
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward understanding CO oxidation on high-entropy alloy electrocatalysts†
María Paula
Salinas-Quezada
a,
Jack K.
Pedersen
a,
Paula
Sebastián-Pascual
a,
Ib
Chorkendorff
b,
Krishanu
Biswas
c,
Jan
Rossmeisl
 *a and
María
Escudero-Escribano
*ade
*a and
María
Escudero-Escribano
*ade
aDepartment of Chemistry, Center for High Entropy Alloy Catalysis, University of Copenhagen, 2100 Copenhagen, Denmark. E-mail: maria.escudero@icn2.cat; jan.rossmeisl@chem.ku.dk
bDepartment of Physics, Surface Physics and Catalysis, Technical University of Denmark, 2800 Lyngby, Denmark
cDepartment of Materials Science and Engineering, Indian Institute of Technology Kanpur, 208016 Kanpur, India
dCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and Barcelona Institute of Science and Technology, UAB Campus, 08193 Bellaterra, Barcelona, Spain
eCatalan Institution for Research and Advanced Studies (ICREA), Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 24th April 2024
Abstract
Understanding the catalytic activity of high-entropy alloys (HEAs) toward the conversion of small molecules such as carbon monoxide (CO) can provide insight into their structure–property relations. The identification of specific descriptors that govern the CO oxidation on HEAs is crucial to design new materials with customized compositions and structures. Herein, we have rationally assessed the CO oxidation mechanism on an extended AgAuCuPdPt HEA electrocatalyst under an acidic electrolyte. We compare the HEA performance with respect to platinum (Pt), palladium (Pd), and gold (Au) monometallic surfaces for CO oxidation. We also evaluated the same reaction on a binary AuPd alloy and a quaternary AuCuPdPt polycrystalline alloy with the aim of understanding the surface composition effects of the HEA. To provide insights into the descriptors controlling the CO oxidation mechanism and overpotential of the different alloy chemistry, we have combined cyclic voltammetry, surface-sensitive characterisation techniques and density functional theory (DFT) simulations. We show that silver (Ag) can improve the catalytic oxidation of CO by perturbing the *OH adsorption energy of Pd, leading to a lower onset potential. Additionally, we observed that Au segregates on the surface and that Cu is not stable at high applied potentials after CO oxidation. We highlight that HEA electrocatalysts are a valuable platform for designing more active and selective electrocatalyst surfaces.
Broader contextIn the last decade, the field of catalysis has witnessed a paradigm shift with the emergence of high entropy alloys (HEAs) as a versatile platform for designing novel catalytic materials. HEAs are promising electrocatalysts for reactions including the electrochemical conversion of small molecules such as carbon monoxide (CO). Electrocatalytic CO oxidation is a key reaction in sustainable energy conversion technologies such as low-temperature fuel cells. Herein, we combine electrochemical methods, ex situ surface-sensitive characterisation and theoretical calculations to investigate CO oxidation on an extended AgAuCuPdPt HEA electrocatalyst. This combination reveals that Ag can enhance CO oxidation. Moreover, we observe Au segregation and Cu instability at high applied potentials after CO oxidation. Insights from our work could help to explore the potential of HEAs in the critical realm of electrocatalysis. This is in line with the broader goal of finding sustainable energy solutions and highlights the significant role that materials science can play in addressing pressing environmental, scientific, and technological challenges. |
1. Introduction
The development of a decarbonised economy based on renewable energy conversion and storage is a prospective solution for the green transition toward the replacement of fossil fuels by sustainable energy and clean technologies.1,2 Electrocatalysts are crucial in the efficient conversion of energy in fuel cells by converting chemical energy directly into electricity through electrochemical reactions.3–5 However, one major drawback of low-temperature fuel cells such as direct formic acid or direct alcohol fuel cells (DFAFC or DAFC, respectively) is the formation of CO during the oxidation reactions taking place at the fuel cell device (oxidation of formic acid, methanol or ethanol), which poisons the anode.6 The poisoning occurs when CO molecules bind tightly to the catalytically active sites, causing a decrease in the fuel cell performance.6,7 Pt has extensively been studied as an electrocatalyst for low-temperature fuel cells, and its high affinity for CO adsorption decreases its activity in low-potential alcohol oxidation.8,9 As a result, there is a renewed interest in finding and developing advanced electrocatalysts with higher CO tolerance.10,11 To design catalysts that actively oxidize CO, it is crucial to understand the mechanisms and descriptors that govern this reaction on different surface structures and compositions.The oxidation of CO on platinum-group metals has been extensively studied and is best described by a Langmuir–Hinshelwood (LH) mechanism proposed by Gilman.12 In acidic media, the co-adsorption of carbon monoxide COads and surface oxygen-containing species such as OHads, derived from water, react to form CO2, as is shown in the following equations:
| H2O + * ↔ *OH + H+ + e− |
| *CO + *OH → CO2 + H+ + e− + 2* |
Understanding the adsorption of *OH and not only of *CO on bimetallic and multimetallic alloys such as high-entropy alloys (HEAs), is key to designing more active and stable catalysts. This is because *OH is a key descriptor for the catalytic activity of CO oxidation that can be selectively tuned by tailoring the chemical environment in the surface alloy.24,25 Yeh et al.26 defined a HEA as an alloy that consists of at least five metals with molar fractions ranging from 5–35%. HEAs have been claimed to possess promising and tunable electrocatalytic properties due to the possibility of selectively tuning the binding site energies through rational combination of elements.27–31 HEAs differ from traditional alloys because they contain multiple elements, leading to an increased number of binding sites. This enhances the likelihood of an electrocatalytic active site being present, which can be achieved by adjusting the alloy chemistry.29,32 To understand the relationship between the surface structure and the electrocatalytic activity and selectivity of electrocatalysts, it is crucial to carry out benchmarking of the new surfaces with model-extended surfaces such as single-crystalline or polycrystalline electrodes, which have widely been characterized.33,34 From a fundamental perspective, studying the electrooxidation of CO on extended HEAs could provide a better understanding of the correlation between CO electrocatalysis and the active site distribution of the surface.
In this work, we rationally assess the oxidation of carbon monoxide on an extended AgAuCuPdPt HEA electrocatalyst. We have compared the electrocatalytic activity of the HEA with respect to monometallic surfaces (Pt, Pd, Au), bimetallic AuPd alloy, and the quaternary AuCuPdPt alloy in acidic media. These monometallic and alloy surfaces have been selected to assess the role or contribution of each metal present in the HEA towards the CO oxidation. To describe how the surface composition affects the CO oxidation mechanism and overpotential, we combine electrochemical measurements and surface-sensitive techniques with DFT calculations. Additionally, we investigate surface segregation after electrochemistry with ex situ X-ray photoelectron spectroscopy (XPS) and ion scattering spectroscopy (ISS), providing a detailed insight into the HEAs surface composition before and after the electrochemistry. This research provides a key to design new strategies and materials to selectively tune the *CO and *OH descriptors for active CO oxidation on metal alloys through the combination of multiple elements.
2. Experimental
2.1. Preparation of the extended high-entropy alloy electrocatalysts
The extended AgAuCuPdPt high-entropy alloy and AuCuPdPt quaternary alloy were synthesized by vacuum arc melting under a high-purity argon atmosphere, where the purity of the metals Cu, Ag, Au, Pt, and Pd was 99.99 atomic percent (at%), from Alfa Aesar, USA. More specific details on the synthesis process can be found in a previous work by Katiyar et al.352.2. Surface pretreatment
The working electrodes used for the electrochemical measurements were an extended HEA (AgAuCuPdPt; 5 × 5 mm), a quaternary alloy (AuCuPdPt; 5 × 5 mm), a polycrystalline Pd disk (Mateck, 99.999% purity; 5 × 3 mm), a polycrystalline Au disk (Mateck, 99.999% purity; 5 × 3 mm), a polycrystalline Pt disk (Mateck, 99.999% purity; 3 × 3 mm), and a PdAu alloy (Mateck, 99.999% purity; 5 × 3 mm). All the working electrodes have a geometric area of 0.1963 cm2 except for the Pt disk, which is 0.0707 cm2. Before the electrochemical measurements, the working electrodes were manually polished using alumina slurries (Struers, Germany) of progressively smaller particle sizes (1, 0.3, and 0.05 μm) to achieve a mirror-like finish. Subsequently, the working electrodes were cleaned in an ultrasonic bath with Milli-Q water (18.2 MΩ cm, TOC < 3 ppm) three times for 10 minutes. The working electrodes underwent an electrochemical pre-treatment cleaning process. In the case of Pt and Au, they were cycled from 0.05 to 1.3 V vs. RHE, while in the case of Pd, they were cycled from 0.2 to 1.3 V vs. RHE, at a rate of 500 mV s−1 in 0.1 M HClO4 until a stable cyclic voltammogram was observed. However, the AuPd, AuCuPdPt and AgAuCuPdPt electrodes were cycled from −0.05 to 1 V vs. RHE 500 mV s−1 in 0.1 M HClO4. The blank cyclic voltammograms of the HEA before and after the surface pre-treatment are shown in Fig. S3 of the ESI.†All glassware was cleaned by soaking them in a saturated potassium permanganate solution (KMnO4) overnight and rinsing it with Milli-Q water (18.2 MΩ cm, TOC < 3 ppm) and a solution consisting of a mixture of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 30% H2O2 and 96% H2SO4. These cleaning steps were followed by multiple heating runs and rinsing with Milli-Q water (18.2 MΩ cm, TOC < 3 ppm) to remove any sulphate traces.
:1 30% H2O2 and 96% H2SO4. These cleaning steps were followed by multiple heating runs and rinsing with Milli-Q water (18.2 MΩ cm, TOC < 3 ppm) to remove any sulphate traces.
2.3. Electrochemical measurements
All the experiments were carried out at room temperature. As electrolyte, we used 0.1 M HClO4 (Sigma Aldrich 70%, 99.999% trace metals basis), which was prepared with Milli-Q water (18.2 MΩ cm, TOC < 3 ppm). Argon (Airliquid, Ar ≥ 99.999%) was used to deoxygenate the solutions, and CO (Airliquid, 99.97%) was used to dose CO. The electrochemical experiments were carried out in a single-compartment three-electrode setup and were executed using a VSP-300 potentiostat (Bio-Logic instruments) controlled by a computer. The working electrodes were tested in a meniscus configuration; a Pt mesh was used as a counter electrode, and a reversible hydrogen electrode (RHE) was used as a reference electrode and placed in a Luggin capillary.Blank cyclic voltammograms were performed at 50 mV s−1 in an Ar-saturated 0.1 M HClO4 electrolyte. Prior to the CO oxidation measurements, the cell was bubbled with CO for 10 min while keeping the working electrodes at open circuit potential to make sure the cell environment was saturated with CO. Then, the catalytic measurements were recorded in a CO-saturated 0.1 M HClO4 electrolyte. All the CO oxidation cyclic voltammograms were carried out at 50 mV s−1.
2.4. Ion scattering spectroscopy (ISS) and X-ray photoelectron spectroscopy (XPS) analysis
ISS and XPS measurements were performed by a Theta Probe instrument (Thermo Scientific) using an Al anode X-ray source (Kα line = 1486.6 eV, pass energy 50 eV). The ISS base pressure was <5.9 × 10−9 mbar, and the ISS spectra were acquired with He+ ions at 1 keV energy. The scattering angle for ISS was 125°, and the incidence angle of the ion beam was 45°. Initially, 20 scans were performed in order to clean the surface from impurities collected during the transfer, but we only considered the fifth cycle. Each scan took approximately one minute. The XPS survey spectra are represented in Fig. S5 of the ESI.† The XPS spectra were collected with a pass energy of 50 eV with a step size of 0.1 eV and a dwell time of 50 ms for 50 scans. For the XPS data analysis, we used CasaXPS software.2.5. Density functional theory (DFT) simulations
DFT simulations were performed with the GPAW code (version 22.1.0)36,37 for obtaining energies and ASE (version 3.22.1)38 for manipulating and optimizing atomic geometries. Energies were calculated with the RPBE functional39 using periodic planewave wavefunctions with an energy cut-off of 400 eV. The periodically repeated surface slabs were constructed as (111) terminated face-centered cubic surfaces sized 3 × 2 atoms with 5 layers of atoms and 10 Å of vacuum above and below the slab. The three bottom layers were fixed during the geometry optimization to simulate a rigid bulk crystal. For alloys, disordered arrangements of the surface were simulated by selecting the elements at random. The lattice constant of the slabs was set as the average of the constituent elements of the top layer.40 The Brillouin zone of the surface slabs was sampled on a uniformly spaced grid of 4 × 6 × 1 k-points. Molecules were simulated in periodically repeated unit cells sized 25 × 25 × 25 Å. All structures were relaxed to a force below 0.03 eV Å−1. For ease of reproducibility, structures and scripts used in the simulations in the current work have been made publicly available at https://nano.ku.dk/english/research/theoretical-electrocatalysis/katladb/co-oxidation-on-agaucupdpt.3. Results and discussion
To understand the voltammetric fingerprint of the electrode–electrolyte interface, it is crucial to report the base cyclic voltammograms.4Fig. 1A shows the base cyclic voltammograms (CVs) of the extended AgAuCuPdPt HEA in contact with 0.1 M HClO4 under two different potential windows. In the top CV, the upper and lower potential limits are 1.0 V vs. RHE and 0.05 V vs. RHE, respectively. The blank CV shows two main voltammetric regions, the hydrogen adsorption/desorption region below <0.3 V vs. RHE and the double layer or pseudocapacitive region between 0.4 and 0.9 V vs. RHE. The CV profile for the extended AgAuCuPdPt HEA in Fig. 1A shows reduced charges in the hydrogen adsorption/desorption region compared to polycrystalline Pt. The charge for HEAs was 160 μC cm−2, whereas the charge reported for a monolayer on polycrystalline Pt is 210 μC cm−2, showing a lower coverage of adsorbates on the HEAs.41 By enlarging the potential limits of the CV to 1.3 V vs. RHE, new features in the anodic and cathodic regions appear, as at potentials more positive than 0.9 V vs. RHE, the oxide growth region appears. The cyclic voltammogram displays the voltammetric fingerprint of the HEA through its applied potential and surface-sensitive features, such as the hydrogen adsorption/desorption and the oxide regions that distinguish it from its individual metal components.42–45 Notably, the oxide formation on the HEA starts near 0.9 V vs. RHE, which in comparison with Pd or Pt, is a shift to higher potentials due to the influence of the other metals.46,47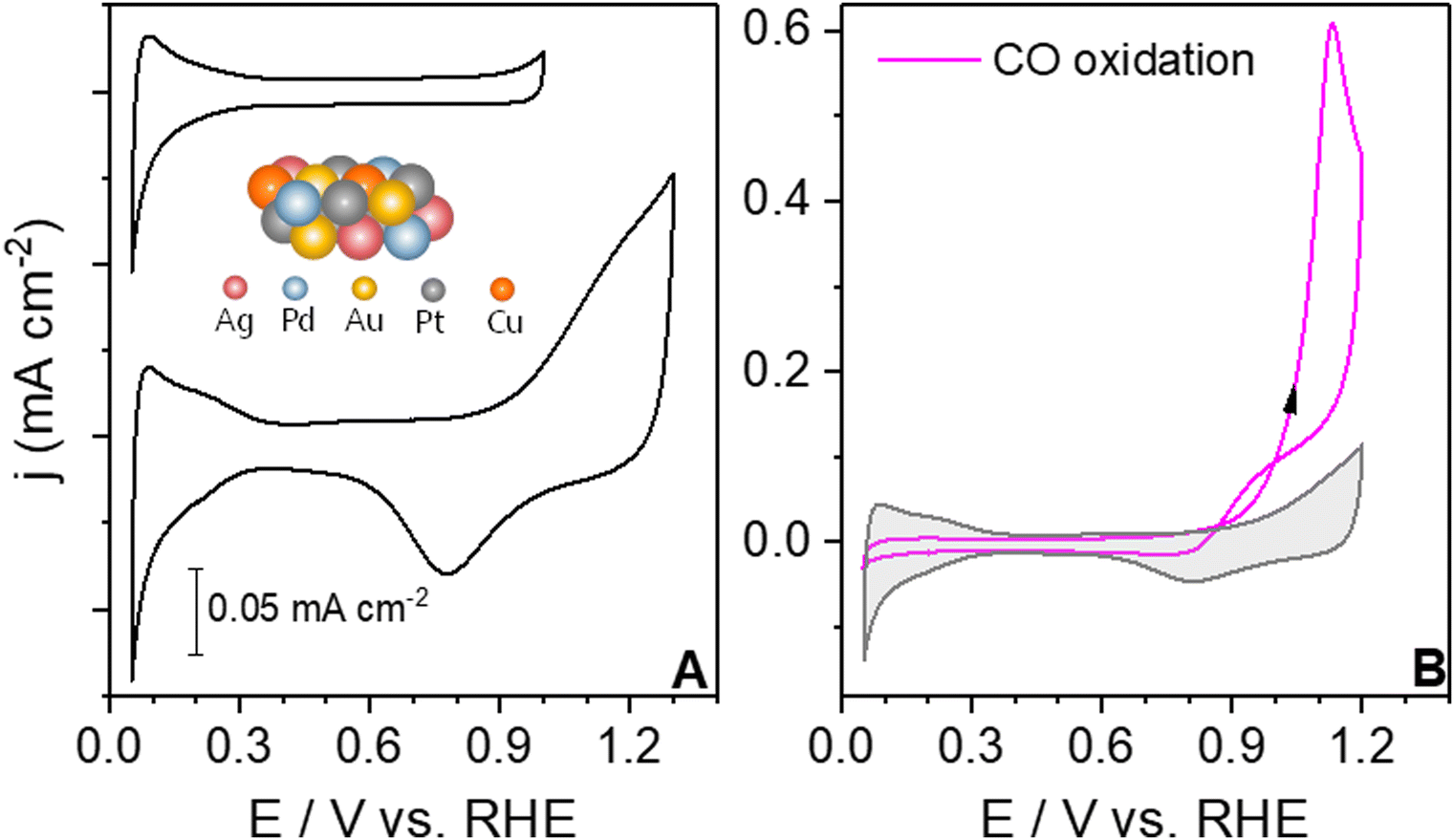 | ||
| Fig. 1 (A) Blank cyclic voltammograms of an extended AgAuCuPdPt HEA in contact with 0.1 M HClO4 saturated in an Ar atmosphere at 50 mV s−1 at two different potential windows. (B) The magenta solid line corresponds to the CO oxidation curve in CO-saturated atmosphere at 50 mV s−1 and the grey shadow to the blank cyclic voltammogram on the extended AgAuCuPdPt HEA. | ||
After ensuring the reproducibility of the blank CV, we carried out the CO oxidation experiments on the HEA. By studying the CO oxidation under mass-limited conditions, we aimed to identify the potential at which the absence of OH suppresses the CO oxidation according to the LH mechanism.12 Additionally, we have compared the various extended alloy surfaces with differing compositional surfaces under the same reaction conditions to gain insight into their CO oxidation reactivity. Fig. 1B shows the CO oxidation voltammogram for the extended AgAuCuPdPt HEA in 0.1 M HClO4. During the positive sweep, CO oxidation starts around 0.95 V vs. RHE, followed by a sharp increase in current. During the negative sweep, mass transport of CO still controls the process and then the current drops down at around 0.98 V vs. RHE, displaying a slight current hysteresis between the forward and backward scans. Below this potential, electrooxidation of CO on the surface ceases.
To shed some light on the parameters governing the overpotential for the CO oxidation reaction and understand how surface composition affects the CO oxidation mechanism, we analysed the CO oxidation reaction on monometallic polycrystalline Pd, Au and Pt electrodes in combination with DFT calculations. Fig. 2 shows both the blank CVs (A), (B), and (C) and CO oxidation curves (D), (E) and (F) on these three monometallic electrodes. The black dashed lines represent the equilibrium potentials obtained with DFT simulations. Additionally, the modelled onset, denoted as the modelled equilibrium potentials for *OH adsorption at high *CO coverage, is represented with vertical blue lines. The modelled extinction, which is the lowest potential at which the CO oxidation could theoretically be performed, is shown with vertical orange lines. The latter concepts will be discussed in detail in the final paragraphs. Fig. 2A displays a typical base cyclic voltammogram of a polycrystalline platinum electrode.46,48 The platinum cyclic voltammogram is measured between 0.05 and 1.0 V vs. RHE, and it is divided into three regions: the hydrogen and anion adsorption/desorption region, double layer, and oxide formation region. In the hydrogen and anion adsorption/desorption region, between 0.05–0.4 V vs. RHE, two prominent peaks are observed. The first peak at ca. 0.15 V vs. RHE is related to the presence of (110) sites, and the second one at ca. 0.3 V vs. RHE is attributed to (100) sites.49–52Fig. 2B shows the voltammetric profile for a polycrystalline Pd electrode recorded in the 0.2–1.2 V vs. RHE potential range. Three main features are revealed in the CV transient.47,53,54 These features correspond to the following electrode processes: from 0.2 to 0.4 V vs. RHE, the hydrogen adsorption/desorption region, from 0.45 to 0.65 V vs. RHE, the double layer region, and from 0.65 V to 1.2 V vs. RHE the surface oxide formation/reduction. The blank CV of polycrystalline Au appears in Fig. 2C, where the double-layer region, the oxide formation/reduction and hydrogen adsorption/desorption regions can be easily distinguished. Two characteristic peaks are observed in the monolayer oxidation region at 1.3 V and 1.45 V vs. RHE corresponding to the Au(100) and Au(110) faces, respectively.55,56Fig. 2C shows a dashed orange line representing the double layer zoomed in 15 times. A small voltammetric feature is observed near 0.5 V vs. RHE, which could be associated with some capacitive adsorption processes, such as the formation of surface-bonded hydroxyls.57
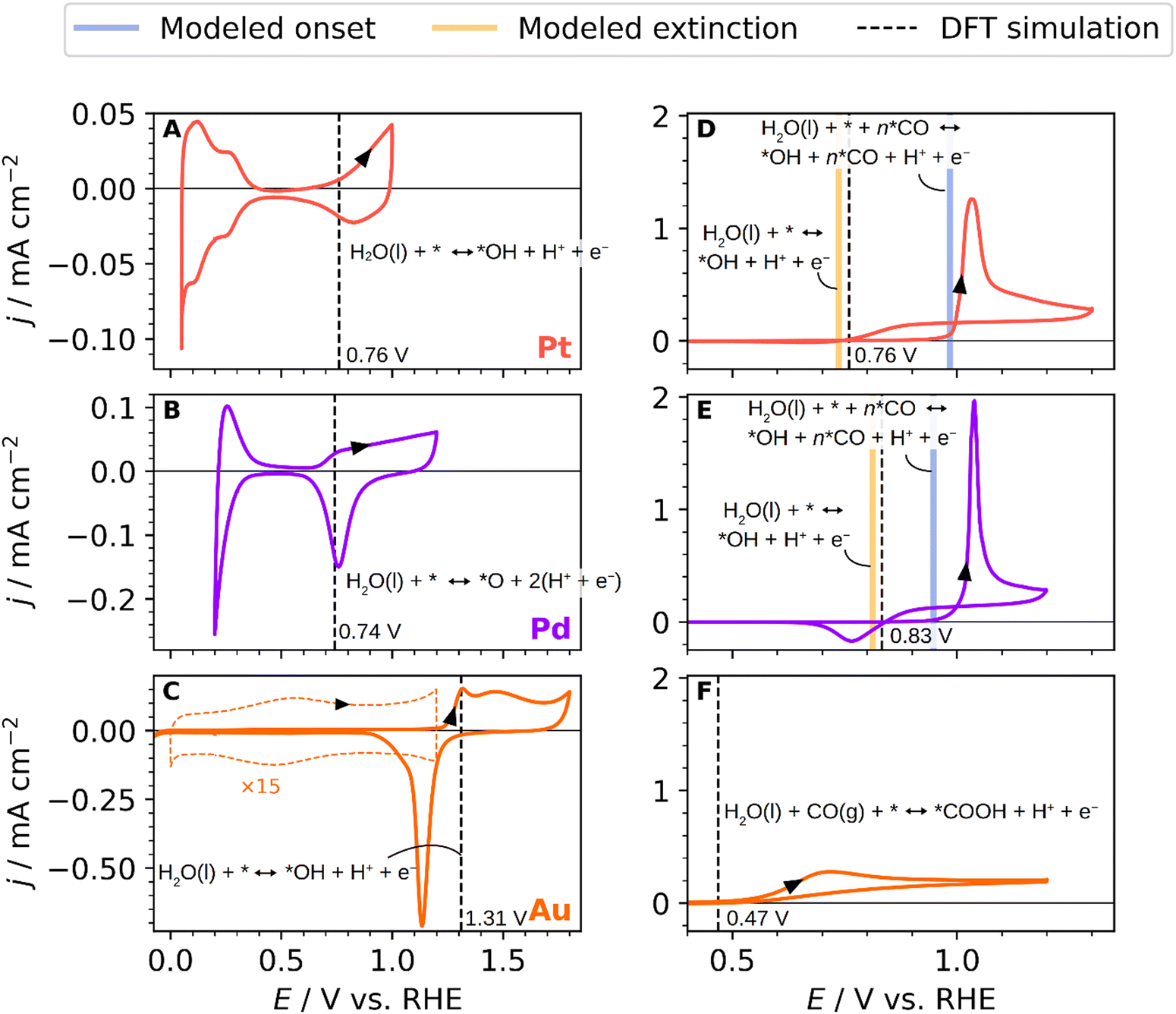 | ||
| Fig. 2 Blank stable cyclic voltammograms of (A) Pt (poly), (B) Pd (poly), and (C) Au (poly) in contact with 0.1 M HClO4 saturated in an Ar atmosphere at 50 mV s−1 and (D)–(F) corresponding CO oxidation curves in CO-saturated atmosphere at 50 mV s−1. Equilibrium potentials obtained with DFT simulations (black dashed lines) as well as modelled equilibrium potentials for *OH adsorption at low (vertical orange line) and high (vertical blue line) *CO coverage are shown. | ||
The CV profile for the extended AgAuCuPdPt HEA in Fig. 1A shows that the onset potential for the formation of oxygenates on the surface is observed near 0.9 V vs. RHE, that is at more positive potential than the onset potential of 0.7 V vs. RHE observed for Pt and Pd. We infer that these differences in the CV profile are due to the co-existence of alloying elements that affect the interactions between the surface and electrolyte, changing the energetics and coverage of the adsorbates on the surface.43,58
Fig. 2D–F show the continuous CO oxidation on Pt, Pd and Au polycrystalline electrodes. The voltammetric profile for the CO oxidation on the platinum surface is shown in Fig. 2D, displaying the qualitative characteristics of the curve predicted for the continuous CO oxidation model proposed by Koper et al.59 Starting from a low potential, the system follows the low current branch, where the onset of CO oxidation occurs at a higher potential near 1.0 V vs. RHE than the stripping peak of an adsorbed CO monolayer in the absence of CO in the solution, that starts around 0.8 V vs. RHE (see Fig. S1 of the ESI†).60 The sluggishness in the ongoing CO oxidation process can be attributed to the presence of adsorbed CO, which blocks the surface and hinders the bulk CO oxidation. In the reverse scan, and after stripping the CO from the surface, fresh active surface sites are generated, but they start to become occupied by CO molecules from the solution at potentials beyond 0.75 V vs. RHE.61,62 As shown in Fig. 2D, the onset of the CO adlayer oxidation starts around 1.0 V vs. RHE. The current settles down around 1.05 V vs. RHE where the system becomes diffusion-limited in CO. A flat current plateau can be observed during the negative reverse scan, suggesting that the system is still being controlled by mass transport.62 Afterwards, the current drops to zero at around 0.75 V vs. RHE, a potential value that we ascribe to the potential at which adsorbed OH formation ceases. Fig. 2E shows the CO oxidation reaction on Pd, which follows the same current transient behaviour as that on Pt. Unlike Pt, on Pd the CO oxidation current in the reverse scan drops down at higher potential values of 0.83 V vs. RHE, suggesting that the OH coverage on Pd, necessary to oxidize CO, also drops down at those potential values with subsequent surface poisoning by CO. This is surprising because Pd shows the presence of oxygen species at similar potentials as Pt at around 0.7 V vs. RHE. Using DFT simulations, we attribute the higher potential of extinction of the CO oxidation on Pd to the absence of *OH, while the observed oxidation potential in the blank CV is attributed to the more stably adsorbed *O. Additionally, in the reverse scan, we also record a negative current between 0.7–0.85 V vs. RHE, which we ascribe to the reduction of the adsorbed *O adatoms on the Pd surface54 or an anion displacement from the surface,61 meaning that oxides and *OH groups co-exist on the surface in the upper potential window of Pd.
The voltammetric profile for the continuous CO oxidation on Au is shown in Fig. 2F. The onset of the CO oxidation is kinetically controlled and starts around 0.5 V vs. RHE, and this potential value agrees with previous reports,63,64 which is followed by a maximum CO oxidation current density at 0.7 V vs. RHE. Compared with Pt and Pd, Au shows the lowest current for CO oxidation in the forward or anodic scan. This is due to the weak adsorption energy of CO on gold in acidic media,65i.e., CO does not poison the surface. However, the activity achieved is similar to the one achieved on Pt and Pd in the backward scan, i.e., under diffusion control, reaching a current value of 0.17 mA cm−2. The potential at which the CO current is suppressed on Au is considerably lower than on Pt and Pd, around 0.49 V vs. RHE. As OH-specific adsorption/desorption does not occur on gold at this potential value, this result indicates that CO oxidation follows a different mechanism than on Pt and Pd. On Pt and Pd, the specific adsorption or onset adsorption of *OH on the surface controls the potential window at which CO is oxidized in the backward or diffusion-limited region. Previous works have suggested that water or OH anions in solution react with CO to form CO2 at such low overpotential on gold.57,63,65,66 We find with DFT simulations, however, that the oxidative adsorption of the *COOH species could happen at a potential of 0.47 V vs. RHE as illustrated in Fig. 2F with the vertical, dashed line. This intermediate would then readily oxidize to CO2.
Aiming to address how the combination of different elements or surface compositions affects the CO oxidation reaction mechanism, we have also investigated the CO oxidation on three different alloys: AuPd, AuCuPdPt, and the extended AgAuCuPdPt HEA. The aim of these experiments is to gain a better understanding of the electrochemical response of AgAuCuPdPt HEAs. Au and Ag do not strongly bind CO. Thus, with this study, we aim to experimentally assess the influence of Au (when comparing pure Pd with AuPd) and Ag (when comparing AuCuPdPt with AgAuCuPdPt) on the surface *OH adsorption and its effect on the CO oxidation mechanism. Fig. 3A shows the blank CV of the AuPd alloy. The CV exhibits lower current density in the hydrogen adsorption/desorption region than pure Pd, and the potential region for the Pd hydride formation and formation of hydrogen is shifted to lower values, indicating a different arrangement of Pd atoms on the surface.67 The oxide region features appear at a higher potential than on pure Pd due to the influence of enriched Au atoms on the alloy surface.68 The voltammogram of the continuous oxidation of CO on the AuPd alloy is shown in Fig. 3D. The onset potential for CO oxidation starts near 1 V vs. RHE and displays a much smoother current increase compared to the sharp CO stripping peak of pure Pd in Fig. 2D. The maximum current is reached at about 1.2 V vs. RHE. The CO oxidation peak is broadened and shifted to more anodic potential values, in comparison with pure Pd, due to the increase of Au content on the surface.20 We suggest that the broadening of the CO oxidation peak is due to a lack of *OH sites that make it difficult to remove the CO or oxidize more CO unless a higher potential is applied.
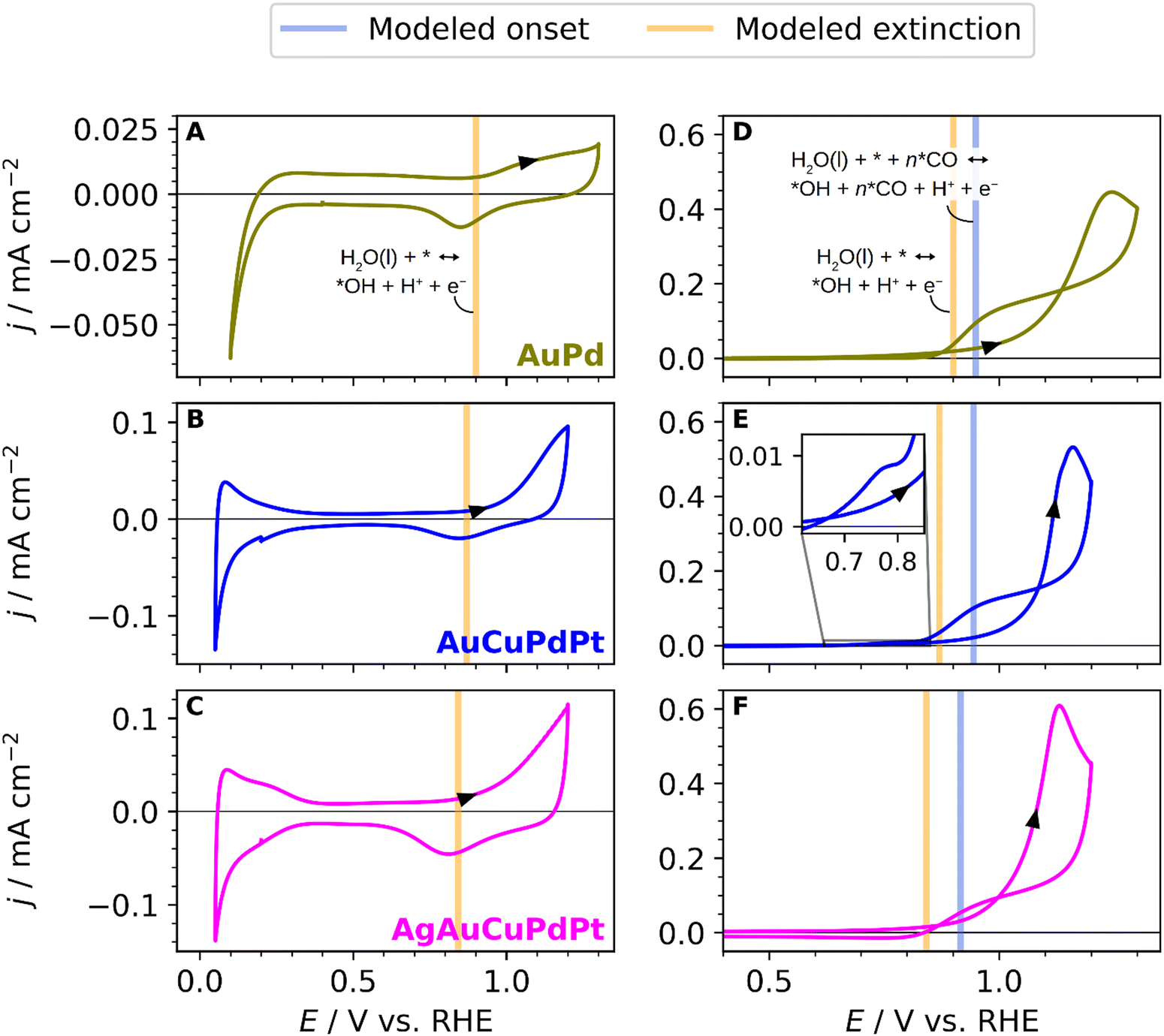 | ||
| Fig. 3 (A)–(C) Blank stable cyclic voltammograms of extended polycrystalline PdAu alloy, quaternary AuCuPdPt alloy and extended AgAuCuPdPt HEA in contact with 0.1 M HClO4 saturated in an Ar atmosphere at 50 mV s−1. (D)–(F) CO oxidation curves in a CO saturated atmosphere at 50 mV s−1. Modelled equilibrium potentials are shown for *OH adsorption at low (vertical orange line) and high (vertical blue line) *CO coverage for surface compositions suggested by the simulations: Au0.5Pd0.5 (D), Au0.4Pd0.6 (E), and Ag0.2Au0.3Pd0.5 (F). The inset in E shows a magnification of a miniscule positive current at low extinction potentials, presumably caused by surface Pt. | ||
Fig. 3B shows the voltammetric characteristics of the AuCuPdPt alloy, highlighting the hydrogen adsorption/desorption range between 0.05 and 0.3 V vs. RHE, the double-layer capacitance region between 0.3 and 0.9 V vs. RHE, and the oxide region starting around 1.0 V vs. RHE. We can observe that the oxide region in the quaternary alloy occurs at a higher potential than on pure Pd or Pt, reflecting the influence of the other metals on the alloy surface. The voltammetric profile for the continuous CO oxidation on the quaternary alloy is shown in Fig. 3E. The current–potential transient follows the same trend as the AuPd alloy. However, the CO oxidation peak is reached at lower potential values than on AuPd. We ascribe this result to a possibly higher presence of Pd on the surface. We have plotted the blank CV of the AgAuCuPdPt HEA and the CO oxidation curve in Fig. 3C and F, respectively, for a better comparison with the other alloys. The onset potential for CO oxidation starts at ca. 0.95 V vs. RHE, and the general shape of the voltammogram is very similar to the previous quaternary and bimetallic alloys. The CO oxidation curve displays a notable variation, particularly in the negative scan where the current drops and the hysteresis is less pronounced due to the lower onset potential for the CO oxidation. We infer that this variation is likely caused by the presence of Ag on the surface, as it is not present in the other two alloys.
To investigate the surface composition after electrochemistry and get a deeper insight into the role of the different elements on the catalytic properties of the HEA alloy, we have conducted ion scattering spectroscopy (ISS) measurements. ISS provides high surface sensitivity and has been used to determine which elements are present at the surface. Fig. 4 shows the ISS spectra of an extended AgAuCuPdPt HEA alloy before and after continuous CO oxidation. Three peaks are observed in both ISS spectra: one at around 775 eV, which corresponds to Cu, the second at about 845 eV, which corresponds to Pd or Ag or both, and the last peak near 890 eV, which corresponds to Pt or Au or both. Since the atomic masses of Pd and Ag and of Pt and Au are relatively similar, respectively, it is not possible to distinguish their peaks by ISS using He+ ions. The use of ISS is qualitative because the ISS equipment has not been standardized and the use of reference samples is needed.69,70
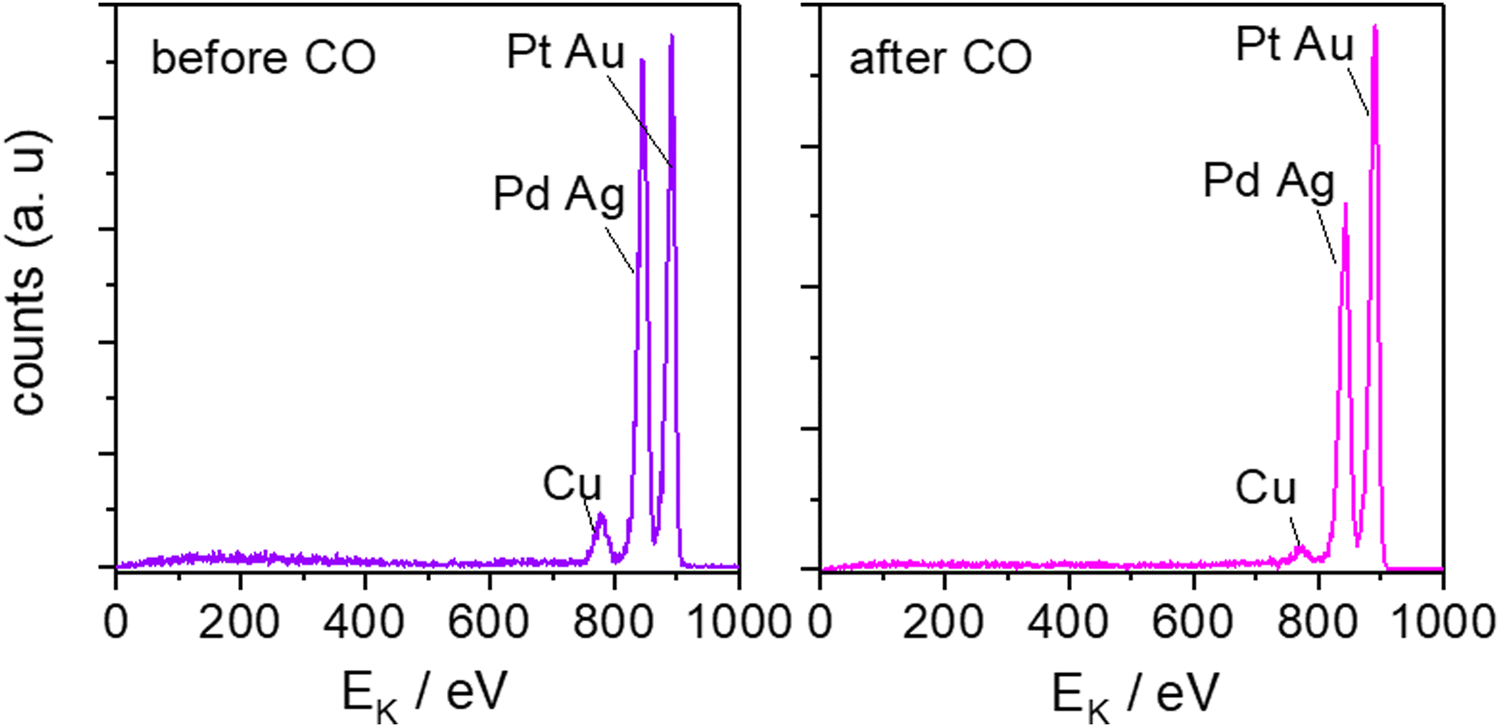 | ||
| Fig. 4 ISS spectra of an extended AgAuCudPt. HEA before and after continuous CO oxidation, after five scans. | ||
To quantify the composition of the active surface phase of the HEA, we carried out ex situ XPS measurements before and after the CO oxidation reaction. The aim of this analysis is to evaluate changes in surface composition induced by applying high potentials at which CO oxidizes. The XPS spectra of the HEA are shown in Fig. 5. The position of the adventitious carbon (C 1s) is shown in Fig. S4 of the ESI.† The two platinum peaks in Fig. 5G correspond to the Pt 4f7/2 at 70.4 eV and 4f5/2 at 73.7 eV, that are consistent with the metallic character of platinum.71 After the continuous CO oxidation, the Pt 4f7/2 and 4f5/2 binding energies shift 0.2 eV negatively, indicating a possible electron transfer from the neighboring atoms to Pt.72 Similarly, Au 4f7/2 at 83.9 eV and 4f5/2 at 87.6 eV binding energies are consistent with the metallic state.73 The negative shift by 0.1 eV on the Au 4f binding energies after CO oxidation follows the same trend as Pt 4f.74 In Fig. 5E, three peaks are observed, corresponding to Au 4d3/2 at 353.2 eV, Pd 3d3/2 at 340.5 eV and the overlap of the Au 4d5/2 and Pd 3d5/2 peaks near 335 eV, which are in good agreement with the literature.75 After CO oxidation, the Pd 3d peaks are 0.2 eV negatively shifted, which could be induced by hybridization changes with Ag and Au atoms,76 while the Au 4d peaks remain at the same binding energies. Before the CO oxidation, Ag 3d5/2 and 3d3/2 peaks at 367.9 eV and 373.9 eV are observed, with a slightly negative shift of 0.3 eV in comparison with the metallic character of silver,73,75 which could be attributed to the presence of metallic and Ag2O species.77 After CO oxidation, the Ag 3d binding peaks are 0.2 eV negatively shifted, which could be attributed to the synergistic effect with Au in CO oxidation.78Fig. 5F shows the binding energy of the Cu 2p3/2 and 2p1/2 peaks that appear near 931.9 eV and 951.7 eV. Compared with metallic Cu, the peaks are shifted to lower binding energy by 0.7 eV, characteristic of Cu2O species.79 We infer that we have a combination of Cu0 and Cu2O species.80 After CO oxidation, the Cu 2p peaks have shifted negatively by 0.2 eV. We attribute this displacement to the synergetic effect between Ag and Cu.81
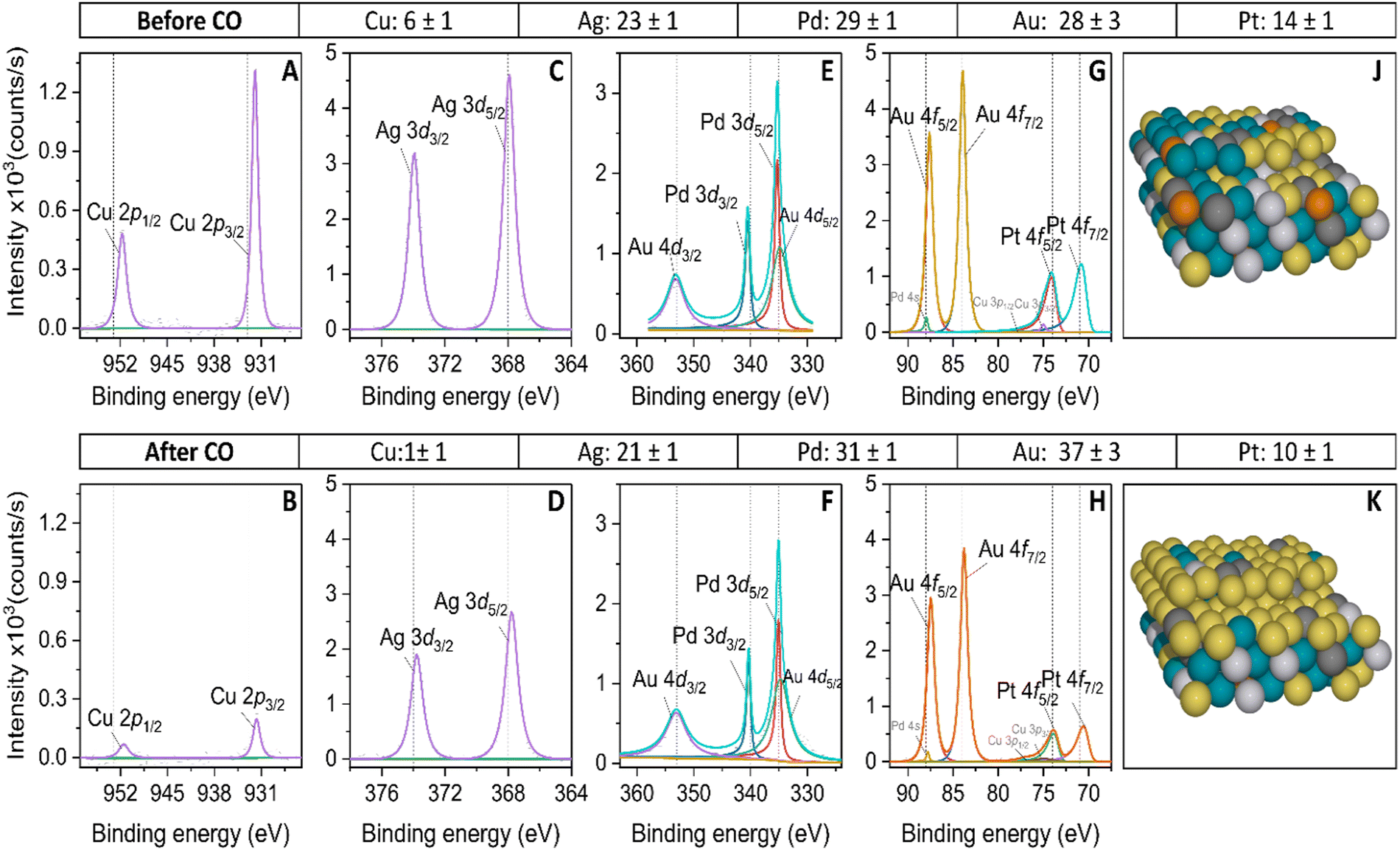 | ||
| Fig. 5 XPS spectra of scan of the (A) and (B) Cu2p, (C) and (D) Ag 3d, (E) and (F) Au4dPd3d, and (G) and (H) Au4fPt4f peaks of the extended AgAuCuPdPt HEA before and after continuous CO oxidation. All spectra have been background-subtracted (Shirley-type background). (J) and (K) Schematic representation of the HEA surface before and after continuous CO oxidation. | ||
Additionally, we have quantified the binding energy peaks of the XPS spectra to estimate the atomic percentages of the metals before and after the continuous CO oxidation. We have observed significant changes in the Cu intensity peaks after the CO oxidation peak. The decrease in these peaks indicates that the Cu atomic percentage has reduced from 6% to 1%, confirming its lower presence on the surface and likely lower participation in the CO oxidation. Besides, the angle-resolved XPS spectra on Cu 2p confirm the lower concentration of Cu on deeper layers of the HEA (see Fig. S6 of the ESI†). Likewise, the atomic percentage of Pt has decreased from 14% to 10% after CO oxidation. Interestingly, Au increased its atomic percentage significantly from 28% to 37%, making it the most abundant exposed metal on the extended HEA surface. It has the highest oxidation onset (see Table S4 in the ESI†) compared to other elements. This makes Au the most stable metal at the applied potential and less likely to dissolve because of its oxophobicity. The high amount of Au at the surface of the HEA explains the broad features of the voltammetric CO oxidation profile, which is more similar to the gold-rich AuPd CV than to single Pd or Pt. Additionally, the delayed onset for oxide species formation observed on the HEA surface relative to Pt and Pd can also be explained by the high presence of Au at the surface. On the other side, the atomic percentage of Ag has slightly decreased from 23% to 21%, and its presence on the surface is significant reinforcing the idea that Ag plays a role in activating the CO oxidation on the HEA, promoting the binding sites of oxygenates or OH groups on Pd or Pt sites. Pd experienced a slight increase in atomic percentage after CO oxidation and, like Ag, remained on the surface highly contributing to the CO oxidation process. It is important to mention that the XPS measurements were taken at open circuit potential and may differ from those under potential control. The atomic percentages for binary AuPd and quaternary AuCuPdPt alloys are represented in Table S1 of the ESI.†
To investigate how Ag and the other elements influence the CO oxidation onset and extinction potentials, we developed a predictive model based on DFT simulations. Assuming a reaction pathway where *CO and *OH need to react on the surface through an LH mechanism, we simulated hundreds of *CO and *OH adsorption energies on face-centered cubic (111) surfaces at various surface compositions and *CO coverages within the quaternary AgAuPdPt composition space. These simulations allowed us to construct a model with which the adsorption energy of *CO and *OH at an arbitrary surface composition and *CO coverage could be predicted (see Fig. S7–S12 and Tables S2 and S3 in the ESI†). The presumably most abundant close-packed (111) surface facets provide an initial model system to obtain trends for. We have not considered other surface facets in the current study, but future studies may consider investigating the catalytic trends of these. By first generating a randomly constructed surface with a desired composition, then equilibrating it with CO, and finally probing the *OH adsorption energies at the available surface sites, we were able to construct the map of the onset potentials for CO oxidation shown in Fig. 6A. More specifically, the onset potential is modelled as the *OH adsorption energy at the first per mille of the most strongly adsorbing surface sites (Fig. S13 in the ESI†). The underlying hypothesis behind this assumption is that as soon as the first *OH is able to adsorb, and oxidize away surface adsorbed *CO, the surface becomes slightly more open to *OH adsorption, which now will adsorb a bit more strongly due to the lower *CO coverage. This process repeats until all *CO has been stripped away.
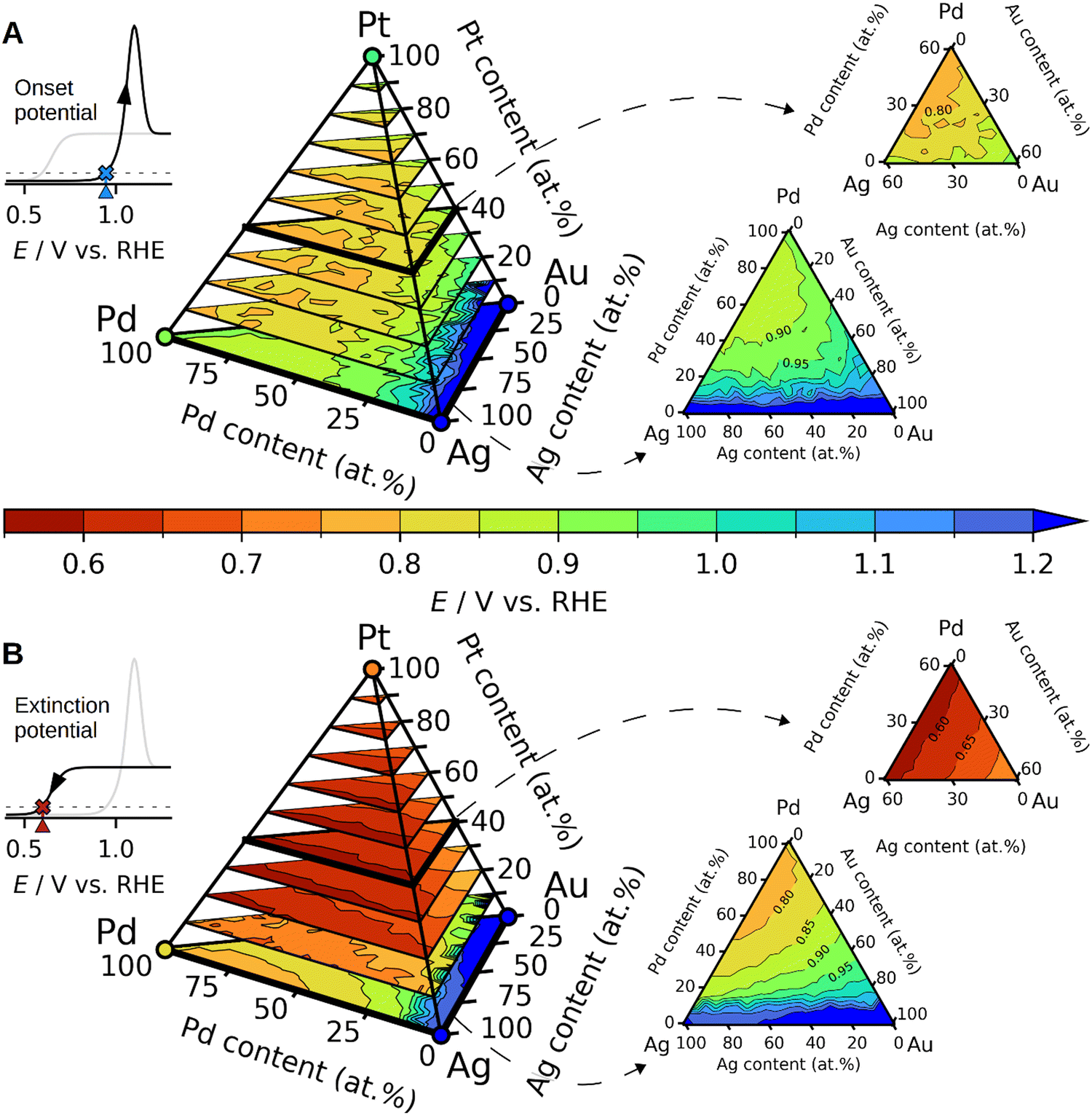 | ||
| Fig. 6 Simulated CO oxidation potentials as a function of the composition in the quaternary AgAuPdPt composition space. (A) Onset potentials simulated as the potential at which *OH can adsorb to a surface equilibrated with *CO. (B) Extinction potentials, i.e. where the CO oxidation ceases, simulated as the potential at which *OH can adsorb to a surface without adsorbed *CO. Both A and B are slices of the quaternary composition space highlighted corresponding to constant Pt concentrations of 0 and 40 at%. | ||
By probing the adsorption energies of *OH without any *CO adsorbed at arbitrary surface compositions, we were able to model the lowest potentials at which the CO oxidation could theoretically be performed. We have denoted this potential as the extinction potential, and the resulting map in composition space is shown in Fig. 6B. The *OH adsorption energy used in this map is the 10th percentile of the most strongly adsorbing surface sites; the assumption being that an appreciable amount of *OH must be present to prevent surface poisoning by CO.
The maps presented in Fig. 6 allow us to propose some trends in the CO oxidation on disordered alloys composed of Ag, Au, Pd, and Pt. For instance, an increased concentration of Ag is predicted to lower the onset of CO oxidation when replacing Au atoms, as can be seen by tracing the contour plots at lines of constant Pd and Pt concentration in Fig. 6A. According to our model, the reason for this behaviour is a ligand effect in which adsorption of *OH is slightly stronger on Pd and Pt atoms when they have neighbouring Ag surface atoms compared to neighbouring Au atoms (ESI,† Table S3). More generally, increased concentrations of Au are predicted to increase both the onset and extinction potentials, as can be seen by going towards the Au corners in the tetrahedrons in Fig. 6.
The validity of the model is confirmed to some extent by the predictions on the tested compositions in Fig. 2 and 3, where the model predictions for onset and extinction potentials are shown with blue and orange vertical lines, respectively. The model reproduces the onset potentials of pure Pt and Pd to happen just below 1 V vs. RHE, and the predicted extinction potentials are also very much in agreement with observation and DFT simulations, as shown in Fig. 2D and E. The experimentally observed differences in the onset and extinction potentials of the alloys are discernible yet subtle amounting to not more than 0.1 V. In comparison with experiments, the model proves useful as a tool for qualitatively comparing the alloys by the addition or removal of an element. Performing a quantitative comparison between the experimental data and the model presents challenges due to the unknown composition of the electrode's surface layer. Therefore, Fig. 3D–F displays proposed alloy compositions aimed at approximating the electrochemical response.
One salient prediction that the model presents is that the presence of Pt on the surface crucially lowers the extinction potential as seen in Fig. 6B. Hence, using the model to support the determination of the surface composition, we surmise that Pt is largely absent in the surface of the sampled alloys in Fig. 3, since the predicted Pt extinction potentials below ∼0.8 V vs. RHE are largely absent. Indeed, we presume that the feature observed in the cathodic scan of AuCuPdPt shown in the inset of Fig. 3E is an identifying characteristic of a low Pt surface concentration.
In addition to explaining trends, the reactivity maps in Fig. 6 allow us to propose new optimal compositions to validate in future studies. Inspection of Fig. 6A shows that the lowest onset potentials are predicted to be observed on the ternary face of Ag–Pd–Pt compositions for near-equimolar ratios of Ag, Pd, and Pt. Likewise, these compositions are also predicted to show the lowest extinction potentials, as shown in Fig. 6B.
Conclusion
We have shown the CV profiles and characterization of an extended AgAuCuPdPt HEA for CO oxidation in acidic media. The blank CV of the HEA provides limited information, only indicating the onset of the oxide region at around 0.9 V vs. RHE and a suppressed hydrogen adsorption/desorption region, with a lower charge compared to pure Pt. During the CO oxidation experiments, the addition of Ag to the HEA enhances the effectiveness of CO oxidation by reducing the onset potential and minimizing the hysteresis. These results are supported by a predictive model using DFT simulations where *CO and *OH need to react on the surface through an LH mechanism. Using the model, we suggest that an increased concentration of Ag lowers the onset of CO oxidation when replacing Au atoms due to strengthening of the *OH binding on neighboring Pd or Pt atoms, enhancing the CO oxidation. Surface-sensitive ISS and XPS methods show the existence of the five metals following electrochemical CO oxidation. Additionally, we observed Au segregating at the HEA surface and the instability of Cu after CO oxidation. The current data do not allow us to draw conclusions about the changes in the composition of Pd and Ag. However, it is evident that they are less present on the surface. Future characterization techniques, such as inductively coupled plasma mass spectrometry (ICP-MS), are necessary to better understand the changes that have occurred on the surface of the HEA. Our findings propose a novel approach to designing electrocatalysts by tailoring the surface composition and electronic structure and thus fine-tuning the binding energies for CO oxidation on multimetallic alloys.Author contributions
M. P. S. Q: writing – original draft and editing, investigation, methodology, visualization, data curation. J. K. P: writing – original draft, investigation, visualization, review and editing, methodology, data curation, software. P. S. P: methodology, supervision, writing – review and editing. I. C: resources, writing – review and editing. K. B: resources, writing – review and editing. J. R: conceptualization, resources, supervision, writing – review and editing, funding acquisition. M. E. E: conceptualization, resources, supervision, methodology, writing – review and editing, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the support from the Danish National Research Foundation Center for High-Entropy Alloy Catalysis (CHEAC, DNRF-149). We gratefully acknowledge the funding by Villum Fonden, part of the Villum Center for the Science of Sustainable Fuels and Chemicals (V-SUSTAIN grant 9455).References
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- O. Hoegh-Guldberg, D. Jacob, M. Taylor, T. Guillén Bolaños, M. Bindi, S. Brown, I. A. Camilloni, A. Diedhiou, R. Djalante, K. Ebi, F. Engelbrecht, J. Guiot, Y. Hijioka, S. Mehrotra, C. W. Hope, A. J. Payne, H. O. Pörtner, S. I. Seneviratne, A. Thomas, R. Warren and G. Zhou, Science, 2019, 365, eaaw6974 CrossRef CAS PubMed.
- N. Dyantyi, J. C. Calderón Gómez, L. Mekuto, P. Bujlo and G. Pattrick, Electrocatalysts: Selectivity and utilization, INC, 2021 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: fundamentals and applications, 2001 Search PubMed.
- G. Zoski Cynthia, Handbook of Electrochemistry, Elsevier, 2007 Search PubMed.
- S. Gottesfeld and J. Pafford, J. Electrochem. Soc., 1988, 135, 2651–2652 CrossRef CAS.
- E. Christoffersen, P. Liu, A. Ruban, H. L. Skriver and J. K. Nørskov, J. Catal., 2001, 199, 123–131 CrossRef CAS.
- R. Adzic and N. Marinkovic, Platinum Monolayer Electrocatalysts, 2020 Search PubMed.
- Á. Cuesta and C. Gutiérrez, Catalysis in Electrochemistry: From Fundamentals to Strategies for Fuel Cell Development, 2011, pp. 339–373 Search PubMed.
- H. A. Gasteiger, N. Markovic, P. N. Ross. Jr and E. J. Cairns, Electrochim. Acta, 1994, 39, 1825–1832 CrossRef CAS.
- X. Chen, L. P. Granda-Marulanda, I. T. McCrum and M. T. M. Koper, Nat. Commun., 2022, 13, 1–11 CAS.
- S. Gilman, J. Phys. Chem., 1963, 67, 1898–1905 CrossRef CAS.
- A. V. Petukhov, W. Akemann, K. A. Friedrich and U. Stimming, Surf. Sci., 1998, 402–404, 182–186 CrossRef CAS.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Electroanal. Chem., 2002, 524, 242–251 CrossRef.
- F. Maroun, F. Ozanam, O. M. Magnussen and R. J. Behm, Science, 2001, 293, 1811–1814 CrossRef CAS PubMed.
- S. R. Brankovic, N. S. Marinkovic, J. X. Wang and R. R. Adžić, J. Electroanal. Chem., 2002, 532, 57–66 CrossRef CAS.
- J. B. Xu, T. S. Zhao and Z. X. Liang, J. Power Sources, 2008, 185, 857–861 CrossRef CAS.
- J. H. Choi, K. J. Jeong, Y. Dong, J. Han, T. H. Lim, J. S. Lee and Y. E. Sung, J. Power Sources, 2006, 163, 71–75 CrossRef CAS.
- M. Baldauf and D. M. Kolb, J. Phys. Chem., 1996, 100, 11375–11381 CrossRef CAS.
- K. Gossner and E. Mizera, J. Electroanal. Chem., 1982, 140, 35–45 CrossRef CAS.
- A. Piednoir, M. A. Languille, L. Piccolo, A. Valcarcel, F. J. C. S. Aires and J. C. Bertolini, Catal. Lett., 2007, 114, 110–114 CrossRef CAS.
- F. Gao, Y. Wang and D. W. Goodman, J. Am. Chem. Soc., 2009, 5734–5735 CrossRef CAS PubMed.
- E. Plaza-Mayoral, I. J. Pereira, K. Nicole Dalby, K. D. Jensen, I. Chorkendorff, H. Falsig, P. Sebastián-Pascual and M. Escudero-Escribano, ACS Appl. Energy Mater., 2022, 5, 10632–10644 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- V. A. Mints, J. K. Pedersen, A. Bagger, J. Quinson, A. S. Anker, K. M. Ø. Jensen, J. Rossmeisl and M. Arenz, ACS Catal., 2022, 12, 11263–11271 CrossRef CAS.
- J. W. Yeh, S. K. Chen, S. J. Lin, J. Y. Gan, T. S. Chin, T. T. Shun, C. H. Tsau and S. Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- B. Cantor, I. T. H. Chang, P. Knight and A. J. B. Vincent, J. Mater. Sci. Eng. A, 2004, 375–377, 213–218 CrossRef.
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Mater., 2019, 4, 515–534 CrossRef CAS.
- T. A. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3, 834–845 CrossRef CAS.
- T. Löffler, A. Ludwig, J. Rossmeisl and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 2–12 CrossRef.
- H. J. Qiu, G. Fang, Y. Wen, P. Liu, G. Xie, X. Liu and S. Sun, J. Mater. Chem. A, 2019, 7, 6499–6506 RSC.
- J. K. Pedersen, T. A. A. Batchelor, D. Yan, L. E. J. Skjegstad and J. Rossmeisl, Curr. Opin. Electrochem., 2021, 26, 100651 CrossRef CAS.
- A. Cuesta, M. Escudero, B. Lanova and H. Baltruschat, Langmuir, 2009, 25, 6500–6507 CrossRef CAS PubMed.
- C. L. Bentley, M. Kang and P. R. Unwin, J. Am. Chem. Soc., 2019, 141, 2179–2193 CrossRef CAS PubMed.
- N. Kumar, C. S. Tiwary and K. Biswas, J. Mater. Sci., 2018, 53, 13411–13423 CrossRef CAS.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 1–11 CrossRef.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- A. Hjorth Larsen, J. JØrgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. SchiØtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- C. M. Clausen, J. K. Pedersen, T. A. A. Batchelor, J. Rossmeisl, F. Pg, I. Pg, C. Fonbsl, D. Gvmmz, V. F. Uif, O. F. X. Dbubmzujd, P. Pg, B. S. F. Qvmmfe, C. Uif, F. Mfwfm, B. O. E. Uijt, S. Jo and B. X. Cpoe, Nano Res., 2022, 15, 4775–4779 CrossRef CAS.
- S. Trasatti and A. Petrii, J. Electroanal. Chem. Interfacial Electrochem., 1992, 327, 353–376 CrossRef CAS.
- A. Visintin, J. C. Canullo, W. E. Triaca and A. J. Arvia, J. Electroanal. Chem., 1988, 239, 67–89 CrossRef CAS.
- X. Chen, C. Si, Y. Gao, J. Frenzel, J. Sun, G. Eggeler and Z. Zhang, J. Power Sources, 2015, 273, 324–332 CrossRef CAS.
- S. Nellaiappan, N. K. Katiyar, R. Kumar, A. Parui, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, ACS Catal., 2020, 10, 3658–3663 CrossRef CAS.
- V. Márquez, J. S. Santos, J. G. Buijnsters, S. Praserthdam and P. Praserthdam, Electrochim. Acta, 2022, 410, 139972 CrossRef.
- J. Solla-Gullón, P. Rodríguez, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2008, 10, 1359–1373 RSC.
- J. P. Hoare, J. Electrochem. Soc., 1964, 111, 610 CrossRef CAS.
- Q. S. Chen, J. Solla-Gullón, S. G. Sun and J. M. Feliu, Electrochim. Acta, 2010, 55, 7982–7994 CrossRef CAS.
- J. Clavilier, R. Faure, G. Guinet and R. Durand, J. Electroanal. Chem., 1980, 107, 205–209 CrossRef CAS.
- A. Rodes and J. Clavilier, J. Electroanal. Chem., 1993, 344, 269–288 CrossRef CAS.
- F. J. Vidal-Iglesias, R. M. Arán-Ais, J. Solla-Gullón, E. Herrero and J. M. Feliu, ACS Catal., 2012, 2, 901–910 CrossRef CAS.
- M. J. Janik, I. T. Mccrum and M. T. M. Koper, J. Catal., 2018, 367, 332–337 CrossRef CAS.
- L. H. Dall’Antonia, G. Tremiliosi-Filho and G. Jerkiewicz, J. Electroanal. Chem., 2001, 502, 72–81 CrossRef.
- M. Grdeń, M. Łukaszewski, G. Jerkiewicz and A. Czerwiński, Electrochim. Acta, 2008, 53, 7583–7598 CrossRef.
- S. Strbac, R. R. Adzic and A. Hamelin, J. Electroanal. Chem., 1988, 249, 291–310 CrossRef CAS.
- C. Jeyabharathi, P. Ahrens, U. Hasse and F. Scholz, J. Solid State Electrochem., 2016, 3025–3031 CrossRef CAS.
- B. B. Blizanac, C. A. Lucas, M. E. Gallagher, M. Arenz, P. N. Ross and N. M. Marković, J. Phys. Chem. B, 2004, 108, 625–634 CrossRef CAS.
- C. M. Clausen, T. A. A. Batchelor, J. K. Pedersen and J. Rossmeisl, Adv. Sci., 2021, 8, 1–7 Search PubMed.
- M. T. M. Koper, S. J. Thomas, N. M. Marković and P. N. Ross, J. Phys. Chem. B, 2001, 105, 8381–8386 CrossRef CAS.
- A. Couto, A. Rincón, M. C. Pérez and C. Gutiérrez, Electrochim. Acta, 2001, 46, 1285–1296 CrossRef CAS.
- M. T. M. Koper, S. C. S. Lai and E. Herrero, Fuel Cell Catalysis: A Surface Science Approach, 2008, pp. 159–207 Search PubMed.
- C. A. Angelucci, E. Herrero and J. M. Feliu, J. Solid State Electrochem., 2007, 11, 1531–1539 CrossRef CAS.
- P. Rodriguez, N. Garcia-araez and M. T. M. Koper, Phys. Chem. Chem. Phys., 2010, 12, 9373–9380 RSC.
- B. E. Hayden, D. Pletcher, M. E. Rendall and J. P. Suchsland, J. Phys. Chem. C, 2007, 111, 17044–17051 CrossRef CAS.
- P. Rodriguez and M. T. M. Koper, Phys. Chem. Chem. Phys., 2014, 16, 13583–13594 RSC.
- P. Rodriguez, Y. Kwon and M. T. M. Koper, Nat. Chem., 2012, 4, 177–182 CrossRef CAS PubMed.
- M. Łukaszewski and A. Czerwiński, J. Solid State Electrochem., 2008, 12, 1589–1598 CrossRef.
- M. Łukaszewski and A. Czerwiński, Electrochim. Acta, 2003, 48, 2435–2445 CrossRef.
- H. Niehus, W. Heiland and E. Taglauer, Surf. Sci. Rep., 1993, 17, 213–303 CrossRef CAS.
- H. H. Brongersma, M. Draxler, M. de Ridder and P. Bauer, Surf. Sci. Rep., 2007, 62, 63–109 CrossRef CAS.
- J. Z. Shyu and K. Otto, Appl. Surf. Sci., 1988, 32, 246–252 CrossRef CAS.
- J. B. Goodenough, R. Manoharan, A. K. Shukla and K. V. Ramesh, Chem. Mater., 1989, 1, 391–398 CrossRef CAS.
- C. D. Wagner, NIST X-ray Photoelectron Spectrometry Database, 1991 Search PubMed.
- W. Ye, H. Kou, Q. Liu, J. Yan, F. Zhou and C. Wang, Int. J. Hydrogen Energy, 2012, 37, 4088–4097 CrossRef CAS.
- J. R. Rumble, D. M. Bickham and C. J. Powell, Surf. Interface Anal., 1992, 19, 241–246 CrossRef CAS.
- I. A. Abrikosov, W. Olovsson and B. Johansson, Phys. Rev. Lett., 2001, 87, 2–5 CrossRef PubMed.
- J. F. Weaver and G. B. Hoflund, Chem. Mater., 1994, 6, 1693–1699 CrossRef CAS.
- J. H. Liu, A. Q. Wang, Y. S. Chi, H. P. Lin and C. Y. Mou, J. Phys. Chem. B, 2005, 109, 40–43 CrossRef CAS PubMed.
- N. Pauly, S. Tougaard and F. Yubero, Surf. Sci., 2014, 620, 17–22 CrossRef CAS.
- D. Tahir and S. Tougaard, J. Phys.: Condens. Matter, 2012, 24, 175002 CrossRef PubMed.
- L. Rout, A. Kumar, R. S. Dhaka and P. Dash, RSC Adv., 2016, 6, 49923–49940 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00023d |
| This journal is © The Royal Society of Chemistry 2024 |