
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the charge transfer dynamics in 3D–1D nanocomposites over solar driven synergistic selective valorization of lignocellulosic biomass: a new sustainable approach†
Arpna
Jaryal
a,
Ajit Kumar
Singh
b,
Shivali
Dhingra
a,
Himanshu
Bhatt
a,
Manvi
Sachdeva
a,
Hirendra N.
Ghosh
*c,
Arindam
Indra
 *b and
Kamalakannan
Kailasam
*a
*b and
Kamalakannan
Kailasam
*a
aAdvanced Functional Nanomaterials Group, Institute of Nano Science and Technology (INST), Knowledge City, Sector 81, SAS Nagar, Manauli PO, Mohali, 140306, Punjab, India. E-mail: kamal@inst.ac.in; kkamal17@gmail.com
bDepartment of Chemistry, Indian Institute of Technology (BHU), Varanasi, 221005, Uttar Pradesh, India. E-mail: arindam.chy@iitbhu.ac.in
cSchool of Chemical Sciences, National Institute of Science Education and Research (NISER), Bhubaneswar, 752050, Odisha, India. E-mail: hnghosh@niser.ac.in
First published on 25th May 2024
Abstract
Photocatalytic redox valorization of lignocellulosic biomass to fine chemicals is in its infancy stages where it can be effectively utilized for sustainable energy conversion. In this direction, an effective 3D–1D (Aeroxide P25 TiO2 and CdS) nanocomposite has been demonstrated to upgrade several biomass-derived platform chemicals (e.g. HMF, FFaL, vanillyl alcohol) in a selective and synergistic redox pathway under visible light irradiation for the first time. The successful utilization of the photocatalytic system resulted in the visible light-driven selective hydrogenation of HMF to BHMF along with the coproduction of H2 without the addition of any reducing agent under natural sunlight. In addition, the simultaneous production of valuable commodity chemical, i.e. vanillin, through oxidation has also been earmarked. The intimate interfacial contact between CdS as a visible light active photocatalyst and P25 TiO2 as an active hydrogenation site assists the facile migration of photogenerated electrons towards P25 TiO2. The coupling of electrons with in situ generated protons led to 95% yield of BHMF whereas oxidative photogenerated holes yielded 35% vanillin, thus abolishing the need for extra redox additives. The synergistic effect bestowed by the semiconductor heterojunction manifested excellent photoredox activity accompanying strong inter-particle interactions which were thoroughly investigated by employing electrochemical, PL, XPS and transient absorption spectroscopy (TAS). Thus, a new sustainable “biomass-based photo-refinery” and cost-effective low carbon-intensity approach has been elucidated for visible light-based hydrogenation activity of TiO2 unveiling a fabrication strategy of photocatalysts with efficient solar spectrum harvesting.
Broader contextTo cope with increasing energy demand and to substitute non-renewable fossil energy sources, solar driven valorization of non-edible biomass laid forwards a sustainable and economical approach. The idea of “biomass photo-refinery” by harvesting natural sunlight and second-generation lignocellulosic biomass can meet the ultimate goal of carbon-neutral bio-chemical production. To meet this demand, sufficient efforts are being attempted for synergistic utilization of electrons and holes to derive H2 or CO2 reduced products through photoreduction along with photo oxidation of biomass precursors to fine chemicals. Nevertheless, the key challenge of the simultaneous photoredox cycle for biomass-based precursor reduction and oxidation to fine chemicals is still elusive with a surplus amount of biomass feedstocks available. For this purpose, our initial report of implementing a visible light active semiconductor for upgrading various biomass-derived platform chemicals, e.g. reducing HMF to BHMF and furfural to furfuryl alcohol, while oxidizing vanillyl alcohol to vanillin, has been achieved through photoredox conversions with the co-production of H2. This selective transformation that utilizes the charge carriers in a more effective manner is a much-needed effort to achieve carbon neutrality and an industrially feasible process to be attempted. Thus, here we propose a new, complete biomass-based photorefinery process for sustainable chemical production. |
Introduction
The effective utilization of renewable energy sources for wide applications is necessary to cope with exhausting fossil fuel reservoirs and increasing energy demands.1,2 The constructive harvesting of solar energy to produce green fuels and valuable chemicals presents one such approach. Sunlight driven biomass valorization to value-added products by photorefinery has advantages over energy intensive thermal routes due to its benign reaction conditions and sustainable pathways.3,4 The complex and rigid structure of raw lignocellulosic biomass makes its usage in photorefinery a challenging task; therefore, the upconversion of biomass-derived platform chemicals is being primarily studied. Lignocellulosic biomass is a widely available carbon rich source of lignin (phenols), cellulose (hexose) and hemicellulose (hexose and pentose) that can depolymerize to various platform chemicals.5,6 For example, 5-hydroxymethylfurfural (HMF) and furfurals (FFaL) are C6 and C5 furanics-based platform molecules derived from glucose and xylose-based monosaccharides, respectively.7 Along with these, rigid lignin components can also be cleaved to supply guaiacol and phenol-based monomers.8 Furthermore, these biomass-derived platform molecules can be upgraded to valuable products with vast applications in the polymer industry, pharmaceutics and energy sector.9To carry out these upgradations, heterogeneous photocatalysis has shown superiority over expensive and harsh thermocatalytic routes by means of green and inexpensive technology for environmental remediation.10 The implementation of visible light active semiconductors as a heterogeneous photocatalyst offers a sustainable and cost-effective method to perform selective chemical conversions. Early studies by Baur and Neuweiler have suggested the utilization of photogenerated holes and electrons on semiconductor surfaces for oxidation and reduction, respectively.11 Based on this principle, we have recently reported the effective employment of Aeroxide P25 TiO2 nanoparticles for the selective hydrogenation of HMF to bis-hydroxymethylfuran (BHMF) with the simultaneous oxidation of anisyl alcohol to p-anisaldehyde in UV light irradiation but in relatively lower yields.12 Thus the selective production of BHMF from HMF presents a challenging task as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogenation of the furan ring is thermodynamically favoured over CO hydrogenation.13 However, the CO based hydrogenated product of HMF with active hydroxyl functionalization bestows numerous potential applications in functional polymers, artificial fibers and resins, and it also serves as a valuable biopolymer precursor.14 Considering the thermodynamic difficulty in selective BHMF production, there exist a few reports on photocatalytic HMF reduction in the presence of expensive catalytic systems with an additional hydrogenating source and/or operating under basic conditions.15–18 However, the coupled redox transformations over semiconductor photocatalysts by means of photogenerated excitons exhibit the advantage of benign reaction conditions without extra reducing agents, thus boosting the chemical economy.
C hydrogenation of the furan ring is thermodynamically favoured over CO hydrogenation.13 However, the CO based hydrogenated product of HMF with active hydroxyl functionalization bestows numerous potential applications in functional polymers, artificial fibers and resins, and it also serves as a valuable biopolymer precursor.14 Considering the thermodynamic difficulty in selective BHMF production, there exist a few reports on photocatalytic HMF reduction in the presence of expensive catalytic systems with an additional hydrogenating source and/or operating under basic conditions.15–18 However, the coupled redox transformations over semiconductor photocatalysts by means of photogenerated excitons exhibit the advantage of benign reaction conditions without extra reducing agents, thus boosting the chemical economy.
TiO2 has proved itself as one of the versatile photocatalysts employed in water and air purification,19 H2O splitting,20 CO2 reduction21 and organic conversions.22,23 However, the wide optical band gap absorbing UV (∼5%) restrains the maximum accumulation of sunlight as the higher part (∼45%) of the solar spectrum consists of visible light. Taking into account the advantages of TiO2 as a non-toxic, cheap, photostable semiconductor, intensive research is being carried out to activate TiO2-based photocatalytic systems in visible light.24 The decoration of TiO2 with photosensitizers, noble metals and heteroatom doping, and its heterostructures with narrow optical band gap-based semiconductors have improved visible region absorption of the solar spectrum.25 Among these techniques, TiO2 coupling with visible light active photocatalytic systems not only increases the sunlight harvesting ability but also improves the separation and migration of photogenerated charge carriers.26 Talking about visible light active semiconductors, cadmium sulphide (CdS) has been preferred over various photocatalysts due to its excellent visible light absorption ability and sufficient band potentials suitable for proton reduction.27,28 The incorporation of CdS with TiO2 has been reported for its successful activation in visible light for catalysing dye degradation,29 H2 generation30 and oxidation of organic molecules,31 whereas no report has been published on its applicability for biomass-derived substrate hydrogenation.
Therefore, in this work we have modified P25 TiO2 nanoparticles by making its heterojunction with CdS based nanostructures. The resulting interfacial contact has facilitated the photogenerated electron migration (e−) from the CdS surface towards the active catalytic center of TiO2, hence boosting its hydrogenation activity even under visible light. The TiO2–CdS nanocomposite has successfully upgraded useful biomass-based platform molecules, i.e. HMF and FFaL, to their hydrogenated products BHMF and furfuryl alcohol (FFoL), respectively, with the synergistic production of vanillin (Van) from vanillyl alcohol (VanOL) utilizing photogenerated holes (h+) of CdS in a more selective manner for the first time. The collected photocatalyzed products present vast application potential in various industries ranging from polymers to pharmaceuticals, fibers and flavors.32–34 Further to our extended efforts for effective utilization of e− and h+ in a photocatalytic system, we were able to show the effective H2 generation along with reduced and oxidized products with the robust photocatalytic TiO2–CdS nanocomposites under visible light. Therefore the present work demonstrates a new photo-refinery system for the generation of fuels and chemicals providing a sustainable system integrated with renewable energy utilization.
Results and discussion
The CdS nanorods were synthesized via a solvothermal route and further integrated with Aeroxide P25 TiO2 nanoparticles via oppositely charge assisted coulombic interactions as CdS has a negatively35 and TiO2 has a positively charged surface.36 The detailed synthesis procedure is provided in the ESI.† For the heterostructures, three different weight ratios, i.e. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1 and 1:2, of P25 TiO2 and CdS were synthesized and the superior photocatalytic activity observed with P25 TiO2 (2) : CdS (1) labelled as TiO2–CdS was further characterized in detail.
:1, 1:1 and 1:2, of P25 TiO2 and CdS were synthesized and the superior photocatalytic activity observed with P25 TiO2 (2) : CdS (1) labelled as TiO2–CdS was further characterized in detail.
The crystallographic phase examination of CdS, P25 TiO2 and TiO2–CdS was done using powder X-ray diffraction (PXRD) pattern measurements (Fig. 1a). PXRD patterns confirmed the formation of hexagonal phase in CdS nano rods (JCPDS #41-1049).37 The presence of multiple phases, i.e. anatase (JCPDS #21-1272) and rutile (JCPDS #21-1276), in the P25 TiO2 nanoparticles was also confirmed with crystallographic measurements.38 In addition there exists a broad peak in the range 17°–35° (2θ) that relates to the amorphous phase in TiO2. The presence of diffraction planes corresponding to TiO2 and CdS in all three ratios of TiO2–CdS, i.e. (2:1), (1:1) and (1:2), confirms the successful formation of a heterostructure (Fig. S1, ESI†).
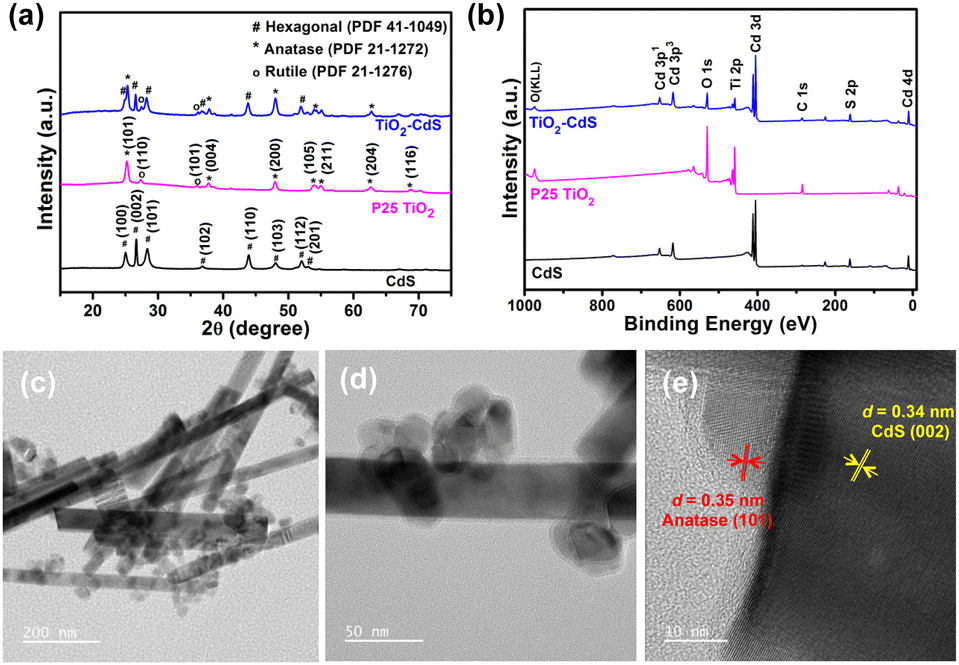 | ||
| Fig. 1 (a) PXRD patterns and (b) XPS survey spectra of CdS, P25 TiO2 and TiO2–CdS; (c) and (d) TEM images of the TiO2–CdS nanocomposite having nanoparticles of P25 TiO2 covered CdS nanorods (magnification scale of 200 nm in image (c) and 50 nm in image (d)); and (e) HRTEM image of TiO2–CdS with 10 nm magnification scale. | ||
The composite heterojunction formation was further supported by X-ray photoelectron spectroscopy (XPS) analysis. Fig. 1b presents the XPS survey scan of individual systems and the TiO2–CdS heterostructure with binding energy corresponding to elements Cd, S, Ti, and O. The morphology and nanostructures of CdS, TiO2 and TiO2–CdS were analyzed through FESEM, TEM and HRTEM. Fig. S2a (ESI†) displays the FESEM image of CdS with rod-like structures and nanoparticles of TiO2 can be observed in Fig. S2b (ESI†). In Fig. S2c (ESI†), the presence of both nanostructures with respective morphologies can be observed clearly in the nanocomposite. Further analysis of the TiO2–CdS nanocomposite by TEM reveals the uniform distribution of TiO2 nanoparticles over the CdS nanorods (Fig. 1c and d). The high-resolution TEM (HRTEM) image (Fig. 1e) clearly shows the intimate contact between the two nanostructures with lattice fringes of 0.34 nm and 0.35 nm attributed to the (002) and (101) diffraction planes of hexagonal CdS and anatase TiO2, respectively.38,39 The nanocomposite formation between P25 TiO2 and CdS was further validated from TEM-EDAX analysis. Fig. S3 (ESI†) shows TEM images of the nanocomposite and the selected area elemental mapping with Ti, O, Cd, and S elements present in the TiO2–CdS heterostructure.
The CdS, TiO2 and TiO2–CdS systems were further analyzed for their optical characteristics by UV-vis absorption spectroscopy that shows TiO2 absorbance in the UV region, whereas CdS and TiO2–CdS have absorbance in the visible region (Fig. 2a). To study the photocatalytic behaviour of a semiconductor based catalyst for organic transformations, the band structure with comparable band potential values is required. In order to determine the band potentials, different characterization techniques were adopted. From the Tauc plot (Fig. 2b) measured from UV-vis absorption data, optical band gap (Eg) values of 2.4 eV and 3.2 eV were calculated for CdS and TiO2, respectively. From valence-band XPS (Fig. 2c, d), the valence band (EVB) potentials of 1.28 eV and 2.5 eV were calculated for CdS and TiO2, respectively. The conduction band (ECB) potential values were calculated using the relation ECB = EVB − Eg, and estimated to be −1.12 eV and −0.70 eV for CdS and TiO2, respectively. Fig. 2e depicts the band potential diagram of the synthesized CdS nanorods and P25 TiO2 nanoparticles with sufficient negative potential of photogenerated electrons suitable to catalyse reduction reactions.
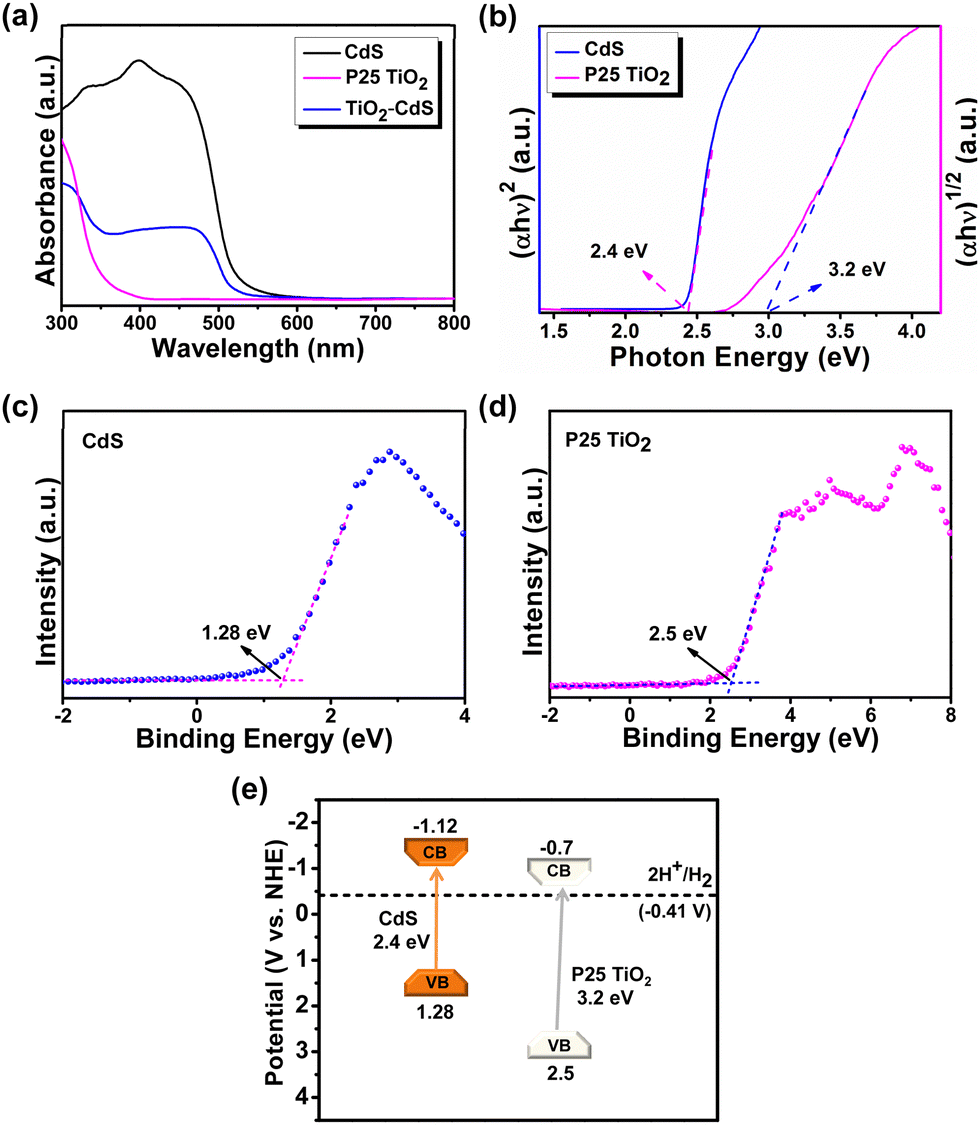 | ||
| Fig. 2 (a) Solid state DRS UV-vis absorption spectra of CdS, TiO2 and TiO2–CdS; (b) Tauc plot of CdS and P25 TiO2; valence band XPS of (c) CdS and (d) P25 TiO2; and (e) band energy diagram of CdS and P25 TiO2. | ||
The change in electronic structure for the TiO2–CdS heterostructure was further analysed through high-resolution XPS measurements. Fig. S4a and b (ESI†) compare Cd 3d and S 2p based high-resolution XPS for CdS and TiO2–CdS where a prominent shift in binding energy towards higher values indicates decreased electron density in CdS at the heterojunction with P25 TiO2.38,40 Meanwhile, a slight negative shift in binding energy for Ti 2p and O 1s was observed after the heterojunction formation (Fig. S4c and d, ESI†). These results suggest the existence of electronic interactions at the interface with electron transfer from CdS to TiO2 upon hybridization in TiO2–CdS.
Furthermore, the better charge mobility in TiO2–CdS was evaluated from EIS Nyquist plots shown in Fig. S5a (ESI†). The EIS curves reflect a smaller arc radius for TiO2–CdS compared to CdS and TiO2 demonstrating lower resistance and thus higher charge transfer efficiency in the nanocomposite.41 The Randles circuit was fitted (Fig. S5a inset, ESI†) with components Rs, Rp and CPE representing bulk solution resistance, charge-transfer resistance and double-layer capacitance, respectively. The improved charge carrier separation under light was investigated by photoelectrochemical characterizations where the highest photocurrent was observed for TiO2–CdS compared to bare CdS and TiO2 (Fig. S5b, ESI†). In addition, higher current density was recorded for the heterostructure under light irradiation as recorded in the linear scan voltammetry (LSV), as shown in Fig. S5c (ESI†).
To further elucidate the enhanced charge separation in the TiO2–CdS heterostructure, an extensive spectroscopic analysis was conducted using photoluminescence (PL), time-resolved PL, and transient absorption (TA) spectroscopy. Fig. 3a shows the PL spectra of CdS, P25 TiO2 and TiO2–CdS after the photoexcitation at 440 nm. The PL spectrum of CdS exhibits two luminescent features. First, a narrow peak around 550 nm originated due to the recombination of the band edge charge carriers. Second, a broad and highly intense peak in the red region of the spectra assigned to the trap-mediated recombination.42 On the other side, P25 TiO2 exhibits a broad PL band having a peak maximum around 530 nm, which is attributed to the recombination of electrons of the conduction band (CB) and holes of the trap state near the valence band (VB).43 Fig. S6a and b (ESI†) schematically illustrates the origin of different types of PL in both CdS and P25 TiO2, respectively. For the TiO2–CdS heterostructure, a significant reduction in the PL intensity of TiO2 was observed. This is possibly due to the migration of the holes from the TiO2 trap states to CdS VB, as shown in Scheme 1. In addition to that, the intensity of CdS trap PL was also observed to be decreased in the heterostructure as compared to bare CdS, which suggests the transfer of the trapped electron from CdS to TiO2 CB. Next, to explore the charge transfer characteristics, we have utilized the time-correlated single photon counting (TCSPC) technique using 440 nm laser excitation. The average lifetime for the bare CdS and P25 TiO2 was calculated to be ∼15.67 ns and ∼10.18 ns, respectively, by fitting their PL decay traces (Fig. 3b) in the tri-exponential decay functions (Table S1, ESI†). The average lifetime for the TiO2–CdS heterostructure (18.54 ns) was found to be longer than both of the bare systems. Owing to the type II band alignment, the photo-generated electrons and holes would migrate to the opposite direction at the interface, i.e., electron towards TiO2 and hole towards CdS, enhancing the charge separation lifetime and reducing the recombination.
 | ||
| Fig. 3 (a) Photoluminescence (PL) spectra and (b) time resolved decay traces of CdS, P25 TiO2 and TiO2–CdS after the photoexcitation of 440 nm. (c) Transient absorption (TA) spectra of CdS at different pump probe time delays after the photoexcitation of 440 nm. (d) Comparative TA kinetics of CdS monitored at 500 nm and 590 nm. (e) TA spectra of TiO2–CdS at different pump probe time delays after the photoexcitation of 440 nm. (f) Comparative TA kinetics of CdS and TiO2–CdS monitored at the probe wavelength of 500 nm and categorized into the four time domains: (I) growth time range of both CdS and TiO2–CdS (up to 1 ps), (II) short decay time scale (1–5 ps), (III) tens of ps time scale (5–100 ps), and (IV) the longest time decay range (100–5000 ps). | ||
 | ||
| Scheme 1 Schematic representation of the charge transfer processes at the interface of CdS and TiO2; (1) hot electron transfer, (2) band edge electron transfer, (3) trap state electron transfer from CdS to TiO2, and (4) trap state hole transfer from TiO2 to CdS. | ||
It is well-known that the excited state charge carrier dynamics play a crucial role in heterostructure-based photocatalytic applications.44 In this manner, TA spectroscopy has emerged as a powerful and versatile tool to monitor the carrier dynamics at the interface of the heterostructure. Henceforth, we have performed ultrafast transient measurements by exciting the samples with a visible pump laser of wavelength 440 nm to explore the interfacial charge carrier dynamics in the TiO2–CdS heterostructure. Fig. 3c shows the TA spectra of CdS, probing in the visible region spanning a broad time range (from 0.1 ps to 5 ns). The TA spectra of CdS exhibit a characteristic photoinduced bleach signal peaking at 500 nm, which is assigned to state filling of the conduction band edge 1σ states (1Σ transition) of CdS.42 As the effective mass of holes is much higher than that of electrons in CdS and the VB possesses a higher density of states, the bleach signal is largely contributed by electrons.45 Apart from the characteristic strong bleach (1Σ transition), a weak bleach feature is also observed in the red region of the spectra, which is due to the filling of inter-bandgap states with photo-excited electrons.46 Furthermore, to evaluate the temporal population and depopulation of the band edge (BE) as well as trap states (TS), the TA kinetics were monitored at their respective bleach positions (for BE: 500 nm and for TS: 590 nm) (Fig. 3d). The faster growth, as well as recovery of the TS bleach signal as compared to the BE bleach signal was ascribed to the faster trapping and de-trapping of electrons in these trap states, which confirms their shallow nature.47
Next, to understand the charge transport mechanism at the interface of CdS and TiO2, we carried out transient investigation towards the TiO2–CdS heterostructure. The TA spectra of TiO2–CdS (Fig. 3e) exhibit a similar spectral shape and peak maxima as that of bare CdS, which implies that the CdS nanorods maintain a similar size and shape in the heterostructure. However, its TA kinetics were found to be different, as shown in Fig. 3f. To explain the charge migration dynamics at the interface, we have categorized this comparative TA kinetics in four-time domain regions; (I) growth time range of both CdS and TiO2–CdS (up to 1 ps), (II) short decay time scale (1–5 ps), (III) tens of ps time scale (5–100 ps), and (IV) the longest time decay range (100–5000 ps). However, a visible region photoexcitation (pump energy 2.82 eV; 440 nm) can only excite the CdS electrons from VB to the higher energy CB states in the heterostructure as TiO2 has a larger band gap (3.2 eV). (I) In the pristine CdS system, the higher energy CB state electrons slowly cooled down to the CB edge state with a time constant of ∼0.30 ps (Table S2, ESI†). In the case of the heterostructure, we observed a faster growth of the band edge signal, which was ascribed as the hot electron transfer (HET) from the higher energy CB states to the TiO2 with the HET rate (kHET) of ∼6.67 ps−1 (τHET ∼0.15 ps). (II) In the bare CdS, the band edge electrons were further trapped in the mid-band gap trap states with a time constant of ∼1.69 ps (Table S2, ESI†). But in the presence of TiO2, the faster decay of the CdS band edge signal was observed, which validates the band edge electron transfer (BET) from CdS to TiO2 with the transfer rate (kBET) of ∼0.41 ps−1 (τBET ∼ 2.44 ps); detailed calculations are provided in the ESI† file. (III) Next, the tens of ps time scale kinetics of bare CdS with the time constant of 28 ps (Table S2, ESI†) was assigned to the transport of electrons in the trap state of CdS.47 For the heterostructure, it was found to be faster than bare CdS, which suggests the possibility of migration of the trap state electrons to the TiO2 CB edge. This was further validated by the faster decay kinetics observed for CdS trap state electrons (590 nm) in the presence of TiO2, as shown in Fig. S6b (ESI†). (IV) Finally, the longest time scale (100–5000 ps) of the TA kinetics reflects the recombination dynamics of the photocatalyst. This time domain kinetics showed the opposite behaviour (slower kinetics) to the earlier time region kinetics for the heterostructure, which is also reflected in the higher average lifetime of the heterostructure than bare CdS in TCSPC analysis (Table S1, ESI†). The possible reason for this is that the hot, band edge and trapped electron transfer from CdS to TiO2 collectively slow down the recombination process in the CdS region and enhance the electron population in the TiO2 CB. Overall, our detailed spectroscopic investigation concludes that the collective contribution of all four charge transfer processes (Scheme 1): (1) hot electron transfer from high energy CB states of CdS to TiO2 CB states, (2) band edge electron transfer from CB edge of CdS to TiO2 CB edge, (3) electron transfer from CdS trap states to TiO2 CB edge and (4) trapped hole transfer from TiO2 trap states to CdS VB edge, enhanced the charge separation at the interface of CdS and TiO2, which would be beneficial in the visible light-based biomass conversion by the heterostructure.
After characterizing the semiconducting systems, the photocatalysts were scrutinized to perform biomass-derived platform chemical upgradation to valuable products (Table 1). In this work, we have carried out the photocatalytic hydrogenation of various biomass-derived platform chemicals to their upgraded products having industrial value, without the aid of external H2 as a reducing agent. Generally, the hydrogenation of biomass derived platform chemical requires external H2 gas as a hydrogenating agent that in turn increases the operational cost of the process. However, the photocatalytic oxidation of HMF coupled with H2 production results in a liquid phase oxidized product and gas phase reduced product that have been reported recently.48,49 Here, an initial report on P25 TiO2-based hydrogenation of HMF coupled with biomass-based substrate oxidation generating two biomass-derived organic products along with H2 is presented, unveiling a chemically economical and futuristic approach. The photocatalytic activity of P25 TiO2 has been improved dramatically under visible light by incorporating CdS as a narrow band-gap based semiconductor. The maximum BHMF yield of 95% along with 15.6 μmol g−1 H2 simultaneously with 35% vanillin (Table 1, entry 6) was recorded even without any reducing agents. The inactivity of bare CdS and P25 TiO2 to catalyse HMF hydrogenation under visible light (Table 1, entries 1 & 2) highlights the importance of both the systems i.e. CdS for redox active photogenerated excitons and TiO2 for providing active hydrogenation sites supported by the Lewis acidic surface.50 The importance of the TiO2–CdS heterostructure as a visible light active photocatalyst was further verified by the reaction carried out under controlled conditions i.e. without catalyst (Table 1, entry 7) and in the dark (Table 1, entry 8).
| Entry | Catalyst | Substrates | BHMF yield (%) | Vanillin yield (%) | H2 (μmol g−1) |
|---|---|---|---|---|---|
| Reaction conditions: catalyst: 10 mg; HMF: 50 μmol & VanOL: 250 μmol; ACN: 5 mL; time: 6 h; 100 mW cm−2 intensity: (>420 nm) xenon lamp; 25 °C.a Time: 12 h.b Dark.c H2 atmosphere.d No VanOL.e No HMF.f Sunlight, 31 °C: 50 mW cm−2 intensity: conversion (HMF = 87% VanOL = 29%).g Reactant = furfural (FFaL): product = furfuryl alcohol (FFoL).h Reactant = diformylfuran (DFF): product = hydroxymethylfurfural (HMF).i Mechanical mixture of TiO2 and CdS; ND = not detected; and NA = not applicable. | |||||
| 1 | CdS | HMF:VanOL | ND | ND | ND |
| 2 | P25 TiO2 | HMF:VanOL | ND | ND | ND |
| 3 | TiO2–CdS (2:1) |
HMF:VanOL | 58 | 17 | 3.3 |
| 4 | TiO2–CdS (1:1) |
HMF:VanOL | 44 | 22 | 7.34 |
| 5 | TiO2–CdS (1:2) |
HMF:VanOL | 36 | 24 | 15 |
| 6a | TiO2–CdS (2:1) |
HMF:VanOL | 95 | 35 | 15.6 |
| 7 | Without catalyst | HMF:VanOL | ND | ND | ND |
| 8b | TiO2–CdS | HMF:VanOL | ND | ND | ND |
| 9c | TiO2–CdS | HMF:VanOL | 53 | 13 | NA |
| 10d | TiO2–CdS | HMF | ND | NA | ND |
| 11e | TiO2–CdS | VanOL | NA | 22 | 23.6 |
| 12f | TiO2–CdS | HMF:VanOL | 64 | 26 | 2.4 |
| 13g | TiO2–CdS | FFaL:VanOL | 23 | 14 | 28 |
| 14h | TiO2–CdS | DFF:VanOL | 8 | 11 | ND |
| 15i | TiO2–CdS | HMF:VanOL | 12 | 12.4 | 2.6 |
Furthermore, to get insights into the reaction mechanism, the photocatalytic reactions were performed under H2 atmosphere, without hole scavenger (VanOL) and without HMF. The reduced activity under external H2 (Table 1, entry 9) signifies the idle nature of molecular hydrogen for HMF hydrogenation and further emphasizes the valued contribution of in situ generated protons and electrons as a hydrogenation source. Additionally, the efficient contribution of biomass-based alcohol, i.e. vanillyl alcohol, as a hole scavenger to produce protons by undergoing oxidation over photogenerated holes was verified as no BHMF yield was recorded for reaction without VanOL (Table 1, entry 10). However, vanillin production was recorded along with an increased amount of H2 generation in the absence of HMF, which shows the utilization of photogenerated charge carriers i.e. holes (h+) and electrons (e−) for VanOL oxidation and proton reduction to molecular H2, respectively (Table 1, entry 11). Further testing the photocatalytic efficiency of the present catalytic system, reaction was performed under natural sunlight (Table 1, entry 12). The increased product yield as well as higher turnover number extends the efficient utilization of solar energy as an environmentally sustainable approach for green chemical production. In order to study the wide applicability of the present heterostructure for biomass-derived substrate hydrogenation, two other biomass-derived substrates were tested, i.e. furfural (FFaL) and diformylfuran (DFF). The successful hydrogenation of FFaL generated furfuryl alcohol (FFoL) and DFF yielded hydroxymethylfurfural (HMF) by reducing one carbonyl group (Table 1, entries 13 and 14). In addition, the photocatalytic activity of the TiO2 and CdS mixture obtained after simple physical mixing was also carried out (Table 1, entry 15). The noticeable product formation further supports the existence of electrostatic interactions between oppositely charged surfaces of individual semiconducting systems. Also sufficient vanillin production was recorded simultaneously with the hydrogenated products projecting the practical relevance of the current nanocomposite system.
On the basis of charge migration study and reactions carried out under controlled conditions, a plausible mechanistic pathway was proposed (Scheme 2). Under visible light irradiation, charge carriers (h+ and e−) are induced on the CdS surface (Eg = 2.4 eV) where positively charged holes initiate the oxidation of surface adsorbed vanillyl alcohol (VanOL) with successive deprotonation to generate vanillin and protons (H+) via a two-electron transfer (TET) process.51 The photogenerated electrons are excited to a higher potential conduction band edge (−1.12 eV), which can migrate to the lower potential conduction band of P25 TiO2 (−0.7 eV) through a semiconductor − metal like interfacial Schottky junction.52 The advanced charge carrier separation in CdS after interface formation has already been furnished by electrochemical, photoelectrochemical and spectroscopic (XPS, TAS) studies as discussed above. Finally, the negatively charged electrons transferred to the P25 TiO2 surface transform HMF into BHMF via a proton-coupled electron transfer process (PCET) utilizing in situ generated protons (H+).17 The puzzle over the selective hydrogenation of only HMF and oxidation of only VanOL was investigated using electrochemical measurements. Fig. S8a and b (ESI†) show the cyclic voltammetry profiles with marked on-set potentials of HMF and VanOL oxidation and the higher oxidation potential value (1.9 V) for HMF advocates the preferred VanOL oxidation over HMF.
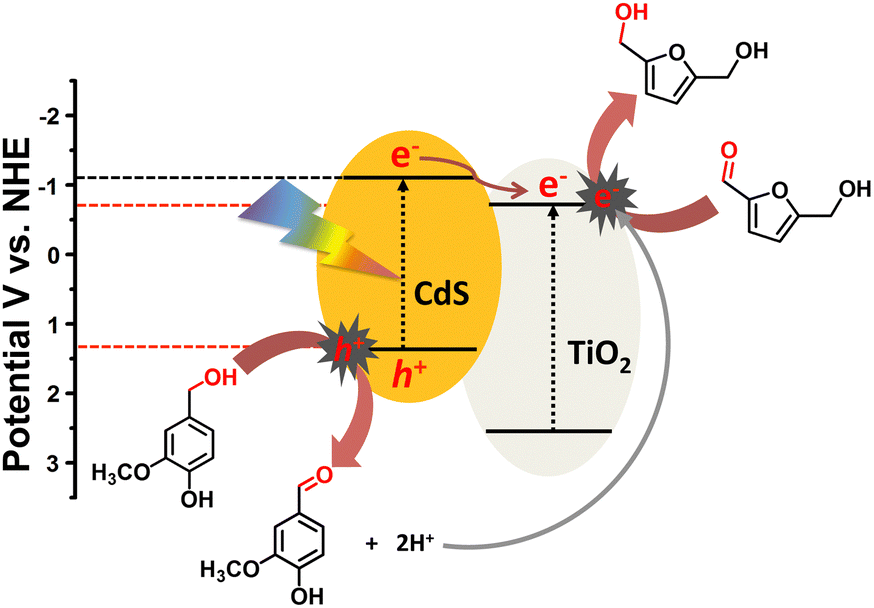 | ||
| Scheme 2 Schematic of a plausible mechanism followed for the oxidation of VanOL and reductive hydrogenation of HMF over TiO2–CdS under visible light. | ||
After thorough investigation of the catalytic activity and the underlying mechanism, the reproducibility study of the TiO2–CdS heterostructure was carried out for four cycles. Fig. 4 shows good photocatalytic performance of the present heterostructure after each catalytic cycle, signifying its stable and reproducible nature for simultaneous organic conversions under visible light. However, a slight decrease in product yield can be assigned to slight loss of the photocatalyst during washing after each cycle or adsorption of impurities during the reaction. Furthermore, the physical characterizations of the recovered photocatalyst were performed using PXRD and XPS techniques. Fig. S9a and b (ESI†) show similar diffraction patterns as well as overlapping XPS spectra for fresh and recycled TiO2–CdS that further support the extension of the present catalytic system as a potential candidate for possible large-scale applications.
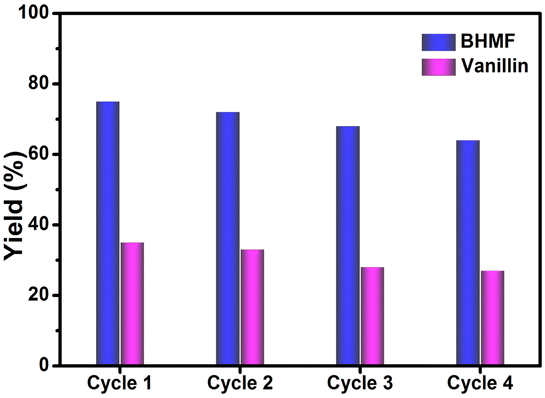 | ||
| Fig. 4 Recyclability study of the TiO2–CdS photocatalyst. Reaction conditions: catalyst, 100 mg: HMF, 50 μmol: VanOL, 250 μmol: ACN, 25 mL: time, 6 h: 100 mW cm−2 intensity, (>420 nm) xenon lamp: and 25 °C. | ||
Conclusions
In summary, an intimate interfacial contact between nanorods of CdS and nanoparticles of P25 TiO2 was fabricated by an electrostatic self-assembly strategy. The effective photogenerated electron transfer from excited CdS towards P25 TiO2 in a type-II manner significantly inhibits the recombination of charged carriers and further enhances its lifetime. Consequently, visible light driven selective hydrogenation of biomass-derived platform molecules has been achieved for the first time over wide band-gap P25 TiO2 owing to migrated photogenerated electrons from narrow band-gap CdS towards multi-phasic titania. The synergistic application of photogenerated charge carriers and in situ generated protons yielded superior photocatalytic activity of 95% BHMF, simultaneously with 35% vanillin and H2 over the P25 TiO2:CdS (2:1 by weight) heterostructure for the first time. The present dual-functional TiO2–CdS nanocomposite thus advanced previously reported photocatalytic systems to perform selective redox transformations without needing expensive co-catalysts, extra additives or additional reducing agents. In addition, recyclability studies up to four catalytic cycles showed excellent photostability implying its large-scale viability. Overall, the proposed approach put forward a comprehensive study to perform organic transformations over inexpensive catalytic systems utilizing renewable energy sources. Thus it offers an economical and sustainable route of synergistic production of value-added chemicals without additional chemical squander.
Author contributions
Arpna Jaryal: synthesis, investigations, methodology, data curation, writing – original draft. Ajit Kumar Singh: synthesis, data validation. Shivali Dhingra: analysis and validation of experiments. Himanshu Bhatt and Manvi Sachdeva: TAS investigations, methodology, data curation, writing. Hirendra N. Ghosh: conceptualization, review. Arindam Indra: conceptualization, writing – review and editing. Kamalakannan Kailasam: conceptualization, supervision, writing – review and editing, funding acquisition. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Arpna Jaryal thanks CSIR, New Delhi for the fellowship under File No. 09/1129(0010)/2017-EMR-I. Shivali Dhingra thanks INST Mohali for the research fellowship. Ajit Kumar Singh acknowledges IIT (BHU) for providing a senior research fellowship. Himanshu Bhatt and Manvi Sachdeva thank Institute of Nano Science and Technology (INST), Mohali, India and CSIR, New Delhi, respectively, for providing the research fellowship. Dr Hirendra N. Ghosh is grateful to DST, Govt. of India for J C Bose Fellowship (JCB/2018/000047). Dr Arindam Indra acknowledges the financial support from DST-SERB, India, for the Core Research Grant (Grant no.: CRG/2023/002395). Dr Kamalakannan Kailasam thanks INST Mohali and Department of Science and Technology, India (DST) for the generous support.References
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- J. A. Melero, J. Iglesias and A. Garcia, Environ. Sci., 2012, 5, 7393–7420 CAS.
- Y. Xu, L. Wang, Z. Shi, N. Su, C. Li, Y. Huang, N. Huang, Y. Deng, H. Li, T. Ma, X. Y. Kong, W. Lin, Y. Zhou and L. Ye, Energy Environ. Sci., 2023, 16, 1531–1539 RSC.
- J. C. Colmenares, Curr. Opin. Green Sustainable Chem., 2019, 15, 38–46 CrossRef.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- H. Li, S. Yang, A. Riisager, A. Pandey, R. S. Sangwan, S. Saravanamurugan and R. Luque, Green Chem., 2016, 18, 5701–5735 RSC.
- R. J. Van Putten, J. C. Van Der Waals, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC.
- C. Li, J. Li, L. Qin, P. Yang and D. G. Vlachos, ACS Catal., 2021, 11, 11336–11359 CrossRef CAS.
- E. Baur and C. Neuweiler, Helv. Chim. Acta, 1927, 10, 901–907 CrossRef CAS.
- A. Jaryal, B. V. Rao and K. Kailasam, ChemSusChem, 2022, e202200430 CrossRef CAS PubMed.
- N. A. Endot, R. Junid and M. S. S. Jamil, Molecules, 2021, 26, 6848 CrossRef CAS PubMed.
- S. A. F. Cavani and A. G. F. Basile, Chemicals and Fuels from Bio-Based Building Blocks, Wiley, Hoboken, 2016 Search PubMed.
- Y. Guo and J. Chen, RSC Adv., 2016, 6, 101968 RSC.
- S. Dong, Z. Liu, R. Liu, L. Chen, J. Chen and Y. Xu, ACS Appl. Nano Mater., 2018, 1, 4247–4257 CrossRef CAS.
- S. Qiao, Y. Zhou, H. Hao, X. Liu, L. Zhang and W. Wang, Green Chem., 2019, 21, 6585–6589 RSC.
- G. Hu, Z. P. Huang, C. X. Hu, Z. Q. Zhang, R. T. Liu, X. Y. Li, L. Zhang, Q. Wang and H. L. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 8288–8294 CrossRef CAS.
- X. Liu, G. Zhu, X. Wang, X. Yuan, T. Lin and F. Huang, Adv. Energy Mater., 2016, 1600452 CrossRef.
- Z. Yin, Z. Wang, Y. Du, X. Qi, Y. Huang, C. Xue and H. Zhang, Adv. Mater., 2012, 24, 5374 CrossRef CAS PubMed.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987 CrossRef CAS PubMed.
- A. Khan, M. Goepel, A. Kubas, D. Lomot, W. Lisowski, D. Lisovytskiy, A. Nowicka, J. C. Colmenares and R. Glaser, ChemSusChem, 2021, 14, 1351 CrossRef CAS PubMed.
- C. Li, J. Li, L. Qin, P. Yang and D. G. Vlachos, ACS Catal., 2021, 11, 11336–11359 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- V. Etacheri, C. D. Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol. C: Photochem. Rev., 2015, 25, 1–29 CrossRef CAS.
- R. Shi, Z. Li, H. Yu, L. Shang, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu and T. Zhang, ChemSusChem, 2017, 10, 4650–4656 CrossRef CAS PubMed.
- L. Shang, B. Tong, H. Yu, G. I. N. Waterhouse, C. Zhou, Y. Zhao, M. Tahir, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1501241 CrossRef.
- A. K. Singh, A. Jaryal, S. K. Patel, D. Kumar, E. S. S. Iyer, K. Kailasam and A. Indra, J. Mater. Chem. A, 2023, 11, 16724–16733 RSC.
- W. Dong, F. Pan, L. Xu, M. Zheng, C. H. Sow, K. Wu, G. Q. Xu and W. Chen, Appl. Surf. Sci., 2015, 15, 279–286 CrossRef.
- H. Zhao, M. Wu, J. Liu, Z. Deng, Y. Li and B. L. Su, Appl. Catal., B, 2016, 184, 182–190 CrossRef CAS.
- S. Liu, N. Zhang, Z. R. Tang and Y. J. Xu, ACS Appl. Mater. Interfaces, 2012, 4, 6378–6385 CrossRef CAS PubMed.
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS.
- Y. R. Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef PubMed.
- D. Scholz, C. Aellig and I. Hermans, ChemSusChem, 2014, 7, 268–275 CrossRef CAS PubMed.
- Y. Wang, Z. Hu, W. Wang, H. He, L. Deng, Y. Zhang, J. Huang, N. Zhao, G. Yu and Y.-N. Liu, Chem. Sci., 2021, 12, 16065 RSC.
- Q. Tan, K. Li, Q. Li, Y. Ding, J. Fan, Z. Xu and K. Lv, Mater. Today Chem., 2022, 26, 101114 CrossRef CAS.
- Y. Wang, Z. Hu, W. Wang, H. He, L. Deng, Y. Zhang, J. Huang, N. Zhao, G. Yu and Y.-N. Liu, Chem. Sci., 2021, 12, 16065 RSC.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed.
- W. Dong, F. Pan, L. Xu, M. Zheng, C. Haur, S. Kai, W. Guo, Q. Xu and W. Chen, Appl. Surf. Sci., 2015, 349, 279–286 CrossRef CAS.
- D. Zeng, L. Xiao, W.-J. Ong, P. Wu, H. Zheng, Y. Chen and D.-L. Peng, ChemSusChem, 2017, 10, 4624–4631 CrossRef CAS PubMed.
- S. Zhang, H. Yang, H. Huang, H. Gao, X. Wang, R. Cao, J. Li, X. Xu and X. Wang, J. Mater. Chem. A, 2017, 5, 15913–15922 RSC.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jäckel, J. Feldmann, F. Würthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862–869 CrossRef CAS.
- F. J. Knorr and J. L. McHale, J. Phys. Chem. C, 2013, 117, 13654–13662 CrossRef CAS.
- K. L. Corp and C. W. Schlenker, J. Am. Chem. Soc., 2017, 139, 7904–7912 CrossRef CAS PubMed.
- K. Wu, H. Zhu, Z. Liu, W. R.-Córdoba and T. Lian, J. Am. Chem. Soc., 2012, 134, 10337–10340 CrossRef CAS PubMed.
- T. Goswami, H. Bhatt, D. K. Yadav and H. N. Ghosh, J. Phys. Chem. B, 2022, 126, 572–580 CAS.
- T. Goswami, H. Bhatt, D. K. Yadav, R. Saha, K. J. Babu and H. N. Ghosh, J. Chem. Phys., 2022, 156, 034704 CrossRef CAS PubMed.
- G. Han, Y.-H. Jin, R. A. Burgess, N. E. Dickenson, X.-M. Cao and Y. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CrossRef CAS PubMed.
- V. R. Battula, A. Jaryal and K. Kailasam, J. Mater. Chem. A, 2019, 7, 5643–5649 RSC.
- X. Liu, Y. Zhou, D. Zeng, H. Wang, S. Qiao, L. Zhang and W. Wang, ChemistrySelect, 2021, 6, 8074–8079 CrossRef CAS.
- T. Ohno, S. Izumi, K. Fujihara, Y. Masaki and M. Matsumura, J. Phys. Chem. B, 2000, 104, 6801–6803 CrossRef CAS.
- H. J. W. Li, H. Zhou, K. Chen, K. Liu, S. Li, K. Jiang, W. Zhang, Y. Xie, Z. Cao, H. Li, H. Liu, X. Xu, H. Pan, J. Hu, D. Tang, X. Qiu, J. Fu and M. Liu, Sol. RRL, 2019, 1900416 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedures and characterization techniques of the catalyst. PXRD patterns of different weight ratio TiO2–CdS heterostructures. FESEM images, TEM EDX mapping, high-resolution XPS, EIS, photocurrent of CdS, P25 TiO2 and TiO2–CdS, and LSV of TiO2–CdS under dark vs. light. TA kinetics of CdS and TiO2–CdS probed at 500 and 590 nm. Cyclic voltammetry profiles of organic substrates (HMF and VanOL). Recyclability studies up to 4 cycles, physical and chemical characterizations, i.e. PXRD and XPS measurements of the recovered catalyst. See DOI: https://doi.org/10.1039/d4ey00077c |
| This journal is © The Royal Society of Chemistry 2024 |