
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Closing the loop for poly(butylene-adipate-co-terephthalate) recycling: depolymerization, monomers separation, and upcycling†
Mohamed
Ismail
 *a,
Adel
Abouhmad
ab,
Niklas
Warlin
ac,
Sang-Hyun
Pyo
a,
Oliver Englund
Örn
a,
Basel
Al-Rudainy
d,
Cecilia
Tullberg
a,
Baozhong
Zhang
c and
Rajni
Hatti-Kaul
*a
*a,
Adel
Abouhmad
ab,
Niklas
Warlin
ac,
Sang-Hyun
Pyo
a,
Oliver Englund
Örn
a,
Basel
Al-Rudainy
d,
Cecilia
Tullberg
a,
Baozhong
Zhang
c and
Rajni
Hatti-Kaul
*a
aDivision of Biotechnology, Department of Chemistry, Center for Chemistry and Chemical Engineering, Lund University, SE-221 00 Lund, Sweden. E-mail: mohamed.ismail@biotek.lu.se; rajni.hatti-kaul@biotek.lu.se
bDepartment of Microbiology and Immunology, Faculty of Pharmacy, Al-Azhar University, Assiut 71524, Egypt
cCenter for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden
dDepartment of Chemical Engineering, Center for Chemistry and Chemical Engineering, Lund University, SE-221 00 Lund, Sweden
First published on 21st February 2024
Abstract
Efficient recycling and upcycling strategies to retain the material in the economy and away from the ecosystems are important to achieve a sustainable plastic system. Poly(butylene adipate-co-terephthalate) (PBAT) is a biodegradable polyester that has gained considerable interest for various applications. Here, we report a study on enzymatic depolymerization of PBAT, recovery and purification of its monomers, and feasible routes for their recycling/upcycling. PBAT films (15 g L−1) were completely hydrolysed employing a leaf-branch compost cutinase variant (LCC-WCCG, 1.4 mg per gram polymer) to its monomers at a rate of 0.49 g L−1 h−1. LCC-WCCG kinetics were superior to that of other enzymes engineered for PBAT hydrolysis; the data were supported by in silico investigations. The released monomers were separated using membrane filtration and precipitation techniques and recovered with purity exceeding 95%. To close the loop, the monomers were re-polymerized and successfully cast into PBAT films. Moreover, adipic acid was reacted with hexamethylene diamine using Novozym®435 to form a polyamide, while 1,4-butanediol was oxidized to 4-hydroxybutyrate using Gluconobacter oxydans cells. The current study exemplifies a high-impact scientific approach toward a circular plastics economy.
Introduction
The role and importance of plastics in our economy and society have been consistently growing over the past century. With a global production of about 400 million metric tonnes in 2022,1 plastic has become the most abundant anthropogenic material besides steel and concrete, and the market is expected to double over the next 20 years.2 Despite the multiple and immense benefits plastic brings to society, there has been increasing concern worldwide about the negative impact of the current linear plastic system on ecosystems and climate change.3–7 Among the largest contributing factors are the fossil origin of most plastics, the short-term use of about 44% of all plastics, and the low rate of recycling (on average only 8%).8,9 The dominant fraction of the post-consumer plastic is incinerated for energy generation, while the remainder goes for landfilling, or ends up as litter on land and in oceans.5 The recycled plastic is invariably of lower quality than the primary plastic, and used for making products with less stringent qualities. Currently, negotiations are ongoing for a legally binding global plastics treaty to promote sustainable production and consumption of plastics using measures across their life cycle.10 A general recommendation to achieve a sustainable circular plastics economy is to decouple production from fossil resources, improve product design to facilitate reuse and recycling of plastics, and implement an effective plastic collection, sorting, and recycling system.10–16 With stricter regulations coming into place in many countries, there is on one hand increasing ongoing research in developing chemical and biocatalytic recycling methods and on the other hand a growing demand for biodegradable plastics from various end-use sectors with good mechanical properties.10,17 Polyesters constitute a large group of thermoplastic polymers, which could be recyclable or biodegradable depending on their monomer composition.18,19 Theoretically, all polyesters are considered biodegradable (or biologically/chemically recyclable) as the ester bond linking the monomers is susceptible to the action of hydrolytic enzymes. Most aliphatic ester groups in polyesters are chemically/enzymatically cleavable at varying rates in specific environments like composting, whereas polyesters with mixed aromatic and aliphatic ester groups can be relatively tough to degrade, and the aromatic polyesters are known to be recalcitrant to biodegradation or chemical/biological cleavage.20,21Enzymatic hydrolysis of polyesters, especially PET, has been a subject of many reports in the last four decades.18,22–25 Almost all efficient PET hydrolases have been found to be cutinases,26,27 and only few enzymes with high catalytic activity and thermal stability have been identified;28–30 Among the most efficient cutinases is the thermostable leaf-branch compost cutinase (LCC),28 with 33-fold higher activity than the enzymes from Thermobifida fusca, Fusarium solani pisi, and Ideonella sakaiensis PETase.31 Various protein engineering techniques have been used to improve polymer degrading efficiency of the enzymes.31–34 Protein engineering of LCC has resulted in highly thermostable and efficient variants, LCC-WCCG and LCC-ICCG, exhibiting at least 90% depolymerization of PET (300 g L−1) within 10 hours.31
In recent years, the flexible co-polyester PBAT has become increasingly popular for applications in packaging and mulch films due to its favourable material and biodegradability properties, provided by the aromatic fraction interspersed with long aliphatic chains.35 Its production volume in 2021 was estimated to approach 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes, with a CAGR of 15.4% until 2028.36 PBAT is produced by several companies including BASF (Germany), Novamont (Italy), and Kingfa and Tunhe (China).19 Microbial and enzymatic degradation and/or depolymerization of PBAT have been reported earlier although at low degradation rates,37–43 however, reports on how the degrading bacteria can depolymerize or metabolize PBAT degradation products are scarce.39 Various PBAT degrading enzymes have been identified such as carboxylesterase (EC 3.1.1.1), triacylglycerol lipase (EC 3.1.1.3), cutinases (EC 3.1.1.74) and arylesterase (EC 3.1.1.2).43 Recently, a study on the efficient degradation of PBAT to terephthalic acid (TPA) and other oligomers using an engineered cutinase from Thermobifida fusca has been reported.44 Furthermore, crystal structures of the wild type and the generated mutant cutinases soaked with substrate analogs were revealed and used for illustrating the binding mode of PBAT to the enzyme active site.44
000 tonnes, with a CAGR of 15.4% until 2028.36 PBAT is produced by several companies including BASF (Germany), Novamont (Italy), and Kingfa and Tunhe (China).19 Microbial and enzymatic degradation and/or depolymerization of PBAT have been reported earlier although at low degradation rates,37–43 however, reports on how the degrading bacteria can depolymerize or metabolize PBAT degradation products are scarce.39 Various PBAT degrading enzymes have been identified such as carboxylesterase (EC 3.1.1.1), triacylglycerol lipase (EC 3.1.1.3), cutinases (EC 3.1.1.74) and arylesterase (EC 3.1.1.2).43 Recently, a study on the efficient degradation of PBAT to terephthalic acid (TPA) and other oligomers using an engineered cutinase from Thermobifida fusca has been reported.44 Furthermore, crystal structures of the wild type and the generated mutant cutinases soaked with substrate analogs were revealed and used for illustrating the binding mode of PBAT to the enzyme active site.44
This report presents a study on the scaled depolymerization of PBAT using the thermostable variant of LCC enzyme produced by recombinant Escherichia coli, followed by downstream processing to separate the released monomers, TPA, AA, and BDO in pure forms. Kinetics and profile of the LCC-WCCG catalysed depolymerization of PBAT were investigated and supported by in silico analysis of the enzyme structure and its interaction with the polymer. Furthermore, we demonstrated re-polymerization of the obtained monomers into PBAT films, as well as upcycling strategies of the aliphatic monomers (Fig. 1). AA was subjected to enzyme catalysed polymerization with hexamethylenediamine to form a polyamide, while BDO was selectively oxidized to 4-hydroxybutyric acid (4-HBA) using the bacterium Gluconobacter oxydans.
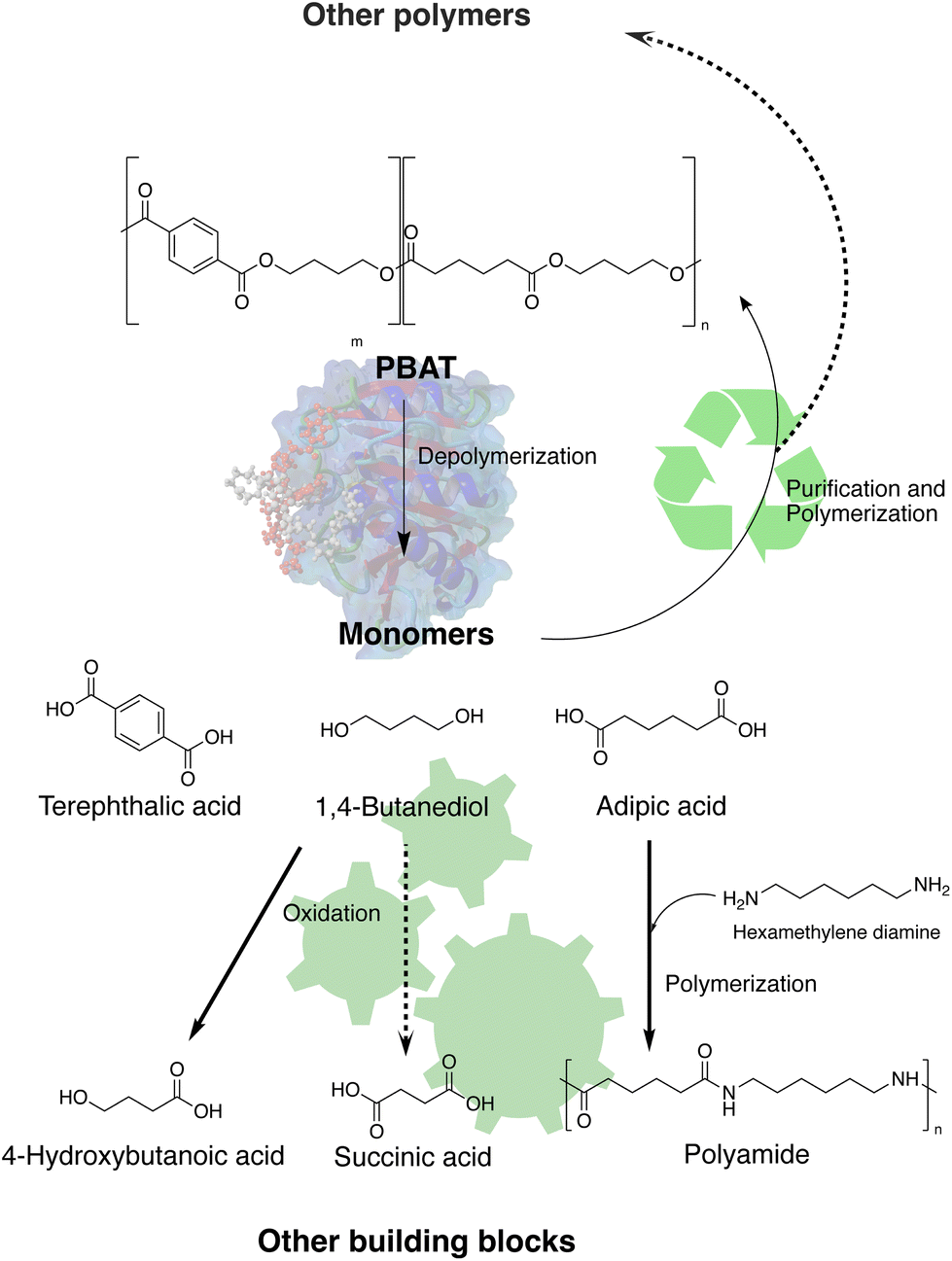 | ||
| Fig. 1 “Closing the loop” of PBAT, starting with the enzymatic hydrolysis into its monomer components (terephthalic acid, 1,4-butanediol, and adipic acid), the subsequent separation of the monomers, recycling, and potential upcycling routes. | ||
Experimental
Materials
Commercial PBAT film (Mn 9100 g mol−1, measured in this study) was provided by BioMi, Matulji, Croatia. NcoI, XhoI restriction enzymes as well as T4 DNA ligase enzyme were purchased from Thermo Fisher Scientific (Waltham, MA, USA). pET28a(+) expression vector and Escherichia coli BL21(DE3) expression host were procured from Novagen (Madison, WI, USA). Dimethyl terephthalate (DMT, 99%), chloroform-d (99.8% atom D), 1,4-butanediol (99%), and dibutyltin oxide (DBTO, 98%) were purchased from Sigma Aldrich. AA (99%) was purchased from Honeywell Fluka™, USA, xylene (99%) was purchased from Scharlau, Turkey, chloroform (99.8%) from Honeywell, USA, and methanol was from VWR chemicals, Sweden.Enzymatic hydrolysis of PBAT film
To 100 mM potassium phosphate buffer pH 8 containing 5 mg mL−1 PBAT films (1.0 × 1.0 cm) in 5 mL glass vials, the purified LCC-WCCG was added at different concentrations (0.01, 0.05, 0.1, 0.2, 0.5, 1, 1.5 μM) in a final volume of 1 mL. For initial screening, the reactions were maintained at 60 °C, 500 rpm in a thermomixer (Hettich, Germany). The effect of temperature on the enzymatic hydrolysis of PBAT was evaluated at different temperatures (30, 50, 70, 80, 90, and 100 °C) using 0.5 μM LCC-WCCG, while maintaining the aforementioned reaction conditions. After 24 hours, the reactions were terminated and the reaction mixtures were kept at 4 °C, the released monomers were analysed by HPLC. All reactions were run in triplicates.Scaled depolymerization experiments were performed at 70 °C as this was the maximum operational temperature possible in the lab scale bioreactor. Initial small-scale reaction was conducted in 50 mL volume containing 100 mM potassium phosphate buffer pH 8, 253 mg PBAT film (1.0 × 1.0 cm pieces) at 70 °C, 150 rpm, and was initiated by adding 1 μM (0.03 mg mL−1) of the purified LCC-WCCG. The reaction was monitored visually through the disintegration of the PBAT films and by HPLC to determine the concentration of the released monomers. After one day, a new batch of PBAT (375 mg) and an additional 0.5 μM fresh pure enzyme were added, while on the third day, fresh 0.156 μM of LCC-WCCG was added to the reaction. The pH of the reaction was monitored and adjusted to 8 using 1 M NaOH.
PBAT depolymerization reaction was then conducted in a 2 L stirred bioreactor (Labfors, Infors AG, Switzerland) with a water jacket for temperature control, containing 100 mM potassium phosphate buffer pH 8, 15 g PBAT films (1.0 × 1.0 cm) in a total reaction volume of 1 L. The reaction was initiated by adding 10.6 mg of the purified LCC-WCCG and incubated at 70 °C, 250 rpm. The pH of the reaction was maintained at pH 8 using 1 M NaOH or 2 M H3PO4, respectively, and samples were taken at intervals for analysis of the released monomers. After 48 hours, a new batch of LCC (11 mg) was added. The reaction was continued for 96 hours to ensure full depolymerization.
The extent of PBAT depolymerization was determined by measuring the weight loss of the polymer and the molar concentration of the monomers released.
Kinetic analysis of LCC-WCCG on PBAT – Michaelis–Menten (MM) kinetics
The conventional MM kinetics were calculated based on HPLC analysis of the released monomers (BDO, TPA, AA) to measure the initial reaction rate for LCC-WCCG (1 μM) activity against PBAT at concentrations of 0.5, 1.3, 2, 4, 6.6, 8, 15, 20, 25 mg mL−1, respectively.The Inverse MM kinetics were calculated following the same procedure described for the conventional kinetics, however, with fixed PBAT load (10 mg mL−1) and variable LCC-WCCG concentrations (0.01, 0.05, 0.1, 0.5, 1.0, 1.5 μM).
All reactions were run in triplicates in 5 mL volume using 100 mM potassium phosphate buffer pH 8. Reactions were incubated at 75 °C (to avoid the fluctuation observed at higher temperatures due to liquid evaporation) in a thermoshaker (Hettich Benelux) at 500 rpm. Samples were taken at intervals over 48 hours, and the released monomers (AA, BDO, and TPA) were quantified by HPLC analysis against a standard curve for each compound. The data were fitted to the Michaelis–Menten equation for calculating the kinetic parameters (eqn (S1)–(S3)†).45
In silico investigation of LCC-WCCG
Crystal structure of LCC mutant S165A was retrieved from the protein data bank (pdb entry: 6THS). YASARA structure program was used to remove the water molecules and other ligands (1,4-diethylene dioxide), and to add the missing hydrogen from the structure. In order to generate the WCCG mutant (WCCG) the residues F243, D238 S283, Y127 in the LCC wild type were swapped, and A165 was also swapped to serine residue. The structure was energy minimised and saved as pdb file before using in further experiments. The generated WCCG variant was subjected to molecular dynamic simulation using AMBER 15IPQ as forcefield, the simulation cell was defined around all atoms by 5 Å, filled with water molecules (0.99 g mL−1), and MD-simulation was run for 3 ns. Thereafter, deviation of the carbon backbone (RMSD-Ca) and fluctuation of the amino acids were determined. Three simulation snapshots were selected to conduct the docking studies on PBAT and PET oligomer. PBAT was built using YASARA structure, composed of two units of TPA-BDO and AA-BDO, respectively, in different arrangements. Similarly, PET was built composed of 4 units of TPA-EG. The energy of binding and dissociation constant were measured, and docking results were evaluated based on the correct binding pose within the active site and the energy of binding. The best pose(s) were used for running MD-simulation to evaluate the deviation of the enzyme and ligand.Downstream processing to separate PBAT degradation products (TPA, AA, BDO)
Purification of TPA, AA, and BDO from the PBAT hydrolysate was initiated by increasing the pH to over 9 to ensure the solubilization of all the monomers. This was followed by centrifugation at 9000 rpm (Sorvall Lynx 4000 centrifuge, Thermo Scientific) to remove any debris of denatured proteins. Subsequently, ultrafiltration using a 10 kDa cut-off membrane filter (RC70PP, Alfa Laval Nakskov A/S) at a pressure of 1 bar at room temperature was carried out for protein separation. The filtration was stopped when the volume reduction reached around 90%. Samples were taken both from retentate and permeate for analysis of monomers. The pH of the ultrafiltration permeate was decreased stepwise to monitor the precipitation of TPA. The precipitate was washed with cold acidified water, dried, and analysed by 1H-NMR spectroscopy in DMSO-d6.The solution obtained after TPA precipitation was subjected to nanofiltration for separation of AA and BDO using a membrane filter with a molecular-weight cut-off of 200 Da (MPF-34, Koch Separation Solutions, Wilmington, Ma, USA), at room temperature, constant transmembrane pressure of 15 bar and a crossflow velocity of 0.5 m s−1.46 The filtration was stopped when the volume reduction was 80%. The concentrations of AA and BDO in both retentate and permeate were analysed by HPLC.
To further purify AA in the retentate fraction, the pH was lowered to 1–2, the precipitate obtained was washed and analysed by HPLC and 1H-NMR spectroscopy in deuterated dimethyl sulfoxide (DMSO-d6).
Alternatively, AA and BDO in the nanofiltration retentate or permeate fractions were concentrated by evaporation using the Rotavapor® R-300 (BUCHI, Switzerland) at 50 °C and 20 mbar, the precipitated AA was washed and analysed to confirm the purity. The BDO purity was verified by 1H-NMR spectroscopy in deuterium oxide (D2O).
PBAT synthesis from the recovered monomers
For synthesis of rPBAT, the purified TPA obtained above was first converted to dimethyl terephthalate, for which TPA (2.35 g, 14.2 mmol), methanol (100 mL), and H2SO4 (1.1 g, 11 mmol) were added to a 250 mL round bottomed flask equipped with a magnetic stirring bar. The mixture was then stirred for 24 h under reflux. Afterwards, the reaction was quenched by addition of aqueous NaHCO3 (200 mL, 0.1 M), and the methanol was subsequently evaporated. The aqueous phase was then extracted with dichloromethane (4 × 100 mL) and finally, the combined organic phase was washed with H2O (100 mL), dried with Na2SO4 (3.65 g, 25.7 mmol), and concentrated in vacuo to give pure DMT as white crystals (2.36 g, 12.2 mmol, 86%). For synthesis of vPBAT, commercial dimethyl terephthalate was used instead.To a 50 mL three necked round bottomed flask equipped with a mechanical stirrer, nitrogen inlet, and outlet was added DBTO (16 mg, 0.060 mmol), dimethyl terephthalate (0.787 g, 4.05 mmol), BDO (1.19 g, 13.2 mmol), and AA (0.591 g, 4.04 mmol). Xylene (1 mL) was used to rinse solid residues stuck on the edges of the flask. The reaction was then heated at 145 °C for 3 h under nitrogen atmosphere, and then at 160 °C for another 2 h to form oligomers (transesterification). After that, a strong nitrogen flow was applied to facilitate the evaporation of excess BDO (polycondensation). After 2 h, the temperature was increased to 180 °C, and the reaction was stirred for another 16 h. Finally, mesitylene (1 mL) was added, and the temperature was increased to 200 °C for 10 h. Afterwards, the crude polymer was cooled to room temperature, dissolved in chloroform (12 mL), and precipitated into methanol (120 mL) to obtain the pure polymer as a white solid.
PBAT film casting
The polymer was dissolved in chloroform (0.6 mL, 100 mg mL−1) and evenly spread onto a Teflon mould (Ø = 28 mm). A glass funnel was placed over the mould to slow down evaporation. The chloroform was allowed to evaporate overnight.Polymerization of adipic acid with hexamethylenediamine
The commercial lipase Novozym®435, CalB (Novozymes, Bagsværd, Danmark) was employed for the amidation reaction of adipic acid (AA) with hexamethylenediamine (HDMA). To a mixture of 5 g L−1 AA and 4 g L−1 HDMA, 10% (w/w) Novozym®435 and 10% (w/v) 3 Å molecular sieves (Thermo Scientific) were added in 1 mL of pre-dried toluene. To dry the toluene, molecular sieves were first dried for 24 h at 100 °C before adding to the solvent, which was then left standing for 24 h prior to use in the reaction. The reaction was performed in a thermomixer (Hettich, Germany) at 70 °C, 500 rpm, and purged with nitrogen. Ten microliter samples were removed for analysis by Fourier-transform infrared spectroscopy (FTIR) and the solvent was removed from the remaining reaction mixture, through evaporation overnight in a fume hood. The dry precipitate was redissolved in water to prepare for HPLC analysis.The reaction was scaled to 50 mL volume of dried toluene in a round-bottom flask; 0.2 M of pure AA and HMDA were mixed by stirring at 250–300 rpm using IKA RCT Classic heated plate magnetic stirrer (IKA-Werke GmbH, Germany) and purged with nitrogen for 30 min at room temperature. The reaction flask was transferred to an oil bath at 85 °C under ambient pressure and connected to a condenser before adding 10% (w/w) of CalB to the total monomers used in the reaction. The reaction was followed by FTIR and 1H-NMR spectroscopy (in trifluoroacetic acid-d) and compared to the NMR of the pure starting material (400 MHz NMR, Bruker, UltraShield Plus 400, Germany).
Oxidation of 1,4-butanediol by G. oxydans
The reactions with BDO from the PBAT hydrolysate after ultrafiltration (5 g L−1) and nanofiltration (4 g L−1) were performed at pH 7 in 20 mL reaction volume with G. oxydans cells (OD600 of 10). Samples were withdrawn at different time intervals for product analysis and pH was adjusted to 7. After 26 hours, the cells were removed, and the reaction mixture was freeze dried for NMR analysis in deuterium oxide (D2O).
LCC-WCCG cloning, expression and purification as well as detailed analyses methods are described in the ESI file.†
Results and discussion
Enzymatic hydrolysis of PBAT
Based on our preliminary investigations, the thermostable LCC variant, LCC-WCCG, previously developed for degradation of PET,31 was selected for depolymerization of PBAT in the present study. Initial treatment of PBAT films (1.0 × 1.0 cm, 5 mg mL−1) with varying LCC-WCCG concentrations (0–1.6 μM) at 60 °C, for 24 h showed the highest release of TPA with 0.8 μM (12 μg mL−1) of the enzyme (Fig. 2A). Screening of the reaction at different temperatures for 24 h revealed the highest release of TPA (0.9 mg mL−1) around 80 °C (Fig. 2B), which was higher than the previously reported optimal operating temperature of 72 °C for PET hydrolysis by the same variant.31 The structure of PBAT is not affected by increase in temperature (the polymer having a Tg of −30 °C, annealing temperature of 0 °C). As the enzyme activity increases with temperature, so does the efficiency of polymer degradation and the decrease in degree of depolymerization above 90 °C, as seen in Fig. 2B, is related to loss of the enzymatic activity due to denaturation and not to the polymer properties. Enzymatic depolymerization of PBAT films could be followed visually with time going from particulate, transparent films, and finally to a clear solution (Fig. 2C). Scanning electron microscopy of the polymer sample treated with the enzyme at a lower temperature (50 °C) revealed the change in the topology of PBAT film with eroded surface, especially with prolonged reaction time (Fig. 2D). These changes occurred at a much higher rate with increase in temperature.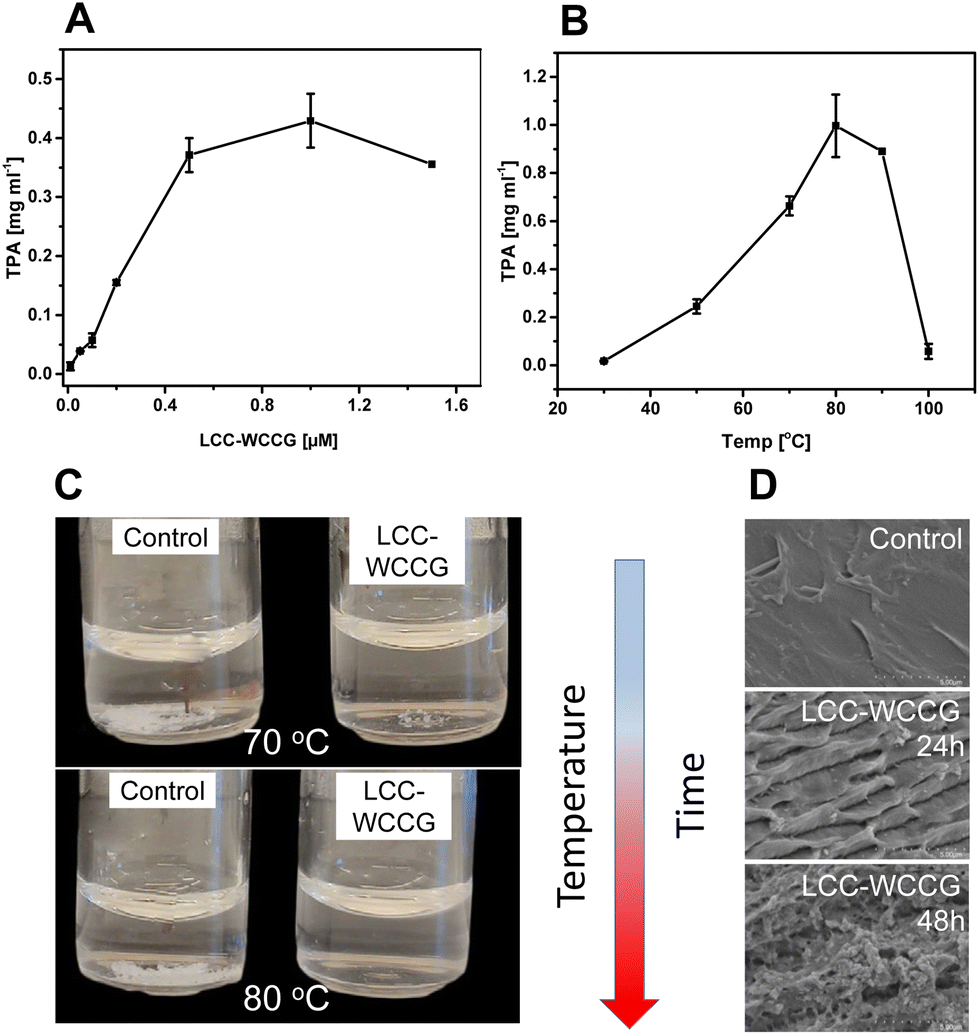 | ||
| Fig. 2 LCC-WCCG catalysed hydrolysis of PBAT (5 mg mL−1) for 24 h in 100 mM potassium phosphate buffer, pH 8. The outcome of the reaction is shown as: (A) concentration of released TPA using 0–1.5 μM enzyme concentration, (B) concentration of released TPA at different reaction temperatures 30–100 °C, (C) visual demonstration of PBAT degradation with respect to the controls without any enzyme, and (D) SEM images of the treated polymer at different incubation times (24 and 48 hours) at 50 °C, with respect to the control without any enzyme treatment. The reactions were performed in triplicates, error bars represent the standard deviation of the measurements. | ||
The kinetic parameters and pattern of hydrolysis of PBAT by LCC-WCCG were then investigated using conventional and inverse Michaelis–Menten (MM) kinetic models by either having an enzyme saturation (conventional model, eqn (S1)†) or a substrate saturation scenario (inverse model, eqn (S2) and (S3)†).45,47,48 The total concentration of all released monomers in the experiments conducted for the respective models was used to calculate the initial reaction rates of LCC-WCCG against PBAT (Fig. 3A and Fig. S1A, B†). In both approaches the MM plots followed a saturation behaviour at a certain point (Fig. S1C and D†). However, in the case of the conventional model, almost linear regression was observed that only reached a plateau at high substrate load (Fig. S1C†). This was not the case with the inverse kinetic model which better fitted the MM curve (Fig. S1D†), which aligns with a previous study by Kari et al. on enzymatic hydrolysis of insoluble cellulose.45 The affinity (Km) between the enzyme and the substrate calculated for the conventional and inverse models were found to be 11.7 g L−1 of PBAT and 273 nM of LCC-WCCG, respectively (Fig. S1E†). Additionally, the reactive site density (Γ) derived from both kinetic models was 59 nmol g−1, which was higher than that reported using the same model for Ideonella sarkiensis PETase variants against amorphous and crystalline PET (Fig. S1E†).47
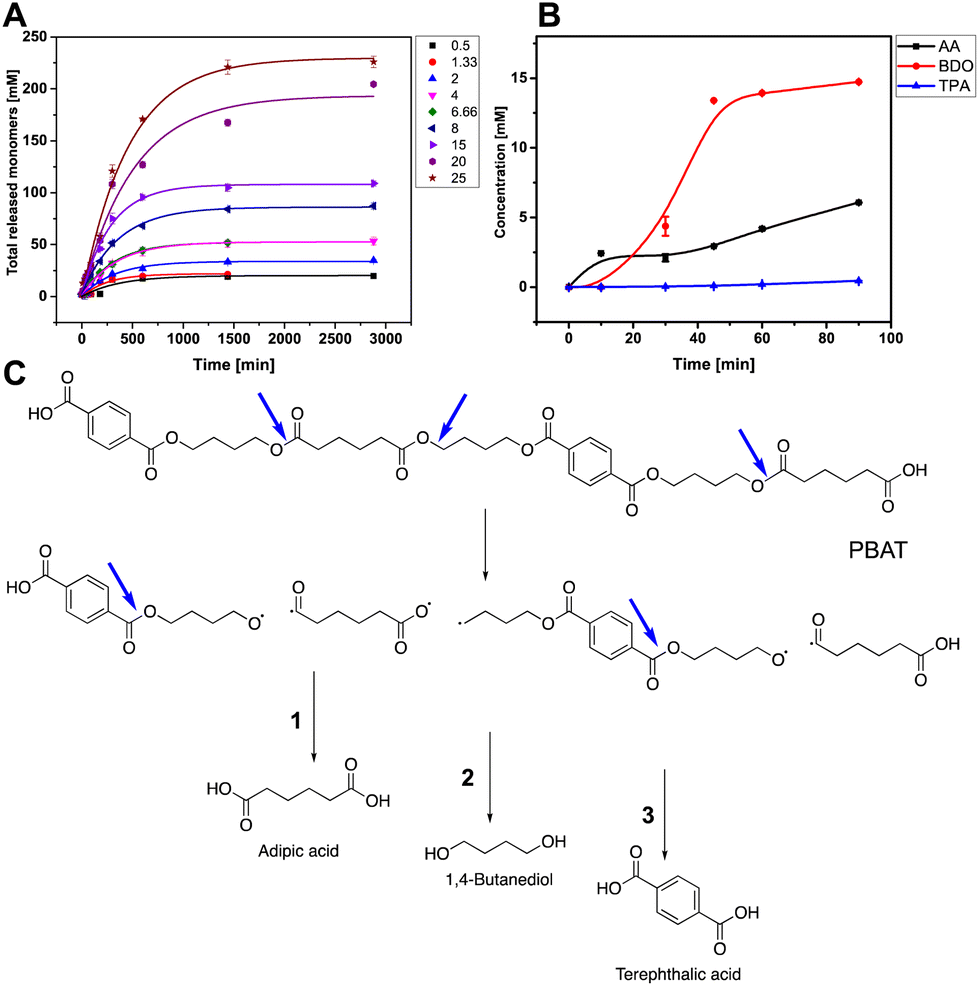 | ||
| Fig. 3 Hydrolytic pattern of PBAT on treatment with LCC-WCCG at 75 °C: (A) total concentration of monomers released on treatment of different concentrations of PBAT (0.5–25 g L−1) with LCC-WCCG (1 μM) as a function of time. (B) Initial depolymerization rate of PBAT at 8 g L−1 polymer concentration. (C) Postulated LCC-WCCG depolymerization pattern of PBAT, starting with hydrolysis of the linear chain ester bonds releasing AA, followed by BDO and TPA, the latter being released at a significantly lower rate. The reactions were performed in triplicates, error bars represent the standard deviation of the measurements. | ||
The specific activity of LCC-WCCG exceeded that of recently reported Thermobifida fusca and Thermobifida cellulosilytica cutinases, engineered for efficient PBAT hydrolysis, by 42 and 43 times, respectively,44 and its catalytic efficiency (Kcat/Km) surpassed PET hydrolase Ple629 by 1.4 × 107 fold (the inverse kinetics of Ple629 were calculated based only on TPA and TPA-BDO dimer released from PBAT hydrolysed film).49 Hence, inspite of the fact that LCC-WCCG variant is engineered for efficient PET degradation, its activity kinetics against PBAT surpassed the enzyme variants developed and tested so far for PBAT hydrolysis.
With all tested concentrations of PBAT (0.5–25 g L−1), AA was the first monomer to be released, followed by BDO, and finally TPA at 7.6 and 22-fold lower initial rates than AA and BDO, respectively (Fig. 3B). This preference of LCC-WCCG toward the linear chain of the polymer is consistent with the previous reports revealing the preference of different cutinases (like Humicola insolens cutinase) to cleave BDO-AA over BDO-TPA ester bond (Fig. 3C).50 Moreover, the hydrolysis rate of the polyester is influenced by its melting temperature; Tm of BDO-AA fraction is 60.4 °C (compared to 223 °C for BDO-TPA) which makes it the primary susceptible linkage for enzymatic attack (the limiting factor being the enzyme thermal stability).21,51 Since PBAT contains 2 molar equivalent BDO and 1 equivalent each of TPA and AA, the released BDO concentration is approximately twice that of AA (as illustrated below).
For a better understanding of the enzyme action on PBAT, in silico analysis of the LCC-WCCG structure was performed. First, the crystal structure was subjected to molecular dynamics (MD)-simulation for 3 ns at 25 and 70 °C to regain the dynamic model of the enzyme. A slight deviation in the overall structure of LCC-WCCG was observed at 70 °C compared to the simulation run at 25 °C (Fig. S2A†). Interestingly, the fluctuation of the amino acid residues indicated a high deviation of the surface residues (R124 and F125) at the interface of the active site pocket that might play a role in catalytic efficiency or substrate specificity of the enzyme by controlling the substrate binding within the active site groove (Fig. S2B and C†).
Based on the deviation of the structure backbone, three snapshots were selected for running the docking experiments (0.4, 1, and 2.5 ns, Fig. S2A†). The oligomers used for the docking study were two repeats of TPA-BDO and AA-BDO units in two different arrangements (PBAT C1, PBAT C2; Fig. S3†). The energy of binding for PBAT oligomer was 1.1 times higher for the binding poses with the aliphatic polyester chains (AA-BDO) in proximity to the catalytic triad (S165, D210, H242) within the active site cavity compared to the aromatic ones (TPA-BDO) (Fig. 4). However, for PBAT-C2 with a longer linear chain, the overall binding energy was lower compared to PBAT-C1. This can be the result of higher flexibility of the aliphatic chain, which makes it more difficult to accommodate within the active site groove (PBAT-C2 being flexible, tends to fold and form loop like structures; Fig. 4). MD-simulation of the best binding modes with PBAT showed deviation of PBAT away from the binding cavity. Nevertheless, the dissociation constants were high (1.4 × 108–1.2 × 109 picomolar of PBAT-C1 and C2, respectively), indicating tight binding to the active site, and hence low reaction rates would be foreseen (Fig. 4 and S4†). Running the simulation at 70 °C revealed a conformational change in W190 and Y95 residues (in contrast to the simulation at 25 °C), which are key residues for the enzyme activity (Fig. S5 and Table S1†). Overall, the in silico analysis supports the observed pattern of monomers release from PBAT during its depolymerization.
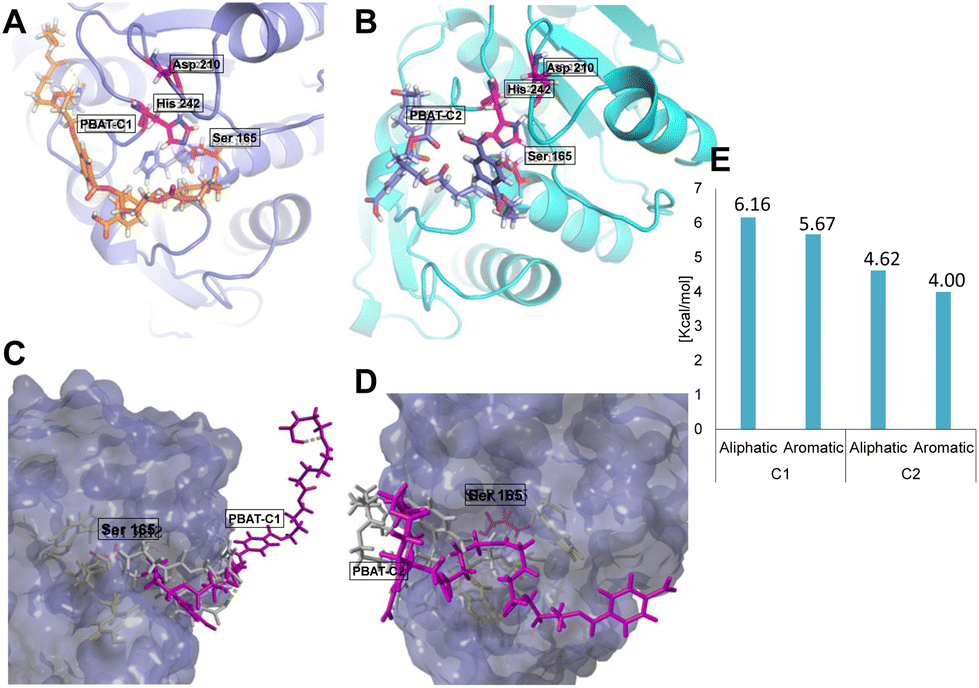 | ||
| Fig. 4 Docking of PBAT oligomer with different arrangements (C1 and C2) on LCC-WCCG. (A) PBAT-C1 (orange) with shorter linear chain within the active site groove of the enzyme with catalytic triad in red, where aliphatic ester bond is close to the catalytic S165 residue. (B) PBAT-C2 (blue) with longer linear chain within the active site of LCC-WCCG (catalytic triad in red). (C and D) Surface view (blue) of LCC-WCCG after MD simulation for the best docked poses of PBAT (magenta) superimposed to the initial docked structure (grey) showing the deviation of the oligomer from the active site pocket. (E) Energy of binding in Kcal mol−1 calculated for the best binding poses for PBAT-C1 and C2 showing preference towards the linear chain of the structure over the aromatic region. | ||
In comparison, the competitive docking revealed preference of LCC-WCCG for PET oligomer with higher binding energy, and significantly lower dissociation constant compared to PBAT (1.7 × 106 picomolar, Fig. S4†), hence allowing the enzyme to be more efficient for depolymerization of PET. The reaction rates of LCC-WCCG against PBAT (Fig. S1E†) are in agreement with the in silico observations; the enzyme exhibited 1.15 times lower specific activity against PBAT (65.77 mg h−1 mg−1) compared to that reported earlier against PET (75.9 mg h−1 mg−1) (Fig. S6†);31 increasing the enzyme efficiency against PBAT could perhaps be improved by protein engineering of the binding site to allow less tight binding to the polymer chain.
Small-scale depolymerization was first done using 12.5 g L−1 PBAT in 50 mL volume at 70 °C, without pH control, which resulted in severe inhibition of the reaction due to pH drop caused by accumulation of the acidic monomers. Based on the HPLC analysis of the released monomers, the reaction stopped at a certain point on the third day when the TPA concentration reached around 15 mM and the pH had dropped to 5 (Fig. S7†). In accordance with the previous report,31 inhibition of LCC-WCCG under acidic conditions could be overcome by readjusting the pH to 8 and adding a fresh batch of the LCC variant. The reaction led to 97.5% weight loss of the PBAT film, over a period of 7 days, with high concentration (totally 1.65 μM, 2.5 mg) of LCC-WCCG added in increments (equivalent to 4 mg enzyme per gram PBAT, Fig. S7†).
The depolymerization reaction was then carried out in 1 L scale in a bioreactor with pH control (Fig. 5A). Fifteen grams of PBAT film in 1L phosphate buffer, pH 8 was completely hydrolysed within 4 days using 1.4 mg LCC-WCCG per gram of the polymer film at an overall rate of 0.49 g L−1 h−1. After a linear increase in the release of monomers during the initial 24 hours, the depolymerization continued at a 33-fold lower rate for 68 hours (Fig. 5B and D). The molar concentration of the released monomers was 23.9, 56.3, and 33.8 mM for TPA, BDO, and AA, respectively (Fig. 5C). The underestimation of the released TPA is attributed to its low aqueous solubility (17 mg L−1 water at 25 °C), and the ratio was restored upon purification (final ratio of the monomers 1:2:1), which agrees well with the ratio of the monomers in PBAT polymer (Table S2†).
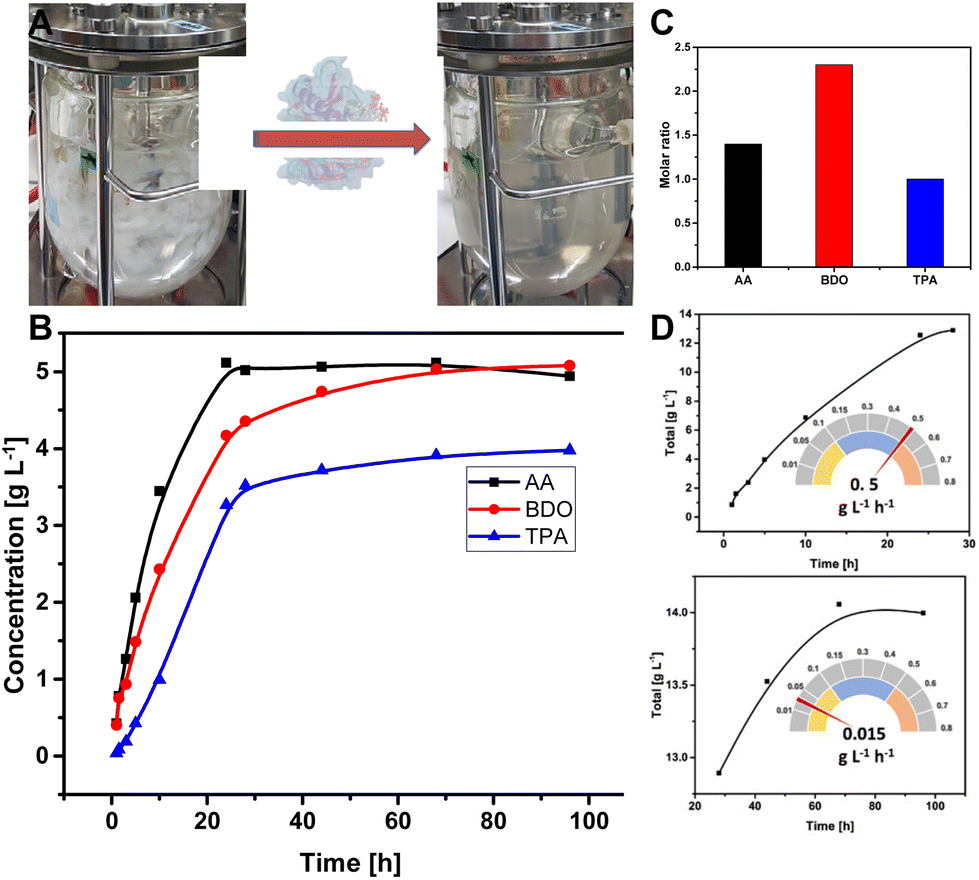 | ||
| Fig. 5 Depolymerization of PBAT in a litre scale using LCC-WCCG enzyme variant (1.4 mg g−1 polymer) at 70 °C: (A) Visual observation of the depolymerization of PBAT films in a bioreactor, (B) profile of the release of monomers during depolymerization as quantified by HPLC, (C) the final ratio of the released monomers, and (D) the calculated reaction rate during different time intervals of the process. The reaction was maintained at pH 8 using 1M NaOH. | ||
Purification of monomers from the PBAT hydrolysate
Subsequent to the depolymerization, separation of the released monomers was initiated by increasing the pH of the hydrolysate to >9 to ensure solubilization of all the components including TPA, followed by ultrafiltration (UF) over a 10 kDa cutoff membrane filter to remove the biocatalyst. The pH of the UF-permeate was then decreased incrementally to selectively precipitate TPA (aided by HPLC analysis of the supernatant) without affecting the levels of BDO and AA (Fig. S8†). At pH 2.5, majority of the TPA (1.34 g from 261 mL of the reaction mixture) was recovered as a white powder (representing a ration of 1:1 with AA), which was freeze-dried and shown to have purity of >95% by 1H-NMR analysis (Fig. S8C†).
Separation of BDO and AA turned out to be relatively challenging; after initial unsuccessful trials using precipitation and ion exchange adsorption techniques, separation by nanofiltration (NF) using a 200 Da cut-off membrane filter was attempted. Interestingly, decent separation of AA and BDO was obtained via a single filtration step at pH 6 (Table 1 and Fig. S9†). About 70% of AA was recovered in the retentate fraction with 69% purity, whereas 65% of BDO was recovered in the permeate with a purity exceeding 74%. The higher purity of BDO is due to minor loss of AA, by adsorption to the membrane, which can be recovered by washing the membrane with water. Noteworthy, the NF-membrane showed reusability with no fouling up to 3 times (no washing was required).
| Sample | AA [mg mL −1] | BDO [mg mL−1] | Volume [mL] | Total AA [g] | Total BDO [g] |
|---|---|---|---|---|---|
| Before NF | 4.9 | 5.0 | 237 | 1.16 | 1.17 |
| NF-permeate | 1.3 | 4.0 | 193 | 0.26 | 0.77 |
| NF-retentate | 18.4 | 8.3 | 44 | 0.81 | 0.36 |
From the retentate, highly pure AA was obtained by precipitation at pH 1–2 (verified by 1H-NMR spectrum; Fig. S10A†). AA has limited solubility in water at a concentration >20 g L−1, which can be further reduced by increasing the concentration, lowering the pH, and/or decreasing the solution temperature. Around 66.8% of the AA was recovered as highly pure crystals suitable for direct use in polymer synthesis (discussed later in the re-polymerization of monomers), while the remaining 33.2% of AA remained in a mixture with BDO. As the monomer concentration in the permeate was low, this fraction was concentrated under vacuum for enabling the precipitation of AA aided by acidifying the solution. About 55% of the AA was recovered as precipitate, which was separated by filtration, at the same time yielding a highly pure solution of BDO with 58% recovery (verified by 1NMR spectrum; Fig. S10B†). Most of the AA and BDO from retentate and permeate fractions can be recovered in pure form by repeating the concentration and precipitation steps. Moreover, the recovery will be easier if higher polymer concentration is subjected to depolymerization yielding monomers at higher concentration, thus minimizing the need for concentration.
Recycling and upcycling of PBAT monomers
| Polymer | SEC | TGA | DSC | ||||
|---|---|---|---|---|---|---|---|
| M n (Da) | M w (Da) | [η] (dL g−1) | T 5 (°C) | T g (°C) | T m (°C) | T c (°C) | |
| a Molecular weight was determined by triple detection SEC in chloroform. The SEC was run in duplicates and the average value is presented. T5 is the temperature at 5% weight loss. Tg is the glass transition temperature and Tm is the melting point, measured from the second heating curves. Tc is the crystallization temperature. | |||||||
| vPBAT | 7600 | 14600 |
0.69 | 305 | −26 | 136 | 92 |
| rPBAT | 7500 | 10600 |
0.54 | 328 | −28 | 137 | 102 |
Furthermore, the thermal properties of rPBAT and vPBAT were investigated by DSC and TGA. DSC analyses showed similar Tg and Tm values for rPBAT and vPBAT (Fig. S15† and Table 2). TGA results indicated that both polymers were rather thermally stable (T5 > 300 °C) (Fig. S16†). Interestingly, the derivative TGA curve profile of vPBAT showed a second thermal decomposition rate maximum at slightly lower temperature (320 °C) (Fig. S16B†), which somewhat lowered the thermal stability compared to rPBAT and cPBAT. The precise reason of this observation remains to be unravelled, but we believe that it might be related to the existence of trace amounts of impurities in the commercial monomers used for the synthesis of vPBAT (below the detection limit of NMR spectroscopy). For instance, the commercial AA contained trace amounts of iron, arsenic, lead, sulphate, etc. It has been reported that the thermal stability of PBAT was lowered by the presence of a few percent of ZnO nanoparticles.54
Polymerization of adipic acid with hexamethylenediamine (HDMA)
The most important application of adipic acid is the synthesis of polyamide Nylon 6,6.55 Currently, Nylon 6,6 is synthesized by melt polycondensation at elevated temperatures which is an energy-intensive process.56 Enzymatic polymerization provides a less energy-demanding alternative; there are several reports on the synthesis of polyesters57,58 and fewer reports describing the synthesis of polyamides using enzymes, in particular lipases, as catalyst.59–63 Novozym®435, immobilized Candida antarctica lipase B (CalB), was tested for catalysing polycondensation of the recovered pure AA with HMDA in toluene or acetonitrile at 70 °C and ambient pressure. HPLC analysis showed the highest reduction in AA concentration in the amidation reaction performed in toluene for 6 days at 70 °C. The amide bond formation was further confirmed by FTIR analysis that showed the removal of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond from a carboxyl group at 1760 1 cm−1 and the formation of an amide bond at 1635 1 cm−1 (Fig. S17†). The reaction was further scaled to 50 mL volume using equimolar ratio of pure AA and HMDA in toluene at higher temperature (85 °C), and the formation of the amide bond was confirmed by FTIR spectroscopy. Furthermore, after the reaction, the crude product was collected by filtration and analysed by 1H-NMR spectroscopy confirming successful biosynthesis of the polyamide (Nylon 6,6) with a number average molecular weight Mn of around 3500 g mol−1 (calculated by end-group analysis, based on 1H-NMR peak integrals) (Fig. S18†). The characterization of polyamides by gel permeation chromatography is limited by their restricted solubility in common GPC solvents like chloroform and THF.64 While monomers with longer alkyl chains are preferred as substrates for enzyme catalysed polymerization, the molecular weight of the polyamide obtained from AA and HMDA was in the same range as that reported previously (Nylon 8,10) under similar and/or modified reaction conditions from commercially available monomers.63,65
O bond from a carboxyl group at 1760 1 cm−1 and the formation of an amide bond at 1635 1 cm−1 (Fig. S17†). The reaction was further scaled to 50 mL volume using equimolar ratio of pure AA and HMDA in toluene at higher temperature (85 °C), and the formation of the amide bond was confirmed by FTIR spectroscopy. Furthermore, after the reaction, the crude product was collected by filtration and analysed by 1H-NMR spectroscopy confirming successful biosynthesis of the polyamide (Nylon 6,6) with a number average molecular weight Mn of around 3500 g mol−1 (calculated by end-group analysis, based on 1H-NMR peak integrals) (Fig. S18†). The characterization of polyamides by gel permeation chromatography is limited by their restricted solubility in common GPC solvents like chloroform and THF.64 While monomers with longer alkyl chains are preferred as substrates for enzyme catalysed polymerization, the molecular weight of the polyamide obtained from AA and HMDA was in the same range as that reported previously (Nylon 8,10) under similar and/or modified reaction conditions from commercially available monomers.63,65
Oxidation of 1,4-butanediol by G. oxydans
BDO serves as a building block not only for PBAT but also for other polyesters like PBT and PBS.66–68 Other important C4 building blocks for polyesters are 4-hydroxybutanoate (4-HB), gamma-butyrolactone, and succinic acid. Here, the bioproduction of 4-HB from BDO was evaluated using Gluconobacter oxydans, a Gram-positive strictly aerobic bacteria known for its ability for partial oxidation of sugars, polyols, aldehydes, and alcohols to produce the corresponding acids and ketones.69,70 The bacterium is used in large scale for the production of vitamin C, dihydroxyacetone, acetic acid and amino acids.71G. oxydans has been recently used in our laboratory for the production of adipic acid from 1,6-hexanediol,72 and earlier for selective oxidation of 5-hydroxymethyl furfural (5-HMF) to 5-hydroxymethyl-2-furan carboxylic acid (HMFCA) with high selectivity.73Initial experiments using pure BDO (10 g L−1) showed the diol to be completely oxidized by the whole cells of G. oxydans, to 4-hydroxybutyrate (4-HB) and/or succinic acid (SA) depending on the starting pH of the reaction (Fig. 6). The highest production of SA and 4-HB was obtained in the reactions with initial pH of 5 and 7, respectively (Fig. S19†). While 4-HB was gradually oxidized to SA with time (maximum yield of 50%) when the pH was not controlled (Fig. 6B), only 4-HB was produced when the pH was maintained at 7 with a maximum yield of 88% at 24 h and productivity of 0.55 g L−1 h−1 (Fig. 6A).
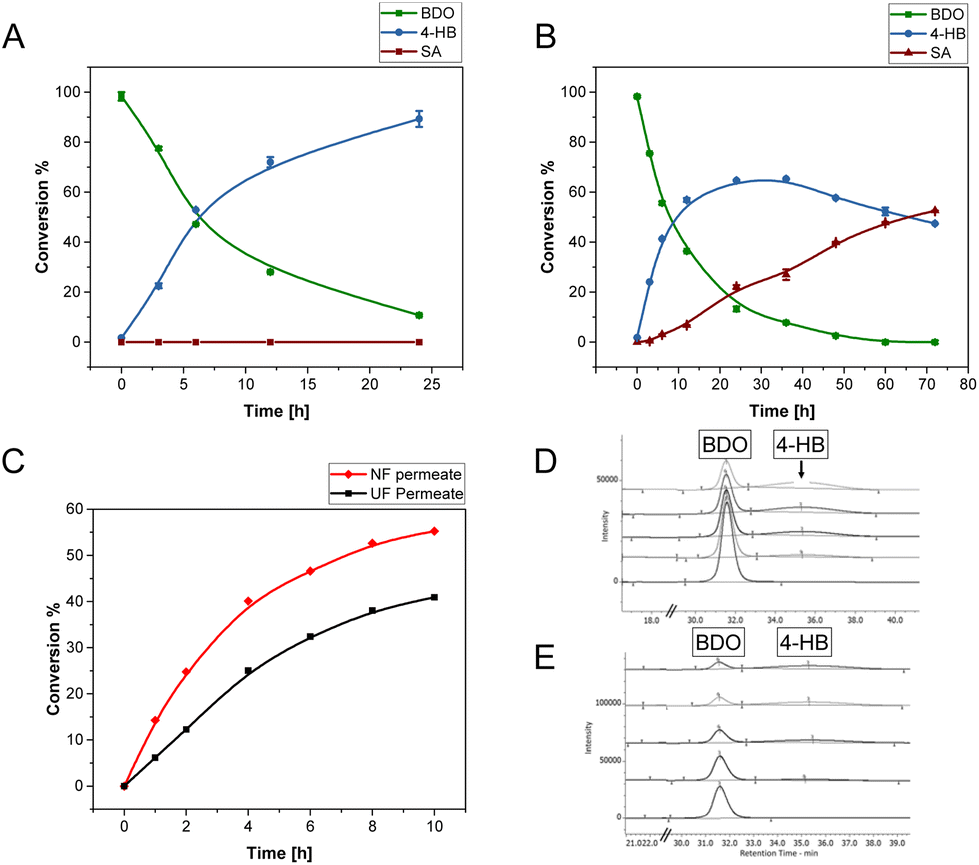 | ||
| Fig. 6 Production of 4-HB from BDO using G. oxydans resting cells in 100 mM sodium phosphate buffer at 30 °C, (A) pH of the reaction with pure BDO (10 g L−1) controlled at 7, (B) pH of the reaction with pure BDO controlled at 5, (C) reactions using ultrafiltration and nanofiltration permeate fractions, with different proportions of AA and BDO, and (D and E) HPLC chromatograms for NF and UF permeate fractions, respectively. Conversion percentage calculated from 4-HB and SA formed with respect to the oxidized BDO. The reactions in A and B were performed in duplicates, error bars represent the standard deviation of the measurements. The reactions in C were performed once. | ||
In order to determine the feasibility of using not completely pure BDO for the microbial oxidation, the permeates from ultrafiltration (UF) of the depolymerized PBAT mixture containing TPA, AA, and BDO, and from nanofiltration (NF) containing BDO and AA, were used as substrates in the reactions maintained at pH 7. 4-HB was the only product observed with yields of 50 and 37% from BDO in NF- and UF-permeate, respectively (Fig. 6C–E and S18†), but with a nearly similar productivity (0.2 g L−1 h−1) during 10 hours of reaction. The lower yield and productivity compared to the oxidation of pure BDO are attributed to the inhibitory effect of TPA (UF-permeate) and AA in the (UF/NF-permeate). The production of 4-HB was further verified by running crude 1NMR that indicated the product formation (Fig. S20†).
Conclusions
The present study demonstrates a scalable circular and green approach for the depolymerization of PBAT as well as recycling of the monomers. We demonstrated the potential of the enzyme LCC-WCCG for complete depolymerization of PBAT at 1.5 wt% (and recently even up to 5 wt% (unpublished data)). In silico analysis of the enzyme structure and its interaction with the oligomer revealed preference towards the aliphatic ester bonds over the aromatic chains, evidenced also by the binding energy and dissociation constants, which was further confirmed by the release of AA and BDO first followed by TPA. Although LCC-WCCG was engineered to depolymerize PET, its activity against PBAT was only marginally lower than that on PET and seems to be due to the tighter binding to PBAT and the resulting higher dissociation constant. Comparison of this study with the previous reports on enzymatic depolymerization of PBAT shows clearly that catalytic efficiency of LCC-WCCG surpasses that of the other enzymes used (Table S3†).A simple downstream processing scheme, using low energy-demanding separation techniques, was developed to obtain the constituent monomers of PBAT at high purity for subsequent valorization. Besides using the monomers for repolymerization to PBAT, other routes were demonstrated for upcycling of the aliphatic monomers.
Overall, this study presents a possible solution for recycling of a compostable polymer because recycling will potentially increase resource efficiency, lower the greenhouse gas emissions, and retain the material value. Since the economic feasibility of biocatalytic recycling is an important factor for its practical implementation, further work is planned using even higher polymer concentrations, optimizing reactor design for depolymerization, maximizing monomer recovery, and techno-economic assessment of the process. Even life cycle assessment of the recycling approach in comparison to composting the polymer after use would be interesting to investigate.
Author contributions
RHK conceived the project idea and was responsible for funding acquisition and project administration. MI, AA, OEÖ, SHP and RHK planned and designed the study. MI and AA performed production and purification of the enzyme, PBAT depolymerization, purification of monomers, oxidation of BDO, polymerization of AA, and analysis of the results. BA-R helped with nanofiltration while CT with product analysis. NW performed repolymerization of PBAT and characterized the polymers made in the study, while BZ supervised the work on polymer synthesis and characterization. OEÖ, MI and AA performed the experiments with the oxidation of BDO and polymerization of AA. MI, NW, AA, OEÖ, SHP, BZ and RHK wrote and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the EU Horizon 2020 (grant no. 953073, for the project sUstainable PLastIcs for the Food and drinks packaging indusTry, UPLIFT) and the Swedish Foundation for Strategic Environmental Research (Mistra, grant no. 2016/1489, for the research program Sustainable Plastics and Transition Pathways, STEPS). We thank Filip Miketa, Bio-mi, Matulja, Croatia for providing us PBAT for the study, and Mats Galbe at Chemical Engineering, Lund University is acknowledged for his support in running the bioreactor.References
- Plastics – the fast Facts 2023·Plastics Europe, https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2023/, (accessed 27 November 2023).
- R. Gould, ISO - Rethinking the future of plastics, https://www.iso.org/news/ref2792-1.html, (accessed 6 July 2023).
- L. Lebreton, B. Slat, F. Ferrari, B. Sainte-Rose, J. Aitken, R. Marthouse, S. Hajbane, S. Cunsolo, A. Schwarz, A. Levivier, K. Noble, P. Debeljak, H. Maral, R. Schoeneich-Argent, R. Brambini and J. Reisser, Sci. Rep., 2018, 8, 1–15 CrossRef CAS PubMed.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- N. Aurisano, R. Weber and P. Fantke, Curr. Opin. Green Sustainable Chem., 2021, 31, 100513 CrossRef CAS.
- C. Álvarez-Hernández, C. Cairós, J. López-Darias, E. Mazzetti, C. Hernández-Sánchez, J. González-Sálamo and J. Hernández-Borges, Mar. Pollut. Bull., 2019, 146, 26–32 CrossRef PubMed.
- H. K. Webb, J. Arnott, R. J. Crawford, E. P. Ivanova, B. Ruj, V. Pandey, P. Jash, V. K. Srivastava, Plastics Europe, B. S. Huyhua, EU DG, J. Claerbout, S. M. Al-Salem, P. Lettieri and J. Baeyens, Sci. Environ. Policy, In-Depth Reports, 2015, 4, 41 Search PubMed.
- Plastics - the Facts 2022·Plastics Europe, https://plasticseurope.org/knowledge-hub/plastics-the-facts-2022/, (accessed 6 July 2023).
- A new global treaty to tackle plastic pollution? - Geneva Solutions, https://genevasolutions.news/sustainable-business-finance/a-new-global-treaty-to-tackle-plastic-pollution, (accessed 1 July 2023).
- European Commission, A Eur. Strateg. Plast. a Circ. Econ., https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1516265440535&uri=COM:2018:28:FIN, (accessed 4 August 2023).
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50–67 CrossRef CAS PubMed.
- S. K. Das, S. K. Eshkalak, A. Chinnappan, R. Ghosh, W. A. D. M. Jayathilaka, C. Baskar and S. Ramakrishna, Mater. Circ. Econ., 2021, 3, 1–22 CrossRef.
- M. G. Davidson, R. A. Furlong and M. C. McManus, J. Cleaner Prod., 2021, 293, 126163 CrossRef CAS.
- D. E. MacArthur, Rethinking the future of plastics RETHINKING THE FUTURE OF PLASTICS THE NEW PLASTICS ECONOMY, https://ellenmacarthurfoundation.org/the-new-plastics-economy-rethinking-the-future-of-plastics, (accessed 9 October 2023).
- M. Bachmann, C. Zibunas, J. Hartmann, V. Tulus, S. Suh, G. Guillén-Gosálbez and A. Bardow, Nat. Sustain., 2023, 6, 599–610 CrossRef.
- PlasticsEurope, Plastics Europe position on the Global Plastics Pollution Agreement objectives, scope, obligations and measures ahead of INC2 A. Requirement to sustainably produce and consume plastics and for environmentally sound management of all plastics waste, https://plasticseurope.org/media/plastics-europe-position-on-the-global-plastics-pollution-agreement-objectives-scope-obligations-and-measures-ahead-of-inc2/, (accessed 6 July 2023).
- S. M. Satti and A. A. Shah, Lett. Appl. Microbiol., 2020, 70, 413–430 CrossRef CAS PubMed.
- J. Jian, Z. Xiangbin and H. Xianbo, Adv. Ind. Eng. Polym. Res., 2020, 3, 19–26 Search PubMed.
- S. S. Ali, T. Elsamahy, E. Koutra, M. Kornaros, M. El-Sheekh, E. A. Abdelkarim, D. Zhu and J. Sun, Sci. Total Environ., 2021, 771, 144719 CrossRef CAS PubMed.
- E. Marten, R. J. Müller and W. D. Deckwer, Polym. Degrad. Stab., 2005, 88, 371–381 CrossRef CAS.
- Y. Tokiwa and T. Suzuki, Nature, 1977, 270, 76–78 CrossRef CAS PubMed.
- M. Sato, Sen'i Gakkaishi, 1983, 39, 209–219 CrossRef.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- F. Kawai, T. Kawabata and M. Oda, ACS Sustainable Chem. Eng., 2020, 8, 8894–8908 CrossRef CAS.
- F. Kawai, T. Kawabata and M. Oda, Appl. Microbiol. Biotechnol., 2019, 103, 4253–4268 CrossRef CAS PubMed.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- S. Sulaiman, S. Yamato, E. Kanaya, J. J. Kim, Y. Koga, K. Takano and S. Kanaya, Appl. Environ. Microbiol., 2012, 78, 1556–1562 CrossRef CAS PubMed.
- Å. M. Ronkvist, W. Xie, W. Lu and R. A. Gross, Macromolecules, 2009, 42, 5128–5138 CrossRef.
- R. J. Müller, H. Schrader, J. Profe, K. Dresler and W. D. Deckwer, Macromol. Rapid Commun., 2005, 26, 1400–1405 CrossRef.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- Y. Cui, Y. Chen, X. Liu, S. Dong, Y. Tian, Y. Qiao, R. Mitra, J. Han, C. Li, X. Han, W. Liu, Q. Chen, W. Wei, X. Wang, W. Du, S. Tang, H. Xiang, H. Liu, Y. Liang, K. N. Houk and B. Wu, ACS Catal., 2021, 11, 1340–1350 CrossRef CAS.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662–667 CrossRef CAS PubMed.
- W. Zeng, X. Li, Y. Yang, J. Min, J. W. Huang, W. Liu, D. Niu, X. Yang, X. Han, L. Zhang, L. Dai, C. C. Chen and R. T. Guo, ACS Catal., 2022, 12, 3033–3040 CrossRef CAS.
- U. Witt, R. J. Müller and W. D. Deckwer, J. Environ. Polym. Degrad., 1995, 3, 215–223 CrossRef CAS.
- Frost & Sullivan, Global Bioplastic Market - Growth Opportunities, Analysis, Forecast, Global, 2023, https://store.frost.com/global-bioplastic-growth-opportunities.html, (accessed 9 October 2023).
- I. Kleeberg, C. Hetz, R. M. Kroppenstedt, R. J. Müller and W. D. Deckwer, Appl. Environ. Microbiol., 1998, 64, 1731–1735 CrossRef CAS PubMed.
- A. Kanwal, M. Zhang, F. Sharaf and C. Li, Polym. Bull., 2022, 79, 9059–9073 CrossRef CAS.
- H. Jia, M. Zhang, Y. Weng, Y. Zhao, C. Li and A. Kanwal, J. Environ. Sci., 2021, 103, 50–58 CrossRef CAS PubMed.
- H. Xiang-dong, G. Yan, L. Qing, Z. Jun, Z. Tao, C. Min, Y. Hong-mei, S. Ying-wu, W. Bin, S. Jiu-sheng and W. Jin-xin, Xinjiang Agric. Sci., 2017, 54, 2086–2091 Search PubMed.
- M. Aarthy, P. Puhazhselvan, R. Aparna, A. S. George, M. K. Gowthaman, N. Ayyadurai, K. Masaki, T. Nakajima-Kambe and N. R. Kamini, Polym. Degrad. Stab., 2018, 152, 20–28 CrossRef CAS.
- F. Muroi, Y. Tachibana, P. Soulenthone, K. Yamamoto, T. Mizuno, T. Sakurai, Y. Kobayashi and K.-i. Kasuya, Polym. Degrad. Stab., 2017, 137, 11–22 CrossRef CAS.
- P. W. Wallace, K. Haernvall, D. Ribitsch, S. Zitzenbacher, M. Schittmayer, G. Steinkellner, K. Gruber, G. M. Guebitz and R. Birner-Gruenberger, Appl. Microbiol. Biotechnol., 2017, 101, 2291–2303 CrossRef CAS PubMed.
- Y. Yang, J. Min, T. Xue, P. Jiang, X. Liu, R. Peng, J. W. Huang, Y. Qu, X. Li, N. Ma, F. C. Tsai, L. Dai, Q. Zhang, Y. Liu, C. C. Chen and R. T. Guo, Nat. Commun., 2023, 14, 1645 CrossRef CAS PubMed.
- J. Kari, M. Andersen, K. Borch and P. Westh, ACS Catal., 2017, 7, 4904–4914 CrossRef CAS.
- B. Al-Rudainy, M. Galbe, F. Lipnizki and O. Wallberg, Membranes, 2019, 9, 99 CrossRef CAS PubMed.
- E. Erickson, T. J. Shakespeare, F. Bratti, B. L. Buss, R. Graham, M. A. Hawkins, G. König, W. E. Michener, J. Miscall, K. J. Ramirez, N. A. Rorrer, M. Zahn, A. R. Pickford, J. E. McGeehan and G. T. Beckham, ChemSusChem, 2022, 15, e202101932 CrossRef CAS PubMed.
- J. A. Bååth, K. Borch, K. Jensen, J. Brask and P. Westh, ChemBioChem, 2021, 22, 1627–1637 CrossRef PubMed.
- I. E. Meyer Cifuentes, P. Wu, Y. Zhao, W. Liu, M. Neumann-Schaal, L. Pfaff, J. Barys, Z. Li, J. Gao, X. Han, U. T. Bornscheuer, R. Wei and B. Öztürk, Front. Bioeng. Biotechnol., 2022, 10, 930140 CrossRef PubMed.
- V. Perz, K. Bleymaier, C. Sinkel, U. Kueper, M. Bonnekessel, D. Ribitsch and G. M. Guebitz, N. Biotechnol., 2016, 33, 295–304 CrossRef CAS PubMed.
- L. C. Arruda, M. Magaton, R. E. S. Bretas and M. M. Ueki, Polym. Test., 2015, 43, 27–37 CrossRef CAS.
- S. V. Mankar, J. Wahlberg, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2023, 11, 5135–5146 CrossRef CAS.
- Anton Paar GmbH, Polymers | Intrinsic Viscosity Measurements for Quality Control of PET, https://www.anton-paar.com, (accessed 1 July 2023).
- T. Wang, Y. Shi, Y. Li and L. Z. Liu, J. Polym. Eng., 2021, 41, 835–841 CrossRef CAS.
- J. B. J. H. Van Duuren and C. Wittmann, in Bioprocessing of Renewable Resources to Commodity Bioproducts, John Wiley & Sons, Ltd, 2014, vol. 9781118175, pp. 519–540 Search PubMed.
- L. L. Estes and M. Schweizer, Ullmann’s Encycl. Ind. Chem., 2011 DOI:10.1002/14356007.a10_567.pub2.
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. Dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC.
- V. Hevilla, A. Sonseca, C. Echeverría, A. Muñoz-Bonilla and M. Fernández-García, Macromol. Biosci., 2021, 21, 2100156 CrossRef CAS PubMed.
- A. Todea, D. M. Dreava, I. C. Benea, I. Bîtcan, F. Peter and C. G. Boeriu, Processes, 2021, 9, 646 CrossRef CAS.
- K. Nasr, J. M. Raquez, P. Zinck and A. Favrelle-Huret, Biopolym. Synth. Prop. Emerg. Appl, 2023, 21–71 Search PubMed.
- A. Torres-Gavilán, E. Castillo and A. López-Munguía, J. Mol. Catal. B: Enzym., 2006, 41, 136–140 CrossRef.
- M. Galmés, E. García-Junceda, K. Świderek and V. Moliner, ACS Catal., 2020, 10, 1938–1946 CrossRef.
- L. Ragupathy, U. Ziener, R. Dyllick-Brenzinger, B. Von Vacano and K. Landfester, J. Mol. Catal. B: Enzym., 2012, 76, 94–105 CrossRef CAS.
- K. Weisskopf and G. Meyerhoff, Polymer, 1983, 24, 72–76 CrossRef CAS.
- S. J. Cooper, E. D. T. Atkins and M. J. Hill, Macromolecules, 1998, 31, 8947–8956 CrossRef CAS.
- U. Witt, T. Einig, M. Yamamoto, I. Kleeberg, W. D. Deckwer and R. J. Müller, Chemosphere, 2001, 44, 289–299 CrossRef CAS PubMed.
- K. Changwichan, T. Silalertruksa and S. H. Gheewala, Sustainability, 2018, 10, 952 CrossRef.
- U. Witt, J. Environ. Polym. Degrad., 1996, 4, 9–20 CrossRef CAS.
- S. Macauley, B. McNeil and L. M. Harvey, Crit. Rev. Biotechnol., 2001, 21, 1–25 CrossRef CAS PubMed.
- G. Keliang and W. Dongzhi, Appl. Microbiol. Biotechnol., 2006, 70, 135–139 CrossRef PubMed.
- A. Gupta, V. K. Singh, G. N. Qazi and A. Kumar, J. Mol. Microbiol. Biotechnol., 2001, 3, 445–456 CAS.
- S. H. Pyo, M. Sayed, O. E. Örn, J. A. Gallo, N. F. Ros and R. Hatti-Kaul, Microb. Cell Fact., 2022, 21, 1–10 CrossRef PubMed.
- M. Sayed, S. H. Pyo, N. Rehnberg and R. Hatti-Kaul, ACS Sustainable Chem. Eng., 2019, 7, 4406–4413 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04728h |
| This journal is © The Royal Society of Chemistry 2024 |