
Spatiotemporal dynamics of cellulose during enzymatic hydrolysis studied by infrared spectromicroscopy†
Tina
Jeoh
 *a,
Jennifer Danger
Nill‡§
a,
Wujun
Zhao§¶
b,
Sankar Raju
Narayanasamy§||
b,
Liang
Chen**
b and
Hoi-Ying N.
Holman
*b
*a,
Jennifer Danger
Nill‡§
a,
Wujun
Zhao§¶
b,
Sankar Raju
Narayanasamy§||
b,
Liang
Chen**
b and
Hoi-Ying N.
Holman
*b
aBiological and Agricultural Engineering, University of California Davis, Davis, CA, USA. E-mail: tjeoh@ucdavis.edu
bBerkeley Synchrotron Infrared Structure Biology (BSISB), Lawrence Berkeley National Lab (LBNL), Berkeley, CA, USA. E-mail: hyholman@lbl.gov
First published on 28th November 2023
Abstract
Cellulose, a sustainable source of fermentable glucose and nanomaterials, exists as highly-ordered, solvent-inaccessible fibrils held by intra- and intermolecular hydrogen bonds. Cellulose hydrolysis rates and extents are limited by the availability and accessibility of productive cellulase binding sites at the water–cellulose interface. There is a need to understand how spatial heterogeneity of celluloses impacts hydrolysis kinetics. We report a real-time, in situ infrared spectromicroscopy study of enzymatic cellulose hydrolysis in buffer with micrometer-scale spatial mapping. Algal cellulose depletion by a purified cellobiohydrolase Cel7A was tracked as time-resolved decreases in the absorption peak intensities of the glycosidic bond (1161 cm−1), C2–O2 (1112 cm−1), C3–O3 (1059 cm−1), and C6–O6 rotamers (1034 cm−1, 1013 cm−1, and 997 cm−1). Depletion kinetics varied spatially, with peak intensities decreasing to zero in some areas and plateauing in others. Hydration impacted cellulose ordering where C6–O6 and C3–O3 peaks were sharp, narrow, and centered at higher frequencies when hydrated, but broader, less well-defined and centered at lower frequencies when dried. Temporal trends of the Lateral Order Index (LOI), Total Crystallinity Index (TCI), and Hydrogen Bonding Index (HBI) indicated that hydrolysis by Cel7A preferentially removed less extensively hydrogen bonded cellulose, without significantly affecting the overall crystallinity of highly ordered cellulose.
1. Introduction
The circular bioeconomy—wherein renewable feedstocks are sustainably utilized to offset energy and valuable materials conventionally produced with petroleum—is key to achieving worldwide carbon neutrality.1,2 Cellulose from lignocellulosic biomass is the most abundant biopolymer on the planet that can be a renewable, sustainable feedstock for fuels and chemical production. On the one hand, complete depolymerization of cellulose yields soluble glucose that can be fermented into biofuels or biochemicals. On the other hand, cellulose can be processed into nanofibrils and nanocrystals with superior mechanical properties for use with advanced biomaterials.3–5 Environmentally sustainable and tunable processes to achieve saccharification and/or nanomaterial production are best achieved by enzyme-mediated reactions.6–8 However, controlling enzyme catalyzed reaction kinetics has long been a challenge. Cellulose hydrolysis is characterized by rapidly declining reaction rates despite the remaining abundance of cellulose in the reaction, which has been postulated to be limited by the accessibility of cellulose to cellulases.9–20 The interaction of cellulase enzymes with cellulose is an interfacial reaction on structurally heterogeneous fibrils.13,18,21 An understanding of spatial and temporally resolved changes of cellulose structural properties during enzyme hydrolysis reaction is needed for controlling bulk reaction kinetics.Advances in in situ, spatial and time-resolved analytical methods have provided insights into cellulase–cellulose and cellulase–lignocellulosic biomass interactions. For example, atomic force microscopy (AFM) has advanced knowledge in microstructural changes in cellulose and the dynamics of cellulolytic enzymes at cellulose interfaces;21–23 super-resolution single molecule imaging by total internal reflectance fluorescence (TIRF) microscopy revealed interaction dynamics of cellulases at cellulose interfaces, quantifying enzyme off-rates and processivities;24,25 coherent Raman spectroscopy contributed to understanding of the role of plant cell wall architecture in limiting enzyme accessibility to cellulose.26 However, an unaddressed knowledge gap hindering full understanding of the mechanism of enzymatic cellulose and lignocellulose hydrolysis is the role of cellulose supramolecular structure in limiting productive cellulase interactions.
Fourier transform infrared (FTIR) spectromicroscopy is one of the most sensitive, label-free, physical techniques for studying spatially resolved changes in biomolecular structure and composition.27 FTIR has been used to characterize cellulose crystallinity, crystalline morphology and intrafibril hydrogen bonding attributed to enzymatic action; however, these studies have primarily been on dried, post-treated, bulk cellulosic samples subjected to varying sample preparation and measurement environments (e.g. surface tension, concentration, humidity, temperature).14,28–32 In one example, Corgié et al. reported evidence of decrystallization and preference for Iα or Iβ crystalline morphology by Thermobifida fusca Cel5A, Cel6B and Cel9A cellulases;28 but a follow-up study of the same reactions using different sample preparation methods by Kruer-Zerhusen et al. reported no conclusive impact of the enzyme action on cellulose crystallinity.29 One strategy to circumvent such artifacts is by in situ, time- and spatial-resolved FTIR spectromicroscopy of cellulose undergoing enzymatic hydrolysis. However, the need for buffered reactions is a barrier to FTIR measurements because of strong mid-IR absorption by water. Closed microfluidics have been used to conduct in situ FTIR spectromicroscopy of enzymatic treatment of plant biomass for bioenergy production.27,33–35 In this study, we used a novel open microfluidic device that enabled us to meet the need for on-demand access to the sample and the need for sustainable detection of FTIR spectral signals from the infrared hydride-OH region and the molecular fingerprint region.
This work was motivated by the hypothesis that local molecular ordering of cellulose can limit the accessibility of these so-called ‘productive binding sites’ where cellulases can successfully form complexes for hydrolysis, and that spatial variability in cellulose influences and is influenced by cellulase hydrolysis. Here, we present the first real-time, micrometer-scale, spatially resolved investigation of cellulose undergoing enzymatic hydrolysis by synchrotron radiation-based FTIR (SR-FTIR) spectromicroscopy. Using a broadband, high-brightness synchrotron infrared source instead of the conventional thermal source enabled wavelength-dependent diffraction-limited spatial resolution with ∼200 times increase in signal-to-noise ratio. This superior resolution allows for identification of changes in structures within crystalline cellulose in space and in time, which is unavailable using a Globar source.
Using well characterized, highly crystalline cellulose fibrils from the green algae Cladophora aegagropila, we demonstrated the impact of hydration on molecular ordering in cellulose fibrils by noting changes in the vibration bands of C2–O, C3–O, and C6–O of cellulose that participate in intra- and intermolecular hydrogen bonding. Spatial and temporal heterogeneity in changes in cellulose abundance and molecular order during hydrolysis by a purified Trichoderma reesei Cel7A (Cel7A) at 37 °C in buffer were recorded and analyzed.
2. Experimental
2.1. Preparation of cellulose and cellulase
Algal cellulose fibrils were isolated from the green algae Cladophora aegagropila as described previously.18 Briefly, marimo balls (Plants for Pets, Amazon, LLC) were pulled apart and rinsed with tap water to remove excess sand before subjecting to alternating alkali (1 M sodium hydroxide, 4 °C) and bleach (0.3% sodium hypochlorite, 100 mM sodium acetate, pH 5, 70 °C) treatments to isolate pure cellulose. Cellulose fibrils were washed extensively then stored at 4 °C in nanopure water with 0.02% sodium azide. Algal cellulose fibrils were previously characterized to be ∼90% crystalline, ∼70% cellulose Iα, with a reducing end content of ∼4 μmol g−1,18 and a productive Cel7A binding capacity of ∼2 μmol g−1.10,13Trichoderma reesei Cel7A (TrCel7A) was purified from Celluclast 1.5 L (Sigma C2730) and stored in 5 mM sodium acetate, pH 5 as previously described.212.2. Real-time synchrotron radiation (SR)-FTIR monitoring of cellulose in buffered systems
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of elastomer and curing agent followed by curing at 80 °C for 3 h. The microchannels were 20 μm wide and their thickness was measured to be 10 μm by a profilometer (Veeco Instruments) (Fig. 1C). The bottom structures (Structure 3, Fig. 1B) were for producing a passive vertical imbibition of buffer solution from the microchannel below through the porous membrane to the sample on the membrane surface. The top layer (Structure 1) is a humidity-control microfluidic structure with four microchannel arrays to provide a horizontal flow of moisture that blankets the sample chamber to minimize evaporation or condensation on the porous membrane during the experiment (Fig. 1C). A vertical through-square was carved out using a craft cutter system (Silhouette America), followed by plasma bonding to the top microchannel. The plasma treatment forms a sealed two-channel fluid system with individually accessible top and bottom microchannel. Silicon-based PSA tape was used as an intermediate layer to create PDMS-silicone sheet constructs.
:1 ratio of elastomer and curing agent followed by curing at 80 °C for 3 h. The microchannels were 20 μm wide and their thickness was measured to be 10 μm by a profilometer (Veeco Instruments) (Fig. 1C). The bottom structures (Structure 3, Fig. 1B) were for producing a passive vertical imbibition of buffer solution from the microchannel below through the porous membrane to the sample on the membrane surface. The top layer (Structure 1) is a humidity-control microfluidic structure with four microchannel arrays to provide a horizontal flow of moisture that blankets the sample chamber to minimize evaporation or condensation on the porous membrane during the experiment (Fig. 1C). A vertical through-square was carved out using a craft cutter system (Silhouette America), followed by plasma bonding to the top microchannel. The plasma treatment forms a sealed two-channel fluid system with individually accessible top and bottom microchannel. Silicon-based PSA tape was used as an intermediate layer to create PDMS-silicone sheet constructs.
 | ||
| Fig. 1 Method for controlling relative humidity (RH) and imbibition flow of thin liquid film in an open microfluidics membrane device for real-time synchrotron FTIR imaging of cellulose ordering and composition during hydrolysis in situ. (A) A photograph of the three-structure stacked PDMS microfluidic membrane device. Scale bar = 5 mm. (B) A schematic showing construction of the open-channel microfluidics membrane device consisting of the top humidity control microchannel plate and the middle sample support plate and the nanopore SiNx membrane sealed by a pressure sensitive polymer layer. The two stacked structures and the bottom buffer reservoir plate were sealed by plasma treatment. (C) An exploded schematic view of the micro-channel capillary arrays controlling the RH in the sample chamber. Arrows show the direction of the water and moisture flows. | ||
 | ||
| Fig. 2 Experimental and data analytic procedure for continuous monitoring of cellulose hydrolysis by Cel7A by SR-FTIR in the open-channel microfluidics membrane device. | ||
To image the spatiotemporal dynamics of cellulose during enzymatic hydrolysis, we typically divided the entire view-field of the cellulose fibrils into equal-sized 5 × 5 μm squares before raster scanning, collecting full SR-FTIR spectra at each pixel. Time-elapsed mapping of sample areas were conducted using in-house software written in the Python programming language (version 3.6). The master program controlled the movement of the microscope stage, spectra collection of FTIR spectrometer and data storage in the OMNIC software program (version 9.8, Thermo Fisher Scientific, Inc.) using the Dynamic Data Exchange (DDE) protocol. The source code of our python programs for controlling the microscope and FTIR bench is available upon request. Raw spectral data reported in this study is available for download.38
3. Results
3.1 Time-resolved SR-FTIR spectra of cellulose in varying hydration states
FTIR spectra of cellulose in the literature are primarily of dried cellulose; thus, the assignment of the peaks unique to cellulose are of dry cellulose. Since our experiments are with hydrated cellulose in buffer, we examined the impact of hydration on the signature peaks of cellulose. Time-resolved spectra of wet cellulose undergoing evaporative drying were recorded over the course of ∼37 minutes (Fig. 3). Cellulose remained fully hydrated for about the first ∼6 minutes, after which a sharp drop in the absorption amplitudes of the water peaks at 1650 cm−1 (H–O–H bending mode) and 2150 cm−1 (H–O–H vibration mode), respectively, were observed (Fig. 3B). A second sharp drop in the amplitude of the 1650 and 2150 cm−1 peaks to zero at about 15 minutes further indicated full removal of moisture.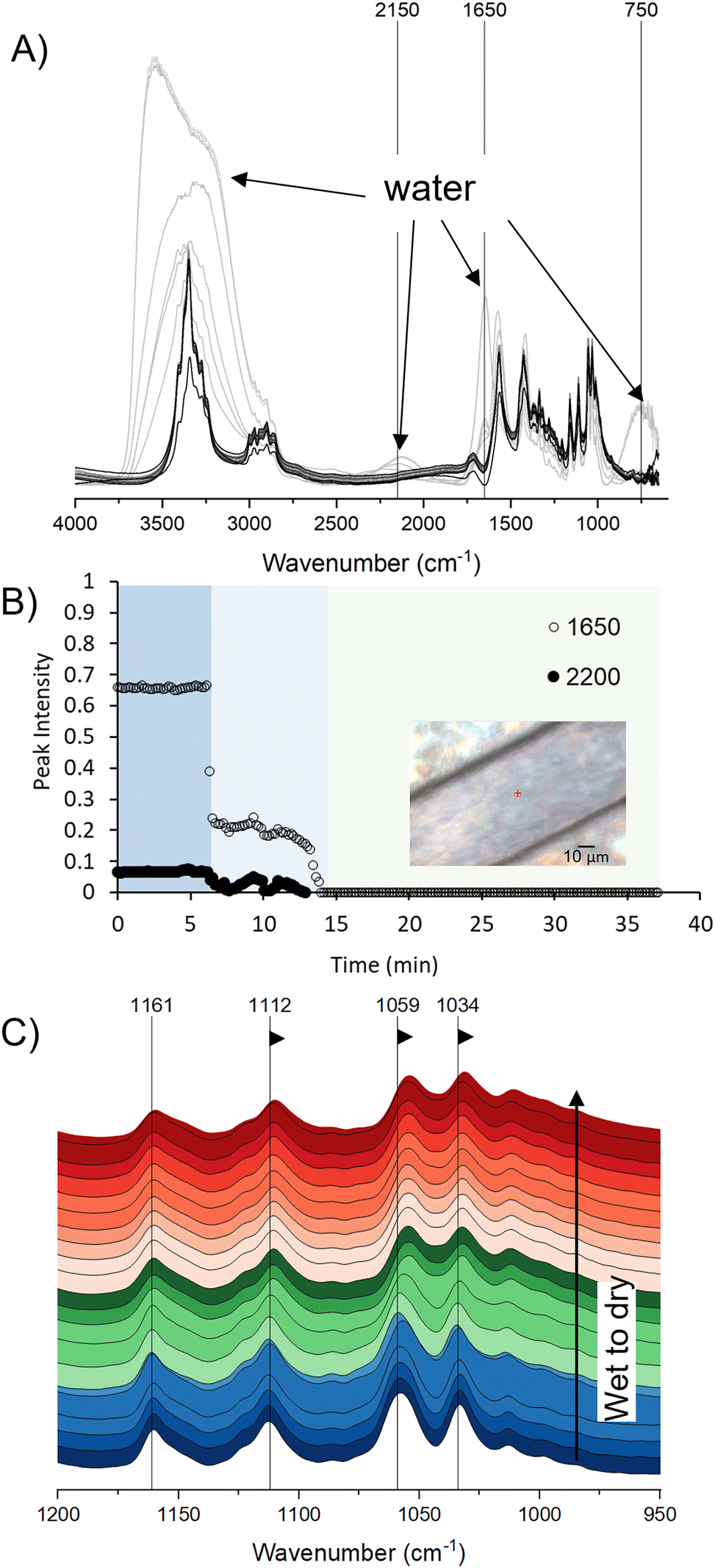 | ||
| Fig. 3 (A) Time-resolved SR-FTIR spectra of cellulose undergoing evaporative dehydration at 25% RH at 37 °C. Absorption peaks at ∼3000–3700 cm−1, 2150 cm−1, 1650 cm−1 and 750 cm−1 associated with water molecules are indicated. Lighter to darker grayscale gradient indicate progression from hydrated to dry spectra. (B) Intensities of the 1650 and 2150 cm−1 absorption bands of water over the course of the experiment. Inset shows brightfield image of the location on the cellulose fibril where the spectra were recorded. (C) A closer view of the spectra between 950–1200 cm−1 showing shifts to lower frequencies (arrows) by the νC2–O2 (1112 cm−1), νC3–O3 (1059 cm−1) and the primary νC6–O6 (1034 cm−1) peaks due to drying. Color-coding indicate fully hydrated spectra in blue, spectra during drying in green, and spectra after drying in red. Spectra recorded every ∼2 min are shown. | ||
Strong absorption of mid-IR energy by water can be an impediment to successful FTIR imaging of biological reactions in situ. Strong water absorption peaks centered around 2150 cm−1, 1650 cm−1, and 750 cm−1 wavenumbers obscure less prominent absorbance signals from other constituents in these regions. Moreover, water molecules can form hydrogen bonds with neighboring molecules with a spectrum of different bond lengths and stretching vibrational frequencies, resulting in the OH functional groups of water forming continuous vibrational frequencies and giving an extensively broad and dominant infrared absorption in the hydrogen bonding (X–H) 3000–3700 cm−1 region39,40 (light gray spectra in Fig. 3A). The absorption due to water, however, generally avoids the fingerprint region (∼900–1400 cm−1) where vibration peaks of the C–O bonds of cellulose dominate. Under controlled hydration levels, the presence of absorbed water at 1650 cm−1 and the absorption peaks in the fingerprint region of cellulose can be clearly distinguished (Fig. 3A).
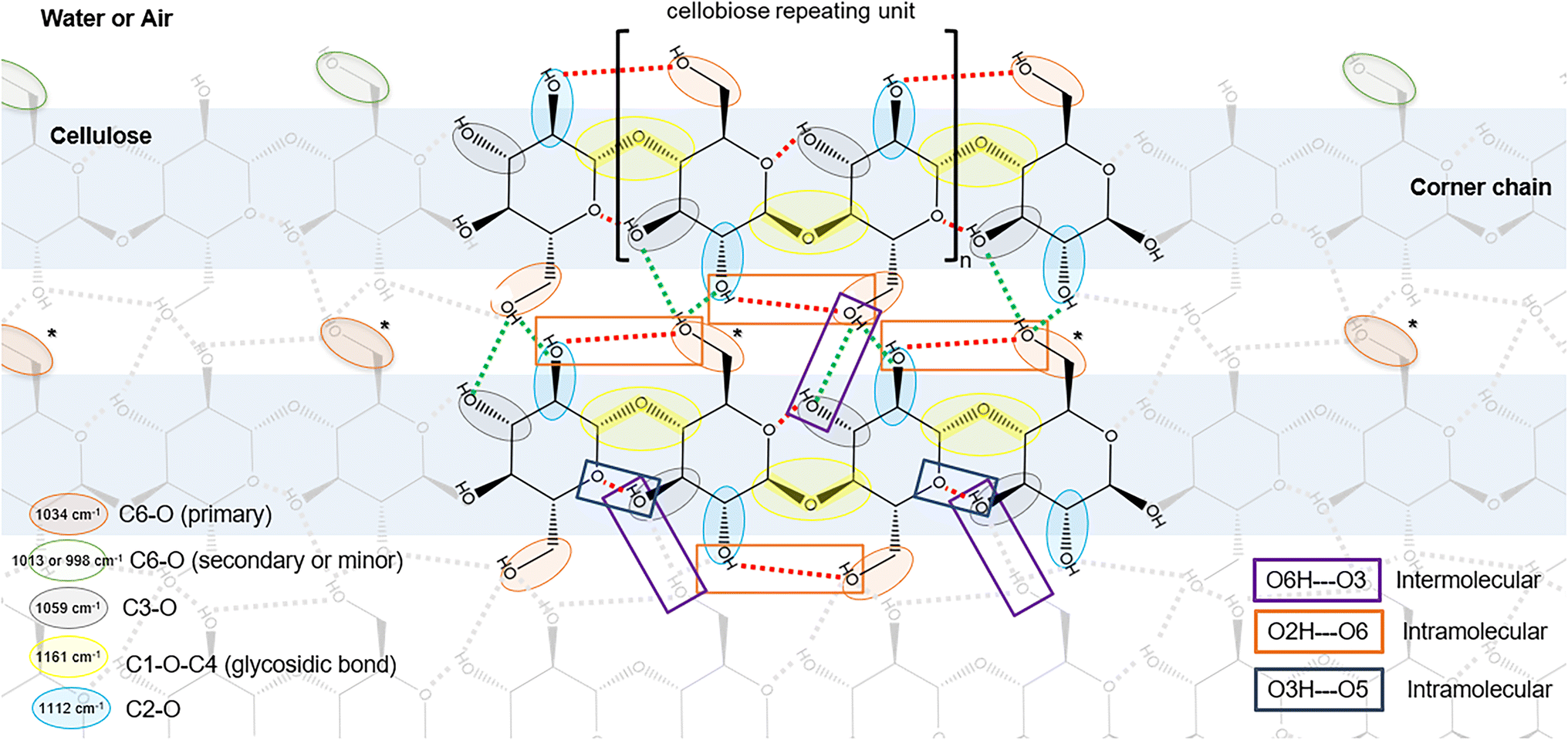 | ||
| Fig. 4 Illustration of top down view of the hydrophobic face of cellulose, highlighting C–O bonds and hydrogen bonds characteristic of cellulose I. Red dotted lines indicate intramolecular hydrogen bonds; green dotted lines indicate intermolecular hydrogen bonds. The C6–O bond can occupy secondary or minor rotamers at cellulose/air interface but likely occupies the primary rotamer within the cellulose structure. C6–O6 bonds in the primary rotameric forms would likely covert to secondary or minor rotamers when exposed to the interface, e.g. if the corner chain is hydrolyzed by Cel7A (indicated by asterisks). | ||
| Cellulose peaks in the fingerprint region | Wavenumber at peak centera (cm−1) | Peak shift when dried | Peak intensity (A.U.)a | Intensity change when dried | ||
|---|---|---|---|---|---|---|
| Dry | Hydrated | Dry | Hydrated | |||
| Peak intensities and positions tracked over time are given in ESI 2. Peak assignments for all cellulose-associated peaks are given in ESI 1 and Table S1.1†a Averaged over the 0–5 minutes and 25–35 min for dry and hydrated states as shown in ESI 2.†b Ratio of secondary to minor C6–O6 peak intensities.c Ratio of primary to minor C6–O6 peak intensities. | ||||||
| ν AS O–C1–O | 1161 ± 0.2 | 1161 ± 0.3 | No change | 0.19 ± 0.0023 | 0.22 ± 0.0052 | −17% |
| ν S O–C1–O | 1207 ± 0.3 | 1207 ± 0.3 | No change | 0.04 ± 0.0010 | 0.04 ± 0.0018 | No change |
| νC1–O1 | 1152 ± 1.6 | 1152 ± 0.3 | No change | 0.11 ± 0.011 | 0.11 ± 0.0052 | No change |
| νC2–O2 | 1111 ± 0.4 | 1113 ± 0.3 | −2.2 cm−1 | 0.21 ± 0.0027 | 0.25 ± 0.0045 | −16% |
| 1123 ± 0.3 | 1124 ± 0.2 | −0.4 cm−1 | 0.08 ± 0.0026 | 0.1 ± 0.0033 | −19% | |
| νC3–O3 | 1057 ± 1.0 | 1059 ± 0.9 | −2.5 cm−1 | 0.31 ± 0.011 | 0.38 ± 0.013 | −22% |
| Minor νC6–O6 (gt) | 997 ± 0.8 | 998 ± 0.7 | −0.7 cm−1 | 0.15 ± 0.0042 | 0.06 ± 0.0035 | +46% |
| Secondary νC6–O6 (gg) | 1012 ± 0.5 | 1013 ± 0.3 | −0.8 cm−1 | 0.22 ± 0.0026 (1.5:1)b |
0.12 ± 0.0034 (2:1)b |
+60% |
| Primary νC6–O6 (tg) | 1032 ± 0.6 | 1034 ± 0.7 | −1.4 cm−1 | 0.34 ± 0.0040 (2:1)c |
0.34 ± 0.0064 (6:1)c |
No change |
Interestingly, the ratios of the average peak intensities of the primary, secondary, and minor absorption bands of C6–O6 shift from ∼6:2:1 in the hydrated state to ∼2:1.5:1 in the dry state (Table 1, and ESI 2†). Yet, to our knowledge, peak intensities of C6–O6 bonds are not known to change with the orientation of polarized light. The change in intensity ratio is driven by the increase in the intensities of the 1013 cm−1 (secondary) and 997 cm−1 peaks (minor) when dried, while the intensity of the 1034 cm−1 peak (primary) remained unchanged. In crystalline cellulose I, trans–gauche (tg) rotamers of C6–O6 dominate within the crystalline structure, facilitating both the C6–O6⋯HO2 intramolecular hydrogen bond across the glycosidic bond, and the C6–O6H⋯O3 intermolecular hydrogen bond with neighboring cellulose41,42 (Fig. 4). Thus, negligible impact of dehydration on the intensity suggests that the primary C6–O6 bonds are not solvent accessible and likely correspond to tg rotamers. Intensity increases in the secondary and minor C6–O6 peaks due to dehydration suggest that these are solvent accessible C6–O6 bonds, likely gauche–gauche (gg) and gauche–trans (gt) rotamers, respectively.49,50
The absorption peaks associated with the anomeric carbon, both as part of the glycosidic bond (1161 and 1207 cm−1) and as free reducing end (1152 cm−1) were minimally impacted by moisture (Fig. 3C, Table 1, and ESI 2†). The absorption peak centers did not shift from a wet to dry state, and only a slight decrease in intensity (17%) was observed at 1161 cm−1 upon drying. It is no surprise that the stiffness of the glycosidic bonds in cellulose I, braced by intramolecular hydrogen bonds (C3O3H⋯O5 and C2O2⋯HO6) on either side, is not influenced by water. The C1–O1 bond of the free reducing-end, already absorbing at a lower frequency (i.e. less stiff than the glycosidic bond), was also not influenced by water.
In general, the impact of moisture appears to be stiffening of the C–O bonds of cellulose involved in intra- and intermolecular hydrogen bonds. At the cellulose/air interface, C2–O6H, C3–O3H and C6–O6H are less likely to engage in hydrogen bonds (intrafibril or with water) than at the cellulose/water interface. Cellulose has been shown to exhibit lower crystallinity indices (CI) in a dry state than in a hydrated state with similar explanations on the role of water in imposing surface ordering.51 The impact of moisture on C–O vibration peaks of cellulose are summarized in Table 1. The full set of peak assignments for the hydrated IR spectra of cellulose in the 900–3500 cm−1, and further discussion on the impacts of hydration/dehydration are provided in ESI 1 and 2.†
3.2. Time- and spatial-resolved SR-FTIR spectra of cellulose undergoing enzyme hydrolysis
IR spectral evolution of algal cellulose undergoing hydrolysis by a purified Cel7A in pH 5 buffer at 37 °C was recorded in an 8 × 8 grid every ∼9.5 minutes over 15-hours. The imaged region consisted of a thick deposition of cellulose in the bottom half and a sparser deposition of fibrils in the top half (Fig. 5). | ||
| Fig. 5 (A) Brightfield image of the algal cellulose region examined in this study. IR spectra were recorded at 64 locations every ∼10 min in an 8 × 8 grid as shown. (B) The imaged area is enlarged, showing the numbering of the locations where spectra were recorded. | ||
The characteristic cellulose peaks in the fingerprint region, particularly centered around 1161 cm−1 (glycosidic bond), 1112 cm−1 (C2–O2), 1059 cm−1 (C3–O3), and 1034 cm−1 (C6–O6) are clearly distinguishable in the time-resolved spectra throughout the reaction (Fig. 6B). During the 15-hour reaction, a ∼3 μm water film was maintained for the first ∼5 hours, after which the water film thickness increased to ∼12 μm (ESI 3†). The time courses of the intensities of cellulose-associated peaks in the fingerprint region (998, 1013, 1034, 1059, 1112, 1124, 1150, 1161, and 1205 cm−1) confirmed negligible interference of water levels on peak intensities in the mapped region (Fig. 6B and ESI 4†). Thus, the full time course data could be used to assess changes to cellulose due to enzyme action. Unfortunately, increased water film thickness at >5 h obscured peaks in the 2700–3900 cm−1 range hydride-OH region (Fig. 6C and ESI 4†).
 | ||
| Fig. 6 (A) Full range IR spectra of cellulose undergoing enzymatic hydrolysis collected over 0–5 h and at 10 h from the circled location in the inset brightfield sample image. Expansions of the (B) 850–1850 cm−1 range showing characteristic peaks of cellulose and adsorbed enzymes, and the (C) 2700–3900 cm−1 range showing C–H vibration and the hydrogen bonding regions of cellulose. The full set of peak assignments for the hydrated IR spectra of cellulose in the 900–3500 cm−1 are provided in Table S.1 in ESI.† | ||
Characteristic amide I and amide II peaks of the enzyme (∼1650 cm−1 and 1550 cm−1, respectively) were clearly visible at low water levels, but replaced by a strong adsorbed water peak at ∼1650 cm−1 in the presence of excess water (Fig. 6B). A large fraction of enzymes on the surface was most likely washed away; moreover, the dominance of the adsorbed water signal at 1650 cm−1 obscured signal from any enzymes that remained bound to cellulose.
 | ||
| Fig. 7 (A) Heat map of the evolution of the peak intensity of the glycosidic bond peak at ∼1161 cm−1 over 15 hours in the imaged region in Fig. 5B. Highest peak amplitudes are mapped as red, with decreasing amplitudes from orange to yellow to green to blue. (B) The 0 h heat map from (A) overlaid on the imaged region. (C) Time course of the amplitudes of the cellulose vibration bands of C6–O6 at 1034 cm−1, C3–O3 at ∼1059 cm−1, C2–O2 at ∼1112 cm−1 and O–C1–O glycosidic bond at ∼1161 cm−1. Plots are arranged in an 8 × 8 grid corresponding to locations in Fig. 5B where spectra were recorded. The x-axes are time in hours (0 to 15 hours); y-axes are absorbances/peak intensities (0–0.8). The 0 h heat map from (A) is overlaid to show relative intensities of the 1161 cm−1 peak at the start of the time course. | ||
Based on the temporal trends of the cellulose peak intensities, the imaged area can be grouped into four regions (i, ii, iii, and iv in Fig. 7C). The cellulose peak intensities in region i do not decrease to zero suggesting incomplete cellulose hydrolysis during the 15 hours; furthermore, some points within the region exhibit increased peak intensities in the first 0–5 hours. Cellulose peak intensities in region ii decreased throughout the 15 hours. Cellulose peaks in region iii similarly decreased over the course of hydrolysis, with peak intensities decreasing to below detection (reaching zero intensity) within the 15 hours, indicating complete hydrolysis of cellulose in this region. The rates of intensity decrease in regions ii and iii reflected trends reminiscent of typical cellulose hydrolysis kinetics with an initial rapid decrease followed by significantly slower rates of depletion. Region iv, where cellulose was sparser, had low intensities, noisier data, and show a baseline shift with increased water into the sample at ∼5 hours. Thus, region iv will not be discussed further.
 | ||
| Fig. 8 FTIR spectra recorded every ∼9.5 min over the course of 5 hours in the range of 950–1075 cm−1 corresponding to locations within regions i and ii. Plot numbers (1–4, 9–12, and 17–20) correspond to locations shown in Fig. 5B and 7C. The spectra are arranged in increasing time order from bottom to top as indicated in plot 17. The bar at the bottom right of each plot and blue shading highlight spectra recorded within the first 1 hour. | ||
In contrast, the C3–O3 (1059 cm−1) and primary C6–O6 (1034 cm−1) peaks in region ii followed expected trends of decreasing intensities throughout the hydrolysis reaction (Fig. 7C). Time resolved spectra in region ii indeed revealed narrow and sharp C3–O3 and primary C6–O6 peaks from the first spectrum at 0 h, indicating fully hydrated cellulose in this region from the start of the reaction (Fig. 8). The C3–O3 and primary C6–O6 peaks decreased in intensity over time with minimal changes to the peak shapes. The time courses of the cellulose peaks in region ii thus show that Cel7A action depleted cellulose without appreciably affecting molecular order.
3.2.2.1. Assessing spatiotemporal changes in molecular order by comparing marker peaks in the cellulose spectra – lateral order index (LOI), total crystallinity index (TCI) and hydrogen bonding index (HBI). Metrics that have been used to assess changes in cellulose molecular ordering from IR spectra include the lateral order index (LOI), total crystallinity index (TCI), and hydrogen bonding index (HBI). The LOI compares the peak intensity at ∼1429 cm−1 attributed to δOH of C6–O6H in crystalline regions to the peak intensity at ∼896 cm−1 associated with amorphous cellulose.55 The TCI compares the peak intensity at 1372 cm−1 associated with δOH with that of the 2860 cm−1 peak due to νCH,56 and the hydrogen bonding index (HBI) compares the ratio of intensities of the peaks at 3345 cm−1νOH due to intra and intermolecular hydrogen bonding57 and 1337 cm−1 due to δOH of O2H and O3H.58 We assessed LOI, TCI and HBI during the first ∼5 hours of the reaction where peaks in the hydride-OH region were clearly distinguishable (Fig. 6).
The algal cellulose used as a substrate in this study was a highly crystalline cellulose with CI ∼ 90%.18 Confirming the high degree of crystallinity of algal cellulose, almost no absorbance peak was detected around 896 cm−1 such that the LOI values were large (≫1) (ESI 5†). Further, the continued absence of signal at 896 cm−1 indicated no increase in amorphous content of the cellulose throughout hydrolysis.
The TCI (1372 cm−1/2860 cm−1) hovered around a ratio of ∼1.5, with some areas fluctuating between ∼1 and 2 (Fig. 9B). Generally tracking the C–O vibration peaks, the intensities of the peaks at 1372 cm−1 and 2860 cm−1 both decreased over time at similar rates (Fig. 9A). At some locations, e.g. at the bottom 2/3 of region iii where cellulose depletes at longer times, TCI appear to decrease from ∼2 to ∼0.5–1 over time. Overall, however, the analysis of the TCI indicates minimal impact on total crystallinity by enzyme action, consistent with the expectation that Cel7A, a reducing-end specific cellobiohydrolase acting on accessible productive binding sites at the fibril surfaces, should not significantly alter bulk molecular ordering of cellulose.
 | ||
| Fig. 9 (A) Peak intensities at 1372 cm−1 and 2860 cm−1, and the (B) total crystallinity index (TCI) 1372 cm−1/2860 cm−1 over the first 5 hours of enzymatic hydrolysis by Cel7A in regions i, ii, and iii. (C) Peak intensities at 3345 cm−1 and 1337 cm−1, and the (D) hydrogen bonding index (HBI) 3345 cm−1/1337 cm−1 over the first 5 hours of enzymatic hydrolysis by Cel7A in regions i, ii, and iii. The 0 h heat map from (Fig. 7A) is overlaid. | ||
The HBI (3345 cm−1/1337 cm−1) ranged from ∼2–20, with an average of ∼5 (Fig. 9D). Both the 3345 cm−1 and the 1337 cm−1 peaks decreased over time (Fig. 9C), but depending on the location within regions i and ii, the ratio of the two peaks (HBI) either remained constant or increased with time. Increases in HBI suggests a more rapid loss of cellulose than of hydrogen bonding; i.e. preferential removal of cellulose that is not participating in intermolecular hydrogen bonds. Accessible productive Cel7A binding sites are likely cellulose reducing ends located at microfibril corners and edges that are less extensively hydrogen-bonded within the fibril structures.59 Though the trends are subtle, the HBI suggest that productive Cel7A binding sites are associated with less extensively hydrogen bonded cellulose.
Taken together, the three indices, LOI, TCI and HBI, indicated that the algal cellulose is highly ordered, and hydrolysis by Cel7A minimally impacts the overall crystallinity of the cellulose, but preferentially removes less extensively hydrogen bonded cellulose. These trends support previous observations of algal cellulose as a highly crystalline cellulose that erode from the surface due to Cel7A hydrolysis,18 and deplete in accessible productive binding sites during hydrolysis by Cel7A.13
3.2.2.2. C6–O6 rotamer ratio as an indicator of molecular order. In highly ordered cellulose I, C6–O6 bonds exist predominantly as tg rotamers that hydrogen bond with –O2H2 of the adjacent glucose residue as a hydrogen acceptor (Fig. 4). The gg rotamer preferentially hydrogen bonds with water, while the gt rotamer is favored in air or within non-native cellulose morphologies such as cellulose II and III. In highly ordered cellulose I fibrils, gg or gt rotamers likely dominate at the aqueous or air interface, and tg rotamers would dominate within the fibril (illustrated in Fig. 4). The ratio of the C6–O6 rotamers at any location over the course of the enzymatic hydrolysis reaction can provide insights into the impact of Cel7A on molecular ordering of cellulose.
The peaks at 1034 cm−1 (primary), 1013 cm−1 (secondary), and 998 cm−1 (minor) are assigned to tg, gg, and gt rotamers, respectively, based on the analysis of hydrated and dehydrated cellulose above (section 3.1.1). The intensities of the secondary 1013 cm−1, and minor 998 cm−1 peaks generally tracked that of the primary 1034 cm−1 peak during enzymatic hydrolysis (Fig. 10A). However, the ratio of the primary:secondary:minor peak intensities (herein referred to as ‘C6 ratios’) revealed interesting patterns (Fig. 10B). For example, there is a noticeable and abrupt change in the temporal trends in the C6 ratios at around 5 hours, coinciding with increased water into the reaction. As discussed above, molecular ordering of cellulose in region i increased due to hydration during the first 5 hours; here, manifesting as increasing C6-ratios in Fig. 10B. In region ii, the C6 ratios appeared to decrease during the first 5 hours (Fig. 10B). During this time, of the three C6–O6 peaks, the primary peak at 1034 cm−1 exhibited the steepest decrease in intensity, suggesting that an accelerated loss of the tg rotamer drove the decreasing C6–O6 ratio (Fig. 10A). A more rapid loss of the tg rotamer of cellulose is consistent with removing cellulose from the corners of the fibrils. For example, as Cel7A processively hydrolyzes a corner chain of cellulose, it removes both interior C6–O6 groups (likely tg) and C6–O6 groups at the aqueous interface (more likely gg). This alone would preserve the original C6 ratio; but removing a corner chain exposes C6–O6 of a previously interior chain, converting some tg rotamers to gg, thereby decreasing the C6 ratio (asterisks in Fig. 4).
 | ||
| Fig. 10 (A) Peak intensities of the minor (998 cm−1), secondary (1013 cm−1), and primary (2860 cm−1) peaks over 15 hours. (B) Ratio of peak intensities of the primary:secondary:minor C6–O6 absorption bands. | ||
Within the first 5 hours, C6 ratios decreased to ∼3:2:1. Just after 5 hours, coinciding with the introduction of excess water into the reaction (ESI 3†), the C6 ratios increased abruptly to ∼5:2:1–∼6:2:1 (Fig. 10B). A possible explanation is the removal of surface enzymes (Fig. 6B). It is possible that the presence of enzymes causes surface disorder of cellulose, and replacing the enzymes with water restores ordering. Where cellulose is fully depleted due to hydrolysis (e.g. region iii), the excessive scatter in the C6 ratios render them meaningless.
4. Discussion
Spatial heterogeneity in the hydrolysis of cellulose by cellulases is posited to contribute strongly to the overall recalcitrance of cellulose,13,60,61 but is challenging to characterize. IR spectromicroscopy enables highly resolved spatial mapping of material composition by bond vibrational modes. In this work, IR spectromicroscopy enabled spatial mapping of changes to cellulose abundance and molecular ordering during enzyme action. The use of a microfluidics system to control temperature, moisture and humidity levels facilitated data collection over 15 hours in an aqueous buffered reaction.Moisture has been a major impediment to IR spectromicroscopy because of strong infrared absorption by water. While water is a critical component of most biological reactions including cellulose hydrolysis, water has also been implicated in imparting changes to molecular ordering of cellulose crystalline fibrils.62,63 The extent of intrafibril hydrogen bonding within cellulose fibrils contribute to the crystalline morphology and crystallinity of cellulose. In this study, spatially resolved changes in localized molecular ordering of cellulose was assessed from examining the key peaks of cellulose in the IR fingerprint region. Glycosidic bonds along the backbone of cellulose are braced on either side by the O5–HO3 and O2H–O6 intramolecular hydrogen bonding (Fig. 4). The bracing imparts rigidity to the cellulose molecule64 such that cello-oligosaccharides decrease in solubility with increasing degrees of polymerization (DP), and chains with DP ≥ 6 are insoluble in water.65 Moreover, the O2H–O6 hydrogen bond, occurring on alternating sides of the glycosidic bonds along the molecule, is suggested to strain the molecule and cause the right-handed twist observed in cellulose fibrils.66 Previous studies have reported physical manifestation of the release of stress within the fibrils during enzymatic hydrolysis as spontaneous untwisting and kinking of fibrils.25,67,68 Water–cellulose and cellulose–cellulose hydrogen bonding at the surface of cellulose not only impact ordering of surface chains, but disorder could propagate into the cellulose fibril structure.59,62 Here, we found that cellulose peaks associated with intra- and intermolecular hydrogen bonds in the IR fingerprint region decreased in frequency when dried, implying that these C–O bonds (C2–O2, C3–O3, and C6–O6) are less stiff at the air–cellulose interface than the water–cellulose interface. Furthermore, the ratios of the peak intensities of the three rotameric forms of C6–O6 tg:gg:gt shifted from ∼6:2:1 in ordered cellulose and ∼2:1:1 in disordered cellulose. Frequency shifts in the C–O bonds that participate in intra- and intermolecular hydrogen bonding between the wet and dried state are likely due to differences in hydrogen bonding patterns at the water–cellulose and air–cellulose interface. Thus, frequency shifts of the C2–O2, C3–O3, and C6–O6 peaks, and intensity ratios of the C6–O6 rotamers are useful indicators assessing changes to surface ordering of cellulose.
The precise role of ‘crystallinity’ in the resistance of cellulose to hydrolysis has been difficult to elucidate. Most often, the crystallinity index (CI), a bulk measure of the fraction of ordered cellulose in a given sample, is used to query the relationship between crystallinity and cellulose digestibility. However, cellulose CI, including those estimated from peak ratios in the FTIR spectra of cellulose, remains an ambiguous measure of cellulose digestibility.18,28,29 This is not to say that cellulose crystallinity, or molecular ordering of cellulose molecules, particularly at the water–fibril interface is irrelevant to enzymatic hydrolysis. Cellulase hydrolysis of cellulose is an interfacial reaction where cellulases must decrystallize and complex to isolated cellulose molecules at the surface of the fibrils. Or, cellulases must locate a decrystallized cellulose molecule at the surface of the fibrils to complex with. Thus, a reasonable hypothesis is that localized surface disorder in molecular arrangement offers more productive binding sites.69 The algal cellulose used in this study was a highly crystalline and recalcitrant cellulose. In our previous studies, we showed that Cel7A is insensitive to the number of reducing ends (or degree of polymerization) and total crystallinity of algal cellulose;18 however, initial cellulose hydrolysis rates by Cel7A correlates with initial binding capacity of cellulose,10 and Cel7A rapidly depletes available productive binding sites on the cellulose, leaving a substrate that is essentially impenetrable to the cellobiohydrolase.13 Indeed, spatial mapping of FTIR spectra in this study showed that the algal cellulose was not uniformly resistant to Cel7A hydrolysis. Some regions appeared to be completely hydrolyzed by Cel7A while others did not appear to deplete significantly.
An unanswered question from this study is whether cellulase action creates localized disorder at the cellulose surface or, does localized disorder at cellulose surfaces invite active cellulase complexation? A more detailed study with higher spatial resolution is necessary to adequately query these questions, to help inform future efforts in enzyme engineering to improve enzyme's ability to decrystallize and complex to cellulose, and biomass pretreatment strategies targeting desired cellulose surface properties.
5. Conclusions
In situ, spatial mapping of the infrared spectra of cellulose during enzymatic hydrolysis in buffer was successfully achieved using an open-channel humidity-controlled microfluidics device. The spatial heterogeneity in hydrolysis rates and changes in molecular ordering of cellulose over the course of hydrolysis by Cel7A was documented. To our knowledge, this is the first documented in situ study of spatiotemporal changes in cellulose during enzymatic hydrolysis in buffer by IR spectral mapping. A major advantage of in situ measurements is tracking of both peak amplitudes and peak centers, to assess depletion rates and changes in molecular ordering of cellulose.Here, we established a label-free, spatial and time-resolved infrared spectroscopic method towards filling the knowledge-gap of how cellulose supramolecular ordering contributes to hydrolysis mechanism and ultimately hydrolysis kinetics. The insights gained with work on isolated cellulose paves the way for future analyses of cellulose within plant cell wall matrices (i.e. lignocellulosic biomass), towards fulfilling the vision for low-cost bioconversion of lignocellulosic materials to sustainable fuels, chemicals and materials.
Author contributions
Tina Jeoh: conceptualization, validation, investigation, resources, writing – review & editing, visualization, supervision, project administration, funding acquisition; Jennifer Danger Nill: conceptualization, validation, investigation, methodology, formal analysis, visualization, writing – review & editing; Wujun Zhao: investigation, methodology; Sankar Raju Narayanasamy: investigation, methodology, visualization, writing – review & editing; Liang Chen: methodology, formal analysis, visualization; Hoi-Ying N. Holman: conceptualization, methodology, formal analysis, validation, investigation, resources, writing – review & editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Hans Bechtel and ALS Beamline 1.4.3 staff for their instrumentation support, and Alex Hitomi and Lucy Knowles for their contributions to cellulose and cellulase preparation. This research used resources of the Berkeley Synchrotron Infrared Structural Biology (BSISB) program and the Advanced Light Source, a U.S. Department of Energy (DOE) Office of Science (OS) User facility under Contract No. DE-AC02-05CH11231. W. Z., L. C., and H.-Y. N. H. were supported by the DOE Office of Science, Office of Biological and Environmental Research under Contract No. DE-AC02-05CH11231. J. D. N. was supported in part by the DOE SCGSR program (Contract No. DE-SC0014664). This work was supported in part by the DOE BER Bioimaging Science Program (Award No. DE-SC0019228).References
- J. L. Field, T. L. Richard, E. A. H. Smithwick, H. Cai, M. S. Laser and D. S. LeBauer, et al., Robust paths to net greenhouse gas mitigation and negative emissions via advanced biofuels, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(36), 21968 CrossRef CAS PubMed.
- N. Scarlat and J. F. Dallemand, Chapter Ten - Future Role of Bioenergy, in The Role of Bioenergy in the Bioeconomy: Resources, Technologies, Sustainability and Policy, ed. C. Lago, N. Caldés and Y. Lechón, Academic Press, 2019, pp. 435–547. Available from: https://www.sciencedirect.com/science/article/pii/B9780128130568000108 Search PubMed.
- D. Klemm, E. D. Cranston, D. Fischer, M. Gama, S. A. Kedzior and D. Kralisch, et al., Nanocellulose as a natural source for groundbreaking applications in materials science: Today's state, Mater. Today, 2018, 21(7), 720–748 CrossRef CAS.
- T. C. Mokhena and M. J. John, Cellulose nanomaterials: new generation materials for solving global issues, Cellulose, 2020, 27(3), 1149–1194 CrossRef CAS.
- X. Yang, M. S. Reid, P. Olsén and L. A. Berglund, Eco-Friendly Cellulose Nanofibrils Designed by Nature: Effects from Preserving Native State, ACS Nano, 2020, 14(1), 724–735 CrossRef CAS.
- P. B. Filson, B. E. Dawson-Andoh and D. Schwegler-Berry, Enzymatic-mediated production of cellulose nanocrystals from recycled pulp, Green Chem., 2009, 11(11), 1808 RSC.
- C. L. Pirich, G. F. Picheth, A. M. Fontes, M. Delgado-Aguilar and L. P. Ramos, Disruptive enzyme-based strategies to isolate nanocelluloses: a review, Cellulose, 2020, 27(10), 5457–5475 CrossRef CAS.
- C. Moreau, S. Tapin-Lingua, S. Grisel, I. Gimbert, S. Le Gall and V. Meyer, et al., Lytic polysaccharide monooxygenases (LPMOs) facilitate cellulose nanofibrils production, Biotechnol. Biofuels, 2019, 12(1), 156 CrossRef PubMed.
- J. Jalak and P. Väljamäe, Multi-Mode Binding of Cellobiohydrolase Cel7A from Trichoderma reesei to Cellulose, PLoS One, 2014, 9(9), e108181 CrossRef PubMed.
- N. Karuna and T. Jeoh, The productive cellulase binding capacity of cellulosic substrates, Biotechnol. Bioeng., 2017, 114(3), 533–542 CrossRef CAS PubMed.
- N. Cruys-Bagger, K. Alasepp, M. Andersen, J. Ottesen, K. Borch and P. Westh, Rate of Threading a Cellulose Chain into the Binding Tunnel of a Cellulase, J. Phys. Chem. B, 2016, 120(25), 5591–5600 CrossRef CAS PubMed.
- J. Nill, N. Karuna and T. Jeoh, The impact of kinetic parameters on cellulose hydrolysis rates, Process Biochem., 2018, 74, 108–117 CrossRef CAS.
- J. D. Nill and T. Jeoh, The Role of Evolving Interfacial Substrate Properties on Heterogeneous Cellulose Hydrolysis Kinetics, ACS Sustainable Chem. Eng., 2020, 8(17), 6722–6733 CrossRef CAS.
- Y. Horikawa, N. Konakahara, T. Imai, A. Kentaro, Y. Kobayashi and J. Sugiyama, The structural changes in crystalline cellulose and effects on enzymatic digestibility, Polym. Degrad. Stab., 2013, 98(11), 2351–2356 CrossRef CAS.
- P. Bansal, B. J. Vowell, M. Hall, M. J. Realff, J. H. Lee and A. S. Bommarius, Elucidation of cellulose accessibility, hydrolysability and reactivity as the major limitations in the enzymatic hydrolysis of cellulose, Bioresour. Technol., 2012, 107, 243–250 CrossRef CAS.
- T. Jeoh, C. Ishizawa, M. F. Davis, M. E. Himmel, W. S. Adney and D. K. Johnson, Cellulase Digestibility of Pretreated Biomass is Limited by Cellulose Accessibility, Biotechnol. Bioeng., 2007, 98(1), 112–122 CrossRef CAS PubMed.
- T. Jeoh, M. J. Cardona, N. Karuna, A. R. Mudinoor and J. Nill, Mechanistic kinetic models of enzymatic cellulose hydrolysis—A review, Biotechnol. Bioeng., 2017, 114(7), 1369–1385 CrossRef CAS.
- P. J. O’Dell, A. R. Mudinoor, S. J. Parikh and T. Jeoh, The Effect of Fibril Length and Architecture on the Accessibility of Reducing Ends of Cellulose Iα to Trichoderma reesei Cel7A, Cellulose, 2015, 22(3), 1697–1713 CrossRef.
- V. Arantes and J. N. Saddler, Cellulose accessibility limits the effectiveness of minimum cellulase loading on the efficient hydrolysis of pretreated lignocellulosic substrates, Biotechnol. Biofuels, 2011, 4(1), 3 CrossRef CAS.
- P. Väljamäe, V. Sild, G. Pettersson and G. Johansson, The initial kinetics of hydrolysis by cellobiohydrolases I and II is consistent with a cellulose surface− erosion model, Eur. J. Biochem., 1998, 253(2), 469–475 CrossRef.
- M. Santa-Maria and T. Jeoh, Molecular-scale investigations of cellulose microstructure during enzymatic hydrolysis, Biomacromolecules, 2010, 11(8), 2000–2007 CrossRef CAS.
- K. Igarashi, A. Koivula, M. Wada, S. Kimura, M. Penttilä and M. Samejima, High Speed Atomic Force Microscopy Visualizes Processive Movement of Trichoderma reesei Cellobiohydrolase I on Crystalline Cellulose, J. Biol. Chem., 2009, 284(52), 36186–36190 CrossRef CAS.
- M. Eibinger, P. Bubner, T. Ganner, H. Planck and B. Nidetzky, Surface structural dynamics of enzymatic cellulose degradation, revealed by combined kinetic and atomic force microscopy studies, FEBS J., 2014, 281(1), 275–290 CrossRef CAS PubMed.
- J. Jung, A. Sethi, T. Gaiotto, J. J. Han, T. Jeoh and S. Gnanakaran, et al., Binding and movement of individual Cel7A cellobiohydrolases on crystalline cellulose surfaces revealed by single-molecule fluorescence imaging, J. Biol. Chem., 2013, 288(33), 24164–24172 CrossRef CAS PubMed.
- A. R. Mudinoor, P. M. Goodwin, R. U. Rao, N. Karuna, A. Hitomi and J. Nill, et al., Interfacial molecular interactions of cellobiohydrolase Cel7A and its variants on cellulose, Biotechnol. Biofuels, 2020, 13(1), 10 CrossRef CAS.
- S. Y. Ding, Y. S. Liu, Y. Zeng, M. E. Himmel, J. O. Baker and E. A. Bayer, How Does Plant Cell Wall Nanoscale Architecture Correlate with Enzymatic Digestibility?, Science, 2012, 338(6110), 1055–1060 CrossRef CAS.
- H. Y. N. Holman, R. Miles, Z. Hao, E. Wozei, L. M. Anderson and H. Yang, Real-Time Chemical Imaging of Bacterial Activity in Biofilms Using Open-Channel Microfluidics and Synchrotron FTIR Spectromicroscopy, Anal. Chem., 2009, 81(20), 8564–8570 CrossRef CAS PubMed.
- S. C. Corgié, H. M. Smith and L. P. Walker, Enzymatic transformations of cellulose assessed by quantitative high-throughput fourier transform infrared spectroscopy (QHT-FTIR), Biotechnol. Bioeng., 2011, 108(7), 1509–1520 CrossRef.
- N. Kruer-Zerhusen, B. Cantero-Tubilla and D. B. Wilson, Characterization of cellulose crystallinity after enzymatic treatment using Fourier transform infrared spectroscopy (FTIR), Cellulose, 2018, 25(1), 37–48 CrossRef CAS.
- Y. Cao and H. M. Tan, Effects of cellulase on the modification of cellulose, Carbohydr. Res., 2002, 337(14), 1291–1296 CrossRef CAS.
- D. L. Sills and J. M. Gossett, Using FTIR to predict saccharification from enzymatic hydrolysis of alkali-pretreated biomasses, Biotechnol. Bioeng., 2012, 109(2), 353–362 CrossRef CAS PubMed.
- I. Spiridon, C. A. Teacă and R. Bodîrlău, Structural changes evidenced by FTIR spectroscopy in cellulosic materials after pre-treatment with ionic liquid and enzymatic hydrolysis, BioResources, 2010, 6(1), 400–413 Search PubMed.
- M. F. Devaux, F. Jamme, W. André, B. Bouchet, C. Alvarado and S. Durand, et al., Synchrotron Time-Lapse Imaging of Lignocellulosic Biomass Hydrolysis: Tracking Enzyme Localization by Protein Autofluorescence and Biochemical Modification of Cell Walls by Microfluidic Infrared Microspectroscopy, Front. Plant Sci., 2018, 9, 200–200 CrossRef.
- K. Loutherback, G. Birarda, L. Chen and H. Y. N. Holman, Microfluidic Approaches to Synchrotron Radiation-Based Fourier Transform Infrared (SR-FTIR) Spectral Microscopy of Living Biosystems, Protein Pept. Lett., 2016, 23(3), 273–282 CrossRef CAS.
- N. Gierlinger, L. Goswami, M. Schmidt, I. Burgert, C. Coutand and T. Rogge, et al., In situ FT-IR microscopic study on enzymatic treatment of poplar wood cross-sections, Biomacromolecules, 2008, 9(8), 2194–2201 CrossRef CAS.
- H. Y. Holman, W. Zhao and S. R. Narayanasamy, Devices and methods for spectroscopy of biomaterials and live cells, WO2022226148A1, 2022.
- H.Y. Holman, W. Zhao, J. Nill, L. Chen, S.R. Narayanasamy and T. Jeoh, An open-channel microfluidic membrane device for in situ hyperspectral mapping of enzymatic cellulose hydrolysis, 2021 Search PubMed.
- W. Zhao, H. Y. Holman and T. Jeoh, Time and Spatially-Resolved Fourier-Transform Infrared (FTIR) Spectromicroscopy of Cellulose in Buffered Reactions with Trichoderma reesei Cel7A [Dataset], Dryad, 2023, DOI:10.25338/B8NP9P.
- K. Hofstetter, B. Hinterstoisser and L. Salmén, Moisture uptake in native cellulose – the roles of different hydrogen bonds: a dynamic FT-IR study using Deuterium exchange, Cellulose, 2006, 13(2), 131–145 CrossRef CAS.
- A. M. Olsson and L. Salmén, The association of water to cellulose and hemicellulose in paper examined by FTIR spectroscopy, Carbohydr. Res., 2004, 339(4), 813–818 CrossRef CAS PubMed.
- Y. Nishiyama, Structure and properties of the cellulose microfibril, J. Wood Sci., 2009, 55(4), 241–249 CrossRef CAS.
- Y. Nishiyama, H. Chanzy, M. Wada, J. Sugiyama, K. Mazeau and V. T. Forsyth, Synchrotron and X-ray Neutron Fiber Diffraction Studies of Cellulose Polymorphs, Adv. X-Ray Anal., 2002, 45, 385–390 CAS.
- J. J. Cael, K. H. Gardner, J. L. Koenig and J. Blackwell, Infrared and Raman spectroscopy of carbohydrates. Paper V. Normal coordinate analysis of cellulose I, J. Chem. Phys., 1975, 62(3), 1145–1153 CrossRef CAS.
- R. H. Marchessault and P. R. Sundararajan, Chapter 2 - Cellulose, in The Polysaccharides, ed. G.O Aspinall, Academic Press, 1983, pp. 11–95. Available from: https://www.sciencedirect.com/science/article/pii/B9780120656028500078 Search PubMed.
- M. C. McCann, N. J. Stacey, R. Wilson and K. Roberts, Orientation of macromolecules in the walls of elongating carrot cells, J. Cell Sci., 1993, 106(4), 1347–1356 CrossRef CAS PubMed.
- L. Chen, R. H. Wilson and M. C. McCann, Infra-red microspectroscopy of hydrated biological systems: design and construction of a new cell with atmospheric control for the study of plant cell walls, J. Microsc., 1997, 188(1), 62–71 CrossRef CAS.
- M. Makarem, C. M. Lee, K. Kafle, S. Huang, I. Chae and H. Yang, et al., Probing cellulose structures with vibrational spectroscopy, Cellulose, 2019, 26(1), 35–79 CrossRef CAS.
- R. H. Wilson, A. C. Smith, M. Kačuráková, P. K. Saunders, N. Wellner and K. W. Waldron, The Mechanical Properties and Molecular Dynamics of Plant Cell Wall Polysaccharides Studied by Fourier-Transform Infrared Spectroscopy1, Plant Physiol., 2000, 124(1), 397–406 CrossRef CAS PubMed.
- R. H. Newman and T. C. Davidson, Molecular conformations at the cellulose–water interface, Cellulose, 2004, 11(1), 23–32 CrossRef CAS.
- R. J. Viëtor, R. H. Newman, M. A. Ha, D. C. Apperley and M. C. Jarvis, Conformational features of crystal-surface cellulose from higher plants, Plant J., 2002, 30(6), 721–731 CrossRef PubMed.
- S. Park, D. K. Johnson, C. I. Ishizawa, P. A. Parilla and M. F. Davis, Measuring the Crystallinity Index of Cellulose by Solid State 13C Nuclear Magnetic Resonance, Cellulose, 2009, 16(4), 641–647 CrossRef CAS.
- J. P. Olsen, B. S. Donohoe, K. Borch, P. Westh and M. G. Resch, Interrelationships between cellulase activity and cellulose particle morphology, Cellulose, 2016, 23(4), 2349–2361 CrossRef CAS.
- M. Schwanninger, J. C. Rodrigues, H. Pereira and B. Hinterstoisser, Effects of short-time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose, Vib. Spectrosc., 2004, 36(1), 23–40 CrossRef CAS.
- N. Kotov, P. A. Larsson, K. Jain, T. Abitbol, A. Cernescu and L. Wågberg, et al., Elucidating the fine-scale structural morphology of nanocellulose by nano infrared spectroscopy, Carbohydr. Polym., 2023, 302, 120320 CrossRef CAS PubMed.
- A. M. A. Nada, S. H. Mohamed, S. Abd El Mongy and R. Seoudi, Preparation, vibrational structure and dielectric properties studies of cotton linter and its derivatives, J. Non-Cryst. Solids, 2009, 355(52–54), 2544–2549 CrossRef CAS.
- M. L. Nelson and R. T. O'Connor, Relation of certain infrared bands to cellulose crystallinity and crystal lattice type. Part II. A new infrared ratio for estimation of crystallinity in celluloses I and II, J. Appl. Polym. Sci., 1964, 8(3), 1325–1341 CrossRef CAS.
- C. M. Lee, J. D. Kubicki, B. Fan, L. Zhong, M. C. Jarvis and S. H. Kim, Hydrogen-Bonding Network and OH Stretch Vibration of Cellulose: Comparison of Computational Modeling with Polarized IR and SFG Spectra, J. Phys. Chem. B, 2015, 119(49), 15138–15149 CrossRef CAS PubMed.
- Y. Marechal and H. Chanzy, The hydrogen bond network in Iβ cellulose as observed by infrared spectrometry, J. Mol. Struct., 2000, 523(1–3), 183–196 CrossRef CAS.
- G. T. Beckham, J. F. Matthews, B. Peters, Y. J. Bomble, M. E. Himmel and M. F. Crowley, Molecular-Level Origins of Biomass Recalcitrance: Decrystallization Free Energies for Four Common Cellulose Polymorphs, J. Phys. Chem. B, 2011, 115(14), 4118–4127 CrossRef CAS PubMed.
- J. Jalak and P. Valjamae, Mechanism of Initial Rapid Rate Retardation in Cellobiohydrolase Catalyzed Cellulose Hydrolysis, Biotechnol. Bioeng., 2010, 106(6), 871–883 CrossRef CAS.
- M. Kurasin and P. Valjamae, Processivity of Cellobiohydrolases Is Limited by the Substrate, J. Biol. Chem., 2011, 286(1), 169–177 CrossRef CAS PubMed.
- J. F. Matthews, C. E. Skopec, P. E. Mason, P. Zuccato, R. W. Torget and J. Sugiyama, et al., Computer simulation studies of microcrystalline cellulose I beta, Carbohydr. Res., 2006, 341(1), 138–152 CrossRef CAS PubMed.
- R. J. Maurer, A. F. Sax and V. Ribitsch, Molecular simulation of surface reorganization and wetting in crystalline cellulose I and II, Cellulose, 2013, 20(1), 25–42 CrossRef CAS.
- X. H. Qian, The effect of cooperativity on hydrogen bonding interactions in native cellulose I beta from ab initio molecular dynamics simulations, Mol. Simul., 2008, 34(2), 183–191 CrossRef CAS.
- J. B. Taylor, The Water Solubilities and Heats of Solution of Short Chain Cellulosic Oligosaccharides, Trans. Faraday Soc., 1957, 53(9), 1198–1203 RSC.
- L. Bu, M. E. Himmel and M. F. Crowley, The molecular origins of twist in cellulose I-beta, Carbohydr. Polym., 2015, 125, 146–152 CrossRef CAS.
- A. J. Bowling, Y. Amano, R. Lindstrom and M. B. J. Brown, Rotation of Cellulose Ribbons During Degradation with Fungal Cellulase, Cellulose, 2001, 8, 91–97 CrossRef CAS.
- P. N. Ciesielski, R. Wagner, V. S. Bharadwaj, J. Killgore, A. Mittal and G. T. Beckham, et al., Nanomechanics of cellulose deformation reveal molecular defects that facilitate natural deconstruction, Proc. Natl. Acad. Sci. U. S. A., 2019, 201900161 Search PubMed.
- T. Uchiyama, T. Uchihashi, T. Ishida, A. Nakamura, J. V. Vermaas and M. F. Crowley, et al., Lytic polysaccharide monooxygenase increases cellobiohydrolases activity by promoting decrystallization of cellulose surface, Sci. Adv., 2022, 8(51), eade5155–eade5165 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1 Cellulose spectra and peak assignments; 2 Cellulose dehydration; 3 Water levels during enzymatic hydrolysis; 4 IR peak intensities over time; 5 LOI and HBI. See DOI: https://doi.org/10.1039/d3gc03279e |
| ‡ Current address: Amyris, Emeryville, CA, USA. |
| § These authors contributed equally to this manuscript. |
| ¶ Current address: Genus PLC, DeForest, Wisconsin, USA. |
| || Current address: Biosciences and Biotechnology Division, Physical and Life Science, Lawrence Livermore National Laboratory, Livermore, CA, USA. |
| ** Current address: TikTok, Mountain View, California, USA. |
| This journal is © The Royal Society of Chemistry 2024 |