-Marzá
*
Institute of Advanced Materials (INAM), Universitat Jaume I, 12006 Castelló, Spain. E-mail: jmata@uji.es; emas@uji.es
First published on 6th June 2024
Abstract
This study introduces an electro-dehydrogenation method for converting amines into nitriles in aqueous environments, simultaneously releasing two moles of H2 using nickel electrodes. This eco-friendly process is selective and operates at room temperature, in contrast to traditional thermal methods that require high temperatures and where condensation products are usually observed. Detailed impedance spectroscopy analysis reveals that the dehydrogenation of amines in aqueous media is facilitated by efficient charge transfer, with the diffusion of amines to the electrode surface identified as the kinetically slowest step. Our study underscores the potential of electrochemistry to enhance the reversible dehydrogenation and hydrogenation of the amine/nitrile pair. By demonstrating the practicality and efficiency of this approach, we highlight the amine/nitrile pair as a promising candidate for liquid organic hydrogen carriers. This has significant implications for the future of hydrogen storage and transport technologies, paving the way for more sustainable and efficient energy solutions.
Introduction
The global climate crisis has intensified the need to adopt renewable energy sources to reduce greenhouse gas emissions and mitigate climate change. Recent energy transition plans, adopted by most countries, emphasize the importance of integrating renewable energies into large-scale industrial applications. However, one of the most significant challenges associated with renewable energy sources is their intermittent nature, which poses issues of stability and reliability in energy supply.1Hydrogen has emerged as a promising energy vector due to its favorable properties, including a high energy density and the potential to generate energy without pollutant emissions. However, the storage and transportation of hydrogen present considerable challenges. In this context, Liquid Organic Hydrogen Carriers (LOHCs) emerge as a viable solution for storing hydrogen safely, efficiently, and non-toxically. LOHCs enable the chemical storage of hydrogen in liquid organic compounds, facilitating its handling and large-scale transportation.2–5
Water electrolysis is a well-established technology for producing pure hydrogen safely and cleanly, through the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). However, the oxygen evolution reaction, stemming from water splitting, is kinetically and thermodynamically unfavorable, limiting the overall efficiency of the process.6 To improve the energy balance of the reaction, a promising strategy is to replace oxygen evolution with the oxidation of less energy-intensive organic substrates, obtaining value-added products in the same process.7,8
Among the systems with potential to replace the OER, oxidation of primary amines represents an attractive option. The oxidation of amines can produce various species, such as aldehydes, ketones, imines, and nitriles (Scheme 1). Specifically, the selective oxidation of primary amines to nitriles is of great interest due to the importance of nitriles as intermediates in chemical synthesis.9–14 Additionally, this process can be energy-efficient and operable at low temperatures through electrocatalytic processes.
 | ||
| Scheme 1 Possible products for the dehydrogenation of amines. | ||
Different and prestigious research groups in the field of electrochemistry have reported the dehydrogenation of amines using nickel as the electrocatalyst. For example, B. Zhang et al., have described the use of NiSe nanorods as electrodes for the oxidation of aromatic and aliphatic primary amines in basic media.15 However, this electrocatalyst requires a complex synthetic process, hindering its scalability for industrial use. Similarly, Z. Yan, in collaboration with the research group of Z. Xu, recently reported a hybrid water electrolyzer based on a Ni/Co metal–organic framework for the oxidation of aromatic and aliphatic amines, but only with a single amine functional group in the system.16 More recently, Choi K.-S. has reported the electro-oxidation behavior of propylamine and benzylamine using NiOOH electrodes onto fluorine-doped tin oxide (FTO), analyzing in detail the effect of different parameters on the reaction, such as the amine concentration and the pH of solution among other factors.17
Despite the advances in amine oxidation, most catalysts developed so far require complex synthetic processes and high temperatures, making scalability for industrial applications challenging. In this work, we propose the use of nickel foam electrodes decorated with Ni (NiNF) as catalysts for the oxidation of primary amines to nitriles in aqueous media without additives, producing molecular hydrogen as the only byproduct. Detailed impedance spectroscopy analysis reveals mechanistic insight related to the response of our nickel electrode. Building on the significant catalytic performance observed in the electrochemical dehydrogenation of amines to nitriles, as well as the hydrogen storage capacity (HSC)18 of the pair amine/nitriles, we investigated the reverse process: the thermocatalytic hydrogenation of nitriles using molecular H2 as the hydrogen source. This study demonstrates the potential of the amine/nitrile pair as the liquid organic hydrogen carrier (LOHC) (Fig. 1).
 | ||
| Fig. 1 Electrodehydrogenation/hydrogenation cycle for the use of the pair amine/nitrile as the LOHC. | ||
Results and discussion
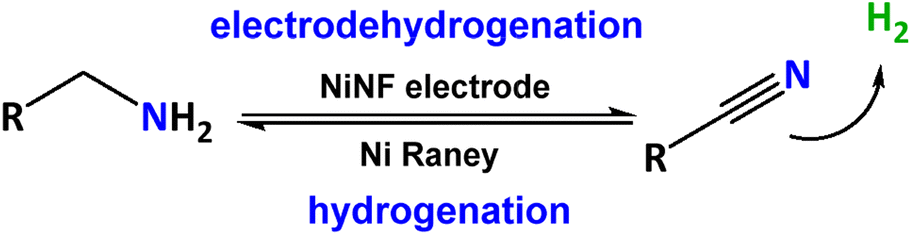
Our initial focus involved examining the catalytic dehydrogenation of amines to nitriles through electrochemical methods (electro-dehydrogenation) in aqueous basic media at ambient temperature. Various nickel-based electrodes were tested as anodes, leveraging the exceptional electrochemical oxidation performance of nickel for organic molecules under alkaline conditions.14–16 The Ni-based electrodes employed in this work include Ni foil, Ni foam (NF), Ni foam decorated with nickel (NiNF), pencil graphite rods decorated with Ni (NiPGR) and Ni45, prepared by powder metallurgy following a previous procedure developed in our research group.18 The deposition of nickel on PGR and Ni foam was performed by electrodeposition. Details for the preparation of the electrodes are described in the ESI,† including microscopy characterization (see section S1.2†).Prior to use, all the electrodes were activated via cyclic voltammetry (CV), in 1 M KOH, to increase the amount of Ni3+ active species at the surface of the electrode (see ESI Fig. S2†). The reversible redox peak observed during the activation corresponds to the transformation of α/β-Ni(OH)2 (Ni2+) to catalytically active γ/β-NiOOH (Ni3+), reported in previous studies.19 The current density associated with the Ni2+/Ni3+ peak increases for each cycle as a result of increasing the concentration of active species on the electrode surface.
Fig. 2a depicts the response of all electrodes in 1 M KOH after activation. As anticipated, NiPGR and NiNF demonstrate elevated current densities at the Ni2+/Ni3+ redox peak, attributed to a higher concentration of nickel active species on their surfaces compared to Ni foil, NF, and Ni45. The comparison of cyclic voltammogram curves between NF and NiNF emphasizes a notable increase in current density resulting from the incorporation of nickel species through electrodeposition on Ni foam (Fig. 2b).
 | ||
| Fig. 2 (a) Cyclic voltammetry curves at a scan rate of 10 mV s−1 of all Ni electrodes in 20 mL of 1 M KOH. (b) Details of (a) for a better comparison of NF and NiNF cyclic voltammetry curves. | ||
The electrocatalytic activity of Ni electrodes was assessed in the dehydrogenation (oxidation) of amines using 1,6-hexanediamine (1a) as the model substrate. The use of this compound is convenient for hydrogen storage purposes as it contains two amino functional groups (-CH2-NH2) capable of generating two nitrile groups (-CN), with the concomitant formation of 4 moles of hydrogen, representing a hydrogen storage capacity (HSC) value of 6.9 wt% of hydrogen.
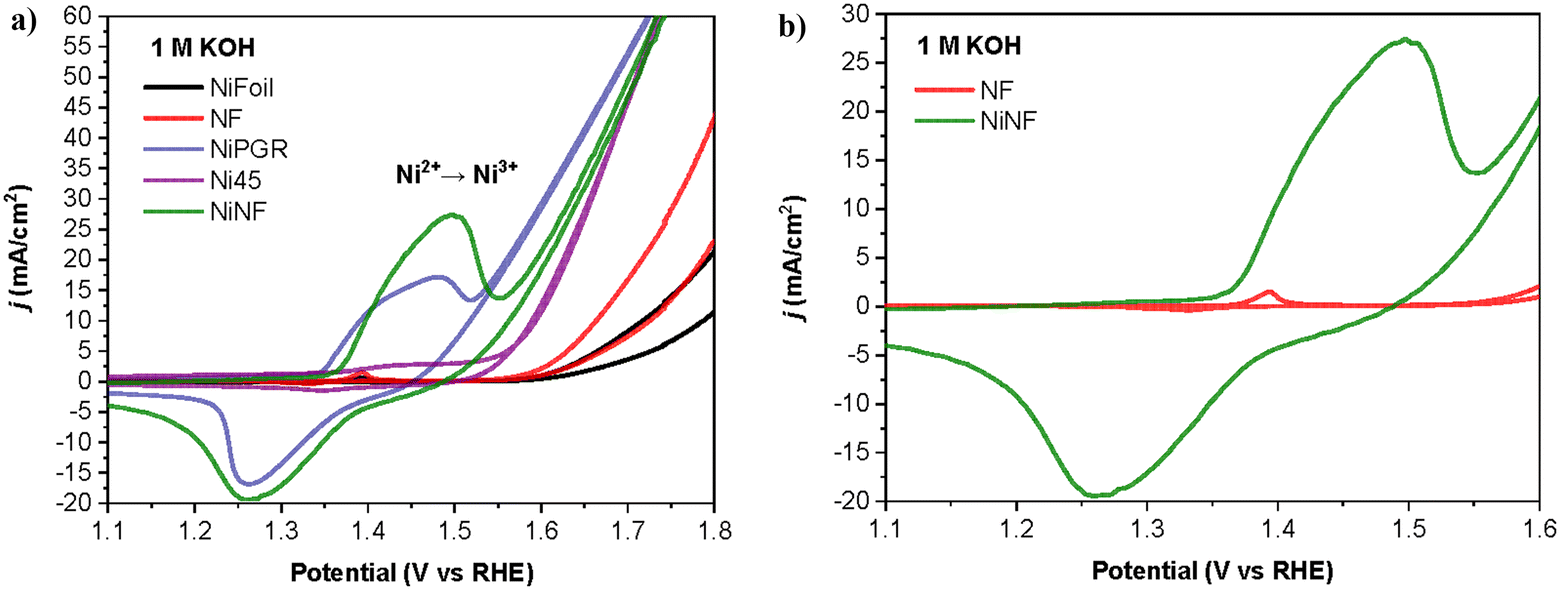
To gain insight into how the electrodes operate in the presence and absence of 1a, we first analyzed the cyclic voltammetry (CVs) curves of the different electrodes under study. Fig. 3 shows a representative example using a NiNF electrode in the presence and absence of 25 mM of 1a. As can be seen, these CVs are very different and there is a noticeable shift in the onset of the Ni2+/Ni3+ peak towards a higher potential in the presence of 1a that may be attributed to the amount of diamine adsorbed onto the electrode surface. Due to the overlap between the oxidation of the diamine 1a and the oxidation of Ni2+ to Ni3+, the peak extends over a longer voltage range. Additionally, there is an increase in the current density which is very clear in the range between 1.5 V and 1.7 V. Besides, the positive current obtained at these potentials in the reverse direction indicates the irreversible oxidation of 1a, which also consumes some Ni3+ species yielding a smaller reduction peak observed for Ni.
 | ||
| Fig. 3 Comparison of cyclic voltammetry curves of NiNF, in 20 mL of 1 M KOH, with and without 1a. | ||
Then, we explored the performance of our different Ni electrodes in the electrochemical dehydrogenation of 1a through chronocoulometric experiments. This process involves applying the theoretical charge needed to convert completely 1a into 2a at 1.55 V vs. RHE, considering a total of eight electrons involved in the process (see the ESI† for details). The experimental set-up consists of a two-compartment cell, utilizing a Nafion 117 membrane to prevent potential reduction of oxidation products by the counter electrode (Fig. 4). Both the cathodic and anodic chambers were filled with a water solution of KOH as the electrolyte, and the required amount of 1a was introduced in the anodic chamber, under magnetic stirring. The Ni electrodes, with an active area of 2 cm2, were immersed in the cell for the duration of the experiments. A platinum electrode was used as a cathode.
 | ||
| Fig. 4 Scheme of a two-compartment cell. | ||
The performance of our Ni-based electrodes in the chronocoulometric dehydrogenation of 1a at room temperature is shown in Table 1. The total charge passed during the process was 392 C, corresponding to the theoretical charge required for the oxidation of 1a into 2a. Detection and quantification of reactants and products were carried out through 1H NMR spectroscopy, with ethylene glycol (EG) serving as an internal integration standard. A common problem found in amine dehydrogenation is selectivity. In fact, under thermal catalytic conditions, the formation of condensation products (imines and related products) is generally observed20,21 and only a few ruthenium homogeneous catalysts induce complete selectivity towards the formation of nitriles.22,23 The electro-dehydrogenation of 1,6-hexanediamine (1a) proceeds through the intermediates 1-amino-5-cyanopentane (i) and 1-amino-6-imino-hexane (ii) (Fig. 5). However, these intermediates have not been observed during our experiments due to their high reactivity in comparison to the parent diamine (1a).
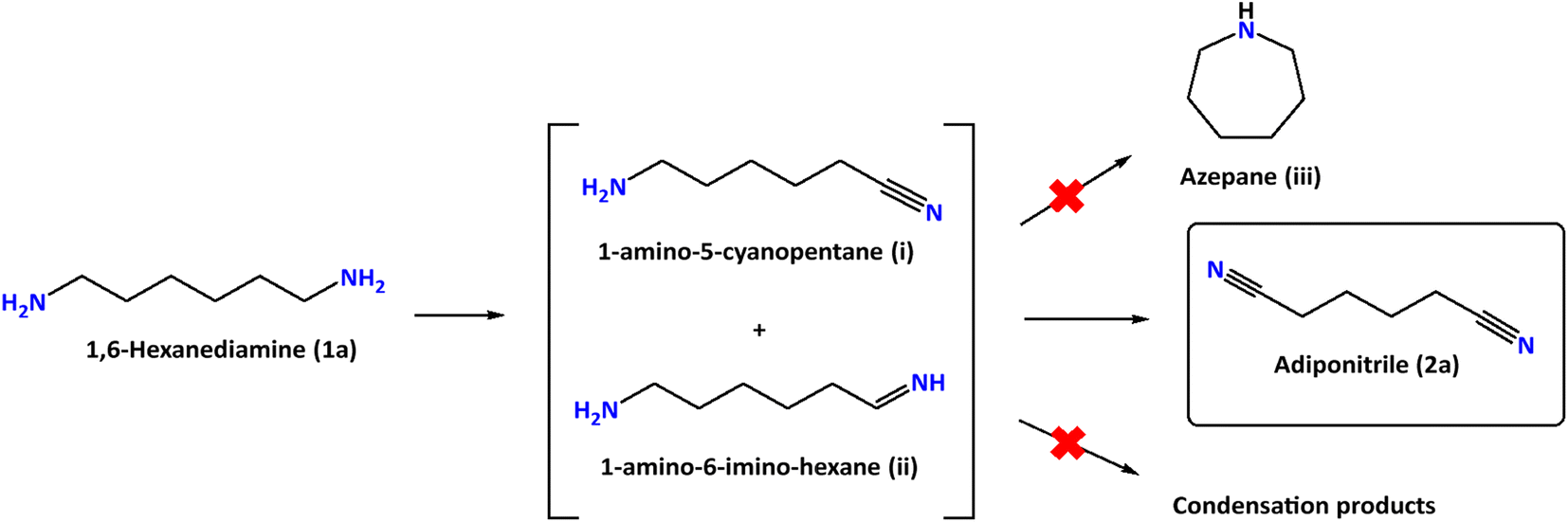 | ||
| Fig. 5 Selective electro-oxidation of 1a targeted in this work. | ||
| Entry | Electrode | J (mA cm−2) | t (h) | FE (%) | Conver. (%) | Yield (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: 1,6-hexanediamine (1a) (0.5 mmol, 25 mM), 1 M KOH (20 mL), 25 °C at 1.55 V vs. RHE. Conversions and yields were determined by 1H NMR spectroscopic analysis using ethylene glycol as an external standard. J represents the current density at the beginning of the chronocoulometry experiment. | ||||||
| 1 | Ni foil | 8 | 150 | 0 | 0 | 0 |
| 2 | NF | 20 | 20 | 59 | 90 | 60 |
| 3 | NiPGR | 15 | 12 | 51 | 99 | 52 |
| 4 | Ni45 | 55 | 6 | 36 | 94 | 37 |
| 5 | NiNF | 77 | 3.5 | 77 | 99 | 78 |
The dehydrogenation of 1a did not occur with Ni foil as the electrode even after 150 hours of reaction, most likely due to the limited active area of this electrode in comparison to the other nickel catalysts (entry 1, Table 1). All the charge in this experiment was consumed in the competing oxygen evolution reaction (OER). A notable enhancement in electro-dehydrogenation of 1a was evident when using the other electrodes, particularly those containing electrodeposited nickel. In entry 5, it is demonstrated that the NiNF electrode exhibited the shortest time to complete conversion of 1a (∼3.5 hours), with the highest faradaic efficiency (FE) and yield for the target product 2a. Conversely, NF, NiPGR, and Ni45 displayed high conversions but suffered yield losses. This discrepancy between conversion and yield stemmed from the instability of 2a in strong basic media (Fig. S16 and 17†). Interestingly, reducing the KOH concentration from 1 M to 0.5 M enabled nearly quantitative yield for 2a, suggesting the prevention of degradation into undesired species under these conditions (Table 2). However, further reduction in KOH concentration to 0.25 M did not improve the reaction performance, as the current density sharply decreased, and the oxidation reaction time increased significantly (entry 3, Table 2). This decline in reaction performance might be attributed to a less efficient generation of the Ni redox pair required for the electrochemical oxidation of 1a when utilizing only 0.25 M KOH, aligning with our previous investigations on the oxidation of organic compounds with Ni-based electrodes.24 Interestingly, in all our chronocoulometric experiments, we have observed that electro-dehydrogenation of 1a is completely selective to the formation of the corresponding adiponitrile (2a) avoiding the formation of undesired intermediates such as cyclization species or condensation products. This observation highlights that the electro-dehydrogenation of amines by nickel species is 100% selective towards the formation of nitriles.
| Entry | Electrolyte | J (mA cm−2) | t (h) | FE (%) | Conver. (%) | Yield (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: 1,6-hexanediamine (1a) (0.5 mmol, 25 mM), 0.25–1 M KOH (20 mL), 25 °C at 1.55 V vs. RHE. Conversions and yields were determined by 1H NMR spectroscopic analysis using ethylene glycol (EG) as an external standard. J represents the current density at the beginning of the chronocoulometry experiment. | ||||||
| 1 | 1 M KOH | 77 | 3.5 | 77 | 99 | 78 |
| 2 | 0.5 M KOH | 77 | 3.25 | 95 | 99 | 96 |
| 3 | 0.25 M KOH | 20 | 24 | 59 | 99 | 60 |
When the reaction of 1a was performed in a one-compartment configuration, we obtained a 93% yield and FE for 2a, with 94% conversion of 1a, suggesting that there is no significant difference between using a two or one compartment cell for the electrooxidation of 1a. There are studies that utilize membrane-free setups, as previously reported for the electrooxidation of benzylamine;16 however, to avoid possible nitrile reduction, we conducted all our experiments using a two-compartment cell.
We assessed the influence of temperature on the oxidation of 1a using NiNF as the working electrode and 0.5 M KOH as the electrolyte. The oxidation of 1a proceeds similarly to that at room temperature or 40 °C providing quantitative yields and FE. However, when the reaction temperature was increased to 60 °C, although complete conversion was observed, only low yields or FE were observed (Table S1†). The low yield and FE at high temperatures has been attributed to degradation (predominantly hydrolysis) of the nitriles under basic pH and high temperature conditions, as noted from NMR studies. Higher temperatures promote side reactions and decomposition of intermediate species, which in turn reduce the selectivity towards the desired product.
The formation of H2 during the dehydrogenation of 1a was confirmed by gas chromatography (Fig. S18 and S19†). When the reaction was performed in the absence of an amine, as expected, H2 and only traces of O2 gas were detected due to the OER, confirming that this process is competing with amine dehydrogenation. However, in the presence of an amine, H2 gas was detected in qualitatively amounts and the intensity increased in comparison when performing the reaction without an amine. These results suggest that in the presence of 1a, the OER is suppressed and all the increase in the intensity of the signal corresponding to the H2 gas formed comes from the –CH2NH2 group of the amine.
To investigate the stability of NiNF, we conducted a series of chronocoulometry experiments using the same electrode five times for the electrooxidation of 25 mM 1a in 0.5 M KOH observing no significant losses in FE and yield for the formation 2a (Table S2†), thus demonstrating the high stability of our NiNF.
Reaction scope
Once we establish the optimal reaction conditions for the electrochemical dehydrogenation of 1a, the efficiency of NiNF electrodes was tested with different primary amines (Table 3). The results showed good versatility with various substrates, including aromatic and aliphatic amines.|
|
|||
|---|---|---|---|
| Entry | Substrate | FE (%) | Yield (%) |
| Reaction conditions: amine (a–e) (0.5 mmol, 25 mM), 0.5 M KOH (20 mL), 25 °C at 1.55 V vs. RHE, NiNF electrode. Yields of nitrile were determined by 1H NMR spectroscopic analysis using ethylene glycol (EG) as an external standard. | |||
| 1 | (a) 1,6-Hexanediamine | 95 | 96 |
| 2 | (b) n-Hexylamine | 89 | 90 |
| 3 | (c) Benzylamine | 75 | 77 |
| 4 | (d) p-Trifluoromethylbenzylamine | 80 | 80 |
| 5 | (e) p-Methoxybenzylamine | 99 | 98 |
Importantly, all substrates were selectively converted to the corresponding nitrile derivatives in almost quantitative yields. Substrates 1a and 1b exhibit similar yields, suggesting that in the case of the diamine, both functional groups operate completely independently despite belonging to the same molecule. Regarding the aromatic substrates, a slight effect is observed depending on the substituent groups at the para-position of the aromatic rings, with an increase in the yield value as the nucleophilic substituent strengthens (entries 3–5, Table 3). The bests results were obtained with 1,6-hexanediamine (1a) and p-methoxybenzylamine (1e) which provided yields of 96% and 98%, respectively and high faradaic efficiencies (95% and 99%, respectively).
Detailed electrochemical analysis
The cyclic voltammetry profile of the electrode closely resembled prior investigations involving nickel oxo-hydroxo (NiOOH) electrodes employed for the oxidation of organic substrates.25 For the sample without an amine, the redox peak is attributed to the Ni+2/Ni+3 redox process.19 In the presence of 1a, the oxidation current displaces the oxidation peak and the baseline upwards. At the same time, the reduction peak decreases indicating that part of the Ni+3 has been already reduced during the reduction cycle of the voltammetry of nickel. The current density (J) achieved at the redox peaks associated with the oxidation of Ni and 1a increased with the concentration of the amine, until reaching a value of ∼80 mA cm−2 in the forward direction (∼60 mA cm−2 in the reverse direction) at a 25 mM concentration of 1a.As can be seen in Fig. 6, Ni is initially oxidized from Ni(OH)2 (Ni2+) to NiOOH (Ni3+), the latter species being the oxidizing agent responsible for the amine oxidation. As previously reported, the amine oxidation mechanism involves a rate-limiting hydrogen transfer step where a hydrogen radical from the α-C of the amine is transferred to the NiOOH, leading to the reduction of NiOOH to Ni(OH)2 and amine oxidation.17 The second oxidation peak observed in the CV, which is overlapped with the Ni(OH)2/NiOOH redox couple, is associated with amine oxidation. This second peak increases with higher amine concentrations. Additionally, the onset and both oxidation peaks (Ni2+/Ni3+ and amine/nitrile) shift towards higher potentials with increasing amine concentration. This shift is likely due to more amine adsorbed on the electrode, which partially blocks the Ni surface and causes an additional potential loss for Ni oxidation, shifting the amine oxidation reaction by several tens of millivolts.24 If we focus now on the reduction direction of the CV, the reduction peak at 1.3–1.4 V for the Ni3+/Ni2+ redox couple decreases with increasing amine concentration. At 1.5 V in the reduction direction, the current remains positive in the presence of an amine, indicating ongoing amine oxidation. As Ni3+ (or NiOOH) is consumed during this reaction, the height of the Ni3+ reduction peak at 1.3–1.4 V diminishes with higher amine concentrations.24
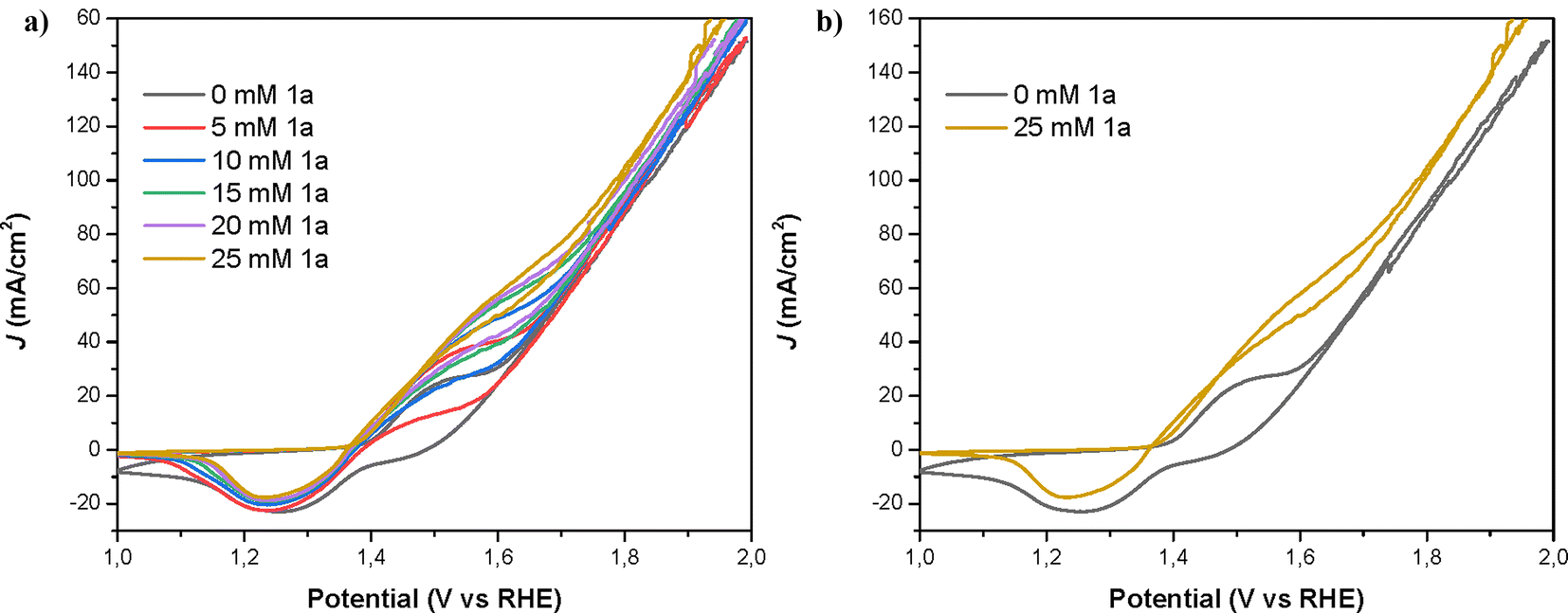 | ||
| Fig. 6 (a) Cyclic voltammetries for the oxidation of 1a (0–25 mM), using NiNF as the working electrode, Pt wire as the counter electrode and Ag/AgCl as the reference (RHE potential scale), in 0.5 M KOH as the electrolyte at 5 mV s−1 scan. (b) Zoom cyclic voltammetries for the oxidation of 1a (0 and 25 mM). | ||
To further examine the performance of NiNF in the electrochemical dehydrogenation of amines, we conducted current density vs. voltage (J–V) and impedance spectroscopy (IS) studies. The advantage of measuring stationary J–V curves lies in the absence of artifacts associated with capacitance charging/discharging as it occurs in other techniques like linear sweep or cyclic voltammetries (LSV or CV), therefore the obtained results are more reliable. Thus, the J–V curve for a solution of 25 mM of 1a illustrates that the onset potential (1.375 V vs. RHE) is displaced 0.15 V vs. the onset of water oxidation without artifacts (Fig. 7). In addition, the current increases linearly until reaching a narrow plateau of 52 mA cm−2 at 1.75V vs. RHE, indicating the limit for efficient amine dehydrogenation. At higher potentials, the OER becomes dominant and therefore if we apply these potentials, the FE towards 1a will be reduced.
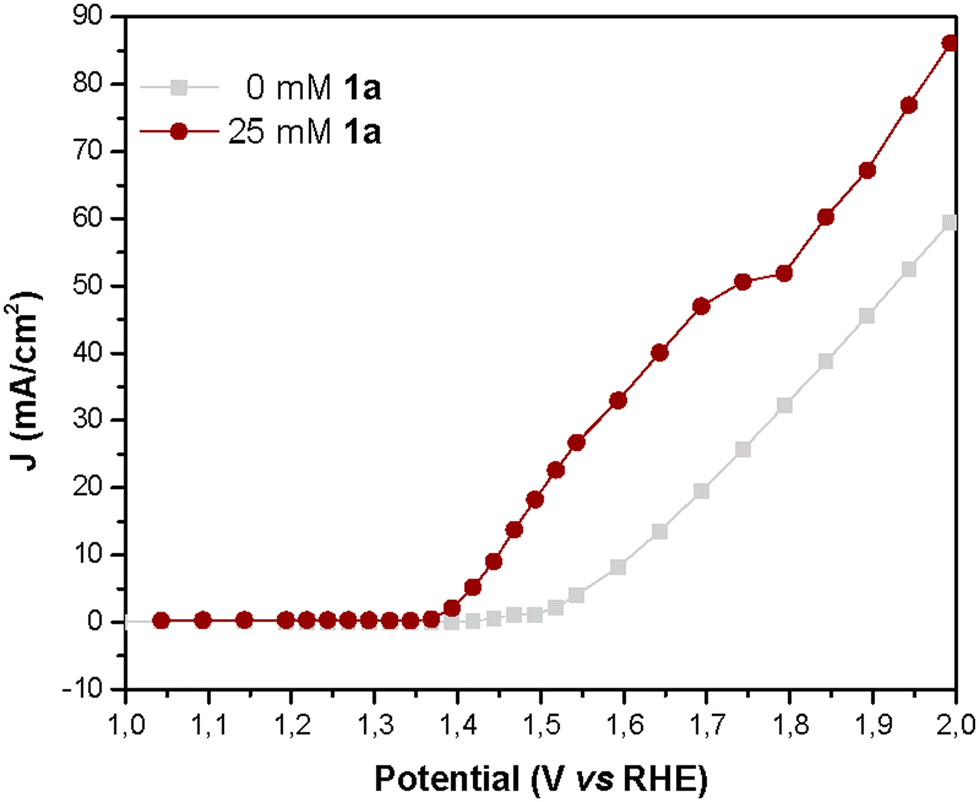 | ||
| Fig. 7 J–V curves obtained during IS measurement of NiNF electrodes in a 0.5 M KOH aqueous solution with and without 1a. | ||
Impedance spectra for samples with and without 1a are plotted in Fig. 8. In the first approach, three distinct arcs are evident for each system. The small arc at high frequencies is associated with the electrical contact with the nickel foam electrode. The large, deformed arc at middle frequencies is linked to charge transport, transfer, and accumulation in the NiNF electrode, which is modelled using the transmission line-based equivalent circuit.19 Finally, the small arc at low frequencies is connected to the diffusion of active species in the solution.
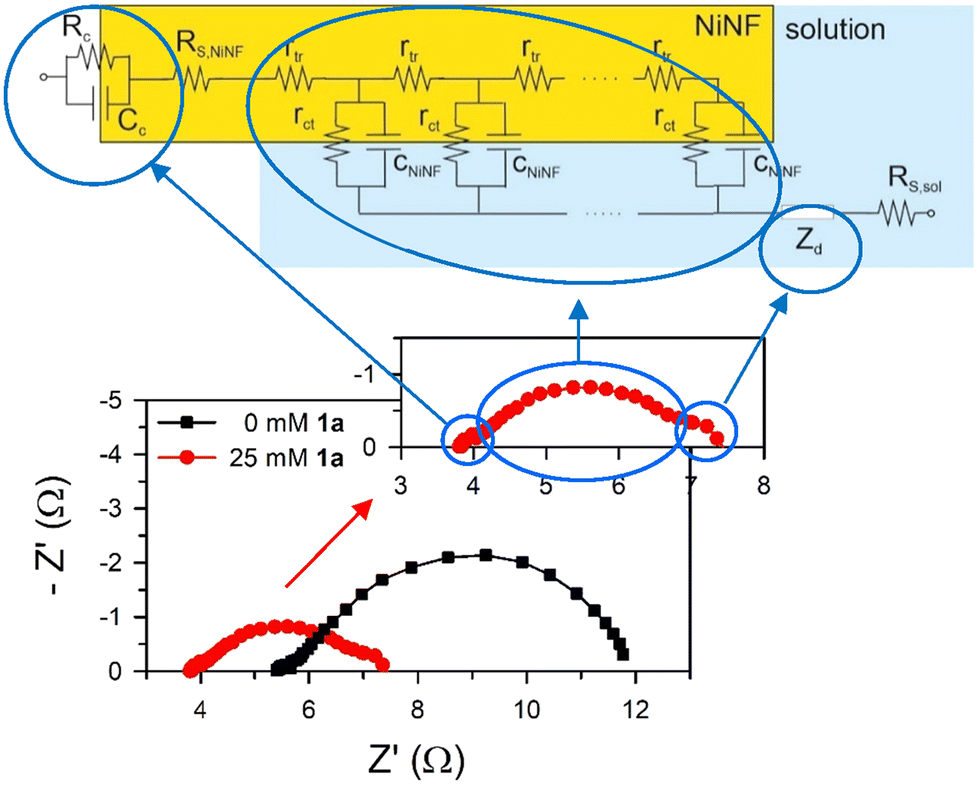 | ||
| Fig. 8 Nyquist plot for the NiNF electrode with and without 1a, in 0.5 M KOH at 1.5 V vs. RHE. The inset shows the IS with 25mM 1a. The figure shows how the equivalent circuit used to fit the data (top) is related to the different arcs. The low frequency arc is associated with diffusion, Zd. The high frequency arc is associated with the nickel foam electrode contact and the large middle-frequency arc is related to a transmission line that includes the transport resistance in the Ni foam, (Rtr = rtr·L), the charge transfer resistance from the Ni to the solution, (Rct = rct/L), and the total capacitance of the electrode, (CNiNF = cNiNF·L), L being the length of the Ni foam immersed in the solution. The displacement of the arcs from the X-axis origin is given by the series resistance, (RS). | ||
Impedance spectroscopy reveals that the displacement of the arcs along the X-axis and their size are smaller in the presence of 1a (Fig. 8). The X-axis displacement is associated with the series resistance of the sample, RS, which arises from multiple contributions, including the NiNF section out of the electrolyte (RS, NiNF), the solution bulk resistance (RS, sol), and the negligible contributions from contacts and wires, all leading to RS ≈ RS, NiNF + RS, sol. In all cases, the length of the NiNF outside the solution remains constant. Therefore, we attribute the variance in series resistance among the samples to the increased conductivity of the solution following the addition of 1a.
As Fig. 8 shows, the impedance is dominated by the largest arc at intermediate frequencies, associated with the transmission line which is determined by the overall transport resistance of charges in the NiNF (Rtr), the charge transfer resistance from the surface of the electrode towards the solution (Rct), and the total contribution to the capacitance of the electrode (CNiNF), which includes contributions from double layer (Helmholtz) capacitance, the capacitance associated with the absorbance of 1a and its derivatives, and the capacitance associated with the different redox states of Ni. In the case shown in Fig. 9a, the variation in the size of the intermediate arc when 1a is added to the solution is associated with the reduction of Rct.
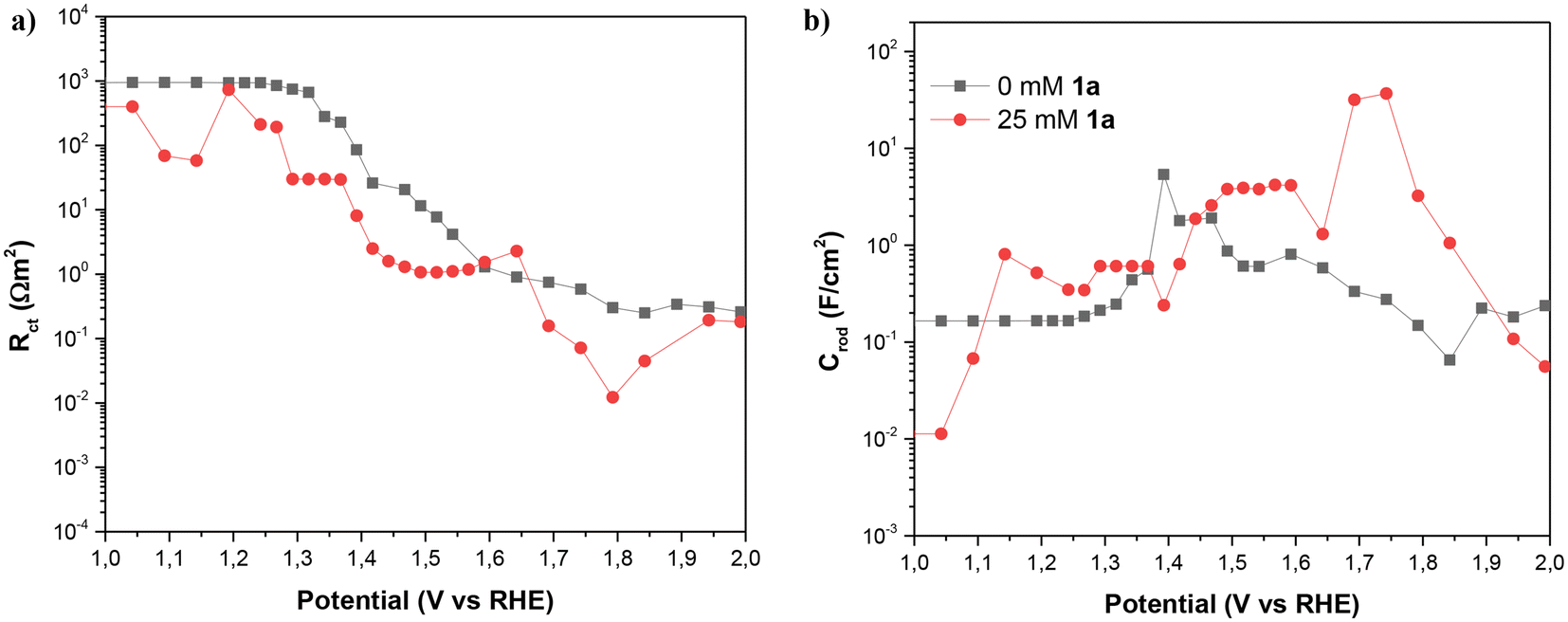 | ||
| Fig. 9 (a) Charge transfer resistance and (b) capacitance of NiNF as a function of the potential vs. RHE with and without 1a, in 0.5 M KOH. Figures (a) and (b) hold the same legend. | ||
Understanding the capacitance and charge transfer of the NiNF electrode is crucial for unraveling the mechanisms influencing the device's performance. As can be observed in Fig. 9, this capacitance and charge transfer are closely related. In Fig. 9b, the NiNF capacitance is depicted as a function of potential vs. RHE. In the absence of 1a, the capacitance of the NiNF electrode increases in the region of 1.35–1.5 V vs. RHE, corresponding to the Ni+2/Ni+3 redox peak observed in cyclic voltammograms. The shape of this peak suggests that both Ni-hydrated and dehydrated phases are present.19 Subsequently, the capacitance decreases, exhibiting another small peak at 1.6 V vs. RHE, closely aligned with the onset of the OER. This peak has previously been attributed to the Ni+3/Ni+4 redox transition, with Ni+4 identified as the species involved in the OER,19 matching very well with the drop in Rct below 10 Ω cm−2 seen in Fig. 9a, indicating an acceleration in the kinetics of the process and the onset of the OER shown in Fig. 7.
In the presence of 1a, a broad CNiNF peak is observed between 1.4 and 1.6 V vs. RHE, which corresponds to the combination of the Ni+2/Ni+3 redox transition peak and the oxidation peak of 1a. Notably, this peak aligns well with the onset of the J–V curve in Fig. 7 and the drop of Rct below 10 Ω cm−2, showing the strong correlation between both phenomena. Between 1.65 and 1.8 V vs. RHE, a significant peak is observed, followed by the dominance of the OER in the J–V curve. Two plausible explanations for this peak could be considered: (i) an oxidation process of intermediates to adiponitrile, or (ii) the Ni+3/Ni+4 redox transition. The completion of reactions at 1.55 V vs. RHE and the absence of overoxidation products favor the likelihood of the second option. The substantial difference in the height of the Ni+3/Ni+4 peaks may be attributed to the competition between the oxidation process of 1a and the OER. In the absence of 1a, once Ni+4 is formed, it promptly oxidizes water, resulting in a small charge accumulation in these states. Conversely, with the addition of 1a, most of the charge transfer is directed toward this species and its intermediates, contributing to the observed high faradaic efficiency. As charge transfer for OER is more challenging, Ni+4 may accumulate, leading to higher capacitance. A further Rct decrease is observed in Fig. 9a for the same solution.
Another noteworthy observation derived from capacitance measurements is the shift of the two Ni redox peaks towards more positive potentials in the presence of 1a. This shift is attributed to the interaction of this species with the surface of the NiNF electrode. Describing it as a coating of the surface is challenging, as the capacitance of NiNF at potentials below 1.3 V vs. RHE in the presence of 1a is greater than in its absence. Conversely, the adsorption of 1a could lead to an increase in capacitance, akin to what has been previously observed for hydrogen absorption on Pd-decorated electrodes.26 Finally, at the higher potentials, Rct becomes so small that Rtr and Rs dominate the J–V curve both for the samples with and without 1a.
Summarizing the impedance results, the substantial changes in the capacitance observed in the NiNF electrode upon the addition of 1a facilitates to understand the potentials at which Ni and 1a redox processes occur and even distinguishing the number of oxidation species accumulated in the electrode. The observation of a large accumulation of Ni+4 on the electrode with 1a in solution, together with the shifts in redox peaks observed, suggests the absorption of 1a species and its derivatives on the electrode surface that consequently promotes the dehydrogenation reaction. Through resistance analysis, the underlying mechanisms governing the behaviour of J–V curves at all measured potentials are identified. Charge transfer predominates for most potentials, except at higher potentials where limitations due to the series resistance control the system's response.
Hydrogenation (reduction) experiments
To complete the dehydrogenation/hydrogenation studies for the utilization of the amine/nitrile pair as the LOHC, we explored the reverse process namely the hydrogenation of nitriles (Fig. 10). Several catalysts and different conditions were tested for the hydrogenation of adiponitrile (2a) to 1a, including Pd, Ru and Ni, using molecular hydrogen (H2) as the hydrogen source (Table S3†). The best performance in terms of conversion, yield and selectivity was achieved with RANEY® Ni that afforded quantitative yields in short reaction times (6 h).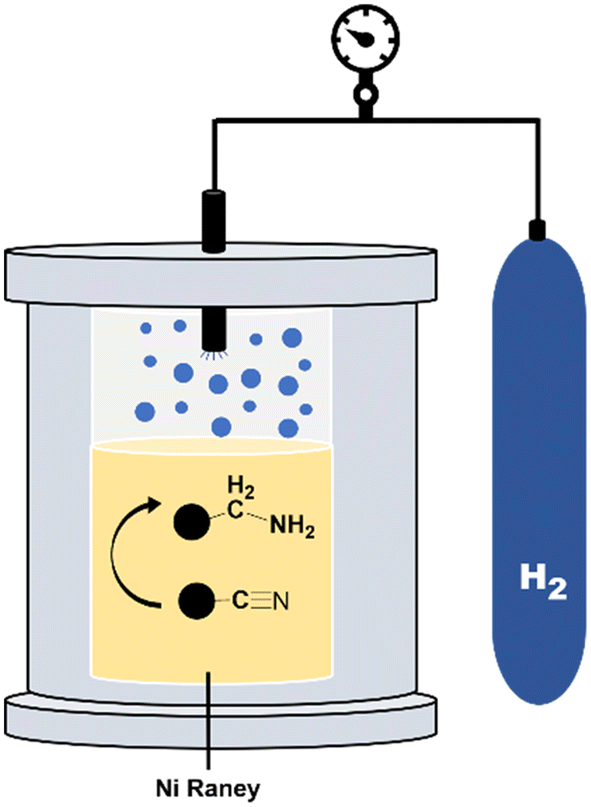 | ||
| Fig. 10 Scheme of a high-pressure reactor. | ||
Using this heterogeneous catalyst, among the different products that could be obtained in the hydrogenation of 2a, 1,6-hexanediamine was obtained in 94% yield, observing very low or null formation of cyclization species and condensation products (Table 4).
| Entry | Substrate | Yield (%) |
|---|---|---|
| Reaction conditions: adiponitrile (0.15 mmol), iPrOH (1 mL + 100 μL of 28% aqueous NH4OH), 20 H2 bar, 80 °C, 6 h. Yields determined by 1H NMR spectroscopy and GC/FID. | ||
| 1 | 1,6-Hexanediamine ( 1a ) | 94 |
| 2 | 1-Amino-5-cyanopentane (i) | 0 |
| 3 | 1-Amino-6-iminohexane (ii) | 0 |
| 4 | Azepane (iii) | 5 |
| 5 | Condensation products | 1 |
Conclusions
In this study, we have showcased the effectiveness of Ni electrodes in the selective electrochemical dehydrogenation of both aromatic and aliphatic amines, yielding the corresponding nitriles and molecular hydrogen. Exceptional yields and high faradaic efficiencies were achieved in the dehydrogenation of 1,6-hexanediamine using easily prepared Ni-based electrodes, underscoring the remarkable efficiency of this electrocatalyst in the reaction. Rigorous impedance analysis has provided valuable insights into the reaction mechanism, elucidating the pivotal role of amine adsorption on the electrode surface in facilitating dehydrogenation. Additionally, the completion of the reaction is contingent upon the diffusion of the amine, highlighting its significance as the decisive step in the process. Our investigations suggest that the electrocatalytic dehydrogenation of primary amines with NiNF electrodes and the catalytic hydrogenation of nitriles by RANEY® Ni in aqueous media represent a promising solution for utilizing this pair as a liquid organic hydrogen carrier. This reversible pathway offers a safe and environmentally friendly method for storing hydrogen. Furthermore, the mild conditions of this method render it suitable for potential large-scale industrial applications.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This study forms part of the Advanced Materials programme and was supported by MCIN with funding from the European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana under the project MFA/2022/043. The authors want to thank project UJI-B2022-33, funded by University Jaume I and Servicio Central de Instrumentación Científica (SCIC) from Universitat Jaume I. N. G. acknowledges PROMETEO/2020/028, funded by Generalitat Valenciana.References
- S. Chu and A. Majumdar, Opportunities and Challenges for a Sustainable Energy Future, Nature, 2012, 294–303, DOI:10.1038/nature11475.
- P. Preuster, C. Papp and P. Wasserscheid, Liquid Organic Hydrogen Carriers (LOHCs): Toward a Hydrogen-Free Hydrogen Economy, Acc. Chem. Res., 2017, 50(1), 74–85, DOI:10.1021/acs.accounts.6b00474.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, A Future Energy Supply Based on Liquid Organic Hydrogen Carriers (LOHC), Energy Environ. Sci., 2011, 2767–2773, 10.1039/c1ee01454d.
- R. H. Crabtree, Nitrogen-Containing Liquid Organic Hydrogen Carriers: Progress and Prospects, ACS Sustainable Chem. Eng., 2017, 5(6), 4491–4498, DOI:10.1021/acssuschemeng.7b00983.
- R. H. Crabtree, Hydrogen Storage in Liquid Organic Heterocycles, Energy Environ. Sci., 2008, 134–138, 10.1039/b805644g.
- P. C. Rao and M. Yoon, Potential Liquid-Organic Hydrogen Carrier (LOHC) Systems: A Review on Recent Progress, Energies, 2020, 13(22), 6040, DOI:10.3390/en13226040.
- D. Ventura-Espinosa, A. Carretero-Cerdán, M. Baya, H. García and J. A. Mata, Catalytic Dehydrogenative Coupling of Hydrosilanes with Alcohols for the Production of Hydrogen On-Demand: Application of a Silane/Alcohol Pair as a Liquid Organic Hydrogen Carrier, Chem. – Eur. J., 2017, 23(45), 10815–10821, DOI:10.1002/chem.201700243.
- D. Ventura-Espinosa, S. Sabater, A. Carretero-Cerdán, M. Baya and J. A. Mata, High Production of Hydrogen on Demand from Silanes Catalyzed by Iridium Complexes as a Versatile Hydrogen Storage System, ACS Catal., 2018, 8(3), 2558–2566, DOI:10.1021/acscatal.7b04479.
- R. H. Crabtree, Hydrogen Storage in Liquid Organic Heterocycles, Energy Environ. Sci., 2008, 134–138, 10.1039/b805644g.
- J. Andersson and S. Grönkvist, Large-Scale Storage of Hydrogen, Int. J. Hydrogen Energy, 2019, 11901–11919, DOI:10.1016/j.ijhydene.2019.03.063.
- R. Chamoun, U. B. Demirci and P. Miele, Cyclic Dehydrogenation-(Re)Hydrogenation with Hydrogen-Storage Materials: An Overview, Energy Technol., 2015, 100–117, DOI:10.1002/ente.201402136.
- Q. L. Zhu and Q. Xu, Liquid Organic and Inorganic Chemical Hydrides for High-Capacity Hydrogen Storage, Energy Environ. Sci., 2015, 478–512, 10.1039/c4ee03690e.
- D. Ventura-Espinosa, A. Marzá-Beltrán and J. A. Mata, Catalytic Hydrogen Production by Ruthenium Complexes from the Conversion of Primary Amines to Nitriles: Potential Application as a Liquid Organic Hydrogen Carrier, Chem. – Eur. J., 2016, 22(49), 17758–17766, DOI:10.1002/chem.201603423.
- J. Cho, B. Kim, S. Venkateshalu, D. Y. Chung, K. Lee and S. Choi, Il. Electrochemically Activatable Liquid Organic Hydrogen Carriers and Their Applications, J. Am. Chem. Soc., 2023, 145(31), 16951–16965, DOI:10.1021/jacs.2c13324.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Boosting Hydrogen Production by Anodic Oxidation of Primary Amines over a NiSe Nanorod Electrode, Angew. Chem., Int. Ed., 2018, 57(40), 13163–13166, DOI:10.1002/anie.201807717.
- M. Xiang, Z. Xu, Q. Wu, Y. Wang and Z. Yan, Selective Electrooxidation of Primary Amines over a Ni/Co Metal-Organic Framework Derived Electrode Enabling Effective Hydrogen Production in the Membrane-Free Electrolyzer, J. Power Sources, 2022, 535, 231461, DOI:10.1016/j.jpowsour.2022.231461.
- M. T. Bender and K. S. Choi, Electrochemical Dehydrogenation Pathways of Amines to Nitriles on NiOOH, JACS Au, 2022, 2(5), 1169–1180, DOI:10.1021/jacsau.2c00150.
- N. Guenani, M. Barawi, I. J. Villar-García, J. Bisquert, V. A. De La Peña O'Shea and A. Guerrero, Highly Porous Ti-Ni Anodes for Electrochemical Oxidations, Sustainable Energy Fuels, 2020, 4(8), 4003–4007, 10.1039/d0se00242a.
- R. Arcas, Y. Koshino, E. Mas-Marzá, R. Tsuji, H. Masutani, E. Miura-Fujiwara, Y. Haruyama, S. Nakashima, S. Ito and F. Fabregat-Santiago, Pencil Graphite Rods Decorated with Nickel and Nickel-Iron as Low-Cost Oxygen Evolution Reaction Electrodes, Sustainable Energy Fuels, 2021, 5(15), 3929–3938, 10.1039/d1se00351h.
- G. P. Lu, X. Li, L. Zhong, S. Li and F. Chen, Ru@UiO-66(Ce) Catalyzed Acceptorless Dehydrogenation of Primary Amines to Nitriles: The Roles of Lewis Acid-Base Pairs in the Reaction, Green Chem., 2019, 21(19), 5386–5393, 10.1039/c9gc02181g.
- K. N. T. Tseng, A. M. Rizzi and N. K. Szymczak, Oxidant-Free Conversion of Primary Amines to Nitriles, J. Am. Chem. Soc., 2013, 135(44), 16352–16355, DOI:10.1021/ja409223a.
- I. Dutta, S. Yadav, A. Sarbajna, S. De, M. Hölscher, W. Leitner and J. K. Bera, Double Dehydrogenation of Primary Amines to Nitriles by a Ruthenium Complex Featuring Pyrazole Functionality, J. Am. Chem. Soc., 2018, 140(28), 8662–8666, DOI:10.1021/jacs.8b05009.
- T. Achard, J. Egly, M. Sigrist, A. Maisse-François and S. Bellemin-Laponnaz, Easy Ruthenium-Catalysed Oxidation of Primary Amines to Nitriles under Oxidant-Free Conditions, Chem. – Eur. J., 2019, 25(58), 13271–13274, DOI:10.1002/chem.201902557.
- D. Carvajal, R. Arcas, L. Gouda, F. Fabregat-Santiago and E. Mas-Marzá, Electrochemical Valorization of HMF Using Ni/Graphite Electrodes, Mater. Chem. Phys., 2024, 311, 128510, DOI:10.1016/j.matchemphys.2023.128510.
- L. Gouda, L. Sévery, T. Moehl, E. Mas-Marzá, P. Adams, F. Fabregat-Santiago and S. D. Tilley, Tuning the Selectivity of Biomass Oxidation over Oxygen Evolution on NiO-OH Electrodes, Green Chem., 2021, 23(20), 8061–8068, 10.1039/d1gc02031e.
- D. Carvajal, R. Arcas, C. A. Mesa, S. Giménez, F. Fabregat-Santiago and E. Mas-Marzá, Role of Pd in the Electrochemical Hydrogenation of Nitrobenzene Using CuPd Electrodes, Adv. Sustainable Syst., 2022, 6(4), 2100367, DOI:10.1002/adsu.202100367.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization by IS, NMR, GC and SEM. See DOI: https://doi.org/10.1039/d4gc01275e |
| This journal is © The Royal Society of Chemistry 2024 |