
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved chemical recyclability of 2,5-furandicarboxylate polyesters enabled by acid-sensitive spirocyclic ketal units†
Nitin G.
Valsange
a,
Niklas
Warlin
 a,
Smita V.
Mankar
a,
Nicola
Rehnberg
b,
Baozhong
Zhang
*a and
Patric
Jannasch
*a
a,
Smita V.
Mankar
a,
Nicola
Rehnberg
b,
Baozhong
Zhang
*a and
Patric
Jannasch
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: patric.jannasch@chem.lu.se; baozhong.zhang@chem.lu.se
bBona Sweden AB, Box 210 74, 200 21 Malmö, Sweden
First published on 22nd January 2024
Abstract
Incorporating hydrolytically sensitive functional groups into polymer backbones provides a feasible strategy to trigger their degradation to the starting monomers, thus enabling chemical recycling of the material. Here, we present two series of copolyesters in which a biobased spirocyclic ketal-functional diester monomer was incorporated into poly(butylene 2,5-furandicarboxylate) (PBLF) and poly(hexamethylene 2,5-furandicarboxylate) (PHLF), respectively. A two-step melt polycondensation resulted in copolyesters with moderate to high molecular weights, as confirmed by intrinsic viscosity and size exclusion chromatography data. Thermogravimetric analysis showed a thermal stability up to 275 °C, and increasing char yields upon incorporation of the spirocyclic monomer. The crystallinity and melting points of the copolyesters decreased with an increasing content of the spirocyclic ketal units in the backbone. Copolyesters containing up to 15% of the spiro-ketal units were semicrystalline, while those containing 20 and 50% spiro-ketal units were completely amorphous. The hydrolytic degradation of copolyesters from the PHLF series was investigated using 3–12 M aq. HCl, and were found to degrade faster than the corresponding homopolyesters. Acid-catalyzed cleavage of the randomly distributed spiro ketal units promoted the rapid fragmentation of the polymer chain into small oligomers, which were subsequently hydrolyzed to the original chemical building blocks. The ketone-terminated telechelic oligomers obtained after the degradation of spirocyclic ketal units were also investigated in a direct polymerization with pentaerythritol. The initial results implied that the oligomers can be re-polymerized into the original polymer. Hence, this work demonstrated a feasible pathway towards chemically recyclable 2,5-furandicarboxylate polyesters with a tuneable degree of crystallinity.
1. Introduction
Over the last two decades, the development of biobased plastics has gained significant interest as an alternative to fossil-based plastics to mitigate the environmental concerns of the latter, including the depletion of fossil resources and the generation of greenhouse gas (GHG) emissions.1,2 Biobased plastics can tackle these problems by reducing the demand for fossil fuels used for their production.3,4 In addition, they help to lower the total GHG emissions because the carbon emissions during the production and end-of-life treatment (e.g. recycling, degradation or decomposition) can be absorbed by plants through photosynthesis to reproduce biomass.5 Although biobased plastics are interesting from the consumer point of view, still most of these materials suffer from the same end-of-life issues as conventional fossil-based plastics. Therefore, there is an increased focus on end-of-life recycling of biobased polymers. Mechanical recycling is one of the common and industrially adopted recycling strategies. However, it typically results in a substantial quality loss, and the recycled material is often not suitable for high-performance applications. Lately, chemical recycling has emerged as an attractive alternative, in which polymer wastes are depolymerized to obtain the starting monomers/building blocks or oligomers, which are then repolymerized to obtain virgin or new polymeric materials with a similar quality as the original.6–11 This circular use of plastics offers not only a feasible solution to the end-of-use issue of materials, but also provides a closed-loop approach towards the circular economy.12 As a result, there have been huge demands for newly designed biobased polymers to enable and facilitate their chemical recycling.Polyesters are an important class of polymers mainly used in packaging, textiles, and biomedical applications. The chemical recycling of polyesters involves solvolysis of the functional ester group in the polymer backbone by either hydrolysis, methanolysis or glycolysis to recover the corresponding monomers and oligomers.13,14 All of these methods have been employed commercially for the chemical recycling of poly(ethylene terephthalate) (PET) waste.15 Hydrolysis of PET, which yields high purity terephthalic acid (TPA), is particularly attractive due to the relatively low cost compared to other methods.16 However, a complete hydrolysis of PET usually requires harsh degradation conditions such as elevated temperature and pressure with the use of highly concentrated acid or base solutions due to the high hydrophobicity and degree of crystallinity of the aromatic polyester.17,18 For example, Yoshioka et al. reported 100% hydrolysis of PET into TPA and ethylene glycol using 10 M sulfuric acid solution at 150 °C.19 Another study reports on TPA recovery from PET scrap material using 18 M sulfuric acid (87%) at 60–93 °C.20 Substantial drawbacks of such hydrolysis methods are the significantly high production cost and the generation of large quantities of inorganic salts and aqueous wastes, which makes the process expensive and less green. Therefore, there is a strong driving force for the development of biobased polyesters that are able to degrade at a high rate under mild conditions.21,22
Incorporation of labile acid-sensitive functional groups (e.g., acetal or ketal) as weak link into the polymer structure is an attractive strategy to enable chemical recycling, because they can be conveniently hydrolyzed with enhanced hydrolysis rate.23,24 Several acetal and ketal monomers have been utilized to prepare rapidly hydrolyzable polymers.25,26 In particular those with rigid cyclic structures have attracted considerable attention because of their ability to simultaneously improve the thermo-mechanical properties and chemical recyclability of the polymers. For instance, a commercially available rigid spirocyclic diol (SPG) with acetal units has been incorporated into poly(butylene terephthalate), and the resulting copolyesters displayed enhanced glass transition temperature (Tg) and toughness in addition to an accelerated hydrolysis rate under acidic conditions.27 The improved hydrolysis was related to the acid-triggered degradation of the acetal groups, which caused efficient scission of the long polymer chains into shorter chains. Subsequently, the shorter oligoester chains degraded faster due to the reduced steric hindrance for water molecules approaching the ester group. In another study, a cyclic diol with ketal units was synthesized and copolymerized with ethylene glycol and dimethyl terephthalate to obtain a series of copolyesters containing varying contents of the cyclic diol.28 The copolyesters demonstrated tuneable degradation under acid conditions where the susceptibility towards hydrolysis increased with the content of the spiro diol in the polymer. In addition, many other acetal and ketal building blocks have been investigated to develop chemically recyclable polymers, including polyesters, polyurethanes, polycycloacetals, and others.29–33 Besides accelerating the degradation of the polymers into the starting building blocks, the use acetal and ketal bonds have also been extended to achieve recycling via the telechelic oligomer/polymer approach. For example, the acetal bonds in poly(acetal-ester)s have been selectively hydrolyzed under acidic conditions to yield aldehyde end-capped telechelic polyesters, which were repolymerized to poly(acetal-ester)s via cycloacetalization with pentaerythritol.8 Since the process requires only fewer bonds to be broken and reformed, it was presented as short-loop recycling and energy efficient as large fraction of initial bond-forming energy was retained.
Sugar is a valuable resource in the production of new biobased monomers for polyester development. 2,5-Furan dicarboxylic acid (FDCA) is the most well-known sugar-based monomer and is obtained by oxidation of 5-hydroxymethylfurfural (HMF).34,35 FDCA has been identified as one of the top value-added chemicals derived from biomass by the U.S. Department of Energy.36 The physical and chemical properties of FDCA mimic those of terephthalic acid (TPA) to a large extent. Thus, it is projected to become a potential replacement of fossil-based TPA in poly(ethylene terephthalate) (PET) production, yielding a 100% biobased poly(ethylene 2,5-furandicarboxylate) (PEF). Additionally, several polyesters of FDCA with various diols have been reported.37–39 Among them, poly(butylene 2,5-furandicarboxylate) (PBF) is viewed as a suitable counterpart of poly(butylene terephthalate) (PBT), especially for fiber and packaging applications. Another benchmark example is poly(hexamethylene 2,5-furandicarboxylate) (PHF), the biobased counterpart of poly(hexamethylene terephthalate) (PHT). In principle, FDCA-based polyesters have a similar tendency towards hydrolysis to those of TPA-based polymers. Therefore, the inclusion of acid-labile acetal/ketal structures into FDCA-based polyesters is expected to enhance their hydrolytic degradation under acidic conditions, which may enable chemical recycling.
In the current paper, we present two series of copolyesters where a cyclo ketal functional spirocyclic diester monomer has been incorporated into the PBF and PHF-base polymers, respectively. The spirocyclic diester was obtained by an acid-catalyzed ketalization of biobased ethyl levulinate and pentaerythritol.40 Using optimized polymerization conditions, two series of copolyesters with various contents of spiro-diester units, respectively, were prepared. The materials were then evaluated with regard to thermal and mechanical properties. Acid hydrolysis experiments were performed on selected copolyesters, and the results indicated that the presence of ketal-functional spiro monomer in the copolyesters significantly enhanced their hydrolytic degradation rate compared to the PHF polyester. Hence, ketal unit containing spiro-diesters are an important building block for the preparation of polyesters and other condensation polymers with improved chemical recyclability.
2. Experimental section
2.1. Materials
Ethyl levulinate (99%), 1,4-butanediol (1,4-BD, >99%), 1,6-hexanediol (1,6-HD, 97%), diethyl 2,5-furandicarboxylate (DEFDC, 98%), dibutyltin(IV)oxide (DBTO, 98%), potassium carbonate (≥99%), and chloroform-d (99.8% atom D) were purchased from Sigma Aldrich. Sulfuric acid (H2SO4, 95%), hydrochloric acid (HCl, 37%), sodium sulfate (anhydrous, ≥99% ACS), ethyl acetate (≥99%), chloroform (99.1%, stabilized with 0.6% ethanol), heptane, toluene, and methanol were purchased from VWR chemicals. Hexafluroisopropanol (HFIP) was purchased from Nanjing Confidence Chemical Co., Ltd. Biobased pentaerythritol (Voxtar™, 99%) was kindly supplied by Perstorp AB. Monomer L was prepared according to the previously published method.40 All chemicals and reagents were used as received.2.2. Polyester synthesis
Two series of copolyesters, denoted PBLFs and PHLFs were prepared by copolymerization of Monomer L and diethyl 2,5-furandicarboxylate together with 1,4-BD and 1,6-HD, respectively. For convenience, the copolyesters are denoted as PBLF-x and PHLF-x, respectively, where x represents the mole% of Monomer L compared to the total diethyl ester content. A two-step melt polymerization protocol involving transesterification and polycondensation steps was used for polyester synthesis. To compensate for the loss of diol via evaporation during the polymerizations, a 10 mol% excess of 1,4-BD with respect to the diester monomers (DEFDC and Monomer L) was used for the copolymers in the PBLF series, while a 5 mol% excess of 1,6-HD was used for the PHLF series. The corresponding homopolyesters PBF and PHF were also synthesized as benchmark materials (synthesis described in the ESI†).1H NMR (400.13 MHz, CDCl3, δ, ppm): 1.35 (s, 6H), 1.64–1.85 (m, 4H), 1.85–1.95 (m, 4H), 1.95–2.03 (m, 4H), 2.38–2.46 (m, 4H), 3.56 (s, 4H), 3.75 (d, J = 11.8 Hz, 2H), 3.93 (d, J = 11.7 Hz, 2H), 4.10 (q, J = 7.6, 8.9 Hz, 4H), 4.39 (q, J = 3.2 Hz, 4H), 7.19 (s, 2H).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 19.65, 19.69, 25.32, 25.37, 25.41, 28.43, 32.22, 33.94, 34.00, 63.83, 63.88, 63.96, 64.98, 65.11, 99.06, 118.49, 118.56, 146.84, 146.92, 158.06, 173.74.
The other copolyesters in the PBLF series (PBLF-10, 15, 20, and 50) were prepared in the same manner as PBLF-5.
1H NMR (400.13 MHz, CDCl3, δ, ppm): 1.37 (s, 6H), 1.38–1.53 (m, 8H), 1.61–1.68 (m, 4H), 1.71–1.84 (m, 4H), 1.96–2.05 (m, 4H), 2.38–2.46 (m, 4H), 3.58 (s, 4H), 3.76 (d, J = 11.8 Hz, 2H), 3.93 (d, J = 11.8 Hz, 2H), 4.02–4.09 (m, 4H), 4.28–4.37 (m, 4H), 7.19 (s, 2H).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 19.63, 25.55, 25.61, 28.38, 28.50, 32.14, 33.90, 63.77, 63.81, 64.32, 65.44, 65.49, 98.99, 118.34, 146.89, 158.12, 173.78.
The other copolyesters in the PHLF series (PHLF-5, 12.5, 15, and 20) were prepared in the same manner as PHLF-10.
2.3. Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TFA mixture. Chemical shifts were reported as δ values (ppm).
:TFA mixture. Chemical shifts were reported as δ values (ppm).
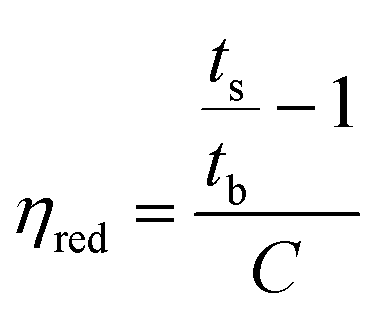 | (1) |
 | (2) |
Differential scanning calorimetry (DSC) measurements were performed on a DSC Q2000 analyzer from TA Instruments. Glass transition temperatures (Tgs), melting temperatures (Tms) and heats of fusion (ΔHms) were determined from the second heating cycle where the samples were heated from −50 to 200 °C. Crystallization temperatures (Tcs) measured from the DSC cooling curve.
2.4. Acid hydrolysis and chemical recycling
The obtained t-PHF was investigated in a direct polymerization with pentaerythritol. Into a 50 ml three-neck round bottom flask equipped with a mechanical stirrer, a nitrogen inlet, and a vacuum distillation outlet were placed t-PHF (1.00 g, 1.24 mmol of ketone end groups), pentaerythritol (85 mg, 0.62 mmol), and p-TSA (8 mg). Subsequently, the reaction mixture was degassed by three successive vacuum-nitrogen cycles at room temperature. The reaction mixture was initially heated at 170 °C under nitrogen until a homogenous melt was obtained. Afterwards, a vacuum was applied to the flask over 30 min, and the reaction was stirred for an additional 2.5 h. Next, the highly viscous reaction mixture was dissolved in chloroform (10 mL), and the product was precipitated in 200 mL cold methanol. The precipitate was washed with fresh methanol (2 × 50 mL) and dried under vacuum to obtain the repolymer (r-PHLF-8.3) as a white solid (0.74 g, 69.0% yield).
3. Results and discussion
3.1. Polyester synthesis
The ketal-containing spiro-diester monomer (Monomer L, Scheme 1) was incorporated in the backbone structures of polyesters based on 2,5-furandicarboxylic acid to investigate its influence on the physical properties and the hydrolytic degradation behaviour of the polyesters. Two series of copolyesters, denoted PBLF and PHLF, were prepared by reacting mixtures of Monomer L and diethyl 2,5-furandicarboxylate (DEFDC) with 1,4-BD and 1,6-HD, respectively, using DBTO catalyst (Scheme 1). For convenience, the copolyesters are denoted as PBLF-x and PHLF-x, respectively, where x represents the mole% of Monomer L compared to the total diethyl ester content.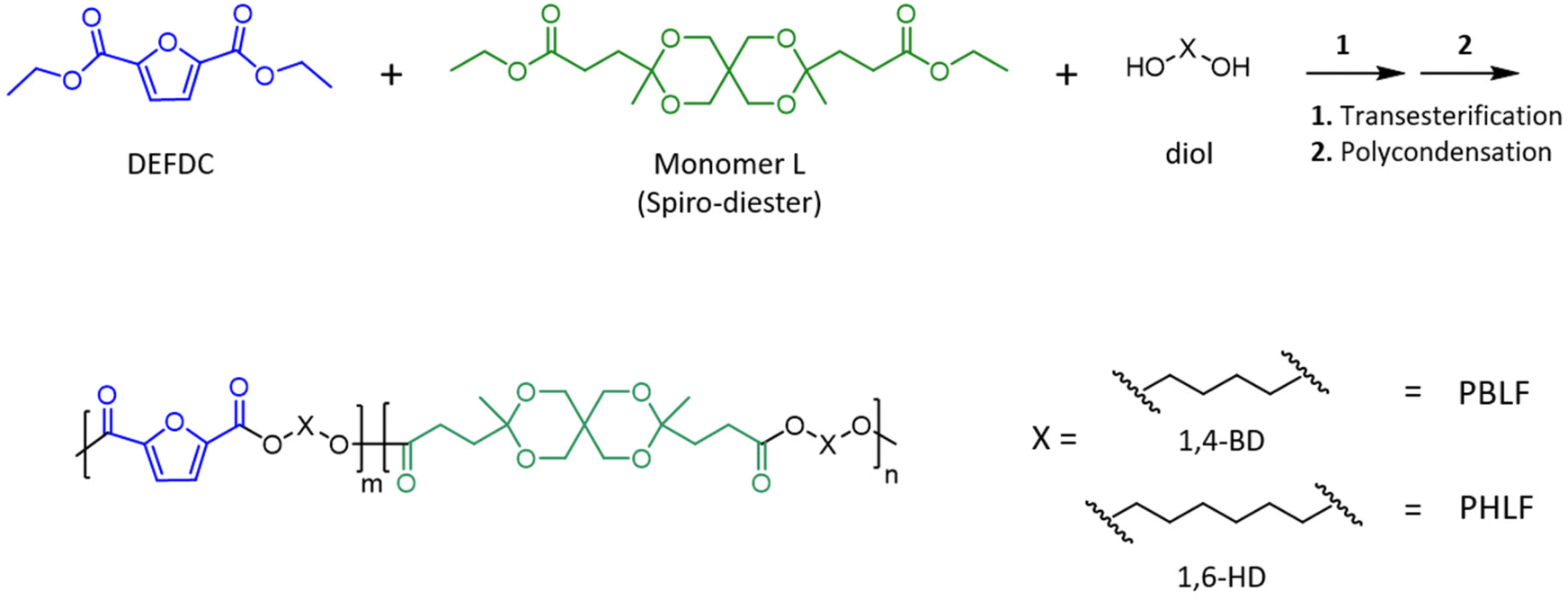 | ||
| Scheme 1 Synthesis of the copolyesters in the PBLF and PHLF series via a two-step melt polycondensation method. | ||
As a starting point, the homopolyesters PBF and PHF were prepared as benchmark materials. A two-step melt polycondensation method involving transesterification and polycondensation steps was developed to prepare the polyesters. A 10 mol% and 5 mol% excess of the diols (1,4-BD and 1,6-HD, respectively) was used in the preparation of PBF and PHF, respectively, in order to compensate for the loss of diol through evaporation during the polymerizations. In both cases, the transesterification was performed at 180 °C during 5 h under a nitrogen atmosphere, and the polycondensation was conducted at 200 °C for 3 h using a high vacuum. At the beginning of the polycondensation step, a vacuum was applied slowly over a period of 15 min to avoid oligomer sublimation at the higher temperature.
The copolymerizations involving the spiro-diester could not be performed under the conditions used for the homopolyesters due to the hydrolytic degradation of the ketal units of Monomer L at higher temperatures. We have previously studied the thermal stability of Monomer L and found it to be extremely sensitive to residual moisture.40 It showed degradation even below 100 °C. On the other hand, after the removal of the water by azeotropic distillation using toluene or high vacuum drying over 3 h, Monomer L showed almost no degradation up to 130 °C, and only very slight degradation between 140–160 °C. Thus, an azeotropic distillation of the reaction mixture was carried out before performing the copolymerizations at elevated temperature. However, copolymerizations at 130 °C resulted in the crystallization of oligomers formed during the transesterification step of the PBLFs syntheses, which gave only low molecular weight products. To avoid this, the temperature was increased stepwise from 130 to 150 °C during the 5 h period of the transesterification step under a nitrogen atmosphere. Thereafter, the polycondensation was performed at 160 °C under a high vacuum for 3 h. Still, it was observed that the reaction mixtures of PBLF-5 and PBLF-10 partially crystallized at the end of the polycondensation due to the high melting temperatures of these copolyesters, close to the polycondensation temperature. In the case of the PHLF copolymers, the transesterification was carried out at 130 °C for 5 h, followed by the polycondensation at 150 °C during 3 h.
After the polycondensations, the viscous reaction mixtures were dissolved in chloroform or hexafluroisopropanol (HFIP), and the products were precipitated in methanol to give the PBLF and PHLF copolyesters in 74–88% and 87–98% isolated yields, respectively (Table 1). The intrinsic viscosity values measured using an Ubbelhode viscometer were in the range 0.47–1.04 and 0.57–0.94 dL g−1 for the PBLF and PHLF series, respectively, indicating the formation of moderate to high molecular weight polymers. This was further confirmed by SEC measurements in chloroform, which showed Mn values in the range 11.4–18.7 and 14.5–42.7 kg mol−1 for the PBLF and PHLF series, respectively. The data of PBF could not be measured by SEC due to insolubility in chloroform. Consequently, the Mn value of this sample was calculated to be 29.5 kg mol−1 by end group analysis from 1H NMR data. The corresponding homopolyesters PBF and PHF showed higher molecular weights than the corresponding copolyesters. The reason behind this is most probably the lower temperature employed during the copolymerizations that hindered an efficient removal of the diol during the polycondensation, which is necessary to reach high molecular weights.
| Polyester | Isolated yielda (%) | [η]b (dL g−1) | M n (kg mol−1) | M n (kg mol−1) | Đ | Spiro-diester (L) contentd | Microstructure analysise | |||
|---|---|---|---|---|---|---|---|---|---|---|
| In feed (%) | In copolymer (%) | n F | n L | R | ||||||
| a Calculated from the weight of the polymer sample obtained after purification. b Intrinsic viscosity [η] was measured in HFIP by using an Ubbelohde viscometer at 25 °C. c Measured by SEC in chloroform against polystyrene standards. d Incorporated content (mol%) of the spiro-diester (L) in the copolymer was estimated by 1H NMR spectroscopy using eqn (3). e Number average sequence length (nF and nL) and degree of randomness (R) were calculated from the 13C NMR spectrum using eqn (4)–(6). n.d. – not determined. | ||||||||||
| PBF | 88 | 1.04 | n.d | n.d | n.d | — | — | — | — | — |
| PBLF-5 | 74 | 0.57 | 12.9 | 40.1 | 3.1 | 5 | 5 | 21.0 | 1.0 | 1.1 |
| PBLF-10 | 79 | 0.47 | 11.4 | 31.1 | 2.7 | 10 | 9.5 | 12.1 | 1.1 | 1.0 |
| PBLF-15 | 83 | 0.57 | 13.7 | 35.6 | 2.6 | 15 | 14 | 7.1 | 1.2 | 1.0 |
| PBLF-20 | 80 | 0.72 | 18.7 | 63.2 | 3.4 | 20 | 20 | 4.3 | 1.2 | 1.1 |
| PBLF-50 | 79 | 0.50 | 15.0 | 39.0 | 2.6 | 50 | 49 | 2.2 | 2.3 | 0.9 |
| PHF | 98 | 0.94 | 42.7 | 83.0 | 1.9 | — | — | — | — | — |
| PHLF-5 | 93 | 0.64 | 17.5 | 43.2 | 2.5 | 5 | 5 | 15.3 | 1.1 | 0.9 |
| PHLF-10 | 94 | 0.66 | 15.7 | 44.7 | 2.8 | 10 | 10 | 8.7 | 1.2 | 1.0 |
| PHLF-12.5 | 87 | 0.64 | 14.5 | 52.4 | 3.6 | 12.5 | 12.3 | 7.1 | 1.2 | 1.0 |
| PHLF-15 | 96 | 0.57 | 14.7 | 43.2 | 2.9 | 15 | 15 | 6.6 | 1.2 | 1.0 |
| PHLF-20 | 90 | 0.58 | 15.3 | 42.2 | 2.8 | 20 | 20 | 4.9 | 1.3 | 1.2 |
The molecular structure of the polyesters was confirmed by 1H NMR spectroscopy and was further corroborated by 13C and 2D NMR investigations (i.e., COSY, HMBC, HMQC and HSQC). The representative 2D NMR spectra of PBF, PBLF-10, PHF, and PHLF-10 are shown in Fig. S3–S18.† In the 1H NMR spectra of the PBLF and PHLF copolymers (Fig. 1), signals in common with the spectra of the PBF and PHF homopolymers were observed, including the signals of the aromatic furanoate residues (signal 1) and the CH2 signals arising from the diol chain segments (signal 2–4). As expected, the signals corresponding to spiro monomer units showed increasing intensity with increasing content in the copolyesters, including the CH2 signals (c and d), the CH3 signal (e) and the CH2 signals of the ketal ring (f, g, and h). In addition, the CH2 signals of the diol chain segments (signals i, j, and k) attached to the spiro monomer units were also observed. Notably, the signals of the CH2 groups in the butylene chain segments closest to the ester links in the PBLF copolymers (2 and i) were observed as two fused triplets individually, giving rise to a total of four triplets corresponding to the four possible dyad fractions of furanoate and spiro monomer units along the polymer chain. Similarly, the remaining CH2 signals of the butylene chain segment (3 and j) were observed as a cluster of signals between 1.64–1.95 ppm due to the multiple resonances from the dyad structures. It should be noted that the signals of butylene furanoate units (signal 1–3) in PBF were shifted slightly downfield due to the different solvent systems (CDCl3:TFA) used for the NMR measurements. Similar behaviour was also observed in the 13C NMR spectrum.
 | ||
| Fig. 1 1H NMR spectra of PBLF and PHLF polyesters produced via melt polycondensation. | ||
In the 1H NMR spectra of the PHLF copolymers, the corresponding CH2 signals (2 and i) of the hexylene chain segment each appeared as a triplet. However, a close inspection revealed that they are originally multiplets resulting from the overlapping of two triplets, demonstrating the presence of possible dyad fractions along the polymer chain. This discrepancy of the dyad signals observed for the PHLF copolymers may be related to the reduced inductive and electronic effects in the longer hexylene chain, as compared to the shorter butylene chain in the PBLF series.
The spiro monomer content in the copolyesters was calculated by comparing the integral of the CH2 signals of the diol chain (2 and i) attached to the ester group of furanoate and spiro monomer:
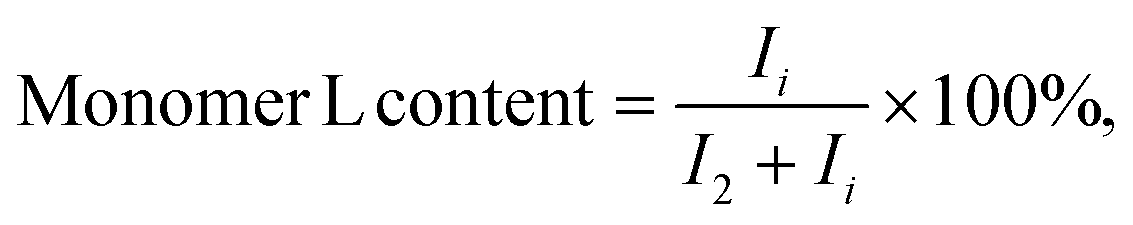 | (3) |
The molecular structure of the copolyesters was further supported by 13C NMR spectroscopy. Fig. S1 and S2† shows stacked 13C NMR spectra of both the PBLFs and PHLF copolyesters, along with the spectra of corresponding homopolyesters. All the signals could be assigned to the carbons in the PBLF and PHLF series of copolyesters. Just like in the 1H NMR spectra, the carbons in the diol chain segments showed chemical sensitivity at the dyad sequence level. In particular, the α-CH2 of the butylene and hexylene chain attached to the oxygen of the ester group split into four recognizable signals (Fig. 2). In addition, the carbons in the furan ring (2, 3) and in the spiro monomer unit (b, e) showed sensitivity to the dyad sequence and split into more than one signal in the two series.
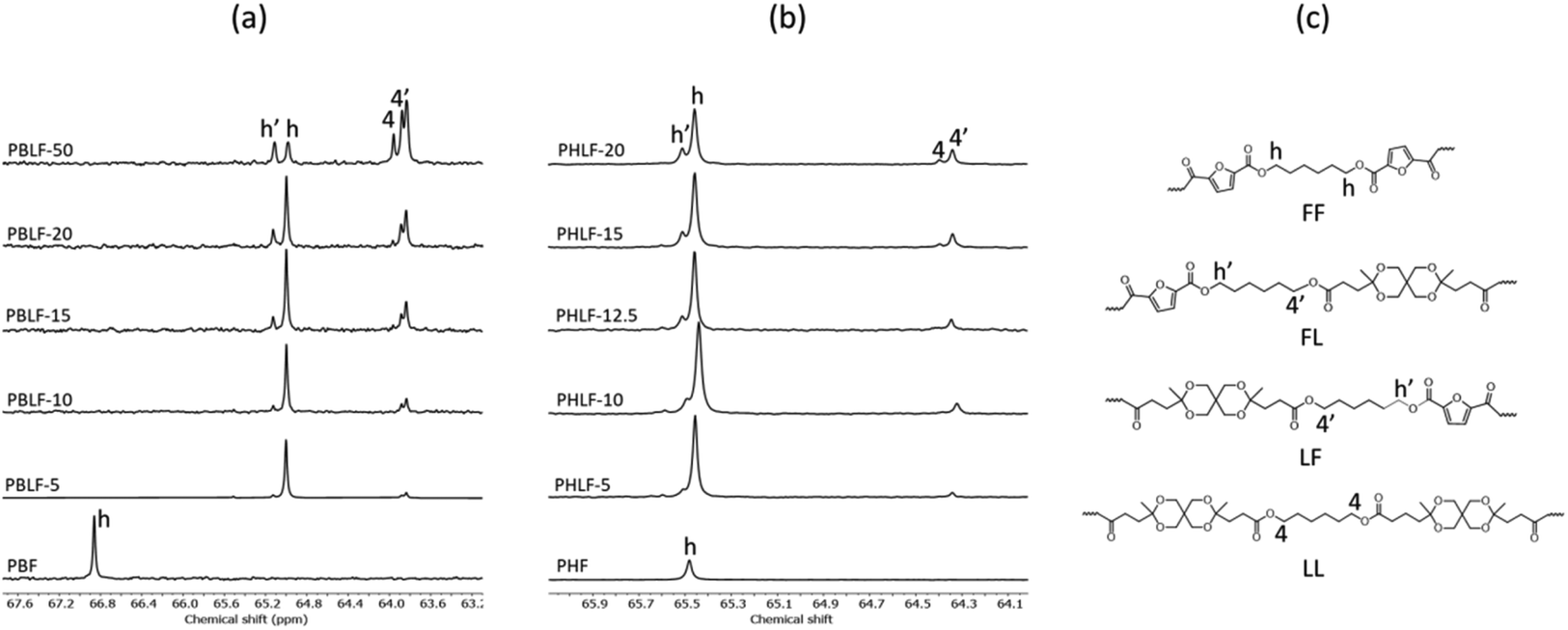 | ||
| Fig. 2 Enlarged 13C NMR spectra of the copolyesters in the PBLF (a) and PHLF (b) series showing the presence of different dyad units (FF, FL, LF, and LL) in the polymer structure (c). | ||
The microstructure of the copolyesters was elucidated by 13C NMR analysis, taking advantage of the sensitivity of the α-carbon of the diol chain segments to the distribution sequence at the dyad level. The four possible dyads, FF, FL, LL, and LF in the PBLF and PHLF copolyesters appeared in the region 63.8–65.2 and 64.2–65.6 ppm, respectively, as four well-resolved signals (h, h′, 4, and 4′) whose intensities were integrated for each copolyesters composition (Fig. 2). The resulting values were used in eqn (4)–(6) to calculate the corresponding number average sequence length and degree of randomness (Table 1):
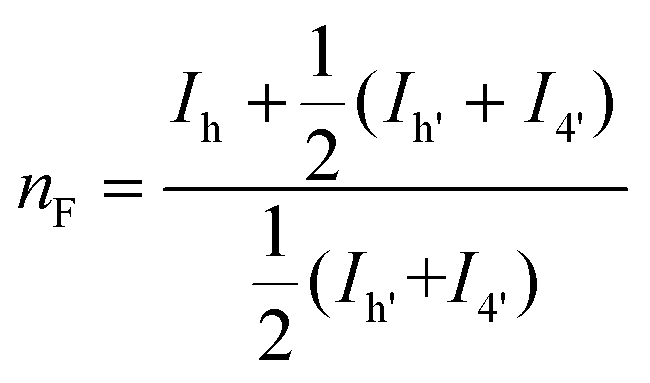 | (4) |
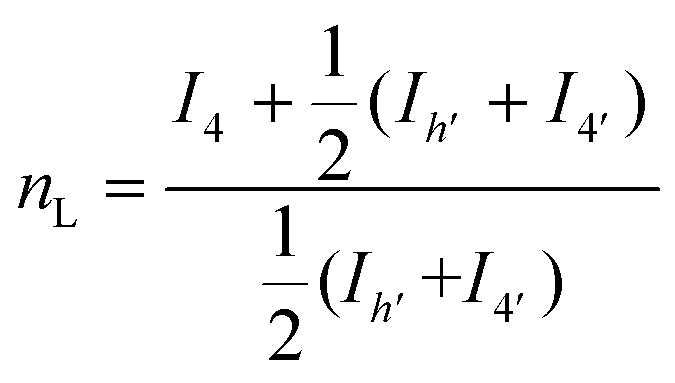 | (5) |
 | (6) |
It must be noted that signal h′ and 4′ should theoretically show nearly the same intensities because of the non-distinguishable structures of FL and LF dyads. However, in PBLF-50 the values significantly deviated from each other due to the overlapping of a neighbouring signal (carbon f, Fig. S1†) with signal 4′. Therefore, in the calculations for PBLF-50, the value of Ih′ was used for signal 4′ instead of I4′.
As can be seen, the sequence length of the furanoate units (nF) in both PBLF and PHLF series decreased with increasing content of the spiro monomer units in the copolyesters, as expected. On the other hand, the sequence length of the spiro monomer units (nL) did not change significantly until 20 mol% of the spiro monomer was incorporated in the copolyester; the nL values ranged between 1.0–1.3. The value of nL increased slightly when the spiro monomer content was increased to 50 mol% in PBLF-50. Expectedly, the values of nF (2.2) and nL (2.3) were almost the same at equimolar amounts of the furanoate and spiro monomer units in the copolymer structure. The calculated randomness (R) value was close to 1 for all copolyesters in the two series, suggesting a random chemical microstructure and a highly statistical distribution of the spiro monomer in the copolymer chains.
3.2. Thermal properties and crystallization behaviours of copolyesters
The thermal stability of the polymers was evaluated by thermogravimetric analysis (TGA) under nitrogen (Fig. S19† and Table 2). For PBF and PHF, a single decomposition rate maximum at Td ∼375 °C was observed, consistent with literature data.41 The copolyesters in the PBLF series showed two decomposition rate maxima (Td ∼300 and 365 °C). The first Td at ∼300 °C was attributed to the degradation of the spirocyclic ketal units in the PBLF backbone, and its intensity increased with spiro monomer content in the backbone. The second decomposition rate maximum at ∼365 °C was ascribed to the degradation of the PBF units in the copolyesters. In contrast, the copolyesters in the PHLF series showed three decomposition rate maxima. The decomposition rate maxima at Td ∼320 and 370 °C were consistent with degradation spirocyclic ketal units and the PHF units in the copolyester backbone, respectively. The observation of a third decomposition rate maxima at Td ∼435 °C may be attributed to the degradation of crosslinked structures formed during the thermal degradation of the spirocyclic ketal unit.42 Oddly, the PBLF copolyesters did not show this feature despite containing spirocyclic ketal units in the backbone. Furthermore, Td,5 (the temperature at 5% weight loss) was above 275 °C for all the copolyesters, indicating the possibility for melt processing. The Td,5 values showed a general decreasing trend as increasing contents of spiro units were incorporated in the copolyesters. On the other hand, the char yields (CY) increased with the spiro monomer contents, which was consistent with the results for polyesters with rigid building blocks.43| Polyester | T d, max (°C) | T d,5 (°C) | Char yieldc (wt%) | T g (°C) | T m (°C) | ΔHmf (J g−1) | T c (°C) | T cc (°C) |
|---|---|---|---|---|---|---|---|---|
| a The temperature at which a local maximum of the rate decomposition was observed by TGA. b The temperature at 5% weight loss by TGA. c Measured by TGA. d Glass transition temperature measured from the second heating DSC scan. e Crystalline melting point measured from the second heating DSC scan. f Enthalpy of melting measured from the second heating DSC scan. g Crystallization temperature measured from the first cooling DSC scan. h Cold-crystallization temperature measured from the second heating DSC scan. | ||||||||
| PBF | 375 | 342 | 2 | 39 | 168 | 33 | 104 | 101 |
| PBLF-5 | 285, 360 | 313 | 10 | 37 | 165 | 36 | 113 | — |
| PBLF-10 | 290, 367 | 306 | 11 | 34 | 158 | 29 | 99 | 108 |
| PBLF-15 | 306, 365 | 294 | 13 | 34 | 150 | 2 | — | 121 |
| PBLF-20 | 304, 363 | 294 | 13 | 34 | — | — | — | — |
| PBLF-50 | 323, 367 | 291 | 20 | 28 | — | — | — | — |
| PHF | 372 | 335 | 5 | 17.5 | 143 | 42 | 94 | — |
| PHLF-5 | 311, 372, 436 | 317 | 10 | 13 | 136 | 46 | 73 | 69 |
| PHLF-10 | 317, 369, 436 | 310 | 12 | 12.5 | 129 | 38 | 67 | 75 |
| PHLF-2.5 | 316, 370, 436 | 308 | 12 | 13 | 125 | 16 | — | 90 |
| PHLF-15 | 316, 372, 434 | 308 | 13 | 13.5 | 122 | 4 | — | 95 |
| PHLF-20 | 325, 372, 438 | 308 | 13 | 13 | 118 | 0.4 | — | — |
The thermal properties and melt crystallization behavior of the polyesters were evaluated by differential scanning calorimetry (DSC), and the corresponding second heating and first cooling curves are shown in Fig. 3a–d, and the results are presented in Table 2. The homopolyesters PBF and PHF are well-known semicrystalline polymers44,45 and exhibited melt transitions during heating as well as exothermic crystallization during cooling from the melt. As can be seen, PBF showed a melting point at Tm = 168 °C and crystallization temperature at Tc = 104 °C, while PHF showed the Tm and Tc at 143 and 94 °C, respectively. These values were comparable to those reported in the literature (PBF, Tm = 170 °C and Tc = 103 °C; PHF, Tm = 147 °C and Tc = 95 °C).46,47 PHF possessed higher melt enthalpy than PBF, demonstrating a higher degree of crystallinity. Furthermore, the crystallization exotherm of PBF was much broader as compared to that of PHF, indicating its slower melt crystallization under the experimental conditions. As a result of that, PBF exhibited an exothermic cold crystallization peak during the subsequent heating cycle. Therefore, the higher crystallizability of PHF is most probably attributed to the presence of the more flexible hexamethylene chain, as compared to the shorter butylene chain in PBF. In both copolyester series, as expected, the incorporation of the spiro monomer suppressed the crystallinity in the copolyesters. This can be seen from the decreasing values of both the melting temperature (Tm) and heat of enthalpy (ΔHm) values with the increasing spiro monomer content. This is explained by the irregular structure of the statistical copolymers, and that the close-packing of the polymer chains was disrupted by the rigid spiro monomer. PBLF-5 and PHLF-5 deviated from this trend and possessed melt enthalpies of 36 and 46 J g−1, respectively, which were slightly higher than for the corresponding homopolyesters. The higher crystallinity of these copolyesters was most probably due to the combined effect of their lower molecular weight compared to the corresponding homopolyesters, and the low spiro monomer content in relation to the other copolyesters in the series. The reduction of the crystallinity with the spiro monomer content was comparatively less pronounced in the case of PHLF series compared to the PBLF one. This may again relate to the higher crystallinity of PHF as compared to PBF. For instance, copolyesters with up to 20% of spiro monomer exhibited crystallinity in the PHLF series, although the endotherm for PHLF-20 was very minor (Tm = 118 °C and ΔHm = 0.4 J g−1). In the PBLF series, the crystallinity was completely lost after the incorporation of more than 15% of spiro diester monomer and thus PBLF-20 was an amorphous polymer. Although copolyesters with up to 15% or 20% spiro diester monomer showed the presence of crystallinity, only those containing 5 and 10% of that monomer were able to crystallize from the melt, as indicated by the appearance of crystallization peak during the cooling cycle. Still, during subsequent heating they tended to cold crystallize (except PBLF-5), demonstrating some crystallization from the melt. This indicated that the incorporation of the spiro monomer retarded the rate of crystallization in copolyesters due to its rigid and non-planar nature. Thus, the observed endothermic melt peaks in other copolyesters (i.e., PBLF-15, 20 and PHLF-15, 20) resulted mainly from cold crystallization during heating since the corresponding melt and cold crystallization enthalpies were nearly the same.
 | ||
| Fig. 3 Second heating (a and c) and first cooling (b and d) DSC traces of the copolyesters in the PBLF and PHLF series (exo up). | ||
The Tg of PBF and PHF were 39 and 17.5 °C, respectively. The higher Tg of PBF compared to PHF was expected due to the shorter alkylene chain in the PBF backbone. Moreover, the Tg value of PBF was exactly the same as the value reported in the literature (39 °C vs. 39 °C).48 The Tg of PHF differed from those reported in various studies (17.5 °C vs. 7–28 °C), which may be due to different molecular weights of the PHF samples.45 However, the Tg value of PHF in the present study was close to that reported by Wu et al. (15 °C).47 Concurrently with the reductions in Tm and crystallinity, a depression of the glass transition temperature (Tg) was also observed with increasing spiro monomer content. However, the effect was less pronounced and the Tg was only reduced by 4 to 5 °C in both series (except PBLF-50) upon the inclusion of the spiro monomer. Moreover, the Tg values of the copolyesters in each series were nearly equal, indicating that the backbone rigidity of the homopolyesters was not significantly affected by the spiro monomer structure in the polyester chain. Here it must be noted that the molecular weights of the copolyesters were considerably lower than for the homopolyesters. Hence, the observed decrease in Tg might be caused by the combined effect of differences in molecular weight and the content of the spiro diester unit in the copolyesters.
As can be seen, the Tgs of the polymers were quite close to room temperature, i.e., below room temperature in the case of PHLF copolyesters and slightly above for PBLF ones. It is generally known that polymers with Tgs close to room temperature (or below) often continue to slowly crystallize over a period of time at ambient storage. This occurs because the polymer chains in the amorphous regions have sufficient molecular mobility to rearrange into the existing crystals or to form new crystallites. This process is known as secondary crystallization or progressive crystallization in polymers. In order to investigate if the current polyesters continued to crystallize during ambient storage, samples obtained from the DSC study (cooled from the melt at 10 °C min−1) were left for two weeks at room temperature before recording a DSC heating scan (Fig. S21†). The results revealed an additional melting peak in the PHLF copolymers at around 50 °C, corresponding to the melting of defect-rich or small-sized secondary crystallites formed upon storage. The melt enthalpy of these crystallites increased with the spiro diester content in the series, indicating an increasing hindrance of the crystallization during the cooling of the sample during the initial DSC study, and/or a higher segmental mobility due to the replacement of the more rigid furan units by the aliphatic spiro monomer units. In addition to the appearance of the low-melting crystallites, the enthalpy values of the primary melting peaks increased, hinting a thickening of the primary crystallites formed during the cooling from the melt state in the initial DSC experiment. This increase was most pronounced for PHLF-12.5 and PHLF-15, which showed several fold increased crystallinity despite the higher content of spiro monomer. Also, in the case of PHLF-20, the observed increase in crystallinity was remarkable as this polymer did not crystallize at all from the melt, but demonstrated a melting peak at 112 °C with an enthalpy value of 32 J g−1 after storage for two weeks. The significant degree of secondary crystallization of PHLF-12.5–20 was due to a slower crystallization rate. As a result, most of the crystallization occurred during storage. This was further validated by the absence of a cold crystallization peak, confirming that crystallization occurred entirely during the storage. In contrast, PBLFs did not undergo any secondary crystallization during storage, presumably because their Tgs were above room temperature (Fig. S20†). Hence, secondary crystallization was observed when these polyesters were kept above their Tg at 50 °C during 40 h. Similar to what was observed for the PHLF series, two melting endotherms appeared in the subsequent heating cycle; the first peak at around 75 °C corresponded to the melting of the defect-rich or small crystallites formed during the annealing process, and the second to the primary crystallites which showed increased melt enthalpy values to hint crystal thickening after annealing. The exception was PBLF-20 which exclusively crystallized during the annealing at 50 °C.
Finally, thermal annealing of the polymers within the melt interval (∼10–15 °C below Tm) was performed to investigate the effect on the crystallinity and the melting temperature. Such a treatment is expected to melt and recrystallize small and imperfect crystallites formed during cooling and cold crystallization to form more perfect and thick crystallites. Polyester samples previously cooled from the melt at 10 °C min−1 were annealed during 4 h below their respective melting temperature, and then cooled, before recording their heating scans. As can be seen in Fig. S22,† comparatively narrow and sharp melting endotherms were observed after the annealing, especially for polyesters which crystallized from melt (PBF, PHF, PBLF-5-10 and PHLF-5-10), suggesting the formation of nearly homogenous crystallites. Broader endotherms with a shoulder was observed for PBLF-15-20 and PHLF-12.5-15, hinting the presence of different crystallite population. Still, the appearance of such a large melt endotherm after the annealing is remarkable because these polymers could not crystallize from the melt during cooling due to a very slow crystallization rate. For all polyesters, the enthalpy of melting was significantly increased upon annealing, in parallel with and consequently, the melting temperature (Table S1†). The improvement was more pronounced for copolyesters than for homopolyesters, presumably due to the molecular weight differences. Copolyesters PBLF-15-20 and PHLF-12.5-15 showed several-fold increased enthalpies because the chains had sufficient mobility and time to crystallize more effectively.
3.3. Dynamic mechanical properties
The dynamic mechanical properties of PBLFs and PHLFs were studied by DMA analysis of hot-pressed samples. PBLF-50 and PHLF-20 could not be analysed due to their soft and sticky character. The temperature dependence of the storage (E′) and loss (E′′) moduli are shown in Fig. 4a–d for PBLF and PHLF series, respectively. As can be seen, all the samples exhibited two molecular relaxations. The β relaxation appearing between −100 to −50 °C in both series was attributed to the reorientation and local motions of the carboxyl groups in the amorphous phase.49 At higher temperatures, the α relaxation from −5 to 125 °C in PBLF series and −10 to 80 °C in PHLF series can be attributed to the glass transition of the amorphous phase. The E′ values were taken at 0 °C where all the polymers were in the glassy state, and the values of the PBLF and PHLF series were found to be in the narrow range of 2.0–3.0 and 2.3–2.5 GPa, respectively (Table 3). PBF (2.71 GPa) showed higher E′ than that of PHF (2.33 GPa), probably due to the more rigid backbone structure.50 On the other hand, no obvious trend was observed based on the spiro monomer content, although a slight decreasing trend was seen in the PBLF series, except for PBLF-5 and PBLF-15. PBLF-5 showed a higher E′ than PBF, which can be explained by its higher degree of crystallinity due to its lower molecular weight compared to PBF. However, the higher E′ of PBLF-15 as compared to PBLF-10 was more difficult to explain. The reduction in modulus may be related to the relatively flexible nature of the spiro monomer in contrast to the stiff 2,5-furan dicarboxylate monomer. For all the polyesters in both series, the storage modulus showed an upturn after passing the glass transition region, suggesting a stiffening effect caused by cold crystallization of the sample during the heating process. This may be attributed to the increased chain mobility in the rubbery state, and the lower heating rate (3 °C min−1) compared with the DSC scans, which favoured the crystallization. | ||
| Fig. 4 DMA data showing the (a and c) storage modulus and (b and d) loss modulus of the polyesters in the PBLF and PHLF series (1 Hz, 0.1% strain). | ||
| Polyester | E′a (GPa) | T g (°C) | tanδmaxc |
T
gd (°C) |
|---|---|---|---|---|
|
a Storage modulus taken at 0 °C.
b Determined at the peak maximum in the E′′ curve.
c Determined as the peak maximum in the tanδ curve.
d Determined by DSC.
|
||||
| PBF | 2.71 | 51 | 66 | 39 |
| PBLF-5 | 3.04 | 41 | 57 | 37 |
| PBLF-10 | 2.31 | 39 | 55 | 34 |
| PBLF-15 | 2.67 | 34 | 52 | 34 |
| PBLF-20 | 2.03 | 29 | 36 | 34 |
| PHF | 2.33 | 27 | 40 | 18 |
| PHLF-5 | 2.39 | 26 | 37 | 13 |
| PHLF-10 | 2.36 | 25 | 37 | 13 |
| PHLF-12.5 | 2.51 | 23 | 43 | 13 |
| PHLF-15 | 2.32 | 24 | 57 | 13 |
The Tg values were taken at the local maximum of E′′ at the glass transition (Fig. 4b, d and Table 3). As can be seen, the DMA values were slightly higher than those determined by the DSC method. This discrepancy might be related to the different measurement principles, probing the thermal and mechanical response, respectively. Unlike the DSC results, the Tg values decreased slightly with increasing content of the spiro diester in the polymer chain, which was expected because of the less rigid nature of the spiro monomer. Still, the reduction in backbone stiffness was not very significant, and the resulting values of E′ and Tg of the copolyesters were quite comparable to the respective homopolyesters. This suggests that the insertion of the spiro monomer into the PBF and PHF polyesters did not significantly affect the thermomechanical characteristics. However, the presence of the chemically sensitive ketal structure can be expected to facilitate the hydrolytic degradation to enable chemical recycling.
3.4. Acid hydrolysis and chemical recycling
The typical hydrolysis of aromatic polyesters proceeds by the random chain scission of polymer chains in the amorphous phase domain into low molecular weight chain segments (oligomers).51,52 Subsequently follows the hydrolysis of oligomers into the starting monomers. The initial fragmentation into oligomers is usually slow and rate-determining, which leads to a long induction period before the degradation can accelerate. The most important reason for this is associated with the high degree of crystallinity, the high hydrophobicity of the aromatic polyesters and poorly electrophilic ester bond.53,54 The insertion of oxygen-rich spiro-ketal units in the polyester backbone can significantly enhance the hydrophilicity, reduce the degree of crystallinity, and offer weak links to facilitate polymer chain scission. This increases the water penetration into the polymer matrix. Subsequently, the acid catalyzed cleavage of the randomly distributed spiro-ketal units in the copolyesters can trigger accelerated hydrolysis through an efficient fragmentation of the polymer chain into small oligomers.A high hydrolysis rate generally implies an efficient degradation into the initial building blocks, and thus opens up for chemical recyclability. Here, the scope was centred on an initial study of the hydrolytic degradation and recyclability of the ketal-containing polyesters. In order to assess how the spiro-ketal units influence the degradability of the copolyesters, the hydrolytic degradation under acidic conditions was investigated. The degradation experiments were performed on semi-crystalline polyesters from the PHLF series. As-obtained powder samples (after isolation) of PHF and the two copolyesters PHLF-10 and PHLF-20 were selected in order to study the influence of the spiro-monomer content and the degree of crystallinity on the degradability. The melt enthalpies of PHF, PHLF-10 and PHLF-20 powders were 50, 43, and 26 J g−1. The hydrolysis experiments were conducted at 60 and 85 °C during 24 h using aqueous solutions of 3, 6 and 12 M HCl, respectively. After the hydrolysis, the reaction mixture was neutralized with K2CO3, and the pH was then adjusted to between 8–9 in order to remove any monomers that precipitated from the unhydrolyzed polymer residue. Next, the mixture was filtered to obtain a solid polymer residue and the filtrate, which were separately analyzed by 1H NMR spectroscopy as described further below. The rate of the hydrolytic degradation was followed by measuring the weight loss (%) of the sample, which was calculated by comparing the masses of the polymer sample before and after degradation. As expected, the weight loss increased with the concentration of the acid and temperature (Fig. 5a and b). Under each testing condition, the observed weight loss of PHF was lower than for the copolyesters PHLF-10 and PHLF-20, suggesting a slow degradation rate. The homopolyester showed almost no weight loss at 60 °C in both 3 and 6 M HCl, and only a slightly higher weight loss of 9% in 12 M HCl. Upon raising the temperature to 85 °C, the weight loss in 3 and 6 M HCl increased to some extent (2 and 8%, respectively). On the other hand, when the concentration was raised to12 M, a remarkably increased weight loss of 42% was obtained, highlighting the synergistic effect of temperature and acid concentration on the hydrolysis rate. Compared to PHF, the copolyesters exhibited higher weight loss, indicating that these samples were more prone to hydrolysis. The weight loss of PHLF-10 at 60 °C in 3, 6, and 12 M HCl were 18, 26, and 41%, respectively, and at 85 °C the loss increased to 28, 44, and 85% under the same conditions. PHLF-20 showed improved weight loss compared to PHLF-10 under each test condition. The values were 33, 45, and 54% in 3, 6, and 12 M HCl, respectively, at 60 °C, and further increased to 44, 70, and 91%, respectively, at 85 °C. The higher weight loss of PHLF-20 correlated with the higher content of spiro-ketal units in the polymer backbone. In any case, the copolyesters displayed enhanced hydrolysis than PHF, especially at 85 °C in 12 M HCl, where PHLF-10 and PHLF-20 degraded to 85 and 91% of their original weight. These values were significantly higher than that obtained for PHF (42%) under similar conditions. These results clearly show that the presence of the acid-sensitive ketal units in the polymer chain facilitated an efficient fragmentation into low molecular weight polymer or oligomer chains, which then degraded much faster than the longer polymer chains of PHF. This may be described by the reduced hydrophobicity and improved water penetration of samples containing the short polymer or oligomeric chains. The initial chain fragmentation due to the cleavage of ketal units was confirmed by 1H NMR analysis polymer samples at an early stage of degradation. The 1H NMR spectra of residual PHLF-10 solid samples were recorded after 30 min of degradation in 6 and 12 M HCl at 60 °C (Fig. 6). In both spectra, the signals of the spirocyclic ketal unit (c–h) were completely absent, suggesting complete chain scission at the ketal units to give oligomeric structures containing both hydroxyl and levulinate ester end groups (Fig. 6, structure I), or only levulinate ester end groups (Fig. 6, structure II). As seen in Fig. 6, the spectrum recorded after degradation in 12 M HCl showed a prominent signal originating from –CH2OH end groups, indicating the subsequent hydrolysis of the levulinate end group to obtain oligomer structure III.
 | ||
| Fig. 5 Weight loss (%) of PHF, PHLF-10, and PHLF-20 during hydrolytic degradation at 60 (a) and 85 °C (b) in aqueous hydrochloric acid (3, 6, and 12 M) for 24 h. | ||
 | ||
| Fig. 6 Stacked 1H NMR spectra of PHLF-10 before hydrolysis (a), the residual solid after hydrolysis in 6 M (b) and 12 M (c) HCl for 30 min. | ||
Both the polymer residue and the hydrolyzed products released in the aqueous filtrate were studied using 1H NMR spectroscopy. The spectra of the solid polymer residues are shown in Fig. S23–S28.† Although PHF showed almost negligible weight losses at almost all conditions except in 12 M HCl at 85 °C, a significant reduction in molecular weight was observed. This was concluded after comparison of the signal intensities of the end group (−CH2OH) and the main chain protons. The calculated molecular weights of the PHF residues ranged between 2.2–27.6 kg mol−1, notably lower than the molecular weight of the untreated sample (49.8 kg mol−1). As expected, the molecular weights of the residual powders decreased with increasing weight loss in the polymer samples, which was directly correlated to the acid concentration and temperature employed. For instance, the molecular weight of the PHF sample hydrolyzed in 3 M HCl at 60 °C was 27.6 kg mol−1, and this was reduced to 2.2 kg mol−1 when hydrolysis was performed in 12 M HCl at 85 °C, corresponding to oligomers containing hexamethylene 2,5-furandicarboxylate units. For the residues of PHLF-10 and PHLF-20 copolyesters, the spectra showed the complete disappearance of the spirocyclic ketal signals, which confirmed the chain scission into oligomers of hexamethylene 2,5-furandicarboxylate. However, the signals of the levulinate ester group were still present in most of the solids, most clearly in those obtained after hydrolytic treatment in the 3 and 6 M acid solutions. This suggested the presence of both hydroxyl and ester end groups in the oligomers. The content of levulinate groups in the PHLF-10 and PHLF-20 residues after hydrolysis in 3 M HCl at 60 °C were estimated to 7 and 10 mol%, respectively, which almost corresponded to 50–70% of starting levulinate ester units present in the original polymer sample. The content of levulinate groups gradually decreased when increasingly stronger conditions were employed, and was completely eliminated after hydrolysis of PHLF-10 and PHLF-20 in 6–12 M HCl at 85 °C to give hydroxyl-terminated oligomers of hexamethylene 2,5-furandicarboxylate. As can be seen in Fig. 5, 1H shifts from hydroxyl-terminated residues were only observed under the conditions that resulted in high weight losses. Hence, the presence of levulinate groups in the residual solids from the experiments at low acidic conditions may imply that there was no hydrolysis of furanic ester groups, because hydrolysis of aliphatic ester groups is generally expected to be faster than the aromatic ester groups.55 However, the observed weight losses were higher than the overall weight fraction of the spirocyclic ketal ester units in the polymer backbones. Thus, it is reasonable to assume that hydrolysis of the furanic ester group also occurred during the treatment in lower acidic conditions.
Finally, the crude product obtained from the aqueous filtrate of PHLF-20 was analyzed by 1H NMR spectroscopy (Fig. S29†). The spectrum showed characteristic signals attributed to 2,5-furandicarboxylic acid, 1,6-hexanediol, and levulinic acid, which confirmed the hydrolysis of the polyesters into the original monomers or building blocks. Notably, pentaerythritol was not recovered due to its higher solubility in the aqueous phase. The spectrum also showed some additional small signals (indicated by *). After careful analysis, they were ascribed to the chlorinated products (1,6-dichlorohexane or 1-chlorohexanol) resulting from the reaction of HCl with 1,6-hexanediol. This finding suggested that it is important to strike a balance between the use of harsh conditions to facilitate rapid degradation and the use of mild conditions to avoid the formation of chlorinated side products. Hence, a more comprehensive investigation is needed to identify optimum conditions for a high degradation rate with a minimum of side reactions.
Despite being less environmentally friendly than HCl, hydrolysis experiments were also carried out using sulfuric acid (H2SO4) to assess the efficiency compared to HCl. Copolyester PHLF-20 was subjected to hydrolytic degradation at 85 °C during 24 h using aqueous solutions of 6 and 12 M H2SO4, respectively. The hydrolysis in 12 M H2SO4 resulted in the formation of a black-coloured solution, suggesting further degradation of the hydrolyzed products in the highly concentrated acid solution (Fig. S30†). In contrast, no such observation was made during the hydrolysis in 6 M acid solution. The measured weight loss of the polymer (87%) was higher than that in 6 M HCl (70%) and almost comparable to that in 12 M HCl (91%) under similar conditions. Furthermore, the 1H NMR spectrum of the crude product obtained from this aqueous filtrate mainly displayed signals corresponding to hydrolyzed products, with no identifiable signals from any side products (Fig. S31†). Hence, moderately concentrated H2SO4 appears to be effective in minimizing the occurrence of side reactions during the hydrolytic degradation of 2,5-furandicaboxylate polyesters.
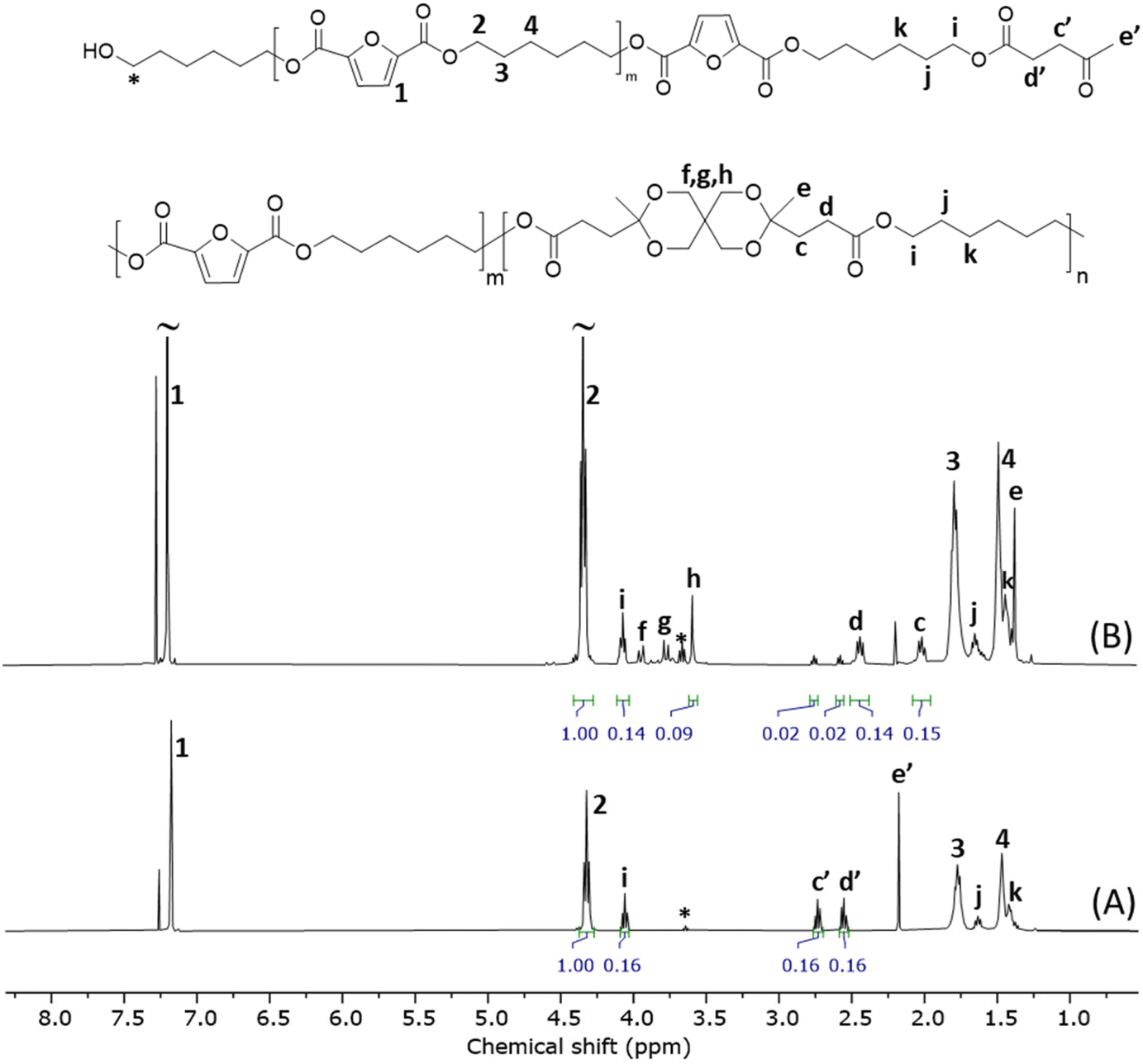 | ||
| Fig. 7 Stacked 1H NMR spectra of the telechelic t-PHF (A) and copolyester (r-PHLF-8.3) obtained after the polymerization of t-PHF (B). | ||
Next, the telechelic oligomer was investigated in a direct polymerization with pentaerythritol using p-TSA as a catalyst. The reaction was performed in the melt state at 170 °C for 3 h, under a high vacuum to drive off the water formed. The 1H NMR spectrum (Fig. 7, spectrum B) of the purified product showed signals (c–h) from the spirocyclic ketal rings (L units), indicating the formation of the PHLF copolyester backbone. However, the conversion did not reach completion as small signals of ketone end groups were still present. The overall conversion was 88% and did not increase upon further stirring till 5 h. Stirring for a longer time (15 h) led to the formation of a crosslinked product. The spectrum also contained small signals beside the main signals from the spirocyclic ring, which could be attributed to the partially ketalized (monocyclic) structures present at the chain ends. The estimated molar content of spiro units in the product was 8.3%, suggesting the formation of copolyester r-PHLF-8.3. The intrinsic viscosity and Mn values of r-PHLF-8.3 were 0.34 dL g−1 and 6.6 kg mol−1, respectively. The lower values of the repolymer (r-PHLF-8.3) compared to the original PHLF copolyesters also resulted in a lower polymerization yield (69%). The photographs of the initial PHLF-20, t-PHF and r-PHLF-8.3 samples are shown in Fig. S32.† TGA analysis of the product obtained in the initial repolymerization experiment showed a Td,5 (311 °C) as PHLF-10, implying similar thermal stabilities. Moreover, DSC showed a slightly lower Tg (10 °C) and Tm (121 °C) than PHLF-10, presumably due to the lower molecular weight of the repolymer. The possible reason behind the lower yield and molecular weight obtained may be due to the high melt viscosity of the polymerization mixture that hindered efficient mixing in the present setup. Therefore, a thorough optimization of the polymerization conditions is required, with a particular focus on achieving reduced melt viscosity to facilitate improved mixing and mass transfer. This may lead to higher conversion and molecular weight to provide a more efficient repolymerization. Nevertheless, this initial investigation demonstrated that copolyesters with spirocyclic ketal units may be recycled via a telechelic approach, which in practice fulfil closed-loop recycling.
4. Conclusions
A biobased ketal-functional spirocyclic diester monomer was successfully incorporated to various contents in PBF and PHF to obtain the PBLF and PHLF series of copolyesters, respectively. The calculated spiro monomer contents were in excellent agreement with the monomer feed, demonstrating a high reactivity and stability of the monomers during the melt polycondensation, thus allowing a high level of structural control. All the polyesters were thermally stable up to 275 °C, and the Tg values of the polyesters were essentially independent of the spirocyclic monomer content. In contrast, Tm and the degree of crystallinity decreased markedly with increasing contents of the spiro monomer. However, the effect was not significant for copolyesters containing up to 10 mol% of spiro monomer. DMA measurements showed no apparent relationship between the storage moduli at 0 °C and the spiro monomer content, and the value of the moduli of each of the polyester series fell into a quite narrow span to indicate that the backbone rigidity of the polyesters was not significantly affected by the spiro monomer. Investigations of the hydrolytic degradation of the polyesters in concentrated HCl solutions showed that the degradation rate was significantly higher for copolyesters than the homopolyesters, and increased with the spiro monomer content. Careful NMR analysis revealed that the spiro ketal units were very sensitive towards hydrolysis, which triggered the loss of molecular weight and the overall fragmentation of the polyesters into hexamethylene 2,5-furandicarboxylate oligomers. Additional analysis revealed that the samples further degraded to form the original monomers or building blocks of the polyesters. The preliminary assessment of the direct polymerization of the ketone-terminated telechelic oligomers suggested an alternative recycling possibility for the polyesters, although a thorough optimization of the polymerization is still needed. Consequently, the overall study demonstrated that the straightforward incorporation of the biobased ketal-functional spirocyclic diester monomer can be used to regulate the Tm and crystallinity of 2,5-furandicarboxylate polyesters, and also significantly accelerates the hydrolytic degradation to enable a viable pathway towards chemical recycling of this class of biobased plastics.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Swedish Foundation for Strategic Environmental Research (Mistra) through the “STEPS” project (No. 2016/1489), the Swedish Research Council for Sustainable Development FORMAS (No. 2021-01107), the Crafoord Foundation (No. 20160774 and 20180939), and the Royal Physiographic Society in Lund. Nilson Group AB is acknowledged for financial support. B. Z. thanks the EIT Climate-KIC flagship eCircular for the kind support.References
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS.
- T. Iwata, Angew. Chem., Int. Ed., 2015, 54, 3210–3215 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- E. Feghali, L. Tauk, P. Ortiz, K. Vanbroekhoven and W. Eevers, Polym. Degrad. Stab., 2020, 179, 109241 CrossRef CAS.
- S. V. Mankar, J. Wahlberg, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2023, 11, 5135–5146 CrossRef CAS.
- A. R. Rahimi and J. M. Garciá, Nat. Rev. Chem., 2017, 1(16), 1–11 Search PubMed.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y. X. Chen, Science, 2018, 360, 398–403 CrossRef CAS PubMed.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef CAS PubMed.
- M. Hong and E. Y.-X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- M. Han, in Recycling of Polyethylene Terephthalate Bottles, Elsevier, 2019, pp. 85–108 Search PubMed.
- G. P. Karayannidis, A. P. Chatziavgoustis and D. S. Achilias, Adv. Polym. Technol., 2002, 21, 250–259 CrossRef CAS.
- S. Mishra, V. S. Zope and A. S. Goje, Polym. Int., 2002, 51, 1310–1315 CrossRef CAS.
- T. Yoshioka, T. Sato and A. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353–1355 CrossRef CAS.
- G. E. Brown and Jr. R. C. O'Brien, US3952053, 1976.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 1494–1506 CrossRef CAS PubMed.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2018, 10, 172–186 RSC.
- J. E. Park, D. Y. Hwang, G. H. Choi, K. H. Choi and D. H. Suh, Biomacromolecules, 2017, 18, 2633–2639 CrossRef CAS PubMed.
- H. Hu, Y. Tian, Z. Kong, W. B. Ying, C. Chen, F. Li, R. Zhang and J. Zhu, ACS Sustainable Chem. Eng., 2021, 9, 2280–2290 CrossRef CAS.
- A. Hufendiek, S. Lingier and F. E. Du Prez, Polym. Chem., 2019, 10, 9–33 RSC.
- B. Kost and M. Basko, Polym. Chem., 2021, 12, 2551–2562 RSC.
- Y. Tian, J. Li, H. Hu, C. Chen, F. Li, W. B. Ying, L. Zheng, Y.-L. Zhao, J. Wang, R. Zhang and J. Zhu, J. Hazard. Mater., 2022, 430, 128392 CrossRef CAS PubMed.
- J. E. Park, D. Y. Hwang, G. H. Choi, K. H. Choi and D. H. Suh, Biomacromolecules, 2017, 18, 2633–2639 CrossRef CAS PubMed.
- N. Warlin, E. Nilsson, Z. Guo, S. V. Mankar, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, Polym. Chem., 2021, 12, 4942–4953 RSC.
- S. Hayashi, Y. Tachibana, N. Tabata and K.-i. Kasuya, Eur. Polym. J., 2021, 145, 110242 CrossRef CAS.
- S. Ma, J. Wei, Z. Jia, T. Yu, W. Yuan, Q. Li, S. Wang, S. You, R. Liu and J. Zhu, J. Mater. Chem. A, 2019, 7, 1233–1243 RSC.
- R. Sedrik, O. Bonjour, S. Laanesoo, I. Liblikas, T. Pehk, P. Jannasch and L. Vares, Biomacromolecules, 2022, 23, 2685–2696 CrossRef CAS PubMed.
- A. G. Pemba, M. Rostagno, T. A. Lee and S. A. Miller, Polym. Chem., 2014, 5, 3214–3221 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–555 RSC.
- B. Robles-Hernández, M. Soccio, I. Castrillo, G. Guidotti, N. Lotti, Á. Alegría and D. E. Martínez-Tong, Polymer, 2020, 204, 122825 CrossRef.
- G. Guidotti, M. Soccio, M. C. García-Gutiérrez, T. Ezquerra, V. Siracusa, E. Gutiérrez-Fernández, A. Munari and N. Lotti, ACS Sustainable Chem. Eng., 2020, 8, 9558–9568 CrossRef CAS PubMed.
- Z. Terzopoulou, N. Kasmi, V. Tsanaktsis, N. Doulakas, D. N. Bikiaris, D. S. Achilias and G. Z. Papageorgiou, Materials, 2017, 10, 801 CrossRef PubMed.
- N. G. Valsange, M. N. Garcia Gonzalez, N. Warlin, S. V. Mankar, N. Rehnberg, S. Lundmark, B. Zhang and P. Jannasch, Green Chem., 2021, 23, 5706–5723 RSC.
- M. Soccio, M. Costa, N. Lotti, M. Gazzano, V. Siracusa, E. Salatelli, P. Manaresi and A. Munari, Eur. Polym. J., 2016, 81, 397–412 CrossRef CAS.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S.-H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- C. Japu, A. Alla, A. Martínez De Ilarduya, M. G. García-Martín, E. Benito, J. A. Galbis and S. Muñoz-Guerra, Polym. Chem., 2012, 3, 2092–2101 RSC.
- H. Hu, R. Zhang, J. Wang, W. B. Ying, L. Shi, C. Yao, Z. Kong, K. Wang and J. Zhu, Green Chem., 2019, 21, 3013–3022 RSC.
- M. Chen, Z. Jiang and Z. Qiu, Eur. Polym. J., 2021, 161, 110860 CrossRef CAS.
- H. Hu, R. Zhang, J. Wang, W. B. Ying, L. Shi, C. Yao, Z. Kong, K. Wang and J. Zhu, Green Chem., 2019, 21, 3013–3022 RSC.
- H. Xie, L. Wu, B.-G. Li and P. Dubois, Ind. Eng. Chem. Res., 2018, 57, 13094–13102 CrossRef CAS.
- E. Bianchi, M. Soccio, V. Siracusa, M. Gazzano, S. Thiyagarajan and N. Lotti, ACS Sustainable Chem. Eng., 2021, 9, 11937–11949 CrossRef CAS PubMed.
- H. Hu, R. Zhang, A. Sousa, Y. Long, W. B. Ying, J. Wang and J. Zhu, Eur. Polym. J., 2018, 106, 42–52 CrossRef CAS.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1026–1036 CrossRef CAS.
- C. Sammon, J. Yarwood and N. Everall, Polym. Degrad. Stab., 2000, 67, 149–158 CrossRef CAS.
- D. A. S. Ravens, Polymer, 1960, 1, 375–383 CrossRef CAS.
- D. Kint and S. Muñoz-Guerra, Polym. Int., 1999, 48, 346–352 CrossRef CAS.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50–67 CrossRef CAS PubMed.
- S. Peng, B. S. Wu, L. Wu, B. G. Li and P. Dubois, J. Appl. Polym. Sci., 2017, 134, 44674 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03099g |
| This journal is © The Royal Society of Chemistry 2024 |