 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilization of lithium metal in concentrated electrolytes: effects of electrode potential and solid electrolyte interphase formation†
Anusha
Pradhan
a,
Shoma
Nishimura
a,
Yasuyuki
Kondo
a,
Tomoaki
Kaneko
 b,
Yu
Katayama
a,
Keitaro
Sodeyama
c and
Yuki
Yamada
*a
b,
Yu
Katayama
a,
Keitaro
Sodeyama
c and
Yuki
Yamada
*a
aSANKEN, Osaka University, 8-1, Mihogaoka, Ibaraki, Osaka 567-0047, Japan. E-mail: yamada@sanken.osaka-u.ac.jp
bDepartment of Computational Science and Technology, Research Organization for Information Science and Technology (RIST), 1-18-16, Hamamatsucho, Minato-ku, Tokyo 105-0013, Japan
cCenter for Basic Research on Materials, National Institute for Materials Science (NIMS), 1-1, Namiki, Tsukuba, Ibaraki 305-0044, Japan
First published on 16th April 2024
Abstract
Lithium (Li) metal negative electrodes have attracted wide attention for high-energy-density batteries. However, their low coulombic efficiency (CE) due to parasitic electrolyte reduction has been an alarming concern. Concentrated electrolytes are one of the promising concepts that can stabilize the Li metal/electrolyte interface, thus increasing the CE; however, its mechanism has remained controversial. In this work, we used a combination of LiN(SO2F)2 (LiFSI) and weakly solvating 1,2-diethoxyethane (DEE) as a model electrolyte to study how its liquid structure changes upon increasing salt concentration and how it is linked to the Li plating/stripping CE. Based on previous works, we focused on the Li electrode potential (ELi with reference to the redox potential of ferrocene) and solid-electrolyte-interphase (SEI) formation. Although ELi shows a different trend with DEE compared to conventional 1,2-dimethoxyethane (DME), which is accounted for by different ion-pair states of Li+ and FSI−, the ELi-CE plots overlap for both electrolytes, suggesting that ELi is one of the dominant factors of the CE. On the other hand, the extensive ion pairing results in the upward shift of the FSI− reduction potential, as demonstrated both experimentally and theoretically, which promotes the FSI−-derived inorganic SEI. Both ELi and SEI contribute to increasing the Li plating/stripping CE.
Introduction
Lithium (Li) metal negative electrodes have been broadly employed in advanced rechargeable batteries. The basic reason for this is that Li (an alkali metal) bears the lowest atomic number among all metal elements, and hence a high theoretical capacity of 3860 mA h g−1 can be achieved with its plating and stripping reactions. At the same time, a high battery voltage can be attained due to its low electrode potential of −3.0 V vs. the standard hydrogen electrode (SHE). However, Li metal shows a lower plating/stripping coulombic efficiency (CE), which has hampered its practical applications.1,2 The poor CE is due to the strong reducing ability of Li metal. In general, the electrode potential of Li is set far outside the potential window of organic electrolytes, which accelerates the reductive decomposition of the electrolytes. Practically, the reduction products accumulate on the negative electrode to form an interphase known as the solid electrolyte interphase (SEI).3 This SEI is Li+-conducting but electron-insulating and thus kinetically retards further electrolyte decompositions by blocking direct contact between the electrode and electrolyte.4,5 Hence, the nature of the SEI is an important factor that dominates the CE of Li metal electrodes.To minimize the reductive decomposition of the electrolytes, various electrolyte concepts have been proposed. The state-of-the-art concepts are (a) concentrated electrolytes,6–11 (b) localized concentrated electrolytes,12,13 (c) electrolyte additives,14,15 (d) weakly solvating electrolytes,16–18 and (e) liquefied gas electrolytes,19,20etc. Among them, concentrated electrolytes are one of the most fundamental concepts that have suggested the importance of liquid structures, from which various electrolyte design concepts have been developed. We reported in 2014 that concentrated electrolytes with extensive ion pairing promote the preferential reduction of salt anions, thus leading to an anion-derived SEI, which may contribute to stabilization of the Li metal/electrolyte interface.9,21 This mechanism is widely accepted and applied to various electrolyte systems, including aqueous electrolytes.22,23 On the other hand, we discovered in 2022 that the extensive ion pairing induced by, for example, increasing salt concentration can remarkably upshift the Li electrode potential, ELi, which decreases the ELi–potential-window gap, thus suppressing electrolyte reduction and leading to a higher CE of Li plating/stripping.24 As a result, there are several factors to be discussed for the stabilization mechanism of Li metal electrodes in concentrated electrolytes, namely (i) liquid structure, (ii) ELi, and (iii) SEI formation.
Here, we have chosen a combination of LiN(SO2F)2 (LiFSI) and 1,2-diethoxyethane (DEE) as a model electrolyte to discuss how each factor contributes to increasing the CE of Li plating/stripping. Compared to conventional 1,2-dimethoxyethane (DME), DEE is known as a weakly Li+-solvating solvent due to the steric hindrance effect of the bulkier ethyl groups.17,25 The LiFSI/DEE system exhibits a high CE of Li plating/stripping even at a low 1 mol dm−3 (M) concentration and a further increased CE at a higher salt concentration.25 Here we compared LiFSI/DEE with LiFSI/DME to highlight the effects of Li+-solvation ability as well as salt concentration on (i) liquid structure, (ii) ELi, and (iii) SEI formation. Based on our previous work that highlights ELi,24 we first evaluated the ELi in LiFSI/DEE with reference to the ferrocene redox potential (Fc/Fc+) and discussed its shift based on the liquid structures. Next, we evaluated the CE of Li plating/stripping in LiFSI/DEE compared to LiFSI/DME and discussed its relationship with ELi. In addition, with an eye to the SEI formation, we also investigated the reduction potential of the electrolyte with reference to Fc/Fc+ and discussed its variation in salt concentrations based on density functional theory-based molecular dynamics (DFT-MD) simulations.
Results and discussion
Li electrode potential (ELi)
E Li was evaluated with reference to the Fc/Fc+ redox potential on a Pt electrode as an internal standard of electrode potentials recommended by IUPAC.26,27 For this, a three-electrode cell with Pt as the working electrode and Li metal as the counter and reference electrodes was used (Fig. 1a). Fc (1 mM) was introduced to the LiFSI/DEE electrolyte. Fig. 1b shows cyclic voltammetry (CV) profiles of the Fc/Fc+ redox reaction at various LiFSI salt concentrations (units of mol kg−1are hereafter denoted as m). The CV profiles are close to that of a fully reversible system (a peak separation of 59 mV at 25 °C) except for the lowest concentration of 0.12 m that shows high electrolyte resistance. The CV profiles show that the Fc/Fc+ redox potential with reference to Li/Li+ was shifted to lower potentials at higher salt concentrations. If it is supposed that the electrode potential of Fc/Fc+ is constant and independent of the electrolyte used, then the different CV redox potentials result from the Li reference electrode, suggesting that ELi (with reference to Fc/Fc+) changes depending on the electrolyte used. We extracted the Fc/Fc+ redox potential (with reference to Li/Li+) from the half-wave potential at the centre point between the oxidation and reduction peaks in the CV and then converted it to ELi (with reference to Fc/Fc+) by just adding a negative sign (e.g., a Fc/Fc+ potential of 3 V vs. Li/Li+ corresponds to ELi = −3 V vs. Fc/Fc+). For accurate assessment of ELi, we prepared two or three cells for each electrolyte and obtained average ELi values (Fig. S1 and Table S1†). Fig. 1c and Table S2† summarize the relationship between ELi and salt concentration. As a comparison, the data for LiFSI/DME reproduced from our previous publication are also shown.24 For both DEE and DME systems, ELi is upshifted concomitantly with increasing salt concentration, suggesting that the reducing ability of the Li metal is weakened at high salt concentration.24,28 For LiFSI/DEE, ELi was the highest (−2.95 V vs. Fc/Fc+) for the highest salt concentration (10 m, corresponding to approximately 5 M), and it was the lowest (−3.42 V vs. Fc/Fc+) for the lowest salt concentration (0.12 m, corresponding to 0.1 M).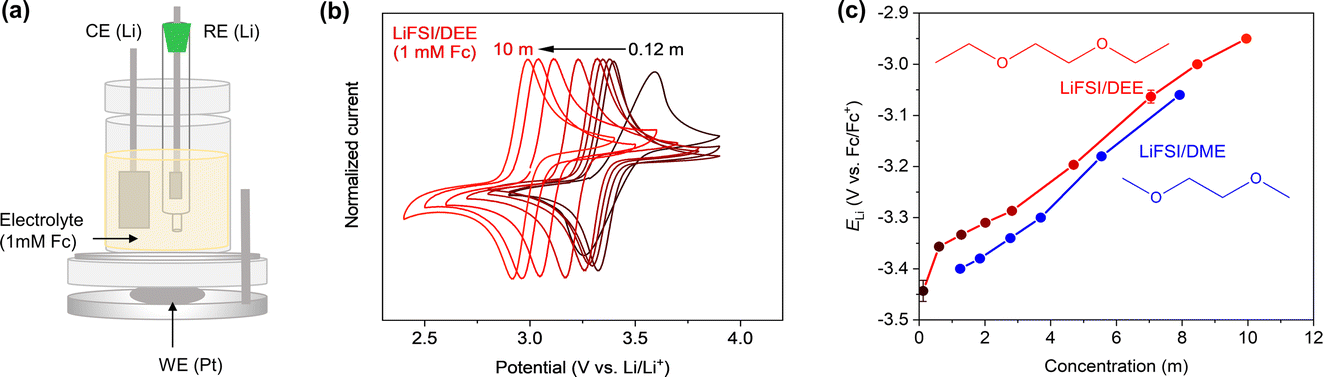 | ||
| Fig. 1 Evaluation of ELi with reference to the Fc/Fc+ redox potential. (a) Schematic of a three-electrode cell with Pt as a working electrode (WE) and Li metal as counter/reference electrodes (CE/RE). (b) Cyclic voltammograms (scan rate: 5 mV s−1) of the Fc/Fc+ redox reaction in LiFSI/DEE at various LiFSI concentrations (mol kg−1 = m) of 0.12, 0.60, 1.3, 2.0, 2.8, 4.7, 7.1, 8.5, and 10 m. 0.12 m and 10 m correspond to 0.1 M and 5 M, respectively. (c) ELi at various LiFSI concentrations evaluated from the cyclic voltammograms. Average ELi values obtained with two or three cells for each concentration are plotted with error bars (standard deviations). The data for LiFSI/DME are reproduced from ref. 24 as a comparison. | ||
A comparison of DEE and DME systems enables us to discuss the results from the viewpoint of the Li+-solvation abilities. It was reported that DEE has a weaker Li+-solvation ability than DME due to the steric hindrance effect of its bulkier ethyl groups.17,25 In the low concentration region below 4 m, LiFSI/DEE showed meaningfully higher ELi values than LiFSI/DME, suggesting that a weak solvation environment of Li+ leads to a high ELi. Similar correlations are also reported for dimethoxymethane (DMM), DME, and diglyme (G2), whose solvation abilities are in the order of DMM < DME < G2 and ELi values are in the order of DMM > DME > G2.24 To verify the effect of Li+-solvation abilities, we further measured ELi in 1.0 M LiFSI/DEE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene, in which toluene with almost no Li+-solvation ability was introduced at different molar ratios (DEE:toluene = 10:0, 7:3, and 4:6). As shown in Fig. S2,† the three variations of 1.0 M LiFSI/DEE:toluene showed different ELi values, even at the fixed 1.0 M concentration. The highest ELi (−3.26 V vs. Fc/Fc+) was achieved by introducing the largest amount of toluene, suggesting that the weakly solvating environment of Li+ leads to a higher ELi.
:toluene, in which toluene with almost no Li+-solvation ability was introduced at different molar ratios (DEE:toluene = 10:0, 7:3, and 4:6). As shown in Fig. S2,† the three variations of 1.0 M LiFSI/DEE:toluene showed different ELi values, even at the fixed 1.0 M concentration. The highest ELi (−3.26 V vs. Fc/Fc+) was achieved by introducing the largest amount of toluene, suggesting that the weakly solvating environment of Li+ leads to a higher ELi.
On the other hand, increasing the salt concentration above 4 m, both LiFSI/DEE and LiFSI/DME showed similar ELi values at similar molalities. Hence, ELi in the concentrated region is not related to the Li+-solvation abilities of the solvent molecules. In such a concentrated region, free solvent molecules that can solvate Li+ are remarkably decreased in number, which induces ion pairing of Li+ and FSI−. Hence, the commonly high ELi values are due to similar ion-pair states at high concentrations, as discussed later.
In essence, the upward shift of ELi is achieved by (i) increasing salt concentrations or (ii) employing weakly Li+-solvating solvents. The resultant high ELi can decrease the ELi–potential-window gap, which can weaken the driving force of electrolyte reduction on Li metal. Therefore, concentrated electrolytes and weakly solvating electrolytes are inherently less susceptible to its reduction in conjunction with Li metal. The influence of ELi on the CE of Li plating/stripping will be discussed in the following sections.
Liquid structure
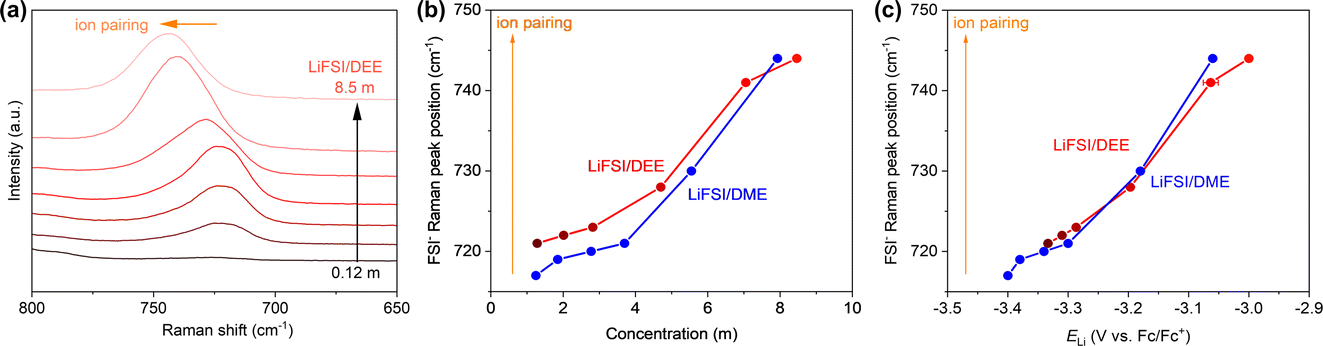
Next, we discuss how ELi is related to the liquid structure of electrolytes. A theoretical consideration shows that ELi is linearly correlated with the Li+ chemical potential in the electrolyte (μLi+),where F is the Faraday constant.24 It should also be noted that the Li+ chemical potential in the SEI is cancelled out during the derivation of the equation; hence, the SEI chemistry does not theoretically affect ELi.24 Basically, μLi+ indicates to what extent the Li+ is stable in its environment. Hence, μLi+ should be closely related to its coordination environment in the electrolyte. To this end, we studied the liquid structure of LiFSI/DEE.
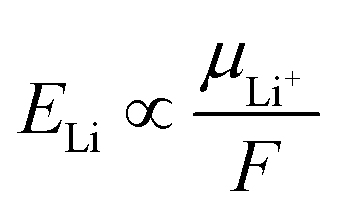
The dissolution of LiFSI in DEE (an aprotic solvent) is illustrated as competitive coordination of DEE and FSI− (both being Lewis bases) towards the Li+ (a Lewis acid). There are several random and driven interactions of Li+–DEE and Li+–FSI− taking place in the solution structure, which generates various LiFSI–DEE solvates, such as (a) solvent-separated ion pairs (SSIPs), (b) contact ion pairs (CIPs, FSI− coordinating to a single Li+ ion), and (c) aggregates (AGGs, FSI− coordinating to two or more Li+ ions) depending on the salt concentrations.29 Broadly, an increase in the salt concentration results in (i) a reduction in free solvent molecules and (ii) a surge in the ionic association (CIPs and then to AGGs).29 Focusing on the coordination environment of Li+, it is mainly coordinated by solvent molecules at low concentrations but is forced to be paired with FSI− at high concentrations. To identify such Li+ coordination environments, we study the ion-pair states in LiFSI/DEE at different concentrations using Raman spectroscopy (Fig. 2a). We focused on the S–N–S vibration peak of FSI− in the range of 650–800 cm−1, and the wavenumber of this peak is sensitive to the ion-pair states of Li+ and FSI−.29 A flat profile was observed for low concentrations (0.12 m and 0.60 m) because of the insufficient amount of FSI− for detection. Over 1.3 m (corresponding to 1.0 M), the S–N–S vibration peak of FSI− was observed, and it was shifted to a higher wavenumber with increasing salt concentration. This higher wavenumber shift has been attributed to more extensive ion pairing of Li+ and FSI− from SSIPs to CIPs and AGGs.29 At low concentrations, SSIPs and CIPs are dominant; Li+ is solvated by DEE molecules or partially coordinated by an FSI− anion. Considering the low ELi (i.e., low μLi+) at low concentrations, Li+ is highly stable in such solvent-dominant coordination environments. On the other hand, with an increase in the LiFSI concentration, CIPs and AGGs become dominant; Li+ is coordinated by more FSI− anions in the –Li+–FSI−–Li+–FSI−–Li+– aggregate with partial DEE solvation. This situation results in high ELi (i.e., high μLi+), thus Li+ being highly unstable in such anion-dominant coordination environments.
 | ||
| Fig. 2 Spectroscopic analysis on the liquid structures of LiFSI/DEE. (a) Raman spectra of LiFSI/DEE at various LiFSI concentrations of 0.12, 0.60, 1.3, 2.0, 2.8, 4.7, 7.1, 8.5, 10 m. The Raman peak in the range of 650–800 cm−1 is derived from the S–N–S vibration of FSI−. The wavenumber resolution was 1 cm−1. (b) Raman peak positions of FSI− in LiFSI/DEE and LiFSI/DME at various LiFSI concentrations. The Raman peak position is an indicator of how extensively FSI− is paired with Li+. The data for LiFSI/DME are reproduced from ref. 24 as a comparison. (c) Raman peak positions of FSI− plotted versus ELi. | ||
To theoretically support the spectroscopic analysis, DFT-MD was applied to the dilute (LiFSI:DEE = 1:10 by mol, corresponding to 0.85 m) and concentrated (LiFSI:DEE = 1:2 by mol, corresponding to 4.2 m) electrolytes. Fig. 3 shows the supercells and representative coordination environments of Li+ in dilute and concentrated electrolytes. In the dilute electrolyte (Fig. 3b), Li+ is mainly solvated by DEE molecules, though an FSI− anion is partially coordinated to Li+, suggesting that the coordination states are SSIPs and CIPs. On the other hand, in the concentrated electrolyte (Fig. 3e), Li+ is more coordinated by multiple FSI− anions, suggesting the presence of AGGs. To quantitatively discuss the local coordination environment of Li+, we analyzed the pair distribution functions, g(r), in the DFT-MD (Fig. 3c and f). The integrals of the pair distribution function, shown as N(r), give the coordination number of each atom to Li+. We found that, in the dilute electrolyte, Li+ is coordinated by four O atoms of DEE and less than one O atom of FSI− on average (Fig. 3c). In contrast, the average Li+ environment in the concentrated electrolyte is that of Li+ coordinated by three O atoms of DEE and one O atom of FSI− (Fig. 3f). All these results are consistent with the Raman spectroscopic analysis (Fig. 2a).
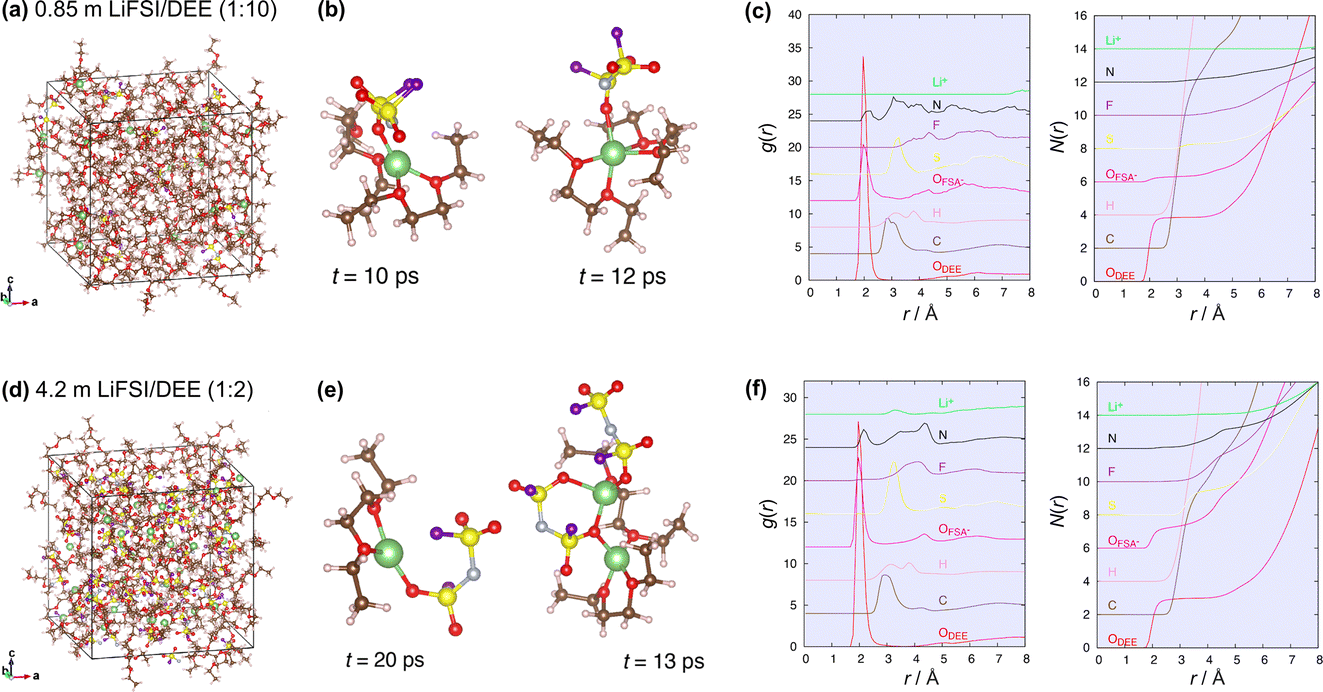 | ||
| Fig. 3 DFT-MD simulations on the liquid structures of (a–c) dilute (0.85 m, LiFSI:DEE = 1:10 by mol) and (d–f) concentrated (4.2 m, LiFSI:DEE = 1:2 by mol) electrolytes. (a and d) Supercells used. (b and e) Representative local coordination states of Li+. (c and f) Pair distribution functions (g(r)) from Li+ and integrated coordination numbers (N(r)) to Li+. Atom colors: Li, green; C, brown; H, light pink; O, red; N, blue; S, yellow; F, purple. Li atoms are magnified in size. | ||
Next, we compared the ion-pair states in the LiFSI/DEE and LiFSI/DME electrolytes to discuss the effects of the Li+-solvation abilities. Fig. 2b and Table S2† show the Raman peak positions of the S–N–S vibration of FSI−, which is an indicator of how extensively Li+ is paired with FSI− to form SSIPs, CIPs, or AGGs.24,29 Both electrolytes showed higher wavenumber shifts of the FSI− vibration with increasing salt concentrations, suggesting progressive formation of ion pairs. However, when compared at similar concentrations, LiFSI/DEE showed more extensive ion pairing (i.e., a higher wavenumber above a resolution error of 1 cm−1) than LiFSI/DME, except at a high concentration region of around 8 m. This indicates that the ion pairing is promoted in a weakly Li+-solvating solvent, which may be a reason for the higher ELi values for LiFSI/DEE than LiFSI/DME at similar concentrations. To demonstrate this, we made a plot of ELivs. the Raman peak position of FSI− for LiFSI/DEE and LiFSI/DME (Fig. 2c).24 The plots of LiFSI/DEE and LiFSI/DME are overlapping, and both represent a linear correlation. This suggests that the ion-pair state of Li+, which can be controlled by modifying the salt concentrations or Li+-solvation abilities, subdues μLi+, and hence ELi, in the various electrolyte systems.
Li plating/stripping reaction
Having found the link between ELi and liquid structure, Li plating/stripping in LiFSI/DEE was studied in a Cu/Li half-cell configuration. The charge–discharge voltage profiles are presented in Fig. S3.† To accurately evaluate the CE of Li plating/stripping at various salt concentrations, we prepared three cells for each concentration. Based on the charge–discharge profiles, we evaluated the average CE of Li plating/stripping (Fig. 4a, Tables S2 and S3†). Here we extracted the CE values of the 2nd to 20th cycles for the average CE. The CE in the 1st cycle was excluded from the average CE because it is mostly affected by the irreversible capacity for SEI formation, which is not suitable for discussing the stability of Li metal after SEI formation. In both LiFSI/DEE and LiFSI/DME, the average CE increased concomitantly with the increase in salt concentration (Fig. 4a). Such trends have been widely reported in various electrolytes.24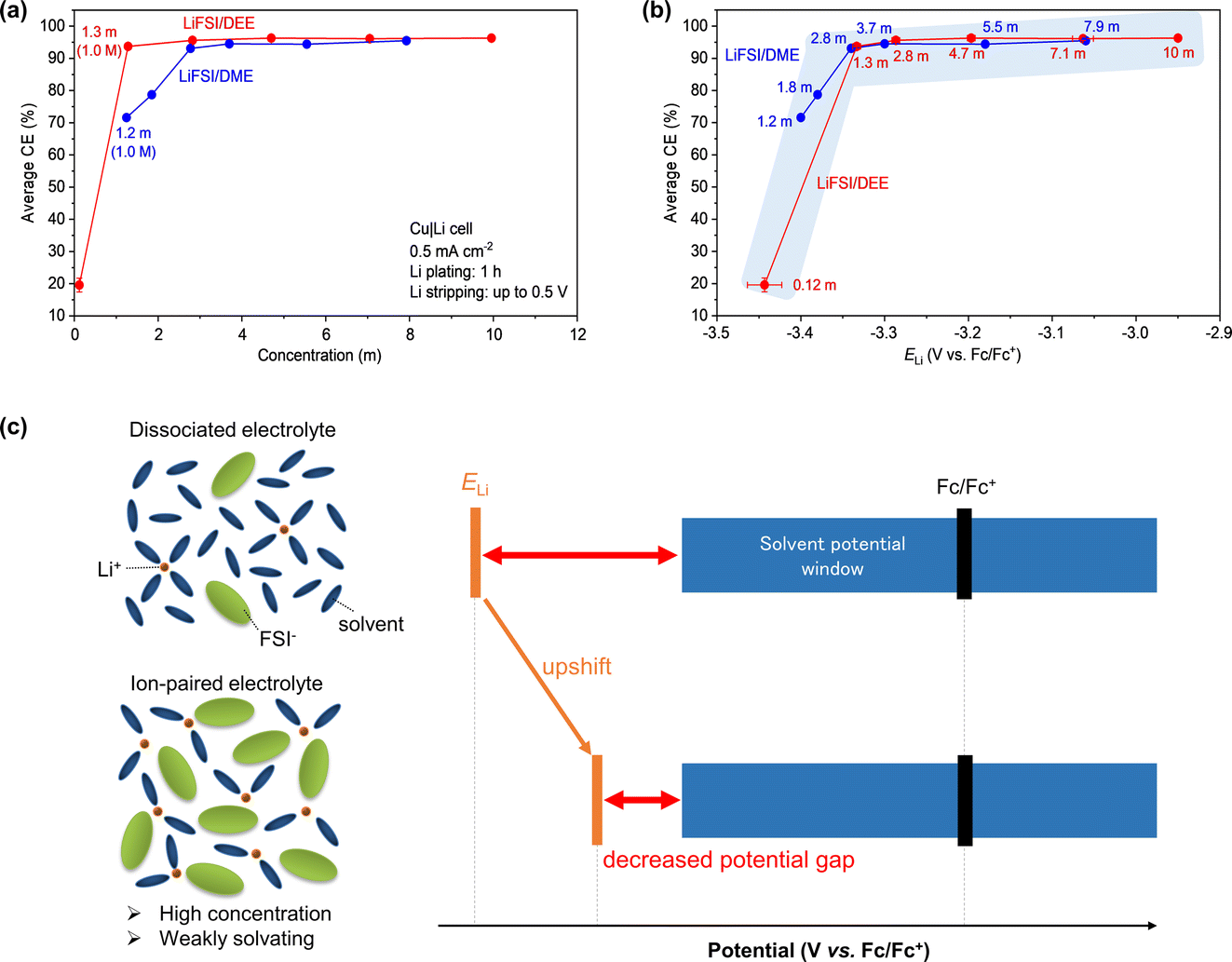 | ||
| Fig. 4 Average CE of Li plating/stripping in LiFSI/DEE and LiFSI/DME plotted versus (a) LiFSI concentrations and (b) ELi. The average CE was evaluated from the 2nd to 20th cycles in three Cu|Li cells for each LiFSI/DEE electrolyte. The error bars are standard deviations. The current density was 0.5 mA cm−2. Li plating on Cu was performed for 1 h, followed by Li stripping up to 0.5 V. The data for LiFSI/DME, evaluated under a similar condition, are reproduced from ref. 24 as a comparison. (c) Potential diagrams and liquid structures of dilute (dissociated) and concentrated (ion-paired) electrolytes. The upward shift of ELi can decrease the ELi–potential-window gap. | ||
Comparing LiFSI/DEE and LiFSI/DME, we found a significant difference in CE in the low-concentration region of <2 m. As shown in Fig. 4a, LiFSI/DEE showed a high CE of 93.8% at 1.3 m (corresponding to 1.0 M), while LiFSI/DME showed a remarkably lower CE of 71.6% at a similar concentration of 1.2 m (corresponding to 1.0 M). Such a difference was also reported previously, highlighting the usefulness of weakly Li+-solvating solvents, but its mechanism was not clear.25 Inspired by our previous paper,24 here we focused on ELi as an influential factor and replotted the average CE versus ELi in both electrolyte systems (Fig. 4b). We found that the plots of LiFSI/DEE and LiFSI/DME are overlapping with each other; different electrolytes with similar ELi values result in similar average CE values. It should also be noted that, even for weakly Li+-solvating DEE, further dilution from 1.3 m (1.0 M) to 0.12 m (0.1 M) LiFSI/DEE resulted in a much lowered CE of only 19.6%. This can also be accounted for by the ELi-CE correlation because ELi is significantly lower at 0.12 m (−3.44 V vs. Fc/Fc+) than at 1.3 m (−3.33 V vs. Fc/Fc+). All the results suggest that ELi is one of the dominant factors of the CE for Li plating/stripping.
Next, we discuss how ELi affects the Li metal CE or the stability of the Li metal/electrolyte interface (Fig. 4c). ELi is usually far below the cathodic limit of the potential window of organic electrolytes. However, when ELi is upshifted by forming extensive Li+–FSI− pairs in the electrolyte, the gap between ELi and the potential window can be decreased.24 Since the ELi–potential-window gap corresponds to the driving force for the reductive decomposition of the electrolyte (or the reducing ability of Li), the decreased gap can prevent the unnecessary reductive decomposition of the electrolyte, thus leading to a higher CE of Li plating/stripping. This way, the Li loss is minimized in an electrolyte with high ELi, which brilliantly paves the path for longer cycling life of Li metal batteries.
It is also worth noting that there is a change in trend in the ELi-CE correlation. Below ELi = −3.33 V vs. Fc/Fc+, the CE is drastically increased with increasing ELi, whereas above ELi = −3.33 V vs. Fc/Fc+, the CE is only gradually increased. As a result, there should be at least two mechanisms that describe the ELi-CE correlation. At present, however, it is an open question what the two mechanisms are and why there is a trend change at the ELi value of −3.33 V vs. Fc/Fc+.
Electronic structure and SEI formation
Having established ELi as an influential factor, we next discuss other factors, namely (i) Li deposition morphology and (ii) SEI formation. The two factors have been widely studied for various dilute and concentrated electrolytes, including the specific cases of LiFSI/DEE and LiFSI/DME (both 1 M and 4 M).25 For (i), Li is deposited in a rounded shape in both LiFSI/DEE and LiFSI/DME, and there is no remarkable difference between low (1 M) and high (4 M) salt concentrations.25 Hence, (i) Li deposition morphology may not be a major factor that accounts for the high CE in weakly solvating solvents as well as at high concentrations. As for (ii), a widely accepted notion is that, in concentrated electrolytes, the Li-salt anion is preferentially reduced over the solvent to generate an inorganic-rich SEI, which can stabilize the Li metal/electrolyte interface to prevent unfavorable electrolyte decomposition.9 We proposed this mechanism in 2014 based on the unique electronic structure at high concentrations, in which the lowest unoccupied molecular orbital (LUMO) energy level of the Li-salt anion is shifted downward to be more susceptible to reduction.9,21 Hence, when discussing the concentration effect, we need to consider the electronic structure and resulting SEI chemistry as well.For LiFSI/DEE, the SEI on Li has been reported in detail at low (1 M) and high (4 M) concentrations.25 A unique feature of this specific electrolyte is that, even at the low (1 M) concentration, the SEI is derived primarily from LiFSI, thus being rich in inorganic species with Li, F, O, and S elements.25 As a result, there is no remarkable difference in SEI chemistries in 1 M and 4 M LiFSI/DEE. Hence, the observed gradual increase in the CE (93.8% to 96.3%, Fig. 4a) from 1.3 m (1.0 M) to 10 m (over 4 M) LiFSI/DEE cannot be accounted for by the SEI chemistries. The increased CE may result from the upshift of ELi that can decrease the ELi–potential-window gap.
To understand this similar SEI formation process in dilute and concentrated LiFSI/DEE, DFT-MD was applied to the dilute (LiFSI:DEE = 1:10 by mol, corresponding to 0.85 m) and concentrated (LiFSI:DEE = 1:2 by mol, corresponding to 4.2 m) electrolytes. Fig. 5a and b shows the projected density of states (PDOS) of the equilibrium trajectories. The curves in blue and red denote the density of electronic states of LiFSI and DEE, respectively. To discuss the electrolyte reduction, we focus on the lowest edge of the conduction bands (i.e., unoccupied orbitals), which corresponds to the LUMO and directs the nature of the reduction reactions. Notably, the LUMO structures of the PDOS profiles are not remarkably different in dilute and concentrated LiFSI/DEE; in both electrolytes, the LUMO is mainly composed of FSI−. This means that under a reducing atmosphere (e.g., on Li metal), FSI− receives an electron and thus is reduced to form an FSI−-derived SEI. The similar LUMO structures at low and high concentrations result from two factors. First, there is only a small difference in the ion-pair states of FSI− (SSIPs and CIPs in both cases) owing to the weakly solvating nature of DEE (Fig. 3). Second, the inherently high unoccupied orbital level of ethers (i.e., high reduction stability) makes the unoccupied orbital of FSI− the lowest energy level at any concentration. This accounts for the similar SEI chemistries observed for dilute and concentrated LiFSI/DEE. This theoretical study indicates that weakly solvating solvents are useful in promoting the preferential reduction of FSI− for inorganic-rich SEI formation as well as in increasing ELi, both of which are beneficial to stabilizing the Li metal/electrolyte interface.
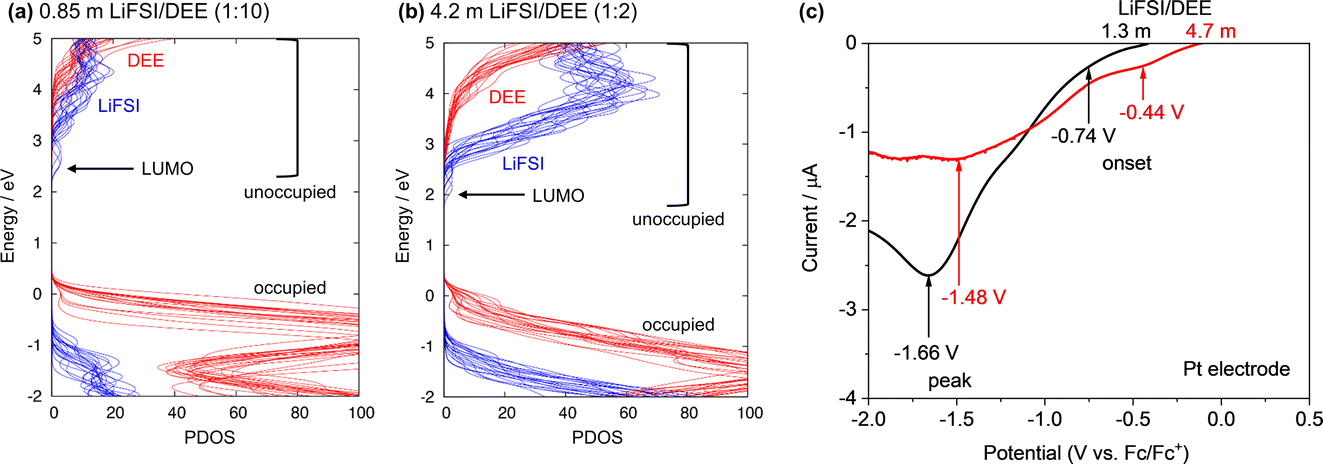 | ||
| Fig. 5 Theoretical and experimental investigations on the FSI− reduction potential. PDOS profiles of (a) dilute (0.85 m, LiFSI:DEE = 1:10 by mol) and (b) concentrated (4.2 m, LiFSI:DEE = 1:2 by mol) electrolytes. (c) LSV curves of a Pt electrode in 1.3 m and 4.7 m LiFSI/DEE at 1 mV s−1. The onset and peak potentials of the reduction reactions are indicated. The onset potential was defined at the cathodic current flow of 0.25 μA. | ||
Another notable feature of the PDOS profiles is that the lowest edge of the unoccupied orbitals of FSI− is shifted downward at high concentrations, which is also observed in other concentrated electrolytes.9,21 This downward shift is qualitatively explained by the coordination of Li+ (a strong Lewis acid) to FSI−, which results in partial electron donation from FSI− to Li+. This electronic feature indicates that the reduction potential of FSI− is upshifted at higher concentrations, thus promoting the reduction of FSI− to generate an inorganic SEI. To experimentally identify the reduction potential, linear sweep voltammetry (LSV) was performed for a Pt electrode in 1.3 m (1.0 M) and 4.7 m (3.0 M) LiFSI/DEE using a three-electrode cell with Li metal counter and reference electrodes. Fig. 5c shows LSV curves, in which the potential is shown with reference to Fc/Fc+. Here we focus on the reduction onset potential, which results from the reduction of FSI− to form an inorganic SEI. We found that the reduction onset potential of FSI− was −0.74 V vs. Fc/Fc+ in 1.3 m LiFSI/DEE but was upshifted to −0.44 V vs. Fc/Fc+ in 4.7 m LiFSI/DEE. This FSI− reduction potential (well over 2 V vs. Li/Li+) seems to be quite high but it was also observed in FSI-based ionic liquids with LiFSI salt.30,31 Otherwise, focusing on the reduction current peak, it was also shifted from −1.66 V to −1.48 V vs. Fc/Fc+. This upshift in the FSI− reduction potential is consistent with the PDOS profiles.
Discussion
An important attribute of concentrated electrolytes and weakly solvating electrolytes is the extensive ion pairing of Li+ and FSI−. This extensive ion pairing provides two features to the concentrated electrolytes: (i) an upshift of ELi, resulting from more unstable Li+ ions (i.e., high μLi+), and (ii) an upshift of FSI− reduction potential, resulting from partial electron donation from FSI− to Li+. Feature (i) weakens the reducing ability of Li metal, while feature (ii) promotes the formation of an FSI−-derived inorganic SEI, both of which contribute to stabilizing the Li metal/electrolyte interface and leading to higher Li plating/stripping CE. However, there is a requirement to assess each contribution in a quantitative manner.A thing to note is that the observed ELi-CE correlation may hold true in the presence of a similar good SEI. In this study as well as the previous paper, we used LiFSI in all electrolytes, in which LiFSI more or less contributes to the SEI formation in various solvents and at various concentrations, thus highly stabilizing the Li metal/electrolyte interface.24 On the other hand, when using LiPF6, although a similar ELi-CE correlation was observed, the CE values were much lower than those for LiFSI in the same solvents.24 This suggests that the SEI chemistry is undoubtedly important to increase the CE. However, even in the FSI−-derived good SEI, the interface is only kinetically stabilized outside the potential window. In this situation, the upward shift of ELi plays a vital role in decreasing the ELi–potential-window gap to weaken the driving force of electrolyte reduction.
To achieve 100% CE for Li plating/stripping, an ultimate goal is to enable a greater upward shift of ELi inside the potential window of the electrolyte. This situation causes no decomposition of Li salt or solvent, leading to SEI-free Li metal electrodes. In this regard, a further question arises to the two upshifts of ELi and the FSI− reduction potential; which upshift is larger? To answer this question, the overall potential diagram is presented in Fig. 6. Comparing 1.3 m (1.0 M) and 4.7 m (3.0 M) LiFSI/DEE, ELi is upshifted by +0.13 V, while the FSI− reduction potential is further upshifted by +0.30 V for the onset and by +0.18 V for the peak. This implies that we cannot shift ELi beyond the FSI− reduction potential; hence, the reduction of FSI− is inevitable on Li metal. This difficulty lies essentially in the fact that both (i) upshift of ELi and (ii) upshift of the FSI− reduction potential are currently achieved by the same strategy of forming extensive ion pairs. To overcome this situation, the upshift of ELi (i.e., higher μLi+) must be achieved independently of ion pairing. This requires a full reconsideration of the design concept of both anion and solvent but may be a promising step forward.
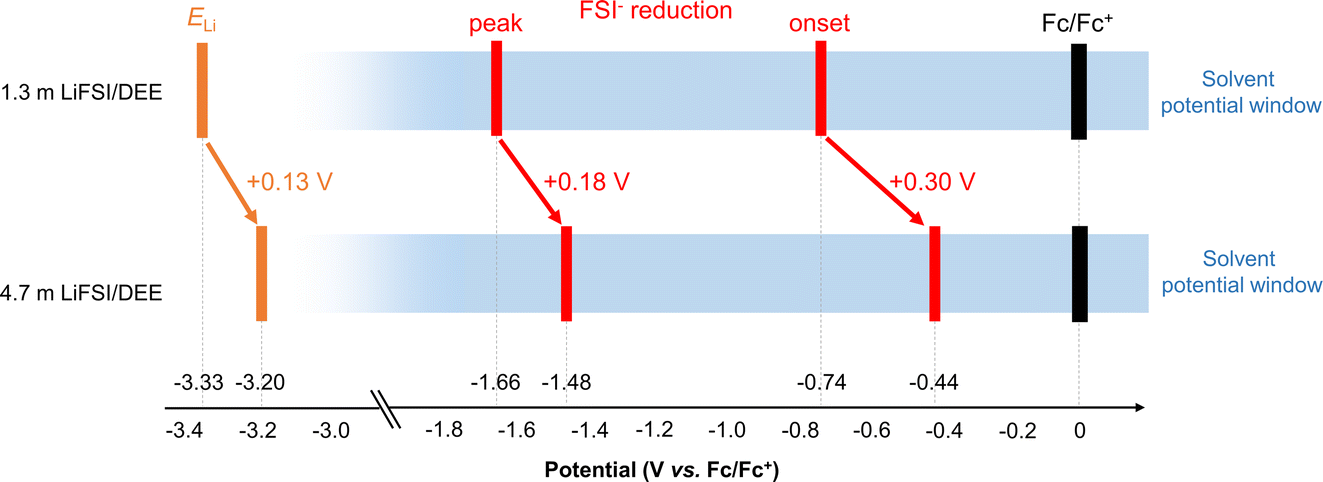 | ||
| Fig. 6 Potential diagram of dilute (1.3 m) and concentrated (4.7 m) LiFSI/DEE that highlights the upward shift of ELi and FSI− reduction potential. | ||
Conclusions
Using LiFSI/DEE as a model electrolyte, we studied the effects of salt concentration and solvation ability on the liquid structure, the shift of ELi, and the shift of FSI− reduction potential, and discussed their contributions to stabilizing the Li metal/electrolyte interface and increasing the Li plating/stripping CE. Generally, increasing the salt concentrations or employing weakly Li+-solvating solvents leads to more extensive ion pairing of Li+ and FSI− (i.e., from SSIPs to CIPs and AGGs). This structural feature stabilizes the Li metal/electrolyte interface and thus increases the Li plating/stripping CE in two ways. First, the extensive ion pairing decreases the LUMO level of FSI− owing to the strong Lewis acidity of Li+, which results in the upward shift of the FSI− reduction potential to promote inorganic SEI formation. Second, increasing the extent of ion pairing can shift ELi upward owing to the unstable Li+ (i.e., increased μLi+), which decreases the ELi–potential-window gap and weakens the driving force of electrolyte reduction. As a result, there is a clear correlation between ELi and CE in various solvents.There are still many open questions as listed below.
(1) To what extent do the two factors (ELi and SEI) contribute to stabilizing the Li metal/electrolyte interface?
(2) Why is the FSI−-derived SEI good? Is there any alternative for FSI−?
(3) How does the solvent reduction potential change at high concentrations?
(4) How is ELi or μLi+ theoretically described and linked to the liquid structure?
(5) Is it possible to independently shift ELi and the FSI− reduction potential without controlling the ion-pair state?
(6) Can ELi be moved into the potential window to achieve an SEI-free Li metal electrode?
Experimental
Materials
LiFSI was provided by Nippon Shokubai. Ferrocene (Fc) and DEE were purchased from Sigma Aldrich and Tokyo Chemical Industry, respectively. DEE was dried to a H2O content of 16 ppm (Karl Fischer titration) with activated molecular sieves. Super dehydrated toluene (H2O content of 6 ppm) was purchased from Fujifilm. All the electrolytes were prepared by adding LiFSI to the solvent in an argon-filled glove box. For the evaluation of the Li electrode potential (ELi), 1 mM Fc was added to the electrolyte.Electrochemical measurement
All the electrochemical measurements were performed in an argon atmosphere. For the ELi measurement, a three-electrode cell, with Pt as the working electrode and Li metal as the counter and reference electrodes, was used. The temperature of the cell was maintained at 25 °C for 2 hours using a thermostatic oven. The cell was then subjected to cyclic voltammetry (CV) with a Celltest 1470E system from Solartron. The redox potential of the Fc/Fc+ couple was measured with reference to the Li reference electrode, and ELi of the various electrolytes was evaluated given that the redox potential of Fc/Fc+ is constant.Electrochemical Li plating/stripping tests were performed using Cu|Li coin cells with various electrolytes (not containing Fc). The surface area of the Cu electrode was 1.13 cm2. The coin-cell parts were purchased from Hohsen. Cu foil and Li foil were purchased from Hohsen and Honjo Metal, respectively. A glass fiber (GC-50, Advantec) was used as the separator. The Li plating/stripping tests were conducted with a charge–discharge unit (TOSCAT-3100, Toyo System) at a constant current density of 0.5 mA cm−2 for 1 h during Li plating on Cu and up to a cut-off voltage of 0.5 V during Li stripping. The average CE (2nd–20th cycle) was calculated, excluding the first cycle because it is mostly affected by the SEI-formation process.
The FSI− reduction potential was estimated using linear sweep voltammetry (LSV) with a VMP3 system from Biologic, using a similar cell to that used for the ELi measurement. A Pt plate was used as the working electrode and Li metal foil was used as the counter and reference electrodes. Using the ELi value in each electrolyte, the observed potential vs. Li/Li+ was converted to that vs. Fc/Fc+ to show the LSV curves.
Material characterization
Raman spectroscopy was applied to understand the liquid structure of the electrolytes with a laser excitation wavelength of 532 nm and a resolution of 0.8 cm−1 employing an NRS-5100 spectrometer from JASCO. The instrument was calibrated with a standard Si peak with a wavelength value of 520 cm−1. The electrolytes were properly sealed in quartz cells in an argon-filled glove box and were excited using the 532 nm laser.Computational details
DFT-MD simulations were performed using the CP2K code.32 DZVP-MOLOPT-SR-GTH-type mixed Gaussian and plane-wave basis sets were used where the cutoff energy of the plane wave was chosen as 400 Ry. The PBE functional33 with a D3-type semi-empirical van der Waals correction34 and GTH norm-conserving pseudopotentials35 were employed. DFT-MD simulations were performed in the NVT ensemble with a time step of 1 fs using a Nosé–Hoover chain thermostat36–38 with a chain length of three. DEE and LiFSI molecules were randomly distributed in the cubic unit cell whose lattice constants were determined by the experimental densities of the electrolytes. The number of molecules and atoms are summarized in Table S4.† Firstly, we roughly optimized the atomic positions using the Hellman–Feynman's force. Secondly, the liquid structures were annealed by the DFT-MD simulations for 30 ps at 450 K. Using the annealed structures, we performed DFT-MD simulation for 30 ps at 300 K. The trajectories of the last 20 ps were used for the calculations of the pair distribution functions. Structures obtained at every 1 ps time step from 10 ps to 30 ps were chosen to calculate the projected density of states (PDOS).Author contributions
Y. Y. proposed and supervised the project. A. P., S. N., Y. Kondo, Y. Katayama, and Y. Y. designed the experiments. A. P. and S. N. conducted the experiments. T. K. and K. S. designed and conducted the theoretical calculations. All authors contributed to the discussion. A. P., Y. Kondo, and Y. Y. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JSPS KAKENHI Grant-in-Aid for Transformative Research Areas (B) (23B207, 23H03824, 23H03827) and JST-GteX (JPMJGX23S3).References
- J. G. Zhang, W. Xu, J. Xiao, X. Cao and J. Liu, Chem. Rev., 2020, 120, 13312–13348 CrossRef CAS PubMed.
- B. Acebedo, M. C. Morant-Miñana, E. Gonzalo, I. Ruiz de Larramendi, A. Villaverde, J. Rikarte and L. Fallarino, Adv. Energy Mater., 2023, 13, 2203744 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- S. K. Jeong, H. Y. Seo, D. H. Kim, H. K. Han, J. G. Kim, Y. B. Lee, Y. Iriyama, T. Abe and Z. Ogumi, Electrochem. Commun., 2008, 10, 635–638 CrossRef CAS.
- Y. Yamada, Y. Takazawa, K. Miyazaki and T. Abe, J. Phys. Chem. C, 2010, 114, 11680–11685 CrossRef CAS.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J. G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- Y. Zhang, Y. Wu, H. Li, J. Chen, D. Lei and C. Wang, Nat. Commun., 2022, 13, 1297 CrossRef CAS PubMed.
- X.-Q. Zhang, X.-B. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef.
- Y. X. Yao, X. Chen, C. Yan, X. Q. Zhang, W. L. Cai, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090–4097 CrossRef CAS PubMed.
- T. D. Pham, A. Bin Faheem, J. Kim, H. M. Oh and K. K. Lee, Small, 2022, 18, 2107492 CrossRef CAS PubMed.
- Z. Jiang, J. Mo, C. Li, H. Li, Q. Zhang, Z. Zeng, J. Xie and Y. Li, Energy Environ. Mater., 2023, 6, e12440 CrossRef CAS.
- C. S. Rustomji, Y. Yang, T. K. Kim, J. Mac, Y. J. Kim, E. Caldwell, H. Chung and Y. S. Meng, Science, 2017, 356, eaal4263 CrossRef PubMed.
- Y. Yang, Y. Yin, D. M. Davies, M. Zhang, M. Mayer, Y. Zhang, E. S. Sablina, S. Wang, J. Z. Lee, O. Borodin, C. S. Rustomji and Y. S. Meng, Energy Environ. Sci., 2020, 13, 2209–2219 RSC.
- K. Sodeyama, Y. Yamada, K. Aikawa, A. Yamada and Y. Tateyama, J. Phys. Chem. C, 2014, 118, 14091–14097 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- S. Ko, T. Obukata, T. Shimada, N. Takenaka, M. Nakayama, A. Yamada and Y. Yamada, Nat. Energy, 2022, 7, 1217–1224 CrossRef CAS.
- Y. Chen, Z. Yu, P. Rudnicki, H. Gong, Z. Huang, S. C. Kim, J. C. Lai, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2021, 143, 18703–18713 CrossRef CAS PubMed.
- R. R. Gagne, C. A. Koval and G. C. Lisensky, Inorg. Chem., 1980, 19, 2854–2855 CrossRef CAS.
- G. Gritzner and J. Kůta, Electrochim. Acta, 1984, 29, 869–873 CrossRef.
- S. C. Kim, X. Kong, R. A. Vilá, W. Huang, Y. Chen, D. T. Boyle, Z. Yu, H. Wang, Z. Bao, J. Qin and Y. Cui, J. Am. Chem. Soc., 2021, 143, 10301–10308 CrossRef CAS PubMed.
- S.-D. Han, O. Borodin, D. M. Seo, Z.-B. Zhou and W. A. Henderson, J. Electrochem. Soc., 2014, 161, A2042–A2053 CrossRef CAS.
- S. Kato, N. Serizawa and Y. Katayama, J. Electrochem. Soc., 2022, 169, 076509 CrossRef CAS.
- S. Kato, N. Serizawa and Y. Katayama, J. Electrochem. Soc., 2023, 170, 056504 CrossRef CAS.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00038b |
| This journal is © The Royal Society of Chemistry 2024 |