
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydride-free reduction of propargyl electrophiles: a nickel-catalyzed photoredox strategy for allene synthesis†
Tingjun
Hu
a,
Vincent
Fagué
a,
Didier
Bouyssi
a,
Nuno
Monteiro
a and
Abderrahmane
Amgoune
 *ab
*ab
aUniversite Lyon 1, Institut de Chimie et Biochimie Moléculaires et Supramoléculaires, UMR 5246 du CNRS, 1, rue Victor Grignard, 69100 Villeurbanne, France. E-mail: abderrahmane.amgoune@univ-lyon1.fr
bInstitut Universitaire de France (IUF), 1 rue Descartes, 75231 Paris, France
First published on 2nd April 2024
Abstract
Herein we report a catalytic reduction of propargyl carbonates to allenes mediated by a nickel molecular catalyst. The catalytic system uses light as the driving force and amine as the sole hydrogen source. Contrary to other catalytic approaches, the process proceeds without the intermediacy of metal hydride species. The commonly observed pathway in transition metal catalyzed reductive processes is replaced by a pathway involving a sequence of electron transfer and proton transfer. Using this catalytic approach, a wide range of allenes could be obtained under mild conditions. Experimental investigations support the dual role of trialkylamine as a reductant and a proton source and have revealed the key mechanistic features of the reaction. A key protodenickelation step of the Ni(II) allenyl intermediate is proposed to account for the reduction process. Finally, we also demonstrate that the selective SN2′ reduction process can be also efficiently driven by an electrochemical approach.
Introduction
Allenes are key intermediates in organic synthesis, offering a rich palette of chemical reactivity and serving as essential building blocks in the construction of complex molecules. They are important structural motifs found in natural products and pharmaceuticals, and used to design advanced functional materials as well, making them interesting target molecules.1 In this regard, transition metal-catalyzed reactions of propargyl electrophiles with nucleophilic partners stand among the most attractive strategies for constructing allene motifs featuring otherwise difficult-to-access substitution patterns and functionalities. A long-standing challenge in the development of such methods has been the difficulty in predicting and controlling regioselectivity (SN2vs. SN2′ selectivity), and thus preventing the formation of isomeric propargylic by-products.2In this domain, catalytic hydride reductions of propargyl halides and propargyl alcohol derivatives have emerged as established methodologies since the mid-80s, serving as practical tools for the synthesis of mono- to tri-substituted allenes. Traditional methods rely essentially on palladium3 or copper4 catalysis in combination with conventional hydride-based reducing agents, i.e. aluminium or boron hydrides,3b,4e formates,3c,4b and silanes.4a,c,d In these processes, transition metal hydrides serve as key intermediates (Fig. 1A). However, the reliance on stoichiometric Al, B, and Si-based metalloid hydrides often in conjunction with a noble transition metal can limit broader applications with respect to functional group tolerance, safety, and sustainability concerns.
 | ||
| Fig. 1 Synthetic methods for allenes involving transition metal (TM)-catalyzed reduction of propargyl electrophiles. | ||
Complementary to conventional thermal protocols, photo- or electrocatalytic processes provide practical and efficient routes for catalytic reduction reactions, harnessing electrons and protons as redox equivalents, and thus obviating the use of hydrogen gas or hydride donors.5 From a mechanistic standpoint, all these methodologies rely on the formation of metal hydrides as catalytically relevant intermediates. More recently, alternative pathways exempt of hydride intermediates have attracted increasing interest as sustainable and cost-effective strategies for hydrogenation/reduction processes.5a,b The reaction manifolds involve a reduction of the transition metal catalyst that activates the substrate followed by protonation. In this context, we present a novel nickel-catalyzed photoredox reduction strategy for a selective, sustainable and robust access to diversely substituted allenes under mild reaction conditions from propargyl electrophiles.6 Unlike traditional methods, our approach eliminates the requirement for hydride nucleophiles, instead relying on protodemetalation of an allenylmetal intermediate. Notably, we demonstrate that a trialkylamine plays the roles of both a redox mediator, as a reductive quencher of the photocatalytic cycle, and a proton source, via proton transfer from its photochemically generated amine radical cation (Fig. 1B).7,8
Results and discussion
Development and optimization studies
Preliminary studies have focused on the reduction of alk-2-ynyl carbonate 1a as a model substrate (Table 1). Extensive experimentation led to the identification of nickel(II) nitrate, 1,10-phenanthroline (2), and 9,10-diphenylanthracene (DPA, 3a) as an effective and inexpensive nickel source, ligand and organo-photocatalyst, respectively. DPA is commonly used as an organic semi-conductor in OLEDs and in blue glow sticks; however, its potential as an effective photocatalyst has only been illustrated very recently.9 Acetone, a sustainable and non-toxic solvent, was also identified as an appropriate medium for the reaction. Thus, when the reduction of 1a was performed in the presence of triethylamine (1 equiv.) with 5 mol% of catalyst and ligand each under purple light (405 nm) irradiation, the corresponding allene 6 was produced exclusively within 3 h, and in almost quantitative spectroscopic yield (96%, 19F NMR) (Table 1, entry 1). The reaction also proceeded but to a lesser extent with the air-sensitive Ni(COD)2/1,10-Phen catalytic system (Table 1, entry 2). Control experiments confirmed that the dual catalytic system, light irradiation, and triethylamine are essential for this transformation (Table 1, entries 3–7). 1,2,3,5-Tetrakis(carbazol-9-yl)-4,6-dicyano-benzene (4-CzIPN, 4) as well as the iridium complex Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 (5) proved to be competent alternative photocatalysts. However, the reaction efficiency is compromised by concomitant formation of several unidentified by-products, most likely generated from photo-degradation of the desired allenic product, leading to lower yields (Table 1, entries 9 and 10; and Table S6, ESI†). Furthermore, the reaction proceeded well with lower loadings of DPA, down to 1 mol%, provided that the reaction time was extended to 24 h (Table 1, entry 11). Other DPA derivatives were also investigated. Remarkably, both 9-(4-methoxyphenyl)-10-phenyl-anthracene (DPA-OMe, 3b) and 9-(4-methoxycarbonylphenyl)-10-phenylanthracene (DPA-CO2Me, 3c) had a beneficial effect on the reaction, affording quantitative yields of 6 (Table 1, entries 12 and 13). Interestingly, a carboxylic acid derivative (DPA-CO2H, 3d) also showed significant photocatalytic activity, although the reaction was hampered by slower reaction rates (Table 1, entry 14).10 Under the improved reaction conditions, a nickel catalyst loading of 2 mol% was still sufficient for excellent catalytic activity within 24 h, especially with DPA-CO2Me as the photocatalyst (Table 1, entries 15 and 16). Finally, we identified the OBoc group as a competent alternative leaving group, where OAc proved less efficient (Table 1, entries 17 and 18).|
|
||
|---|---|---|
| Entry | Deviation from the above reaction | Yieldb |
| a Reactions were performed on a 0.25 mmol scale using an EvoluChem™ PhotoRedOx Box device equipped with one 18 W purple LED lamp (405 nm). Dtbbpy = 4,4′-di-tert-butyl-2,2′-dipyridyl. b Yields (%) were determined by 19F NMR spectroscopy using fluorobenzene as an internal standard. c 1.1 equiv. of Et3N were used. Approximately 20% of 1a remained unreacted. d 38% conversion. | ||
| 1 | None | 96 |
| 2 | Ni(COD)2/1,10-Phen as catalyst | 56 |
| 3 | No nickel salt | 0 |
| 4 | No ligand | 0 |
| 5 | No photocatalyst (light only) | 0 |
| 6 | No light (dark) | 0 |
| 7 | w/o Et3N | 0 |
| 8 | Quinuclidine instead of Et3N | 0 |
| 9 | [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (1 mol%) as PC | 66 |
| 10 | 4-CzIPN (2 mol%) as PC | 53 |
| 11 | 1 mol% DPA (24 h) | 78 |
| 12 | 1 mol% DPA-OMe (24 h) | 99 |
| 13 | 1 mol% DPA-CO2Me (24 h) | 99 |
| 14 | 1 mol% DPA-CO2H (48 h)c | 80 |
| 15 | 1 mol% DPA-OMe, 2 mol% [Ni/L] (24 h) | 81 |
| 16 | 1 mol% DPA-CO2Me, 2 mol% [Ni/L] (24 h) | 89 |
| 17 | R = Boc | 78 |
| 18 | R = Ac | 36d |

|
||

Scope and limitations
The substrate scope was subsequently explored under our standard set of reaction conditions using a broad range of propargylic carbonates featuring diverse substitution patterns. Structurally diverse mono- to tri-substituted allenes could be delivered regioselectively in good to high yields. Tolerated substituents include electron rich or poor (hetero)aromatic rings, cycloalkyl and short or long chain alkyl groups bearing a variety of functional groups such as CF2, alkene, carboxylic ester, as well as protected ketone and diol, amine, or alcohol that can facilitate the post-reduction derivatization to nitrogen- and oxygen-containing molecules of higher complexity (Table 2).
Regarding access to internal allenes, the alkyne substitution pattern had no observable impact on the outcome of the reaction. Internal alkynes with alkyl (15–19 and 25–26) or (hetero)aryl (6–14 and 20–23) substituents performed equally well. Moreover, ortho substituents on the aryl rings had no detrimental effect on the reaction efficiency and selectivity (11 and 12). Additionally, a propargylic silyl ether did not interfere with the reaction (19), and a glycosyl ester (20) resisted interchange of the alkoxy moiety with methoxide generated in the reaction medium. On the other hand, the substitution mode at the carbon center adjacent to the carbonate leaving group had a notable impact on the profile of the reaction. While tertiary (6–20) as well as secondary (21–23 and 26) propargyl carbonates featuring alkyl groups delivered the desired allenes with satisfactory yields, α-aryl propargylic carbonates proved to be less suitable substrates for the transformation. Hence, 1,3-bisarylallenes (e.g., 24) were not accessible by this method, only providing degradation products. Accordingly, monoaryl 1,3-disubstituted allenes were only provided in moderate yields starting from α-aryl propargylic carbonates. However, these monoaryl allenic compounds may still be afforded in good yields by simply inverting the substitution pattern of the propargyl substrate (e.g.25, path a vs. path b).
Regarding access to terminal allenes (27–29), these were delivered in satisfactory yields from primary propargyl carbonates, although alkynyl derivatives were also formed as minor regioisomeric products.11 Alternatively, terminal allenes can be obtained from terminal propargylic carbonates, as illustrated by the reduction of the mestranol carbonate derivative (30). The generality of the method was further showcased by the successful reduction of enyne carbonates under our catalytic conditions, thus opening a new synthetic entry to valuable vinylallenes (31, 32). Notably, electrophiles that are both allylic and propargylic offer several potential sites for the reduction to take place and therefore raise additional selectivity issues, i.e. competition between the allylic and propargylic functionalities. Remarkably, the 1,4-enyne substrate delivered the desired vinyl allene (32) chemoselectively as the sole reaction product. Finally, cyclic carbonates have also been shown to be competent electrophiles which further expand the diversity of accessible structures to include unprotected allenol derivatives, which have become ubiquitous as building blocks for downstream methodologies (33).12 Importantly, in addition to proceeding in a sustainable fashion under very mild reaction conditions, the reduction process was easily amenable to simple scale up. For instance, a gram-scale synthesis of allene 6 could be achieved in high yield without the need for reoptimizing reaction parameters (Scheme 1).
 | ||
| Scheme 1 Gram-scale synthesis. | ||
Mechanistic investigations
We then turned our attention to studying the pathway of this hydride-free Ni/photoredox reduction process and the exact role of Et3N (Fig. 2). We envisioned that the delivery of the hydrogen atom would proceed through the following pathway (Fig. 2A): (i) photooxidation of Et3N by the excited DPA* (E* = +1.19 V vs. SCE in DMSO); (ii) deprotonation of the α-amino C–H (pKa = 14.7 in MeCN) of the triethylamine radical cation species by methanolate, which is formed upon spontaneous decarboxylation of the Ni–OCO2Me intermediate,13 and finally, (iii) protonation of the Ni–allenyl bond by the generated methanol to form the desired allene product. This was confirmed by a deuterium labelling experiment using fully deuterated triethylamine (Et3N-d15), which led to 90% deuterium incorporation into the nonexchangeable allenylic position. Interestingly, when the reaction was carried out in the presence of deuterated methanol, the desired product was obtained with a deuteration yield of 68%. This experiment indicates that methanol can serve as a protonolysis shuttle agent. The lower deuterium incorporation ratio is likely due to the presence of in situ generated CH3OH through deprotonation of the Et3N radical cation species by methanolate. Interestingly, when iPr2NMe was used, we could detect the formation of hemiaminal ether species 34, most likely ensuing from the capture of an iminium intermediate by methanolate (Fig. 2B). We speculated that the resulting N-α-carbon radical intermediate may undergo spontaneous single electron oxidation to provide this iminium intermediate.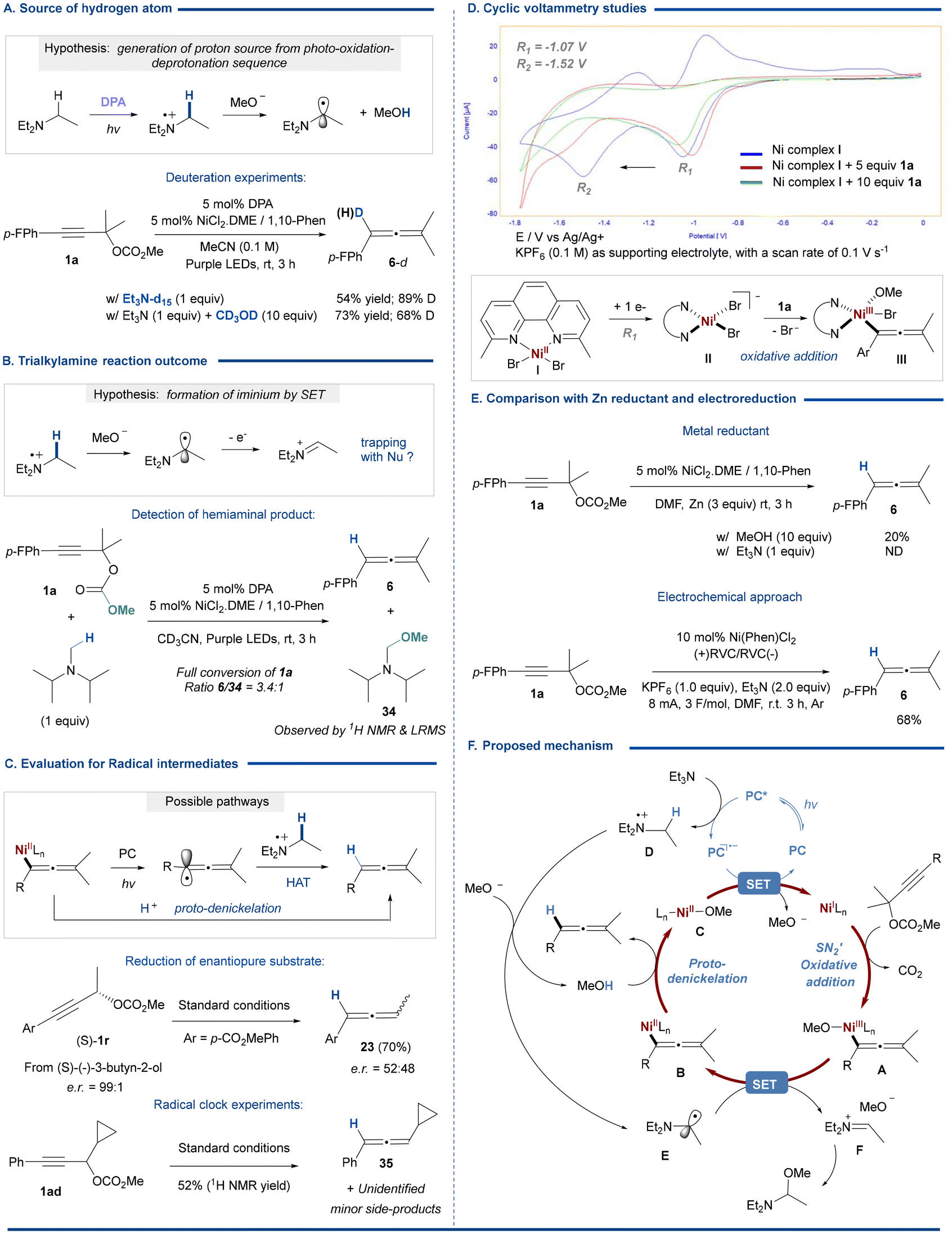 | ||
| Fig. 2 Mechanistic experiments and comparison with alternative reductive methodologies. | ||
Next, we aimed to evaluate whether propargyl/allenyl radical intermediates may be formed under photoredox conditions (Fig. 2C). As anticipated from a pathway involving radical species, the reaction was inhibited in the presence of 1 equivalent of 2,2,6,6-tetramethylpiperidinyloxy (TEMPO).14 In addition, we observed a loss of stereochemistry when exposing enantiopure (S)-1r to catalytic conditions. The racemization process may involve either propargyl/allenyl radicals6b or metallotropic rearrangements of the oxidative addition nickel complex, possibly via η3-propargyl/allenyl nickel intermediates.15 Noteworthily, post-reduction photochemical erosion of allene chirality may also proceed (see the ESI†).16 Finally, the reaction of the cyclopropyl derivative 1ad resulted in the formation of the non-opened cyclopropyl allene 35 as a major product, alongside minor byproducts that could not be isolated or identified. This result argues against the formation of the free allenyl radical intermediate as a main pathway. All these experiments support the direct protonolysis of the oxidative addition allenyl–Ni complex involving a proton transfer process from the amine to the allene product. The detection of the hemiaminal ether 34 indicates that the trialkylamine reagent is converted into iminium species, most likely through a SET process from the α-aminoalkyl radical.
Finally, we were also interested to delineate the nature of low valent nickel species that promotes the oxidative addition of the propargyl carbonate, and particularly its oxidation state.17 For that purpose, we conducted cyclic voltammetry (CV) measurement of the prepared Ni(II) complexes bearing the neocuproine ligand (Fig. 2D). This ligand, which is also effective for catalysis (see Table S3, ESI†), was chosen because it provides more interpretable CVs for the nickel complexes.18 Indeed, the CV of complex I exhibited two reversible reduction waves at −1.07 V and −1.52 V (vs. Ag/AgNO3) associated with Ni(II)/N(I) and Ni(I)/Ni(0) reductions (Fig. 2D). Upon the addition of several equivalents of propargyl carbonate 1a (5 and 10 equiv.), an increase in current is observed at the first reduction peak associated with Ni(II)/N(I) reduction along with a complete loss of reversibility. A slight shift of the reduction peak is also observed, most likely due to pre-coordination of the substrate to nickel (Fig. S20, ESI†). This experiment is consistent with a facile oxidative addition of the propargyl carbonate to the electrogenerated Ni(I) species (II) to give a Ni(III)–allenyl species (III). Furthermore, a new reduction peak is observed at lower potential (−1.6 V). We speculate that it may be associated with the reduction of a Ni(II)–allenyl intermediate presumably formed by rapid reduction of the Ni(III) transient species.
With this information in mind, we were interested in evaluating the effect of the reduction strategy on the catalytic efficiency (Fig. 2E). Notably, the use of zinc as a reductant, instead of the DPA/Et3N system under photoredox conditions, resulted in very low conversion to allene, thus indicating the significant influence of the photoredox promoted electron transfer process using homogeneous reductants.19 We reasoned that electrochemical reduction may also stand as a reliable technology to promote the electron transfer in a controlled and smooth manner. Remarkably, the reaction also proceeded well under electrochemical conditions.20 The desired allene could be obtained in good yield using RVC electrodes in an undivided cell under 8 mA constant current electrolysis. Satisfactorily, the use of Et3N, playing the roles of both a hydrogen source and a reductant also affords the possibility of avoiding the use of a sacrificial anode.21
Based on these investigations, a plausible mechanism for this nickel catalyzed, selective and hydride-free reduction of propargyl carbonates into allenes can be proposed, as shown in Fig. 2F. The pathway involves oxidative addition of the propargyl carbonate to low valent ligated Ni(I) species, which are generated via photocatalytic (or electro-) reduction, to give a Ni(III)–allenyl intermediate A, after spontaneous decarboxylation of the carbonate substituent. In parallel, photo- (or electro-) oxidation of the trialkylamine would generate the radical cation intermediate D that may undergo rapid deprotonation by the in situ generated methanolate anion. The resulting N-α-carbon radical intermediate E can participate in single electron transfer with the Ni(III)–allenyl intermediate A to give an iminium species F and the Ni(II)–allenyl intermediate B. A final protonolysis step with in situ generated methanol will deliver the desired allene product, and the Ni(II) species C can be reduced to regenerate the Ni(I) species.
Conclusions
In conclusion, our study presents a new strategy for the synthesis of allenes through the development of a photocatalytic hydride-free protocol for the regioselective reduction of propargyl carbonates. This method offers several key advantages, including the utilization of low loadings of inexpensive catalysts and ligands, alongside a sustainable and non-toxic solvent, acetone. Furthermore, our investigation unravels a novel mechanistic pathway involving hitherto undocumented protodenickelation of allenylnickel species,22 shedding light on new avenues for catalytic hydrogen transfer processes. The use of trialkylamine as an inexpensive reductant and a source of hydrogen atoms further underscores the versatility and efficiency of our approach.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CNRS, the French Ministry of Research, the ICBMS, and the Université Claude Bernard Lyon 1. The NMR and Mass Centers of the Université Claude Bernard Lyon 1 are gratefully acknowledged for their contribution. T. H. thanks the Chinese Council Scholarship for a doctoral fellowship. A. A. thanks the Institut Universitaire de France (IUF) for its support. We thank Maurice Médebielle and Jérémy Merad for their help with cyclic voltammetry experiments and HPLC determination of enantiomeric ratios, respectively.Notes and references
- For representative books and reviews on allene chemistry, see: (a) Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) S. Ma, Chem. Rev., 2005, 105, 2829–2871 CrossRef PubMed; (c) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef PubMed; (d) P. Rivera-Fuentes and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 CrossRef CAS PubMed.
- Representative reviews: (a) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384–5418 RSC; (b) C. Q. O'Broin and P. J. Guiry, J. Org. Chem., 2020, 85, 10321–10333 CrossRef PubMed; (c) S. Du, A.-X. Zhou, R. Yang, X.-R. Song and Q. Xiao, Org. Chem. Front., 2021, 8, 6760–6782 RSC.
- (a) For an overview of early discoveries: J. Tsuji and T. Mandai, Synthesis, 1996, 1–24 CrossRef CAS Selected papers: (b) Y. Colas, B. Cazes and J. Gore, Tetrahedron Lett., 1984, 25, 845–848 CrossRef CAS; (c) J. Tsuji, T. Sugiura, M. Yuhara and I. Minami, J. Chem. Soc., Chem. Commun., 1986, 922–924 RSC; (d) T. Mandai, T. Matsumoto, Y. Tsujiguchi, S. Matsuoka and J. Tsuji, J. Organomet. Chem., 1994, 473, 343–352 CrossRef CAS; (e) H. Ohmiya, M. Yang, Y. Yamauchi, Y. Ohtsuka and M. Sawamura, Org. Lett., 2010, 12, 1796–1799 CrossRef CAS PubMed; (f) Y. Zheng, B. Miao, A. Qin, J. Xiao, Q. Liu, G. Li, L. Zhang, F. Zhang, Y. Guo and S. Ma, Chin. J. Chem., 2019, 37, 1003–1008 CrossRef CAS.
- (a) C. Deutsch, B. H. Lipshutz and N. Krause, Angew. Chem., Int. Ed., 2007, 46, 1650–1653 CrossRef CAS PubMed; (b) C. Zhong, Y. Sasaki, H. Ito and M. Sawamura, Chem. Commun., 2009, 5850–5852 RSC; (c) C. Deutsch, B. H. Lipshutz and N. Krause, Org. Lett., 2009, 11, 5010–5012 CrossRef CAS PubMed; (d) H. Reeker, P.-O. Norrby and N. Krause, Organometallics, 2012, 31, 8024–8030 CrossRef CAS; (e) Y. Kim, H. Lee, S. Park and Y. Lee, Org. Lett., 2018, 20, 5478–5481 CrossRef CAS PubMed See also: (f) K. Guo and A. W. Kleij, Angew. Chem., Int. Ed., 2021, 60, 4901–4906 CrossRef CAS PubMed.
- (a) G. Durin, M.-Y. Lee, M. A. Pogany, T. Weyhermüller, N. Kaeffer and W. Leitner, J. Am. Chem. Soc., 2023, 145, 17103–17111 CrossRef CAS PubMed; (b) F. Arcudi, L. Đorđević, N. Schweitzer, S. I. Stupp and E. A. Weiss, Nat. Chem., 2022, 14, 1007–1012 CrossRef CAS PubMed; (c) C. Casadevall, D. Pascual, J. Aragon, A. Call, A. Casitas, I. Casademont-Reig and J. Lloret-Fillol, Chem. Sci., 2022, 13, 4270–4282 RSC; (d) T. Hu, M. Jaber, G. Tran, D. Bouyssi, N. Monteiro and A. Amgoune, Chem. – Eur. J., 2023, e202301636 CrossRef CAS PubMed.
- Nickel-catalyzed reactions of propargyl electrophiles have been documented to provide effective ways of accessing allenic compounds. However, to the best of our knowledge, Ni-catalyzed reduction protocols remain unexplored to this date. For a review, see: (a) X. Liu, C. Jiao, S. Cui, Q. Liu, X. Zhang and G. Zhang, ChemCatChem, 2023 DOI:10.1002/cctc.202300601 For selected nickel-catalyzed approaches towards allene synthesis from propargyl electrophiles, see: (b) R. Soler-Yanes, I. Arribas-Alvarez, M. Guisan-Ceinos, E. Bunuel and D. J. Cardenas, Chem. – Eur. J., 2017, 23, 1584–1590 CrossRef CAS PubMed; (c) Y. Jin, H. Wen, F. Yang, D. Ding and C. Wang, ACS Catal., 2021, 11, 13355–13362 CrossRef CAS; (d) Z.-Z. Zhou, X.-R. Song, S. Du, K.-J. Xia, W.-F. Tian, Q. Xiao and Y.-M. Liang, Chem. Commun., 2021, 57, 9390–9393 RSC.
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- For recent examples of metallaphotoredox transformations involving tertiary amines as proton transfer reagents, see: Z.-Y. Gu, W.-D. Li, Y.-L. Li, K. Cui and J.-B. Xia, Angew. Chem., Int. Ed., 2023, e202213281 CAS See also ref. 5d.
- For physical properties and previous (rare) uses of DPA as a photocatalyst, see: (a) Y. Chen, X. Wang, X. He, Q. An and Z. Zuo, J. Am. Chem. Soc., 2021, 143, 4896–4902 CrossRef CAS PubMed; (b) M. Neumeier, U. Chakraborty, D. Schaarschmidt, V. de la Pena O'Shea, R. Perez-Ruiz and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2020, 59, 13473–13478 CrossRef CAS PubMed; (c) K. Xu, J. Zhao, X. Cui and J. Ma, J. Phys. Chem. A, 2015, 119, 468–481 CrossRef CAS PubMed; (d) V. Gray, D. Dzebo, A. Lundin, J. Alborzpour, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Mater. Chem. C, 2015, 3, 11111–11121 RSC; (e) S. K. Chattopadhyay, C. V. Kumar and P. K. V. Das, Chem. Phys. Lett., 1983, 98, 250–254 CrossRef CAS.
- An additional advantage of incorporating a functional group into the diphenylanthracene structure lies in the large increase in polarity facilitating separation of these photocatalysts from aliphatic allenes featuring polarities similar to that of DPA.
- Primary propargylic carbonates have been shown to generate mixtures of terminal allenes and propargylic reduced compounds due to steric reasons: T. Mandai, T. Matsumoto, Y. Tsujiguchi, S. Matsuoka and J. Tsuji, J. Organomet. Chem., 1994, 473, 343–352 CrossRef CAS.
- J. M. Alonso and P. Almendros, Chem. Rev., 2021, 121, 4193–4252 CrossRef CAS PubMed.
- For a seminal paper on transition metal-catalyzed decarboxylative reactions of propargyl electrophiles, see: J. Tsuji, H. Watanabe, I. Minami and I. Shimizu, J. Am. Chem. Soc., 1985, 107, 2196–2198 CrossRef CAS For recent applications, see ref. 2b.
- See the ESI† for details.
- Similarly to related Pd-catalyzed processes, racemization may occur through interconversion of η1-allenyl, η1-propargyl and η3-allenyl/propargyl ligands on the metal: (a) K. Mikami and A. Yoshida, Angew. Chem., Int. Ed. Engl., 1997, 36, 858–860 CrossRef CAS; (b) H. Wang, H. Luo, Z.-M. Zhang, W.-F. Zheng, Y. Yin, H. Qian, J. Zhang and S. Ma, J. Am. Chem. Soc., 2020, 142, 9763–9771 CrossRef CAS PubMed The racemization of an optically active η1-allenyl–palladium(II) species has been reported to occur via an η3-allenyl/propargyl dimetallic complex: (c) S. Ogoshi, T. Nishida, T. Shinagawa and H. Kurosawa, J. Am. Chem. Soc., 2001, 123, 7164–7165 CrossRef CAS PubMed.
- For photosensitized racemization of allenes, see: O. Rodriguez and H. Morrison, J. Chem. Soc. D, 1971, 679 RSC.
- T. Kerackian, D. Bouyssi, G. Pilet, M. Médebielle, N. Monteiro, J. C. Vantourout and A. Amgoune, ACS Catal., 2022, 12, 12315–12325 CrossRef CAS.
- This ligand minimizes speciation issues with Ni(II) and undesired disproportionation reactions with Ni(I): T. Tang, A. Hazra, D. S. Min, W. L. Williams, E. Jones, A. G. Doyle and M. S. Sigman, J. Am. Chem. Soc., 2023, 145, 8689–8699 CAS.
- The poor yield obtained with the Zn reductant may be attributed to the harsher electron transfer with a heterogeneous metal reductant and/or to undesired pathways involving organometallic intermediates.
- For previous metal-catalyzed electroreductive cleavages of propargyl compounds, see. (a) S. Olivero and E. Duñach, Tetrahedron Lett., 1997, 38, 6193–6196 CrossRef CAS; (b) H. Tanaka, H. Takeuchi, Q. Ren and S. Torii, Electroanalysis, 1996, 8, 769–772 CrossRef CAS.
- For other recent examples of electrochemical strategies using Et3N as a sacrificial reductant, see Ni-catalyzed processes: (a) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338 CrossRef CAS PubMed; (b) R. J. Perkins, A. J. Hughes, D. J. Weix and E. C. Hansen, Org. Process Res. Dev., 2019, 23, 1746–1751 CrossRef CAS; (c) A. Claraz and G. Masson, ACS Org. Inorg. Au, 2022, 2, 126–147 CrossRef CAS PubMed , (Review) For specific examples of reduction processes, see: (d) B. Huang, L. Guo and W. Xia, Green Chem., 2021, 23, 2095–2103 RSC; (e) J. Ke, H. Wang, L. Zhou, C. Mou, J. Zhang, L. Pan and Y. R. Chi, Chem. – Eur. J., 2019, 25, 6911–6914 CrossRef CAS PubMed.
- For a recent mechanistic investigation of protodenickelation of Ni(II)-aryl species, see: P. E. Piszel, B. J. Orzolek, A. K. Olszewski, M. E. Rotella, A. M. Spiewak, M. C. Kozlowski and D. J. Weix, J. Am. Chem. Soc., 2023, 145, 8517–8528 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00984c |
| This journal is © The Royal Society of Chemistry 2024 |