
Development of a high-efficiency and environmentally friendly melamine–formaldehyde resin-based solid phase microextraction fiber for enhanced extraction of polycyclic aromatic hydrocarbons from environmental water
Pengfei
Li
ab,
Yehong
Han
a,
Dandan
Han
a and
Hongyuan
Yan
 *ab
*ab
aHebei Key Laboratory of Public Health Safety, College of Public Health, College of Chemistry and Materials Science, Hebei University, Baoding, 071002, China. E-mail: yanhy@hbu.edu.cn
bState Key Laboratory of New Pharmaceutical Preparations and Excipients, Key Laboratory of Medicinal Chemistry and Molecular Diagnosis of Ministry of Education, Hebei University, Baoding 071002, China
First published on 6th February 2024
Abstract
An innovative solid-phase microextraction (SPME) fiber coating, melamine–formaldehyde resin (MFR), was synthesized with a green cross-linker (paraformaldehyde), and it exhibited high heat resistance (up to 350 °C). Seven polycyclic aromatic hydrocarbons (PAHs) were chosen as analytes, and the results demonstrated the MFR-based SPME fiber's exceptional enrichment capabilities and efficient extraction, with high enrichment factors and significantly improved efficiency compared to the conventional polydimethylsiloxane (PDMS) fibers. The unique superiority of the MFR was attributed to its strong π–π interactions and amino-associated enhancement, as well as its porous structure and rough surface, which provided abundant adsorption sites and facilitated rapid mass transport of analytes. Furthermore, the preparation conditions of the MFR-coated SPME fiber were compared with those of commercially available and reported fibers, emphasizing the advantages of the MFR-coated fiber, such as its simplicity, cost-effectiveness, reproducibility, and eco-friendliness. The MFR-based SPME fiber exhibited significant advantages, including high enrichment factors (ranging from 1906- to 7153-fold) for PAHs and good fiber-to-fiber reproducibility (8.7–14.6%), outperforming commercially available PDMS fibers by a factor of 4.9–82.8-fold. This study also considered important factors affecting the extraction process, such as ionic strength, temperature, and time, and optimized these parameters for the best extraction efficiency. The greenness of the developed method was assessed using the Analytical Eco-Scale, and the results showed that it was green in terms of reagent dosage, energy consumption, and waste. The developed headspace solid phase microextraction–gas chromatography–tandem mass spectrometry (HS-SPME-GC-MS/MS) method based on the MFR coating exhibited excellent precision, accuracy, and repeatability, making it a reliable method for the determination of trace PAHs in environmental water samples.
Introduction
There is growing concern about the impact of the chemical industry on the environment and people; although the amount of toxic and harmful byproducts produced in analytical chemistry is insignificant compared to the waste from chemical production, the hidden threat to the environment and humans cannot be ignored.1,2 Green analytical chemistry was introduced in 1999 to guide the reduction or elimination of laboratory waste or emissions from analytical processes, including numerous steps in sample preparation and analytical methods, and to reduce adverse impact on the environment and people.3,4 The establishment of fast, accurate and sensitive analytical methods has always been the primary task of analytical chemists; green analytical chemistry reminds chemists to reduce the impact of analytical processes on the environment and humans, while meeting the needs of production and life.Sample pretreatment technology plays an important role in analytical chemistry; its purpose is to eliminate the interference of matrix components in complex samples, and to enrich and concentrate trace analytes, which can promote the establishment of accurate and sensitive detection methods.5 Ordinarily, sample preparation is considered as the least green step in the analytical process.6 Therefore, the development of environmentally friendly sample pretreatment techniques is of great significance for the development of sustainable analytical methods. To address this need, researchers are devoting efforts towards developing preconcentration techniques. To date, the reported sample pretreatment techniques mainly included solid-phase microextraction (SPME), miniaturized solid-phase extraction (SPE), single drop microextraction (SDME) and liquid–liquid microextraction (LLME).7–11 Among these technologies, SPME is an eco-friendly sample pretreatment method that consolidates sampling, extraction, enrichment, and purification into a single step.9 This approach minimizes the consumption of organic reagents and reduces pollution. Furthermore, due to its high efficiency, user-friendly operation, and solvent-free desorption, SPME coupled with gas chromatography has found extensive use in the detection of trace analytes in various matrices.12
In SPME, the fiber coating serves as the fundamental component, influencing extraction efficiency and sensitivity.13 Traditional SPME fiber coating materials, such as polydimethylsiloxane/divinylbenzene (PDMS/DVB), polydimethylsiloxane (PDMS), and polyacrylate (PA), are prone to fouling and have limited tolerance for high temperatures (e.g., PDMS/DVB fibers are limited to 270 °C).14,15 Furthermore, their high cost, instability, and short lifespan restrict their applications. Consequently, new materials, including metal–organic framework (MOF) compounds, covalent organic frameworks (COFs), microporous organic networks (MONs), and graphene materials have been developed as alternative SPME fiber coatings.16–19 However, during the synthesis of these materials, many toxic and harmful organic reagents, as well as conditions requiring high temperatures and pressures, and a long reaction time are often utilized. It contradicts the principle of green analytical chemistry. Therefore, it is of great significance to seek green tactics to synthesize sustainable fiber coatings, that is, use the least or no harmful solvents, operate under mild conditions, and reduce reaction time while maintaining high efficacy.
Resins have been applied as potential adsorbents in SPE, matrix solid-phase dispersion (MSPD), and magnetic solid-phase extraction (MSPE), thanks to their unique characteristics, such as high thermal stability, adjustable size, and excellent adsorption properties.8,20–22 In addition, mild reaction conditions and environmentally friendly reaction solvents make resins promising candidates as novel sustainable fiber coatings for green analytical chemistry. Regrettably, some aldehyde reagents, such as formaldehyde and glutaraldehyde, are often utilized as cross-linkers to synthesize resins, which are often toxic and carcinogenic.23–25 Therefore, alternative cross-linkers with low toxicity or even non-toxicity are of great significance in the synthesis of sustainable SPME fiber coatings. Furthermore, to the best of our knowledge, there is no existing literature on the utilization of the melamine–formaldehyde resin (MFR) as a fiber coating for headspace SPME.
In this study, a novel MFR with uniform particle size, a rough surface, and high heat resistance was synthesized using an environmentally friendly synthesis strategy that employed paraformaldehyde as a green cross-linker. The entire synthesis process was completed in water, without the involvement of harmful solvents under mild reaction conditions within 1.5 h. The MFR was initially utilized as a headspace SPME fiber coating, and the extraction efficiency was evaluated using polycyclic aromatic hydrocarbons (PAHs) as analytes. The potential adsorption mechanism of the MFR-based SPME fiber was also investigated. Finally, the MFR-based SPME, in combination with GC-MS/MS, exhibited a remarkable enrichment capacity for the quantification of trace levels of PAHs in environmental water. This work provides a simple, sustainable, and eco-friendly synthetic method for the development of new SPME fiber coatings.
Experimental
Chemicals and reagents
Paraformaldehyde was acquired from Damao Chemical Co., Ltd (Tianjin, China). Melamine and polyvinylpyrrolidone (PVP) were obtained from Kermel Chemical Co., Ltd (Tianjin, China). Hexene was supplied by Sigma-Aldrich (Shanghai, China, and St Louis, MO, USA). Acenaphthene (Ace), acenaphthylene (Acy), fluorene (Fl), phenanthrene (Phen), anthracene (Anth), fluoranthene (Fluo), and pyrene (Pyr) were all purchased from Aladdin Chemical Co., Ltd (Shanghai, China).Instrumentation and analytical conditions
Scanning electron microscopy (SEM, Phenom Pro, Eindhoven, Netherlands) was used to evaluate the morphological features of the MFR and MFR-based fiber coating. A Vertex70 FTIR spectrometer (Bruker, Karlsruhe, Germany) was used to obtained the Fourier-transform infrared spectra (FT-IR) of the MFR. X-ray photoelectron spectroscopy (XPS, Thermo Scientific™ K-Alpha™+) was used to obtain the functional groups and elemental composition information of the MFR. The pore size distribution of the MFR was acquired by nitrogen adsorption/desorption measurements on a TriStar II 3020 instrument (Micromeritics, USA). Thermogravimetric analysis (TGA, TA instruments Q2000) was employed to evaluate the thermal stability, and the temperature was initially set at 35 °C, increasing to 600 °C at 10 °C min−1 under a high purity nitrogen atmosphere. Gas chromatography was performed using the Thermo Fisher Trace 1300 GC system, and a TG-5MS capillary column (30 m × 0.25 mm, i.d. 0.25 μm) was employed to separate PAHs. Mass spectral detection was conducted using a Thermo Fisher TSQ9000 triple quadrupole mass spectrometer. The instrumental conditions were as follows: splitless mode; ion source temperature, 290 °C; transfer line temperature, 290 °C; inlet temperature, 290 °C; a constant flow rate of 1 mL min−1 helium was used as the carrier gas; temperature program: the column temperature was initially set at 60 °C, increasing to 220 °C at 20 °C min−1, and then increasing to 250 °C at 5 °C min−1 and keeping at 10 min. Data acquisition was performed in the selective reaction monitoring (SRM) mode. Monitored ion pairs and collision energy of seven PAHs are shown in Table 1.| Analytes | Mass/product mass | Collision energy (eV) |
|---|---|---|
| a Quantitative ion pair and collision energy. | ||
| Acenaphthylene | 150.1/98 | 24 |
| 151.1/150.1 | 12 | |
| 152.1/151.1a | 16a | |
| Acenaphthene | 152.1/151.1 | 16 |
| 153.1/152.1 | 18 | |
| 154.1/153.1a | 12a | |
| Fluorene | 165.1/164.1 | 16 |
| 166.1/165.1a | 16a | |
| Phenanthrene | 176.1/150.1 | 22 |
| 178.1/152.1 | 18 | |
| 178.1/152.1a | 16a | |
| Anthracene | 176.1/150 | 22 |
| 178.1/152.1a | 16a | |
| 179.1/177.1 | 26 | |
| Fluoranthene | 200.1/199.1 | 14 |
| 202.1/200.1a | 32a | |
| Pyrene | 200.1/199.1 | 16 |
| 201.1/200.1 | 14 | |
| 202.1/200.1a | 32a | |
Preparation of the MFR and MFR-based SPME fiber
To summarize, 0.74 g of paraformaldehyde and 0.52 g of melamine were dissolved in 10.0 mL of water and stirred at 50 °C for 45 min to create a clear colloid solution. Subsequently, the mixture, along with 0.70 g of PVP, was added to 30.0 mL of dilute hydrochloric acid (pH 3.5). Stirring was maintained for 45 min at 100 °C. Finally, the product was washed with water and freeze-dried.Before the preparation of the MFR-based fiber, a stainless-steel fiber (2 cm in length) underwent an 8-minute corrosion process with nitrohydrochloric acid and was then cleaned by immersion in acetone, methanol, and ultrapure water while being subjected to ultrasonic treatment for 10 min. The fiber was subsequently dried overnight at 65 °C. Physical adhesion was employed to coat the MFR powder, with a silicone sealant diluted in toluene (0.5 g mL−1) serving as the adhesive. To achieve a thin layer, the fiber needle was gently rotated on weighing paper after being vertically immersed in and withdrawn from the silicone sealant three times. The fiber was inserted into the adsorbent and subjected to treatment at 75 °C for 30 min. This process was repeated three times, and a coverslip was used to gently remove the uncoated particles to obtain the MFR-based SPME fiber. Prior to using the MFR-based SPME fiber, it was conditioned at 300 °C for 0.5 h.
Sampling and sample preparation
Environmental water samples were gathered from the Zaozuodian Lake, Gudingdian Lake, Donghu Lake, and Huchenghe River, all situated in the Baoding City. Additionally, tap water from the laboratory and bottled water from the local market were collected for the analysis. These collected water samples were promptly stored at −18 °C. Prior to the analysis, the environmental water samples underwent filtration, and the filtered samples were used for headspace solid-phase microextraction (HS-SPME).HS-SPME of environmental water samples
In brief, 10.0 mL of environmental water containing 1.5 g of NaCl was introduced into a sealed 30.0 mL glass bottle fitted with a polytetrafluoroethylene–silicone composite gasket. The SPME device utilized a 5 μL microinjection needle to draw the fiber into the needle, punctured the septum, and extended the SPME fiber coating, positioning it above the water sample. The extraction was performed at 65 °C for 65 min at 500 rpm. Following the extraction, thermal desorption was carried out at 290 °C for 3 min. Before proceeding to the next step, the SPME fiber was conditioned at 300 °C for 0.5 h.Results and discussion
Preparation and characterization of the MFR-based SPME fiber
The synthesis process is shown in Fig. 1. Clear colloid hydroxymethyl-melamine derivatives (HMDs) were formed after 45 min of stirring at 50 °C. The formation of ether and methylene bridges, as confirmed by FT-IR analysis of the adsorbent, allowed the HMDs to be cross-linked through nucleation and aggregation in the presence of an acidic catalyst. Consequently, macromolecular polymers with three-dimensional network structures were generated. Paraformaldehyde released formaldehyde in hot water; the use of paraformaldehyde as a cross-linker in the MFR synthesis process eliminated the risks associated with traditional aldehyde reagent cross-linkers mentioned in the prior literature.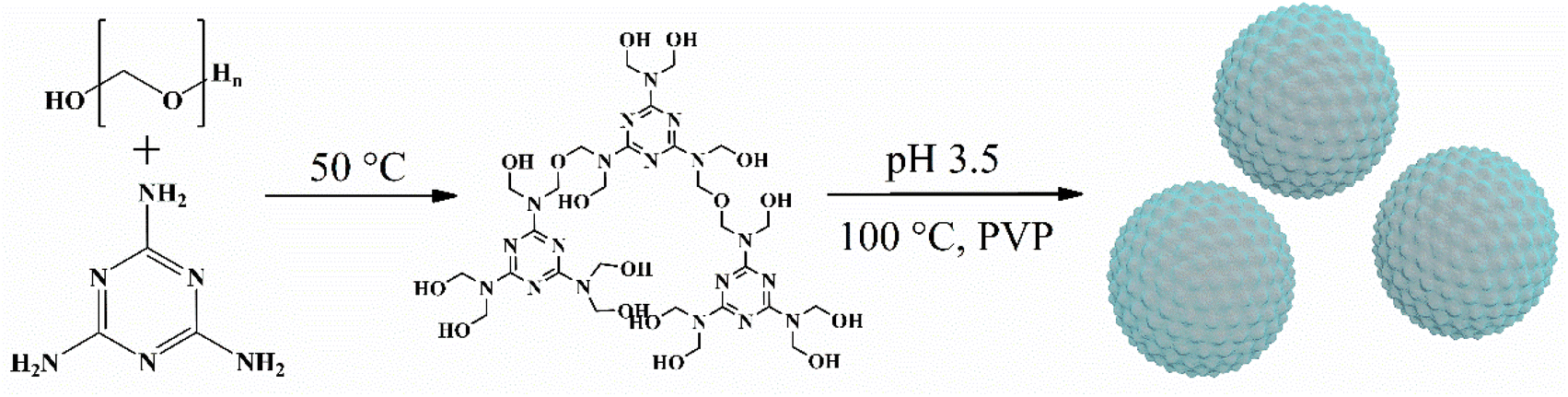 | ||
| Fig. 1 Schematic diagram of the synthesis of MFR. | ||
In principle, the geometric inhomogeneity of a solid adsorbent surface can dramatically change its adsorption properties, and it is critical for adsorbate–adsorbent interactions.26,27 For adsorbents of the same quality, a smaller particle size and rougher surface mean a larger specific surface area, enhancing interfacial adsorption and improving the external mass transfer coefficient.28,29 In this study, PVP was employed to control particle size and surface roughness. Various quantities of PVP (0, 0.10, 0.30, 0.50, and 0.70 g) were investigated to achieve the optimal product. As illustrated in Fig. 2a–e, an increase in the amount of PVP resulted in a slight reduction in particle size, but an increase in surface roughness, ultimately led to a more uniform particle size. This effect may be attributed to PVP, a negatively charged amphiphilic polymer known to adsorb onto various colloids.30 As colloid HMDs formed, they adhered to the PVP chain, forming HMD–PVP clusters. Over time, these clusters aggregated to form microspheres with rough surfaces.31 These rough surfaces provided more adsorption sites, making the combination of a rough surface and uniform particle size more conducive to the reproducible adsorption of PAHs.
 | ||
Fig. 2 SEM images of the MFRs (a–e) with a PVP quality of 0, 0.10, 0.30, 0.50, and 0.70 g, respectively. SEM images of the fiber (f) before and (g) after corroding by using aqua regia; SEM images of the MFR-based SPME fiber at different magnifications: (h) ×500; (i) ×5000; (j) ×25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. 000. | ||
FT-IR spectroscopy was used to confirm the synthesized MFR, as depicted in Fig. 3a. Specific absorption peaks were observed, consistent with prior research.32 Vibrations of the imine appeared at approximately 3370 cm−1, while both 810 and 1556 cm−1 originated from the vibration of the triazine ring. Peaks at 1160 and 1494 cm−1 corresponded to methylene, and the peak at 1008 cm−1 was attributed to the vibration of the hydroxymethyl group. Fig. 3b and c show the N2 adsorption–desorption isotherm and pore size distribution. According to the IUPAC guidelines, the N2-adsorption–desorption isotherm of the MFR exhibited a type III isotherm, indicating that the interactions between MFR and PAHs were relatively weak, and the adsorbed molecules clustered around the most favorable sites on the surface of MFR.33 The pore size distribution confirmed the presence of a mesoporous structure, which was beneficial for adsorbing PAHs. Fig. 3d–g show the XPS analysis results. The XPS C 1s (Fig. 3e) spectra can be fitted into C–H (284.7 eV), C–O (285.4 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (285.6 eV), and C–N (287.1 eV). N 1s spectra (Fig. 3f) could be deconvoluted into –NH– (398.4 eV), CN (399.6 eV), and –NH2 (407.1 eV), while O 1s spectra (Fig. 3g) could be fitted into three peaks of C–O (530.6 eV), CO (531.9 eV), and O–H (532.7 eV). Both FT-IR spectroscopy and XPS analysis confirmed the successful synthesis of the MFR, revealing its strong π conjugated system and abundant amino architectures. Furthermore, the TGA curve (Fig. 3h) confirmed its excellent thermostability. The initial weight loss during the temperature increased from 35 to 150 °C, which was due to the evaporation of physisorbed water. The second weight loss under the temperature range of 150 to 350 °C was attributed to the decomposition of oligomers and the pyrolysis of some polar functional groups (–OH and –NH2) on the surface. The third weight loss under the temperature range of 350 to 600 °C corresponded to the disintegration of the MFR. The MFR's impressive thermostability makes it a promising candidate for use as an SPME fiber coating. In Fig. 2f and g, SEM images of the fiber before and after corrosion by using aqua regia are presented, illustrating the smooth surface that facilitates the formation of a uniform MFR-based SPME fiber. Additionally, the surface morphology of the MFR-based SPME fiber (Fig. 2h–j) showed that the stacking of the MFR created numerous crevices, resulting in numerous macropores that could serve as buffer spaces for the rapid sorption of analytes.34 The thickness of the MFR-based SPME fiber was approximately 70 μm.
N (285.6 eV), and C–N (287.1 eV). N 1s spectra (Fig. 3f) could be deconvoluted into –NH– (398.4 eV), CN (399.6 eV), and –NH2 (407.1 eV), while O 1s spectra (Fig. 3g) could be fitted into three peaks of C–O (530.6 eV), CO (531.9 eV), and O–H (532.7 eV). Both FT-IR spectroscopy and XPS analysis confirmed the successful synthesis of the MFR, revealing its strong π conjugated system and abundant amino architectures. Furthermore, the TGA curve (Fig. 3h) confirmed its excellent thermostability. The initial weight loss during the temperature increased from 35 to 150 °C, which was due to the evaporation of physisorbed water. The second weight loss under the temperature range of 150 to 350 °C was attributed to the decomposition of oligomers and the pyrolysis of some polar functional groups (–OH and –NH2) on the surface. The third weight loss under the temperature range of 350 to 600 °C corresponded to the disintegration of the MFR. The MFR's impressive thermostability makes it a promising candidate for use as an SPME fiber coating. In Fig. 2f and g, SEM images of the fiber before and after corrosion by using aqua regia are presented, illustrating the smooth surface that facilitates the formation of a uniform MFR-based SPME fiber. Additionally, the surface morphology of the MFR-based SPME fiber (Fig. 2h–j) showed that the stacking of the MFR created numerous crevices, resulting in numerous macropores that could serve as buffer spaces for the rapid sorption of analytes.34 The thickness of the MFR-based SPME fiber was approximately 70 μm.
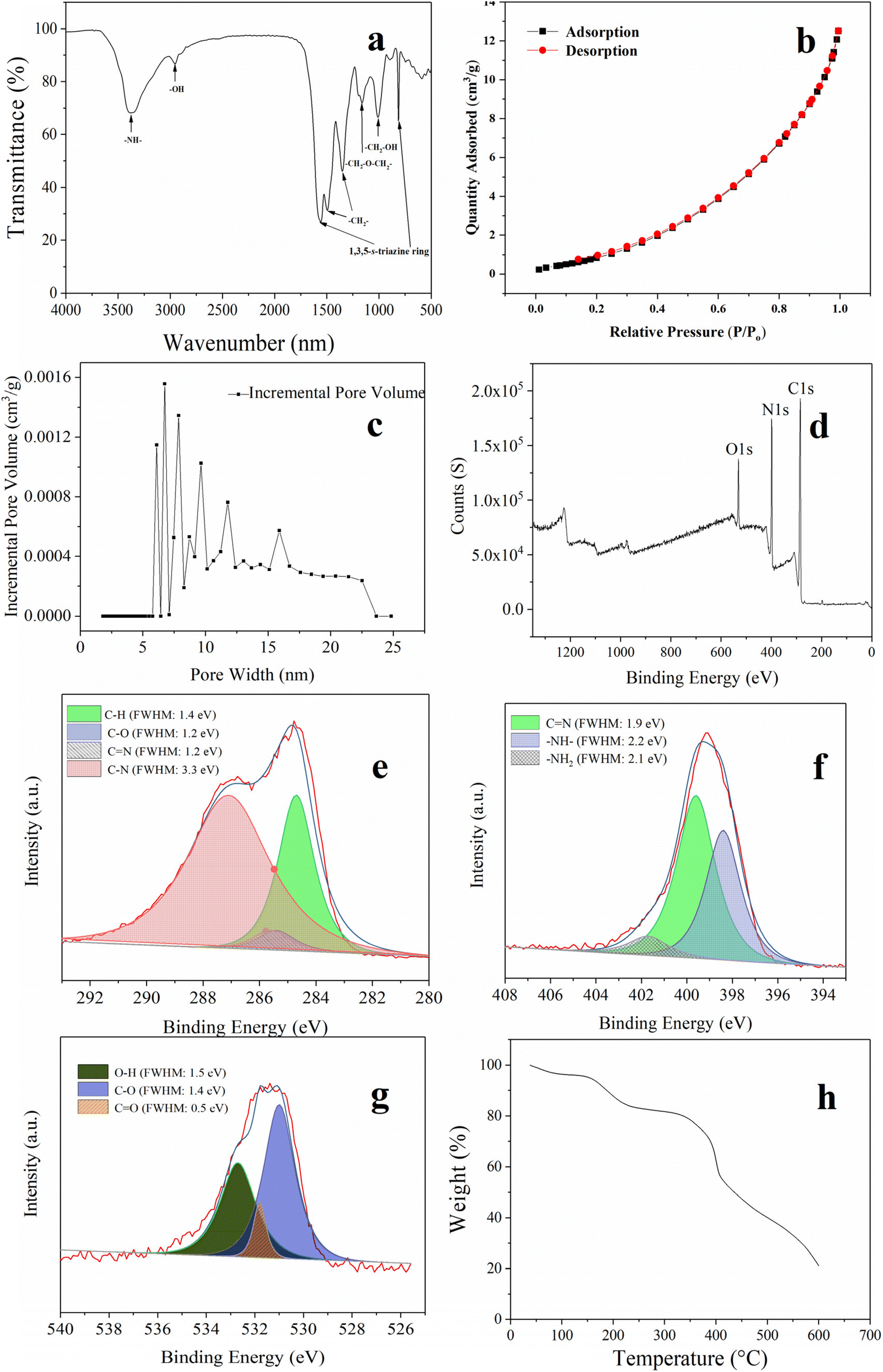 | ||
| Fig. 3 (a) FT-IR spectra; (b) N2 adsorption–desorption isotherms; (c) pore size distribution; (d–g) XPS spectra; and (h) thermogravimetric analysis of the MFR. | ||
Optimization of the HS-SPME process
Typically, factors such as thickness of the fiber coating, environmental water ionic strength, adsorption temperature and time, and desorption temperature and time can all potentially influence extraction efficiency. Therefore, the optimization of these parameters was investigated using the control-variate method. In all optimization experiments, a 10.0 mL spiked sample (5 ng mL−1) was employed.The thickness of the fiber coating significantly influenced the extraction efficiency of PAHs. In this work, the optimal dosage of the MFR was investigated, determined by coating number. Taking into account the limited space between the corroded stainless-steel wire and the needle, the extraction efficiency of one- and two-layer MFR-coated fibers was investigated. The results showed that the extraction efficiency of the fiber was higher when the fiber coating had two layers (2.0 to 2.8 times higher). This is because the increased number of monodisperse MFR microspheres enhances the adsorption sites of the fiber for PAHs, improving the extraction efficiency of the MFR-coated fiber. Hence, a two-layer MFR-coated fiber (approximately 2 mg of MFR powder) was selected for further experiments.
HS-SPME relies on an equilibrium distribution of analytes among the gas, liquid, and fiber coatings. The number of analytes extracted increases until adsorption equilibrium is reached. It is crucial to optimize the extraction time since it significantly impacts the extraction efficiency. Different extraction time profiles (20, 35, 50, 65, and 80 min) were investigated at a constant temperature (55 °C). As indicated in Fig. 4a, the optimal extraction efficiency for Fluo and Pyr was achieved at 65 min. In contrast to Fluo and Pyr, the highest extraction efficiency for the other PAHs was attained in 35 min due to competitive adsorption between these analytes. Fluo and Pyr possess stronger conjugated structures and hydrophobicity, facilitating stronger π–π conjugation with the MFR, resulting in competitive adsorption. Considering the extraction efficiency of all seven PAHs, 65 min was selected for the subsequent experiments.
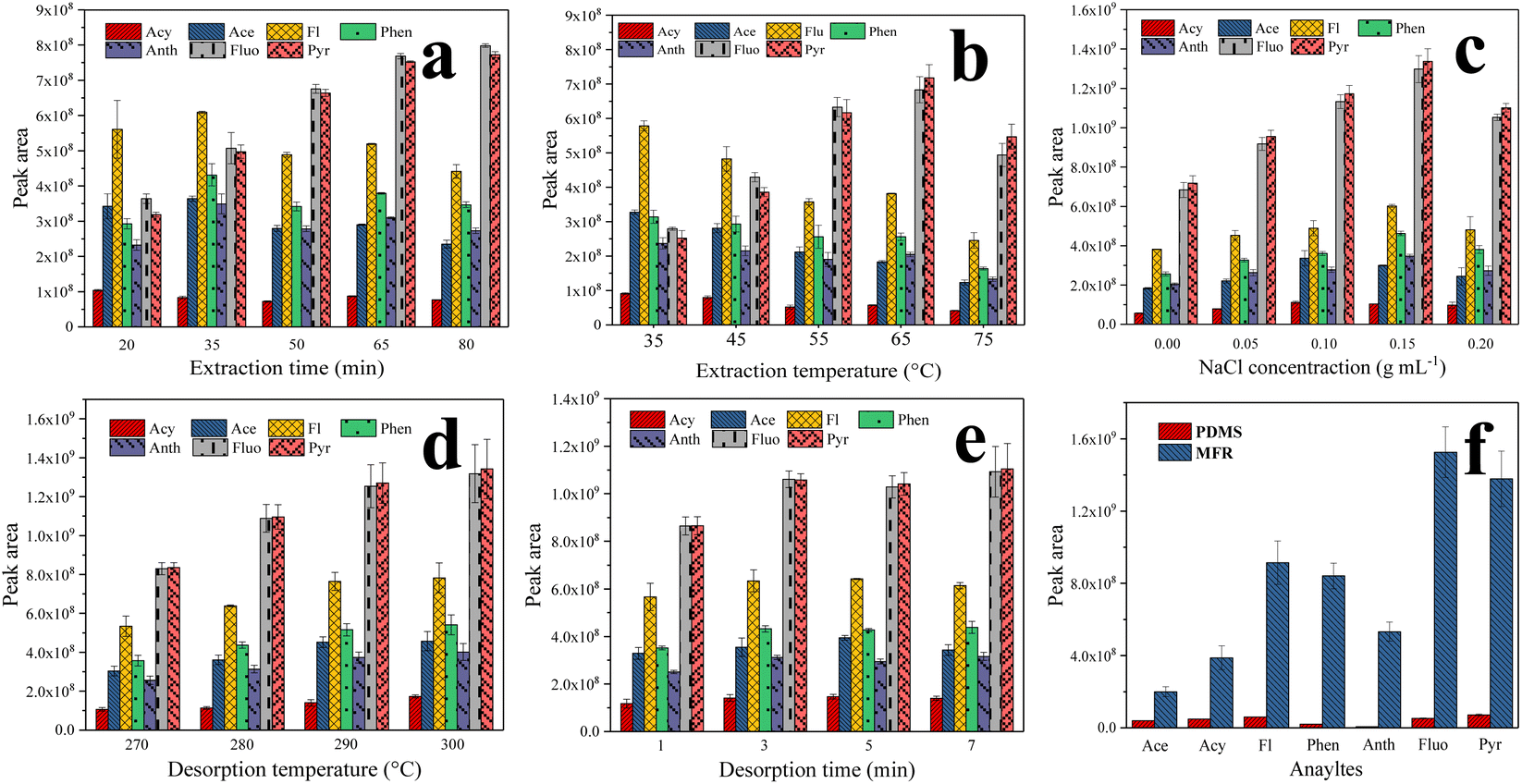 | ||
| Fig. 4 Effects of SPME conditions on the extraction efficiencies: (a) extraction time; (b) extraction temperature; (c) NaCl concentration; (d) desorption temperature; (e) desorption time; and (f) comparison of the enrichment efficiencies of PDMS and the MFR. | ||
The ideal temperature increases the diffusion coefficient, while ensuring a high distribution coefficient of PAHs between water samples and the MFR, influencing the extraction efficiency positively. In this study, the effect of extraction temperatures (35, 45, 55, 65, and 75 °C) was investigated. As depicted in Fig. 4b, Fluo and Pyr obtained the ideal extraction efficiency at 65 °C. However, for the other PAHs, the extraction efficiency decreased with increasing temperature, because the reduction of partition coefficients at high temperatures affected the extraction of PAHs by the MFR coating, negatively impacting the extraction efficiency.35 Given the extraction efficiency of all seven PAHs, 65 °C was chosen for further experiments.
Appropriate ionic strength can enhance extraction efficiency due to the salting-out effect. NaCl concentration, ranging from 0 to 0.20 g mL−1 (w/v), was investigated for ionic strength. Initially, as the NaCl concentration increased, analyte extraction was enhanced due to salting-out. However, when the NaCl concentration was further increased, the interaction between PAHs and salt ions became significant, inhibiting the extraction efficiency by decreasing the activity coefficients of the analytes, as shown in Fig. 4c. Hence, a NaCl concentration of 0.15 g mL−1 was selected for further experiments.
Additionally, desorption temperature and desorption time are critical parameters for SPME. Incomplete desorption can reduce method sensitivity, while excessively high desorption temperature and prolonged desorption time can shorten the lifespan of the SPME fiber. The effects of desorption temperature (270, 280, 290, and 300 °C) and desorption time (1, 3, 5, and 7 min) were examined, as shown in Fig. 4d and e. Given the thermal stability of the MFR-based SPME fiber, thermal desorption was performed at 290 °C for 3 min.
Validation of the HS-SPME-GC-MS/MS method
As the results presented in Table 2, the sensitivity, precision, and accuracy of the developed HS-SPME-GC-MS/MS method were evaluated. The proposed HS-SPME-GC-MS/MS method exhibited a strong correlation coefficient (r > 0.9974). The limits of detection (LODs) (S/N = 3) ranged from 0.04 to 0.15 ng L−1, and the limits of quantitation (LOQs) (S/N = 10) ranged from 0.13 to 0.50 ng L−1. Intra-day precision and inter-day precision were assessed using the relative standard deviation (RSD, %), with values ranging from 1.7% to 6.0% and 5.7% to 7.5%, respectively. Meanwhile, recoveries in the range of 76.5% to 125.2% were calculated at concentrations of 0.50, 1.0, and 10 ng mL−1 (Table 3). The RSDs were 1.7% to 6.0% for single fiber repeatability and 8.7% to 14.6% for fiber-to-fiber reproducibility, respectively. Furthermore, the MFR-based SPME fiber demonstrated an excellent extraction efficiency after 40 cycles (recoveries of seven PAHs ranged from 72.7% to 102.0%). All the results indicate that this method exhibits satisfactory precision, accuracy, and repeatability and can be applied for the separation and efficient enrichment of PAHs in environmental water matrices.| Analytes | Calibration plot (y = ax + b) | r | Line range (ng L−1) | LOQs (ng L−1) | RSD (%) | EFs | |||
|---|---|---|---|---|---|---|---|---|---|
| Intra-day | Inter-day | Single fiber repeatability | Fiber-to-fiber reproducibility | ||||||
| Acy | y = 3.5 × 107x + 7.3 × 106 | 0.9980 | 10–10000 |
0.25 | 4.2 | 7.1 | 4.2 | 11.6 | 7153 |
| Ace | y = 5.8 × 107x + 1.3 × 107 | 0.9997 | 10–10000 |
0.46 | 2.7 | 7.1 | 2.7 | 14.6 | 4609 |
| Fl | y = 1.6 × 108x + 4.3 × 107 | 0.9974 | 10–10000 |
0.30 | 3.2 | 7.5 | 3.2 | 11.5 | 2414 |
| Phen | y = 1.6 × 108x + 4.3 × 107 | 0.9998 | 10–10000 |
0.13 | 6.0 | 6.5 | 6.0 | 9.6 | 3821 |
| Anth | y = 1.0 × 108x + 1.7 × 107 | 0.9999 | 10–10000 |
0.50 | 1.7 | 5.7 | 1.7 | 14.2 | 1906 |
| Fluo | y = 3.0 × 108x + 5.0 × 107 | 0.9996 | 10–10000 |
0.26 | 2.3 | 6.5 | 2.3 | 8.7 | 3028 |
| Pyr | y = 3.0 × 108x + 2.9 × 107 | 0.9982 | 10–10000 |
0.26 | 2.4 | 6.6 | 2.4 | 9.0 | 2957 |
| Analytes | 0.5 ng mL−1 | 1.0 ng mL−1 | 10 ng mL−1 | |||
|---|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | |
| Acy | 99.9 | 11.3 | 86.3 | 2.6 | 85.7 | 3.8 |
| Ace | 125.2 | 0.8 | 109.1 | 5.2 | 96.3 | 4.5 |
| Fl | 88.2 | 12.6 | 77.3 | 2.0 | 76.5 | 3.5 |
| Phen | 89.5 | 10.9 | 78.7 | 7.6 | 78.3 | 1.7 |
| Anth | 105.8 | 11.1 | 99.4 | 4.8 | 100.9 | 1.6 |
| Fluo | 120.6 | 3.9 | 117.4 | 3.7 | 100.8 | 3.4 |
| Pyr | 122.7 | 6.1 | 114.1 | 4.8 | 101.4 | 3.7 |
Application of the HS-SPME-GC-MS/MS method
Four types of environmental water samples (three lake water, one river water, one tap water, and two bottled water) were analyzed to assess its practicality. Among the four types of water samples, the sample from the Huchenghe River exhibited the presence of Anth (13 ng L−1), Fluo (27 ng L−1), and Pyr (82 ng L−1). In comparison with the other water samples, it was observed that the sample from the Huchenghe River flows through an urban area with municipal wastewater discharge, resulting in higher concentrations of organic pollutants, including PAHs, than in the other water samples. This indicates that the developed HS-SPME-GC-MS/MS method is dependable for the determination of trace PAHs in environmental water.Comparison with the reported literature
To verify that the materials synthesized in this work have an eco-friendly preparation process, the types and amounts of the reaction solvent, reaction temperature and reaction time used in the preparation of fiber coatings for the SPME of PAHs were compared, as shown in Table 4. In this study, the MFR offers several advantages, including an eco-friendly preparation process, eliminating the use of harmful organic solvents. It greatly reduces the risk to human health and damage to the environment. In addition, the mild conditions and short time required for the synthesis of the MFR greatly save time and energy. Therefore, the synthesis process of the MFR is more conform the concept of green chemistry and provides a sustainable strategy for the preparation of fiber coatings.| Fiber coating | Reaction solvent and volume | Reaction temperature and time | Linearity | LODs | RSDs of reproducibilitya (%) | EFs | Ref. |
|---|---|---|---|---|---|---|---|
| a Fiber-to-fiber reproducibility. b Sulfuric acid, potassium permanganate and hydrogen peroxide were used in the preparation of graphene with the modified Hummers method. MOF@MON: metal–organic framework@microporous organic network. COF-TAPB-TMC: covalent organic framework from 1,3,5-tris(4-aminophenyl)benzene and trimesoyl chloride. COF/rGO: covalent organic framework/reduced graphene oxide composite. | |||||||
| MOF | N,N-Dimethylformamide (13.5 mL) | 130 °C, 24 h | 0.1–100 ng mL−1 | 20–5570 ng L−1 | 4.3–9.3 | 3104–5980 | 15 |
| COF-TAPB-TMC | 1,4-Dioxane (60 mL) | RT, >25 h | 0.002–2 ng mL−1 | 0.41–0.94 ng L−1 | 8.5–11.0 | 819–2420 | 16 |
| MOF@MON | N,N-Dimethylformamide (15 mL) | 110 °C, >48 h | 0.1–500 ng L−1 | 0.02–0.3 ng L−1 | 6.5–8.7 | 1215–3805 | 17 |
| Toluene (10 mL) | |||||||
| Triethylamine (5 mL) | |||||||
| COF/rGO | o-Dichlorobenzene (2.0 mL), n-butyl alcohol (2.0 mL) | 120 °C, >72 h | 0.5–250 ng mL−1 | 90–590 ng L−1 | 5.0–7.8 | 1288–8816 | 18 |
| Modified Hummersb | |||||||
| MOF | Water (48 mL) | 160 °C, 72 h | 0.010–1 ng mL−1 | 0.07–1.67 ng L−1 | 1.64–9.78 | 130–2288 | 19 |
| TiO2-melamine–formaldehyde | Formaldehyde solution (37%, 5 mL) | 90 °C, >24 h | 0.15–15 ng mL−1 | 10–100 ng L−1 | 3.07–10.55 | 653–1007 | 40 |
| Water (40 mL) | |||||||
| Graphene | Water (50 mL) | 150 °C, 13 h | 10–1000 ng mL−1 | 2–10 ng L−1 | 6.4–11.9 | 842–2458 | 41 |
| MFR | Water (40 mL) | 100 °C, 1.5 h | 0.01–10 ng mL−1 | 0.04–0.15 ng L−1 | 8.7–14.6 | 1906–7153 | This work |
In addition, the MFR-based SPME fiber provides a high enrichment capacity. The fiber was employed for the SPME of PAHs, exhibiting similar repeatability between needles as documented in the literature, while the established method showed lower LODs and satisfactory recoveries. It is worth emphasizing that the proposed method, utilizing MFR as a coating material, delivers high enrichment factors (1906–7153-fold), indicating that the HS-SPME-GC-MS/MS method holds potential for the monitoring of PAHs in environmental water.
Environmental assessment of the HS-SPME-GC-MS/MS method
In the context of analytical chemistry and green analytical chemistry, the development of environment friendly sample pretreatment technologies, including the preparation of sustainable solid phase microextraction fiber coatings through green synthesis strategies, is of great significance to reduce or eliminate negative effects on the environment and population. To assess the environmental effects of the proposed method, the greenness was evaluated using the Analytical Eco-Scale. The analytical ecological scale is based on an ideal green analytical value of 100, and then penalty points for each analytical procedure parameter of the method (chemical consumption, hazard, energy and waste), and the resulting total score is used to evaluate the greenness of the method.36,37 In this work, a total score of 95 was obtained on the Analytical Eco-Scale and this reflected that this work proposes an environment friendly synthesis strategy to prepare a novel fiber coating, which uses headspace solid phase microextraction combined with GC-MS/MS for the sensitive detection of PAHs in environmental water (Table 5). The synthesis strategy has the advantages of using water as the solvent, the synthesis time is short, the reaction conditions are mild, and no toxic and harmful products are produced. Under this premise, the proposed method also showed great improvement in performance, including high accuracy, sensitivity and enrichment factors.| Reagent | PPs |
|---|---|
| Hydrochloric acid | 2 |
| Instruments | PPs |
|---|---|
| Heater | 1 |
| GC-MS/MS | 2 |
| Total PPs: 5 | |
| Analytical Eco-Scale total score: 95 | |
Adsorption capabilities and adsorption mechanism
Enrichment factors (EFs) were calculated to evaluate the adsorption capability of the fiber. It is worth mentioning that the EFs of the proposed method ranged from 1906- (Anth) to 7153-fold (Acy). These were defined as the ratio of the sensitivity of this method after extraction to that before extraction (i.e., direct injection of the standard solution) using peak area for quantification.15–17 To further evaluate the practicality of the MFR-based SPME fiber, the adsorption capability was assessed and compared to that of a commercial fiber (100 μm PDMS). The HS-SPME process for the commercial PDMS fiber was carried out following the procedure outlined in the literature.38 As shown in Fig. 4f, the enrichment efficiencies of the MFR coating were significantly higher than those of PDMS (4.9–82.8 times). This exceptional superiority in capturing PAHs can be attributed to the strong π-conjugated system and abundant amino architectures of the MFR, which give rise to amino enhancement and π–π stacking interactions. Additionally, the porous structure, rough surface of the MFR, and the presence of through macropores in the fiber coating increase the number of adsorption sites and facilitate the rapid mass transport of analytes.39 Consequently, the MFR-based SPME fiber coating exhibited an excellent enrichment performance, demonstrating that the MFR can be effectively used as a potential fiber coating for extracting nonpolar pollutants.Conclusion
In summary, MFR with uniform particle size and adjustable surface roughness was innovatively utilized as a headspace SPME fiber coating. Developed through an environmentally friendly synthesis strategy, the MFR offers the advantages of simplicity, cost-effectiveness, reproducibility, and eco-friendliness. Importantly, the MFR exhibits exceptional thermal stability, withstanding temperatures up to 350 °C, rendering it suitable for HS-SPME with thermal desorption. It is noteworthy that the remarkable adsorption performance of this MFR-based SPME fiber was evident in the extraction of seven PAHs from environmental water samples. In detail, a high enrichment capacity (ranging from 1906- to 7153-fold) was achieved during validation, primarily attributed to the strong π–π interactions and the amino-enhanced affinity between the MFR and analytes, in addition to the porous structure and rough surface of the MFR. In comparison with commercially available fibers (such as PDMS), the enrichment efficiencies of the MFR fiber coating exceeded PDMS by factors ranging from 4.9 to 82.8 times. All these results collectively support the notion that the design concept of the MFR-based SPME fiber is a valuable approach for the pretreatment and analysis of PAHs or similar pollutants in environmental water.Author contributions
Pengfei Li: data curation, visualization, and writing – original draft. Yehong Han: visualization. Dandan Han: methodology. Hongyuan Yan: conceptualization, writing – review and editing, and project administration.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (82073605 and 82373634), the Nature Science Interdisciplinary Research Project of Hebei University (DXK202014), the Innovation Team Program of Hebei University (IT2023A06) and the Post-graduate's Innovation Fund Project of Hebei Province (CXZZBS2023006).References
- R. Naidu, B. Biswas, I. R. Willett, J. Cribb, B. K. Singh, C. P. Nathanail, F. Coulon, K. T. Semple, K. C. Jones, A. Barclay and R. J. Aitken, Environ. Int., 2021, 156, 106616 CrossRef CAS PubMed.
- P. M. Nowak, Green Chem., 2023, 25, 4625–4640 RSC.
- S. S. Nasrollahi and N. S. Moosavi, TrAC, Trends Anal. Chem., 2023, 166, 117163 CrossRef CAS.
- M. Shi, X. Zheng, N. Zhang, Y. Guo, M. Liu and L. Yin, TrAC, Trends Anal. Chem., 2023, 166, 117211 CrossRef CAS.
- L. Xia, J. Yang, R. Su, W. Zhou, Y. Zhang, Y. Zhong, S. Huang, Y. Chen and G. Li, Anal. Chem., 2020, 92, 34–48 CrossRef CAS PubMed.
- M. Tobiszewski, A. Mechlińska, B. Zygmunt and J. Namieśnik, TrAC, Trends Anal. Chem., 2009, 28, 943–951 CrossRef CAS.
- S. Tang, H. Zhang and H. K. Lee, Anal. Chem., 2016, 88, 228–249 CrossRef CAS PubMed.
- P. Li, Y. Lu, J. Cao, M. Li, C. Yang and H. Yan, J. Chromatogr. A, 2020, 1623, 461192 CrossRef CAS PubMed.
- S. Wang, P. Li, Y. Han, H. Liu and H. Yan, Anal. Chim. Acta, 2022, 1227, 340328 CrossRef CAS PubMed.
- S. Tang, T. Qi, P. D. Ansah, J. C. N. Fouemina, W. Shen, C. Basheer and H. K. Lee, TrAC, Trends Anal. Chem., 2018, 108, 306–313 CrossRef CAS.
- Y. Yamini, M. Rezazadeh and S. Seidi, TrAC, Trends Anal. Chem., 2019, 112, 264–272 CrossRef CAS.
- I. Pacheco-Fernández, M. Rentero, J. H. Ayala, J. Pasán and V. Pino, Anal. Chim. Acta, 2020, 1133, 137–149 CrossRef PubMed.
- J. Zheng, Y. Kuang, S. Zhou, X. Gong and G. Ouyang, Anal. Chem., 2023, 95, 218–237 CrossRef CAS PubMed.
- B. Zhang, G. Xu, L. Li, X. Wang, N. Li, R. S. Zhao and J. Lin, Chem. Eng. J., 2018, 350, 240–247 CrossRef CAS.
- S. H. Huo, J. Yu, Y. Y. Fu and P. X. Zhou, RSC Adv., 2016, 6, 14042–14048 RSC.
- X. Yang, J. Wang, W. Wang, S. Zhang, C. Wang, J. Zhou and Z. Wang, Microchim. Acta, 2019, 186, 145 CrossRef PubMed.
- Y. Jia, H. Su, Z. Wang, Y. L. E. Wong, X. Chen, M. Wang and T. W. D. Chan, Anal. Chem., 2016, 88, 9364–9367 CrossRef CAS PubMed.
- C. Yu, X. Luo, F. Wu and J. Zhang, Food Anal. Methods, 2023, 16, 1131–1144 CrossRef.
- Q. L. Li, X. Wang, X. F. Chen, M. L. Wang and R. S. Zhao, J. Chromatogr. A, 2015, 1415, 11–19 CrossRef CAS PubMed.
- Y. Wang, W. Zhao, X. Tian, H. Song, R. Gao, X. Tang, X. Zhang, Y. Hao and Y. Tang, Chem. Eng. J., 2020, 392, 123716 CrossRef CAS.
- Y. Zhao, L. Xu, C. Yang, T. Chen and L. Yu, Appl. Organomet. Chem., 2019, 33, e5112 CrossRef.
- M. Wang, F. Qiao and H. Yan, Green Chem., 2021, 23, 5179 RSC.
- Y. Lu, P. Li, H. Yan and S. Shen, J. Agric. Food Chem., 2022, 70, 1327–1334 CrossRef CAS PubMed.
- D. Zou, P. Li, C. Yang, D. Han and H. Yan, Anal. Chim. Acta, 2022, 1226, 340271 CrossRef CAS PubMed.
- M. Wang, X. Chang, X. Wu, H. Yan and F. Qiao, J. Chromatogr. A, 2016, 1458, 9–17 CrossRef CAS PubMed.
- A. Malijevský, J. Chem. Phys., 2014, 141, 184703 CrossRef PubMed.
- E. Leiva, C. Tapia and C. Rodríguez, Water, 2021, 13, 2960 CrossRef CAS.
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC.
- B. D. Young and B. M. Van Vliet, Int. J. Heat Mass Transfer, 1988, 31, 27–34 CrossRef CAS.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182–1195 CrossRef PubMed.
- W. Zhu, K. Liang and Y. Ren, Ceram. Int., 2021, 47, 17192–17201 CrossRef CAS.
- Y. Wu, Y. Li, L. Qin, F. Yang and D. Wu, J. Mater. Chem. B, 2013, 1, 204–212 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- H. Cheng, Y. Song, Y. Bian, R. Ji, F. Wang, C. Gu, X. Yang, M. Ye, G. Ouyang and X. Jiang, Sci. Total Environ., 2019, 681, 392–399 CrossRef CAS PubMed.
- L. Liu, W. K. Meng, Y. S. Zhou, X. Wang, G. J. Xu, M. L. Wang, J. M. Lin and R. S. Zhao, Chem. Eng. J., 2019, 356, 926–933 CrossRef CAS.
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, TrAC, Trends Anal. Chem., 2012, 37, 61–72 CrossRef.
- M. Papageorgiou, D. Lambropoulou, C. Morrison, J. Namieśnik and J. Płotka-Wasylka, Talanta, 2018, 183, 276–282 CrossRef CAS PubMed.
- A. M. Sulej-Suchomska, Ż. Polkowska, T. Chmiel, T. M. Dymerski, Z. J. Kokot and J. Namieśnik, Anal. Methods, 2016, 8, 4509–4520 RSC.
- N. Zhang, X. Lei, T. Huang, L. Su, L. Zhang, Z. Xie and X. Wu, Chem. Eng. J., 2020, 386, 124003 CrossRef CAS.
- M. Sun, X. Wang, Y. Ding and J. Feng, Microchim. Acta, 2022, 189, 456 CrossRef CAS.
- S. Zhang, Z. Li, X. Yang, C. Wang and Z. Wang, RSC Adv., 2015, 5, 54329–54337 RSC.
| This journal is © The Royal Society of Chemistry 2024 |