
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUrolithin B reduces cartilage degeneration and alleviates osteoarthritis by inhibiting inflammation†
Hong
Xue‡
ab,
Hongyu
Zhou‡
ab,
Qiliang
Lou‡
ab,
Putao
Yuan
ab,
Zhenhua
Feng
ab,
Li
Qiao
ab,
Jiateng
Zhang
ab,
Hongwei
Xie
ab,
Yang
Shen
ab,
Qingliang
Ma
ab,
Shiyu
Wang
ab,
Boya
Zhang
c,
Huali
Ye
ab,
Jiao
Cheng
ab,
Xuewu
Sun
 *ab and
Peihua
Shi
*ab
*ab and
Peihua
Shi
*ab
aDepartment of Orthopaedic Surgery, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, China. E-mail: peihua_shi@zju.edu.cn; xuewu_sun@zju.edu.cn
bKey Laboratory of Musculoskeletal System Degeneration and Regeneration Translational Research of Zhejiang Province, Hangzhou, China
cDepartment of Dermatology, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, China
First published on 28th February 2024
Abstract
Osteoarthritis is the most prevalent degenerative joint disease reported worldwide. Conventional treatment strategies mainly focus on medication and involve surgical joint replacement. The use of these therapies is limited by gastrointestinal complications and the lifespan of joint prostheses. Hence, safe and efficacious drugs are urgently needed to impede the osteoarthritis progression. Urolithin B, a metabolite of ellagic acid in the gut, exhibits anti-inflammatory and antioxidant properties; however, its role in osteoarthritis remains unclear. In this study, we demonstrated that urolithin B efficiently inhibits the inflammatory factor-induced production of matrix metalloproteinases (MMP3 and MMP13) in vitro and upregulates the expression of type II collagen and aggrecan. Urolithin B alleviates cartilage erosion and osteophyte formation induced by anterior cruciate ligament transections. Moreover, urolithin B inhibits the activation of the NF-κB pathway by reducing the phosphorylation of Iκb-α and the nuclear translocation of P65. In summary, urolithin B significantly inhibits inflammation and alleviates osteoarthritis. Hence, urolithin B can be considered a potential agent suitable for the effective treatment of osteoarthritis in the future.
1. Introduction
Osteoarthritis (OA) is a chronic degenerative disease that significantly impairs physical function and quality of life; it predominantly affects weight-bearing and highly mobile joints, such as the knee and hip joints.1–3 Although the precise etiology of OA remains unclear, previous research suggests its association with multiple risk factors, including age, obesity, occupational overuse, sex, trauma, and biomechanical alterations; among these factors, age and obesity have emerged as the most prominent ones.4–7 The major pathologies of OA include cartilage erosion, synovial inflammation, subchondral bone remodeling, and osteophyte formation.8,9 The current clinical approaches for managing OA primarily involve the oral or topical administration of NSAIDs, intra-articular corticosteroid injections, and joint replacement.10–12 However, these methods have inherent limitations, such as gastrointestinal side effects, a limited duration of effectiveness, and a finite lifespan of the prosthetic joints.13,14 Hence, the development of safe and effective therapeutic strategies is crucial for decelerating disease progression and enhancing the overall well-being of patients with OA.Inflammation is involved in the early stages of OA; pro-inflammatory cytokines, including IL1-β, IL6, and TNF-α, play pivotal roles in disease progression.15,16 Elevated levels of IL1-β and TNF-α have been detected in the joint fluid, cartilage, and synovium of osteoarthritis patients.17 These inflammatory mediators bind to specific receptors, such as IL-1 receptor type I (IL-1R) and TNF receptor I (TNFRI), on the surface of chondrocytes and activate intracellular inflammatory pathways, including NF-κB, MAPK, and Wnt signaling cascades. The activated NF-κB pathway triggers the release of nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and chemokines, which intensify chondrocyte catabolism and inhibit anabolism.18–21 In the cartilage microenvironment, type II collagen and proteoglycans are the principal components of the extracellular matrix (ECM). Expression of these components is significantly suppressed in an inflammatory environment.22,23 Concomitantly, chondrocytes increase the production of proteinases involved in ECM degradation, which include matrix metalloproteinases (MMP1, MMP3, and MMP13) and disintegrin-like metalloproteinases with thrombospondin type 1 motifs (ADAMTS4 and ADAMTS5) and contribute to cartilage destruction.24,25 Hence, targeting the functions of IL1-β and TNF-α, or regulating downstream signaling pathways, can potentially represent effective strategies for inhibiting cartilage degradation.
Urolithin B (UB), a monohydroxy-substituted dibenzo[b,d]pyran-6-one derivative, is generated by the gut microbiota through the metabolism of ellagic acid-rich foods, including pomegranates, strawberries, and walnuts.26 UB is generated in the distal colon and exhibits enhanced lipophilicity, favorable absorption, biological activity, and safety.27 Moreover, UB has significant anti-inflammatory, antioxidant, lipid-lowering, and gut microbiota-modulating properties.28–30 Previous reports have highlighted the importance of UB in attenuating myocardial ischemia/reperfusion injury, treating degenerative diseases, and combating osteoporosis.31–33 However, the role of UB in osteoarthritis remains unclear. Considering its potent anti-inflammatory effects, we hypothesized that UB can alleviate inflammation and suppress OA progression. Consequently, this study incorporated a series of experiments to elucidate the protective effects and mechanisms of UB against OA.
2. Methods and materials
2.1 Materials
Urolithin B (Cat# HY-126307) was obtained from MedChemExpress (New Jersey, USA), dissolved in DMSO, and then stored at −20 °C. Fetal bovine serum (Cat# 10099141C), Dulbecco's modified Eagle's medium (DMEM) (Cat# 11965-118), and penicillin/streptomycin (Cat# 15410163) were purchased from Gibco (New York, USA). HumanKine® recombinant human IL-1 beta protein (Cat# HZ-1164) and HumanKine® recombinant human TNF alpha protein (Cat# HZ-1014) were purchased from Proteintech (Chicago, USA). Cell counting kit-8 (CCK-8) (Cat# K1018) was purchased from APExBIO Technology (Houston, Texas, USA). DAPI (Cat# 40728ES03) and Hieff® qPCR SYBR Green Master Mix (Cat# 11201ES03) were obtained from Yeasen (Shanghai, China). Safranin O (Cat# G1371), Alcian blue (Cat# G2541), and toluidine blue (Cat# G2543) were purchased from Solarbio (Beijing, China). 5XEvo M-MLV RT Master Mix (Cat#AG11603) was derived from Accurate Biology (Hunan, China). The enhanced BCA protein assay kit (Cat#P0009), RIPA lysis buffer (Cat# P0013E), phosphatase inhibitor cocktail (Cat# P1091), PMSF (Cat# ST506), and Triton X-100 (Cat# ST797) were supplied by Beyotime Biology (Shanghai, China). PVDF membrane (Cat# 1620256) was purchased from Bio-Rad (California, USA). HRP AffiniPure goat anti-mouse IgG (H + L) (Cat# FDM007), HRP AffiniPure goat anti-rabbit IgG (H + L) (Cat# FDR007), and DyLight 594 AffiniPure goat anti-rabbit IgG (H + L) (Cat#: FD0129) were obtained from Fude Biological Technology (Hangzhou, China). ATDC5 cells (Cat# IM-M049) were obtained from the National Collection of Authenticated Cell Cultures (Shanghai, China). Table 1 shows all antibodies used herein.| Antibodies | Species | Source | Identifier | Dilutions (WB/IF) |
|---|---|---|---|---|
| iNOS | Rabbit | Abcam | ab178945 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 :1000 |
| COX-2 | Rabbit | Abcam | ab179800 | 1:1000 |
| MMP13 | Rabbit | Abcam | ab39012 | 1:1000/1:100 |
| ADAMTS5 | Rabbit | Abcam | ab41037 | 1:1000 |
| β-Actin | Mouse | Abcam | ab8226 | 1:10000 |
| GAPDH | Rabbit | Proteintech | 10494-1-AP |
1:10000 |
| Histone 3 | Rabbit | Proteintech | 17168-1-AP | 1:5000 |
| Aggrecan | Mouse | Abcam | ab3778 | 1:1000 |
| COL2a | Rabbit | Proteintech/Biorbyt | 28459-1-AP/orb182930 | 1:1000/1:100 |
| P65 | Rabbit | Cell signaling technology | #8242 | 1:1000/1:400 |
| Ikb-α/p-Ikb-α(S32) | Rabbit | Cell signaling technology | #4812/#2859 | 1:1000 |
2.2 Chondrocyte culture
Three-day-old C57BL/6 mouse knee joint cartilage was minced and digested with collagenase type II for 8–12 hours. The remaining cartilage was filtered out, and the cells were cultured in 10 cm dishes with DMEM supplemented with 10% serum. The culture medium was refreshed every 48 hours until the density of the cells reached 80–90%.34 Cell harvesting involved removing the culture medium, washing the cells with PBS three times, and trypsinizing for dissociation. On a 6-well plate, chondrocytes were plated at a density of 4 × 105 cells per well. After 24 hours of incubation at 37 °C in a 5% CO2 incubator, the cells were stimulated with 10 ng ml−1 IL-1β and 50 ng ml−1 TNF-α to induce inflammation.35,36 The culture medium was replaced every two days. After four days of cultivation, the cells were collected and used for subsequent analyses such as western blotting or quantitative real-time PCR.It is worth noting that there are significant differences in the physiological functions between aged and young chondrocytes. Compared to young mice, aged mouse chondrocytes are more susceptible to systemic or local inflammation stimuli, making them more representative of the actual disease condition. However, current studies predominantly utilize immature murine articular chondrocytes for in vitro experiments, which have been validated by multiple research groups.37,38 Previous findings indicate that primary chondrocytes from young mice exhibit increased production of PGE2 and NO when stimulated with IL-1β, but this sensitivity decreases with passaging.39,40 Therefore, cultivating primary chondrocytes from immature mice remains an effective method for studying the physiological function and pathogenesis of osteoarthritis in vitro. However, using aged mouse chondrocytes is also equally significant in investigating the mechanisms of osteoarthritis. This requires a reasonable selection based on different experimental conditions.
2.3 Cell viability assay
100 μL mouse chondrocytes were seeded in a 96-well plate at a density of 8 × 103 cells per well. After 24 hours of incubation, different concentrations of UB (0, 0.3125, 0.625, 1.25, 2.5, 5, 10, 20 μM), IL1-β (0, 0.625, 1.25, 2.5, 5, 10, 20, 40 ng ml−1), and TNF-α (0, 3.125, 6.25, 12.5, 25, 50, 100, 200 ng ml−1) were added to the cells and incubated for 48 h, 96 h, or 7 days. The CCK-8 buffer was diluted with a serum-free culture medium, and 100 μL of the diluted solution was added to each well. The plate was then incubated at 37 °C for 2 hours. After incubation, the optical density (OD) at 450 nm was measured using an ELX800 absorbance microplate reader (BioTek Instruments, Winooski, VT, USA) to assess the impact of UB, IL1-β, and TNF-α on chondrocyte viability.2.4 Chondrocyte micromass culture
20 μL of chondrocyte suspension containing 4 × 105 cells were added to the center of the 12-well plate and allowed to settle for 4 hours at 37 °C.41 After the cells had fully adhered to the plate, 1 ml fresh culture medium was added, and the cells were cultured for 24 hours. Thereafter, the cells were treated with 10 ng ml−1 IL-1β or 50 ng ml−1 TNF-α to induce inflammation. The culture medium was changed every two days. Following 7 days of culture, the medium was removed, and the cells were then fixed with 4% paraformaldehyde for 15 minutes and used for safranin O, Alcian blue, and toluidine blue staining.42 The images were captured using an EPSON scanner (Seiko Epson Corporation, Japan).This assay was used to assess the deposition of extracellular matrix at the macroscopic level. The 7-day incubation period allowed chondrocytes to produce a greater amount of extracellular matrix, thereby resulting in more visually prominent staining results.
2.5 Nuclear-cytoplasmic protein separation
ATDC5 cells were seeded into a 6-well plate at a density of 4 × 105 cells per well to ensure uniform distribution. Following a 24-hour incubation, the cells were subjected to a 2 h pre-treatment using a serum-free medium with or without UB. Subsequently, the cells were exposed to a medium containing 10% serum, and supplemented with 10 ng ml−1 IL1-β or 50 ng ml−1 TNF-α for 30 minutes. After removing the medium, the cells were washed three times with PBS. The isolation of nuclear and cytoplasmic proteins was performed according to the instructions provided with the cytoplasmic-nuclear protein separation kit (Beyotime Biology Cat# P0027). The obtained protein samples were quantitatively analyzed using the BCA assay kit. Then, western blotting was used to investigate the distribution of NF-kB P65 protein within the cytoplasm and nucleus.2.6 Western blotting
Chondrocytes were seeded at a density of 4 × 105 cells per well. After 24 hours of cultivation in an incubator at 37 °C and 5% CO2, chondrocytes were treated with IL-1β (10 ng ml−1) or TNF-α (50 ng ml−1) for 4 days.43 Cells were lysed using RIPA lysis buffer supplemented with PMSF and phosphatase inhibitor. After centrifugation, the supernatant was collected as the total protein extract. Protein concentrations were measured using the BCA kit. The extracted proteins were separated by SDS-PAGE and transferred to a PVDF membrane. Non-specific binding was blocked with 5% skimmed milk for 60 minutes at room temperature. Specific primary antibodies were incubated with the membrane overnight at 4 °C. After washing, the membrane was incubated with HRP-conjugated secondary antibodies for 60 minutes at room temperature. Following additional washes, the membrane was visualized using the Amersham Imager 600 (GE, USA). Quantitative analysis of protein bands was performed using ImageJ in a blinded manner.2.7 Quantitative real-time PCR
Mouse chondrocytes were seeded at a density of 4 × 105 cells per well in a 6-well plate and cultured at 37 °C under a 5% CO2 atmosphere for 24 hours to allow for complete adhesion. Subsequently, the cells were treated with IL-β (10 ng ml−1) or TNF-α (50 ng ml−1), along with varying concentrations of UB (0, 5, 10 μM) for 48 h. mRNA was extracted using an mRNA extraction kit (AG, Hunan, China) from treated cells. The concentration was determined using a Nano-Drop2000 spectrophotometer. The RNA purity data are shown in ESI Table 1.† cDNA was synthesized using reverse transcription reagents from AG. For qRT-PCR, the reaction mixture included SYBR Green qPCR Master Mix (5 μL), distilled water (3 μL), cDNA (1 μL), and forward and reverse primers (1 μL). The ABI Prism 7500 system (ABI, Foster City, CA, USA) was used to quantify target gene expression, with β-actin as the reference gene for normalization. See Table 2 for primer sequences of target and reference genes.| Genes | Forward primers (5′-3′) | Reverse primers (5′-3′) |
|---|---|---|
| iNOS | GTTCTCAGCCCAACAATACAAGA | GTGGACGGGTCGATGTCAC |
| COX-2 | TTCAACACACTCTATCACTGGC | AGAAGCGTTTGCGGTACTCAT |
| MMP3 | ACTGTGTCCCAAGGAGAGGAG | AAACCATCTACACAGTTCAGACAC |
| MMP13 | CAAGCAGTTCCAAAGGCTACA | TAGGGCTGGGTCACACTTCT |
| ADAMTS5 | ATGCAGCCATCCTGTTCACC | AAGGCCAAGTAGATGCCCAATTT |
| SOX9 | TAATTCCCCAGGCTCTTGGAT | GCAGCCGGGATTTAAGGCTC |
| COL2a | TGGCTTAGGGCAGAGAGAGA | CGTCGTGCTGTCTCAAGGT |
| Aggrecan | CACTGTCAAAGCACCATGCC | TAGGCTGGCTCCCATTCAGT |
| β-actin | GCAGGAGTACGATGAGTCCG | ACGCAGCTCAGTAACAGTCC |
2.8 Molecular docking
Due to the unavailability of the complete crystal structure of NF-kB p65, we employed Protein Data Bank (PDB ID: 1IKN) and UniProt databases to predict the full-length sequence of NF-kB p65, with the AlphaFold ID: Q04207. This sequence encompasses the DNA-binding domain, dimerization domain, nuclear localization signal, and transcriptional activation domain. We obtained the chemical structure of UB and JSH-23 from PubChem (https://pubchem.ncbi.nlm.nih.gov/compound/5380406#section=Structures) and (https://pubchem.ncbi.nlm.nih.gov/compound/16760588).Molecular docking of the NF-kB p65 protein with JSH-23 and UB was performed using the Dock module in MOE v2022.02.44 We specifically targeted the nuclear localization signal (NLS) motif (301KRKR304) of P65 for molecular docking with UB and JSH-23. MOE v2022.02 was used to determine specific docking sites, interaction modes, binding free energies, and binding images.
2.9 Anterior cruciate ligament transection (ACLT) models
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Zhejiang University and approved by the Animal Ethics Committee of Sir Run Run Shaw Hospital (affiliated with Zhejiang University, Hangzhou, Zhejiang).15 male mice (C57BL/6, 8 weeks old) were obtained from Ziyuan Experimental Animal Technology Co., Ltd (Hangzhou, China). The mice were housed in a controlled barrier facility under pathogen-free conditions with regulated temperature, humidity, and a 12-hour light/dark cycle. They had unrestricted access to food and water. A double-blind method was used to randomly allocate the mice into three groups: Sham, ACLT, and ACLT + UB. Before surgery, the mice were anesthetized with 4% chloral hydrate. Measures were taken to monitor and maintain the mice's temperature and respiration during the procedure. The ACLT group and the ACLT + UB group underwent anterior cruciate ligament transection (ACLT) on their right knee joint, while the Sham group underwent sham surgery. After the surgery, any mice showing severe signs of wound inflammation that affected their vital signs were excluded from the study. One week after the surgery, the ACLT + UB group received intraperitoneal injections of UB at a dose of 50 mg kg−1 every two days.45 The Sham group and the ACLT group were administered a placebo. After 8 weeks, the mice were euthanized, and their knee joints were collected for analysis using micro-CT scanning, immunofluorescence, and safranin O/fast green staining. For counting the osteophytes, we employed a double-blind method to manually count the number of osteophytes per field.
2.10 Histology and immunofluorescence
The knee joint specimens from mice were fixed with 4% paraformaldehyde for 48 hours. Then, a 10% ethylenediaminetetraacetic acid (EDTA) solution was immediately used for decalcification until complete softening of the bone tissue. Subsequently, the softened tissues were embedded in paraffin and cut into 3 μm thick sections. After complete deparaffinization, the sections were stained with the fast green solution for 5 minutes. Afterward, the sections were briefly rinsed with an acid solution for 15 seconds and then subjected to safranin O staining for 5 minutes. The extent of cartilage damage in the mouse knee joints was assessed using a triple-blind method, as per the Osteoarthritis Research Society International (OARSI) scoring criteria (see Table 3).| Grade | Osteoarthritic damage |
|---|---|
| 0 | Normal |
| 0.5 | Loss of safranin-O without structural changes |
| 1 | Small fibrillations without loss of cartilage |
| 2 | Vertical clefts down to the layer immediately below the superficial layer and some loss of surface lamina |
| 3 | Vertical clefts/erosion to the calcified cartilage extending to <25% of the articular surface |
| 4 | Vertical clefts/erosion to the calcified cartilage extending to 25–50% of the articular surface |
| 5 | Vertical clefts/erosion to the calcified cartilage extending to 50–75% of the articular surface |
| 6 | Vertical clefts/erosion to the calcified cartilage extending >75% of the articular surface |
For immunofluorescence assay, paraffin sections were deparaffinized and subjected to antigen retrieval by heating in a 0.01 M sodium citrate solution (pH = 6) at 95–100 °C for 20 minutes. After cooling, permeabilization was achieved by treating the sections with 0.1% Triton X-100 for 30 minutes, followed by blocking with 5% BSA for 60 minutes to minimize non-specific binding. Then, sections were incubated overnight at 4 °C with specific primary antibodies against MMP13 and COL2a. After PBS washing, the sections were incubated with a secondary antibody for 60 minutes at room temperature in the dark. The sections were then rinsed with PBS, stained with DAPI (1:1000 dilution in PBS) for 15 minutes to counterstain cell nuclei, and thoroughly cleaned. The fluorescence intensity of the target proteins was detected using the Magscanner KF-PRO-120 system. ImageJ was employed for quantitative analysis.
2.11 Statistical analysis
The experiments were performed in triplicate to ensure reproducibility. Statistical analysis was carried out using Prism 9 software (GraphPad Software, Inc., San Diego, CA, USA). The results were expressed as mean ± SD. Student's t-test was used to compare two groups, while one-way or two-way ANOVA followed by Tukey's post hoc analysis was applied for comparisons between multiple groups, as appropriate. The threshold for statistical significance was set at P < 0.05. Significance levels were indicated by asterisks (*P < 0.05, **P < 0.01, or ***P < 0.001).3. Results
3.1 Urolithin B reduces the inflammation of chondrocytes in vitro
The chemical structure of UB is depicted in Fig. 1A. Initially, the viabilities of the chondrocytes, treated with various concentrations of UB, IL1-β, and TNF-α, were assessed using the CCK-8 assay. The results revealed no significant effects of UB, IL1-β, and TNF-α on chondrocyte viability, indicating that these factors were safe for further investigations (ESI Fig. 1†). To simulate the inflammatory microenvironment in OA, IL-1β and TNF-α were used. The impact of UB on the inflammation was explored by stimulating chondrocytes with 10 ng ml−1 IL-1β or 50 ng ml−1 TNF-α for 2 or 4 days. QPCR and western blotting analyses revealed that IL-1β and TNF-α dramatically induced the expression of iNOS and COX-2, crucial pro-inflammatory factors involved in anabolic and catabolic processes in chondrocytes, whereas UB significantly inhibited this phenomenon in a dose-dependent manner (Fig. 1B–K).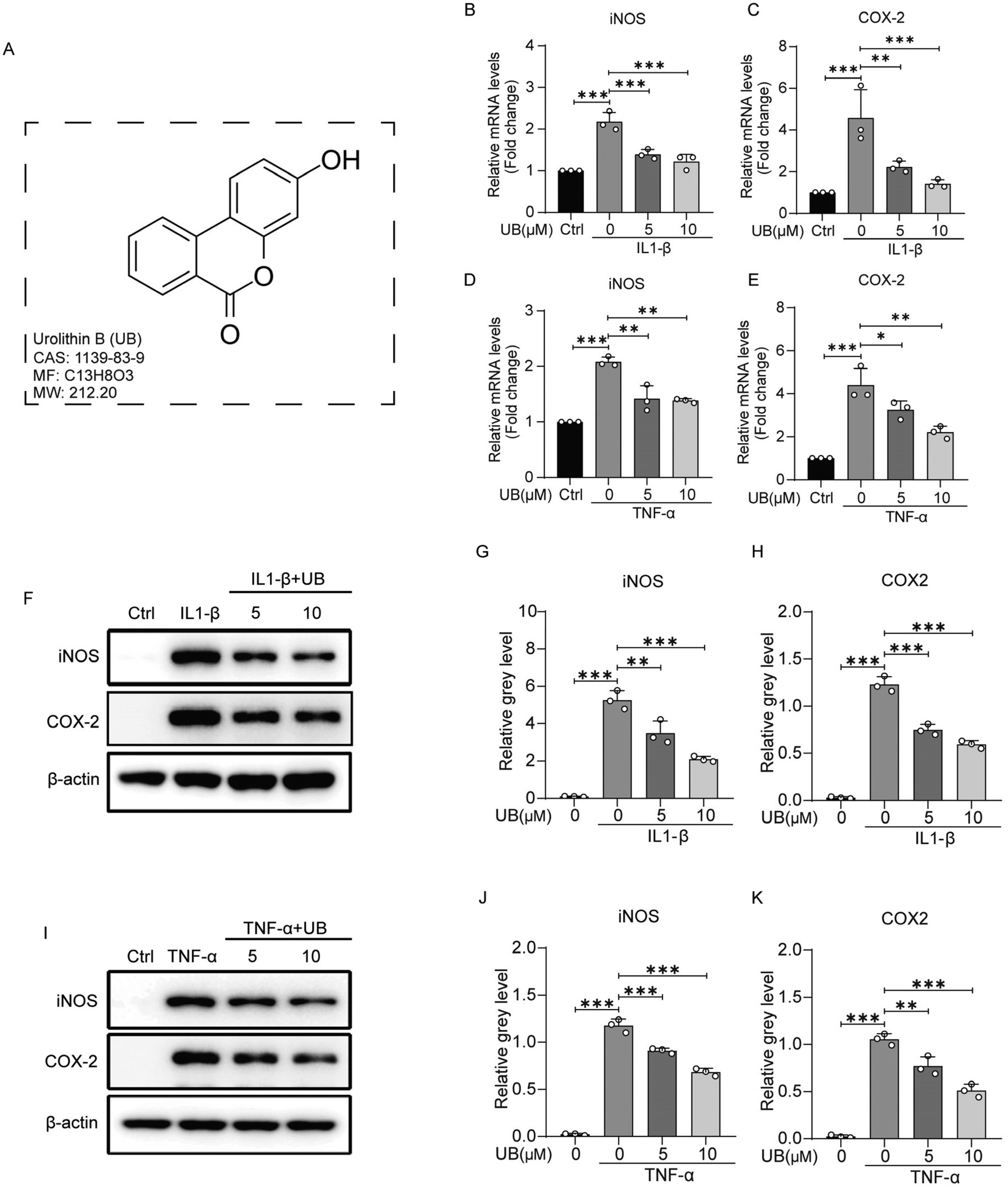 | ||
| Fig. 1 Urolithin B reduces the inflammation of chondrocytes in vitro. (A) The chemical structure of urolithin B. (B–E) Mouse chondrocytes were treated with 10 ng ml−1 IL-1β or 50 ng ml−1 TNF-α in combination with varying concentrations of urolithin B for 48 h. mRNA was extracted, and the transcription levels of iNOS and COX-2 were analyzed using qPCR assays (n = 3). (F and I) Mouse chondrocytes were treated with 10 ng ml−1 IL-1β or 50 ng ml−1 TNF-α in combination with varying concentrations of urolithin B for 4 days. Total protein was extracted, and western blotting was performed to determine the protein levels of iNOS and COX-2 (n = 3). (G, H, J and K) Quantitative analysis of the relative gray value (n = 3). All data are presented as mean ± SD. Statistical significance is denoted as *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
3.2 Urolithin B restrains the degradation of the extracellular matrix
Under inflammatory conditions, the enhanced rate of catabolism and decreased anabolism in chondrocytes lead to ECM degradation. Therefore, it is crucial to explore the effects of UB on the expression of critical genes associated with metabolic processes. As shown in Fig. 2, IL-1β and TNF-α significantly promoted the transcriptional activity of catabolism-related genes, such as MMP3, MMP13, and ADAMTS5, while the expression of anabolism-related genes, including SOX9, COL2a, and aggrecan, was inhibited. However, UB treatment efficiently reversed these changes in a concentration-dependent manner.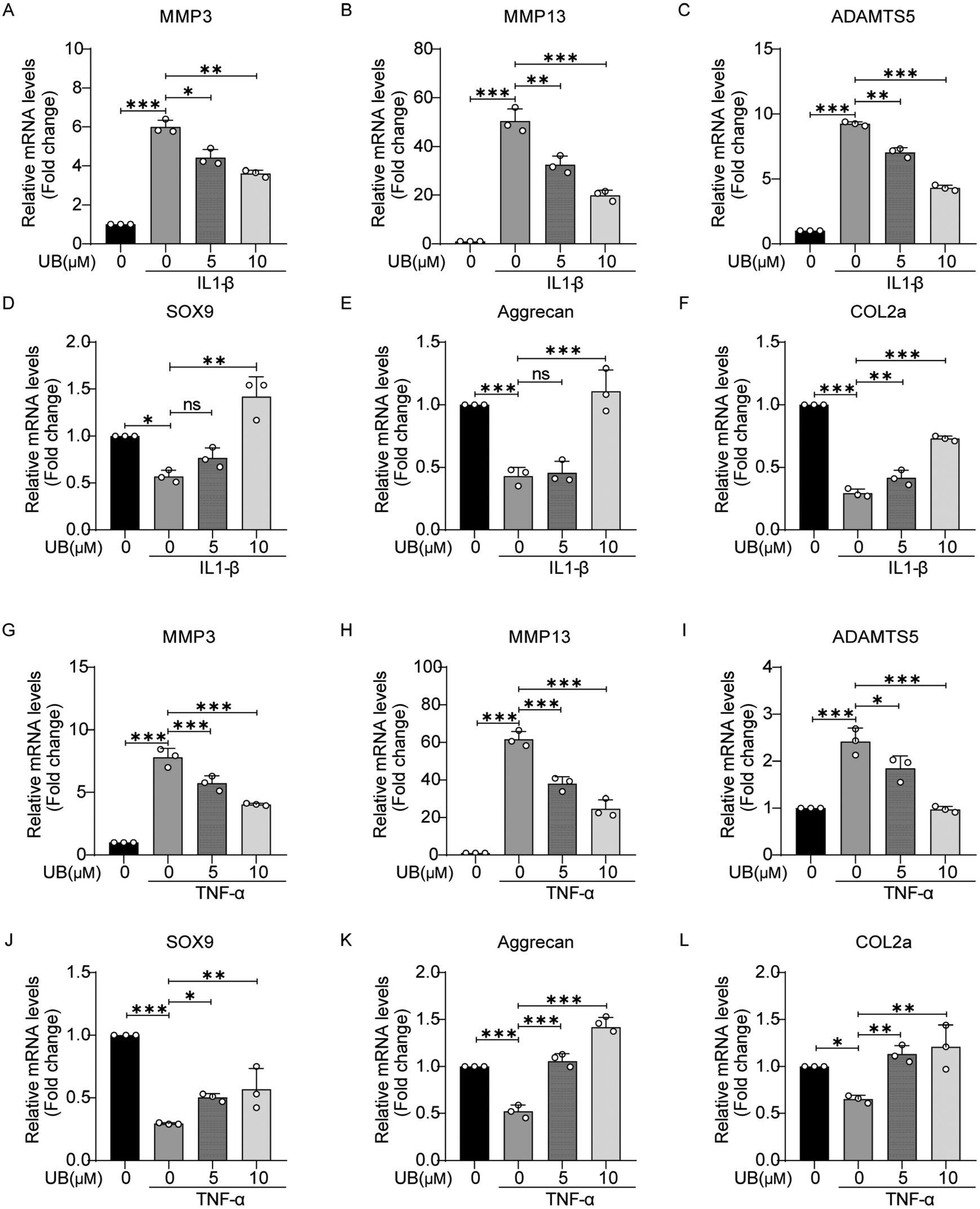 | ||
| Fig. 2 Urolithin B regulates the OA-related gene expression. (A–L) Mouse chondrocytes were treated with 10 ng ml−1 IL-1β or 50 ng ml−1 TNF-α in combination with varying concentrations of urolithin B for 48 h. mRNA was extracted. qPCR was used to detect the transcription levels of MMP3, MMP13, ADAMTS5, SXO9, COL2a and aggrecan (n = 3). All data are presented as mean ± SD. Statistical significance is denoted as *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
The visual assessment of collagen II and proteoglycans in the ECM was performed using safranin O, Alcian blue, and toluidine blue staining which revealed that inflammatory factors significantly suppress the anabolism in chondrocytes, which is characterized by light staining with safranin O, Alcian blue, and toluidine blue. However, UB restrains the degradation of ECM, and this effect was enhanced with increasing concentrations of UB (Fig. 3A and B). Similarly, an immunofluorescence assay demonstrated that UB significantly upregulates COL2a synthesis and inhibits MMP13 expression in an inflammatory microenvironment (Fig. 3C and D, ESI Fig. 2A and B†). Furthermore, UB inhibits the expression of MMP13 and ADAMTS5 and increases the synthesis of COL2a and aggrecan at the protein level. These results are consistent with the qPCR data (Fig. 3E–H). Collectively, these results indicated that UB reduces the degradation of the chondrocyte matrix induced by inflammatory factors.
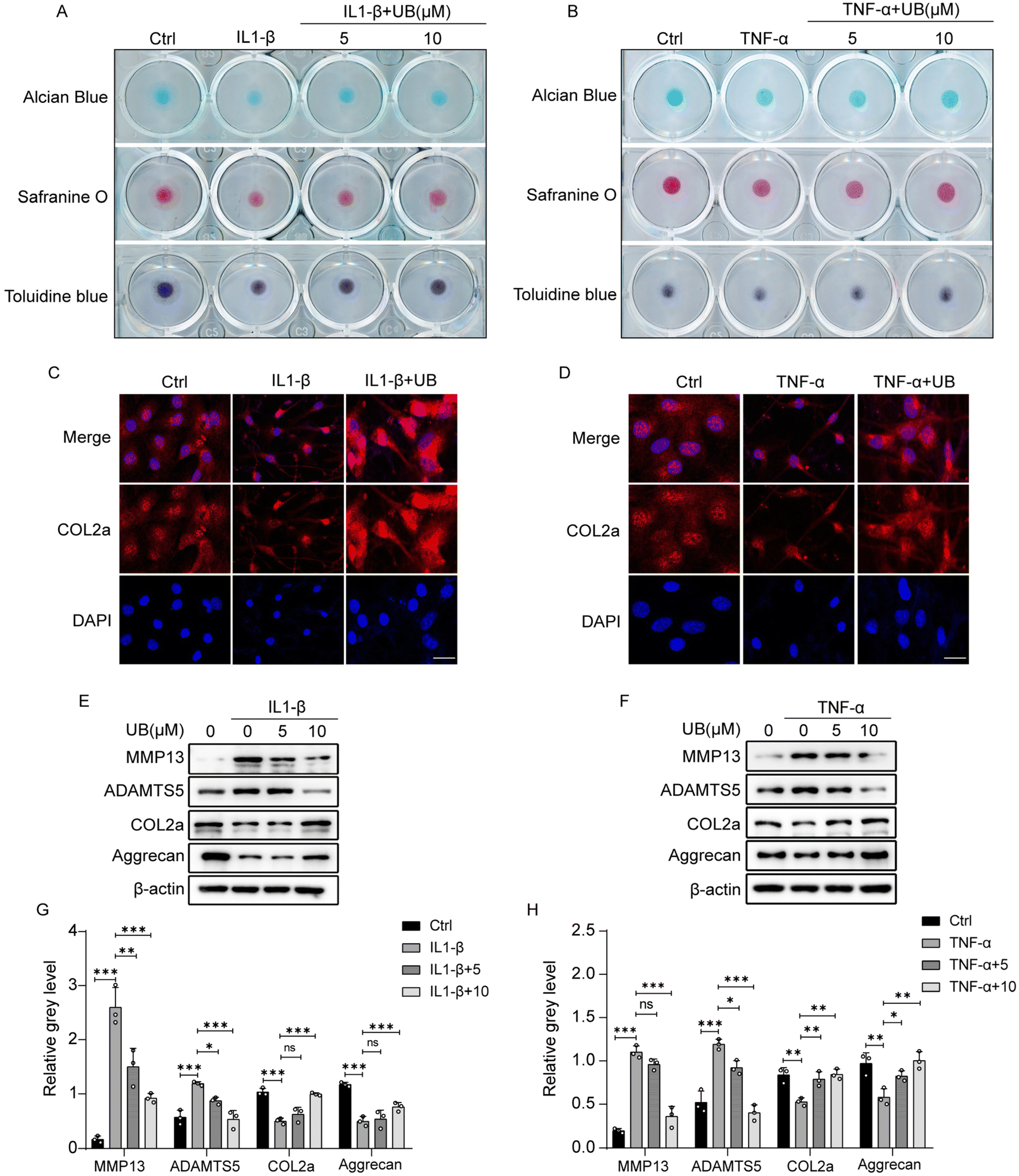 | ||
| Fig. 3 Urolithin B restrains the degradation of the extracellular matrix. (A–B) Mouse chondrocytes were co-treated with IL-1β (10 ng ml−1) or TNF-α (50 ng ml−1) along with various concentrations of UB for 7 days. Staining with safranin O, Alcian blue, and Toluidine blue was performed to visualize the extracellular matrix (n = 3). (C–D) Mouse chondrocytes were co-treated with IL-1β (10 ng ml−1) or TNF-α (50 ng ml−1) and 10 μM UB for 4 days. Immunofluorescence staining was used to assess COL2a expression (n = 3), scale bar = 50 μm. (E and F) Mouse chondrocytes were co-treated with IL-1β (10 ng ml−1) or TNF-α (50 ng ml−1) and 10 μM UB for 4 days. Western blot analysis was performed to evaluate protein expression levels of MMP13, ADAMTS5, COL2a, and aggrecan (n = 3). (G and H) Quantitative analysis of the relative gray value (n = 3). All data are presented as mean ± SD. Statistical significance is denoted as *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
3.3 Urolithin B restrains the NF-κB pathway activation
Phosphorylation of IκB-α majorly regulates the NF-κB signaling pathway. IL1-β and TNF-α stimulation significantly upregulates the phosphorylation of IκB-α, which can be restrained by UB (Fig. 4A and B, ESI Fig. 3A and B†). The nuclear translocation of NF-κB P65 is necessary for its transcriptional activity. Therefore, nuclear-cytoplasmic protein separation assays were conducted to investigate the effects of UB on P65 nucleocytoplasmic shuttling. We found that UB inhibits the inflammatory factor-induced nuclear translocation and the transcriptional activity of NF-κB P65 (Fig. 4C and D, ESI Fig. 3C and D†). Similarly, immunofluorescence assays indicated that UB significantly suppresses the nuclear translocation of NF-κB P65 (Fig. 4E, ESI Fig. 3E†).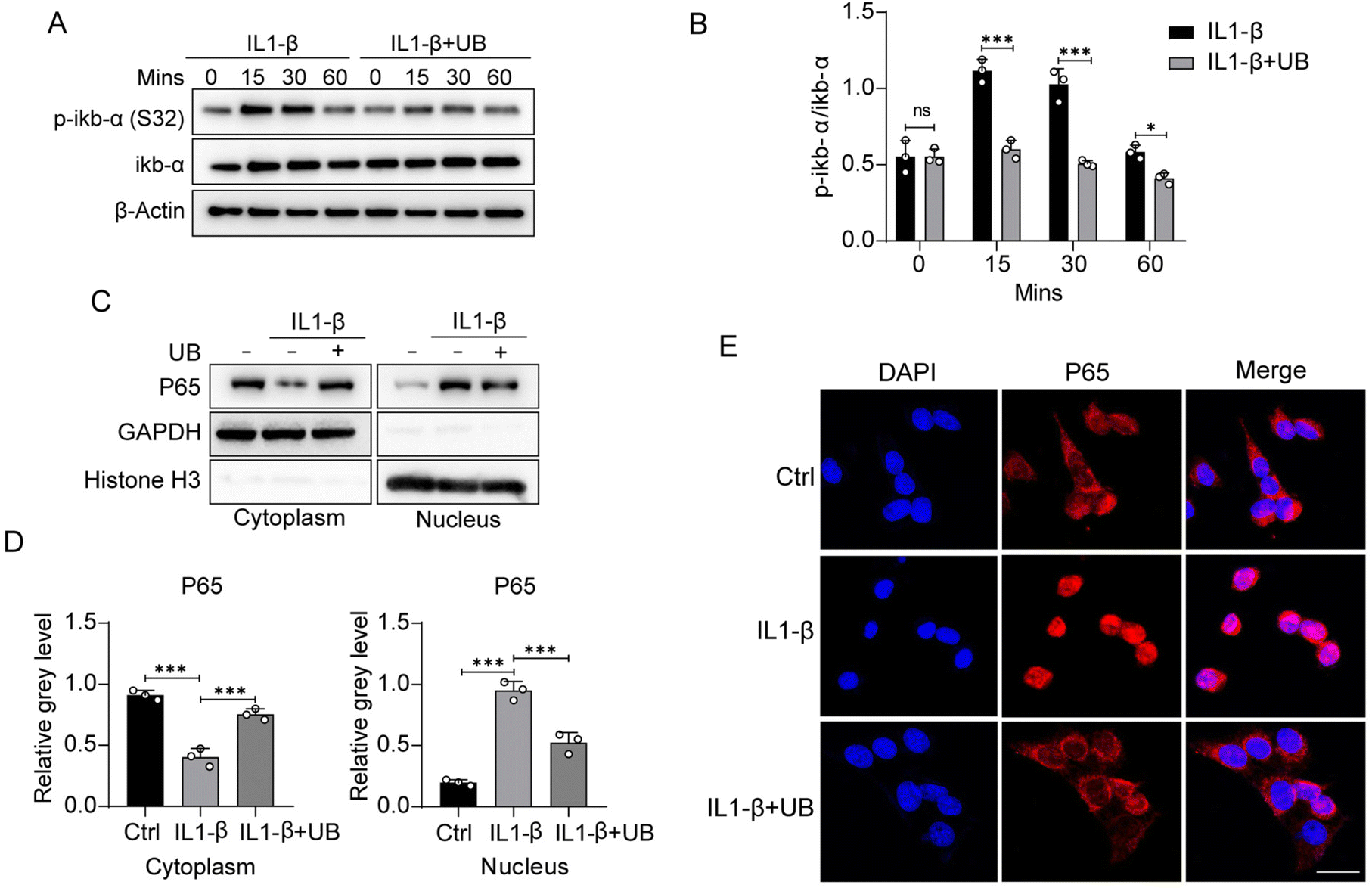 | ||
| Fig. 4 Urolithin B restrains the NF-κB pathway activation induced by IL1-β. (A) ATDC5 cells were pretreated with UB for 2 h, followed by treatment with IL-1β (10 ng ml−1) with or without UB for 0, 15, 30, and 60 min. Total cellular proteins were extracted and subjected to western blotting to assess the phosphorylation levels of IkB-α (n = 3). (B) Quantitative analysis was performed to evaluate the phosphorylation levels of IkB-α (n = 3). (C) ATDC5 cells were pretreated with UB for 2 h, followed by treatment with IL-1β (10 ng ml−1) with or without UB for 60 min. Nuclear and cytoplasmic proteins were separated and analyzed using western blotting to determine the distribution of P65 (n = 3). (D) Quantitative analysis was conducted to measure the grayscale values of the P65 protein bands (n = 3). (E) A cellular immunofluorescence assay was performed to examine the nuclear-cytoplasmic distribution of P65 (n = 3). (F) Molecular docking was carried out to investigate the interaction between UB and P65. All data are presented as mean ± SD. Statistical significance is denoted as *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
In summary, due to the inhibition of the phosphorylation of IκB by UB, ubiquitination-mediated degradation of IκB was avoided. The dissociation of IκB and P50/P65 is restricted, leading to the sequestration of P50/P65 in the cytoplasm, preventing their translocation into the nucleus to activate the transcription of inflammation-related genes. As a result, UB exerts its anti-inflammatory effect by suppressing the phosphorylation of IκB-α and blocking the nuclear translocation of NF-κB P65.
3.4. Urolithin B delays the progression of osteoarthritis induced by anterior cruciate ligament transaction
We constructed a mouse model of ACLT to explore the effects of UB on OA in vivo (Fig. 5A). The micro-CT results indicated significant symptoms, including narrow joint spaces, periarticular hyperosteogeny, and osteophyte formation, in the knee joints of ACLT mice compared to those in the sham group. However, the UB treatment exhibited remarkable efficacy in ameliorating periarticular hyperosteogeny and inhibiting osteophyte formation (Fig. 5B and D). Safranin O–fast green staining revealed significant erosion of articular cartilage in ACLT mice, whereas UB treatment efficiently delayed degradation of the cartilage matrix. Additionally, immunofluorescence staining revealed a substantial expression of MMP13 and a small amount of COL2a in the articular cartilage of the ACLT group. Strikingly, UB reduced MMP13 levels and promoted COL2a expression (Fig. 5C–G). Furthermore, H&E staining of organs demonstrated that UB exhibited excellent safety (ESI Fig. 4A†). These results suggest that UB plays a crucial role in reducing cartilage degradation. In vivo data indicate that UB improves osteoarthritis, which highlights its potential as a therapeutic approach.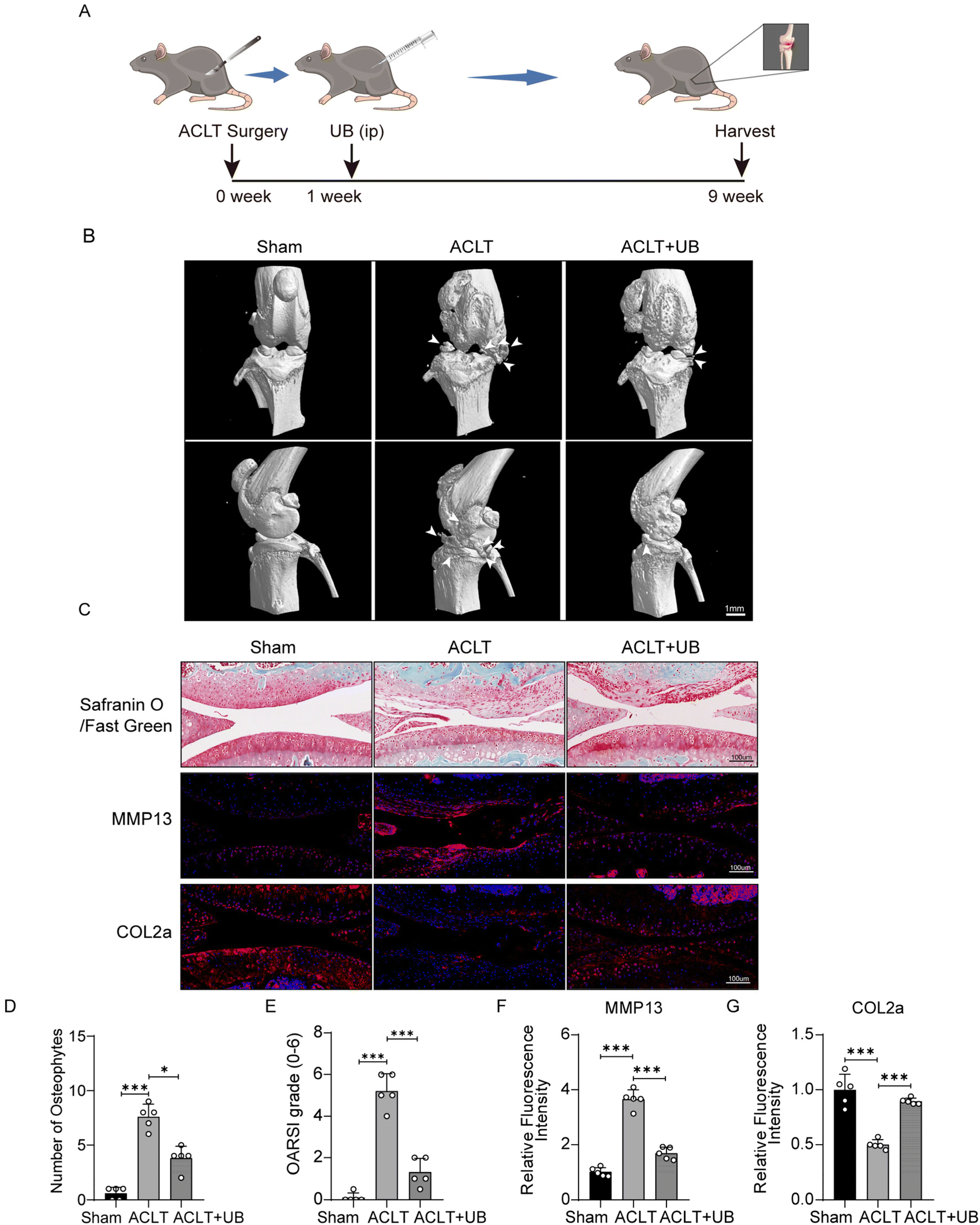 | ||
| Fig. 5 Urolithin B delays the progression of osteoarthritis induced by anterior cruciate ligament transaction. (A) Schematic representation of the anterior cruciate ligament transection model. (B) MicroCT analysis of mouse knee joint lesions (n = 5). (C) Safranin O–fastgreen staining of mouse knee joint specimens, along with immunofluorescence staining for COL2a and MMP13 (n = 5). (D) Quantitative analysis of periarticular osteophyte formation in mouse knee joints (n = 5). (E) Osteoarthritis Research Society International (OARSI) scoring of mouse knee joints (n = 5). (F and G) Quantitative analysis of relative immunofluorescence intensity of COL2a and MMP13 in mouse knee joints (n = 5). All data are presented as mean ± SD. Statistical significance was denoted as *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
4. Discussion
OA is a complex disease with limited therapeutic options. In the advanced stages of the disease, severe pain and joint stiffness significantly affect the quality of a patient's life; moreover, expensive joint replacements pose a substantial economic burden on both patients and society46,47 Therefore, in recent research, the mechanisms underlying OA and the development of safe, efficient, and cost-effective therapeutic strategies have been primarily focused on. In this study, we conducted a series of experiments to validate the anti-inflammatory and anti-osteoarthritic effects of UB and elucidate the underlying mechanisms.Previously, OA has been defined as a degenerative disease caused by cartilage erosion. It is a complex condition involving various joint pathologies, with inflammation playing a key role in these pathologies.48,49 Inflammatory markers, such as CRP, IL-6, and TNF-α, are reported to increase with age. Previous epidemiological research has established a correlation of elevated inflammatory markers with pain and functional limitations in elderly patients with OA.49 In obese populations, the adipose tissue releases adipokines that induce proinflammatory cytokines in the synovium and cartilage, thus intensifying joint inflammation.50 Mechanical stresses on joints activate mechanoreceptors on chondrocyte surfaces, triggering intracellular inflammatory pathways.51 The NF-κB signaling cascade, a downstream pathway stimulated by these inflammatory and mechanical signals, plays a critical role in the production of inflammatory factors. The activation of this pathway accelerates ECM degradation via enhanced expression of iNOS and COX-2. Hence, investigating the association between UB and the NF-κB signaling pathway can significantly aid in understanding the anti-inflammatory mechanism of UB. Moreover, activated chondrocytes enhance catabolism and reduce anabolism, causing disrupted cartilage homeostasis and ECM degradation; thereby, OA progression is promoted.52 Consequently, anti-inflammatory agents can efficiently alleviate these symptoms and suppress joint deterioration in patients with OA. In our study, we have demonstrated that UB significantly inhibits the excessive upregulation of iNOS and COX-2 induced by IL-1β and TNF-α, demonstrating potent anti-inflammatory properties of UB. Furthermore, in vitro analyses revealed that UB significantly suppresses the expression of genes related to cartilage matrix degradation, such as MMP3, MMP13, and ADAMTS5, thereby delaying ECM degradation. Additionally, UB treatment increased the synthesis of ECM components, including type II collagen and proteoglycans, which reflects the ECM repair process. These findings suggest that UB blocks the ECM degradation caused by inflammation in vitro. Notably, the anti-inflammatory and cartilage-protective effects of UB can be attributed to its inhibitory effect on the NF-κB signaling pathway. Our results demonstrate that UB efficiently suppresses the phosphorylation of IκB-α at ser-32 and hampers the nuclear localization of NF-kB P65. Consequently, the release of iNOS and COX-2 from chondrocytes is reduced and the synthesis of MMPs is restrained; hence, the loss of ECM is inhibited.
Although we have proved that UB restrains the activation of the NF-kB pathway, the mechanism has not been illustrated. Based on our current results, we speculate that UB may inhibit the phosphorylation of IκB by suppressing the activation of the NEMO complex (IKKα/IKKβ/IKKγ). By inhibiting the activity of this kinase, UB prevents the transfer of the γ-phosphate of ATP or GTP onto the serine residues of IκB,53 consequently inhibiting the phosphorylation of IκB. Due to this inhibition, ubiquitination-mediated degradation of IκB was limited. The dissociation of IκB from P50/P65 is restricted, leading to the detention of P50/P65 in the cytoplasm, preventing their translocation into the nucleus to activate the transcription of inflammation-related genes.
Furthermore, we conducted a molecular docking experiment between UB and NF-kB P65 to reveal the binding sites and modes, providing supplementary evidence for the inhibition of NF-kB P65 nuclear translocation by UB. In this experiment, we take the nuclear localization signal (NLS) motif of NF-kB P65 as the docking site. The nuclear localization signal (NLS) motif (301KRKR304) plays a crucial role in facilitating NF-kB P65 nuclear localization through binding with importin α3 (ESI Fig. 5A†), an important protein involved in mediating nuclear translocation.54 We compared the docking between UB and NF-kB P65 with the docking between JSH-23 (an inhibitor of NF-kB P65 nuclear translocation) and NF-kB P65.55,56 Notably, our analysis revealed that both UB and JSH-23 can bind to the NLS motif, forming hydrogen bonds with the amino acid residue ARG302 (ESI Fig. 5B–E†). The calculated lowest binding energy was determined as −4.37794542 kcal mol−1 and −4.8580966 kcal mol−1, respectively. UB exhibited slightly weaker binding affinity compared to JSH-23. UB interacts with NLS by forming two hydrogen bonds with bond lengths of 4.30 and 3.57 Å, contributing energies of −0.5 and −0.8 kcal mol−1, respectively. JSH-23 interacts with NLS by forming one hydrogen bond with a bond length of 3.83 Å, contributing an energy of −0.5 kcal mol−1 (ESI Fig. 5D and G†). It is important to highlight that previous studies have indicated that any mutations in the cluster of basic amino acid residues within the NF-kB P65 NLS motif can disrupt its interaction with importin α3, leading to the inhibition of NF-kB P65 nuclear translocation.57,58 Consequently, our results provide evidence that both UB and JSH-23 can interact with the ARG302 residue in the NLS motif. This interaction potentially impedes the binding of importin α3 with NF-kB P65, thereby hindering the nuclear entry of NF-kB P65.
Similarly, the molecular mechanism underlying the impacts of NF-κB on the release of downstream inflammatory factors remains unclear. E74 like ETS transcription factor 3 (ELF3), belonging to the ETS transcription factor family, plays a crucial regulatory role in cell differentiation and proliferation.59 Normally, ELF3 is exclusively expressed in the epithelial tissues; however, in the inflammatory microenvironment, it is expressed in chondrocytes.60 The transcriptional activation of ELF3 depends on the nuclear translocation of NF-κB, and the activated ELF3 can upregulate iNOS and COX-2 genes at the transcript level by directly binding to ETS binding sites of their promoters.61–63 Interestingly, ELF3 acts as an endogenous regulator of MMP13 promoter activity induced by IL-1β and TNF-α. Moreover, activated ELF3 inhibits the transcription of COL2a through downregulated promoter functions.64 Collectively, ELF3, a downstream signaling molecule of NF-κB, plays a significant role in promoting inflammation and regulating the metabolic balance of chondrocytes. Therefore, further research on whether UB affects the expression of ELF3 in chondrocytes is required for the comprehensive knowledge of mechanisms underlying its anti-inflammatory and cartilage-protective effects.
Previous clinical trials have demonstrated that the consumption of ellagic acid-rich foods can lower blood lipids, exert neuroprotective effects, alleviate brain damage, improve cognition, and enhance plasma antioxidant capacity. Unfortunately, there are no direct clinical trials on UB. Most clinical studies have focused on the pharmacological effects of ellagic acid and the microbiota involved in ellagic acid metabolism. We think several difficulties impede the clinical application of UB: (1) UB's biological distribution varies among organs due to the influence of transport proteins. (2) The extraction and purification of UB are costly, making large-scale production challenging in the short term. (3) The metabolic pathways, toxicity, and side effects of UB in the human body have not been fully elucidated. Further research is needed to overcome these issues.
5. Conclusions
In conclusion, in this study, we demonstrated that UB, a metabolite of ellagic acid found in common dietary sources, exhibits potent anti-inflammatory effects and cartilage-protective properties. UB inhibits IL-1β and TNF-α-induced inflammation in vitro and restricts the expression of matrix-degrading enzymes produced by activated chondrocytes. Notably, UB decreases ECM degradation; moreover, it promotes the synthesis of type II collagen and proteoglycans to repair ECM damage. In the mouse model of ACLT, UB significantly ameliorated OA joint pathology by inhibiting articular cartilage erosion, periarticular osteophyte formation, and subchondral bone sclerosis. UB alleviates chondrocyte inflammation by suppressing the activation of the NF-κB signaling pathway and reduces associated adverse events. Considering the safety and efficacy, UB is proposed as a potential therapeutic agent for the effective management of OA.Author contributions
Peihua Shi: conceptualization, methodology, supervision, and funding acquisition. Hong Xue: software, validation, formal analysis, writing – original draft, writing – review & editing, and data curation. Hongyu Zhou: writing – review & editing. Qiliang Lou: data curation. Putao Yuan: formal analysis, writing – original Draft, and writing – review & editing. Zhenhua Feng: investigation. Li Qiao: resources. Jiateng Zhang: visualization. Hongwei Xie: validation. Yang Shen: writing – review & editing. Qingliang Ma: software. Shiyu Wang: software. Boya Zhang: funding acquisition. Huali Ye: validation. Jiao Cheng: data curation. Xuewu Sun: project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (82172458, 82271604, and 82103740) for financial support.References
- J. Martel-Pelletier, A. J. Barr, F. M. Cicuttini, P. G. Conaghan, C. Cooper and M. B. Goldring, et al., Osteoarthritis, Nat. Rev. Dis. Primers, 2016, 2, 16072 CrossRef PubMed.
- S. Glyn-Jones, A. J. R. Palmer, R. Agricola, A. J. Price, T. L. Vincent and H. Weinans, et al., Osteoarthritis, Lancet, 2015, 386, 376–387 CrossRef CAS PubMed.
- D. T. Felson, A. Naimark, J. Anderson, L. Kazis, W. Castelli and R. F. Meenan, The prevalence of knee osteoarthritis in the elderly. the framingham osteoarthritis study, Arthritis Rheum., 1987, 30, 914–918 CrossRef CAS PubMed.
- E. Yusuf, R. G. Nelissen, A. Ioan-Facsinay, V. Stojanovic-Susulic, J. DeGroot and G. Van Osch, et al., Association between weight or body mass index and hand osteoarthritis: A systematic review, Ann. Rheum. Dis., 2010, 69, 761–765 CrossRef PubMed.
- J. N. Katz, K. R. Arant and R. F. Loeser, Diagnosis and Treatment of Hip and Knee Osteoarthritis: A Review, JAMA, J. Am. Med. Assoc., 2021, 325, 568–578 CrossRef CAS PubMed.
- E. Losina, A. M. Weinstein, W. M. Reichmann, S. A. Burbine, D. H. Solomon and M. E. Daigle, et al., Lifetime risk and age at diagnosis of symptomatic knee osteoarthritis in the US, Arthritis Care Res., 2013, 65, 703–711 CrossRef PubMed.
- T. D. Brown, R. C. Johnston, C. L. Saltzman, J. L. Marsh and J. A. Buckwalter, Posttraumatic osteoarthritis: A first estimate of incidence, prevalence, and burden of disease, J. Orthop. Traumatol., 2006, 20, 739–744 Search PubMed.
- V. B. Kraus, F. J. Blanco, M. Englund, M. A. Karsdal and L. S. Lohmander, Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use, Osteoarthritis and Cartilage, W.B. Saunders Ltd, 2015, vol. 23, pp. 1233–1241 Search PubMed.
- D. B. Burr and M. A. Gallant, Bone remodelling in osteoarthritis, Nat. Rev. Rheumatol., 2012, 8, 665–673 CrossRef CAS PubMed.
- W. Zhang, G. Nuki, R. W. Moskowitz, S. Abramson, R. D. Altman and N. K. Arden, et al., OARSI recommendations for the management of hip and knee osteoarthritis. Part III: Changes in evidence following systematic cumulative update of research published through January 2009, Osteoarthr. Cartil., 2010, 18, 476–499 CrossRef CAS PubMed.
- K. Gaffney, J. Ledingham and J. D. Perry, Intra-articular triamcinolone hexacetonide in knee osteoarthritis: Factors influencing the clinical response, Ann. Rheum. Dis., 1995, 54, 379–381 CrossRef CAS PubMed.
- G. N. Lewis, D. A. Rice, P. J. McNair and M. Kluger, Predictors of persistent pain after total knee arthroplasty: A systematic review and meta-analysis, Br J Anaesth., 2015, 114, 551–561 CrossRef CAS PubMed.
- N. K. Arden, I. C. Reading, K. M. Jordan, L. Thomas, H. Platten and A. Hassan, et al., A randomised controlled trial of tidal irrigation vs corticosteroid injection in knee osteoarthritis: the KIVIS Study, Osteoarthr. Cartil., 2008, 16, 733–739 CrossRef CAS PubMed.
- C. Wainwright, J. C. Theis, N. Garneti and M. Melloh, Age at hip or knee joint replacement surgery predicts likelihood of revision surgery, J. Bone Jt. Surg., Br., 2011, 93 B, 1411–1415 CrossRef PubMed.
- D. Scott and A. Kowalczyk, Osteoarthritis of the knee, BMJ Clin Evid., 2007, 2007, 841–848 Search PubMed.
- S. Kaneko, T. Satoh, J. Chiba, C. Ju, K. Inoue and J. Kagawa, Interleukin-6 and interleukin-8 levels in serum and synovial fluid of patients with osteoarthritis, Cytokines, Cell. Mol. Ther., 2000, 6, 71–79 CrossRef CAS PubMed.
- J. Martel-Pelletier, R. Mccollum, J. Dibattista, M.-P. Faure, J. A. Chin and S. Fournier, et al., The interleukin–1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function, Arthritis Rheum., 1992, 35, 530–540 CrossRef CAS PubMed.
- J. A. Roman-Blas and S. A. Jimenez, NF-κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis, Osteoarthr. Cartil., 2006, 14, 839–848 CrossRef CAS PubMed.
- A. B. Blom, S. M. Brockbank, P. L. Van Lent, H. M. Van Beuningen, J. Geurts and N. Takahashi, et al., Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: Prominent role of Wnt-induced signaling protein 1, Arthritis Rheum., 2009, 60, 501–512 CrossRef CAS PubMed.
- M. Kapoor, J. Martel-Pelletier, D. Lajeunesse, J. P. Pelletier and H. Fahmi, Role of proinflammatory cytokines in the pathophysiology of osteoarthritis, Nat. Rev. Rheumatol., 2011, 7, 33–42 CrossRef CAS PubMed.
- S. B. Abramson, M. Attur, A. R. Amin and R. Clancy, Nitric oxide and inflammatory mediators in the perpetuation of osteoarthritis, Curr. Rheumatol. Rep., 2001, 3, 535–541 CrossRef CAS PubMed.
- C. Chadjichristos, C. Ghayor, M. Kypriotou, G. Martin, E. Renard and L. Ala-Kokko, et al., Sp1 and Sp3 transcription factors mediate interleukin-1β down-regulation of human type II collagen gene expression in articular chondrocytes, J. Biol. Chem., 2003, 278, 39762–39772 CrossRef CAS PubMed.
- J. Saklatvala, Tumour necrosis factor α stimulates resorption and inhibits synthesis of proteoglycan in cartilage, Nature, 1986, 322, 547–549 CrossRef CAS PubMed.
- J. A. Mengshol, M. P. Vincenti, C. I. Coon, A. Barchowsky and C. E. Brinckerhoff, Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor κB: Differential regulation of collagenase 1 and collagenase 3, Arthritis Rheum., 2000, 43, 801–811 CrossRef CAS PubMed.
- S. S. Glasson, R. Askew, B. Sheppard, B. Carito, T. Blanchet and H. L. Ma, et al., Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis, Nature, 2005, 434, 644–648 CrossRef CAS PubMed.
- J. Yang, R. Lee, S. M. Henning, G. Thames, M. Hsu and H. Manlam, et al., Soy protein isolate does not affect ellagitannin bioavailability and urolithin formation when mixed with pomegranate juice in humans, Food Chem., 2016, 194, 1300–1303 CrossRef CAS PubMed.
- J. C. Espín, R. González-Barrio, B. Cerdá, C. López-Bote, A. I. Rey and F. A. Tomás-Barberán, Iberian pig as a model to clarify obscure points in the bioavailability and metabolism of ellagitannins in humans, J. Agric. Food Chem., 2007, 55, 10476–10485 CrossRef PubMed.
- G. Lee, J. S. Park, E. J. Lee, J. H. Ahn and H. S. Kim, Anti-inflammatory and antioxidant mechanisms of urolithin B in activated microglia, Phytomedicine, 2019, 55, 50–57 CrossRef CAS PubMed.
- A. O. Abdulrahman, A. Kuerban, Z. A. Alshehri, W. H. Abdulaal, J. A. Khan and M. I. Khan, Urolithins attenuate multiple symptoms of obesity in rats fed on a high-fat diet, Diabetes, Metab. Syndr. Obes., 2020, 13, 3337–3348 CrossRef CAS PubMed.
- D. Bialonska, P. Ramnani, S. G. Kasimsetty, K. R. Muntha, G. R. Gibson and D. Ferreira, The influence of pomegranate by-product and punicalagins on selected groups of human intestinal microbiota, Int. J. Food Microbiol., 2010, 140, 175–182 CrossRef CAS PubMed.
- D. Zheng, Z. Liu, Y. Zhou, N. Hou, W. Yan and Y. Qin, et al., Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway, Pharmacol. Res., 2020, 153, 104655 CrossRef CAS PubMed.
- T. Yuan, H. Ma, W. Liu, D. B. Niesen, N. Shah and R. Crews, et al., Pomegranate's Neuroprotective Effects against Alzheimer's Disease Are Mediated by Urolithins, Its Ellagitannin-Gut Microbial Derived Metabolites, ACS Chem. Neurosci., 2016, 7, 26–33 CrossRef CAS PubMed.
- Y. Li, Q. Zhuang, L. Tao, K. Zheng, S. Chen and Y. Yang, et al., Urolithin B suppressed osteoclast activation and reduced bone loss of osteoporosis via inhibiting ERK/NF-κB pathway, Cell Proliferation, 2022, 55, 1–15 Search PubMed.
- S. Shen, Y. Yang, P. Shen, J. Ma, B. Fang, Q. Wang, K. Wang, P. Shi, S. Fan and X. Fang, circPDE4B prevents articular cartilage degeneration and promotes repair by acting as a scaffold for RIC8A and MID1, Ann. Rheum. Dis., 2021, 80, 1209–1219 CrossRef CAS PubMed.
- X. Feng, J. Pan, J. Li, C. Zeng, W. Qi, Y. Shao, X. Liu, L. Liu, G. Xiao, H. Zhang, X. Bai and D. Cai, Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR, Aging, 2020, 12, 1087–1103 CrossRef CAS PubMed.
- C. Cho, L. J. Kang, D. Jang, J. Jeon, H. Lee, S. Choi, S. J. Han, E. Oh, J. Nam, C. S. Kim, E. Park, S. Y. Jeong, C. H. Park, Y. S. Shin, S. I. Eyun and S. Yang, Cirsium japonicum var. maackii and apigenin block Hif-2α-induced osteoarthritic cartilage destruction, J. Cell. Mol. Med., 2019, 23, 5369–5379 CrossRef CAS PubMed.
- O. Gabay, M. Gosset, A. Levy, C. Salvat, C. Sanchez, A. Pigenet, A. Sautet, C. Jacques and F. Berenbaum, Stress-induced signaling pathways in hyalin chondrocytes: inhibition by Avocado-Soybean Unsaponifiables (ASU), Osteoarthr. Cartil., 2008, 16, 373–384 CrossRef CAS PubMed.
- M. Gosset, F. Berenbaum, C. Salvat, A. Sautet, A. Pigenet, K. Tahiri and C. Jacques, Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis, Arthritis Rheum., 2008, 58, 1399–1409 CrossRef CAS PubMed.
- M. Gosset, F. Berenbaum, S. Thirion and C. Jacques, Primary culture and phenotyping of murine chondrocytes, Nat. Protoc., 2008, 3, 1253–1260 CrossRef CAS PubMed.
- C. Salvat, A. Pigenet, L. Humbert, F. Berenbaum and S. Thirion, Immature murine articular chondrocytes in primary culture: a new tool for investigating cartilage, Osteoarthr. Cartil., 2005, 13, 243–249 CrossRef PubMed.
- S. Eldridge, G. Nalesso, H. Ismail, K. Vicente-Greco, P. Kabouridis, M. Ramachandran, A. Niemeier, J. Herz, C. Pitzalis, M. Perretti and F. Dell'Accio, Agrin mediates chondrocyte homeostasis and requires both LRP4 and α-dystroglycan to enhance cartilage formation in vitro and in vivo, Ann. Rheum. Dis., 2016, 75, 1228–1235 CrossRef CAS PubMed.
- T. Hayami, M. Pickarski, Y. Zhuo, G. A. Wesolowski, G. A. Rodan and L. T. Duong, Characterization of articular cartilage and subchondral bone changes in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis, Bone, 2006, 38, 234–243 CrossRef PubMed.
- X. Feng, J. Pan, J. Li, C. Zeng, W. Qi, Y. Shao, X. Liu, L. Liu, G. Xiao, H. Zhang, X. Bai and D. Cai, Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR, Aging, 2020, 12, 1087–1103 CrossRef CAS PubMed.
- A. Scarpino, G. G. Ferenczy and G. M. Keserű, Comparative Evaluation of Covalent Docking Tools, J. Chem. Inf. Model., 2018, 58, 1441–1458 CrossRef CAS PubMed.
- J. E. Collins, J. N. Katz, E. E. Dervan and E. Losina, Trajectories and risk profiles of pain in persons with radiographic, symptomatic knee osteoarthritis: Data from the osteoarthritis initiative, Osteoarthr. Cartil., 2014, 22, 622–630 CrossRef CAS PubMed.
- E. Losina, A. D. Paltiel, A. M. Weinstein, E. Yelin, D. J. Hunter and S. P. Chen, et al., Lifetime medical costs of knee osteoarthritis management in the United States: Impact of extending indications for total knee arthroplasty, Arthritis Care Res., 2015, 67, 203–215 CrossRef PubMed.
- R. F. Loeser, S. R. Goldring, C. R. Scanzello and M. B. Goldring, Osteoarthritis: A disease of the joint as an organ, Arthritis Rheum., 2012, 64, 1697–1707 CrossRef PubMed.
- J. Sellam and F. Berenbaum, The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis, Nat. Rev. Rheumatol., 2010, 6, 625–635 CrossRef CAS PubMed.
- O. P. Stannus, G. Jones, L. Blizzard, F. M. Cicuttini and C. Ding, Associations between serum levels of inflammatory markers and change in knee pain over 5 years in older adults: A prospective cohort study, Ann. Rheum. Dis., 2013, 72, 535–540 CrossRef CAS PubMed.
- X. Wang, D. Hunter, J. Xu and C. Ding, Metabolic triggered inflammation in osteoarthritis, Osteoarthr. Cartil., 2015, 23, 22–30 CrossRef CAS PubMed.
- T. J. Knobloch, S. Madhavan, J. Nam, S. Agarwal Jr. and S. Agarwal, Regulation of chondrocytic gene expression by biomechanical signals, Crit. Rev. Eukaryotic Gene Expression, 2008, 18, 139–150 CrossRef CAS PubMed.
- J. Saklatvala, Inflammatory Signaling in Cartilage: MAPK and NF-κ B Pathways in Chondrocytes and the Use of Inhibitors for Research into Pathogenesis and Therapy of Osteoarthritis, Curr. Drug Targets, 2007, 8, 305–313 CrossRef CAS PubMed.
- M. Karin and Y. Ben-Neriah, Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity, Annu. Rev. Immunol., 2000, 18, 621–663 CrossRef CAS PubMed.
- T. J. Florio, R. K. Lokareddy, D. P. Yeggoni, R. S. Sankhala, C. A. Ott, R. E. Gillilan and G. Cingolani, Differential recognition of canonical NF-κB dimers by Importin α3, Nat. Commun., 2022, 13, 1207 CrossRef CAS PubMed.
- X. Chen, G. Liu, Y. Yuan, G. Wu, S. Wang and L. Yuan, NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling, Cell Death Dis., 2019, 10, 906 CrossRef CAS PubMed.
- X. Lai, M. Wang, Y. Zhu, X. Feng, H. Liang, J. Wu, L. Nie, L. Li and L. Shao, ZnO NPs delay the recovery of psoriasis-like skin lesions through promoting nuclear translocation of p-NFκB p65 and cysteine deficiency in keratinocytes, J. Hazard. Mater., 2021, 410, 124566 CrossRef CAS PubMed.
- J. Robbins, S. M. Dilworth, R. A. Laskey and C. Dingwall, Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence, Cell, 1991, 64, 615–623 CrossRef CAS PubMed.
- U. Zabel, T. Henkel, M. S. Silva and P. A. Baeuerle, Nuclear uptake control of NF-kappa B by MAD-3, an I kappa B protein present in the nucleus, EMBO J., 1993, 12, 201–211 CrossRef CAS PubMed.
- A. Verger and M. Duterque-Coquillaud, When Ets transcription factors meet their partners alexis verger and martine duterque-coquillaud, BioEssays, 2002, vol. 24, pp. 362–370 Search PubMed.
- H. Peng, L. Tan, M. Osaki, Y. Zhan, K. Ijiri and K. Tsuchimochi, et al., ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes, J. Cell. Physiol., 2008, 215, 562–573 CrossRef CAS PubMed.
- F. Grall, X. Gu, L. Tan, J. Y. Cho, M. S. Inan and A. R. Pettit, et al., Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor α in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor κB-mediated induction of the Ets transcription factor ESE-1, Arthritis Rheum., 2003, 48, 1249–1260 CrossRef CAS PubMed.
- F. T. Grall, W. C. Prall, W. Wei, X. Gu, J. Y. Cho and B. K. Choy, et al., The Ets transcription factor ESE-1 mediates induction of the COX-2 gene by LPS in monocytes, FEBS J., 2005, 272, 1676–1687 CrossRef CAS PubMed.
- S. Rudders, J. Gaspar, R. Madore, C. Voland, F. Grall and A. Patel, et al., ESE-1 is a Novel Transcriptional Mediator of Inflammation that Interacts with NF-κB to Regulate the Inducible Nitric-oxide Synthase Gene, J. Biol. Chem., 2001, 276, 3302–3309 CrossRef CAS PubMed.
- M. B. Goldring, M. Otero, D. A. Plumb, C. Dragomir, M. Favero and K. El Hachem, et al., Roles of inflammatory and anabolic cytokines in cartilage metabolism: Signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis, Eur. Cells Mater., 2011, 21, 202–220 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fo03793b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |