
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeodymium recovery from NdFeB magnets: a sustainable, instantaneous, and cost-effective method†
Sandeep
Bose
 a,
Benilde
Mizero
a and
Parisa A.
Ariya
*ab
a,
Benilde
Mizero
a and
Parisa A.
Ariya
*ab
aDepartment of Chemistry, McGill University, Montreal, Quebec H3A 0B8, Canada
bDepartment of Atmospheric and Oceanic Sciences, McGill University, Montreal, Quebec H3A 0B9, Canada. E-mail: parisa.ariya@mcgill.ca
First published on 23rd February 2024
Abstract
Rare earth elements (REEs) are critical in modern electronics, yet sustainable, effective recovery technologies for REEs are scarce. Herein, we develop a sustainable and cost-effective neodymium (Nd) extraction technology from electronic waste using a carboxylate functionalized nanocellulose (CFNC). We demonstrate a solution-processed synthesis of CFNC from a cost-effective, readily available cellulose precursor without harsh conditions or complicated procedures. We show that Nd precipitates as Nd-CFNC complex, which can be easily separated from the solution by centrifugation. As low as 150 ppm of Nd3+ concentration is sufficient to form the precipitate instantaneously. We observe the removal of ∼252 ± 5 mg of Nd3+ per gram of CFNC, which, to our knowledge, provides the highest removal capacity at the shortest contact time of a few seconds. We explore the effect of ionic strength, pH, and temperature on the performance of CFNC. As a real-time application, we demonstrate Nd recovery from a NdFeB magnet present in waste electronic equipment. The removal using CFNC is speedy, efficient, and selective. Solar-driven electrodeposition is used to recover Nd/Nd2O3. We envision that our material will provide a low-cost, promising, sustainable technology for removing other RREs from e-waste, mines, and industrial wastewater.
Introduction
Neodymium (Nd) is one of the most critical rare earth elements (REEs) whose demand has increased over the decades due to its unique properties.1 It is essential for critical technologies such as wind turbines,1 electric vehicles,2 and next-generation electronics.3,4 Due to rapid urbanization and advancement in science and technology, the consumption of Nd-based electronic equipment has increased exponentially. As a result, a huge amount of electronic waste is generated. The global e-waste generated during 2019 was estimated to be 53.6 million metric tons.5 Yet, merely 17.4% of all global e-waste is collected correctly and recycled.5 The e-waste is a concentrated source of many transition and rare earth metals.6 The content of these metals is significantly higher in the e-waste than in natural minerals.7 Recycling of Nd (or any REE) from e-waste will lead to active use of the valuable metal and result in overall economic benefits.Nd is primarily used in these critical technologies as neodymium–iron–boron magnets (NdFeB). These magnets are important in electronic devices, motors, magnetic resonance imaging (MRI) scanners, and green energy sectors such as wind turbine generators and solar panels.8,9 The demand for Nd has increased significantly due to the rapid growth of these industries.8 Nd has been identified as critical material by the U.S. Department of Energy because of its importance to technology.10 According to a recent United States Geological Survey (USGS) report, nearly 70% of the total mine production of raw rare earths occurs only in China.11
The shortage of diverse supply chains and the ongoing demand for REE have significantly enhanced prices. The average price of Nd has increased from $10 per kg in 2001 to $130 per kg in 2022, a nearly 1200% increase in the last two decades.11–20 The lack of diversification and monopoly in the supply of this element can halt the technological advancement of various countries. A decline in the uninterrupted supply of this critical material could lead to a geopolitical conflict and may negatively impact the global economy.21 As minerals and metals are non-renewable resources, a sufficient supply of rare earths will be challenging. To address these challenges, sustainable recycling technologies are highly desirable. The use of sustainable technologies for the recovery of Nd from e-waste may be a viable alternative to the mining of Nd.22
Several methods such as chemical precipitation,23 liquid–liquid extraction (LLE),24 ion exchange,25 solid–liquid extraction (SLE)26 have been extensively used to separate Nd and other REE. At present, solvent extraction is the widely used technique available for separation.27 However, the technique is expensive, time-consuming, lacks selectivity, and needs to be more sustainable as it requires substantial quantities of toxic organic solvents, that causes a negative impact on the environment.28 To counter these challenges, adsorption processes are the recent focus to achieve affordable, efficient, and environment-friendly techniques that provide better removal capacities. As a result, sustainable material such as cellulose, one of the most abundant biopolymer, has gained remarkable attention.29
Cellulose is a polysaccharide easily obtained from wood pulp that consists of β (1→4) linkage of D-glucose units. The glucose units contain many hydroxyl groups, which may be utilized for chemical modification depending on the requirement of the adsorbates.30 Previous reports suggest cellulose has been modified with poly(hydroxamic acid)/poly(aldoxime), and thiourea to remove Nd3+ from the aqueous solution via adsorption.31–33 Although cellulose-based adsorbents have many advantages, limited accessibility of the inner crystalline region of cellulose reduces the density of functionalities, hindering the evolution of a high-capacity and rapid rare earth adsorbent.34,35 The literature agrees that cellulose nanofibrils consist of alternating crystalline and amorphous cellulose regions.30 Selective oxidation of the amorphous region of cellulose nanofibrils could improve the functional group accessibility of the inner crystalline region.
In this work, we selectively oxidize the amorphous part of the cellulose fibrils, resulting in the formation of two anionic, water-dispersible carboxylate functionalized nanocellulose (CFNC), namely carboxylate functionalized cellulose nanocrystals (CFCNC) and dicarboxylate functionalized cellulose (DFC). The structure of CFCNC is different from that of conventional cellulose nanofibrils. CFCNC has a crystalline body similar to conventional cellulose but sandwiched between the highly functionalized amorphous cellulose chains protruding from each end.35 The structure of DFC consists of an amorphous region of cellulose functionalized with carboxylates at each end. Both these DFC and CFCNC, collectively termed CFNC, has a high density of carboxylate functionalities that forms hydrogen bond with water and describes their water-dispersible nature. Due to the higher magnitude of functional group density, CFNC can adsorb and efficiently remove higher quantities of rare earths from aqueous solution. Herein, we utilize CFNC to remove Nd3+ from an aqueous solution, measure the high removal capacity, and investigate the effects of physicochemical parameters on Nd3+ precipitation. Besides, we have shown the selective extraction of Nd from NdFeB magnet (removed from e-waste) using CFNC as a real-time application.
Methodology
Materials required
The cellulose used for the synthesis of CFNC, neodymium(III) chloride hexahydrate (NdCl3·6H2O, 99.9% trace metal basis), sodium chloride (NaCl, >99.5%), calcium chloride dihydrate (CaCl2·2H2O, ≥99.0%), aluminum chloride (AlCl3), sodium (meta)periodate (NaIO4, >99.0%), sodium chlorite (NaClO2, 80%), hydrogen peroxide (H2O2, 30 wt%), ethylene glycol (Reagent plus >99%), hydroxylamine hydrochloride (NH2OH·HCl), nitric acid (HNO3, 99.9% trace metal basis) and iron(II) sulfate heptahydrate (FeSO4·7H2O, ACS reagent, >99.0%) were all purchased from Sigma-Aldrich. Hydrochloric acid (HCl), sodium hydroxide (NaOH), and dimethyl sulphoxide (DMSO) were obtained from Fischer Scientific. Ethanol (95% vol) was purchased from Commercial Alcohols by Greenfield Global (Canada). All the glassware is cleaned thoroughly with aqua regia, washed with distilled water, and dried before use. All the chemicals are used without further purification. Sample preparation for all the experiments is discussed in detail in ESI.†Experimental procedure
Measurement of aldehyde content of AFNC
0.1 g of AFNC was added to 50 mL of Milli-Q water, and the pH of the solution was adjusted to 3.5 by adding 0.1 M HCl. To this solution, 10 mL of hydroxylamine hydrochloride was added and stirred continuously for 10 min. Due to hydroxylamine-aldehyde reaction, hydrochloric acid was released that resulted in the decrease in pH of the solution. To measure the aldehyde content, the resulting solution was titrated against NaOH (10 mM) until the pH was adjusted to 3.5 (initial value).35Measurement of carboxylate content of CFNC
For carboxylate content measurement, conductometric titrations were performed. 0.02 g of CFNC was added to 140 mL of Milli-Q water. To this, 2 mL of NaCl (20 mM) was added and pH was adjusted to 3 by addition of 0.1 M HCl. The solution was titrated with NaOH (10 mM) until the pH reached 11. The CFNC charge density was calculated based on the volume of NaOH consumed to neutralize the carboxylate group (weak acid).35Neodymium ion (Nd3+) removal experiments
A stock solution of NdCl3 with concentration 10 mM (1442 ppm) was prepared by dissolving NdCl3·6H2O in Milli-Q water. To prepare different concentrations, the stock solution was further diluted. To perform the precipitation experiment, NdCl3 solution was added to the CFNC dispersion which results in the formation of Nd-CFNC precipitate. The precipitate formed is due to the Nd(COO)3 complex formation of the Nd3+ ions with the carboxylate group of CFNC. The precipitation reaction is performed at pH at 5. The supernatant was separated from the Nd-CFNC precipitate by centrifugation at 4000 rpm. The equilibrium concentration (Ce) of Nd3+ ion in the supernatant was measured using inductively coupled plasma mass spectrometry (ICP MS). Each precipitation experiment is performed three times. The Nd3+ removal capacity (qe) in mg g−1 and removal percentage were calculated from the following equation.| Removal percentage = [(C0 − Ce)/C0] × 100 | (1) |
| qe = [(C0 − Ce)/m] × V | (2) |
Effect of contact time on Nd3+ removal
For Nd3+ removal experiment, 500 ppm of CFNC was incubated with 200 ppm of Nd3+ solution. For all the experiments, the concentration of CFNC was taken as 500 ppm. The precipitation reaction is performed at pH at 5. After the immediate addition of Nd3+ solution (200 ppm) into CFNC, the solution was vortexed for 5 s, 10 s, 30 s, 60 s, 120 s, and 300 s and then centrifuged at 4000 rpm for 5 min. The concentration of Nd3+ precipitated can be calculated by subtracting the equilibrium concentration (Ce) of Nd3+ present in the supernatant (using ICP MS) from the initial concentration (C0) of the Nd3+ solution (200 ppm) used for the experiment.Effect of initial concentration (C0) on Nd3+ removal
To 500 ppm of CFNC, varied Nd3+ concentrations of 10 ppm, 50 ppm, 100 ppm, 200 ppm, 500 ppm, 800 ppm, and 1000 ppm were added (pH = 5). The total volume of each of the mixed solution was 5 mL. After mixing, the solution was vortexed for 1 min followed by centrifugation at 4000 rpm for 5 min. The equilibrium concentration (Ce) of the Nd3+ ion in the supernatant was measured by ICP MS.Effect of CFNC concentration on Nd3+ removal
The as-synthesized CFNC solution has a concentration of 500 ppm. To prepare different concentration of CFNC, the as-synthesized CFNC was first freeze dried. The freeze dried sample was then used to prepare CFNC concentrations of 100 ppm, 200 ppm, 300 ppm, 400 ppm, 500 ppm, and 600 ppm (pH = 5). The CFNC solutions were prepared by using Milli-Q water. 200 ppm of Nd3+ solution was used in this experiment. After mixing, the solution was vortexed for 1 min and centrifuged at 4000 rpm for 5 min. Ce of the Nd3+ ion in the supernatant was measured by ICP MS.Effect of ionic strength on Nd3+ removal
The effect of monovalent, divalent, and trivalent ions on CFNC mediated Nd3+ precipitation were studied using NaCl, CaCl2, and AlCl3 salt solutions, respectively. 200 ppm of Nd3+ solution was added to each salt solutions such that the final concentrations of the salt solution become 100, 200, 300, 400 ppm and 500 ppm (pH = 5). CFNC (500 ppm) was added to the final solutions, followed by vortexing for 1 min and centrifuging at 4000 rpm for 5 min. The total volume of the solution was kept at 5 mL and Ce of the supernatant was measured by ICP MS.Effect of temperature on Nd3+ removal
200 ppm of Nd3+ solution was prepared and heated at different temperatures. CFNC (500 ppm) dispersion was added to the Nd3+ solution to make up the total volume 5 mL (pH = 5). Nd3+ solution temperature was adjusted such that addition of CFNC adjust the final temperatures to 10, 20, 30, 40, 50, 60, 70, and 80 °C. The temperature was measured using a thermometer. The mixture was vortexed for 1 min, then centrifuged at 4000 rpm for 5 min. Similar experiments were performed with prior heating of CFNC followed by the addition of Nd3+ solution. The effect of temperature (10 °C–80 °C) was also investigated on the already formed Nd3+-CFNC precipitate. At last, Ce of the supernatant containing Nd3+ ion was measured using ICP MS.Effect of pH on Nd3+ removal
The CFNC-Nd3+ precipitation experiment was carried out using 200 ppm of Nd3+ solution, with pH ranging from 1.5 to 7. The pH of the solution was adjusted by using HCl and NaOH solutions. To this solutions, 500 ppm of CFNC dispersion was added and the mixture was vortexed for 1 min and then centrifuged at 4000 rpm for 5 min. The total volume of the solution was 5 mL and Ce was measured using ICP MS. The pH > 7 was not studied due the formation neodymium hydroxide.Selective removal of Nd3+ from the NdFeB magnet
NdFeB magnet from an electronic waste was crushed, made to powder form and demagnetized at 750 °C using a tube furnace. 0.5 g of the demagnetized powder was added to 30 mL of conc. HNO3 and continuously stirred at room temperature for 40 min to completely dissolve the magnet.36 The pH of the solution was adjusted to 3 because we have observed Nd3+ shows better removal capacity in the pH range 3–7. Therefore, most of our experiments containing Nd3+, Fe2+, B3+, Pr3+ and Dy3+ were conducted at 3. The CFNC dispersion was added to the solution, vortexed for 1 min and centrifuged at 4000 rpm for 5 min. The Ce of Nd3+, Fe2+, B3+, Pr3+ and Dy3+ were measured using ICP MS.Instrumentation
Transmission electron microscopy (TEM) data are obtained using Thermo Scientific Talos F200X G2 (S) TEM equipped with Super-X Windowless Energy Dispersive Spectrometer (four quadrants in-column SDD detector) operating at 200 kV. Scanning electron microscopy (SEM) measurements are performed using FEI Quanta 450 Environmental Scanning Electron Microscope operating at 30 kV equipped with EDAX Octane Super 60 mm2 SDD. X-ray photoelectron spectroscopy (XPS) measurements were carried out using a Thermo Fischer Scientific Nexsa G2 spectrometer with monochromatic Al Kα X-rays (hυ = 1486.6 eV). Dynamic light scattering (DLS) measurements are carried out using Malvern NanoSight NS 500 with a laser wavelength of 532 nm equipped with a CCD camera (temperature, 25 °C; scan, 100; dispersant, water; refractive index, 1.33; viscocity, 0.08872 cP; dielectric constant, 78.5).A PerkinElmer FTIR Infrared spectrometer Spectrum II equipped with attenuated total reflectance (ATR) accessory with diamond crystal, LiTaO3 MIR detector, ZnSe 200 μm windows is utilized for recording the FTIR spectra (range: 4000–400 cm−1; scan: 32; resolution: 4 cm−1). All the photographic images are taken with a Nikon D5100 DSLR camera. A Thermo Finnigan iCapQ Inductively Coupled Plasma Mass Spectrometry (ICP-MS) coupled with an autosampler is employed to determine the concentration of metal ions present in the solution. A silicon solar cell (100 W) obtained from Eco Worthy Solar with dimensions 101 × 3.5 × 46 cm3 is connected to a MotoMaster Eliminator portable power pack and battery booster for power supply. The electrodeposition experiment is performed by using a TENMA digital DC power supply (60 V, 2A).
Results and discussion
Synthesis of CFNC and quantification
For the synthesis of CFNC, the first step was to prepare aldehyde-functionalized nanocellulose (AFNC). For this synthesis, 5 g of cellulose was mixed with 350 mL of Milli-Q water and stirred continuously. To this cellulose dispersion, 21.5 g of NaCl, and 7.15 g of NaIO4 was added and allowed to react for 96 h at room temperature. The reaction was carried out in a 500 mL beaker covered with aluminum foil to stop the deactivation of periodate in presence of light. The reaction was stopped by adding 3 mL of ethylene glycol by quenching the unreacted periodate. AFNC was washed several times with Milli-Q water and vacuum-filtered. To prepare CFNC, the next step was to convert the aldehyde groups of AFNC into the dicarboxylate group. Consequently, 5 g of solid AFNC was added to 275 mL of Milli-Q water. To this, 13.25 g of NaCl, 7.45 g of NaClO2, and 4.8 mL of H2O2 were added and stirred for 24 h at room temperature. During the reaction, the solution turned acidic (pH < 5), which was adjusted to pH 5 for a minimum of 6 h by sporadic addition of 0.5 M NaOH solution. The resulting solution was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min to remove non-disintegrated fibers. CFNC was precipitated by adding ethanol to the supernatant and centrifugation at 3000 rpm for 5 min. The resulting CFNC was dispersed in Milli-Q water and purified by dialysis (Mw cut off 12–14 kDa) for 96 h to remove unwanted chemicals and salts.35,37,38
000 rpm for 10 min to remove non-disintegrated fibers. CFNC was precipitated by adding ethanol to the supernatant and centrifugation at 3000 rpm for 5 min. The resulting CFNC was dispersed in Milli-Q water and purified by dialysis (Mw cut off 12–14 kDa) for 96 h to remove unwanted chemicals and salts.35,37,38
The amorphous and crystalline region of cellulose fibrils and the synthetic procedure used to prepare CFNC are schematically illustrated in Fig. 1A. The chemical structure and possible reaction pathways of cellulose with periodate and chlorite are depicted in Fig. 1B. Periodate allows oxidative cleavage of vicinal diols, breaking the C–C bond, followed by the oxidation of the hydroxyl group to dialdehydes (AFNC). Further, the dialdehyde moieties of AFNC in the presence of chlorite and hydrogen peroxide were selectively oxidized to dicarboxylates (CFNC). The synthesized CFNC is a transparent, water-dispersible, and electrostatically stabilized colloidal cellulose. Fig. S1† shows the image of AFNC and CFNC dispersion.
 | ||
| Fig. 1 (A) Schematic representation of carboxylate functionalized nanocellulose (CFNC) synthesis from cellulose nanofibers. (B) The reaction pathways of cellulose nanofibers with periodate and chlorite. | ||
The amount of aldehyde and carboxylate content is essential to quantify the CFNC required to precipitate a given concentration of Nd3+ ions. The aldehyde group content of AFNC was estimated using a hydroxylamine hydrochloride reaction. The initial pH of the solution before the reaction was 3.5. During the reaction, hydrochloric acid release further decreased the solution's pH below 3.5. The resulting solution was then titrated against the sodium hydroxide (NaOH), and a change in pH was noticed during the titration (Fig. S2†). Based on the amount of NaOH required to bring back the pH to an initial value of 3.5, the aldehyde content of AFNC was calculated. The aldehyde content was found to be 7.67 mmol per gram of AFNC. The carboxylate content of the CFNC was obtained using conductometric titration that shows two equivalence points on the addition of NaOH (Fig. S3†). These two equivalence points correspond to the pKa of dicarboxylates (pKa1–4.9, pKa2–7.8) (Fig. S4†), which is beneficial in understanding the effect of pH on the adsorption of Nd3+. The carboxylate content was found to be 6.1 mmol per gram of CFNC from the conductometric titration.
Characterization of CFNC and Nd-CFNC
CFNC consists of two types of cellulose dicarboxylates: CFCNC and DFC. These CFCNC and DFC can be separated by gradual precipitation in ethanol solvent. Fig. S5† shows the CFNC precipitation as a function of ethanol concentration. With the increase in ethanol concentration, CFNC starts precipitating and continues until an ethanol concentration of 150% w/w. At 150% w/w concentration, CFNC precipitation stops, corresponding to the precipitation of CFCNC. Beyond 150%, the precipitation formation was observed again till ethanol concentration reached 300% w/w, corresponding to the precipitation of DFC. We have separated the CFCNC and DFC to study the structure and morphology of these cellulose-based adsorbents.The TEM images of the dried dispersion of cellulose nanofibers before the oxidation reaction (Fig. 2A). The image depicts a rod-like crystalline morphology of the cellulose nanofibers. The morphology of CFCNC was obtained by TEM, as shown in Fig. 2B. CFCNC presents a rod-like structure, indicating a crystalline structure similar to cellulose nanofiber. The crystalline nature of the CFCNC is supported by the HRTEM image in the inset of Fig. 2B, which displays the crystalline lattices with a lattice distance of 0.39 nm corresponding to the (200) plane of cellulose.39 Besides, selected area electron diffraction (SAED) performed on the sample shows distinct bright spots (Fig. S6A†), indicating the material's crystallinity. This suggests that after two-step oxidation, cellulose does not notably disrupt the crystalline nature of CFCNC. The TEM image of DFC is displayed in Fig. 2C, with the inset unveiling the SAED pattern. No bright or concentric spots on the SAED pattern point out the amorphous nature of DFC (Fig. S6B†). As CFCNC and DFC both contain carboxylate groups and precipitate Nd3+, we have not separated them for our experiments and use the as-synthesized CFNC for the precipitation of Nd3+. However, if required, these can be separated using the ethanol-based precipitation method.
 | ||
| Fig. 2 (A) TEM image of the cellulose fibers before the oxidation reactions. (B) TEM image of CFCNC with insets showing the lattice distance and SAED to confirm the crystalline nature. (C) TEM image of DFC with inset presenting the SAED that verifies the amorphous nature. (D) Photographic image of Nd-CFNC precipitate formation at various Nd3+ initial concentrations. (E) FT-IR spectra of CFNC and Nd-CFNC precipitate. (F) O 1s spectra of CFNC (G) O 1s spectra of Nd-CFNC precipitate. FTIR and XPS were measured at a Nd3+ concentration (Co) of 200 ppm. The CFNC concentration was 500 ppm and pH of the solution was 5. | ||
Neodymium removal is done by a simple method of adding CFNC dispersion to NdCl3 solutions. Fig. 2D shows the images of Nd-CFNC precipitates obtained when CFNC was added to various concentrations of Nd3+ ions; the precipitate forms at an Nd3+ concentration of 150 ppm or above. No deposit is observed below 150 ppm, i.e., at 100 ppm, 50 ppm, and 10 ppm. The possible interactions between the Nd3+ ion and CFNC could explain this. The Nd3+ and CFNC are charged species, and the interaction is likely electrostatically driven, and a stable Nd-CFNC complex in solution does not lead to precipitation. The concentration of Nd3+ that is required to neutralize CFNC (500 ppm) is theoretically ∼166 ppm (based on the molar ratio calculation of COO−:Nd3+ = 3:1). Beyond this concentration, CFNC is completely neutralized, and further addition of Nd3+ will lead to the formation of a visible white color Nd-CFNC precipitate. An increase in Nd3+ concentration leads to an increase in the precipitate formation until CFNC is saturated. We have experimentally observed the precipitate at 150 ppm or above (Fig. 2D), which is slightly lower than the theoretical value and could be due to the self-aggregation of a few CFNC molecules in the dispersion. Self-aggregation of the charged CFNC molecules hinders the carboxylate groups from participating in the precipitate formation and lowers the concentration of CFNC available for complex formation.
The nature of CFNC during the precipitation can be understood in terms of the hydrodynamic size (HDS) of Nd-CFNC aggregates in the supernatant. Fig. S7A† display the DLS measurement that shows the change in HDS of the supernatant at various Nd3+ concentration and Fig. S7B† shows the graph of HDS of Nd-CFNC aggregates in the supernatant against varying Co. The hydrodynamic size does not change significantly until Co ∼ 75 ppm, suggesting no aggregation occurs up to 75 ppm. After 75 ppm, the HDS increases until 150 ppm, reaching a maximum value of 350 nm. Beyond this concentration, significant precipitation takes place. Further increasing Co ∼ 165 ppm decreases the HDS because the larger size aggregates are precipitated out, leaving the smaller aggregates in the solution. When Co ∼ 175 ppm, most of the CFNC precipitated, and the HDS should reach close to zero. However, the HDS value at Co ∼ 175 ppm is 50 nm, and beyond this concentration, the HDS is almost unchanged, corresponding to self-aggregated unreacted CFNC left in the supernatant.
To acquire molecular information on the adsorption of Nd3+ on CFNC, FTIR spectra show the Nd-CFNC complex along with CFNC (Fig. 2E). CFNC shows a peak at 3363 cm−1 due to the vibration stretching of –OH bonds. Similarly, features at 1417 cm−1, 1121 cm−1, 992 cm−1, and 1626 cm−1 correspond to –CH2 bending, C–OH stretching, –CH2–O–CH2 stretching (pyranose ring), and –COO vibration (–COO−Na+), respectively.38,40 After adsorption of Nd3+, the peaks due to –OH stretching, –CH2 bending, C–OH stretching, –CH2–O–CH2 stretching were shifted to 3323 cm−1, 1421 cm−1, 1111 cm−1, and 1001 cm−1, respectively. Interestingly, we have observed the –COO peak which was previously present at 1626 cm−1 (–COO−Na+) is now shifted to 1601 cm−1. The shift in the –COO peak confirms the interaction of Nd3+ with the carboxylate group of CFNC.41
We performed the XPS of CFNC before and after Nd(III) adsorption to observe any changes in the C and O signature. Before complex formation, the XPS feature of O 1s shows peaks at 531.3 eV, 533.0 eV, 533.6 eV, and 535.8 eV due to O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, O–H, O–C, and OH2, respectively. After complex formation, the O 1s of the complex display peaks at 531.5 eV, 533.1 eV, 533.9 eV, and 536.2 eV that could be attributed to CO, O–H, O–C, and OH2, respectively (Fig. 2F).42 The O 1s peaks are slightly shifted to higher binding energies after complex formation. This shift could be due to the increase in electropositive character of the carboxylate oxygen by sharing the electron density to the attached Nd3+ ion. Besides, an addition feature at binding energy 529.5 eV was observed. Such features were obtained when a metal oxide bond exists in the system. Hence, the peak at 529.5 eV could be assigned to the O–Nd bond.43 C 1s peaks show similar shifts towards higher binding energy. C 1s peak of CFNC show C–O and CO features at 286.2 eV and 288.3 eV (Fig. S8A†) which after complex formation with Nd3+ shifts to 286.4 eV and 288.5 eV, respectively (Fig. S8B†).44 The C 1s and O 1s features confirm the attachment of carboxylate oxygen to Nd3+ in the Nd-CFNC complex.
C, O–H, O–C, and OH2, respectively. After complex formation, the O 1s of the complex display peaks at 531.5 eV, 533.1 eV, 533.9 eV, and 536.2 eV that could be attributed to CO, O–H, O–C, and OH2, respectively (Fig. 2F).42 The O 1s peaks are slightly shifted to higher binding energies after complex formation. This shift could be due to the increase in electropositive character of the carboxylate oxygen by sharing the electron density to the attached Nd3+ ion. Besides, an addition feature at binding energy 529.5 eV was observed. Such features were obtained when a metal oxide bond exists in the system. Hence, the peak at 529.5 eV could be assigned to the O–Nd bond.43 C 1s peaks show similar shifts towards higher binding energy. C 1s peak of CFNC show C–O and CO features at 286.2 eV and 288.3 eV (Fig. S8A†) which after complex formation with Nd3+ shifts to 286.4 eV and 288.5 eV, respectively (Fig. S8B†).44 The C 1s and O 1s features confirm the attachment of carboxylate oxygen to Nd3+ in the Nd-CFNC complex.
We also investigated the XPS to know the binding of Nd3+ with CFNC. Fig. S8C† shows the XPS spectra of Nd3+ before adsorption (from NdCl3·6H2O solution) along with the adsorbed ion in the Nd-CFNC complex. Before adsorption, XPS shows a doublet corresponding to 3d5/2 and 3d3/2 core levels of Nd because of the spin–orbit coupling.45 The separation between the doublets is around 22.4 eV. The peaks appearing at binding energies of 983.3 and 1005.7 eV correspond to a characteristic Nd3+ feature.46 The deconvolution of 3d5/2 and 3d3/2 peaks indicate the presence of several peaks. The shoulders observed on the lower binding energy of Nd 3d5/2 and both sides of Nd 3d3/2 correspond to the shake-off and shake-up satellite peaks.47,48 After complex formation, the XPS feature for Nd3+ is slightly shifted to lower binding energy. A shift of ∼0.4 eV in the binding energy of Nd 3d5/2 and Nd 3d3/2 was noticed. This could be due to the attachment to carboxylate groups which shares the electron density and reduces the positive charge on Nd3+. As a result, the binding energy shifts towards a lower value.
Neodymium removal studies using CFNC
The Nd3+ removal capacity of CFNC initial concentrations (Co) varied from 150 ppm to 1000 ppm, as shown in Fig. 3A. At this concentration range, CFNC is saturated and does not show a significant change in the removal capacity upon varying the concentration. The average removal capacity observed was 252 ± 5 mg of Nd3+ per gram of CFNC, represented as 252 ± 5 mg g−1. Fig. 3B displays the Nd3+ removal capacity of CFNC as a function of contact time. Nd3+ addition to CFNC, precipitation occurred instantly in 5 s, presenting a fast and facile Nd3+ removal technology. The equilibrium is achieved in 5 s, beyond which an increase in contact time does not increase the removal capacity significantly and remains almost constant. A maximum removal capacity of 251 ± 8 mg g−1 is observed for a contact time of 5 s. | ||
| Fig. 3 (A) Nd3+ removal capacity of CFNC as a function of initial concentration (Co) (CFNC = 500 ppm, pH = 5). (B) Nd3+ removal capacity of CFNC as a function of contact time (Co = 200 ppm, CFNC = 500 ppm, pH = 5). (C) Nd3+ removal percentage against the concentration of CFNC (Co = 200 ppm, pH = 5). (D) Nd3+ removal percentage of CFNC as a function of initial concentration (CFNC = 500 ppm, pH = 5). | ||
A contact time of 5 s suggest the exceptional precipitating ability of CFNC. A comparison of the solubility product (Ksp) of CFNC with other dicarboxylate such as oxalic acid is necessary to understand the superior precipitating ability CFNC. The solubility product of Nd-CFNC and Nd-oxalate were calculated using the conductivity method at room temperature (295 K). The Ksp of Nd-CFNC was found to be 5.02 × 10−18 mol5 L−5 and Ksp of Nd-oxalate was calculated as 5.73 × 10−10 mol5 L−5. (Ksp)Nd-CFNC < (Ksp)Nd-Oxalate by ∼108 times, suggesting CFNC is a better precipitating agent than oxalate. The main advantage of CFNC relative to other carboxylate-based precipitating agents such as oxalic acid is the lower solubility of CFNC in water. CFNC has a cellulose backbone, a macromolecular structure with large number of hydrophobic units that limit the solubility in water as compared to oxalic acid. Lower solubility favors the gravimetric factor, and it precipitates easily with Nd3+. Therefore, although both forms precipitate with Nd3+, CFNC is more selective towards Nd3+ than oxalate.
Fig. 3C depicts the plot of the removal percentage of Nd3+ as a function of CFNC concentration. 200 ppm of Nd3+ mixed with varying concentrations of CFNC from 100 to 600 ppm. The as-synthesized CFNC has a concentration of ∼500 ppm. The synthesized CFNC (solution phase) is first dried and powdered to prepare different concentrations. The powdered CFNC is utilized to prepare solutions of the desired concentration. From Fig. 3C, it is clear that, as we increase the CFNC concentration, the removal percentage of Nd3+ increases. The increase is linear up to 600 ppm, suggesting a stoichiometric removal of ∼180 ppm (90% removal percentage) of Nd3+ by 600 ppm of CFNC (∼COO−:Nd3+ = 3:1). Beyond a concentration of 600 ppm for CFNC (700 ppm or more), the concentration of Nd3+ (200 ppm) used is not able to saturate the CFNC. As a result, no precipitate formation is observed, and removal percentage cannot be determined. Fig. 3D plots the removal percentage of Nd3+ as a function of initial concentration (Co). Fig. 3D indicates a gradual decrease in the removal percentage with the increase in the initial concentration (Co). As CFNC concentration is constant throughout the experiment (500 ppm), an increase in Nd3+ concentration beyond a certain point saturates all the active sites present in CFNC, decreasing the removal percentage. The maximum removal obtained was 95% at Co ∼ 150 ppm. The removal percentage at Co < 150 ppm was not studied because, below 150 ppm, Nd3+ forms an electrostatically charged and stable Nd-CFNC complex in the solution that does not lead to precipitation.
Fig. 3D is useful to calculate the thermodynamic parameters of the Nd3+ adsorption on CFNC. Fig. 3D shows a maximum removal percentage of 95% at Co ∼ 150 ppm. Substituting these values eqn (1), the equilibrium concentration (Ce) was calculated to be 7.5 ppm which is same as obtained using the ICP MS. To calculate thermodynamic parameters, determination of equilibrium constant (Kc) is essential. Kc can be calculated using the following equation.49–51
| Kc = Cs/Ce = Co − Ce/Ce | (3) |
| ΔG° = −RTlnKc | (4) |
At room temperature (295 K), ΔG° value was calculated to be −7.173 kJ mol−1. As ΔG° for an adsorption system is negative, one can say that adsorption process is favorable. Thus, the larger the Kc value from unity, the system has more negative ΔG° and, consequently, the larger is the tendency of the system to go toward the forward (adsorption) direction. In the case of having an equilibrium constant smaller than unity, the value of ΔG° calculated from eqn (4) would have a positive value. Therefore, it is obvious that having a positive ΔG° (or Kc value lower than 1) does not permit the system to be progressed to the forward direction too much and the amount of adsorbate adsorbed on the adsorbent would not be significant at all. Therefore, in such situations, it is said that the adsorption is not a favorable process. Thus, a negative ΔG° means that the adsorption is thermodynamically favorable to takes place (Kc > 1). On the other hand, the adsorption process would not be favorable when ΔG° has a positive value (Kc < 1).
The equilibrium constant is a temperature dependent property and increase in temperature decreases the equilibrium constant. As the Kc at 295 K is already known, Kc value at different temperature is required to calculate the enthalpy of adsorption (ΔH°). The Kc value at 305 K was found to be ∼15. ΔH° can now be determined using integrated van't Hoff equation.
| ln(K305/K295) = ΔH° (T2 − T1)/RT1T2 | (5) |
Using the above equation, ΔH° value comes out to be −11.93 kJ mol−1. Once ΔG° and ΔH° are known, the entropy of adsorption (ΔS°) can be easily calculated from the equation:
| ΔG° = ΔH° − TΔS° | (6) |
At 295 K, ΔS° value is equal to −16.13 JK−1mol−1. ΔS° value for Nd3+ adsorption on CFNC is negative. A negative value of ΔS° is expected, as the adsorption process results in the decrease of disorder nature or randomness in the system. During the reaction, formation of a solid precipitate from aqueous reactants understandably decreases the entropy of the system. Since ΔS° is negative, TΔS° is also negative and the second term of eqn. (6) becomes positive. For the overall process to be feasible (ΔG° negative), |ΔH°| must be greater than |TΔS°|. From the calculation we observed the same. Thus, one can say that adsorption of Nd3+ on CFNC is thermodynamically feasible and enthalpy driven.
Effects of physicochemical conditions
| Nd3+ + COO− ⇌ Nd(COO)3 | (7) |
| COO− + H+ ⇌ COOH | (8) |
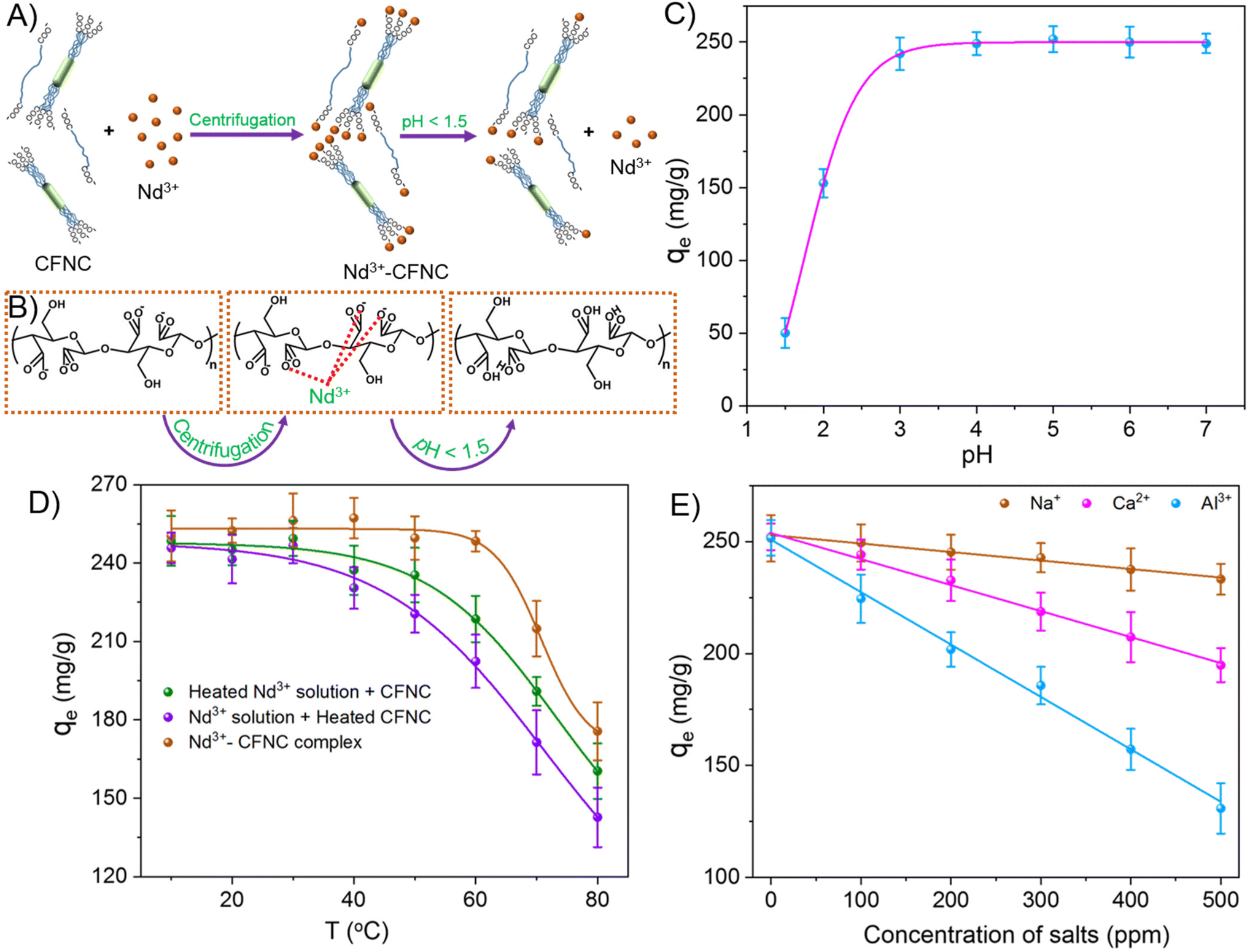 | ||
| Fig. 4 (A) Schematic of Nd3+ removal using CFNC and the effect on precipitation at lower pH (<1.5). (B) Chemical structure of CFNC, Nd-CFNC, and protonated CFNC at lower pH. (C) Effect of pH on the removal capacity of Nd3+ by CFNC. (D) Effect of temperature on the Nd3+ removal capacity of CFNC (pH = 5). (E) Effect of monovalent (Na+), divalent (Ca2+), and trivalent (Al3+) ions on the removal capacity of Nd3+ using CFNC (pH = 5). All the measurements were done at Nd3+ concentration (Co) of 200 ppm, and CFNC concentration of 500 ppm. | ||
The better removal capacity at pH ∼ 2 or 3 is likely due to the high concentration of Nd3+, which will shift the equilibrium in eqn (7) towards the right-hand side, increasing COO− concentration. As a result, the precipitation reaction occurs readily, enhancing removal capacity. At pH < 2, the H+ ion concentration dominates, and an increase in the H+ shifts the equilibrium in eqn (8) towards the right-hand side, increasing protonation and lowering free COO− concentration. Consequently, the adsorption of Nd3+ and removal capacity is reduced. The pH above 7 was not investigated in our experiment owing to the precipitation of neodymium hydroxide in an alkaline medium.52
The second case involves the heating of only CFNC dispersion at various temperatures followed by Nd3+ addition. The addition of Nd3+ shows a sufficient diminution in the average removal capacity (220 mg g−1) compared to the unheated sample until 50 °C. Heating CFNC dispersion before adsorption allowed substantial aggregation, resulting in fewer binding sites available for the adsorption reflected in the removal capacity. After 50 °C, further heating steers to enhance aggregation, and the removal capacity is reduced significantly (Fig. S9B†). In the third case, the already formed Nd-CFNC precipitate was heated at various temperatures to study the effect of temperature after adsorption. When heating was done after adsorption, the removal capacity did not change considerably (up to 60 °C). At higher temperatures (above 60 C), the average removal capacity reduced significantly, possibly due to the temperature-induced desorption of Nd3+ from the Nd-CFNC complex (Fig. S9C†).
Nd3+ removal using seawater, tap water, and DI water
The total volume of water on Earth is estimated to be 1.386 billion km3, out of which 97.5% comes from saline water and only 2.5% is fresh water.56 As per USGS (United States Geological Survey), only 16% of all water used in the U.S.A. in 2015 is saline.57 About 97% of the country's saline water is used for the thermoelectric power industry and the rest 3% is used for mining and other industrial processes.57 Thus, the direct use of seawater as an alternative to freshwater increases the utility of saline water for industrial processes and brings a cost-effective solution for many industries.Sea water, without further purification, if used as a replacement for ultrapure water (Milli-Q), could reduce the overall cost of Nd and other REEs extraction, making it more affordable and sustainable. We have used ultrapure water (Milli-Q) for most of our experiments, which contains very few ions. Our experiments showed that Nd3+ displays selectivity towards CFNC even in a considerable salt concentration. We performed our experiments with artificial seawater to check whether it can be used as an alternative to Milli-Q water for Nd3+ extraction. Artificial seawater contains NaCl (24.53 g L−1), NaF (0.003 g L−1), Na2SO4 (4.09 g L−1), KCl (0.695 g L−1), KBr (0.101 g L−1), MgCl2 (5.20 g L−1), CaCl2 (1.16 g L−1), SrCl2 (0.0025 g L−1), NaHCO3 (0.201 g L−1), H3BO3 (0.027 g L−1), and H2O (988.968 g L−1).58 200 ppm of Nd3+ solution was made using artificial seawater to check the removal capacity of CFNC. Nd3+ removal capacity using artificial seawater was ∼150 mg g−1, about 60% of the removal capacity obtained using Milli-Q water (Fig. S11†). The artificial seawater also gives a reasonable removal capacity and could be used as an alternative to Milli-Q water. We have also tested the removal capacity in the presence of deionized (DI) water and tap water, which shows fairly good values of 230 mg g−1 and 205 mg g−1, respectively (Fig. S13†). The interfering ions content in DI water and tap water is less and therefore shows higher removal capacity than seawater.
Selective extraction of Nd3+ from NdFeB magnet using CFNC
NdFeB magnet contains 20–30 wt% of REEs, 60–70 wt% of Fe, 1 wt% of B, and mixtures of some other metals in minor quantities.59 To show a real-time application, we have used a NdFeB magnet extracted from an unusable Home Theatre speaker (electronic waste) and performed Nd removal experiments using it. The details of the acid digestion procedure to prepare metal ion solution are mentioned in the ESI.† The B3+, Pr3+, and Dy3+ ions do not significantly affect the Nd3+ removal process owing to their minimal concentration in the solution. Therefore, our primary focus was on the selective and sustainable removal of Nd over iron (Fe), which is a severe challenge of recycling the Nd from the magnet.Fig. 5A shows the image of the speaker containing the NdFeB magnet from where the magnet was extracted. Fig. 5B depicts the photographic embodiment of the extracted magnet from the speaker. The same magnet was utilized for preparing the metal ion solution. The as-prepared solution was adjusted to prepare a stock solution containing 1000 ppm of Nd3+. The stock solution is diluted to form various concentrations of Nd3+ for concentration-dependent measurements. The minimum concentration of the sample was adjusted up to 200 ppm of Nd3+ to observe the precipitate formation visibly, as CFNC shows clear precipitation at Co ∼ 150 ppm or above. Therefore, all the measurements were done at Co ≥ 200 ppm. Since each of the concentrations of B3+, Pr3+, and Dy3+ is <500 ppb in the solution, they do not affect the Nd3+ precipitation significantly.
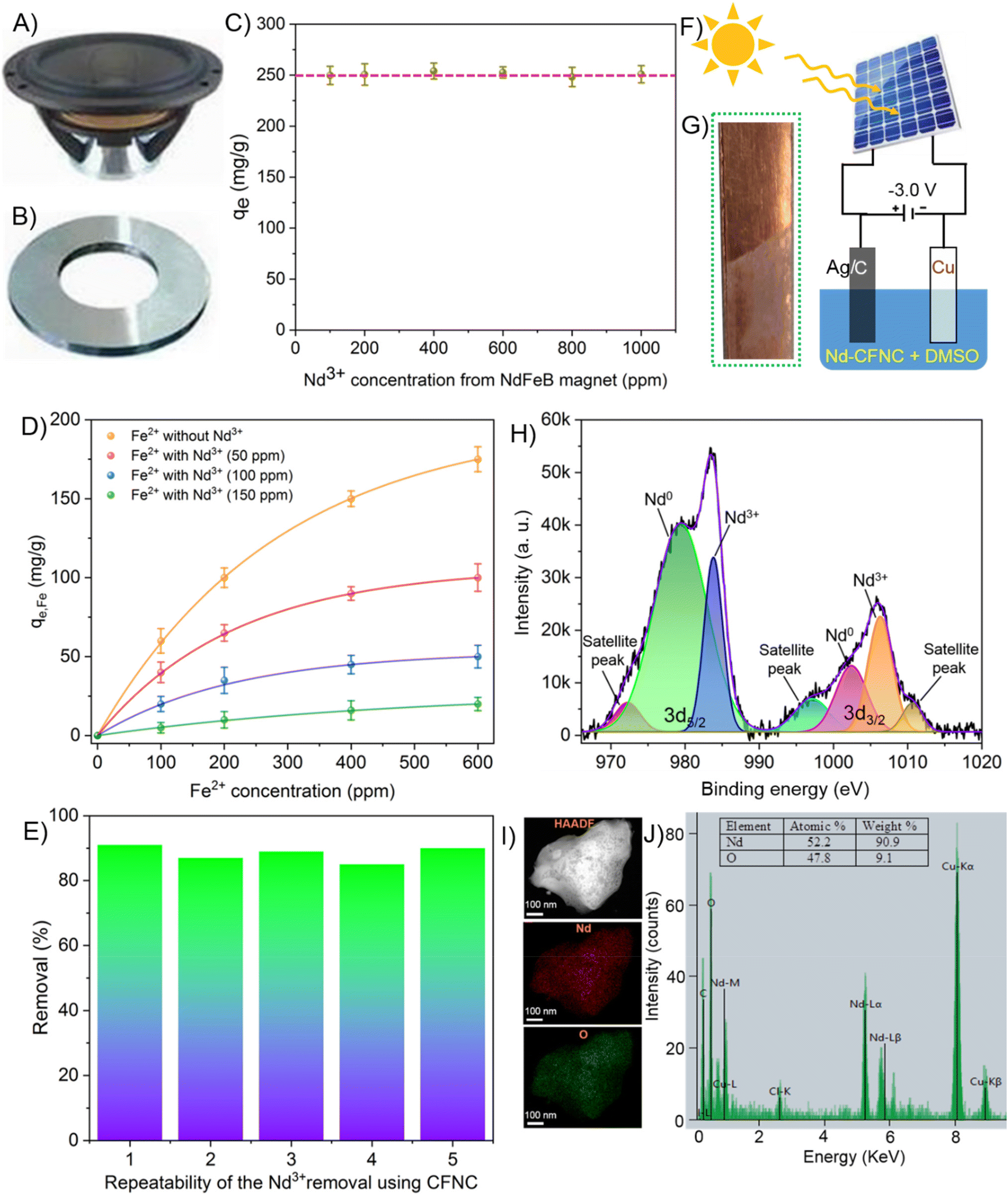 | ||
| Fig. 5 (A) The image of an unused Home Theatre speaker from where the NdFeB magnet was removed. (B) The image of the NdFeB magnet extracted from the speaker. (C) Nd3+ removal capacity of CFNC as a function of Nd3+ concentration. Different concentrations of Nd3+ were prepared from the acid-digested solution of NdFeB powder. (D) Fe2+ removal capacity of CFNC in the absence and presence of various concentrations of Nd3+ (CFNC = 500 ppm, pH = 5). (E) Repeatability experiment of the Nd3+ removal percentage using CFNC. (F) Schematic of the solar-driven electrodeposition performed to extract Nd from Nd-CFNC complex. (G) The image of electrodeposited Nd/Nd2O3 on a copper plate. (H) XPS spectrum of the electrodeposited Nd/Nd2O3. (I) High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of the electrodeposited Nd/Nd2O3 along with the elemental mapping of Nd and oxygen. (J) Energy dispersive spectroscopy (EDS) along with the elemental composition of the electrodeposited material. | ||
The Nd3+ removal capacity as a function of the concentration of Nd3+ ions is presented in Fig. 5C. We performed precipitation experiments with the as-synthesized CFNC (500 ppm). Nd3+ concentration is varied in the range of 200 ppm to 1000 ppm. CFNC is saturated in the operating concentrated range and the removal capacity is unaltered upon varying concentrations. An increase in the concentration of Nd3+ will likely increase the amount of Fe2+ in the solution. The average removal capacity observed is 245 ± 8 mg g−1 which is quite close to the value when no other interfering ions present 252 ± 5 mg g−1 (Fig. 3B), under our experimental conditions. This suggests that the presence of Fe2+ ions in the solution has negligible interference with the removal capacity of Nd3+ and 97% of the removal capacity is retained even in the presence of other interfering ions. Most of the CFNC selectively precipitates Nd3+ leaving Fe2+ ions in the solution. These studies suggest CFNC is a promising material suitable for selective Nd3+ removal from solutions, which may enable recycling of Nd from e-waste and help address environmental issues.
Since the solution is prepared from the magnet, it contains a fixed proportion of Nd3+ and Fe2+, and changing the concentration of one of them automatically changes the concentration of the other. Therefore, it does not give any idea of the removal capacity of Nd3+ up on varying concentrations of Fe2+ and vice versa. To understand the effect, we manually prepared Nd3+ and Fe2+ solutions and mixed the solutions at various Fe2+ concentrations to measure the Fe2+ removal capacity. Fig. 5D presents the variation of removal capacity Fe2+ as a function of Fe2+ concentration. The adsorption properties of Fe2+ on CFNC are tested in the presence and absence of Nd3+. In the absence of Nd3+ in the solution, the removal capacity shows a significant increase with increasing the concentration of Fe2+. The Fe2+ removal capacity reaches from ∼58 mg g−1 at 100 ppm of Fe2+ to ∼185 mg g−1 at 600 ppm of Fe2+ without Nd3+. As the Nd3+ concentration increases from 50 ppm to 150 ppm, the Fe2+ removal capacity decreases gradually. When Nd3+ concentration is 50 ppm, the Fe2+ removal capacity shows ∼35 mg g−1 at 100 ppm of Fe2+ to ∼100 mg g−1 at 600 ppm of Fe2+, whereas at Nd3+ concentration of 150 ppm, the Fe2+ removal is substantially reduced to ∼6 mg g−1 at 100 ppm and almost remains constant at Fe2+ > 100 ppm (∼20 mg g−1 at 600 ppm). The results can be since the carboxylate groups on CFNC preferred Nd3+ over Fe2+ for binding, leaving fewer binding sites for the removal of Fe2+. The higher binding energy between Nd3+ and COO− compared to Fe2+ and COO− could be the reason for such behavior.55 Due to the oxidizing nature of HNO3, there is a possibility that some of the Fe2+ gets oxidized to Fe3+ during the course of digestion. To check the oxidation, we have performed UV-Vis measurement of the leachate solution after 5 min as well as after 120 min digestion with acid. Even after 120 min we have not observed significant changes in the UV-Vis features (Fig. S12†) indicating a very little conversion of Fe2+ to Fe3+ during that period. We compared removal capacity of both the Fe2+ ions (Fig. 5D) and Fe3+ ions (Fig. S13†). However, we noticed a negligible change in the removal capacity on changing from Fe2+ to Fe3+. A slight increase in the removal capacity of Fe3+ is possibly due to its higher charge and binding energy with CFNC as compared to Fe2+ ion. Therefore, we can clearly say that although there is possibility that Fe2+ might get oxidized during acid digestion but does not significantly impact the removal capacity of Nd3+. Repeatability of the Nd3+ removal by CFNC is performed on the solution prepared from the magnet, showing a reasonably consistent removal percentage throughout different experiments (Fig. 5E). The removal percentage was 85–95%, indicating a high reproducibility of the material.
To check whether the precipitation occurs in the absence of CFNC or not, removal capacity was checked in the presence as well as in the absence of CFNC. In presence of CFNC, precipitation occurs at all pH, but the removal capacity is less a lower pH whereas it increase 5 times when the pH is in the range 3–7 similar to that of Fig. 4C. However, in the absence of CFNC, no precipitate formation was noticed due to the absence of Nd-CFNC interaction. Therefore, the removal capacity value is zero as at all pH (Fig. S14†). No pH induced precipitation of the leachate solution was noticed in the absence of CFNC, which also suggest pH (in the range 1–7) has no impact on the stability of the leached NdFeB magnet solution.
To know the electrodeposition potential cyclic voltammetry experiments were performed. During cyclic voltammetry experiments, platinum disk of 1 mm diameter was used as a working electrode. As a counter electrode platinum wire was used. A silver wire was used as pseudoreference electrode and ferrocene was added as internal standard. The ferrocene/ferrocenium (Fc/Fc+) redox potential was recorded against the Pt pseudoreference electrode after the direct dissolution of 10 mM of ferrocene in DMSO and was used as the reference potential. Nd-CFNC precipitate obtained from the leachate solution was dissolved in dry DMSO and Tetrabutylammonium hexafluorophosphate (TBAPF6, 0.1 M) was employed as supporting electrolyte. Fig. S15† presents the cyclic voltammograms recorded with a scan rate of 20 mV s−1 at room temperature for the pure DMSO and Nd-CFNC in DMSO. The cyclic voltammogram of the Nd-CFNC solution in DMSO presents a reductive loop that begins when scanning the potential to values more cathodic than −2.5 V and shapes a peak at −2.8 V against the reference (Fc/Fc+), which is attributed to the reduction of Nd trivalent cations to the zerovalent state. Moreover, an oxidative peak is observed in the reverse scan at −1.5 V that is ascribed to the oxidation of the deposited metallic Nd, implying that the overall reaction is irreversible.
For electrodeposition, the set up was almost identical with the only difference being the use of copper sheet as working electrode and the Ag/C electrode was used as reference electrode. We avoid the use of Fc/Fc+ for the solar electrodeposition due to the toxicity of ferrocene to humans and aquatic life. Since the Nd3+ reduction occurs at −2.8 V against Fc/Fc+ reference and the E1/2 of Ag/Ag+ (pseudoreference electrode) against Fc/Fc+ in DMSO was found to be −211 mV (Fig. S16†), the Nd3+ reduction against Ag/Ag+ will occur at ∼−3.0 V. Therefore, for the electrodeposition experiment a potential of −3.0 V was applied.
Upon precipitation, the centrifuged material (Nd-CFNC complex) is accumulated and dissolved in DMSO, and electrolysis is performed using solar energy to collect metallic Nd.60 The entire experiment was set up inside a glove box under inert conditions to minimize oxygen and moisture contamination. Dry DMSO was chosen as the best solvent to carry out the electrolysis because of the solubility of the material (Nd-CFNC) in it, polarity of the solvent that will help to ionize the material and facilitate electrolysis, stability, high reducing potential. Besides, dry DMSO has negligible moisture that will prevent the oxidation of metallic Nd formed during the electrodeposition. A solar-driven generator can supply power to a DC power supply, eventually used for the electrodeposition. After the electrolysis ends, the cathode is thoroughly rinsed with acetone to remove the electrolyte (DMSO), and the cathode is weighed. The difference in the mass of the cathode before and after the electrolysis is considered the mass of the electrodeposited material.
The schematic of the solar-driven electrodeposition setup for the Nd extraction is displayed in Fig. 5F. The solution is continuously stirred at room temperature during the electrolysis process to obtain better electrodeposition. A potential of −3.0 V (current ∼−2 mA) was applied to the solution for 3 h for Nd electrodeposition.61 After 3 h of electrolysis, 0.25 mg of the material was deposited on the cathode. After electrodeposition, the material was removed from the glove box to perform spectroscopic and microscopic analysis to confirm the metallic nature of the deposited material. However, the contact with ambient conditions results in the surface oxidation of the deposited material forming a greyish colour film as shown in Fig. 5G.60 An SEM image of the deposited thin film shows irregular morphology, porous nature, and non-uniform distribution of material on the copper surface (Fig. S17†).
The metallic nature of the electrodeposit was investigated by XPS measurements. Fig. 5H presents the XPS spectrum of the deposited material. The XPS spectrum shows the presence of Nd 3d5/2 peaks corresponding to metallic Nd and Nd2O3 at 978.6 eV and 983.5 eV, respectively.61 The 1001.3 eV and 1005.9 eV features represent the 3d5/2 peaks of Nd (0) and Nd2O3, respectively. Besides, C 1s spectra show a feature at 284.8 eV that corresponds to the C–C peak from the adventitious carbon (Fig. S18A†). A peak at 530.8 eV of the O 1s spectra suggests an oxide (O2−) feature from the Nd–O bond (Fig. S18B†).43 The survey spectrum suggests the absence of any impurity (Cl−) in the electrodeposited sample, shown in Fig. S18C.†
A high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) is used to investigate the distribution of Nd and O in the electrodeposited samples. The STEM mapping shows that Nd and O are homogeneously distributed throughout the sample (Fig. 5I), suggesting that Nd is not only present as pure metal but also that some Nd gets oxidized under ambient conditions to form Nd2O3. The EDS analysis shows a high mass percentage (90.9%) and high atomic percentage (52.2%) of Nd present in the sample (Fig. 5J). The presence of oxygen is ascribed to oxidation under ambient conditions during the transfer of the material to the TEM chamber despite the use of protective conditions. A slight impurity from chloride could be coming from CFNC and can be neglected. The atomic ratio of Nd and O is 1.09:1 (52.2%/47.8%) in the electrodeposited sample, which is different from that of pure Nd2O3. The atomic ratio of Nd and O in Nd2O3 is 2:3 or 0.6:1. The higher atomic percentage of Nd in the electrodeposited sample suggests that not all the Nd is involved in oxidation but also the metallic form.
We precipitate 10 mL of 200 ppm Nd3+ solution using CFNC, and the Nd-CFNC precipitate is used for electrodeposition. 10 mL of 200 ppm Nd3+ solution contains 2 mg of Nd3+ ion. Assuming that all the Nd3+ ions are electrodeposited on the cathode. Theoretically, 2 mg of Nd should be deposited on the cathode. Upon applying a potential of −3.0 V (−2 mA) and running the electrolysis for 24 h, we observed a deposition of 1.95 mg of the material. Beyond 24 h, no further deposition of Nd is observed (Fig. S19†), suggesting that almost all the Nd is deposited and only CFNC remains in the solution, which was used again for Nd3+ removal. After Nd electrodeposition, the DMSO solvent was removed using a strong vacuum pump. To evaporate DMSO rotary evaporator was kept at 2 mbar pressure to reach a boiling point 30 °C. In such condition, it was kept overnight to remove the solvent and the dried CFNC was again dispersed in water for reuse. We also test the reusability of CFNC for Nd3+ adsorption; we observe the performance of CFNC decreases to a smaller extent after a few cycles. After 5 cycles, nearly 60% of the activity of CFNC is retained (Fig. S20†). The idea behind the recycle technique was to show that CFNC can be further used for Nd extraction and reduce the overall cost enhancing the sustainability of the method. The reduction in adsorption activity after a few successive cycles may be due to the lack of availability of surface binding sites owing to substantial aggregation. Besides, repeated cycles could result in the loss of CFNC, which also reduces adsorption activity. A loss in the quantity of CFNC impacted the electrodeposition of the material, and a gradual decrease in deposition was noticed after successive cycles (Fig. S21†). Although the adsorption properties of CFNC are reduced marginally after a few cycles, it can still be used for industrial applications owing to its biocompatibility, huge availability, and cost-effectiveness. Future work will include removal experiments using industrial permanent magnet leachates to understand the CFNC selectivity towards other rare earths and transition metals.
Comparison of our material with the other reported materials
We compare the Nd3+ removal of CFNC with different adsorbents reported in the literature. Previously, a variety of materials such as charcoal,62 magnetic oxide nanoparticles,63 metallic organic frameworks (MOFs),64 graphene oxide,65 biopolymers,41,66,67 algae,68 and cellulosic materials31–33 are explored for the Nd3+ removal studies. Fig. 6 presents the removal capacity of these materials, which ranges from 16 mg g−1 to 240 mg g−1. To our knowledge, CFNC has the highest removal capacity compared to the materials reported in the literature. Besides, the removal time using our material is considerably lower compared to the reported adsorbents, suggesting the advantages of using CFNC as a sustainable adsorbent for removing Nd3+ due to its high electrostatic charge content and selectivity. Removal of Nd3+ by CFNC opens the possibility of sustainable removal of other rare earths using modified cellulose or related materials.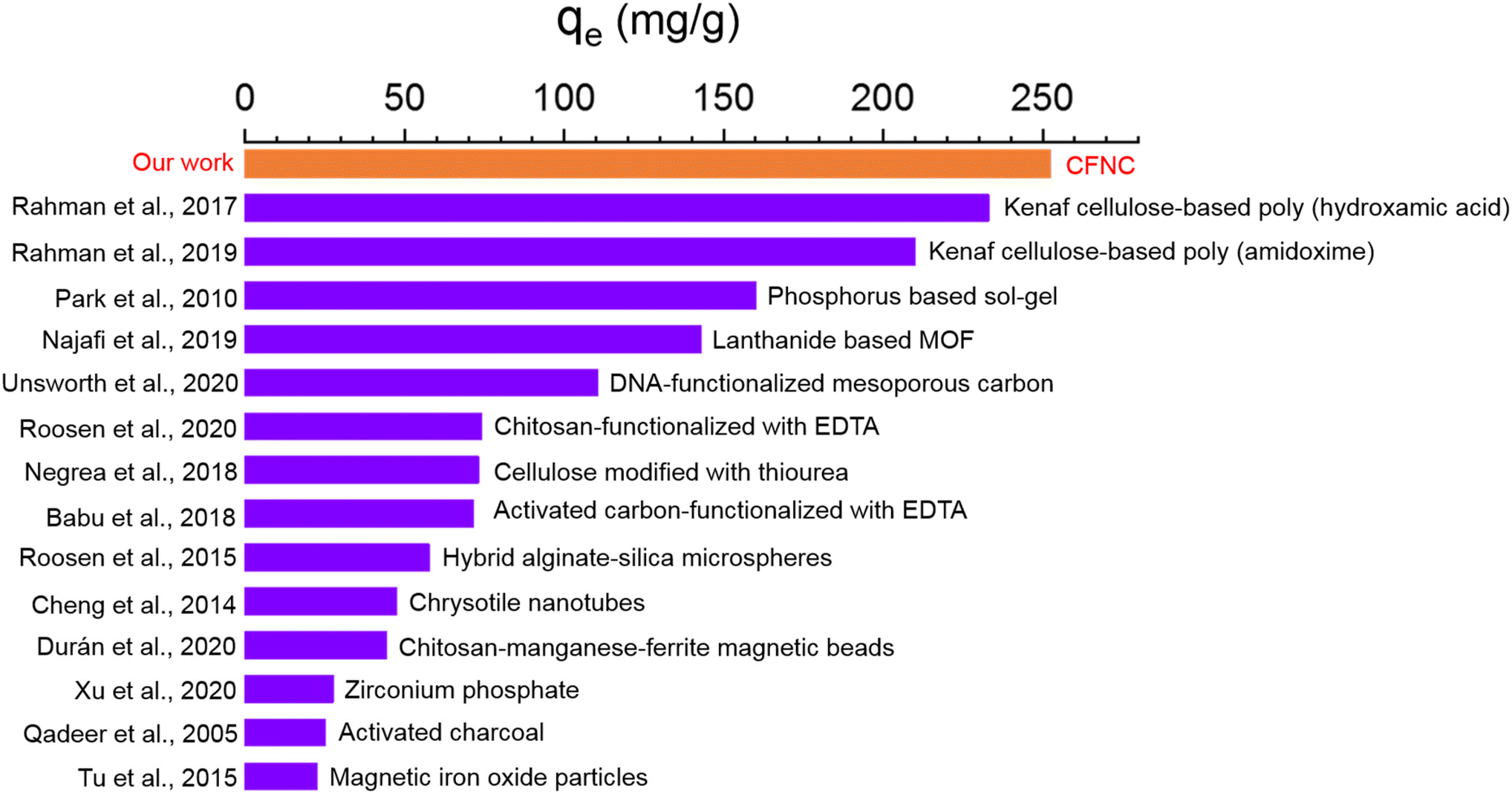 | ||
| Fig. 6 Comparison of removal capacity of our materials with other adsorbents reported in the literature.31–33,41,62–64,66,67,69–73 | ||
Cost analysis
We have performed a cost–benefit analysis that will give an idea about the potential benefits from the proposed approach. Since the electrodeposited Nd surface gets oxidized in presence of air, the electrodeposited material contain both Nd and Nd2O3. Assuming 30% of the electrodeposited Nd get oxidized, a cost analysis was performed for the production of a total 1 ton of electrodeposited material (70% Nd + 30% Nd2O3 = 700 kg of Nd + 300 kg of Nd2O3).From our experimental analysis we found that nearly 500 ppm of CFNC is required for the precipitation of 150 ppm of Nd3+. Taking into consideration, the recyclability and decreasing efficiency after 5 successive cycles, it was calculated that 500 ppm of CFNC can precipitate 630 ppm of Nd3+. As 630 mg of Nd (assuming 100% reduction of Nd3+ to Nd after electrodeposition) requires 500 mg of CFNC, precipitation of 1 ton of Nd roughly require 0.8 ton of CFNC. From lab scale synthesis, it was observed that 5 g of cellulose produces 4.5 g of CFNC. Thus, for the production of 0.8 ton of CFNC, 0.9 ton of cellulose is required as a starting material. The industrial prices of wood pulp cellulose in Canada is $ 752 per ton.74 The cost of 0.9 ton of cellulose is around $ 677. Current industrial production of cellulose in Canada is about 14.2 million tons, by paper and pulp industry alone.75 Since cellulose production and processing are integral parts of the paper and pulp, textile, food processing, and pharmaceutical industries, we can estimate the annual cellulose production by observing output capacities of the aforementioned industries. Hence, our cellulose requirement as a raw material can be met easily. Based on our calculation, the production of 0.8 ton of CFNC from cellulose requires $41384 of other materials for the synthesis. The total cost of equipment that includes apparatus and instrument charges is around $2132. The environmental cost such as vegetation restoration and water treatment combined is approximately $1200. Few other costs include labour ($2500), electricity ($500), water ($50), and combined tax ($2468). The total cost involved in the production of 1 ton of electrodeposited material (700 kg Nd + 300 kg Nd2O3) is $51001. Table S1 in ESI† presents the total production cost breakdown for 1 ton of electrodeposited material. The market value of Nd is $123 kg−1 and Nd2O3 is $50 kg−1. The total market value for 700 kg of Nd and 300 kg of Nd2O3 is $101100. Thus, the profit in the production of 1 ton of the electrodeposited material is $50099. The market value for the material produced is twice as the production cost whereas the net profit is similar to the production cost. The major cost drivers for the production are materials required for the synthesis that constitutes ∼82% of the total cost. Affordable alternatives of the materials could bring the production cost even lower making the proposed project more beneficial and sustainable.
Conclusion
In summary, we demonstrate a solution-processed synthesis of CFNC from a cost-effective, readily available precursor, cellulose, without harsh conditions or complicated procedures. The CFNC precipitates Nd3+ ions of concentration as low as 150 ppm or above and shows an Nd3+ removal capacity of 252 ± 5 mg g−1. The adsorption capacity of CFNC does not show a significant change in temperature up to 60 °C. CFNC acts as an effective adsorbent at pH as low as 3 and shows uniform removal capacity up to pH 7. The Nd3+ removal capacity of CFNC is quite efficacious, even in the presence of ions with high ionic strength. Selective removal of Nd3+ in the presence of Fe2+ and other ions is successfully done from an NdFeB magnet using CFNC. Nd is successfully recovered as Nd/Nd2O3 from the Nd-CFNC complex using solar-driven electrodeposition. The reusability of CFNC for Nd3+ adsorption makes it a suitable candidate for industrial applications. We recommend future works in green ionic liquids (deep eutectic solvents) as a replacement for acids and organic solvents for dissolving metals and metal-CFNC complexes that could enhance sustainability in various domains of green technology.Author contributions
S. B. has performed most of the experiments, analysis, and drafting of the first version of the manuscript. B. M. helped in ICP MS analysis and also in the experimental setup related to electrodeposition. P. A. A., the Principal investigator, proposed the project, supervised all co-authors, and completely revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. B. would like to thank AMURE/PSAC for his fellowship. PAA acknowledges her funding for this project from the Canadian Foundation for Innovation (CFI) Grant 259754 and the Natural Sciences and Engineering Research Council of Canada (NSERC) Grant 223464. She also acknowledges NSERC CREATE PURE for partial funding for BM. We acknowledge PRIMA Quebec and MITACS for support. The authors would like to thank David Liu from the facility of electron microscopy research at McGill University for running our STEM samples.References
- T. Fishman and T. E. Graedel, Nat. Sustain., 2019, 2, 332–338 CrossRef.
- H. Jin, P. Afiuny, S. Dove, G. Furlan, M. Zakotnik, Y. Yih and J. W. Sutherland, Environ. Sci. Technol., 2018, 52, 3796–3802 CrossRef CAS PubMed.
- V. Balaram, Geosci. Front., 2019, 10, 1285–1303 CrossRef CAS.
- L. Omodara, S. Pitkäaho, E.-M. Turpeinen, P. Saavalainen, K. Oravisjärvi and R. L. Keiski, J. Clean. Prod., 2019, 236, 117573 CrossRef CAS.
- H. Ghimire and P. A. Ariya, Sustain. Chem., 2020, 1, 154–182 CrossRef.
- B. Deng, D. X. Luong, Z. Wang, C. Kittrell, E. A. McHugh and J. M. Tour, Nat. Commun., 2021, 12, 5794 CrossRef CAS PubMed.
- R. H. Estrada-Ruiz, R. Flores-Campos, H. A. Gámez-Altamirano and E. J. Velarde-Sánchez, J. Hazard. Mater., 2016, 311, 91–99 CrossRef CAS PubMed.
- Y. Yang, A. Walton, R. Sheridan, K. Güth, R. Gauß, O. Gutfleisch, M. Buchert, B.-M. Steenari, T. Van Gerven, P. T. Jones and K. Binnemans, J. Sustain. Metall., 2017, 3, 122–149 CrossRef.
- S. Massari and M. Ruberti, Resour. Policy, 2013, 38, 36–43 CrossRef.
- D. Bauer, D. Diamond, J. Li, D. Sandalow, P. Telleen and B. Wanner, Critical Materials Strategy, U.S. Department of Energy, 2010 Search PubMed.
- D. J. Cordier, National Minerals Information Center (Rare Earth Statistics and Information), United States Geological Survey, 2023 Search PubMed.
- K. Salazar and M. K. McNutt, Metal Prices in the United States Through 2010, U.S. Geological Survey, 2012 Search PubMed.
- J. Gambogi, Mineral Commodities Summaries 2015 - Rare Earths, U.S. Geological Survey, 2015 Search PubMed.
- J. Gambogi, Mineral Commodity Summaries 2016- Rare Earths, U.S. Geological Survey, 2016 Search PubMed.
- J. Gambogi, Mineral Commodity Summaries 2017 - Rare Earths, U.S. Geological Survey, 2017 Search PubMed.
- J. Gambogi, Mineral Commodity Summaries 2018 - Rare Earths, U.S. Geological Survey, 2018 Search PubMed.
- J. Gambogi, Mineral Commodity Summaries 2019- Rare Earths, U.S. Geological Survey, 2019 Search PubMed.
- J. Gambogi, Mineral Commodity Summaries 2020- Rare Earths, U.S. Geological Survey, 2020 Search PubMed.
- J. Gambogi, Mineral Commodity Summaries 2021- Rare Earths, U.S. Geological Survey, 2021 Search PubMed.
- D. J. Cordier, Mineral Commodity Summaries 2022 - Rare-Earths, U.S. Geological Survey, 2022 Search PubMed.
- N. Das and D. Das, J. Rare Earths, 2013, 31, 933–943 CrossRef CAS.
- B. Sprecher, R. Kleijn and G. J. Kramer, Environ. Sci. Technol., 2014, 48, 9506–9513 CrossRef CAS PubMed.
- K. N. Han, Minerals, 2020, 10 Search PubMed.
- Z. Zhao, X. Sun, Y. Dong and Y. Wang, ACS Sustainable Chem. Eng., 2016, 4, 616–624 CrossRef CAS.
- T. Ogata, H. Narita and M. Tanaka, Hydrometallurgy, 2015, 152, 178–182 CrossRef CAS.
- B. Gao, Y. Zhang and Y. Xu, Hydrometallurgy, 2014, 150, 83–91 CrossRef CAS.
- S. Riaño and K. Binnemans, Green Chem., 2015, 17, 2931–2942 RSC.
- C. Yao, J. Rare Earths, 2010, 28, 183–188 CrossRef.
- M. Ioelovich, BioResources, 2008, 3, 1403–1418 Search PubMed.
- M. Mariano, N. El Kissi and A. Dufresne, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 791–806 CrossRef CAS.
- M. L. Rahman, M. S. Sarjadi, S. E. Arshad, M. M. Yusoff, S. M. Sarkar and B. Musta, Rare Met., 2019, 38, 259–269 CrossRef CAS.
- M. L. Rahman, T. K. Biswas, S. M. Sarkar, M. M. Yusoff, M. S. Sarjadi, S. E. Arshad and B. Musta, J. Mol. Liq., 2017, 243, 616–623 CrossRef CAS.
- A. Negrea, A. Gabor, C. M. Davidescu, M. Ciopec, P. Negrea, N. Duteanu and A. Barbulescu, Sci. Rep., 2018, 8, 316 CrossRef PubMed.
- H. Tao, N. Lavoine, F. Jiang, J. Tang and N. Lin, Nanoscale Horiz., 2020, 5, 607–627 RSC.
- T. G. M. van de Ven and A. Sheikhi, Nanoscale, 2016, 8, 15101–15114 RSC.
- A. Ismailova, G. Akanova and D. Kamysbayev, Eurasian Chem.–Technol. J., 2021, 1–9 Search PubMed.
- A. Sheikhi, H. Yang, M. N. Alam and T. G. M. van de Ven, J. Visualized Exp., 2016, 113 DOI:10.3791/54133.
- H. Yang, A. Tejado, N. Alam, M. Antal and T. G. M. van de Ven, Langmuir, 2012, 28, 7834–7842 CrossRef CAS PubMed.
- X. Zhang, N. Xiao, H. Wang, C. Liu and X. Pan, Polymers, 2018, 10, 614 CrossRef PubMed.
- M. A. Al-Ghouti and S. S. Dib, J. Environ. Health Sci. Eng., 2020, 18, 63–77 CrossRef CAS PubMed.
- S. V. Durán, B. Lapo, M. Meneses and A. M. Sastre, Nanomaterials, 2020, 10 DOI:10.3390/nano10061204.
- Q. Wang, D. Xie, J. Chen, G. Liu and M. Yu, J. Mater. Sci., 2020, 55, 7084–7094 CrossRef CAS.
- S. Maroufi, R. Khayyam Nekouei and V. Sahajwalla, ACS Sustainable Chem. Eng., 2018, 6, 3402–3410 CrossRef CAS.
- V. Kuzmenko, N. Wang, M. Haque, O. Naboka, M. Flygare, K. Svensson, P. Gatenholm, J. Liu and P. Enoksson, RSC Adv., 2017, 7, 45968–45977 RSC.
- A. Kotani and H. Ogasawara, J. Electron Spectrosc. Relat. Phenom., 1992, 60, 257–299 CrossRef CAS.
- D. Zhao, D. Zhang, Y. Liu, G. Hu, Y. Gou and F.-S. Pan, Rare Met. Mater. Eng., 2017, 46, 289–295 CrossRef CAS.
- P. Burroughs, A. Hamnett, A. F. Orchard and G. Thornton, J. Chem. Soc., Dalton Trans., 1976, 1686–1698 RSC.
- C. Suzuki, J. Kawai, M. Takahashi, A.-M. Vlaicu, H. Adachi and T. Mukoyama, Chem. Phys., 2000, 253, 27–40 CrossRef CAS.
- H. N. Tran, S.-J. You, A. Hosseini-Bandegharaei and H.-P. Chao, Water Res., 2017, 120, 88–116 CrossRef CAS PubMed.
- C. Fan and Y. Zhang, J. Geochem. Explor., 2018, 188, 95–100 CrossRef CAS.
- W. Konicki, A. Hełminiak, W. Arabczyk and E. Mijowska, Chem. Eng. Res. Des., 2018, 129, 259–270 CrossRef CAS.
- A. Miraoui, M. A. Didi and D. Villemin, J. Radioanal. Nucl. Chem., 2016, 307, 963–971 CrossRef CAS.
- S. S. Genannt Bonsmann, T. Walczyk, S. Renggli and R. F. Hurrell, Eur. J. Clin. Nutr., 2008, 62, 336–341 CrossRef CAS PubMed.
- O. Drabek, I. Kipkoech Kiplagat, M. Komarek, V. Tejnecky and L. Boruvka, Soil Water Res., 2015, 10, 172–180 CrossRef.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill, 15th edn, 1999 Search PubMed.
- https://www.usgs.gov/faqs/how-much-natural-water-there (accessed 10 September 2023).
- https://www.usgs.gov/special-topics/water-science-school/science/saline-water-use-united-states (accessed 10 September 2023).
- M. Ganguly and P. A. Ariya, ACS Earth Space Chem., 2019, 3, 1729–1739 CrossRef CAS.
- P. Venkatesan, Z. H. I. Sun, J. Sietsma and Y. Yang, Sep. Purif. Technol., 2018, 191, 384–391 CrossRef CAS.
- E. Bourbos, A. Karantonis, L. Sygellou, I. Paspaliaris and D. Panias, Metals, 2018, 8, 803 CrossRef CAS.
- B. Zhang, L. Wang, Y. Liu, Y. Zhang, L. Zhang and Z. Shi, Sep. Purif. Technol., 2021, 276, 119416 CrossRef CAS.
- R. Qadeer, J. Radioanal. Nucl. Chem., 2005, 265, 377–381 CrossRef CAS.
- Y.-J. Tu, S.-C. Lo and C.-F. You, Chem. Eng. J., 2015, 262, 966–972 CrossRef CAS.
- M. Najafi, A. Sadeghi Chevinli, V. Srivastava and M. Sillanpää, J. Chem. Eng. Data, 2019, 64, 3105–3112 CrossRef CAS.
- P. Zhang, Y. Wang, D. Zhang, H. Bai and V. V. Tarasov, RSC Adv., 2016, 6, 30384–30394 RSC.
- C. E. Unsworth, C. C. Kuo, A. Kuzmin, S. Khalid and D. Saha, ACS Appl. Mater. Interfaces, 2020, 12, 43180–43190 CrossRef CAS PubMed.
- J. Roosen and K. Binnemans, J. Mater. Chem. A, 2014, 2, 1530–1540 RSC.
- M. A. Kucuker, N. Wieczorek, K. Kuchta and N. K. Copty, PLoS One, 2017, 12, e0175255 CrossRef PubMed.
- L. L. Cheng, X. D. Wei, X. L. Hao, D. Ruan and S. M. Yu, Adv. Mater. Res., 2014, 881–883 Search PubMed.
- J. Roosen, J. Pype, K. Binnemans and S. Mullens, Ind. Eng. Chem. Res., 2015, 54, 12836–12846 CrossRef CAS.
- C. M. Babu, K. Binnemans and J. Roosen, Ind. Eng. Chem. Res., 2018, 57, 1487–1497 CrossRef CAS.
- H.-J. Park and L. L. Tavlarides, Ind. Eng. Chem. Res., 2010, 49, 12567–12575 CrossRef CAS.
- J. Xu, R. Koivula, W. Zhang, E. Wiikinkoski, S. Hietala and R. Harjula, Hydrometallurgy, 2018, 175, 170–178 CrossRef CAS.
- https://www.indexbox.io/search/wood-pulp-price-canada/ (accessed 5 December 2023).
- https://cfs.nrcan.gc.ca/statsprofile/production-and-investment/ (accessed 5 December 2023).
Footnote |
| † Electronic supplementary information (ESI) available: Containing all the experimental details as well as additional experimental results. See DOI: https://doi.org/10.1039/d3gc03756h |
| This journal is © The Royal Society of Chemistry 2024 |