
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmbedding an esterase mimic inside polyesters to realize rapid and complete degradation without compromising their utility†
Yanfen
Wu
a,
Jing
Tian
ac,
Minmin
Sun
b,
Lizeng
Gao
b,
Jun
Xu
 *a and
Zhiqiang
Niu
*a
*a and
Zhiqiang
Niu
*a
aDepartment of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: niuzq@tsinghua.edu.cn; jun-xu@tsinghua.edu.cn
bCAS Engineering Laboratory for Nanozyme, Key Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing, 100101, China
cSchool of Mechanical Engineering, Ningxia University, Yinchuan 750021, Ningxia, China
First published on 19th January 2024
Abstract
Biodegradable plastics are considered as an alternative to commodity non-biodegradable plastics, but they still degrade slowly in the environment. Current strategies to promote their degradation rates often compromise their utility. Here, we enhance the degradation properties of poly(butylene terephthalate/adipate) (PBAT) by embedding an esterase mimic inside the polyester without compromising its processing and mechanical properties. This esterase mimic contains a binuclear zinc site and catalyzes the depolymerization of PBAT with a turnover frequency (TOF) an order of magnitude higher than that of conventional additives (ZnO). With 1 wt% of this enzyme mimic embedded, PBAT exhibits a 3.7-fold enhancement of the degradation rate under composting conditions. Mechanistic investigation indicates that the binuclear zinc binding center polarizes the ester carbonyl group and increases hydroxide uptake, thus leading to faster degradation. This work provides a possible approach to designing polymers that meet requirements of stability during use and fast degradation after use.
Introduction
The escalating global crisis of plastic pollution is a direct consequence of the substantial generation and inadequate management of plastic waste, resulting in the rapid and extensive accumulation of plastic debris in the biosphere.1–6 In the face of this challenge, considerable efforts have been devoted to the development of biodegradable plastics over the past decades, aiming to substitute persistent commodity plastics.7–10 The design and synthesis of new biodegradable plastics offer promising prospects, but also pose notable complexities, as they must address various life cycle requirements through precise modulation of the molecular structure.11,12 Currently, the prevailing types of biodegradable plastics are mainly polyester-based materials, including polylactide (PLA), PBAT and polycaprolactone (PCL), which are progressively being endorsed and utilized in food packaging, agriculture, pharmaceuticals and other applications.13 Among these, PBAT stands out as a notable example—a widely produced aliphatic–aromatic copolymer composed of terephthalic acid (TPA), butylene glycol (BDO), and adipic acid (AA).8,11,14,15 PBAT is widely considered as a promising alternative to low-density polyethylene (LDPE) due to its remarkable mechanical properties and favorable biodegradability.16However, it is worth noting that the degradation rate of biodegradable plastics depends on the physicochemical characteristics of the plastics and the scenarios at the end of their life, and rapid decomposition can only be observed under specific and favorable conditions.14,17–19 The most favorable treatment is composting, where an abundance of microorganisms, and suitable temperature and humidity levels facilitate the degradation of biodegradable plastics.20 Nevertheless, current industrial composting treatment cycles are generally shorter than the complete decomposition cycle of biodegradable plastics.16,21 This mismatch can lead to tricky microplastics issues and actual landfill disposal.22 Meanwhile, the misinterpretation of the term “biodegradable” by the general public has resulted in a large proportion of plastic wastes being directly discarded into the environment. Numerous studies have demonstrated the remarkably slow degradation rate of biodegradable plastics in the environment. For instance, little visible decomposition is observed after one year in seawater, highlighting the persisting issue of environmental accumulation of these wastes.23 Furthermore, for the aliphatic–aromatic copolyester PBAT, most PBAT-degrading microorganisms cannot directly utilize its monomers and require microbial consortia for complete metabolism to CO2.24,25 Therefore, the development of biodegradable plastics with accelerated degradation under both composting and ambient conditions is important for the mass use of such materials. However, a significant challenge lies in achieving enhanced degradability while ensuring shelf-life requirements.
To address this issue, two categories of strategies have been developed for PBAT, as illustrated in Fig. 1. The first strategy is based on molecular design, including increasing the proportion of aliphatic segments, introducing more hydrolysable monomers (Fig. 1a),26 and designing ionic aggregates of polyester (Fig. 1b).27 However, these approaches often require a longer reaction time and involve higher energy consumption due to the elevated reaction temperatures and vacuum conditions involved.28 Another way to manage the degradation of PBAT is blending other polymers or introducing additives. For instance, the addition of ZnO,29,30 FeCl3,31 clays,32,33 surfactants34 and organic acids35 to PBAT has been reported to accelerate its degradation (Fig. 1c). Considering the widespread use of additives in plastics, the strategy of accelerating PBAT degradation through additives may be more practical and easier to implement. Nevertheless, the accelerated degradability often accompanies reduced tensile properties and poor processibility, which can be attributed to weak interaction of the additives with the polymer and catalytic thermal degradation during processing, respectively.29,36 To ensure practicality and degradability, effective interaction between additives and PBAT chains is essential, which requires additives with enhanced interfacial affinity and structural similarity to PBAT. Recently, several studies have attempted to embed active enzymes directly into polymers to enhance their degradability,37,38 but incorporating natural enzymes through co-blending poses challenges due to enzymes’ poor thermal stability.
 | ||
| Fig. 1 Schematic illustration of the strategies proposed to enhance the degradability of PBAT. (a) Incorporating additional aliphatic co-monomers into PBAT.26 (b) Designing an ionic aggregate of polyester through polycondensation catalyzed by H3PO4.27 (c) Introducing inorganic additives into PBAT, which often compromises the mechanical properties of PBAT.29–31 (d) Embedding a biomimetic Zn2L additive into PBAT to offer both utility and degradability. | ||
Herein, we embedded a molecular mimic of esterase (a binuclear Zn2L complex) into PBAT, resulting in both rapid and complete degradation of the polymer while preserving its utility (Fig. 1d). Upon embedding Zn2L, enhanced mechanical properties of PBAT were observed and the aforementioned thermal degradation problems were circumvented. Furthermore, the PBAT composites exhibited significantly enhanced hydrolysis kinetics at near-neutral pH and accelerated degradation under composting conditions. Mechanistic studies have shown that the embedded Zn2L functions as a chelating agent and very likely favors intramolecular hydrolysis in the polymer matrix. Under composting conditions, the embedding of Zn2L led to enhanced water uptake, facilitating the initial hydrolytic stage of composting and thus accelerating the degradation of PBAT. Taking practical implications into account, cytotoxicity assays were performed and the results revealed that PBAT/Zn2L maintains an equivalent level of biocompatibility to pure PBAT.
Results and discussion
The mismanagement of plastic waste has resulted in the widespread dispersion of post-consumer plastics in the environment, making them difficult to collect, sort, and centralize processing. This problem can be largely circumvented by developing advanced additives that can accelerate their degradation without affecting their use. In our previous studies,39–41 we have demonstrated that a molecular mimic of OpdA esterase (binuclear Zn2L complex) can depolymerize polyethylene terephthalate (PET) and other polyesters under mild conditions in solution. However, whether this esterase mimic is equally effective in a solid polymer matrix remains unknown. Here, we employed tert-butyl substituted Zn2L as the additive to modify PBAT degradability (Fig. 1d) due to its high catalytic activity40 and suitable hydrophilicity in combination with PBAT (Fig. S1†). The compound was synthesized through a Schiff base reaction, involving the condensation of a primary amine with an aldehyde. Its structure was confirmed by proton nuclear magnetic resonance spectroscopy (1H NMR), elemental analysis, infrared spectroscopy, and mass spectrometry (see the ESI†).The intrinsic catalytic activity of this esterase mimic in PBAT hydrolysis (Fig. 2a) was first examined in an aqueous solution. The turnover frequency (TOF) values for Zn2L were 9 and 15 times higher than those of conventional Lewis acid catalysts such as zinc acetate (Zn(OAc)2) and zinc oxide (ZnO), respectively (Fig. 2b). This enhancement can be attributed to the unique structure of Zn2L, which evidently induces a proximity effect, increasing the local effective concentration of the reactants.39–41 In addition, the conversion rate of PBAT with Zn2L reached 37 times that of the blank control (Fig. 2b, inset). Moreover, Zn2L remained stable under the hydrolysis conditions, as supported by 1H NMR and diffuse reflection infrared Fourier transform (DRIFTS) spectra (Fig. S2†).
 | ||
| Fig. 2 Reaction kinetics of PBAT hydrolysis in solution. (a) PBAT hydrolysis over Zn2L via an intramolecular pathway.37,38 (b) Turnover frequency (TOF) of Zn2L, Zn(OAc)2 and ZnO toward hydrolysis of PBAT at pH 8 (NaOH aqueous solution) and 60 °C. The inset shows the conversion rate of PBAT with and without Zn2L (0.2 g L−1). (c) Time course analysis of Mw and polydispersity (Đ) of PBAT hydrolysis. (d) DRIFT spectra of butyl hexanoate (BH) and butyl benzoate (BB) in the presence and absence of Zn2L. (e) The hydrolysis kinetics of PBAT and the proportion of adipate (A) to the sum of adipate and terephthalate (T) in solution over Zn2L at pH 8 (NaOH aqueous solution) and 60 °C. | ||
We then investigated the depolymerization pathway to understand the high catalytic performance of Zn2L. As depicted in Fig. 2c, the weight-average molecular weight (Mw) of PBAT rapidly decreased from 120 kg mol−1 to approximately 35 kg mol−1 and the polydispersity (Đ) also decreased from 3.3 to about 2.0 after 2 days of the reaction. These observations suggest a random chain scission depolymerization: the long chains of PBAT were first fragmented, followed by the catalytic hydrolysis of the resulting short-chain polymers, leading to a further decrease in molecular weight. Further, the interaction between esters and zinc centers was characterized by DRIFTS. Butyl hexanoate (BH) and butyl benzoate (BB) were used as model molecules for the aliphatic and aromatic segments of PBAT, respectively. As shown in Fig. 2d, the peak of ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in BB exhibited a red shift from 1731 cm−1 to 1720 cm−1 in the presence of Zn2L. Similarly, the peak of ν(CO) in BH was red-shifted by 13 cm−1. The red shifts of ν(CO) suggest that the carbonyl groups in the aliphatic and aromatic segments were both weakened by the formation of CO⋯Zn between the esters and the catalyst, resulting in an enhanced reactivity. Fig. S3† illustrates the 1H NMR spectrum of the reaction solution, which exhibited the presence of three monomeric products: terephthalate (T), butanediol (BDO), and adipate (A). The proportion of adipate in relation to the sum of adipate and terephthalate was calculated by integrating the area of the corresponding peaks. This proportion ranged from 0.64 to 0.7 throughout the reaction (Fig. 2e), slightly higher than the proportion of the feedstock PBAT (0.55). This result indicates that the aliphatic segment was preferentially targeted, although Zn2L facilitated the hydrolysis of both aliphatic and aromatic segments.
O) in BB exhibited a red shift from 1731 cm−1 to 1720 cm−1 in the presence of Zn2L. Similarly, the peak of ν(CO) in BH was red-shifted by 13 cm−1. The red shifts of ν(CO) suggest that the carbonyl groups in the aliphatic and aromatic segments were both weakened by the formation of CO⋯Zn between the esters and the catalyst, resulting in an enhanced reactivity. Fig. S3† illustrates the 1H NMR spectrum of the reaction solution, which exhibited the presence of three monomeric products: terephthalate (T), butanediol (BDO), and adipate (A). The proportion of adipate in relation to the sum of adipate and terephthalate was calculated by integrating the area of the corresponding peaks. This proportion ranged from 0.64 to 0.7 throughout the reaction (Fig. 2e), slightly higher than the proportion of the feedstock PBAT (0.55). This result indicates that the aliphatic segment was preferentially targeted, although Zn2L facilitated the hydrolysis of both aliphatic and aromatic segments.
The accelerated PBAT hydrolysis in solution encouraged us to incorporate the esterase mimic (Zn2L) into PBAT and investigate its activity in the solid state. Thanks to its high thermal stability (Fig. S4†), Zn2L was embedded into PBAT (PBAT/Zn2L) using a sequential process involving melt compounding, pelletization and blown film techniques. Fig. S5a† presents visual representations of the resulting composites. PBAT/Zn2L films were evaluated for various properties related to their utilization and processing. The surface morphology and elemental distribution of the films were characterized using scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS), as depicted in Fig. 3a. The prepared PBAT/Zn2L film exhibited a flat surface, and the homogeneous distribution of Zn elements indicated well-dispersed Zn2L within the polymer matrix. Torque monitoring during the compounding process of PBAT and Zn2L shows that it achieved the same stable torque values as neat PBAT (Fig. 3b). This phenomenon suggests that Zn2L does not cause a reduction in the molecular weight or thermal degradation of PBAT under the processing conditions. A decrease in molecular weight would typically manifest as a decrease in polymer viscosity and, consequently, a decrease in torque.32,42 Furthermore, the tensile tests demonstrated that PBAT/Zn2L still retains high tensile strength, akin to those inherent to PBAT (Fig. 3c and Table S1†). Additionally, after Zn2L embedding, the elongation at break, yield strength and elastic modulus all increased by 11–20% (Fig. 3c inset and Table S1†), indicating that this modification enhanced the mechanical properties of PBAT. Specifically, the augmentation in tensile strength serves as further corroboration of the uniform dispersion of Zn2L throughout the polymer substrate.30,43 Meanwhile, the elongation at break of PBAT reached 609%, representing a 17% enhancement after Zn2L incorporation (Fig. 3c). Since the mechanical properties of the material are closely related to the crystallization behavior, differential scanning calorimetry (DSC) was first performed to find out the reason. Fig. 3d shows that the crystallization temperature (Tc) of PBAT/Zn2L shifted to approximately 80 °C, which is 10 °C higher than that of pure PBAT. This shift indicates that the heterogeneous nucleation of Zn2L increased the crystallization rate of PBAT.36,44 The melting curves of PBAT/Zn2L demonstrate that the melting temperature (Tm) of PBAT remained unchanged at around 124 °C even after the addition of Zn2L (Fig. 3e). The calculated crystallinity (Xc) of PBAT and PBAT/Zn2L is approximately 19% (Table S2†). These findings indicate that the addition of Zn2L does not affect crystallinity, but only influences the spherulite dimensions during crystallization. The change in the spherulite dimensions of PBAT after the addition of Zn2L was observed using polarized optical microscopy for further confirmation. With the introduction of Zn2L, the PBAT spherulite size decreases, and the nucleation density increases significantly (Fig. S5b†). Therefore, the increase in elongation at break of PBAT/Zn2L is attributed to the increased toughness of the polymer owing to the reduced spherulite dimension of PBAT/Zn2L. Moreover, the small-angle X-ray scattering (SAXS) results revealed a slight increase in the long period (shift to lower q) upon incorporating Zn2L into the PBAT matrix (Fig. S5c and Table S3†). This observation, along with the negligible difference in crystallinity (Xc), confirms the thickening of the crystalline lamellae in the PBAT/Zn2L composite. Additionally, Fig. S5d† demonstrates that the thermal stability of PBAT/Zn2L is comparable with that of pristine PBAT. This result is consistent with the torque monitoring and indicates that Zn2L does not induce thermal degradation of the polyester, unlike traditional Lewis acid additives.29,31,45 Comparison of Fourier transform infrared spectra also indicates that the chemical structure of PBAT did not undergo appreciable changes after embedding Zn2L (Fig. S5e†).
 | ||
| Fig. 3 Characterization of the PBAT/Zn2L composite. (a) Scanning electron microscopy (SEM) image and the corresponding energy-dispersive spectroscopy (EDS) elemental mappings of the PBAT/Zn2L composite; scale bar: 50 μm. (b) Torque curves during the processing of PBAT and PBAT/Zn2L. (c) Typical stress–strain curves of PBAT/Zn2L and PBAT. The inset shows the elastic deformation phase at the beginning of the tensile test. (d and e) Differential scanning calorimetry (DSC) curves of PBAT and PBAT/Zn2L during cooling (d) and secondary heating (e). | ||
To investigate the catalytic activity of Zn2L in the solid matrix, PBAT/Zn2L composite was subjected to hydrolysis at near-neutral pH in an aqueous solution. The hydrolysis rate of PBAT/Zn2L (5 wt%) was significantly accelerated compared to that of pure PBAT, resulting in a 20% BDO yield after 14 days of reaction. In contrast, the BDO yield for the pure PBAT was less than 1%. The leaching of the embedded Zn2L during the progression of the reaction was monitored by quantifying the Zn content in the solution using inductively coupled plasma optical emission spectroscopy (ICP-OES) (Fig. 4a). The concentration of Zn2L in the solution ranged from approximately 30% to 50% of the theoretically expected value, assuming proportional release of Zn2L into the solution (Fig. S6†). Perhaps this is due to the re-adsorption of the released Zn2L onto the PBAT surface. Then, the catalytic degradation of PBAT can be deconvoluted into two contributions: Zn2L dissolved in the solution and Zn2L not in the solution. We estimated the contribution from the dissolved Zn2L by extrapolating the linear relationship between the catalyst concentration and the conversion rate (Fig. S7†). In this way, we could see that the production of BDO mainly came from the contribution from Zn2L in the solid, as illustrated by the shaded area in Fig. 4b.
 | ||
| Fig. 4 Mechanistic understanding of PBAT/Zn2L hydrolysis. (a) The concentration of Zn2L released in solution during the hydrolysis reaction. (b) BDO yield from the contribution of Zn2L in the solid matrix at pH 8 and 60 °C. (c) XPS of the Zn 2p spectra for Zn2L and embedded Zn2L. (d) Fourier transform of the k3-weighted EXAFS spectra of the Zn K-edge of Zn2L and embedded Zn2L. (e) Schematic illustration of PBAT hydrolysis catalyzed by the embedded Zn2L in the solid phase. Nu denotes a nucleophile. | ||
In previous studies, we have demonstrated that the di-zinc centers will bind to ester carbonyl and hydroxide, respectively, and thus facilitate intramolecular hydrolysis.39,40 However, the role of Zn2L in the solid state is yet to be considered. We compared the structural differences between free Zn2L and Zn2L embedded in PBAT using X-ray photoelectron spectroscopy (XPS) and X-ray absorption fine structure (XAFS). After blending with the polymer, there is a shift in the Zn 2p3/2 binding energy from 1021.95 eV (Zn2L) to 1021.7 eV (embedded Zn2L) (Fig. 4c), indicating an increased electron density of Zn. This phenomenon may be due to the interaction between the zinc sites and the backbones of PBAT. Additionally, the coordination number (CN) of Zn increased from 4.9 for free Zn2L to 6.1 for embedded Zn2L (Fig. 4d and Fig. S8, and Table S4†), further suggesting the formation of a new Zn–O bond. The coordination of the carbonyl group to zinc in solid PBAT could activate the ester bond and thus would promote hydrolysis. We then investigated hydroxide uptake in the solid phase. The pH variations in the solution reflected the enhanced hydroxide uptake capacity of PBAT upon the introduction of Zn2L (Fig. S9†). Specifically, the immersion of PBAT/Zn2L (5 wt%) films resulted in a decrease of the solution's pH from 8.1 to 7.1 after 48 hours, which contrasts starkly with the minimal pH variation observed upon immersing pure PBAT films. The binding of hydroxide ions to the embedded Zn2L may involve the infiltration of the host solution into the internal micropores of the film, followed by the displacement of molecules/ions that are axially coordinated on Zn2L (at the solid–liquid interface) by hydroxide. The above evidence suggests that the embedded Zn2L plays two roles, namely to activate the ester bond and to take up free nucleophiles (e.g., hydroxide) into the polymer matrix via coordination to the zinc sites (Fig. 4e). Therefore, it is probable that the accelerated depolymerization of PBAT occurs through an intramolecular hydrolysis process similar to that in the solution phase.39,40
The degradation of PBAT with and without the Zn2L additive was then investigated under composting conditions using different compost soils. PBAT containing 1 wt% Zn2L exhibited a significantly accelerated degradation rate, resulting in a remaining mass of only 15% after a simulated 40-day composting period using industrial compost soil I, compared to 47% for the blank PBAT (Fig. 5a). When subjected to the same experiment using compost soil II, the composite demonstrated a degradation rate 3.7 times higher than the blank (Fig. 5b). The variation in degradation rates observed across compost soils underscores the strong influence of scenarios on the biodegradation of biodegradable plastics. Water absorption experiments indicate an increase in PBAT's water absorption upon the introduction of Zn2L (Fig. S10†). We further monitored the changes in molecular weight (Mw) and dispersity (Đ) during the composting of PBAT (low Mw) with embedded Zn2L. As shown in Fig. 5c, despite the low mass loss in the initial stages of PBAT composite degradation, Đ significantly increased along with the decrease of Mw, which corresponds to the initial hydrolysis process and long-chain scission. Previous studies have demonstrated that soil microbes can only take up and utilize small PBAT molecules,46 thus the rapid chain-breaking process facilitates subsequent microbial mineralization. Moreover, the surface of PBAT had transformed from smooth to rough after composting, and the PBAT/Zn2L composite exhibited more pronounced surface cracks and roughness compared to pure PBAT (Fig. 5d).
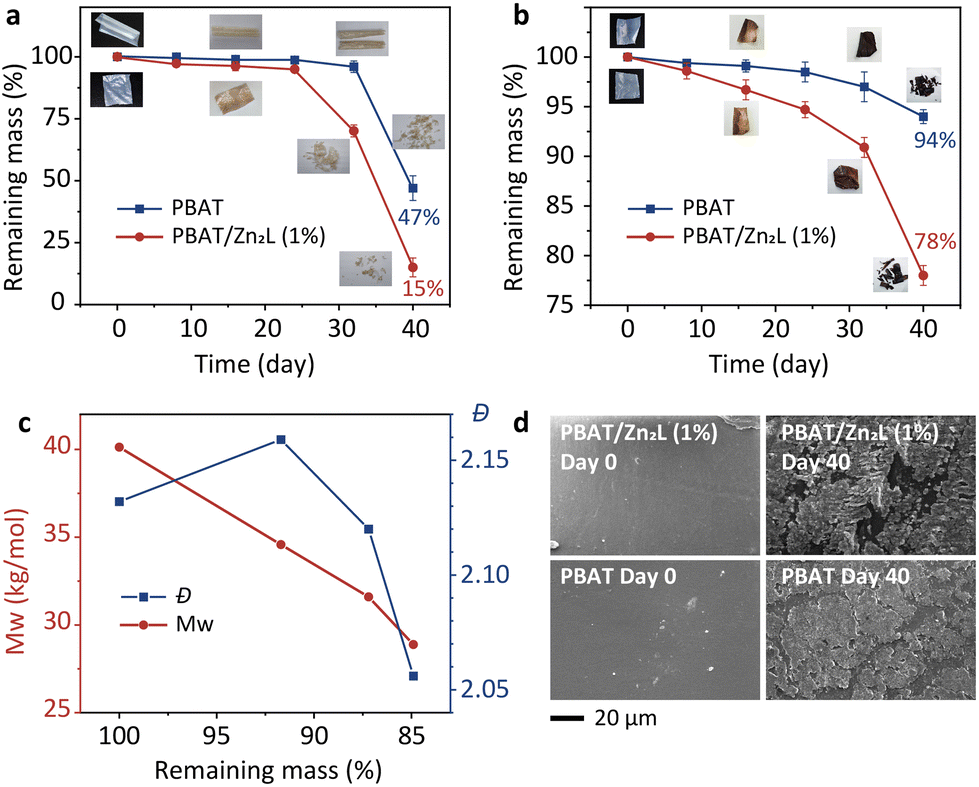 | ||
| Fig. 5 Simulated composting of PBAT and PBAT/Zn2L. (a and b) Remaining mass of PBAT and PBAT/Zn2L (1 wt%) under simulated composting scenarios with composting soil I (a) and soil II (b). (c) Analysis of the molecular weight (Mw) and dispersity (Đ) of PBAT/Zn2L at the initial stage of simulated composting. (d) SEM images of PBAT/Zn2L (1 wt%) and PBAT before and after 40 days of composting; scale bar: 20 μm. | ||
The results presented above demonstrate that embedding Zn2L significantly expedites the degradation of PBAT upon disposal without compromising its processibility and mechanical properties. However, PBAT serves as a primary material for food packaging and agricultural mulch, necessitating its biocompatibility and non-cytotoxicity. In this regard, the cytotoxicity of PBAT/Zn2L was meticulously examined. Three cell lines commonly employed in plastic cytotoxicity studies (fibroblast L929, alveolar epithelial cell A549, and macrophage RAW 254.7) were selected for the investigation.47–50 Cell proliferation was quantified using the highly sensitive cell counting kit-8 assay. After a 24-hour incubation period, the three cell types both maintained their original morphologies (Fig. S11†) and growth on both pure PBAT and PBAT/Zn2L surfaces. Quantitative assessment of cell viability revealed negligible differences in cell growth between PBAT and PBAT/Zn2L for all three types of cells (Fig. 6). Consequently, it can be inferred that Zn2L embedding does not induce cytotoxic effects, indicating that PBAT/Zn2L possesses the same level of biocompatibility as pure PBAT.
 | ||
| Fig. 6 In vitro cytotoxicity assay. Cell viability of A549 cells, L929 cells and RAW 254.7 cells grown for 24 h in the presence of pure PBAT and PBAT/Zn2L (1 and 5 wt%). | ||
Conclusions
In summary, we employed an esterase mimic (binuclear Zn compound) as an alternative additive in PBAT to optimize its utility and degradability. This esterase mimic exhibits a high TOF for PBAT hydrolysis and effectively targets both aliphatic and aromatic segments. The molecular-scale dispersion of Zn2L within the polymer matrix enhances the elongation at break, tensile strength, yield strength and elastic modulus of PBAT. Mechanistic studies showed that Zn2L in the solid state plays a major role in accelerating PBAT depolymerization. The PBAT/Zn2L composite exhibits an enhanced degradation rate compared to PBAT under composting conditions. This enhancement can be attributed to the short hydrolysis induction period as facilitated by Zn2L. Moreover, cytotoxicity studies confirmed that the PBAT/Zn2L composite maintains an equivalent level of biocompatibility to pure PBAT. This work underscores the feasibility of utilizing this esterase mimic as an additive that offers both utility and degradability for biodegradable plastics, which could increase the composting capacity and efficiency in a limited space.Author contributions
Z. N. and J. X. conceptualized and guided this work. Y. W. designed the experiments. Y. W. and J. T. performed the preparation and characterization of the PBAT/Zn2L composite. M. S. and L. G. conducted the cytotoxicity experiments. J. X. guided the preparation of PBAT/Zn2L and simulated the composting experiment. Z. N., Y. W., and J. T. wrote the paper. All the authors participated in the data analysis and commented on the manuscript.Conflicts of interest
Z. N. and Y. W. have filed a patent (CN 114395233 B). The other authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22375113 and 22075162), the National Key R&D Program of China (2019YFA0709200 and 2021YFD1700700), and the Tsinghua University Initiative Scientific Research Program (20221080067). We acknowledge the 4B9A and 1W1B station in the Beijing Synchrotron Radiation Facility (BSRF) for the collection of XAFS data.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- R. Li, W. Zeng, R. Zhao, Y. Zhao, Y. Wang, F. Zhang, M. Tang, Y. Wang, X. Chang, F. Wu and Z. Liu, Nano Res., 2023, 16, 12223–12229 CrossRef CAS.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- S. Zhang, M. Li, Z. Zuo and Z. Niu, Green Chem., 2023, 25, 6949–6970 RSC.
- X. Liu, Z. Fang, D. Xiong, S. Gong, Y. Niu, W. Chen and Z. Chen, Nano Res., 2022, 16, 4625–4633 CrossRef.
- M. Flury and R. Narayan, Curr. Opin. Green Sustainable Chem., 2021, 30, 100490 CrossRef CAS.
- T. D. Moshood, G. Nawanir, F. Mahmud, F. Mohamad, M. H. Ahmad and A. AbdulGhani, Curr. Opin. Green Sustainable Chem., 2022, 5, 100273 CrossRef CAS.
- M. K. M. Smith, D. M. Paleri, M. Abdelwahab, D. F. Mielewski, M. Misra and A. K. Mohanty, Green Chem., 2020, 22, 3906–3916 RSC.
- H. Kim, H. Jeon, G. Shin, M. Lee, J. Jegal, S. Y. Hwang, D. X. Oh, J. M. Koo, Y. Eom and J. Park, Green Chem., 2021, 23, 2293–2299 RSC.
- J. G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- A. C. Albertsson and M. Hakkarainen, Science, 2017, 358, 872–873 CrossRef CAS PubMed.
- M. T. Zumstein, R. Narayan, H. E. Kohler, K. McNeill and M. Sander, Environ. Sci. Technol., 2019, 53, 9967–9969 CrossRef CAS PubMed.
- M. R. Havstad, Plastic Waste and Recycling, Elsevier, 2020, pp. 97–129 Search PubMed.
- P. Skoczinski, L. Krause, A. Raschka, L. Dammer and M. Carus, Methods Enzymol., 2021, 648, 1–26 CAS.
- X. Zhao, K. Cornish and Y. Vodovotz, Environ. Sci. Technol., 2020, 54, 4712–4732 CrossRef CAS PubMed.
- K. L. Law and R. Narayan, Nat. Rev. Mater., 2021, 7, 104–116 CrossRef.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- K. S. Salem, K. Clayson, M. Salas, N. Haque, R. Rao, S. Agate, A. Singh, J. W. Levis, A. Mittal, J. M. Yarbrough, R. Venditti, H. Jameel, L. Lucia and L. Pal, Matter, 2023, 6, 3348–3377 CrossRef.
- S. M. Emadian, T. T. Onay and B. Demirel, Waste Manage., 2017, 59, 526–536 CrossRef CAS PubMed.
- C. DelRe, Y. Jiang, P. Kang, J. Kwon, A. Hall, I. Jayapurna, Z. Ruan, L. Ma, K. Zolkin, T. Li, C. D. Scown, R. O. Ritchie, T. P. Russell and T. Xu, Nature, 2021, 592, 558–563 CrossRef CAS PubMed.
- Y. Sun, B.-Y. Peng, Y. Wang, X. Wang, S. Xia and J. Zhao, Chem. Eng. J., 2023, 464, 142714 CrossRef CAS.
- A. R. Bagheri, C. Laforsch, A. Greiner and S. Agarwal, Global Chall., 2017, 1, 1700048 CrossRef PubMed.
- R.-J. Müller and W.-D. Deckwer, J. Biotechnol., 2001, 86, 87–95 CrossRef PubMed.
- I. E. Meyer-Cifuentes, J. Werner, N. Jehmlich, S. E. Will, M. Neumann-Schaal and B. Öztürk, Nat. Commun., 2020, 11, 5790 CrossRef CAS PubMed.
- H. Hu, Y. Tian, J. Wang, R. Zhang and J. Zhu, Polym. Degrad. Stab., 2022, 195, 109795 CrossRef CAS.
- H. J. Lee, W. Y. Cho, H. C. Lee, Y. H. Seo, J. W. Baek, P. C. Lee and B. Y. Lee, J. Am. Chem. Soc., 2022, 144, 15911–15915 CrossRef CAS PubMed.
- P. K. Samantaray, A. Little, A. M. Wemyss, E. Iacovidou and C. Wan, ACS Sustainable Chem. Eng., 2021, 9, 9151–9164 CrossRef CAS.
- A. Del Campo, E. de Lucas-Gil, F. Rubio-Marcos, M. P. Arrieta, M. Fernández-García, J. F. Fernández and A. Muñoz-Bonilla, Polym. Degrad. Stab., 2021, 185, 109501 CrossRef CAS.
- G. Lu, J. Zhu, H. Yu, M. Jin, S. Y. H. Abdalkarim and Y. Wei, Cellulose, 2021, 28, 7735–7748 CrossRef CAS.
- X. Li, S. Gong, L. Yang, X. Xia, C. Linghu, J. Wang and Z. Luo, Polymer, 2021, 228, 123927 CrossRef CAS.
- G. A. M. Falcão, M. B. C. Vitorino, T. G. Almeida, M. A. G. Bardi, L. H. Carvalho and E. L. Canedo, Polym. Bull., 2017, 74, 4423–4436 CrossRef.
- S. Mohanty and S. K. Nayak, J. Polym. Environ., 2012, 20, 195–207 CrossRef CAS.
- P. H. Camani, A. G. Souza, R. F. S. Barbosa, N. C. Zanini, D. R. Mulinari and D. S. Rosa, Chemosphere, 2021, 269, 128708 CrossRef CAS PubMed.
- R. C. Rebelo, L. P. C. Gonçalves, A. C. Fonseca, J. Fonseca, M. Rola, J. F. J. Coelho, F. Rola and A. C. Serra, Polymer, 2022, 256, 125223 CrossRef CAS.
- T. Wang, Y. Shi, Y. Li and L.-Z. Liu, J. Polym. Eng., 2021, 41, 835–841 CrossRef CAS.
- Q. Huang, S. Kimura and T. Iwata, Polym. Degrad. Stab., 2021, 190, 109647 CrossRef CAS.
- Q. Huang, M. Hiyama, T. Kabe, S. Kimura and T. Iwata, Biomacromolecules, 2020, 21, 3301–3307 CrossRef CAS PubMed.
- S. Zhang, Q. Hu, Y.-X. Zhang, H. Guo, Y. Wu, M. Sun, X. Zhu, J. Zhang, S. Gong, P. Liu and Z. Niu, Nat. Sustain., 2023, 6, 965–973 CrossRef.
- S. Zhang, Y. Xue, Y. Wu, Y.-X. Zhang, T. Tan and Z. Niu, Chem. Sci., 2023, 14, 6558–6563 RSC.
- Z. Sun and W. Chen, Matter, 2023, 6, 2562–2564 CrossRef CAS.
- L. C. Arruda, M. Magaton, R. E. S. Bretas and M. M. Ueki, Polym. Test., 2015, 43, 27–37 CrossRef CAS.
- D. Meng, J. Xie, G. I. N. Waterhouse, K. Zhang, Q. Zhao, S. Wang, S. Qiu, K. Chen, J. Li, C. Ma, Y. Pan and J. Xu, J. Appl. Polym. Sci., 2019, 137, 48485 CrossRef.
- C. Ye, F. Yu, Y. Huang, M. Hua, S. Zhang and J. Feng, Sci. Total Environ., 2022, 832, 155124 CrossRef CAS PubMed.
- M. Murariu, A. Doumbia, L. Bonnaud, A. L. Dechief, Y. Paint, M. Ferreira, C. Campagne, E. Devaux and P. Dubois, Biomacromolecules, 2011, 12, 1762–1771 CrossRef CAS PubMed.
- M. T. Zumstein, A. Schintlmeister, T. F. Nelson, R. Baumgartner, D. Woebken, M. Wagner, H. E. Kohler, K. McNeill and M. Sander, Sci. Adv., 2018, 4, eaas9024 CrossRef CAS PubMed.
- K. Fukushima, A. Rasyida and M.-C. Yang, J. Polym. Res., 2013, 20, 302 CrossRef.
- S. K. Rahimi, R. Aeinehvand, K. Kim and J. U. Otaigbe, Biomacromolecules, 2017, 18, 2179–2194 CrossRef PubMed.
- J. Hwang, D. Choi, S. Han, J. Choi and J. Hong, Sci. Total Environ., 2019, 684, 657–669 CrossRef CAS PubMed.
- K. Savva, X. Borrell, T. Moreno, I. Perez-Pomeda, C. Barata, M. Llorca and M. Farre, Chemosphere, 2023, 313, 137494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04500e |
| This journal is © The Royal Society of Chemistry 2024 |