
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt nanoparticle-catalysed N-alkylation of amides with alcohols†
Rui
Ma
a,
Jie
Gao
a,
Lan
Zhang
 b,
Ning
Wang
b,
Yue
Hu
a,
Stephan
Bartling
a,
Henrik
Lund
a,
Sebastian
Wohlrab
*a,
Rajenahally V.
Jagadeesh
*ac and
Matthias
Beller
*a
b,
Ning
Wang
b,
Yue
Hu
a,
Stephan
Bartling
a,
Henrik
Lund
a,
Sebastian
Wohlrab
*a,
Rajenahally V.
Jagadeesh
*ac and
Matthias
Beller
*a
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: Sebastian.Wohlrab@catalysis.de; jagadeesh.rajenahally@catalysis.de; matthias.beller@catalysis.de
bFaculty of Environment and Life, Beijing University of Technology, 100124 Beijing, China
cNanotechnology Centre, Centre for Energy and Environmental Technologies, VŠB–Technical University of Ostrava, Ostrava-Poruba, Czech Republic
First published on 8th December 2023
Abstract
A protocol for efficient N-alkylation of benzamides with alcohols in the presence of cobalt-nanocatalysts is described. Key to the success of this general methodology is the use of highly dispersed cobalt nanoparticles supported on carbon, which are obtained from the pyrolysis of cobalt(II) acetate and o-phenylenediamine as a ligand at suitable temperatures. The catalytic material shows a broad substrate scope and good tolerance to functional groups. Apart from the synthesis of a variety of secondary amides (>45 products), the catalyst allows for the conversion of more challenging aliphatic alcohols and amides, including biobased and macromolecular amides. The practical applicability of the catalyst is underlined by the successful recycling and reusability.
Introduction
The amide bond is one of the most fundamental functional groups in organic chemistry. It plays a central role in the formation and preservation of biological systems.1 Amides are generally synthesized by condensation of carboxylic acids with amines,2,3 or by the reaction between amides and aryl or aliphatic halides (Fig. 1).3,4 However, these established methods often exhibit a poor atom economy and inevitably lead to the formation of (stoichiometric amounts of) waste. In contrast to organic halides, alcohols are broadly available raw materials including many renewable resources. Hence, the catalytic N-alkylation of primary amides with alcohols attracted significant attention in recent years (Fig. 1).4–10 From a mechanistic point of view, the method involves an initial dehydrogenation of the alcohol to the corresponding aldehyde, followed by condensation with the amide.8 Then, the newly formed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond gets hydrogenated by a metal-hydride to give the N-alkylated amide, and water is formed as the sole by-product. Unfortunately, most of these transformations require relatively harsh reaction conditions.5,9
N bond gets hydrogenated by a metal-hydride to give the N-alkylated amide, and water is formed as the sole by-product. Unfortunately, most of these transformations require relatively harsh reaction conditions.5,9
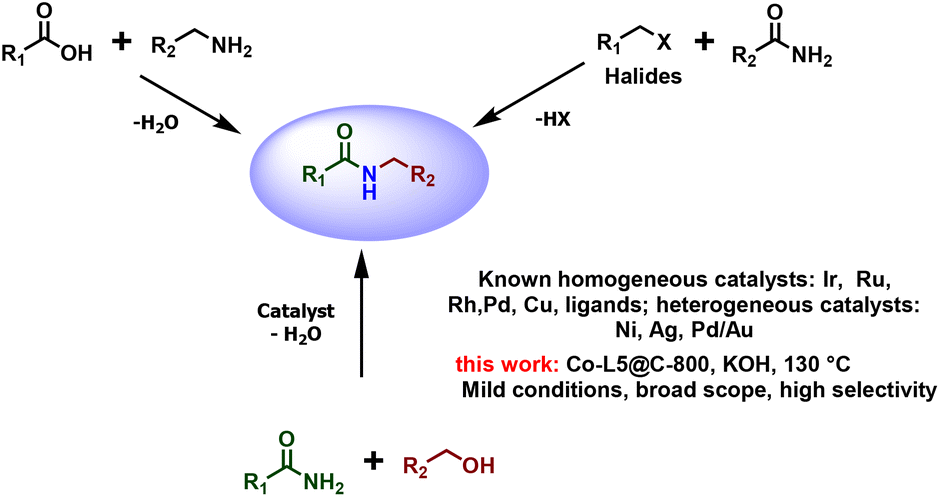 | ||
| Fig. 1 General methods for amide synthesis and our presented approach. | ||
To date, homogeneous metal complexes based on Ir,11 Ru,12 Pd,13 Ni,5 and Cu14 dominate as catalysts for N-alkylation of primary amides with alcohols (Fig. 1). Although these homogeneous systems with suitably designed ligands are usually selective and active, their main drawbacks are the costs and the difficulties in separating and recovering the catalysts from the reaction mixture. In contrast, heterogeneous materials are generally more durable and often easier to separate from products. Over the past decades, only a few heterogeneous systems based on Ag,4 Pd/Au,15 and Ru15 were developed, and all of them required high temperatures (Fig. 1). Therefore, the preparation of metal-based nanomaterials that function under milder conditions continues to be a challenge in the catalytic functionalization of amides.
Here, we show that N-alkylation of primary amides with alcohols proceeds in the presence of non-noble and reusable cobalt-based nanocatalysts under comparably mild conditions. Following an operationally simple protocol, a series of functionalized and structurally diverse secondary amides were synthesized.
Results and discussion
Catalyst preparation and evaluation
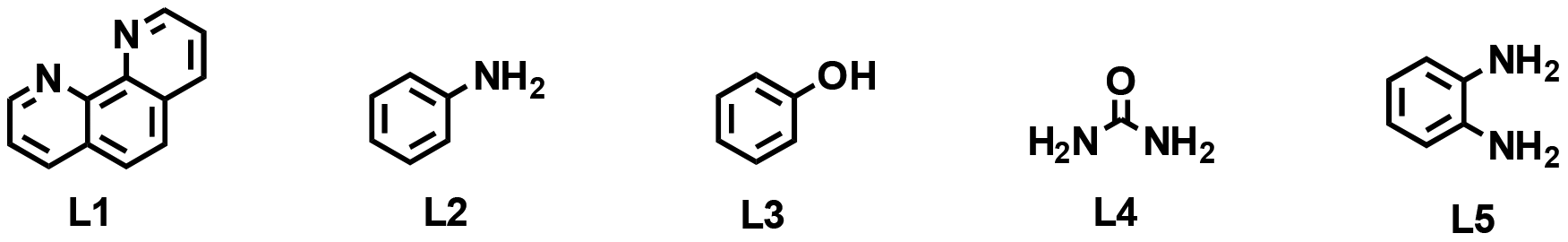
The general synthesis of cobalt catalysts followed our previously developed protocols16–20 and was initiated by dissolving cobalt(II) nitrate hexahydrate and different types of nitrogen ligands in methanol solution in the presence of various supports. After stirring overnight and removing the solvent, the obtained metal complexes were directly immobilized on the surface of the respective support (ZSM-5, TiO2, Al2O3, and carbon). The resulting materials were pyrolyzed under argon at different temperatures to obtain potential catalysts (as shown in Table 1). The detailed catalyst preparation procedure is described in the ESI.† The obtained materials are labelled as Co–L@support-T, where Co, L and T represent the type of metal (cobalt), ligand (L), and pyrolysis temperature (T), respectively.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Metal content (wt%) | Conversion (%) | Yield (%) |
| a Reaction conditions: 0.5 mmol of benzamide, 0.55 mmol of benzyl alcohol, 60 mg of catalyst, 0.3 mmol of KOH, 1 atm argon, 3 mL of toluene, 115 °C, and 24 h. b Same as [a] at 130 °C, 24 h. c Same as [b] without a catalyst. d Same as [b] without a base. Yields were determined by GC based on benzamide using n-hexadecane as the standard. n.r means no reaction. The effect of KOH on the reaction is shown in Fig. S1.† | ||||
| 1a | Co-L1@ZSM-5-800 | n.r | n.r | |
| 2a | Co-L1@TiO2-800 | 7 | 7 | |
| 3a | Co-L1@Al2O3-800 | 40 | 40 | |
| 4a | Co-L1@MgO–Al2O3-800 | 46 | 46 | |
| 5a | Co-L1@C-800 | 0.889 | 80 | 80 |
| 6a | Co@C-800 | 0.921 | n.r | n.r |
| 7a | Co-L2@C-800 | 0.921 | 49 | 49 |
| 8a | Co-L3@C-800 | 0.892 | n.r | n.r |
| 9a | Co-L4@C-800 | 0.876 | 55 | 55 |
| 10a | Co-L5@C-800 | 0.937 | 87 | 87 |
| 11a | Co-L5@C-400 | 0.880 | 53 | 53 |
| 12a | Co-L5@C-600 | 0.880 | 83 | 83 |
| 13a | Co-L5@C-1000 | 0.896 | 66 | 66 |
| 14b | Co-L5@C-800 | 0.937 | 99 | 99 |
| 15c | Without catalyst | n.r | n.r | |
| 16d | Without base | n.r | n.r | |
|
||||

In our initial investigations, we tested the influence of the support on the formation of cobalt nanoparticles for N-alkylation of benzamide with benzyl alcohol as a benchmark reaction. After testing different bases (Table S1†), KOH was chosen as the optimal one and it was applied in all further experiments. Compared with methanol, ethanol, THF, and n-hexane, toluene gave the best results as the reaction solvent (Table S2†). As shown in Table 1, the native properties of the catalyst could strongly alter the reactivity and selectivity.21 The influence of the variation in the ligand and cobalt content is shown in Table S3.† Based on these results, carbon powder was selected as the most suitable carrier material among all the supports tested (Table 1, entries 1–5). Afterwards, the influence of the selected organic ligands was tested since ligand-free material, Co@C-800, showed no activity in the model reaction (Table 1, entry 6). Comparison of the catalytic performance of materials generated using diverse ligands (L1: phenanthroline, L2: aniline, L3: phenol, L4: urea, and L5: o-phenylenediamine) showed that Co-L5@C-800 gave the highest product yield of 87% (Table 1, entries 5 and 7–10). Next, the effect of the pyrolysis temperature on the catalytic performance was studied. Compared to Co-L5@C-800, the materials pyrolyzed at 400, 600 and 1000 °C exhibited low or moderate catalytic activity (Table 1, entries 10–13). Notably, a 99% yield of the desired product was realized by running the model reaction at 130 °C (Table 1, entry 14). As expected, in the absence of a base or catalyst, no product is formed.
To gain a deeper understanding of this transformation, especially the interplay between the catalyst (Co-L5@C-800) and the base (KOH), a kinetic profile of the model reaction was obtained. As shown in Fig. 2a, in the first 5 hours, the two reactants are mainly consumed, and the yield of the desired secondary amide increased to 90%. Then, the reaction rate slows down approaching nearly full conversion of benzyl alcohol after 22 h. Over the course of the reaction, 3–10% of benzaldehyde was observed as an intermediate, which is formed by the dehydration of benzyl alcohol.22 To understand the effect of Co-L5@C-800 and KOH, control reactions were performed (Fig. 2b).
 | ||
| Fig. 2 Kinetic experiment and control reactions. (a) Kinetic profile of the model reaction. Reaction conditions: 0.5 mmol of benzamide, 0.55 mmol of benzyl alcohol, 60 mg of catalyst, 1 atm argon, 3 mL of toluene, 130 °C, and GC yield. (b) Coupling reaction of benzamide and benzaldehyde to form secondary amide. Reaction conditions: same as (a) with 0.5 mmol of benzamide, 0.55 mmol of benzaldehyde, and 2 h. (c) Proposed reaction sequence. | ||
Experiments in the presence of only Co-L5@C-800 or KOH showed no/minor reactivity, while the desired product yield increased to 77% when both base and catalyst were used. The kinetic profile and control experiments are in agreement with the previously proposed mechanism (Fig. 2c).23,24 This N-alkylation reaction of benzamide starts with a base-promoted dehydrogenation of benzyl alcohol, which occurs on the catalyst surface, to produce benzaldehyde and H–[Co]–H species on the surface (first reaction step).25,26 Then, nucleophilic addition of benzamide to benzaldehyde generated the respective N-benzoylimine. This second step is likely promoted by a base, too, and represents the rate-determining reaction step due to the poor nucleophilicity of primary amides. Finally, cobalt-catalyzed reduction of this imide occurs (third reaction step).23
Characterization of selected materials
To elucidate reasons for the markedly different catalytic activities of the prepared Co–L@C-T catalysts, the effects of ligand and pyrolysis temperature on the structures of Co@C-800, Co-L5@C-400, Co-L5@C-600, Co-L5@C-800, and Co-L5@C-1000 were investigated in detail by transmission electron microscopy (TEM), X-ray powder diffraction (XRD), Brunauer–Emmett–Teller (BET) analysis, and X-ray photoelectron spectroscopy (XPS).Although the powder X-ray diffraction (XRD) data show broad peaks of the support material27 (Fig. S2†), Co-containing phases can be suspected by a peak around 37° 2θ, which is assigned to Co-oxide formation.28–30 However, due to low loading and overlapping peaks, its assignment to different Co-oxides was not successful (Fig. S2†). If the pyrolysis temperature is increased to 800 °C, metallic fcc-Co appeared in the material as seen by its most intense Bragg peaks at 44.2 (111), 51.5 (200) and 75.8° (220).31 Notably, there are only negligible differences in diffraction data of Co@C-800 and Co-L5@C-800 (Fig. S3†).
The presence of organic nitrogen ligands during the thermal catalyst formation induces the formation of some carbon protective layers on the outer surface of metallic Co, which make Co-L5@C-800 more stable to oxidation. Although the signal of a metallic Co0 phase is also observed on Co@C-800, TEM analysis and XPS spectra showed that the surface of these particles is oxidized (Fig. 3d–i and S3†). Consequently, the Co@C-800 material is more sensitive to oxidation.32
 | ||
| Fig. 3 TEM analysis of Co@C-800. (a and b) TEM-HAADF images, (c) EDX elemental mapping image of Co, and (d–i) identified crystal planes of the Co nanoparticles. | ||
The HAADF-TEM images of Co@C-800 revealed that cobalt nanoparticles with a size of 45–50 nm are randomly distributed over the surface of the support material (Fig. 3a). EDX mapping images clearly indicate that cobalt nanoparticles are randomly distributed on the surface of the materials (Fig. 3b–c and Table S4†). In the regions at high cobalt magnification (Fig. 3d–i), the nature of the metal nanoparticles is revealed. As shown in the image of Fig. 3d, labeled using red and blue frames, the apparent lattice distance in the red region was found to be 0.204 nm, corresponding to the (111) plane of metallic Co (Fig. 3d and e).33 Besides, the (111) plane of CoO with a distance of 0.254 nm is also found in the blue region (Fig. 3d and e).34 Moreover, the (222) plane of Co3O4 with a d-spacing of 0.235 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 is also found in the yellow region (Fig. 3f–i). These indicate that the surface cobalt particles in Co@C-800 are in the oxidized state, too. Thus, we propose a core–shell structure of the cobalt nanoparticles in Co@C-800, where the core consists of metallic Co with an oxide shell.
35 is also found in the yellow region (Fig. 3f–i). These indicate that the surface cobalt particles in Co@C-800 are in the oxidized state, too. Thus, we propose a core–shell structure of the cobalt nanoparticles in Co@C-800, where the core consists of metallic Co with an oxide shell.
High angle annular dark field (HAADF) images of Co-L5@C-800 show the Co particles as bright dots with a smaller average diameter of about 9 nm compared to the ligand-free produced pendant (Fig. 4a and b). Obviously, the formation of the Co-L5 complex prevented the growth of cobalt nanoparticles during the pyrolysis process.36
 | ||
| Fig. 4 TEM analysis of Co-L5@C-800. (a and b) TEM-HAADF images, (c) EDX elemental mapping image of Co, and (d–i) identified crystal planes of the Co nanoparticles. | ||
High-magnification TEM images (Fig. 4h and i) show lattice fringes with a d-spacing of 0.195 nm, corresponding to the (111) lattice plane of metallic cobalt.33 Besides, cobalt oxides exist in this sample as indicated by the lattice fringes with a d-spacing of 0.186 nm, which correspond to the (331) lattice plane of Co3O4 (Fig. 4f and g).37 Due to the presence of a ligand, some cobalt nanoparticles are encapsulated by few carbon layers (Fig. 4d). The selected area of the nanoparticle core in Fig. 4d shows the lattice fringes with a d-spacing of 0.171 nm, corresponding to the (200) lattice plane of metallic cobalt (Fig. 4e).38 In addition, the shell shown in Fig. 4d possesses lattice fringes with a d-spacing of 0.337 nm, corresponding to the (002) lattice plane of carbon (Fig. 4e).39 Furthermore, the corresponding EDX mapping images of Fig. 4b and c clearly indicate that cobalt nanoparticles are randomly distributed on the surface of the materials, and cobalt species could be clearly detected (Table S5†).
The TEM images of Co-L5@C-400, Co-L5@C-600 and Co-L5@C-1000 are shown in Fig. S5–S7.† Interestingly, the materials prepared at a lower pyrolysis temperature, Co-L5@C-400 and Co-L5@C-600, show the formation of cobalt nanoparticles, too (Fig. S5 and S6†). It is worth mentioning that compared with Co-L5@C-800, the particle size of Co-L5@C-1000 is increased to 20.5 nm due to the relatively higher pyrolysis temperature (Fig. S7†).
Next, we performed XPS investigations to obtain further insights into the surface composition and oxidation state of cobalt in these materials. The surface composition of the different catalysts is shown in Table S6† and reveals C, O, Co, and N as the main elements. Small amounts of S and Si can be found as well, which are typical residues in carbon supports. The Co 2p and N 1s spectra of the Co-L5 temperature series are shown in Fig. S4.† All these materials display the peaks of cobalt oxide (due to the overlap of CoO and Co3O4, CoxOy is used to represent cobalt oxide) at around 780.4 eV.40 For Co-L5@C-400 (Fig. S4a†), the Co 2p3/2 peak is shifted to slightly higher binding energies at about 781.2 eV. Together with the pronounced satellite features, this suggests Co2+ as the main oxidation state.40 For higher pyrolysis temperatures, the main Co 2p3/2 peaks are observed at typical binding energies of oxidized Co at about 780.4 eV.40 For Co-L5@C-600 (Fig. S4a†), the less pronounced satellite features indicate the additional presence of Co3O4.40 For the pyrolysis temperatures 800 and 1000 °C, an additional peak at 778.4 eV appears which is characteristic of metallic cobalt.41 XPS data indicated that the surface concentration of Co species decreases with increasing pyrolysis temperature from 0.3 to 0.1 at% obviously due to an increased carbon coverage (Table S6†). In addition, the relative concentration of metallic cobalt increases, too (Table S7†). In comparison, the reference sample Co@C-800 shows no metallic Co at the surface (see Fig. S4a,†) even though it could be detected in the bulk material by XRD (see the XRD section Fig. S2-3†). These results underline the importance of the added ligand to form and protect near surface Co species. The nitrogen concentration also decreased with increase in pyrolysis temperature from 1.2 at% for 400 °C to 0.1 at% for 1000 °C (Table S6†). The N 1s XPS spectra shown in Fig. S4b† are deconvoluted showing four N species: pyridinic N (398.5 eV), pyrrolic N (399.8 eV), graphitic N (401.2 eV), and oxidized pyridinic-N (403.4 eV).42,43 Going from low to higher pyrolysis temperatures, the observed N species change from predominantly pyridinic-N and pyrrolic-N species44 to graphitic-N, which are incorporated into the support lattice.44
Later, the textures of these materials were measured by N2 physisorption. The specific surface area (SBET) is summarized in Fig. S8.† Among all the samples, Co@C-800 has the largest specific surface area of 201 m2 g−1. With the increase of pyrolysis temperature from 400 to 1000 °C, the surface area of Co-L5@C-T samples increased from 121 m2 g−1 to 164 m2 g−1.
Synthetic applications and substrate scope
Under the optimized reaction conditions, we explored the scope of Co-L5@C-800 catalyzed N-alkylation of primary amides with alcohols. As shown in Scheme 1, several functionalized and structurally diverse alcohols underwent the desired reactions with benzamide to produce the corresponding products in good to excellent yields.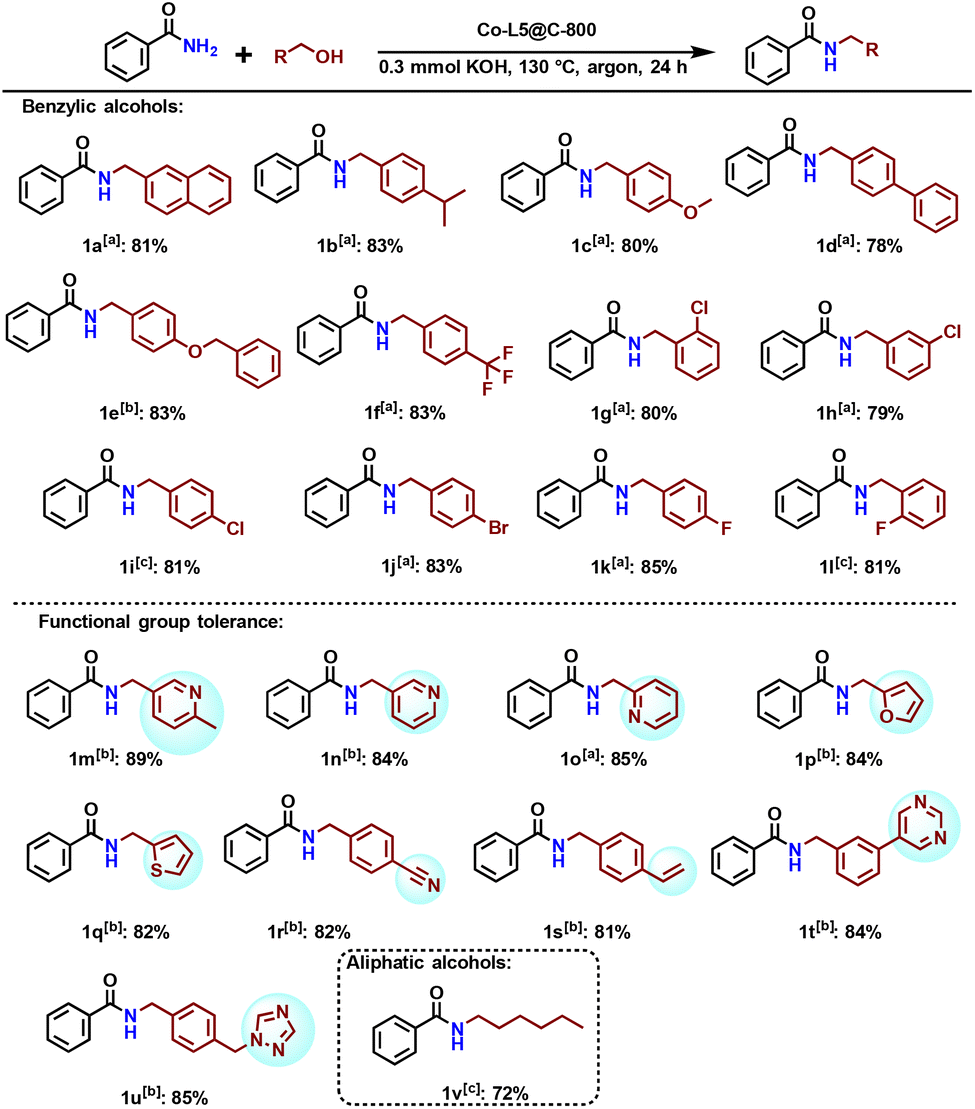 | ||
| Scheme 1 Co-L5@C-800 catalyzed N-alkylation of benzamide with different alcohols. Reaction conditions: a0.5 mmol of benzamide, 0.55 mmol of alcohol, 60 mg of Co-L5@C-800 (1.9 mol% Co), 0.3 mmol of KOH, 3 mL of toluene, 130 °C, 24 h, and 1 atm argon. Isolated yields. bSame as [a] at 140 °C. cSame as [a], GC yield. | ||
The substitution on the aryl ring of benzyl alcohol has no noticeable effect on the efficiency of the reaction. With both electron-withdrawing and electron-donating substituents, the N-alkylated amides are afforded in 78%–85% yield (Scheme 1, 1a–1l). For catalyst applications in advanced organic synthesis, achieving a high degree of chemoselectivity is important, yet often challenging. To showcase this aspect, alcohols containing various functional groups were tested in our protocol. Interestingly, the C–C double bond and nitrile are well tolerated (Scheme 1, 1s and 1r). Furthermore, heterocyclic benzyl alcohols also reacted well and produced the alkylated amides in 81%–89% yield. 1-Hexanol as an example of aliphatic alcohols led to the corresponding amide in good yield, too (Scheme 1, 1v).
The standard reaction protocol is also applicable using diverse amides. Benzamides with both electron-withdrawing and electron-donating substituents reacted well with benzyl alcohol, and the corresponding secondary amides were obtained in 79%–87% yield (Scheme 2, 2a–2i). The biological amides can also react well with benzyl alcohol in the presence of this cobalt system to obtain unreported secondary amides in good yields (Scheme 2, 2j), which provides a method for synthesizing new products. Next, we explored the general applicability for more challenging aliphatic amides. By using this catalytic method, secondary amides could also be produced from less active alkyl amides (Scheme 2, 2k–2l). Similarly, heterocycles were well tolerated, and this system showed excellent regioselectivity in the substrates containing both amino and nitrile groups (Scheme 2, 2m and 2v). Screening of various primary amides and alcohols revealed that the alkylation of amides catalyzed by this cobalt system worked well and provided secondary amides in good to excellent yields (Schemes 1 and 2). To investigate the stability of this novel catalyst, recycling experiments were performed under standard conditions. Indeed, Co-L5@C-800 can be reused conveniently and no significant loss of catalytic activity is observed even after five times (Fig. 5). The recycled catalyst was also characterized to explore the structural differences by XRD. Compared with fresh material, no significant structural difference was seen in the XRD pattern (Fig. S9b†).
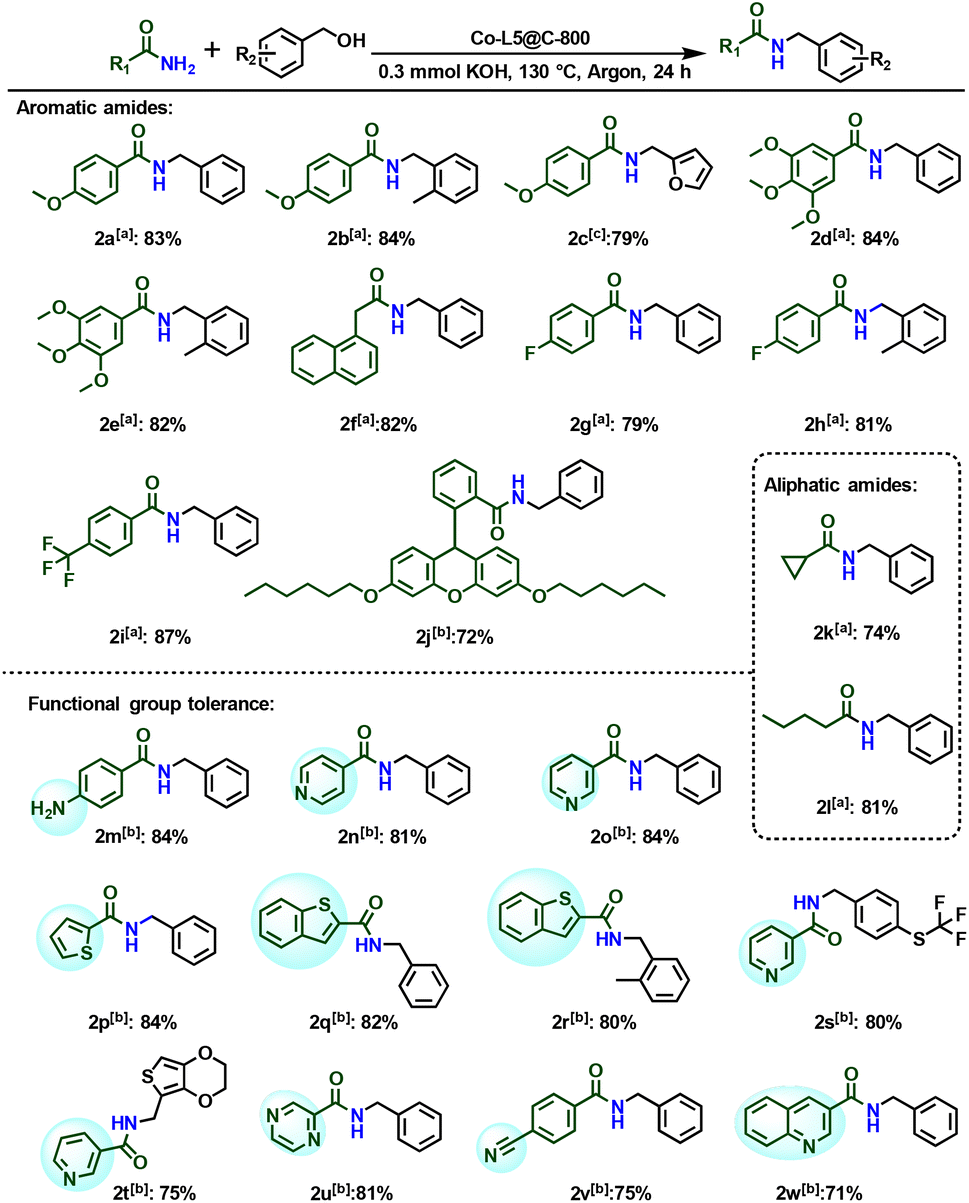 | ||
| Scheme 2 Co-L5@C-800 catalyzed N-alkylation of different amides with benzylic alcohol. Conditions: a0.5 mmol of amide, 0.55 mmol of benzyl alcohol, 60 mg of Co-L5@C-800 (1.9 mol% Co), 0.3 mmol of KOH, 3 mL of toluene, 130 °C, 24 h, and in 1 atm argon. Isolated yields. bSame as [a] at 140 °C. cSame as [a], GC yield. | ||
 | ||
| Fig. 5 Reusability of Co-L5@C-800 in N-alkylation of benzamide with benzyl alcohol. Reaction conditions: 0.5 mmol benzamide, 0.55 mmol benzyl alcohol, 60 mg Co-L5@C-800 (1.9 mol% Co), 0.3 mmol KOH, 3 mL toluene, 130 °C, 24 h, in 1 atm argon. | ||
Conclusions
We reported a novel nano-structured Co-based catalyst for practical and convenient N-alkylation of primary amides with alcohols. Introduction of nitrogen ligands and pyrolysis treatment creates a highly stable and reusable Co-nanoparticles, which activates N-alkylation of primary amides with alcohols under mild conditions. The optimal catalyst (Co-L5@C-800) showed good to excellent activity and selectivity for the synthesis of functionalized and structurally diverse secondary amides.Author contributions
R. V. J., S. W. and M. B. supervised the project. R. M., R. V. J., S. W. and M. B. planned and developed the project. R. M., S. W. and R. V. J. designed the experiments. R. M. and J. G. prepared catalysts and performed all catalytic experiments. J. G. and Y. H. carried out reproducibility experiments. L. Z. and N. W. performed the TEM measurements and analysis. S. B. and H. L. performed XPS and XRD analysis. R. M., J. G., S. W., R. V. J. and M. B. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the European Research Council (EU project 670986-NoNaCat) and the State of Mecklenburg-Vorpommern for financial and general support. We thank the analytical team of the Leibniz-Institut für Katalyse e.V. for their excellent service. We thank Mr Reinhard Eckelt for performing BET. Rui Ma, Jie Gao and Yue Hu thank the China Scholarship Council (CSC) for the fellowship.References
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- X. Dai and F. Shi, Org. Biomol. Chem., 2019, 17, 2044–2054 RSC.
- J. Das and D. Banerjee, J. Org. Chem., 2018, 83, 3378–3384 CrossRef CAS PubMed.
- C. Chen, F. Verpoort and Q. Wu, RSC Adv., 2016, 6, 55599–55607 RSC.
- X. J. Wu, H. J. Wang, Z. Q. Yang, X. S. Tang, Y. Yuan, W. Su, C. Chen and F. Verpoort, Org. Chem. Front., 2019, 6, 563–570 RSC.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed.
- S. Kerdphon, X. Quan, V. S. Parihar and P. G. Andersson, J. Org. Chem., 2015, 80, 11529–11537 CrossRef CAS PubMed.
- A. Wang, Y. Xie, J. Wang, D. Shi and H. Yu, Chem. Commun., 2022, 58, 1127–1130 RSC.
- N. Wang, X. Zou, J. Ma and F. Li, Chem. Commun., 2014, 50, 8303–8305 RSC.
- Y. Watanabe, T. Ohta and Y. Tsuji, Bull. Chem. Soc. Jpn., 1983, 56, 2647–2651 CrossRef CAS.
- X. C. Yu, L. Jiang, Q. Li, Y. Y. Xie and Q. Xu, Chin. J. Chem., 2012, 30, 2322–2332 CrossRef CAS.
- P. Xu, F.-S. Han and Y.-H. Wang, Adv. Synth. Catal., 2015, 357, 3441–3446 CrossRef CAS.
- G. C. Y. Choo, H. Miyamura and S. Kobayashi, Chem. Sci., 2015, 6, 1719–1727 RSC.
- B. Sardar, R. Jamatia, A. Samanta and D. Srimani, J. Org. Chem., 2022, 87, 5556–5567 CrossRef CAS PubMed.
- R. V. Jagadeesh, T. Stemmler, A. E. Surkus, H. Junge, K. Junge and M. Beller, Nat. Protoc., 2015, 10, 548–557 CrossRef CAS PubMed.
- J. Gao, L. Feng, R. Ma, B.-J. Su, A. M. Alenad, Y. Liu, M. Beller and R. V. Jagadeesh, Chem. Catal., 2022, 2, 178–194 CrossRef CAS.
- J. Gao, R. Ma, L. Feng, Y. Liu, R. Jackstell, R. V. Jagadeesh and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 18591–18598 CrossRef CAS PubMed.
- J. Gao, R. Ma, F. Poovan, L. Zhang, H. Atia, N. V. Kalevaru, W. Sun, S. Wohlrab, D. A. Chusov, N. Wang, R. V. Jagadeesh and M. Beller, Nat. Commun., 2023, 14, 5013 CrossRef CAS PubMed.
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M. M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- P. D. Coan, M. B. Griffin, P. N. Ciesielski and J. W. Medlin, J. Catal., 2019, 372, 311–320 CrossRef CAS.
- K. Sun, H. Shan, G. P. Lu, C. Cai and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 25188–25202 CrossRef CAS PubMed.
- A. J. Watson, A. C. Maxwell and J. M. Williams, J. Org. Chem., 2011, 76, 2328–2331 CrossRef CAS PubMed.
- K. Lida, T. Miura, J. Ando and S. Saito, Org. Lett., 2013, 15, 1436–1439 CrossRef PubMed.
- T. Miura, O. Kose, F. Li, S. Kai and S. Saito, Chem. – Eur. J., 2011, 17, 11146–11151 CrossRef CAS PubMed.
- B. J. Hwang, J. Phys. Chem. C, 2007, 111, 15267–15276 CrossRef CAS.
- L. Belles, C. Moularas, S. Smykała and Y. Deligiannakis, Nanomaterials, 2021, 11, 925 CrossRef CAS PubMed.
- K. Deori and S. Deka, CrystEngComm, 2013, 15, 8465–8474 RSC.
- T. Rao Penki, D. Shanmughasundara, B. Kishore and N. Munichandraiah, Adv. Mater. Lett., 2014, 5, 184–190 CrossRef.
- J. Jansson, A. E. C. Palmqvist, E. Fridell, M. Skoglundh, L. Österlund, P. Thormählen and V. Langer, J. Catal., 2002, 211, 387–397 CrossRef CAS.
- A. K. Datye, J. Bravo, T. R. Nelson and L. Pfefferle, Appl. Catal., A, 2000, 198, 179–196 CrossRef CAS.
- X. Deng, Y. Yang, L. Wang, X. Z. Fu and J. L. Luo, Adv. Sci., 2021, 8, 1–9 Search PubMed.
- X. Sun, Y. Lu, T. Li, S. Zhao, Z. Gao and Y.-Y. Song, J. Mater. Chem. A, 2019, 7, 372–380 RSC.
- J. Mu, L. Zhang, G. Zhao and Y. Wang, Phys. Chem. Chem. Phys., 2014, 16, 15709–15716 RSC.
- B. Y. Guan, X. Y. Yu, H. B. Wu and X. W. D. Lou, Adv. Mater., 2017, 29, 1703614 CrossRef PubMed.
- J. O. D. Malafatti, A. J. Moreira, E. C. Paris, L. J. Cardenas Flechas, O. A. P. Pereira and M. Rincón Joya, Catalysts, 2022, 12, 1199 CrossRef CAS.
- X. Peng, L. Wang, L. Hu, Y. Li, B. Gao, H. Song, C. Huang, X. Zhang, J. Fu, K. Huo and P. K. Chu, Nano Energy, 2017, 34, 1–7 CrossRef CAS.
- J. Chen, X. Yuan, F. Lyu, Q. Zhong, H. Hu, Q. Pan and Q. Zhang, J. Mater. Chem. A, 2019, 7, 1281–1286 RSC.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau and A. R. Gerson, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- L. Chen, Y. Zhang, H. Wang, Y. Wang, D. Li and C. Duan, Nanoscale, 2018, 10, 21019–21024 RSC.
- X. Yang, C. Chen, Z. Zhou and S. Sun, Acta Phys.-Chim. Sin., 2019, 35, 472–485 CAS.
- B. J. Matsoso, K. Ranganathan, B. K. Mutuma, T. Lerotholi, G. Jones and N. J. Coville, RSC Adv., 2016, 6, 106914–106920 RSC.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03286h |
| This journal is © The Royal Society of Chemistry 2024 |