 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of surface cations of cleaved mica with water in vapor and liquid forms†
Giada
Franceschi
 *a,
Sebastian
Brandstetter
a,
Jan
Balajka
a,
Igor
Sokolović
a,
Jiří
Pavelec
a,
Martin
Setvín
b,
Michael
Schmid
a and
Ulrike
Diebold
*a
*a,
Sebastian
Brandstetter
a,
Jan
Balajka
a,
Igor
Sokolović
a,
Jiří
Pavelec
a,
Martin
Setvín
b,
Michael
Schmid
a and
Ulrike
Diebold
*a
aInstitute of Applied Physics, TU Wien, Wiedner Hauptstraße 8-10/E134, 1040 Wien, Austria. E-mail: franceschi@iap.tuwien.ac.at; diebold@iap.tuwien.ac.at
bDepartment of Surface and Plasma Science, Charles University in Prague, V Holesovickach 2, 180 00 Praha, Czech Republic
First published on 9th June 2023
Abstract
Natural minerals contain ions that become hydrated when they come into contact with water in vapor and liquid forms. Muscovite mica – a common phyllosilicate with perfect cleavage planes – is an ideal system to investigate the details of ion hydration. The cleaved mica surface is decorated by an array of K+ ions that can be easily exchanged with other ions or protons when immersed in an aqueous solution. Despite the vast interest in the atomic-scale hydration processes of these K+ ions, experimental data under controlled conditions have remained elusive. Here, atomically resolved non-contact atomic force microscopy (nc-AFM) is combined with X-ray photoelectron spectroscopy (XPS) to investigate the cation hydration upon dosing water vapor at 100 K in ultra-high vacuum (UHV). The cleaved surface is further exposed to ultra-clean liquid water at room temperature, which promotes ion mobility and partial ion-to-proton substitution. The results offer the first direct experimental views of the interaction of water with muscovite mica under UHV. The findings are in line with previous theoretical predictions.
Introduction
The interaction of mineral surfaces with water rules a myriad of important natural phenomena, including dissolution and weathering,1,2 which forms soils and removes CO2 from the atmosphere;3 adsorption of toxic ions in groundwater, relevant for environmental remediation;4 and ice nucleation on mineral dust,5–7 which regulates cloud formation and weather patterns. The hydration of the mineral surface ions underpins all these processes. Nowadays, there is great interest in understanding the atomic-scale details of ion hydration at mineral surfaces. Still, only a handful of experimental studies have shared direct views of molecular-level ion hydration structures under pristine (ultra-high vacuum, UHV) conditions.8Muscovite mica (“mica”, in the following) is a common phyllosilicate with alternating K+ and aluminosilicate sheets (see Fig. 1b and c), well suited to model ion hydration at mineral surfaces. Mica cleaves easily and yields virtually step-free, atomically flat surfaces9 ideal for scanning probe microscopies. A recent non-contact atomic force microscopy (nc-AFM) study performed under UHV has shown that cleaving mica along the (001) plane leaves an array of short-range-ordered K+ ions at the surface (see Fig. 1a for a representative image).10 These ions can be easily exchanged with other ions or protons upon immersion in solution without modifying the underlying surface.11–15 Many water-centered studies have been performed as a function of these substitutions, including water adsorption,16–20 the structure of interfacial layers in thin liquid films and bulk liquids,14,20–27 the mobility13 and atomic-scale arrangement21,25,28,29 of hydrated ions, and heterogeneous ice nucleation.30–32
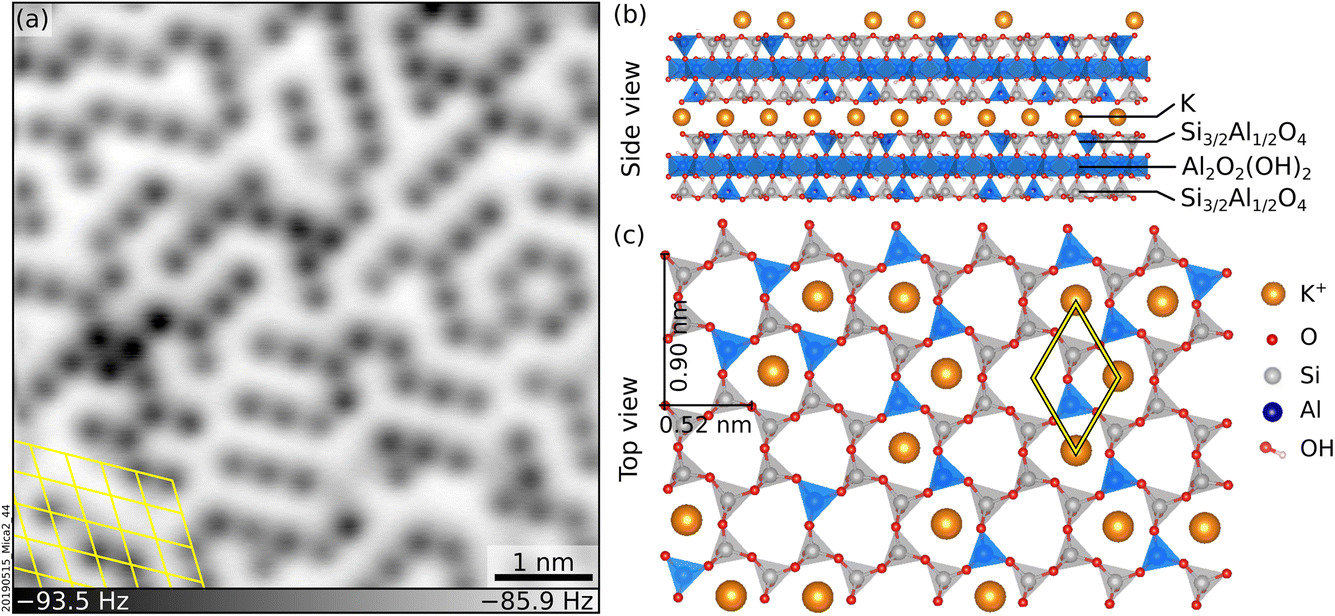 | ||
| Fig. 1 Freshly cleaved mica. (a) Constant-height, nc-AFM image of a UHV-cleaved mica surface acquired at 4.7 K with a qPlus sensor and a metal-terminated tip. 6 × 6 nm2, A = 150 pm, Vs = −9.3 V. K+ ions at the surface appear dark. (b and c) Side and top view of cleaved mica, respectively. (b) K+ layers alternate with aluminosilicate layers consisting of two sheets of Si and Al tetrahedra sandwiching one sheet of Al octahedra plus OH groups. Panel (c) shows the top view after cleaving. The tetrahedra are arranged in rings with quasi-hexagonal symmetry and, in the bulk, K+ ions sit at the center of each ring. After cleaving, roughly 50% of the K+ ions remain on each cleaved surface. | ||
To date, there have been no direct experimental observations of the hydration of the surface K+ ions under UHV. The current molecular-level understanding is based on theoretical studies and simulations.16,17,19–21,31 The lack of experimental data is mainly because the surfaces of clean mica are locally charged and difficult to image with nc-AFM.10,33 The situation differs in ambient or liquid environments, where the interaction with the surrounding environment screens the surface charges and facilitates imaging. Many groups have successfully employed AFM in liquid to resolve the hydration structures of the K+ ions on mica immersed in solution.23,25,26,29,34
At the same time, many theoretical and experimental works have shown that the K+ ions are (at least partially) washed away upon immersion in liquid, and that protons take up the former K+ sites to keep the system locally charge neutral.13,15,35,36 However, there is evidence that potassium carbonate (K2CO3) forms at the surface when mica is cleaved in air due to the reaction of the surface K+ ions with CO2 and H2O, and is dissolved upon immersion in liquid.37 To date, it is unclear under what conditions and to what extent protons substitute the surface K+ ions and whether K2CO3 formation mediates this process.
This experimental study combines constant-height, nc-AFM with X-ray photoemission spectroscopy (XPS) to obtain views of the atomic-scale hydration structures of K+ ions on mica surfaces cleaved under UHV and their behavior upon immersion in liquid water under pristine conditions. Water vapor dosed under UHV at 100 K hydrates these cations through various adsorption geometries. Exposure to ultraclean liquid water in a UHV-based environment partially removes the K+ cations, substituting them with protons even in the absence of CO2(g), i.e., not mediated by K2CO3 formation.
Results and discussion
Exposure to water under UHV
This section investigates the hydration of the K+ ions with atomic precision by dosing sub-monolayer amounts of H2O vapor on UHV-cleaved mica. Initially, water was dosed at room temperature (RT). However, no difference could be detected in nc-AFM between the cleaved surface and a surface dosed with 150 L water at RT (1 Langmuir = 10−6 Torr s). This suggests that water does not adsorb under UHV at RT. Lowering the sample temperature promotes water adsorption (see below). However, warming up the sample to RT after low-temperature water adsorption is not sufficient to restore the surface to its pristine state; as shown in Section S1 ESI,† some of the previously adsorbed water persisted.Fig. 2 shows nc-AFM images of UHV-cleaved mica surfaces after dosing sub-monolayer amounts of water at 100 K. Fig. 2a and d were obtained with a nominal dose of 0.15 L, roughly corresponding to 0.03 H2O/u.c. (see Experimental section for details on dosing and the unit cell size). The images in Fig. 2a and d were acquired in the same area with a metal tip and a CO-functionalized AFM tip, respectively. Both images show species of different heights protruding above the flat surface. On the flat surface in between, the same meandering arrangement of K+ ions of the freshly cleaved surface can be distinguished (the ion lattice here appears fainter compared to Fig. 1a because of the larger tip–sample distance; see Section S2 ESI† for an example of different contrast where the K+ ions of the dry surface are clearly visible). The K+ ions in the background are imaged in dark with both tips. On the other hand, the protruding species – occupying ≈1% of the K+ sites of the dry surface – appear different. With the metal tip (Fig. 2a), they appear as dark (attractive) features at all explored tip–sample distances. Approaching the tip closer to the surface than in Fig. 2a causes a strong interaction with the protruding species inducing surface rearrangements and tip changes without any resolution improvement. With the CO tip (Fig. 2d), the highest species appear as one or more bright (repulsive) round features on top of a dark (attractive) region. When the water dose is larger than 0.2 L, differently protruding species form at the surface and interact with the tip, making it challenging to retain the CO molecule at its apex. Fig. 2b and c correspond to nominal doses of 0.4 L and 0.8 L H2O and were acquired with metal tips.
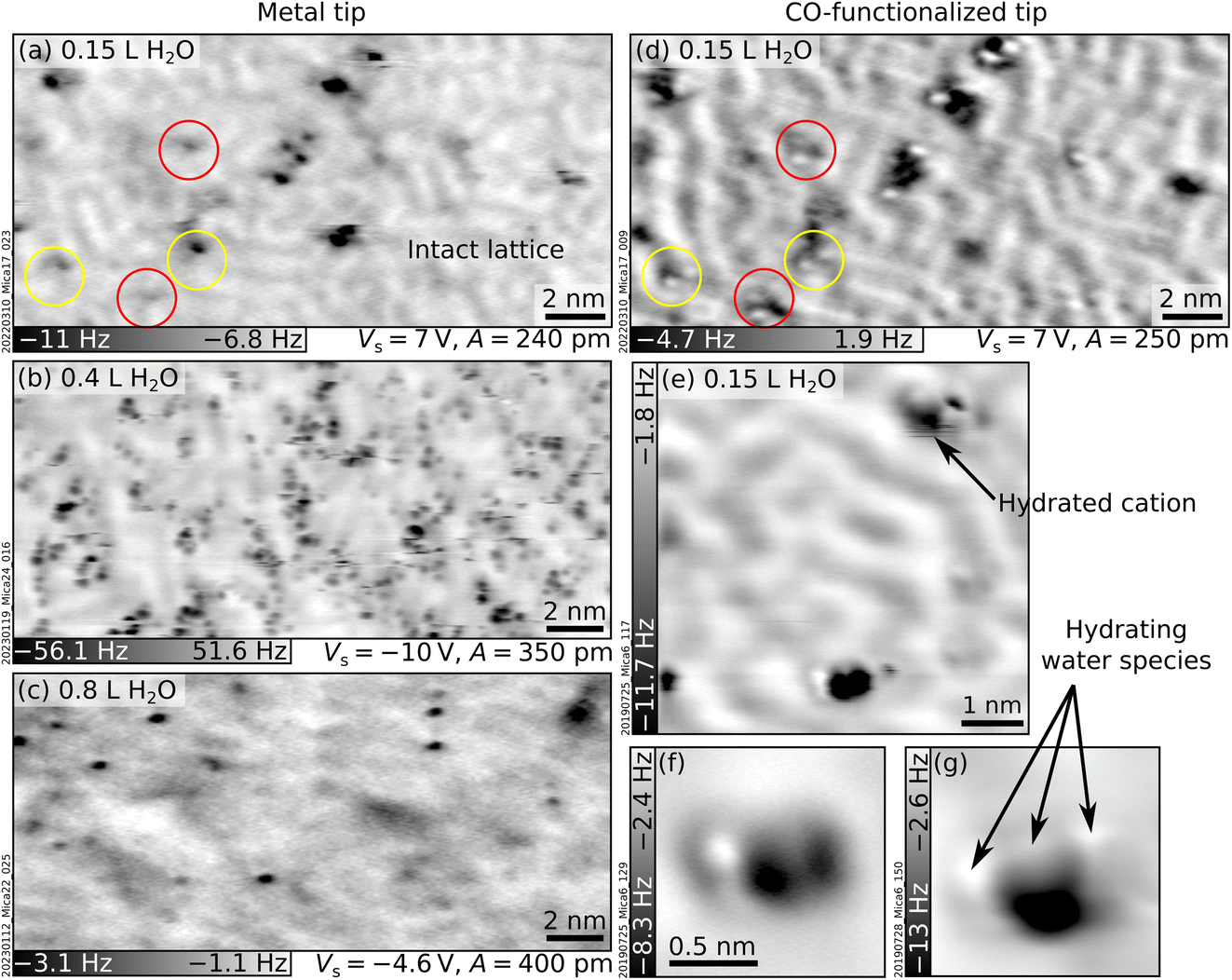 | ||
| Fig. 2 Mica after exposure to water vapor at 100 K. AFM images acquired at T = 4.7 K with metallic (left) and CO-functionalized (right) tips of UHV-cleaved mica plus sub-monolayer amounts of water vapor dosed at 100 K (doses expressed in Langmuir, L). (a and d) 20 × 9.8 nm2 images of the same area after dosing with 0.15 L H2O. Circles highlight the most representative protruding features. (b and c) 20 × 9.3 nm2 images after 0.4 L and 0.8 L H2O, respectively. (e–g) Examples of hydrated cations: (e) 6 × 6 nm2, (f and g) 1.5 × 1.5 nm2, A = 50 pm, Vs = 8 V. | ||
As seen from Fig. 2b and the examples in Fig. 2e–g acquired at the closest possible tip–sample distance, all protruding species imaged with CO-functionalized tips share the common trait of displaying both repulsive (bright) and attractive (dark) parts. However, small differences are always present regarding their height, the number and orientation of bright features, and whether they appear isolated or in small aggregations. A commonly observed species (black arrow in Fig. 2e) consists of an attractive (dark) part plus two repulsive (bright) features oriented along one of the three low-index directions of mica (similar species are indicated by red and yellow circles in Fig. 2d).
The protruding species are identified as hydrated cations based on several pieces of evidence. The first indication comes from previous nc-AFM images of hydrated Na+ ions on NaCl.38 The hydrated cations were measured with a CO-terminated tip and displayed a similar contrast as the protruding species in Fig. 2 (repulsive plus attractive part). Based on a comparison with AFM simulations, the dark and bright features were assigned to the Na+ cation and H2O molecules sensed by the quadrupole-like CO tip.38 The rough calibration of the water doses fits reasonably well with this picture. At 0.15 L, ≈1% of the K+ ions appear as hydrated by ≈2 water molecules. If the H2O molecules only attach to K+, this would correspond to ≈0.01 H2O/u.c., in reasonable agreement with the calibration of 0.03 H2O/u.c. from previous experiments (see Experimental section). A second indication is that DFT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 and MD19 calculations predict solvation forces to dominate the wetting of mica at low temperatures, i.e., H2O molecules should hydrate the K+ ions rather than forming H-bonded networks. Third, MD simulations18–20 predict a large variety of hydrated species (in terms of their size and adsorption geometries of the H2O molecules), consistent with the experimental observations. This variety is expected due to the heterogeneity of the cleaved mica surface. As is visible in Fig. 1a and discussed in more detail in ref. 10, the surface lacks long-range order. Different K+ binding sites are available for H2O molecules. Each site is characterized by a different number and arrangement of the neighboring ions and, hence, a different local electrostatic potential. This potential will determine a unique interaction with the polar H2O molecule and enforce specific adsorption configurations. Fourth, it was observed that surface cations become more mobile with increasing number of hydrating molecules.38,39 This high mobility could be due to weaker bonds of the ions to the surface after hydration, in line with previous predictions that H2O molecules should lift the ions17 (note, however, that the lifting behavior is largely neglected in the literature). The lifting is consistent with the darker appearance (stronger interaction with the tip) of the hydrated cations compared to the dry ions in the background. Ion lifting upon low-temperature water exposure is not unique to mica. Computational studies have predicted a similar pulling effect for Mg ions on MgO substrates40 and Cl ions on NaCl.41
17 and MD19 calculations predict solvation forces to dominate the wetting of mica at low temperatures, i.e., H2O molecules should hydrate the K+ ions rather than forming H-bonded networks. Third, MD simulations18–20 predict a large variety of hydrated species (in terms of their size and adsorption geometries of the H2O molecules), consistent with the experimental observations. This variety is expected due to the heterogeneity of the cleaved mica surface. As is visible in Fig. 1a and discussed in more detail in ref. 10, the surface lacks long-range order. Different K+ binding sites are available for H2O molecules. Each site is characterized by a different number and arrangement of the neighboring ions and, hence, a different local electrostatic potential. This potential will determine a unique interaction with the polar H2O molecule and enforce specific adsorption configurations. Fourth, it was observed that surface cations become more mobile with increasing number of hydrating molecules.38,39 This high mobility could be due to weaker bonds of the ions to the surface after hydration, in line with previous predictions that H2O molecules should lift the ions17 (note, however, that the lifting behavior is largely neglected in the literature). The lifting is consistent with the darker appearance (stronger interaction with the tip) of the hydrated cations compared to the dry ions in the background. Ion lifting upon low-temperature water exposure is not unique to mica. Computational studies have predicted a similar pulling effect for Mg ions on MgO substrates40 and Cl ions on NaCl.41
Finally, the XPS data in Fig. 3 corroborate the overall picture. Fig. 3a and b show the evolution of the O 1s and K 2p peaks with increasing water coverage dosed at 100 K on UHV-cleaved mica, respectively. Fig. 3c and d quantify the evolution of the fitting components. The O 1s peak can be fitted by only two contributions over all coverages: the contribution of the dry surface (given by peaks 1 and 2 in Fig. 3a, assigned to bulk O and bulk OH) and a second contribution, labelled as peak 3, growing larger at increasing doses. At 50 L, where all the water should adsorb molecularly in water ice, the O 1s peak can be fit with component 3 alone, which is thus assigned to molecular H2O. The fact that all other O 1s peaks can be fit by the components of the pristine surface and molecular H2O, supports the hypothesis that water adsorption is molecular at all coverages. Note that dissociative adsorption at very small coverages cannot be completely ruled out: at 0.2 L, a reasonable fit can be obtained without component 3 (however, already at 0.4 L, component 3 is needed). The upper limit for dissociative adsorption at 0.2 L can be derived as roughly 0.1 OH/u.c.
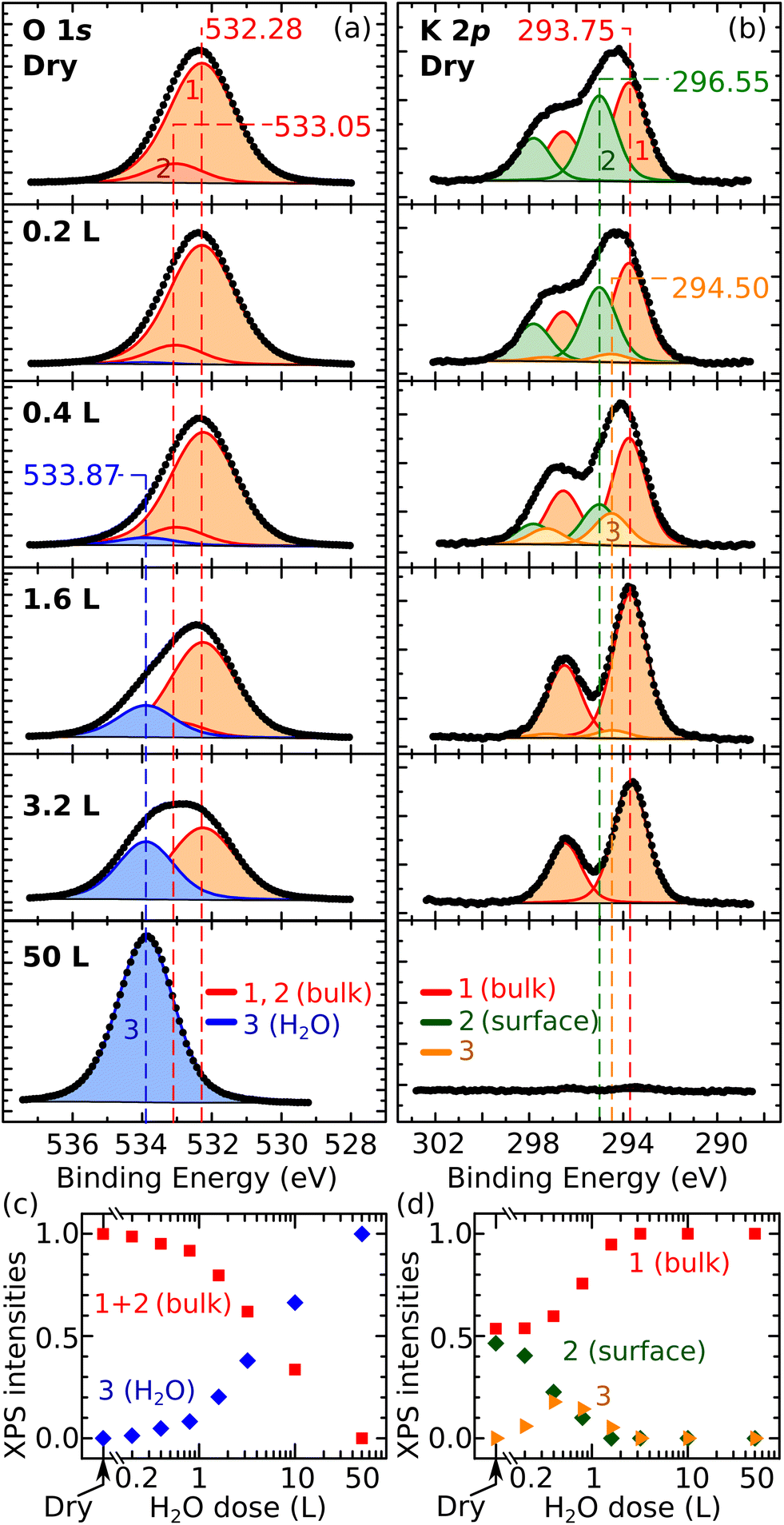 | ||
| Fig. 3 XPS after exposing mica to water vapor at 100 K. (a and b) Experimental data (dots) and fit (solid lines) of O 1s and K 2p core-level peaks of UHV-cleaved mica exposed to water vapor (Al Kα, pass energy 20 eV, 70° grazing emission; doses expressed in Langmuir; XPS data acquired at 100 K). (c and d) Normalized intensities of the fitting components of the O 1s and K 2p core-level peaks as a function of the water dose. Error bars are smaller than the symbol size. The Experimental section provides details about the correction of the energy axes, fitting procedures, and error bar calculations. | ||
The behavior of the K 2p XPS peak is overall consistent. Its shape changes with increasing water dose, featuring a more pronounced separation of the spin-split components. As shown previously,10 the K 2p peak of the dry surface can be fit by two components assigned to different types of K+ ions: the fully coordinated ions in the bulk holding together the aluminosilicate layers, and the lower-coordinated ions at the cleaved surface that produce a core-level-shifted peak at higher binding energy. In Fig. 3b, the bulk and surface components are labeled as 1 and 2, respectively. Above 3 L, the K 2p spectrum is successfully fit by the bulk component alone, suggesting that the K+ ions are hydrated by H2O molecules to again reach full coordination and a bulk-like character. Between 0.2 L and 1.6 L, an additional component (3) at a binding energy between the surface and the bulk components is needed to fit the spectra. This suggests that, at these coverages, the number of H2O molecules hydrating the ions is still insufficient to complete their shell. This agrees with the notion emerging from the AFM data that the K+ ions become progressively more hydrated at larger water doses, and with previous MD studies at room temperature predicting that H2O molecules preferentially adsorb close to the K+ ions to complete their hydration shell.19
Note that the lack of a flat, ordered, ice-like phase emerging from the AFM data is in line with previous MD simulations predicting that 2D ice structures should not form on K-mica.19,32 Consistently, Fig. 2b and c show that increasing water doses do not produce any long-range order expected for a 2D ice layer but only the enlargement of the clusters of hydrated species, which roughly follow the short-range arrangement of the underlying K+ ions. No pattern is visible in the corresponding Fourier-transform images.
A remark about the possible role of intrinsic impurities is due. MD studies suggest that multivalent cations such as Ca2+ and Mg2+ should be better ice nucleators than K+.31 Hence, it cannot be excluded that the few hydrated species present at low coverages are, in fact, hydrated trace impurities. Nonetheless, the behavior of the XPS K 2p peak and the appearance of the surface in AFM at larger H2O doses is consistent with the progressive hydration of surface K+ ions.
Exposure to ultraclean liquid water
To bridge the gap between UHV and environmental conditions where minerals are in contact with liquid water, the UHV-cleaved surface of mica was exposed to ultraclean liquid water using the apparatus described briefly in the Experimental section and in detail in ref. 42. Afterwards, each sample was brought to the main UHV chamber and analyzed either with XPS at room temperature or with nc-AFM at 4.7 K. The surface is clean in XPS except for minor traces of carbon (see inset of Fig. 4c; note that the binding energy of this weak C 1s peak, 286.4 eV, differs significantly from the value of 289.0 eV expected for K2CO3 (ref. 43)). The relative intensities of the main core-level peaks are comparable to those of the UHV-cleaved surface apart from the K peaks, which clearly decrease. Taking the K 2p signal of the cleaved surface as a reference and using the same fits for the bulk and surface components, one can estimate that the liquid-water exposure reduces the surface contribution of the K 2p peak to about 63% of its original value. In large-area AFM images (Fig. 4a), the surface appears covered by several clusters of different heights, one of which is marked with an arrow. Atomic resolution is achieved only in small areas in between the protruding clusters. The black square in Fig. 4a marks such an area, exemplified by Fig. 4b. Round, dark features sit on the same hexagonal grid as on the UHV-cleaved mica. However, pronounced contrast differences are appreciable among the dots, and their overall coverage is substantially smaller than under UHV-cleaved mica (between 50% and 80% of the original value, depending on which of the fainter species are considered for the evaluation). The same behavior is observed after water exposure for ca. 10 minutes instead of one minute.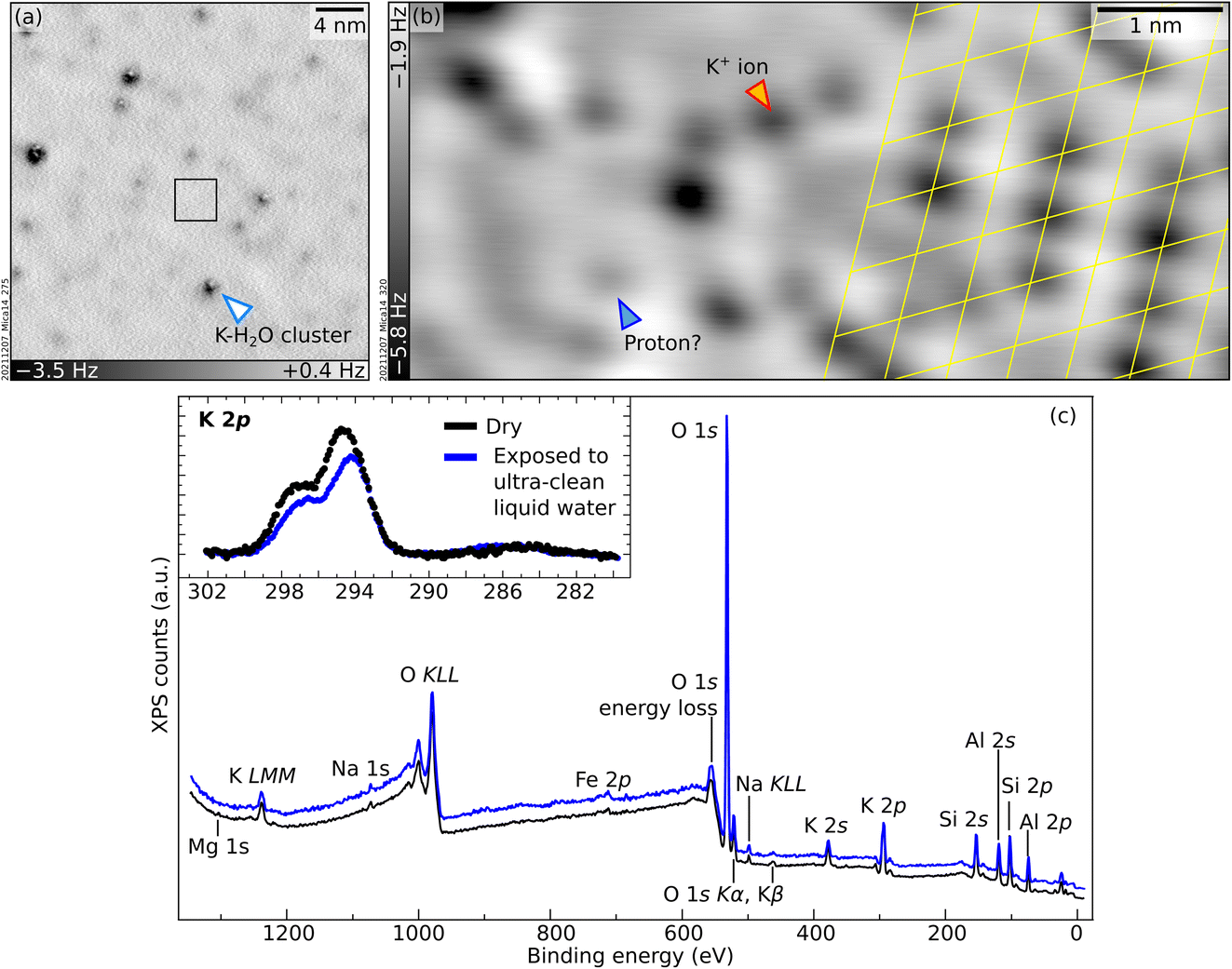 | ||
| Fig. 4 Mica after exposure to clean liquid water. (a and b) Constant-height non-contact AFM images: (a) 30 × 30 nm2, A = 300 pm, Vs = −1.2 V; (b) 6.7 × 3 nm2, A = 100 pm, Vs = −1.2 V. (c) XPS survey and K 2p + C 1s region (inset) measured under UHV at room temperature for an as-cleaved mica surface, before (black) and after (blue) exposure to liquid water (Al Kα, 70° grazing emission, pass energy of 60 eV and 20 eV for the overview and the inset, respectively). In the inset, the Al Kα3 contribution of the K 2p signal has been subtracted. The energy axes were adjusted to account for charging (see Experimental section). | ||
A tentative interpretation is that some K+ ions become mobile upon exposure to liquid water at room temperature, leaving their aluminosilicate tetrahedra rings (see Fig. 1c). To keep the system charge-neutral, the empty rings become occupied by protons – possibly the fainter species visible in AFM. Because the water dries on the sample, the displaced K+ ions are not rinsed away. The clusters visible in AFM could consist of the displaced K+ ions bound by water molecules (and possibly traces of carbon). These three-dimensional clusters should contribute only marginally to the XPS signal, explaining the measured decrease in the surface contribution of the K 2p peak. Another possibility is that K+ ions diffuse into mesoscopic cracks into the sample or to the outer rim of the sample following a “coffee-stain effect” when the water drop dries.
The presented data show that experiments performed under ultraclean conditions result only in a partial substitution of the K+ ions after evaporation of the water. This agrees with previous MD simulations which “do not rule out the possibility that the surface K+ ions remain on the surface after washing”.18 In contrast, previous studies where mica was cleaved in air and then rinsed in water reported the complete substitution of the surface K+ ions.13 The different extent of ion substitution under UHV vs. ambient conditions could be due to the formation of K2CO3 in ambient conditions. There is evidence that K2CO3 forms on air-cleaved mica due to the reaction of K+ ions with water and atmospheric CO2.37 K2CO3 is water-soluble and is thus likely washed away by rinsing, removing all K from the surface, and leaving no choice to the otherwise charged surface, but to adsorb protons from the solution.
Conclusions
This study investigates the interaction of water with the surface K+ ions of cleaved mica under pristine conditions. Hydrated ions are directly visualized with nc-AFM upon dosing water vapor under UHV at 100 K. They appear to be lifted up by water molecules, which orient themselves differently depending on the specific adsorption site. Increasing water coverage causes the clustering of the hydrated species without any apparent ordered ice-like phase. After exposing UHV-cleaved mica to ultraclean water and evaporation of the water, the K+ ions appear to be partially substituted by protons, in contrast with the complete substitution observed after exposure to liquid water under ambient conditions. The difference could be due to the mediation by K2CO3 formed in ambient conditions.Experimental methods and data analysis
Experimental setup
The experiments were carried out in a UHV setup consisting of three interconnected chambers: a preparation chamber for sample cleaving, water vapor dosing, and XPS measurements (base pressure < 1 × 10−10 mbar), an AFM chamber for nc-AFM measurements (base pressure < 2 × 10−11 mbar), and a custom-built compartment (hereafter termed “side chamber”) used for the liquid-drop experiments. The latter is attached to the preparation chamber and has a base pressure < 1 × 10−9 mbar after bakeout.Natural muscovite mica single crystals [(001)-oriented disks of grade V1, with 10 mm diameter and 0.25 mm thickness, from TedPella] were glued on Omicron-style stainless-steel sample plates with epoxy glue. They were cleaved under UHV before each experiment as described elsewhere.10
Water-vapor experiments
Water vapor was dosed by cooling the sample holder on the manipulator to 100 K with liquid nitrogen. Nominal doses are expressed in Langmuir (L), where 1 L corresponds to a dose obtained by an exposure of 1 s at 1.3 × 10−6 mbar (or 100 s at 1.3 × 10−8 mbar, the pressure used for the experiments). Determining effective doses on the sample surface is challenging due to (i) the lack of a 2D adsorption pattern and of a theoretical model for the adsorbed phases, and (ii) the sticking of H2O at the chamber walls, which leads to an inhomogeneous pressure distribution in the vacuum chamber. Previous calibrations in the same setup on Fe2O3 (ref. 44) and Fe3O4 (ref. 45) single crystals, quantified 1 L dosed at 150 K as 0.8–0.9 H2O molecules per nm2. Using the 0.234 nm2 unit cell of mica, this gives ≈0.19 H2O/u.c. (or ≈0.38 H2O/surface K+ ion as there are 0.48 K ions per unit cell10).XPS acquisition and analysis
XPS was acquired with a non-monochromatic dual-anode Mg/Al X-ray source (SPECS XR 50) and a hemispherical analyzer (SPECS Phoibos 100). Spectra were acquired in normal and grazing emission (70° from the surface normal). The insulating nature of the samples (7.85 eV bandgap)46 makes the XPS analysis challenging, both because of binding energy shifts and broadening effects. After cleaving, the spectra shift to higher binding energies (between 5 and 7 eV); the precise shifts are determined by the amount and type of surface contamination, XPS acquisition geometry, and sample thickness.47 In this work, dosing water at 100 K caused progressive shifts to lower binding energies. To correct for the charging, the binding energy axes were calibrated such that the position of the K 2p3/2 component of the bulk corresponded to the literature value of 293.75 eV.47The intensities and positions of the Al-Kα-excited XPS peaks were evaluated with CasaXPS after subtracting a Shirley-type background. Table 1 summarizes the constraints applied for the fits. In detail, the O 1s peaks (Fig. 3a) were fit with two main contributions: a contribution obtained by the sum of peaks 1 and 2, fitting the dry surface, and a second contribution (labelled as peak 3) fitting the surface covered with 50 L H2O, where the signal from the bulk is negligible (see the corresponding K 2p spectrum, Fig. 3b) and all water should be molecular. The peak shapes, FWHM values, and relative positions of components 1 + 2 and 3 were determined from the spectra of the dry surface and of the 50 L-dosed surface, respectively, and were then constrained to fit the other experiments. Peaks 1 and 2 on the dry surface were used to reproduce the asymmetric shape of the corresponding O 1s peak (possibly due to the presence of OH in the subsurface octahedral layer). The position, relative separation, and relative area of components 1 and 2 were constrained to fit the subsequent experiments. One must point out that fitting the O 1s peak poses intrinsic challenges: on the pristine surface, XPS should be sensitive to the OH groups of the subsurface octahedral layer but broadening effects due to charging and different environments of the O atoms do not allow to clearly resolve such an OH component. Broadening is likely present on the water-dosed surface. As this work argues, each surface site is characterized by different adsorption geometries of the water molecules; this will cause different screening effects locally, each corresponding to a slightly shifted peak. The measurements average over all possible configurations. Thus, they produce comparatively broader peaks.
| Identifier | Shape | FWHM | Position (eV) | Area | |
|---|---|---|---|---|---|
| O 1s 1 | O 1s pristine | LA (1.25, 243) | 2.24 | 532.28 | Free |
| O 1s 2 | OH pristine | LA (1.25, 243) | 1.88 | (O 1s 1) + 0.765 | (Area O 1s 1) × 0.134 |
| O 1s 3 | H2O | LA (1.25, 213) | 1.92 | 533.67 | Free |
| K 2p 1 | K 2p bulk | LA (1, 643) | 1.71 | 293.75 (2p3/2) | Free |
| K 2p 2 | K 2p surface | LA (1, 643) | 1.71 | 295.015 (2p3/2) | Free |
| K 2p 3 | K 2p medium coverage | LA (1, 643) | 1.71 | 294.5 (2p3/2) | Free |
The K 2p peaks were fit by multiple sets of 2p peaks. In each set, the separation between 2p3/2 and 2p1/2 was set to 2.8 eV in line with previous work,47 and the area ratio to 2:1. All peaks have the same FWHM. The separation between sets (1) and (2) (bulk and surface components, respectively)10 was set to be the same for normal and grazing emission.
AFM measurements
The AFM measurements were performed at 4.7 K using a commercial Omicron qPlus LT head and a differential cryogenic amplifier.48 The tuning-fork-based AFM sensors (k = 3750 N m−1, f0 = 45 kHz, Q ≈ 50000)49 have a separate wire for the tunnelling current attached to electrochemically etched W tips, which were cleaned in situ by field emission.50 Before each measurement, the tips were further prepared on a clean Cu(110) single crystal by repeated indentation and voltage pulses. CO-functionalized tips51 were used to image the water-exposed samples. After a coarse approach, the tip was approached with the AFM frequency controller, setting a typical frequency shift value of −1 Hz. The controller was switched off, and the tip was approached manually until an AFM contrast was visible. All AFM images were acquired in the constant-height mode. At times, the absolute values of frequency shifts obtained during the acquisition of atomically resolved images were large (up to 100 Hz) and not reproducible from sample to sample or on different regions on the same sample. This is because cleaving can result in domains of trapped charges in insulators,52–54 which can cause long-range electrostatic interactions between the surface and tip.33 The electrostatic fields can be partially compensated by applying a bias voltage between tip and sample. Most of the measurements were performed at a bias voltage (specified in the respective figure captions) such that the surface was measured as close as possible to the local contact potential difference between tip and sample, i.e., at the minimum absolute value of the frequency shift.
Liquid-water experiments
Samples were exposed to liquid water in the side chamber as detailed elsewhere.42 In short, the side chamber is equipped with two water reservoirs that can be cooled to 100 K and are connected to the side chamber through all-metal CF16 angle valves. One reservoir is used to make the water drop, the other as a cryosorption pump to remove the water after the experiment. Each experiment proceeds as follows:• The MilliQ water for the drop is purified by three freeze–pump–thaw cycles. The water supply is partially frozen and kept near 0 °C throughout the experiment to stabilize the vapor pressure at 6 mbar evolving from the water-ice mixture.
• After separating the side chamber with a valve, water vapor is dosed for a fixed time inside the side chamber (typically 7 minutes). The water condenses onto a conical, stainless-steel tip held at 100 K where it forms an icicle.
• The as-cleaved mica sample is transferred under UHV into the side chamber while keeping the tip at 100 K (the vapor pressure of H2O is negligible at this temperature).
• The side chamber is closed off again. The tip is heated to room temperature to let the icicle thaw and a liquid droplet fall onto the sample surface. This results in the complete wetting of the sample surface but no water contact with the sample plate (crucial to avoid contamination).
• After exposure, the liquid and residual water vapor in the side chamber are evacuated using the cryosorption pump.
• The sample is transferred back to the preparation chamber. To make sure that most of the residual water evaporates, the sample is kept at room temperature in the preparation chamber for 20 min (or until the pressure in the chamber drops below 4 × 10−10 mbar). Afterwards, the sample is transferred to the AFM head. Note that the water drop always dries on the sample’s surface, it is not rinsed off like in ex situ experiments. This is because the strongly hydrophilic nature of UHV-cleaved mica prevents the water drop from coming off the sample even when tilted by 90°.
To minimize the contamination displaced from the chamber walls, the side chamber is baked before use. Before the experiment, to further clean the walls of the side chamber and the tip, a few water drops are created, dropped onto the bottom of the side chamber and pumped away.
Author contributions
G. F.: conceptualization, investigation, writing. S. B.: investigation. J. B., I. S., J. P.: investigation, supervision. M. Setvín: supervision, validation. M. Schmid: supervision, validation. U. D.: funding acquisition, supervision, validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 883395, Advanced Research Grant ‘WatFun’). S. B. acknowledges support from the FFG Project No. 23017619 ‘FunPakTrio’. M. Setvín was supported through the FWF project “Super”, project number P32148-N36, and the Czech Science Foundation, project GACR 20-21727X.Notes and references
- G. E. Brown Jr and G. Calas, Geochem Perspect., 2012, 1, 483–484 CrossRef.
- P. Fenter, P. Zapol, H. He and N. C. Sturchio, Geochim. Cosmochim. Acta, 2014, 141, 598–611 CrossRef CAS.
- K. S. Lackner, Science, 2003, 300, 1677–1678 CrossRef CAS PubMed.
- W. Stumm, Chemistry of the Solid-Water Interface: Processes at the Mineral- Water and Particle-Water Interface in Natural Systems, Wiley, 1992 Search PubMed.
- A. Kiselev, F. Bachmann, P. Pedevilla, S. J. Cox, A. Michaelides, D. Gerthsen and T. Leisner, Science, 2017, 355, 367–371 CrossRef CAS PubMed.
- A. Kumar, C. Marcolli and T. Peter, Atmos. Chem. Phys., 2019, 19, 6059–6084 CrossRef CAS.
- J. D. Atkinson, B. J. Murray, M. T. Woodhouse, T. F. Whale, K. J. Baustian, K. S. Carslaw, S. Dobbie, D. O’Sullivan and T. L. Malkin, Nature, 2013, 498, 355–358 CrossRef CAS PubMed.
- J. Peng, J. Guo, R. Ma and Y. Jiang, Surf. Sci. Rep., 2022, 77, 100549 CrossRef CAS.
- W. De Poel, S. Pintea, J. Drnec, F. Carla, R. Felici, P. Mulder, J. A. A. W. Elemans, W. J. P. Van Enckevort, A. E. Rowan and E. Vlieg, Surf. Sci., 2014, 619, 19–24 CrossRef CAS.
- G. Franceschi, P. Kocán, A. Conti, S. Brandstetter, J. Balajka, I. Sokolović, M. Valtiner, F. Mittendorfer, M. Schmid, M. Setvín and U. Diebold, Nat. Commun., 2023, 14, 208 CrossRef CAS.
- G. L. Gaines, J. Phys. Chem., 1957, 61, 1408–1413 CrossRef CAS.
- W. de Poel, S. L. Vaessen, J. Drnec, A. H. J. Engwerda, E. R. Townsend, S. Pintea, A. E. F. de Jong, M. Jankowski, F. Carlà, R. Felici, J. A. A. W. Elemans, W. J. P. van Enckevort, A. E. Rowan and E. Vlieg, Surf. Sci., 2017, 665, 56–61 CrossRef CAS.
- L. Xu and M. Salmeron, Langmuir, 1998, 14, 5841–5843 CrossRef CAS.
- S. S. Lee, P. Fenter, K. L. Nagy and N. C. Sturchio, Langmuir, 2012, 28, 8637–8650 CrossRef CAS.
- A. Prakash, J. Pfaendtner, J. Chun and C. J. Mundy, J. Phys. Chem. C, 2017, 121, 18496–18504 CrossRef CAS.
- M. Odelius, M. Bernasconi and M. Parrinello, Phys. Rev. Lett., 1997, 78, 2855 CrossRef CAS.
- P. J. Feibelman, J. Chem. Phys., 2013, 139, 074705 CrossRef PubMed.
- J. Wang, A. G. Kalinichev, R. J. Kirkpatrick and R. T. Cygan, J. Phys. Chem. B, 2005, 109, 15893–15905 CrossRef CAS.
- R. Debbarma and A. Malani, Langmuir, 2016, 32, 1034 CrossRef CAS.
- X. Ou, X. Wang, Z. Lin and J. Li, J. Phys. Chem. C, 2017, 121, 6813–6819 CrossRef CAS.
- S. Adapa, D. R. Swamy, S. Kancharla, S. Pradhan and A. Malani, Langmuir, 2018, 34, 14472–14488 CrossRef CAS PubMed.
- T. Fukuma, Y. Ueda, S. Yoshioka and H. Asakawa, Phys. Rev. Lett., 2010, 104, 016101 CrossRef.
- K. Kimura, S. Ido, N. Oyabu, K. Kobayashi, Y. Hirata, T. Imai and H. Yamada, J. Chem. Phys., 2010, 132, 194705 CrossRef.
- D. Martin-Jimenez, E. Chacon, P. Tarazona and R. Garcia, Nat. Commun., 2016, 7, 12164 CrossRef CAS.
- T. Arai, K. Sato, A. Iida and M. Tomitori, Sci. Rep., 2017, 7, 4054 CrossRef PubMed.
- K. Kobayashi, N. Oyabu, K. Kimura, S. Ido, K. Suzuki, T. Imai, K. Tagami, M. Tsukada and H. Yamada, J. Chem. Phys., 2013, 138, 184704 CrossRef.
- T. Ichii, S. Ichikawa, Y. Yamada, M. Murata, T. Utsunomiya and H. Sugimura, Jpn. J. Appl. Phys., 2020, 59, SN1003 CrossRef CAS.
- M. Ricci, P. Spijker and K. Voïtchovsky, Nat. Commun., 2014, 5, 4400 CrossRef CAS PubMed.
- D. Martin-Jimenez and R. Garcia, J. Phys. Chem. Lett., 2017, 8, 5707–5711 CrossRef CAS PubMed.
- S. Jin, Y. Liu, M. Deiseroth, J. Liu, E. H. G. Backus, H. Li, H. Xue, L. Zhao, X. C. Zeng, M. Bonn and J. Wang, J. Am. Chem. Soc., 2020, 142, 17956–17965 CrossRef CAS PubMed.
- N. N. Lata, J. Zhou, P. Hamilton, M. Larsen, S. Sarupria and W. Cantrell, J. Phys. Chem. Lett., 2020, 11, 8682 CrossRef CAS PubMed.
- A. Soni and G. N. Patey, J. Phys. Chem. C, 2021, 125, 26927–26941 CrossRef CAS.
- F. Ostendorf, C. Schmitz, S. Hirth, A. Kühnle, J. J. Kolodziej and M. Reichling, Nanotechnology, 2008, 19, 305705 CrossRef CAS.
- K. Pürckhauer, A. J. Weymouth, K. Pfeffer, L. Kullmann, E. Mulvihill, M. P. Krahn, D. J. Müller and F. J. Giessibl, Sci. Rep., 2018, 8, 9330 CrossRef PubMed.
- T. E. Balmer, H. K. Christenson, N. D. Spencer and M. Heuberger, Langmuir, 2008, 24, 1566–1569 CrossRef CAS.
- P. J. Sides, D. Faruqui and A. J. Gellman, Langmuir, 2009, 25, 1475–1481 CrossRef CAS.
- H. K. Christenson and N. H. Thomson, Surf. Sci. Rep., 2016, 71, 367–390 CrossRef CAS.
- J. Peng, D. Cao, Z. He, J. Guo, P. Hapala, R. Ma, B. Cheng, J. Chen, W. J. Xie, X. Z. Li, P. Jelínek, L. M. Xu, Y. Q. Gao, E. G. Wang and Y. Jiang, Nature, 2018, 557, 701–705 CrossRef CAS PubMed.
- S. S. Lee, P. Fenter, C. Park, N. C. Sturchio and K. L. Nagy, Langmuir, 2010, 26, 16647–16651 CrossRef CAS.
- M. Ončák, R. Włodarczyk and J. Sauer, J. Phys. Chem. Lett., 2015, 6, 2310–2314 CrossRef PubMed.
- P. Cabrera-Sanfelix, D. S. Portal, A. Verdaguer, G. R. Darling, M. Salmeron and A. Arnau, J. Phys. Chem. C, 2007, 111, 8000–8004 CrossRef CAS.
- J. Balajka, J. Pavelec, M. Komora, M. Schmid and U. Diebold, Rev. Sci. Instrum., 2018, 89, 083906 CrossRef PubMed.
- A. V. Shchukarev and D. V. Korolkov, Cent. Eur. J. Chem., 2004, 2, 347–362 CAS.
- Z. Jakub, F. Kraushofer, M. Bichler, J. Balajka, J. Hulva, J. Pavelec, I. Sokolović, M. Müllner, M. Setvin, M. Schmid, U. Diebold, P. Blaha and G. S. Parkinson, ACS Energy Lett., 2019, 4, 390–396 CrossRef CAS.
- M. Meier, J. Hulva, Z. Jakub, J. Pavelec, M. Setvin, R. Bliem, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5642–E5650 CAS.
- A. T. Davidson and A. F. Vickers, J. Phys. C: Solid State Phys., 1972, 5, 879 CrossRef CAS.
- K. G. Bhattacharyya, J. Electron Spectrosc. Relat. Phenom., 1993, 63, 289–306 CrossRef CAS.
- F. Huber and F. J. Giessibl, Rev. Sci. Instrum., 2017, 88, 073702 CrossRef.
- F. J. Giessibl, Rev. Sci. Instrum., 2019, 90, 011101 CrossRef.
- M. Setvín, J. Javorský, D. Turčinková, I. Matolínová, P. Sobotík, P. Kocán and I. Ošt’ádal, Ultramicroscopy, 2012, 113, 152–157 CrossRef.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS.
- M. I. Kornfeld, J. Phys. D: Appl. Phys., 1978, 11, 1295–1301 CrossRef CAS.
- J. W. Obreimoff, Proc. R. Soc. London, Ser. A, 1930, 127, 290–297 Search PubMed.
- M. S. Metsik and L. M. Golub, J. Appl. Phys., 1975, 46, 1983–1986 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: S1: water adsorption at room temperature; S2: unusual tip conditions to measure surfaces exposed to water vapor at 100 K. See DOI: https://doi.org/10.1039/d3fd00093a |
| This journal is © The Royal Society of Chemistry 2024 |