
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comprehensive two-step supercritical fluid extraction for green isolation of volatiles and phenolic compounds from plant material†
Veronika
Pilařová
 ,
Kateřina
Plachká
,
Diana
Herbsová
,
Štefan
Kosturko
,
Frantisek
Svec
and
Lucie
Nováková
*
,
Kateřina
Plachká
,
Diana
Herbsová
,
Štefan
Kosturko
,
Frantisek
Svec
and
Lucie
Nováková
*
Department of Analytical Chemistry, Faculty of Pharmacy in Hradec Králové, Charles University, Akademika Heyrovského 1203, 500 03 Hradec Králové, Czech Republic. E-mail: nol@email.cz
First published on 30th April 2024
Abstract
Extraction of compounds with different physicochemical properties from a complex matrix usually involves several individual steps and requires large volumes of organic solvents. In this pioneering study, we propose a comprehensive two-step supercritical fluid extraction using carbon dioxide, ethanol, and water. This novel approach allows the extraction of non-polar and polar analytes within one run in two consecutive steps. Indeed, the first step with a dominant amount of CO2 with only 2% cosolvent allowed the selective extraction of non-polar volatile terpenes only in 20 min. The conditions were then automatically switched. Increasing the cosolvent volume in the extraction solvent up to 44% (v/v) resulted in the extraction of more polar compounds, including flavonoids and phenolic acids, in 60 min. Importantly, switching the supercritical fluid extraction (SFE) conditions does not require any manual intervention but results in two separate fractions containing target compounds with distinctly different physicochemical properties. The novel method was verified in terms of repeatability, accuracy, precision, and greenness. Two-step SFE was applied to seven plant species differing in volatile terpenes and phenolic profiles. The results proved that this concept is suitable for the analysis of complex plant samples. In addition, it enables a reduction in the toxic solvents consumption, extraction time, and manual intervention required for traditional extraction approaches when isolating different groups of metabolites.
Introduction
Plants are an important source of bioactive compounds, including several classes of compounds that differ in physicochemical properties, such as molecular weight, polarity, and acidic-basic properties. Various extraction methods have been developed and optimized to isolate target analytes from plant matrices, but the real challenge arises when different classes of metabolites are to be extracted from the same sample.1–3Conventional methods, such as maceration, decoction, infusion, digestion, percolation, and more advanced Soxhlet extraction and hydrodistillation, are still among the most commonly used methods in many laboratories. They are based on the extraction of the solid plant matrix with water and/or organic solvents of different polarities, e.g., ethanol, methanol, chloroform, and hexane. Inorganic and organic acids, bases, and buffers are also used to improve the extractability of compounds with acidic/basic properties. The selectivity of the method is mainly affected by the extraction solvent. If compounds with different lipophilicity have to be extracted from a matrix, different multi-step extractions are carried out to obtain the fractions, resulting in a time-consuming process. Despite their undoubted simplicity and low demands on laboratory equipment, the classical extraction methods suffer from high consumption of organic solvents and their toxicity, affecting extraction safety, long extraction times, and low extraction yields. Filtration of the extract is also required. In addition, many traditional methods are carried out at elevated temperatures or above the boiling point. This can lead to the degradation of thermolabile compounds and the loss of volatile compounds.2–6 Several instrumental methods have been introduced to overcome these drawbacks and increase the safety of extraction. Ultrasonic-assisted extraction, microwave-assisted extraction, and pulsed electric field extraction significantly reduce the extraction time and solvent consumption. They also increase the extraction yield by accelerating the dissolution and diffusion of the analytes due to the applied mechanical stress. The selectivity of these methods is typically low due to the use of polar solvents (water, alcohols), which, together with mechanical stress, increase the extractability of the entire sample content.2–4 Another method, pressurized liquid extraction (PLE), alters the properties of polar solvents by using elevated pressure. It increases mass transfer and compound solubility, resulting in more effective and faster extraction. Again, PLE primarily uses polar solvents. This makes it the preferred method for the isolation of polar compounds.2,7
In contrast, supercritical fluid extraction (SFE) is based on the use of carbon dioxide under supercritical conditions, p > 74 bar, T > 31 °C. Supercritical CO2 is considered a non-polar solvent due to its extremely low relative permittivity. Thus, SFE is preferred for the extraction of non-polar compounds, and the extraction of polar compounds can be challenging. Nevertheless, the properties of CO2 can be easily tuned due to its high (i) compressibility and (ii) miscibility with different organic solvents. Indeed, (i) the changes in pressure and temperature affect the solvent density, the solubility of the compounds, and thus, their extractability and extraction selectivity targeting different groups of analytes. The increased pressures can result in the extraction of medium-polar compounds. (ii) The addition of 1–20% of polar organic solvents (cosolvents), such as methanol and ethanol, increases the relative permittivity and, thus, the solvent strength of CO2 and the extraction selectivity. Then, the polar compounds can be easily extracted by SFE.2,8,9 It should be emphasized, that selectivity in this regard implies the ability of the extraction method to discriminate against defined unwanted interferences.10 Recently, it has been demonstrated that the amount of cosolvent in CO2 can be increased by more than 20%, resulting in a gas-expanded liquid with different physicochemical properties compared to the liquid state of the organic solvent. The formation of a single-phase liquid must be observed to avoid phase separation and poor extraction repeatability.11
According to the theory of SFE, the use of different conditions to one sample should result in the collection of individual extracts containing compounds with different physicochemical properties. Therefore, we aimed at the optimization of a comprehensive two-step SFE for the extraction of different groups of bioactive compounds from plant materials in two consecutive steps without any sample manipulation. This proof-of-concept extraction method should increase the throughput of the plant samples in laboratories, reduce the use of toxic organic solvents, and be applicable to different plant species. Corymbia citriodora leaves were chosen as a model plant matrix because they are a rich source of various phytochemicals. Our study focused on groups of (i) volatile compounds, including various terpenes, ketones, and alcohols such as 1,8-cineole (eucalyptol), citronellol, citronellal, pinene, etc., (ii) phenolic acids, i.e., ellagic, gallic, and chlorogenic, (iii) flavonoids, i.e., quercetin, rutin, myricetin, quercitrin, etc., and (iv) triterpenoic acids.12–14 SFE has been successfully used in several published papers to extract essential oils15–18 and triterpenoic acids19,20 from leaves and bark of Eucalyptus sp. Typically, the neat CO2 was used at various pressures and temperatures for the extraction of volatiles, while the addition of ethanol (< 5%) was required for the extraction of triterpenoic acids. Phenolic acids and flavonoids are still preferably extracted by simple maceration and Soxhlet extraction.21–24 Nevertheless, several methods using SFE have been published for the extraction of quercetin and related compounds.11,25–28 To the best of our knowledge, only several articles have been published using consecutive steps in SFE, but with different purposes. Chuang demonstrated the application of counter-current extraction under supercritical conditions, including enrichment and esterification, for the extraction of minor components (fatty acid esters, vitamin E, carotenoids) in crude palm oil.29 Multistep fractionation of pine bark has also been carried out under supercritical conditions to extract fatty acids and phenols in different steps.30 Different lipid classes from microalga were extracted in different fractions using SFE and PLE.31 Various conditions, i.e., 150 and 300 bar and 40 °C, using different cosolvent amounts, were used in sequential SFE for the clean-up and removal of ballast compounds in the first step and extraction of carnosic acid and carosol within the second extraction step.32 It is evident that none of the published studies aim to extract different classes of bioactive compounds in a single run as the clean-up was typically carried out within the first step followed by the extraction of the compounds of interest. The workflow we optimized for Corymbia citriodora leaves was successfully applied to different plant species to confirm the ability of the method to selectively extract targeted groups of compounds in two consecutive steps.
Experimental
Chemicals and reagents
Reference standards of 17 volatiles, 3 triterpenoic acids, and 20 phenolic compounds used in this study are summarized in ESI Table S1.† Pressurized liquid CO2 4.5 grade (99.9995%) was purchased from Messer (Prague, Czech Republic). Methanol (MeOH), ethanol (EtOH), acetonitrile (ACN), and heptane in LC/MS grade quality were provided by VWR International (Prague, Czech Republic). Water (Optima LC-MS) was obtained from Fisher (FisherScientific, Loughborough, UK). Ammonia 4 mol L−1 solution in methanol and formic acid (99.9%) for LC/MS were purchased from Sigma-Aldrich (Steinheim, Germany).Standard solutions
The standard stock solutions of all reference standards were prepared by dissolving each compound in ACN, MeOH, or EtOH to obtain a stock solution at a concentration of 1 mg mL−1 as specified in ESI Table S1.† Stock solutions of tamarixetin, isorhamnetin, and ellagic acid were prepared at a concentration of 0.1 mg mL−1 due to their lower solubility. These solutions were stored at −20 °C. Pure ethanol was used for further dilution and preparation of mixed solutions.Plant material
Leaves and stems of lemon-scented gum, cider gum, bay laurel, and common myrtle were collected in the period from November 2020 to January 2021 in the Garden of Medicinal Plants, Hradec Králové. Leaves and stems of lemon grass and tea tree, and rose petals were collected in the period from June to August 2021. The fresh plant material was spread in thin layers on trays and dried by natural air flow (natural convection) in a dark room to avoid material degradation by sunlight. The homogeneous material was ground to powder using an IKA A11 basic analytical mill (IKA-Werke GmnH & Co. KG, Staufen, Germany). It was sieved using a manual set of sieves to obtain a fine homogeneous sample for extraction. The optimization of the extraction method was carried out on Corymbia citriodora leaves (lemon-scented gum, particle size: 0.180–0.315 mm). The final method was applied to Eucalyptus gunii leaves (cider gum, 0.180–0.315 mm), Laurus nobilis leaves (bay laurel, 0.315–0.630 mm), Melaleuca alternifolia stems (tea tree, 0.315–0.630 mm), Rosa hybrida petals (rose, 0.180–0.315 mm), Myrtus communis stems (myrtle, 0.315–0.630 mm), and Cymbopogon citratus leaves (lemon grass, 0.180–0.315 mm). The powdered plant matrix was stored in amber glass bottles in the dark and at room temperature.Supercritical fluid extraction (SFE)
A MV-10 ASFE benchtop analytical system (Waters, Milford, MA, USA) was used for all extraction experiments. It consisted of a fluid delivery module with a high-pressure pump for CO2 cooled by a chiller operating at 5 °C, the pump for the organic cosolvent, a thermostatic oven for holding extraction vessels, an automated back pressure regulator, and a fraction collector module. The dynamic extraction mode was used with the solvent flow rate controlled as a volumetric ratio between CO2 and organic cosolvent. The system was controlled by ChromScope™ software (Waters, Milford, MA, USA). 0.5 g dried homogenized sample was placed in the 5 mL stainless steel extraction vessel between two layers of 3 mm inert glass beads. The system was flushed with a CO2/ethanol mixture for 5 min followed by neat CO2 to remove the residual solvent from the system after each extraction. The collected extract was stored at −20 °C. For the analysis of flavonoids and terpenoic acids, the 1 μL of the sample was diluted with 999 μL of EtOH. The sample was diluted 10 and 100 times for volatiles to cover different concentration levels and to fit the calibration range.Box-Behnken design of experiments (DoE) with three center points was designed in MODDE 12.1 (Sartorius Stedim Data Analytics AB, Umeå, Sweden) to evaluate the effect of specific extraction parameters and their interactions. The first DoE tested EtOH ranging from 2 to 95% as a cosolvent with or without water addition (0–20 vol% in EtOH). Temperature 40–80 °C and pressure 100–320 bar were investigated as listed in ESI Table S2.† The second DoE focused only on the extraction of volatile compounds and tested 0–10% hexane as co-solvent in CO2 while the remaining conditions were in the same ranges in both DoE (ESI Table S3†). The flow rate and extraction time were kept at 2 mL min−1 and 10 min, respectively, in both DoE. A total of 42 experiments were carried out in two different DoE. Multiple linear regression was used to calculate the fitting model and response surface. The optimum values for the tested parameters, which allowed to obtain the maximum peak areas and the highest extraction yield, were obtained by numerical analysis based on the desirability function. The suitability of the model was evaluated by the model validity, model reproducibility, and R2 and Q2 values, where R2 represents the model fit and Q2 represents an estimate of the future prediction precision. The predicted vs. observed plot and coefficient box plots were also used for the model evaluation. The selected optimal conditions suggested by the software were evaluated and compared in terms of extraction yield and are listed in ESI Table S4.† Consequently, 2 and 4 mL min−1 flow rates were tested to evaluate the effect of flow rate on the extraction yield. Extracts were collected at the defined time points during a run, including 5, 10, 15, 30, 45, 60, and 90 min to plot the extraction kinetic curve. EtOH (2 mL) was always added to the collection flask prior the extraction to avoid loss of volatile compounds.
Two-step SFE method
The final SFE procedure combined the following two steps: Step 1, targeting volatile terpenes, used CO2/EtOH + 3% water in a ratio of 98/2 (v/v) as the extraction solvent at 2 mL min−1. The extraction temperature was set at 78 °C and the pressure at the back pressure regulator (BPR) was set at 133 bar. 0.5 mL min−1 EtOH was used as a make-up solvent to avoid the precipitation of extract in the system. The extract was collected for 20 min. The conditions were then changed to Step 2, targeting polar phenolics. Here, 56 vol% CO2 was mixed with 44 vol% of cosolvent, i.e., EtOH and water (99/1, v/v), using the same flow rate, temperature was kept at 80 °C, and pressure at 108 bar. Extraction step 2 was completed in 60 min.Selectivity, repeatability, accuracy, and precision
The selectivities of step 1 and step 2 were verified by analysis of both extracts by each of the analytical methods, and the extracted amounts of compounds were determined. The repeatability was verified by 3 replicates of the Corymbia citriodora sample. Accuracy and precision were determined by standard addition of target compounds to the SFE extracts at 5 different concentration levels, namely 0.1, 0.5, 1, 5, and 10 μg mL−1 for volatile terpenes, and 10, 50, 100, 500, and 1000 ng mL−1 for other compounds. Method accuracy was determined as the agreement between the response in a standard solution at the tested concentration level and in the sample spiked with the same amount of the standard and after the subtraction of the naturally occurring amount. The precision of the method was determined as the relative standard deviation (RSD [%]) between 3 extract replicates spiked with reference standards at the tested concentration levels. For the semi-quantitative determination of plant extracts, the standard calibration curve was used.Extraction completeness was confirmed by ultrasound-assisted extraction (UAE) of the matrix previously extracted by the final SFE method. 10 mL of EtOH was added to the sample and extracted by UAE at 40 °C for 15 min. The sample was filtered through a 0.2 μm PTFE syringe filter and analyzed by UHPSFC-MS/MS methods.
Analysis of SFE extracts
Two ultra-high performance supercritical fluid chromatography-tandem mass spectrometry (UHPSFC-MS/MS) methods published by Plachká et al.33 were used and are briefly summarized here. All experiments were carried out using an Acquity UPC2 supercritical fluid chromatography system (Waters, Milford, MA, USA) with a binary pump, an autosampler, a column thermostat, a BPR, and a PDA detector. The system was coupled to a triple quadrupole mass spectrometer Xevo TQ-XS (Waters) via a commercial SFC-MS dedicated pre-BPR splitter device with an additional binary pump to deliver the make-up solvent (Waters). The autosampler was cooled to 5 °C and the partial loop with needle overfill injection mode was used to inject a 2 and 10 μL sample for methods 1 and 2, respectively. MassLynx Software 4.1 was used for system control, data acquisition, and processing.Method 1 for the analysis of volatile terpenes: A 150 × 3.0 mm Supel Carbon LC column (Merck) packed with 2.7 μm particles at 60 °C and MeOH as organic modifier in gradient elution were used for the separation. The gradient was as follows: 0% for 1.5 min, 0–40% in 1.5–4.0 min, 40–41% in 4.0–6.0 min followed by 1.5 min equilibration at initial conditions. The flow rate was 1.5 mL min−1 and the BPR pressure 3300 psi (227.5 bar). The multimodal ionization source ESCi in positive mode was used with the following settings: corona current – 2 μA, desolvation gas flow rate (N2) – 720 L h−1, cone gas flow rate (N2) – 280 L h−1, nebulizer pressure – 5 bar, probe temperature – 150 °C, cone voltage – 150/5 V, desolvation temperature – 350 °C, capillary voltage – 1 kV.
Method 2 for the analysis of phenolics and triterpenoic acids: Torus Diol column (50 × 3.0 mm, 1.7 μm) from Waters (Milford, MA, USA) was used at 20 °C and 5% water in MeOH as an organic modifier with specific gradient conditions listed in ESI Table S5.† Electrospray ionization in positive and negative mode was used with desolvation gas flow rate (N2) – 500 L h−1, cone gas flow rate (N2) – 250 L h−1, nebulizer pressure – 6 bar, probe temperature – 150 °C, cone voltage – 5 V, desolvation temperature – 200 °C, capillary voltage – 0.5 kV.
Argon was used as collision gas for both methods. The SRM conditions for each target analyte are listed in ESI Table S6.† MeOH containing 10 mmol L−1 formic acid, 10 mmol L−1 ammonia, and 1% H2O was delivered as a make-up solvent at 0.1 mL min−1 and 0.3 mL min−1 for methods 1 and 2, respectively.
Results and discussion
Optimization of key extraction parameters
The DoE enabling fast optimization with the reduced number of experiments compared to one-at-a-time approach was used to optimize of extraction solvent composition, temperature, and pressure as the key extraction parameters. The significance of each parameter and interactions between individual parameters were evaluated.34,35 The ranges of extraction parameters tested were set up with an emphasis on the physicochemical properties of the analytes and the limits of the instrumentation used.The first DoE used green solvents, i.e., a mixture of CO2 and EtOH with or without water addition, which increases the polarity of supercritical CO2 and affects the solubility, and thus the extractability, of a wide range of analytes. Therefore, the EtOH as a cosolvent ranged from 2 to 95% (v/v) with water addition up to 20% (v/v). The single-phase composition of the ternary mixtures used in the method optimization was verified by plotting the individual mixtures in a ternary phase diagram as illustrated in ESI Fig. S1.† As obvious, all solvent compositions fitted in a single-phase liquid region above the dew point/bubble point curve.36 This DoE was preferably aimed at the extraction of flavonoids and phenolic acids defined by log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P < 2.79 and pKa in the range of 4.60–7.49, corresponding to weak acids and neutral compounds. In addition, a higher extraction yield was also expected for the terpenoic acids.
P < 2.79 and pKa in the range of 4.60–7.49, corresponding to weak acids and neutral compounds. In addition, a higher extraction yield was also expected for the terpenoic acids.
Volatile terpenes are characterized as neutral compounds with logP ranging mainly from 2.08 to 6.42. Thus, the nonpolar CO2 with only a small addition of organic cosolvent (EtOH) was expected to be optimal as the extraction solvent. Additionally, the second DoE using the pure nonpolar supercritical CO2 or CO2 with hexane addition up to 10 vol% was examined. Temperature and pressure were tested in the range of 40–80 °C and 100–320 bar, respectively, according to the critical point of CO2 in both DoE designs.
In order to obtain the conditions relevant for different analytes, the compounds were divided into four groups: volatile terpenes, terpenoic acids, phenolic acids, and flavonoids. The choice of organic cosolvent played an important role in the extraction of volatiles. When hexane was used, some compounds such as limonene, linalool, and citronellol were not extracted at all. 4–7% of hexane was beneficial for citronellal and eucalyptol, while no effect was observed for others, such as pinene and terpineol. In addition, the model did not provide sufficient predictive power (Q2 < 0.1) and linearity (R2 < 0.2) for some volatiles. However, the repeatability of the replicates was sufficient, and the model had no lack-of-fit (ESI Fig. S2–4†). For EtOH as organic cosolvent, the proposed model provided sufficient linearity with R2 > 0.7, predictive power with Q2 > 0.45, validity, and repeatability as summarized for selected model compounds in ESI Fig. S5–6.† The extractability of volatiles increased when < 10% of EtOH was used as cosolvent in supercritical CO2. The addition of water to co-solvent was not a statistically significant parameter as the error bar crossed 0 value on the y-axis (see ESI Fig. 5 and 6†). Nevertheless, the addition of a small percentage of water to the ethanolic co-solvent enhanced the extraction yield, as obvious from ESI Fig. 7,† where the red space indicates the highest extraction yield. The effect of temperature and pressure was negligible for both models tested, with factor contributions of 5.4% and 1.0%, respectively. The model for the three terpenoic acids showed that the solvent composition and pressure were the main parameters affecting the extraction yield. The highest extracted amounts of analytes were observed with an increasing percentage of EtOH in the extraction solvent, i.e., > 80% EtOH in CO2, negligible amount of water (2%), and low pressure ranging between 110–130 bar (ESI Fig. S8 and 9†).
The phenolics, including phenolic acids and flavonoids, did not exhibit unambiguous results. The composition of the extraction solvent was evaluated as the most important parameter as shown by the coefficient plots (see ESI Fig. S10 and 11†). However, its effect on the extractability differed depending on the physicochemical properties of the individual analytes. Typically, a higher percentage of EtOH resulted in a higher amount extracted for most target compounds, as evident from the contour plots, for example in the case of caffeic acid. In contrast, for ellagic acid, only 50–60% of EtOH in CO2 was optimal, and for protocatechuic acid, a high extraction yield was obtained with only 10% of EtOH in CO2 (ESI Fig. S12†). On the other hand, an increasing percentage of water could significantly decrease the extractability, probably due to the lower solubility of analytes, such as quercitrin and hirsutrine in the extraction solvents used (ESI Fig. S13–15†).
Based on the models for the analyte target sets, MODDE Optimizer suggested a list of optimal conditions for each analyte target sets (data not shown), which should allow the extraction of the maximum number of target compounds. Of all the proposed conditions, 16 different setups with the lowest probability of failure, listed in ESI Table S4,† were selected and compared experimentally. These conditions were selected based on several facts. (i) For volatile terpenes, only one condition using hexane as a cosolvent was included because the probability of failure varied from 25% to 54% among the proposed experiments. Moreover, the conditions suggested in the 12 experiments using hexane as a cosolvent did not show significant parameter variance. In fact, the amount of hexane varied from 8 to 10% (v/v), the temperature from 45 to 50 °C, and the pressure from 212 to 248 bar. (ii) The probability of failure (up to 90%) of the proposed conditions for extraction of volatile terpenes increased with increasing amount of EtOH and water addition. Pressure and temperature varied in a wide range from 40 to 80 °C and 133 to 315 bar, respectively. Thus, five different conditions were tested in this part of the study. (iii) Three conditions suggested for the extraction of terpenoic acids with a probability of failure < 0.94% were investigated. The EtOH (> 64%) mixed with water (0–5%) and pressure < 150 bar were suggested as the most appropriate conditions. (iv) For phenolics, the proposed conditions covered whole ranges of tested conditions, and no trends were observed. (v) In addition, the conditions suitable for the extraction of all analytes except volatiles, were also tested. Here, the gas-expanded liquid (GXL) was recommended as an extraction solvent containing 93% EtOH with 7% water, a temperature of 80 °C, and a pressure of 320 bar, but with an expected lack of selectivity.
From these tested conditions, the mixture of CO2 with EtOH/H2O (97/3, v/v) in a 98/2 (v/v) ratio provided the highest extracted amounts of volatiles. In addition, based on the preliminary results, no phenolics were extracted in 10 min long extraction using the CO2 with cosolvent in 98/2 (v/v) ratio (data not shown). For the remaining groups of compounds, two conditions using (i) GXL containing 56% CO2 mixed with EtOH/H2O (99/1, v/v) and (ii) GXL composed of EtOH/H2O (91/9, v/v), CO2/cosolvent 5/95, v/v, provided two times higher extraction yields compared to the remaining proposed conditions.
Extraction kinetics
Three selected methods (ESI Table S4† marked in orange) were further optimized in terms of flow rate and extraction time. The first method for the extraction of volatile compounds showed that volatiles were extracted in 15 min using both flow rates. Thus, 20 min was considered as optimal for the final procedure. In fact, no significant differences in extracted amounts were observed between 2 and 4 mL min−1. This means that the extraction was mainly controlled by desorption. In this case, the extractability depends on the diffusion of the analytes from the matrix to the extraction solvent. The same trends were also observed for both methods with GXL aimed at the extraction of phenolic and triterpenoic compounds. The method with 44% EtOH gave the highest extraction yields. The extraction of triterpenoic acids was completed in 40 min. A longer time was required for the complete extraction of phenolic acids and flavonoids, i.e., 60 min.As a result, the final method combined two consecutive steps with a flow rate of 2 mL min−1. The method with 2% of cosolvent was selected as the first step for the extraction of volatile terpenes. The second step used 44% of cosolvent to extract the remaining compounds. Again, the single-phase formation to avoid the phase separation and poor method repeatability was evaluated by the ternary phase diagram based on the data obtained by Lim et al. under 60 °C and 142 bar as the conditions close to our achieved optimum (ESI Fig. S1†).36 Additionally, the optimal temperature and pressure for both steps were close, i.e., 78 °C and 133 bar for step 1 and 80 °C and 108 bar for step 2. Thus, no gap was expected and observed between the two steps due to long equilibration. Similarly, the possibility of failure of the extraction step 2, e.g., by over pressurizing of the system when the much higher pressure would have to be equilibrated before the second step, was minimized.
Final method scheme and method selectivity
The final procedure is summarized in Fig. 1 and took a total of 80 min, including the 20 min step 1 aimed at extraction of volatile terpenes and the 60 min step 2 for the extraction flavonoids and phenolic compounds. The selectivity focused on the target groups of analytes in two consecutive individual steps was evaluated on the Corymbia citriodora sample. Individual extraction steps were carried out and the visual differences between them were observed. For step 1, the extract was yellowish with a strong odor of volatile terpenes, while for step 2, the extract was green with a typical ethanolic odor. | ||
| Fig. 1 Overview of a complete two-step SFE for the extraction of non-polar volatile compounds in a first step extraction followed by second steps with conditions aimed at the extraction of phenolics. EtOH–ethanol. | ||
The obtained extracts were analyzed by UHPSFC-MS/MS methods. The results summarized in Table 1 show the perfect selectivity for the extraction of volatile terpenes. In fact, >88% of terpenes were extracted during SFE step 1 aiming selectively at this type of compounds. Step 2 and UAE extracted only a residual amount, i.e., 2–11% of several terpenes, such as citronellal, geranyl acetate, limonene, linalool, and nerolidol. Flavonoids and phenolic acids were not extracted by SFE step 1 as their extractability increased with increasing EtOH. Only caffeic and protocatechuic acids were partially extracted in the SFE step 1. In fact, 28% of caffeic acid and 43% of protocatechuic acid were observed here. We speculate that this may be due to their higher logP and lower molecular weight when compared to gallic and ellagic acids, which were only extracted in the SFE step 2. Nevertheless, all phenolic acids and flavonoids were also found in the extract from UAE. The residual amount of compounds was high (>50%) despite the plateau obtained during the kinetic experiments. This could be caused by the short extraction time, shallow increased extracted amount of phenolic compounds, channeling effect, and/or tight packing of the material as discussed by Abrahamson for lipids.37 The triterpenoic acids were extracted during the whole extraction procedure. We expected the highest concentration during the SFE step 2 due to the high molecular weight. However, the highest proportion, 79–90%, was extracted in SFE step 1 targeting non-polar compounds and the remaining amounts < 12% were extracted in step 2 and UAE for betulinic and oleanolic acids. For the ursulic acid, the distribution was different compared to other triterpenoic acids, 21–42%.

|
Repeatability, accuracy, and precision
The repeatability of the extraction was verified and determined for neat extracts without any standard addition as RSD for each analyte in 3 replicates on 3 consecutive days (n = 9). The RSD ranged from 4.9 to 23.0% for volatile compounds, 2.4 to 16.6% for flavonoids and phenolic acids, and 18.2 to 28.6% for triterpenoic acids extracted during both extraction steps.Accuracy and precision were evaluated for both SFE steps using the standard addition method, where the known concentration of the compound was added to the sample. Since there is no guideline for the plant analysis, we evaluated both, accuracy and precision, for 5 concentration levels covering the linear calibration range of the UHPSFC-MS/MS from 0.1 to 100 ng mL−1. Fig. 2 shows that most of the results met the criterion of RSD < 20%, which summarizes the error of the two-step SFE and UHPSFC-MS/MS analysis. Only citronellol at a concentration of 100 ng mL−1, ursulic acid at 1000 ng mL−1, and betulinic acid at 10 ng mL−1 showed lower precision expressed as RSD, namely 35%, 29%, and 26%, respectively. For accuracy, the results were mostly in the range of 76–123%. Again, three analytes at different concentration exhibit lower accuracy, namely caryophyllene at the concentration level of 100 μg mL−1 (132%), oleanolic acid at 500 ng mL−1 (135%), and tamarixetin at 50 ng mL−1 (127%).
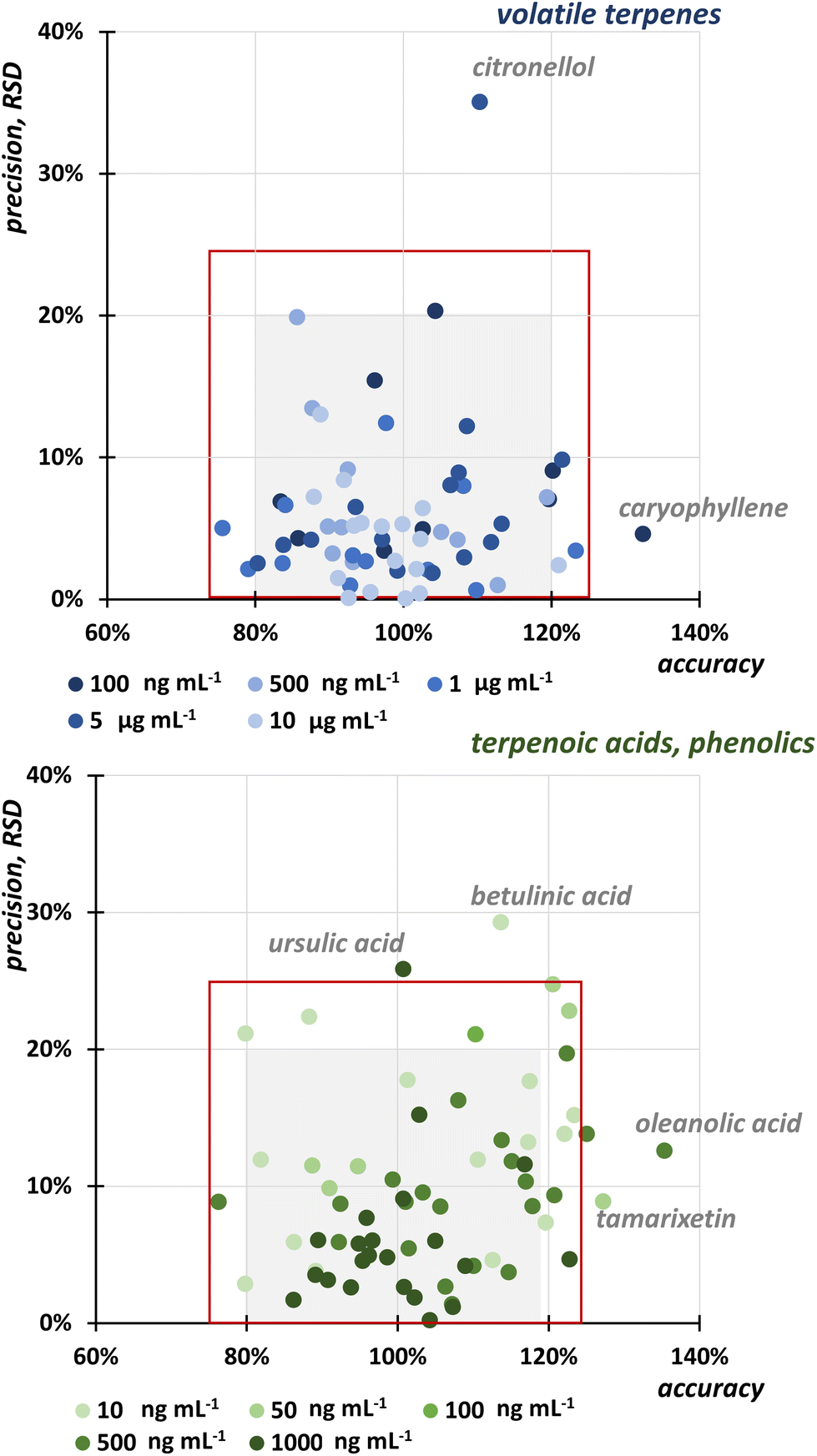 | ||
| Fig. 2 Accuracy and precision determined at 5 concentration levels for individual analytes. The grey square corresponds to the area covering precision in the range of 0–20% expressed as RSD and accuracy in the range of 80–120%. A red square expresses 25% RSD and accuracy in the range of 75–125%. | ||
Method greenness
In the last step, the greenness of the optimized sample preparation method was evaluated. Several metrics have been developed and introduced in recent years to assess method greenness.38 All available metrics are based on the incorporation of different green chemistry criteria. As a result, they provide different method assessments, variable complexity, appearance, and comprehensibility. In 2022, a new metrics tool aiming specifically targeting sample preparation was proposed by Wojnowski et al.39 This freely available software, the Analytical GREEnness Metric Approach for sample preparation (AGREEprep) calculator, based on the 10 principles of green chemistry, was used to estimate the greenness of the newly optimized SFE method. Briefly, these principles include in situ sample preparation (criterion #1), use of safer chemicals (#2), use of sustainable and reusable materials (#3), minimization of waste, sample, and chemicals (#4, #5), maximization of sample throughput (#6), automation (#7), low energy consumption (#8), analytical method (#9), and operator safety (#10). As a flexible tool, AGREEprep translates the criteria into a score range of 0–1. The final score is derived from the evaluation of all tested principles, with score 1 being the greenest and score 0 being the least green. In addition, it is possible to change the weight of each criterion according to its importance for the individual method.The optimized procedure involves external sample collection and transport to the laboratory (#1), onsite sample preparation is not possible as the sample must be dried and homogenized. Toxic materials and chemicals are not required, operator safety is maximized (#2, #10), and sustainable and reusable materials are used (#3). The extracted matrix (< 1 g) is the only waste produced as the extract obtained is not considered as an analytical waste (#4, #5). The entire procedure took 1 h 20 min. However, each step took < 1 h (#6). Here, the weight of the criterion was reduced compared to the standard setup, because the SFE system specifically used within this study did not allow the parallel extraction of several samples, whereas up to 10 samples can be extracted in series without any manual intervention in the conventional SFE systems. In fact, the optimized method is fully automated. The only manual intervention was to place a sample in the extraction cell and extractor and to remove it after the extraction (#7). The total energy consumption per sample (#8), including both extraction steps, was calculated to be 11.81 kW h. No energy consumption was needed for the drying process, as the plant biomass was dried by natural air convection. The UHPSFC-MS/MS analysis also reduced the greenness of the method (#9). The final pictogram is shown in Fig. 3. The total score of the method greenness is 0.62 62 confirming the green method characteristics. Four weak criteria (#1, #6, #8, #9) was not possible to improve in the greenness terms. The detailed report is summarized in ESI Fig. S16.†
 | ||
| Fig. 3 Results of the AGREEprep metrics tool for two-step SFE. | ||
Application to various plant species
The method was applied to the extraction of 7 different plant samples to confirm its suitability for the extraction and analysis of different species, including Corymbia citrioda, Eucalyptus gunii, Melaleuca alternifolia, Cympogon citratus, Myrtus communis, Laurus nobilis, and Rosa hybrida (ESI Fig. S17†).The profiles of the target analytes were compared for all species (Fig. 4). Focusing on volatile compounds (Fig. 4), significant differences were observed between the profiles of all plant species. Overall, M. communis contained the highest amount of volatile terpenes, followed by C. citriodora, and E. gunii. Moreover, the different volatile compounds were dominant in individual extracts. Indeed, pinene was dominant in M. communis, citral (> 95%) in C. citratus, and eugenol (70%) and eucalyptol (20%) dominated in the L. nobilis extract. Different ratios of eucalyptol and geranyl acetate, namely 80/12 (conc%) and 65/30 (conc%), were predominantly observed in E. gunii and M. alternifolia, respectively. For R. hybrida, eugenol (60%), linalool, and citronellal are among the most extracted volatile terpenes. For C. citriodora, the results were different. No dominant volatile compound was observed, and the extract contained a mixture of different target volatiles in very similar amounts, including citronellal, citronellol, menthol, geranyl acetate, linalool, limonene, and terpineol.
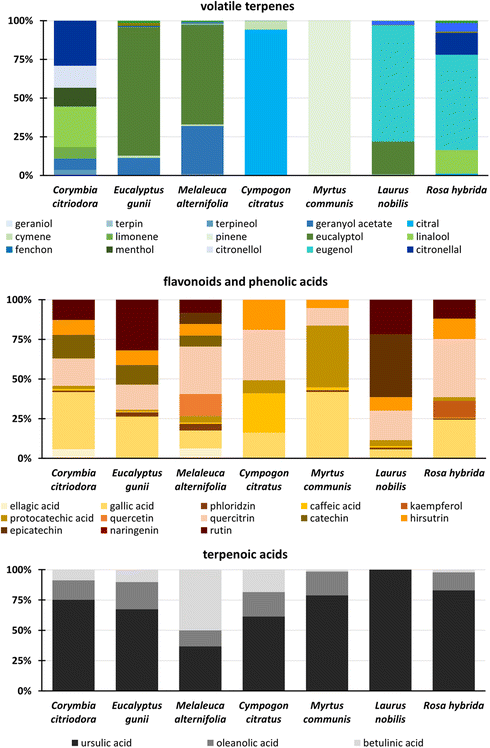 | ||
| Fig. 4 Profiles of target analytes for the group of volatile terpenes, flavonoids, phenolic acids, and triterpenoic acids in SFE extracts of seven different plant species. | ||
The differences in the profiles of SFE extracts targeting on flavonoids and phenolic acids were not so significant, although the extracted amount varied. In fact, the highest amount of flavonoids and phenolic acids, ranging from 1800 to 3000 ng mL−1, was found in R. hybrida, C. citriodora, E. gunii, and M. alternifolia. On the other hand, C. citratus contained ten times lower amount of phenolics. As shown in Fig. 4, gallic acid, quercitrin, rutin, hirsutrin, and catechin were detected and semi-quantified in different ratios in all extracts. C. citriodora and E. gunii provided similar profiles of phenolic acids and flavonoids. No rutin was detected in M. communis, while a high content of protocatechuic acid was found. R. hybrida also contained a high level of kaempferol. L. nobilis had a significantly different profile, with epicatechin, rutin, and quercitrin dominating. M. alternifolia contained mainly quercetin. Gallic acid, caffeic acid, quercitrin, and hirsutrin were dominant in C. citratus. Triterpenoic acids were also found in all samples. Again, the highest amounts (5–11000 ng mL−1) were found in C. citriodora, C. gunii, and M. alternifolia. Ursulic acid dominated most of the extracts (> 60%), typically followed by oleanolic acid with amounts ranging from 10 to 25%. Betulinic acid was typically observed at the lowest concentration. The betulinic acid was dominant (50%) only in M. alternifolia. In addition, betulinic and oleanolic acids were not detected in L. nobilis species.
Based on the described differences, the hierarchical cluster statistical analysis was carried out to create a dendrogram showing the similarities (relationships) between each species. The measured concentrations of target analytes were logarithmically transformed to obtain a normal distribution of the data. Ward linkage was then used to determine the similarity between the plants analyzed. Surprisingly, C. citriodora was found to be the most different species, while E. gunii, M. alternifolia, and L. nobilis were found to be the most related species as shown in Fig. 5. In fact, E. gunii and M. alternifolia provided very similar profiles of target compounds. The second cluster was formed by C. citratus, R. hybrida, and M. communis. Here, the similarity of M. communis and R. hybrida is obvious especially in the ratio of triterpenoic acids. The phenolic profiles of C. citratus and R. hybrida shown in Fig. 4 confirmed a close content of polar compounds, while significant differences were observed for volatile terpenes.
 | ||
| Fig. 5 Hierarchical cluster analysis showing the relationships between the tested species based on the profiles of all target compounds. 1 – Corymbia citrioda, 2 – Eucalyptus gunii, 3 – Melaleuca alternifolia, 4 – Cympogon citratus, 5 – Myrtus communis, 6 – Laurus nobilis, and 7 – Rosa hybrida. | ||
The residual plant matrix was extracted by UAE to evaluate the extraction efficiency of both SFE steps. The results obtained for the 6 plant species confirmed the observations obtained for the model sample of Corymbia citriodora and are presented in the ESI Table S7.†
Conclusions
We developed a comprehensive two-step SFE procedure for the extraction of different target analytes from different plant species. In the first extraction step, the method allowed the extraction of small non-polar volatile terpenes, which was completed in only 20 min, while no target phenolic compound was extracted. The following second extraction step aimed at the extraction of polar phenolic acids and flavonoids. These were successfully extracted in 60 min as confirmed by the kinetic experiments. Although the UAE applied to the residual material extracted residual amounts of target compounds, the repeatability of the SFE extraction was excellent (RSD < 20% for most compounds). This proof-of-concept two-step SFE method demonstrated the feasibility and suitability of SFE in a complex extraction of plant samples when aiming at selective extraction of different target groups of analytes. Traditional approaches typically use large solvent volumes, require long extraction times, usually in hours, and use a wide range of different organic solvents, even toxic ones, to achieve selectivity for polar and non-polar species. In contrast, this holistic SFE method allows rapid extraction of polar and non-polar compounds in less than 1.5 h using green carbon dioxide, ethanol, and water in various ratios as extraction solvents. In addition, no manual intervention is required between individual steps.Author contributions
Veronika Pilařová – conceptualization, methodology, investigation, writing – original draft, Kateřina Plachká – investigation, data curation, visualization, writing – original draft, Diana Herbsová – investigation, validation, Štefan Kosturko – investigation, writing – review & editing, Frantisek Svec – project administration, resources, writing – review & editing, Lucie Nováková – supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the EFSA-CDN project (Reg. No. CZ.02.1.01/0.0/0.0/16_019/0000841) co-funded by ERDF and the Projects of Specific Research SVV 260 662.V. P. thanks to Ing. Anežka Chlebková, the head of the Garden of Medicinal Plants, Hradec Králové, for providing all tested plant material and to Ing. Jan Škop for sample pictures.
References
- M. Ganzera and M. Zwerger, TrAC, Trends Anal. Chem., 2021, 145, 116463, DOI:10.1016/j.trac.2021.116463.
- T. Belwal, S. M. Ezzat, L. Rastrelli, I. D. Bhatt, M. Daglia, A. Baldi, H. P. Devkota, I. E. Orhan, J. K. Patra and G. Das, et al. , TrAC, Trends Anal. Chem., 2018, 100, 82–102, DOI:10.1016/j.trac.2017.12.018.
- J. Azmir, I. S. M. Zaidul, M. M. Rahman, K. M. Sharif, A. Mohamed, F. Sahena, M. H. A. Jahurul, K. Ghafoor, N. A. N. Norulaini and A. K. M. Omar, J. Food Eng., 2013, 117(4), 426–436, DOI:10.1016/j.jfoodeng.2013.01.014.
- Q.-W. Zhang, L.-G. Lin and W.-C. Ye, Chin. Med., 2018, 13(1), 20, DOI:10.1186/s13020-018-0177-x.
- P. K. Mukherjee, Chapter 6 - Extraction and Other Downstream Procedures for Evaluation of Herbal Drugs, in Quality Control and Evaluation of Herbal Drugs, ed. P. K. Mukherjee, Elsevier, 2019, pp. 195–236 Search PubMed.
- F. Bucar, A. Wube and M. Schmid, Nat. Prod. Rep., 2013, 30(4), 525–545, 10.1039/C3NP20106F.
- A. Mustafa and C. Turner, Anal. Chim. Acta, 2011, 703(1), 8–18, DOI:10.1016/j.aca.2011.07.018.
- L. P. Cunico and C. Turner, Chapter 7 - Supercritical Fluids and Gas-Expanded Liquids, in The Application of Green Solvents in Separation Processes, ed. F. Pena-Pereira and M. Tobiszewski, Elsevier, 2017, pp. 155–214 Search PubMed.
- M. Cvjetko Bubalo, S. Vidović, I. Radojčić Redovniković and S. Jokić, Food Bioprod. Process., 2018, 109, 52–73, DOI:10.1016/j.fbp.2018.03.001.
- T. F. Gondo, M. Jönsson, E. N. Karlsson, M. Sandahl and C. Turner, J. Chromatogr., A, 2023, 1706, 464267, DOI:10.1016/j.chroma.2023.464267.
- V. Pilařová, S. Al Hamimi, L. P. Cunico, L. Nováková and C. Turner, Green Chem., 2019, 21(19), 5427–5436, 10.1039/C9GC02140J.
- Surbhi, A. Kumar, S. Singh, P. Kumari and P. Rasane, Adv. Tradit. Med., 2023, 23(2), 369–380, DOI:10.1007/s13596-021-00582-7.
- P. Gullón, B. Gullón, G. Astray, P. E. S. Munekata, M. Pateiro and J. M. Lorenzo, Molecules, 2020, 25(18), 4227, DOI:10.3390/molecules25184227.
- Q. V. Vuong, A. C. Chalmers, D. Jyoti Bhuyan, M. C. Bowyer and C. J. Scarlett, Chem. Biodiversity, 2015, 12(6), 907–924, DOI:10.1002/cbdv.201400327.
- A. Singh, A. Ahmad and R. Bushra, Anal. Methods, 2016, 8(6), 1339–1350, 10.1039/C5AY02009C.
- S. Zhao and D. Zhang, Sep. Purif. Technol., 2014, 133, 443–451, DOI:10.1016/j.seppur.2014.07.018.
- G. Della Porta, S. Porcedda, B. Marongiu and E. Reverchon, Flavour Fragrance J., 1999, 14(4), 214–218, DOI:10.1002/(SICI)1099-1026(199907/08)14:4<214::AID-FFJ814>3.0.CO;2-H.
- M. Topiar, M. Sajfrtova, R. Pavela and Z. Machalova, J. Supercrit. Fluids, 2015, 97, 202–210, DOI:10.1016/j.supflu.2014.12.002.
- M. M. R. de Melo, R. M. A. Domingues, M. Sova, E. Lack, H. Seidlitz, F. Lang Jr., A. J. D. Silvestre and C. M. Silva, J. Supercrit. Fluids, 2014, 95, 44–50, DOI:10.1016/j.supflu.2014.07.030.
- V. H. Rodrigues, M. M. R. de Melo, I. Portugal and C. M. Silva, Sep. Purif. Technol., 2018, 191, 173–181, DOI:10.1016/j.seppur.2017.09.026.
- L. Boulekbache-Makhlouf, E. Meudec, J.-P. Mazauric, K. Madani and V. Cheynier, Phytochem. Anal., 2013, 24(2), 162–170, DOI:10.1002/pca.2396.
- E. Cadahía, E. Conde, M. C. García-Vallejo and B. Fernández de Simón, Phytochem. Anal., 1997, 8(2), 78–83, DOI:10.1002/(SICI)1099-1565(199703)8:2<78::AID-PCA335>3.0.CO;2-O.
- M. M. Okba, R. A. El Gedaily and R. M. Ashour, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1068–1069, 335–342, DOI:10.1016/j.jchromb.2017.10.065.
- A. Nasr, T. Saleem Khan and G.-P. Zhu, Prep. Biochem. Biotechnol., 2019, 49(5), 464–476, DOI:10.1080/10826068.2019.1575860.
- V. Pilařová, L. Kuda, H. K. Vlčková, L. Nováková, S. Gupta, M. Kulkarni, F. Švec, J. Van Staden and K. Doležal, Plant Methods, 2022, 18(1), 87, DOI:10.1186/s13007-022-00919-6.
- M. Hollá, V. Pilařová, F. Švec and H. Sklenářová, Foods, 2023, 12(4), 893, DOI:10.3390/foods12040893.
- M. S. Ekinci, Sep. Sci. Technol., 2022, 57(2), 256–262, DOI:10.1080/01496395.2021.1893333.
- K. G. Martino and D. Guyer, J. Food Process Eng., 2004, 27(1), 17–28, DOI:10.1111/j.1745-4530.2004.tb00620.x.
- M.-H. Chuang and G. Brunner, J. Supercrit. Fluids, 2006, 37(2), 151–156, DOI:10.1016/j.supflu.2005.09.004.
- S. Barbini, J. Jaxel, K. Karlström, T. Rosenau and A. Potthast, Bioresour. Technol., 2021, 341, 125862, DOI:10.1016/j.biortech.2021.125862.
- M. J. Jiménez Callejón, A. Robles Medina, M. D. Macías Sánchez, P. A. González Moreno, E. Navarro López, L. Esteban Cerdán and E. Molina Grima, Algal Res., 2022, 61, 102586, DOI:10.1016/j.algal.2021.102586.
- A. d. P. Sánchez-Camargo, A. Valdés, G. Sullini, V. García-Cañas, A. Cifuentes, E. Ibáñez and M. Herrero, J. Funct. Foods, 2014, 11, 293–303, DOI:10.1016/j.jff.2014.10.014.
- K. Plachká, V. Pilařová, Š. Kosturko, J. Škop, F. Svec and L. Nováková, Anal. Chem., 2024, 96(7), 2840–2848, DOI:10.1021/acs.analchem.3c03599.
- J. Neumann, S. Schmidtsdorff, A. H. Schmidt and M. K. Parr, Sep. Sci. plus, 2024, 2300239, DOI:10.1002/sscp.202300239.
- K. M. Sharif, M. M. Rahman, J. Azmir, A. Mohamed, M. H. A. Jahurul, F. Sahena and I. S. M. Zaidul, J. Food Eng., 2014, 124, 105–116, DOI:10.1016/j.jfoodeng.2013.10.003.
- J. S. Lim, Y. Y. Lee and H. S. Chun, J. Supercrit. Fluids, 1994, 7(4), 219–230, DOI:10.1016/0896-8446(94)90009-4.
- V. Abrahamsson, I. Rodriguez-Meizoso and C. Turner, Anal. Chim. Acta, 2015, 853, 320–327, DOI:10.1016/j.aca.2014.09.052.
- L. P. Kowtharapu, N. K. Katari, S. K. Muchakayala and V. M. Marisetti, TrAC, Trends Anal. Chem., 2023, 166, 117196, DOI:10.1016/j.trac.2023.117196.
- W. Wojnowski, M. Tobiszewski, F. Pena-Pereira and E. Psillakis, TrAC, Trends Anal. Chem., 2022, 149, 116553, DOI:10.1016/j.trac.2022.116553.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00371c |
| This journal is © The Royal Society of Chemistry 2024 |