DOI:
10.1039/D3EY00187C
(Review Article)
EES Catal., 2024,
2, 448-474
Metal halide perovskites for CO2 photoreduction: recent advances and future perspectives
Received
3rd August 2023
, Accepted 4th January 2024
First published on 6th January 2024
Abstract
CO2 emission has inarguably become one of the greatest challenges ever faced by mankind since industrial revolution. Techniques aiming at capture, storage and utilization of CO2 have attracted tremendous interest from both industry and academia. Thermal, electrical and photo-catalytic conversion of CO2 to value-added chemicals and fuels is the most well-known approach for CO2 utilization. In particular, photocatalytic reduction of CO2 (CO2PR) directly employs solar energy as the driving force to activate CO2, yielding various products including CO, CH4 and C2+ hydrocarbons. CO2PR, which mimics photosynthesis occurring in nature, is also regarded as “artificial photosynthesis” and is believed to be a promising approach toward carbon neutral economy. Recently, metal halide perovskites (MHPs) have emerged as potential photocatalysts for CO2PR, owing to their flexible structures and excellent photoelectronic properties. This review presents a comprehensive overview of state-of-the-art developments in MHP-based catalysts for CO2PR. Firstly, the crystal structures and photoelectric properties of MHPs are reviewed in detail, as they are the key factors determining CO2PR catalytic performance. Secondly, design strategies to promote the catalytic efficiency of CO2PR to CO conversion for both lead-based and lead-free MHPs are discussed, including morphological modifications, co-catalyst modifications, ion doping and crystal plane modifications. Thirdly, this review addresses MHP-based CO2PR to CH4 and C2+ products, with special emphasis on approaches adopted to promote specific product selectivity. Lastly, our perspectives and opinions are given on current research challenges and future directions for CO2PR, which we consider are critical for its industrialization.
Broader context
Photocatalytic reduction of CO2 into value-added chemicals (i.e., artificial photosynthesis) is regarded as a fascinating approach to simultaneously solving global warming and energy crisis. As a class of newly emerged semiconductors, metal halide perovskites (MHPs) have attracted great interest in the field of CO2 photoreduction in recent years due to their flexible structures and excellent photoelectronic properties. In this review, we present a comprehensive overview of recent advances in MHP-based catalysts for CO2 photoreduction (CO2PR). We start with a description of the crystal structure and photoelectric properties of MHPs, which are key to CO2PR catalytic performance. Then, strategies (e.g., morphological modifications, co-catalyst modifications, ion doping, crystal plane modifications, etc.) for lead-based and lead-free MHP materials to improve the efficiency of CO2PR to CO conversion are systematically summarized. In particular, a detailed discussion on MHP-based CO2PR to CH4 and C2+ products (with higher economic value) is further presented, with special emphasis on the designed methods for promoting selectivity. Finally, some concluding remarks and an outlook on future challenges and prospects are provided to point out the possible research directions in MHP-based photocatalysts for CO2PR.
|
1. Introduction
Over-reliance on the depleting fossil fuels as the main energy source is one of the major problems hindering the transformation toward sustainable economy, while leading to extensive CO2 emission and causing climate changes at the same time. In response to this daunting challenge, new techniques and process protocols have been developed in various industries to reduce energy consumption, and governments across the globe are promoting the usage of renewable energy sources as clean and green alternatives to conventional fossil fuels. Artificial photosynthesis technology, which utilizes the inexhaustible, green, and pollution-free sunlight to initiate chemical reactions, shows great potential in reducing atmospheric CO2 emission by converting it into valuable solar fuels.1,2 Since CO2 is a thermodynamically stable molecule (the dissociation energy of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is about 750 kJ mol−1),3 the major challenge in CO2 photocatalysis is to find a photocatalyst that can effectively activate and reduce CO2. In 1979, Inoue et al. reported that various semiconductors could act as photocatalysts to reduce CO2 in aqueous solution.4 Since then, many nanostructured materials (e.g., metal oxides,5,6 metal sulfides7 and metal halide perovskites (MHPs)) have been developed for photocatalytic CO2 reduction reaction (referred to as CO2PR in the following discussions).8,9 Among them, extensive research works are focusing on TiO2 and its derivatives, owing to their unique properties including non-toxicity, ease of manufacture, and outstanding photochemical stability.10 Despite these advantages, the application of TiO2 catalysts is limited by their inherent large bandgap, which restricts the ability to harvest solar energy over a wide spectrum. In fact, CO2PR by TiO2 can only occur in the ultraviolet (UV) range,11 greatly limiting the efficiency in utilizing solar energy. Recently, organic–inorganic halide perovskites have drawn great attention in the field of photoelectricity, serving as the key component for solar cells, light-emitting diodes (LEDs), photodetectors, X-rays and lasers.12–22 For example, major breakthroughs have been achieved with perovskite solar cells, with the power conversion efficiency increasing from 3.8% in 2009 to 25.7% in 2023.23 Superior photoelectric properties and favorable carrier mobility are responsible for the impressive performance of perovskite solar cells.24 Moreover, the efficiency of green and red perovskite LEDs also reaches over 20% and 1.1%, respectively.25,26 At the same time, the performance of MHP-based lasers, photodetectors and X-ray detectors is also developing at a rapid pace. Despite numerous advances achieved, the organic groups of most organic–inorganic perovskite materials are extremely sensitive to the environment, limiting their application under industrial conditions.
O is about 750 kJ mol−1),3 the major challenge in CO2 photocatalysis is to find a photocatalyst that can effectively activate and reduce CO2. In 1979, Inoue et al. reported that various semiconductors could act as photocatalysts to reduce CO2 in aqueous solution.4 Since then, many nanostructured materials (e.g., metal oxides,5,6 metal sulfides7 and metal halide perovskites (MHPs)) have been developed for photocatalytic CO2 reduction reaction (referred to as CO2PR in the following discussions).8,9 Among them, extensive research works are focusing on TiO2 and its derivatives, owing to their unique properties including non-toxicity, ease of manufacture, and outstanding photochemical stability.10 Despite these advantages, the application of TiO2 catalysts is limited by their inherent large bandgap, which restricts the ability to harvest solar energy over a wide spectrum. In fact, CO2PR by TiO2 can only occur in the ultraviolet (UV) range,11 greatly limiting the efficiency in utilizing solar energy. Recently, organic–inorganic halide perovskites have drawn great attention in the field of photoelectricity, serving as the key component for solar cells, light-emitting diodes (LEDs), photodetectors, X-rays and lasers.12–22 For example, major breakthroughs have been achieved with perovskite solar cells, with the power conversion efficiency increasing from 3.8% in 2009 to 25.7% in 2023.23 Superior photoelectric properties and favorable carrier mobility are responsible for the impressive performance of perovskite solar cells.24 Moreover, the efficiency of green and red perovskite LEDs also reaches over 20% and 1.1%, respectively.25,26 At the same time, the performance of MHP-based lasers, photodetectors and X-ray detectors is also developing at a rapid pace. Despite numerous advances achieved, the organic groups of most organic–inorganic perovskite materials are extremely sensitive to the environment, limiting their application under industrial conditions.
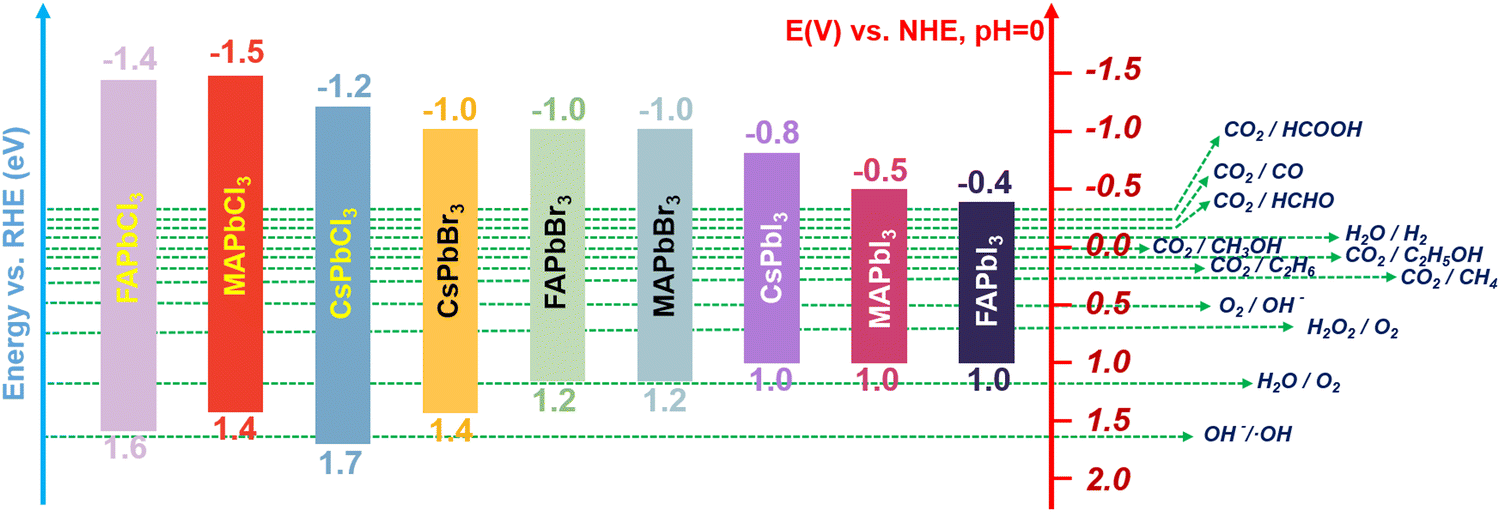
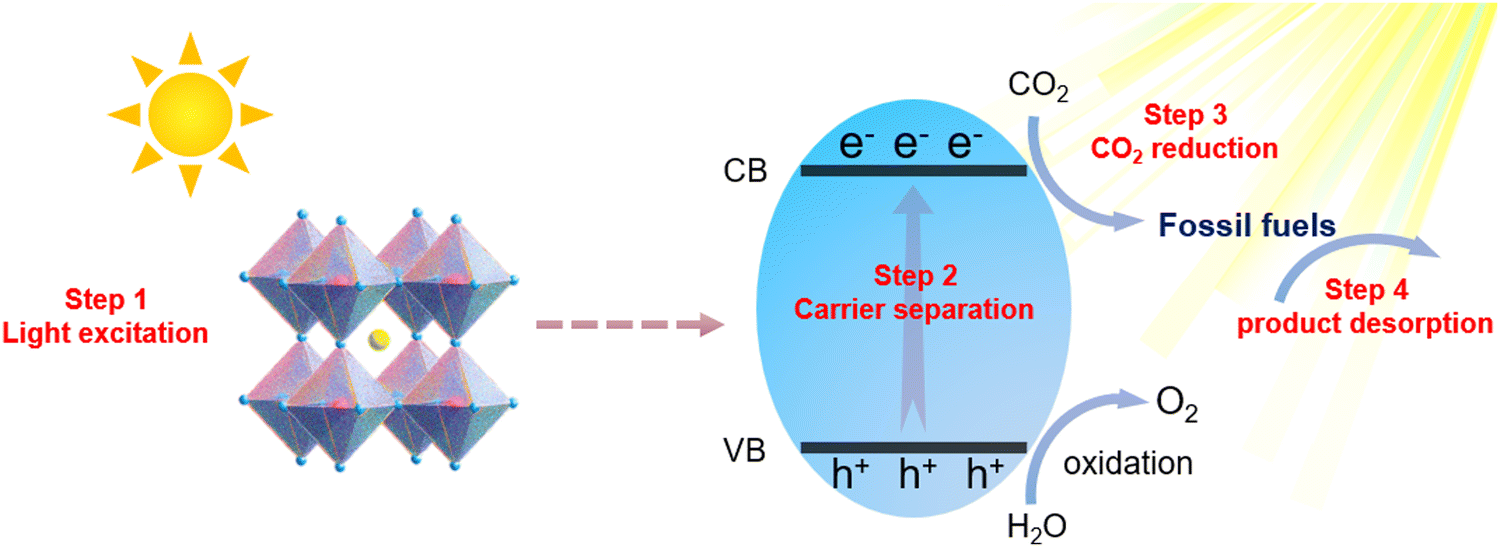
In recent years, MHPs have gradually emerged as intriguing all-inorganic perovskite materials for photoelectronic and energy conversion applications. With metal cations replacing organic cations, MHPs have a greater extinction coefficient than conventional semiconductor nanocrystals and can withstand more defects. Furthermore, the rich structural diversity allows accurate tuning of the bandgap, thus improving the light capture ability. Based on reaction thermodynamics, efficient photocatalysis requires a good match between the electronic energy band (EB) structure of the catalyst and the redox potential of the reaction. In Fig. 1, the relative positions of the conduction band (CB) and valence band (VB) for the majority of halide perovskites are depicted. It is clearly illustrated that the relative positions of the CB of MHPs are sufficiently negative for H2 generation, CO2 reduction, aerobic oxidation and organic matter degradation (via superoxide radicals). On the basis of this plot, halide perovskites are seen as a promising candidate for CO2PR. The CB for most MHPs is more negative than the CO2 reduction potential, which is a prerequisite and essential condition for CO2 reduction to occur.27Fig. 2 demonstrates the four key steps for CO2PR, namely light capture, generation and separation of photogenerated carriers, CO2 adsorption and product desorption.28 By optimizing these key steps, the overall photocatalytic activity for CO2 reduction can be improved. For example, higher molar extinction coefficients of MHPs allow them to capture light more effectively, hence leading to better catalytic performances. The molar extinction coefficient of CsPbX3 (X denotes a halide ion) is nearly 10 times higher than that of CdSe nanocrystals with a similar bandgap. Under the same light conditions, the high molar extinction coefficient means that CsPbX3 nanocrystals can capture more sunlight for photocatalysis. Moreover, MHPs generally have a low exciton binding energy (Eb), implying that excitons are more likely to dissociate into free electrons and holes, further facilitating photocatalytic reactions.29 Nonetheless, strong recombination of photogenerated charge carriers limits the catalytic performance of pristine MHPs, and effective photoreduction of the chemically inert CO2 with high catalytic activity and product selectivity is still difficult to achieve. Development of efficient photocatalysts remains a key challenge and has attracted research interest across the globe. Novel design strategies are in need, together with more detailed understanding of the reaction mechanism, to develop highly active, selective, and stable photocatalytic systems based on MHPs.
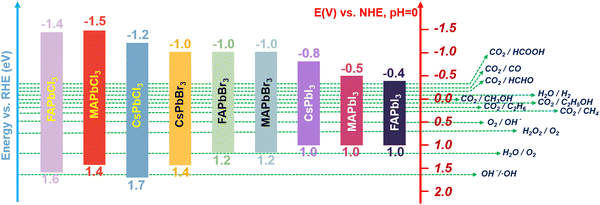 |
| | Fig. 1 Band edge positions of halide perovskites relative to reversible hydrogen electrode (RHE). | |
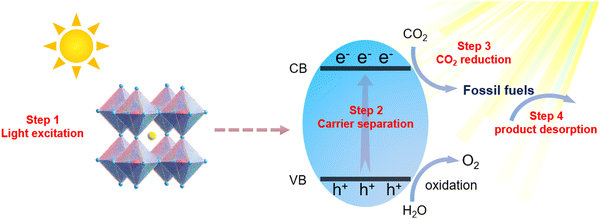 |
| | Fig. 2 Schematic diagram of photoreduction of MHPs. | |
To date, there have been several excellent reviews providing in-depth summaries on MHPs in photocatalysis, discussing the overall photocatalytic performance, stability, and lead toxicity issues.29–37 However, these reviews mainly focus on hydrogen production reaction, pollutant degradation, and other applications. Strategies for the advanced photocatalytic reduction of CO2 to CO are studied to a lesser extent. Moreover, reviews exploring CO2PR toward CH4 and C2+ products with higher added values are rarely summarized. In view of recent advances, we believe it is necessary to give a comprehensive overview of the state-of-the-art materials and design strategies in MHP catalyzed CO2PR. In this review, we first describe the crystal structure and photoelectric properties of MHPs. Following that, detailed discussions are made on the relevant advances accomplished with lead-based and lead-free MHP materials, focusing on strategies to improve the CO2 photoconversion efficiency to CO. More importantly, the CO2 photoreduction to CH4 and C2+ products (which are more economically valuable) based on MHP-catalysts is also discussed. Lastly, our insights into future research directions are provided, addressing the urgent challenges that need to be pursued in order to maximize the potential of MHPs in promoting sustainable development and facilitating their practical applications.
2. Structures and properties of MHPs
2.1 Crystal structures of MHPs
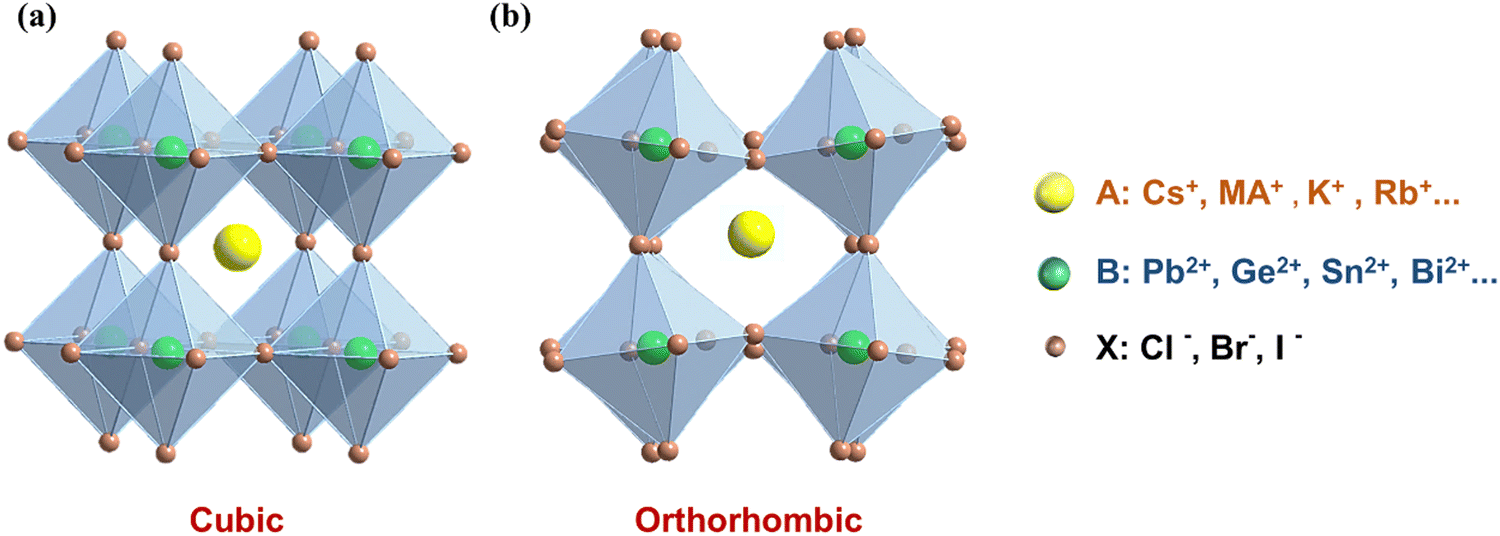
The structure of a material serves as a crucial determinant of its physical and chemical properties, and any variation in its structural arrangement can make a profound impact on its performance. Therefore, a comprehensive understanding of the crystal structure of MHPs is essential to accurately predict and fine-tune their optical, electronic, and catalytic properties. It is generally accepted that the chemical formula of perovskites is ABX3, in which the A-site cation is 12-fold coordinated and the B-site cation is 6-fold coordinated. Typically, perovskite structures are cubic (Fig. 3a) or octahedral in shape (Fig. 3b).38 X represents the halide elements Cl, Br, and I, and B refers to metal ions like Pb2+, Ge2+, Sn2+, and Bi2+. In these ions, the A and B site ions can be partially replaced by ions with similar radius, while the crystal structure remains essentially unchanged. Depending on whether the A-site ions contain organic groups, they can be divided into two categories: organic–inorganic hybrid perovskites (organic ions in the A-site, such as CH3NH3+, methylammonium ion (MA+), formamidine ion (FA+)) and all-inorganic perovskites (only inorganic ions in the A-site, such as Cs+, K+, Rb+). It is worth noting that the majority of research efforts in the realm of MHPs have been devoted to exploring the fascinating properties of lead halide perovskites (LHPs) featuring an APbX3 structure,39,40 due to the exceptional photoelectric properties. Additionally, the remarkable potential of LHPs for various applications such as solar cells, LEDs and photodetectors has made them a focal point of research efforts.41–47 According to the concept of the Goldschmidt tolerance factor, which involves close packing of ions, only MA+, FA+ and Cs+ can stabilize the 3D PbX6 framework, because they are geometrically adapted in the A-site of the twelve-coordinates.40 However, evidence from previous studies has shown that the crystal structure of APbX3 can be irreversibly damaged and decomposed in the presence of moisture, photothermal radiation and polar solvents, which limits their application in many fields.48–51 Therefore, exploring more structurally stable APbX3 or surface modified stable materials is an important research trend in the future.
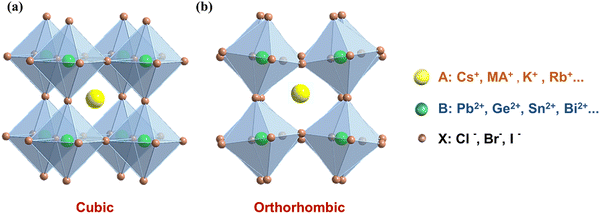 |
| | Fig. 3 (a) Cubic and (b) orthorhombic crystal structures of 3D ABX3 perovskites. | |
2.2 Photoelectric properties of MHPs
The excellent photoelectric properties of MHPs are primarily attributed to their unique structural characteristics. As described above, their highly ordered and symmetrical lattice three-dimensional network provides a favorable environment for efficient charge transport and light absorption, which makes them ideal for photocatalytic applications. These properties include favorable bandgaps for extended absorption of visible light, high charge carrier mobility, and long charge diffusion length that results in a reduced recombination rate of electron–hole pairs. Recent reports52–61 have highlighted these properties, which are important factors supporting the potentially superior photocatalytic performance of MHPs. Specifically, MHPs exhibit (1) a high optical absorption coefficient with easily tunable bandgaps, (2) a long carrier diffusion length and suppressed recombination rate, which improve carrier lifetime, and (3) a well-balanced charge transfer suitable for redox reactions.
2.2.1 Flexible and adjustable bandgap.
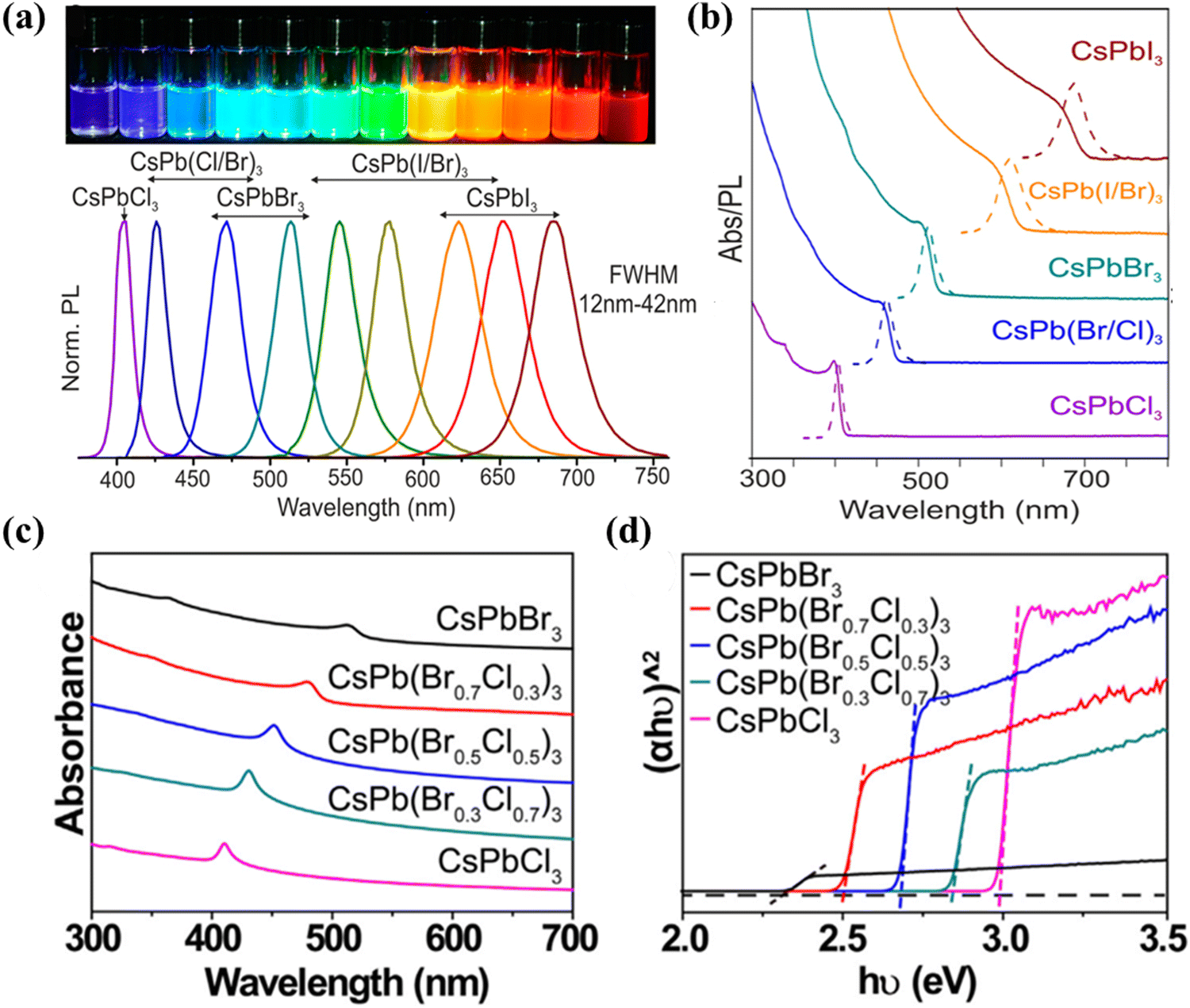
The enhanced light absorption of MHPs compared to conventional semiconductors (e.g., TiO2) has been found to contribute to the improved photo-carrier conversion efficiency, as demonstrated in many perovskite-based solar cells.60 In order to maximize the optical absorption of a photocatalytic material, it is necessary to possess an appropriate bandgap. Theoretically, by reducing the bandgap value, the absorption spectrum of the visible light becomes wider and the efficiency of light utilization can be increased.61 The energy level of MHPs is related to their bandgap, which is composed of the conduction band minimum (CBM) and the valence band maximum (VBM). For MHPs, the VBM consists of an antibonding hybrid state between the 6s orbitals of the B divalent cation and the np orbitals of X (n = 3, 4 and 5 for Cl, Br and I, respectively), with the latter playing a dominant role. The CBM is an antibonding hybrid state of the 6p orbitals of B and the np orbitals of X, and the main contributor is from the 6p orbitals of B.62,63 As A-site ions have little impact on the VBM or CBM, the bandgap of MHPs is frequently adjusted by mixing or modifying B and X-site ions. Adjustable bandgap provides the opportunity to regulate the energy band edge and achieve highly efficient photocatalysis in various applications. Additionally, the absorption spectrum of MHPs can be tuned from visible light to near-infrared (NIR) irradiation utilizing the quantum confinement effect. For example, Protesescu et al. first reported that the optical absorption and emission spectra of colloidal CsPbX3 NCs (Fig. 4a and b) can be tuned by adjusting their composition (proportion of halides in mixed NCs) and particle size (quantum-size effect) to respond across the visible spectral region.64 Guo et al. demonstrated that CsPb(Brx/Cl1−x)3 exhibits a wide absorption range for visible light from 400 to 700 nm, with a gradual shift of the absorption peak from 520 nm (CsPbBr3) to 420 nm (CsPbCl3) by adjusting the value of X (Fig. 4c).65 At the same time, Fig. 4d shows an increase in Eg values from 2.33 to 2.98 eV for CsPb(Brx/Cl1−x)3 (x = 1, 0.7, 0.5, 0.3, 0) with a direct bandgap.65 All of the above evidence suggests that the use of hybrid halides is a viable strategy for modulating the absorption diversity and bandgap in perovskite spectroscopy.
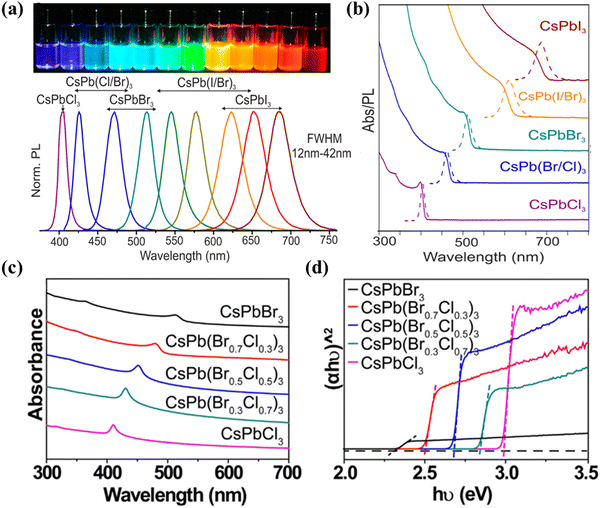 |
| | Fig. 4 (a) Colloidal halide perovskite CsPbX3 NCs (X = Cl, Br, and I) exhibit size- and composition-tunable bandgap energies covering the entire visible spectral region with narrow and bright emission.64 (b) Typical optical absorption and PL spectra of CsPbX3 NCs.64 (c) UV-vis absorption spectra and (d) (αhυ)2versus hυ curves of CsPb(Brx/Cl1−x)3 (x = 1, 0.7, 0.5, 0.3, 0) materials.65 | |
2.2.2 Long carrier lifetime.
Under thermal equilibrium conditions, electrons are continuously excited from the valence band to the conduction band to produce electron–hole pairs. At the same time, electron–hole pairs keep disappearing because the recombination of carriers makes the carriers disappear gradually, and the average existence time of such carriers is called carrier lifetime.64 At equilibrium, the production rate of electrons and holes is equal to the recombination rate, so that the density of carriers in the semiconductor remains constant. Thus, a higher equilibrium carrier concentration favours an increase in carrier lifetime and diffusion length. In the process of photocatalytic reaction, the separation and transport of electrons are the main considerations for the improved catalytic performance. Longer carrier lifetime implies high charge mobility, which is a key advantage for semiconductors. MHPs have been reported to have stable equilibrium carrier concentrations and long carrier lifetime beyond those of commonly used photoelectric materials,65 as well as beneficial interfacial charge transfer kinetics and still undiscovered electron injection kinetics advantages.67
2.2.3 High equilibrium carrier concentration.
Generally, the equilibrium carrier concentration refers to the number of electrons and holes in a material that are present in thermodynamic equilibrium at a given temperature. At equilibrium, the rate of generation of free charge carriers (electrons and holes) through excitation processes is balanced by the rate of recombination of those carriers, resulting in a constant density of free carriers in the material. The equilibrium carrier concentration is a critical parameter in determining the electrical and optical properties of a semiconductor material. It is influenced by factors such as the doping level of the material, its intrinsic carrier concentration, the temperature, and the energy gap between the valence and conduction bands.68 One of the advantages of MHPs is their high equilibrium carrier concentration, which is several orders of magnitude higher than that of conventional semiconductors such as silicon. High carrier concentration originates from the unique crystal structure of MHPs, which consists of alternating layers of metal halide octahedra and organic cations.69 The high carrier concentration in MHPs is beneficial for many photoelectric applications. For example, in solar cells, the high carrier concentration leads to a high open-circuit voltage and short-circuit current, resulting in high power conversion efficiency. In LEDs, the high carrier concentration enables efficient radiative recombination of electrons and holes, leading to bright and efficient emission. Moreover, the high carrier concentration in MHPs also leads to long carrier diffusion lengths, which enable efficient extraction and collection of charge carriers, reducing the recombination losses. Overall, the high equilibrium carrier concentration in MHPs is a significant advantage that makes them promising candidates for various photoelectric applications, offering exceptional performance and potential for future technological advancements.
3. MHP materials for the photoreduction of CO2 to CO as the main product
To date, several technologies have been developed to convert CO2 into hydrocarbons or high value-added chemicals, such as thermocatalysis, biocatalysis, electrocatalysis, and photocatalysis. Different from the conventional thermocatalytic CO2 reduction requiring high temperature and pressure conditions, the photocatalytic CO2 reduction process simulates natural photosynthesis using sunlight and a photocatalyst to catalyze the conversion of CO2 and water, which is also named the artificial photosynthesis. Compared to conventional thermocatalytic CO2 reduction, photocatalytic CO2 reduction has the following advantages: (1) photocatalytic CO2 reduction reaction relies solely on solar energy as the external energy input; (2) the reaction utilizes water and CO2 as raw materials which are abundant and easily accessible; (3) the reaction is free of secondary pollution; and (4) photocatalytic CO2 reduction enables the synthesis of solar fuels and high value-added chemicals at ambient temperature and pressure. Thus, photocatalytic CO2 reduction is often regarded as one of the most promising solutions to solve the energy crisis and environmental problems. Since photocatalytic CO2 reduction is a multi-electron transfer process involving various reaction pathways, it is challenging to produce specific products with high selectivity. Photocatalysis (using single atoms, metal oxides, sulfides, nitrides, etc.) can be performed in a purely gas phase system or solution system, generating the main products including both C1 (e.g., CO, CH4, HCOOH, CH3OH) and C2+ (e.g., C2H4, C2H6, C2H5OH, C3H6) chemicals. The reaction conditions and the corresponding products are displayed in Table 1. When employing MHPs as catalysts, CO2 tends to be reduced to CO and CH4, accompanied by the side reaction of H2 evolution. In a typical CO2PR system, the total electron consumption rate and the electron consumption rate for the reduction of CO2 to carbon products are considered as Rtotal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) electron = 2R(CO) + 8R(CH4) + 2R(H2) and RCO2reduction = 2R(CO) + 8R(CH4), respectively. Thus, the selectivity of CO2 reduction can be calculated as (RCO2reduction)/(Rtotalelectron) × 100%. To evaluate the performance of photocatalysts, electron consumption rate and product selectivity are two important parameters.
electron = 2R(CO) + 8R(CH4) + 2R(H2) and RCO2reduction = 2R(CO) + 8R(CH4), respectively. Thus, the selectivity of CO2 reduction can be calculated as (RCO2reduction)/(Rtotalelectron) × 100%. To evaluate the performance of photocatalysts, electron consumption rate and product selectivity are two important parameters.
Table 1 The theoretical equilibrium potential (E0, V vs. NHE at pH = 7) and redox equation for the conversion of CO2 to different products
| Reaction equation |
Reduction products |
E
0 (V) |
| CO2 + e− = CO2− |
Carbonate anion radical |
−1.90 |
| CO2 + 2H+ + 2e− = CO(g) + H2O |
Carbon monoxide |
−0.53 |
| 2CO2 + 14H+ + 2e− = C2H6(g) + 4H2O |
Ethane |
−0.27 |
| CO2 + 8H+ + 8e− = CH4(g) + 2H2O |
Methane |
−0.24 |
| 2CO2 + 12H+ + 12e− = C2H4(g) + 4H2O |
Ethene |
−0.34 |
| CO2 + 2H+ + 2e− = HCOOH(aq) |
Formic acid |
−0.53 |
| CO2 + 4H+ + 4e− = HCHO(aq) + H2O |
Formaldehyde |
−0.48 |
| CO2 + 6H+ + 6e− = CH3OH(aq) + H2O |
Methanol |
−0.38 |
| 2CO2 + 8H+ + 8e− = CH3COOH(aq) + 2H2O |
Acetic acid |
−0.30 |
| 2CO2 + 10H+ + 10e− = CH3CHO(aq) + 3H2O |
Acetaldehyde |
−0.35 |
| 2CO2 + 12H+ + 12e− = C2H5OH(aq) + 3H2O |
Ethanol |
−0.33 |
| 3CO2 + 16H+ + 16e− = C2H5CHO(aq) + 5H2O |
Propionaldehyde |
−0.32 |
| 3CO2 + 18H+ + 18e− = C3H7OH(aq) + 5H2O |
Propanol |
−0.31 |
| CO2 + 4H+ + 4e− = C(s) + H2O |
Solid carbon |
−0.20 |
As mentioned above, MHPs are extensively applied in photoelectric devices due to their flexible and adjustable bandgap, long carrier lifetime, and high equilibrium carrier concentration. With the maturation of operating systems and the development of characterization techniques, the diversity of structures and intrinsic properties of MHPs have been explored, and their applications in environmental remediation and energy conversion have also received increasing research attention. In particular, photocatalytic CO2 reduction is considered as the utmost promising strategy toward sustainable development with net zero carbon emission. According to numerous studies, the predominance of CO as the main MHP-based reduction product can be attributed to its two-electron transfer mode.70,71 Moreover, CO is an important intermediate product in many chemical synthesis reactions and can be further converted into other organic compounds such as alcohols, ketones and acids.72,73 Therefore, the production of CO by photocatalytic reduction based on MHPs will be discussed in this section with emphasis on the design strategies to improve the efficiency.
3.1 Lead-based MHPs for CO2PR
In 2016, Park's team reported for the first time that an organic–inorganic mixed perovskite (MAPbI3) could drive hydrogen evolution in aqueous hydrogen iodide solution and remain stable throughout the catalytic process.74 As MHPs are unstable in polar solvents, ethyl acetate (EA) and acetonitrile (ACN) are generally considered as suitable media for the CO2PR reaction.8,52 Inspired by this, Xu et al. pioneered the synthesis of more stable CsPbBr3 perovskite quantum dots (CsPbBr3 QDs) in EA and successfully used them for photocatalytic CO2 reduction (Fig. 5a).52 However, the photocatalytic activity and durability of single-component CsPbBr3 QDs are still low, which largely limits their further application. For this reason, they used a simple room-temperature antisolvent precipitation to synthesize a CsPbBr3 QDs/GO composite, which exhibits a higher electron consumption rate of 29.8 μmol g−1 h−1 and the yield is much improved compared to CsPbBr3 QDs (Fig. 5b).52 Moreover, the CsPbBr3 QDs/GO composite shows an enhanced photocurrent response (Fig. 5c) and reduced carrier transfer resistance (Fig. 5d), indicating the more favorable kinetics of the composite photocatalyst compared with CsPbBr3 QDs. This study marks the first application of MHPs in CO2 photoreduction, inspiring subsequent works on MHP-based materials for photocatalytic CO2 reduction. To date, there have been many research works focusing on LHP-based composite photocatalysts, and the product is mainly CO with high selectivity (Table 2).
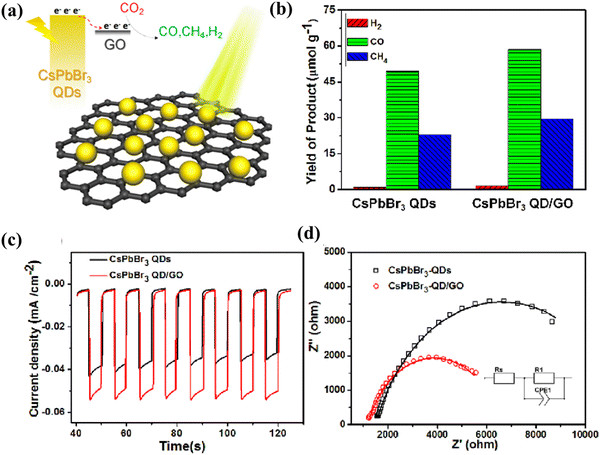 |
| | Fig. 5 (a) Photocatalytic performance: yield of the CO2 reduction products after 12 h of photochemical reaction.52 (b) UV-vis absorption spectra and the external quantum efficiency spectra.52 (c) Amperometric I–t curves plotted at −0.4 V Ag/AgCl under chopped AM 1.5G illumination.52 (d) EIS Nyquist plots recorded under 150-mW cm−2 illumination at a bias of −0.4 V Ag/AgCl; the lines are the corresponding fitted results. The inset is the equivalent circuit model.52 | |
Table 2 LHP-based catalysts for CO2PR with CO-dominated products
| Photocatalyst |
Light source |
System |
R
totalelectron (μmol g−1 h−1) |
Products |
Selectivity (%) |
Ref. |
| CsPbBr3 QDs |
100 W Xe-lamp, AM 1.5G |
EA |
23.7 |
CO, CH4, H2 |
99.3 |
37
|
| CsPbBr3/BP |
200 W Xe-lamp |
EA/H2O |
174.7 |
CO, CH4 |
∼100 |
54
|
| Co2%@CsPbBr3/Cs4PbBr6 |
— |
H2O |
27.1 |
CO, CH4 |
∼100 |
55
|
| CsPbBr3 NC/Pd NS |
150 W Xe-lamp, λ > 420 nm |
H2O (vapor) |
33.79 |
CO, CH4, H2 |
94.8 |
96
|
| CsPbBr3–Au |
100 W Xe-lamp, λ > 420 nm |
ACN/IPA |
47.7 |
CO, CH4 |
∼100 |
98
|
| 3DOM Au–CsPbBr3 |
— |
IPA/H2O/EA |
38.0 |
CO, CH4 |
∼100 |
99
|
| CsPbBr3–Re(600) |
— |
TL/IPA |
73.34 |
CO, H2 |
95 |
101
|
| CsPbBr3–Ni(tpy) |
— |
EA/H2O |
1252 |
CO, CH4 |
∼100 |
102
|
| Mn:CsPb(Br/Cl)3 |
— |
EA |
498.9 |
CO, CH4 |
∼100 |
106
|
| Non-cube CsPbBr3 |
450 W Xe-lamp |
EA/H2O |
182.95 |
CO, CH4 |
∼100 |
107
|
| CsPbBr3–OA/OAm |
— |
H2O (vapor) |
9.6 |
CO |
∼100 |
112
|
| CsPbBr3–glycine |
— |
H2O (vapor) |
55.4 |
CO |
∼100 |
112
|
| CsPbBr3QDs/UiO-66(NH2) |
300 W Xe lamp, λ > 420 nm |
EA/H2O |
101.65 |
CO, CH4 |
∼100 |
115
|
| TiO2/CsPbBr3 |
300 W Xe-lamp (UV-vis light) |
ACN/H2O |
<18.99 |
CO, H2 |
95 |
118
|
| CsPbBr3 QDs/Bi2WO6 |
— |
EA/H2O |
114.4 |
CO, CH4 |
∼100 |
121
|
| (ha)2CsPb2Br7 |
300 W Xe-lamp, full wavelength |
BAC |
377.58 |
CO, CH4 |
∼100 |
161
|
| CsPbBr3/FLG |
Laser diode λ = 405 nm |
EA |
255.4 |
CO, CH4, H2 |
>99 |
162
|
| CsPbBr3–BF4/Co |
— |
EA/IPA |
180 |
CO, CH4 |
∼100 |
163
|
| CsPbBr3 |
150 W Xe-lamp, λ > 380 nm |
EA |
2.7 |
CO, CH4, H2 |
95.2 |
164
|
| CsPbBr3/Pt |
150 W Xe-lamp, λ > 380 nm |
EA |
5.6 |
CO, CH4, H2 |
90.56 |
164
|
| CsPbBr3/Ti3C2Tx MXene |
300 W Xe-lamp, λ > 400 nm |
H2O (vapor) |
112.6 |
CO, CH4 |
∼100 |
165
|
| CsPbBr3 |
300 W Xe-lamp, AM 1.5G |
EA/H2O |
20.9 |
CO, CH4, H2 |
>99 |
166
|
| CsPbBr3/CTF-1 |
— |
EA |
96.4 |
CO |
∼100 |
167
|
| CsPbBr3/CTF-1-Ni |
— |
EA |
173 |
CO |
∼100 |
167
|
| CsPbBr3 NC/Pd NS |
150 W Xe-lamp, λ > 420 nm |
H2O (vapor) |
33.79 |
CO, CH4, H2 |
94.8 |
168
|
| WO3/CsPbBr3/ZIF-67 |
— |
H2O (vapor) |
∼66.25 |
CO |
∼100 |
169
|
| CsPbBr3/WO3 |
300 W Xe-lamp, λ > 400 nm |
EA/H2O |
∼400 |
CO |
∼100 |
170
|
| CsPbBr3/MoS2 |
— |
EA/H2O |
152.4 |
CO, CH4 |
∼100 |
171
|
| P3HT/CsPbBr3 |
— |
ACN/H2O |
475.3 |
CO, CH4 |
∼100 |
172
|
| CsPbBr3@GDY0.3 |
— |
ACN/H2O |
20.4 |
CO |
∼100 |
173
|
| CsPbBr3@GDY0.3–Co |
— |
ACN/H2O |
55.4 |
CO |
∼100 |
173
|
| C60/CsPbBr3 |
— |
ACN/H2O |
90.2 |
CO, CH4 |
∼100 |
174
|
| CsPbBr3@SnO2 |
300 W Xe-lamp |
EA/H2O |
128.2 |
CO, CH4 |
∼100 |
175
|
| CsPbBr3/AgBr |
— |
ACN/H2O |
141.1 |
CO, CH4 |
∼100 |
176
|
| CsPbBr3/BiOBr |
— |
EA/H2O |
72.3 |
CO, CH4 |
∼100 |
177
|
| MIL-100(Fe)–CsPbBr3 |
— |
H2O (vapor) |
82 |
CO, CH4 |
∼100 |
178
|
| 0D CsPbBr3/2D CsPb2Br5 |
— |
H2O (vapor) |
400.62 |
CO, CH4 |
∼100 |
179
|
| NMF/CsPbBr3-NWs |
300 W Xe-lamp, λ > 420 nm |
EA/H2O |
162 |
CO |
∼100 |
180
|
| CsPbBr3 PQDs/PbS |
— |
EA/H2O |
73.1 |
CO, CH4, H2 |
>99 |
181
|
| Mn:CsPbCl3 |
— |
— |
74 |
CO |
∼100 |
182
|
| CsPbBr3@MTB |
300 W Xe-lamp |
EA/H2O |
145.28 |
CO, CH4, H2 |
>99 |
183
|
| CsPbBr3/BiOCl |
300 W Xe-lamp |
EA/H2O |
97.2 |
CO, CH4 |
∼100 |
184
|
| CsPbBr3/CoAl-LDH |
300 W Xe lamp, λ > 420 nm |
EA/H2O |
41.2 |
CO, CH4 |
∼100 |
185
|
| CsPbBr3/NCP |
300 W Xe lamp, λ > 420 nm |
EA/H2O |
77.6 |
CO, CH4 |
∼100 |
186
|
| T-SrTiO3/CsPbBr3 |
— |
H2O (vapor) |
∼120.2 |
CO |
∼100 |
187
|
| Pb-rich Ni:CsPbCl3 NCs |
— |
H2O (vapor) |
339.4 |
CO, CH4 |
∼100 |
188
|
| CsPbBr3/PbSe |
— |
EA/H2O/IPA |
322.4 |
CO |
∼100 |
189
|
| CsPbBr3 QDs/BP NSs |
— |
H2O (vapor) |
76.6 |
CO |
∼100 |
190
|
| MWCNT/CsPbBr3 |
— |
ACN/H2O |
32.96 |
CO, CH4 |
∼100 |
191
|
| CsPbBr3@Cu-TCPP |
300 W Xe lamp, λ > 420 nm |
ACN/H2O |
150.04 |
CO, CH4 |
∼100 |
192
|
| Cs4PbBr6/rGO |
300 W Xe lamp, λ > 420 nm |
EA/H2O |
11.4 |
CO, CH4 |
∼100 |
193
|
| SrTiO3/CsPbBr3 |
300 W Xe lamp |
H2O (vapor) |
∼240.4 |
CO |
∼100 |
194
|
| CsPbBr3/USGO/a-Fe2O3 |
— |
ACN/H2O |
147.6 |
CO |
∼100 |
195
|
| CsPbBr3 NCs/MXene-20 |
— |
EA |
110.64 |
CO, CH4 |
∼100 |
196
|
3.2 Lead-free MHP-based CO2PR
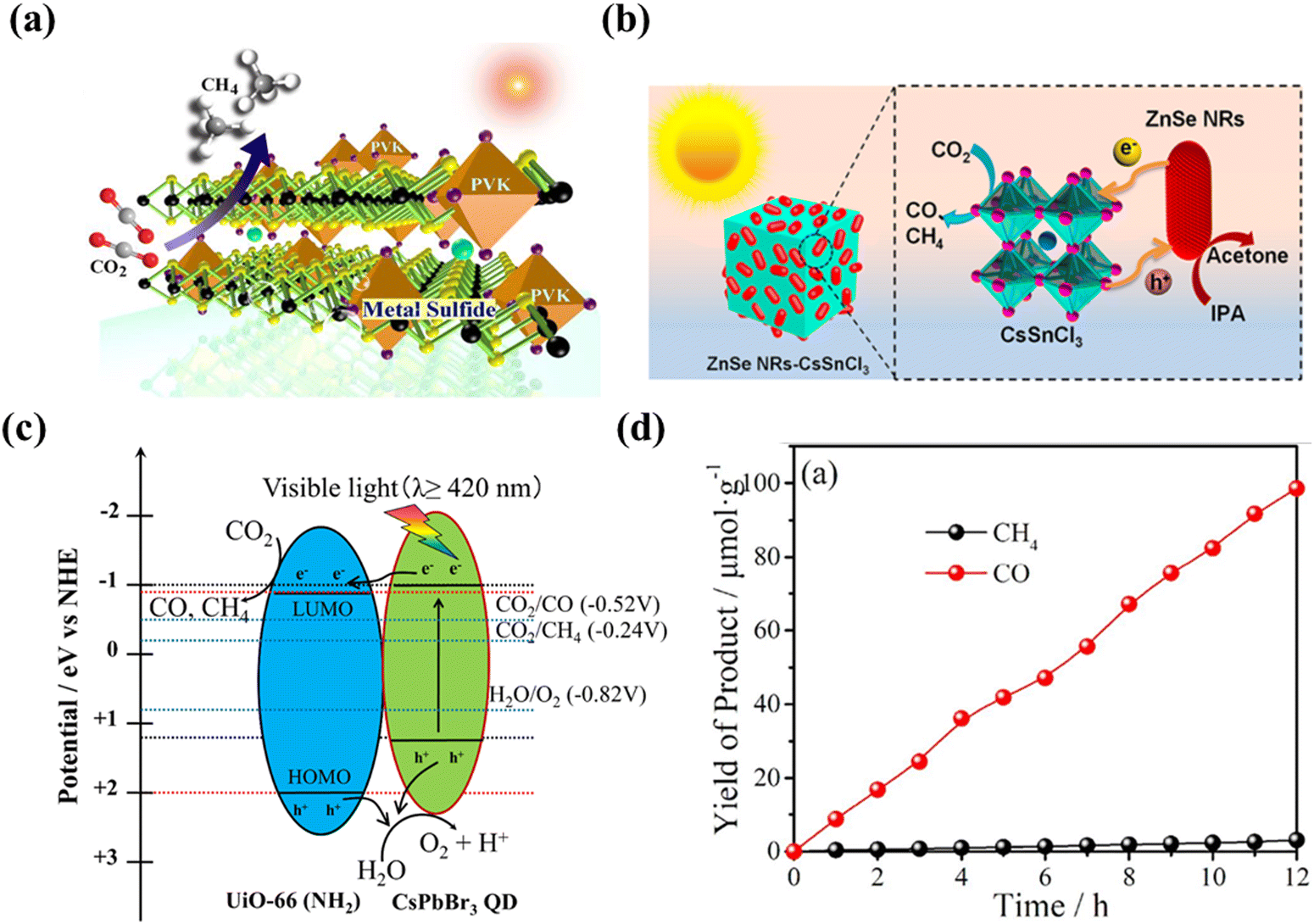
For reasons of environmental considerations and safety issues, the development of green, non-polluting perovskites for CO2 photoreduction is imperative, requiring partial or complete replacement of B-site Pb with atoms of similar radius, thus laying the groundwork for the development of CO2PR in lead-free systems. It has been discovered that the remarkable photoelectric properties of Pb-based perovskite materials can be attributed to their unique electronic configuration, including lone-pair Pb 6s2 electrons and an empty Pb 6p orbital. Moreover, the substantial size and weight of Pb further strengthen these properties, contributing to a robust spin–orbit coupling effect, as elucidated by recent studies.75,76 As such, in order to identify a stable and environmentally benign substitute for Pb, it is necessary to seek elements with similar atomic electronic configurations. Theoretically, the most suitable candidates for Pb replacement are group-14 metal elements, such as tin (Sn) and germanium (Ge), which also possess lone-pair s orbitals akin to Pb.77,78 However, a major challenge arises as Sn- and Ge-based perovskites exhibit poor stability due to the rapid oxidation of Sn2+/Ge2+ to Sn4+/Ge4+. Additionally, the potential toxicity of Sn to human beings upon environmental dispersion may surpass that of Pb, further complicating the quest for an ideal substitute. Hence, the pursuit of a stable and non-toxic Pb substitute demands meticulous investigation and consideration of various factors.
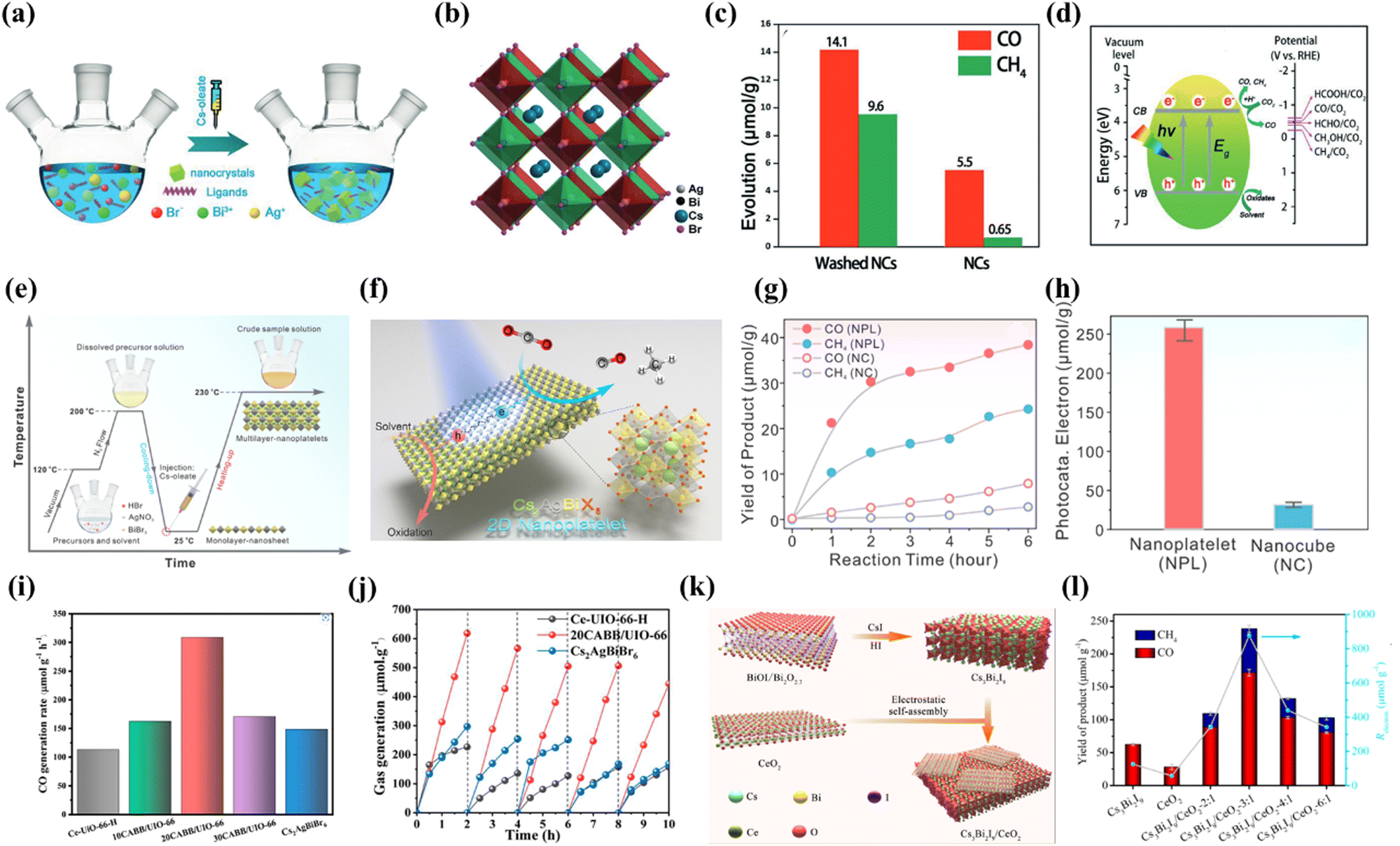
Recently, lead-free double perovskite Cs2AgBiBr6 has led to a research boom due to its non-toxicity, long carrier lifetime and high environmental stability. Taking advantage of these merits, Cs2AgBiBr6 has been adopted for NO removal,79 degradation of organic pollutants,67 H2 evolution,80–83etc. Not long ago, Cs2AgBiBr6 was also explored for photocatalytic reduction of CO2 and promising progress was made. For example, Zhou et al. developed novel lead-free double perovskite Cs2AgBiBr6 nanocrystals (NCs) for CO2 photoreduction by thermal injection (Fig. 6a).84 The corresponding crystal structure is shown in Fig. 6b, where Ag+ and Bi3+ ions located at the B and B′ sites combine with Br− to replace the conventional Pb2+, forming two types of regular octahedra. These two octahedra alternately join to form a rock salt face-centred cubic structure, with the larger Cs+ filling their gaps to form the typical A2BB′X6 structure. Photocatalytic CO2 reduction was carried out in EAC in a Pyrex glass bottle under simulated sunlight (AM 1.5G, 150 mW cm−2) irradiation. Both as-prepared and washed NCs were tested and the photocatalytic performance is displayed in Fig. 6c. After 6 h of continuous irradiation, the CO and CH4 yields are 5.5 and 0.65 μmol g−1, respectively, using Cs2AgBiBr6 NCs without an additional washing process. Fig. 6d presents a preliminary mechanism for the photocatalytic reduction of CO2 by Cs2AgBiBr6 NCs, where Cs2AgBiBr6 NCs have a suitable conduction band to drive CO2 reduction. This is the first example of the successful application of double perovskites for CO2 photoreduction. Based on the above research, scholars began to consider optimizing the dimensions to improve the performance of Cs2AgBiBr6. Very recently, Liu et al. fabricated two-dimensional Cs2AgBiBr6 nanoplatelets (NPLs) and found that Cs2AgBiBr6 NPLs exhibit stronger photocatalytic performance than the corresponding nanocubes (NCs), which was explained by the long in-plane diffusion length and anisotropic charge carryover of the NPLs, as compared to their NC counterparts (Fig. 6e).85Fig. 6f depicts the reaction mechanism of 2D Cs2AgBiBr6 NPLs, which is similar to that of 0D Cs2AgBiBr6 NCs, in which photogenerated charge carriers (electrons and holes) dissociate in the NPLs, with the holes subsequently consumed by solvent molecules and the electrons trapped by CO2. As shown in Fig. 6g, the production rates of both CO and CH4 are significantly higher when NPLs are utilized as catalysts. However, as the reaction time increases, the production rate gradually decreases, which is probably due to the surface poisoning of some surfaces of the photocatalysts. The overall electron consumption of Cs2AgBiBr6 NPLs during the reaction increases by more than 8 times compared to Cs2AgBiBr6 NCs (255.4 μmol g−1vs. 30.8 μmol g−1 in Fig. 6h). However, due to their inherent indirect bandgap, the single-component Bi-based perovskites show insufficient photocatalytic activity in practical applications.
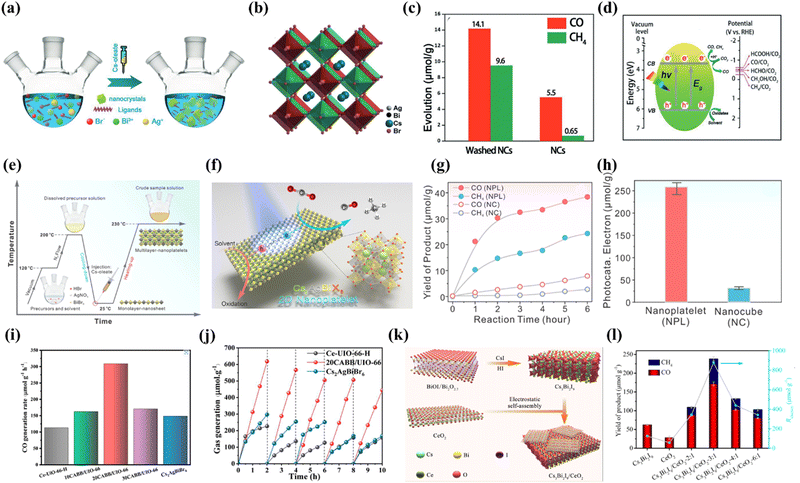 |
| | Fig. 6 (a) Schematic illustration of the solution phase synthesis of Cs2AgBiBr6 NCs (hot-injection route).84 (b) Crystal structure of cubic Cs2AgBiBr6.84 (c) Comparison of the photocatalytic CO2 reduction performance of the as-prepared Cs2AgBiBr6 NCs and washed NCs.84 (d) Schematic diagram of the photoreduction of CO2 on the surface of Cs2AgBiBr6 NCs.84 (e) Schematic illustration of the synthetic procedure of lead-free Cs2AgBiBr6 double perovskite 2D NPLs.85 (f) Schematic illustration of the Cs2AgBiBr6 NPL photocatalyzed CO2 reduction reaction.85 (g) CO (red) and CH4 (blue) product yield evolution as a function of reaction time using Cs2AgBiBr6 double perovskite NPLs (solid) and NCs (open) as catalysts.85 (h) Histogram of the total photocatalytic electron consumption in 6 h for the reactions using Cs2AgBiBr6 double perovskite NPLs (red) and NCs (blue) as catalysts.85 (i) CO generation rate during CO2 reduction under 300 W Xe lamp irradiation with as-prepared samples.45 (j) The recycling test for the photocatalytic activity of Ce-UiO-66-H, 20CABB/UiO-66, and Cs2AgBiBr6.45 (k) The preparation of Cs3Bi2I9 nanosheets and the self-assembly between Cs3Bi2I9 and CeO2 nanosheets.86 (l) The yields of CO and CH4 over Cs3Bi2I9, CeO2 and Cs3Bi2I9/CeO2-x:1 (x = 2–6) in 12 h.86 | |
Encouragingly, recent studies on catalyst growth and optimization of interfacial reactions/conditions are expected to lead to the further development of Bi-based perovskites. Furthermore, by compounding with other materials, building a two-component composite catalyst results in enhanced photocatalytic activity and stability. For instance, Ding's group reported an in situ assembly approach to produce a stable Cs2AgBiBr6/Ce-UiO-66-H composite, in which a tight contact interface is constructed between the two involved components.45 As shown in Fig. 6i, benefiting from the photocatalytic properties and the high adsorption capacity for CO2, the optimized 20Cs2AgBiBr6/Ce-UiO-66-H adsorption-photocatalyst shows excellent performance in CO2 reduction with a rather high CO production rate (309. 01 μmol g−1 h−1), which is 2.1 and 2.7 times higher than that of pure Cs2AgBiBr6 and Ce-UiO-66-H respectively. The excellent photocatalytic conversion of CO2 is attributed to the efficient solar energy harvesting and fast photoexcited carrier separation in the assembled structure. Importantly, due to the in situ synthesis, Cs2AgBiBr6 QDs are intercalated in the Ce-UiO-66-H framework, which leads to better stability and induces a large number of oxygen vacancies in Cs2AgBiBr6/Ce-UiO-66-H, maintaining a constant CO2 conversion during a continuous 10 h cycling test in wet air (illustrated in Fig. 6j). In addition, Feng's group constructed a Type-Z heterojunction composed of Cs3Bi2I9 nanosheets and CeO2 nanosheets (Cs3Bi2I9/CeO2-3:1) as a photocatalyst for CO2 photoreduction and H2O oxidation using an electrostatic self-assembly strategy (Fig. 6k).86 Owing to the well-matched energy levels and tight interfacial contacts, the separation efficiency of photogenerated carriers in the Cs3Bi2I9/CeO2-3:1 composite is significantly improved. The Cs3Bi2I9/CeO2-3:1 composite catalyst shows a high photocatalytic activity in the reduction of CO2 to CO/CH4 with an electron consumption yield of 877.04 μmol g−1, which is 7 and 15 times higher than the yields of pristine Cs3Bi2I9 and CeO2 nanosheets (Fig. 6l). This work exceeds the yields of other currently reported Bi-based perovskites in photocatalytic CO2 reduction.
Overall, MHPs have shown promising results in photocatalytic CO2 reduction to CO due to their high light absorption and efficient charge transfer properties. Particularly, lead-based perovskites have been extensively studied in this area and have achieved favorable conversion efficiencies. However, the toxicity of lead and concerns over stability have led to increased interest in lead-free alternatives. Lead-free perovskites such as Sn-based, Bi-based, and mixed-metal perovskites have been investigated for CO2 reduction and especially Bi-based perovskites have shown comparable or even superior performance to lead-based perovskites. Nonetheless, challenges such as instability under operating conditions and limited understanding of reaction mechanisms remain as major obstacles to their commercial viability. For this reason, the next section will focus on the strategies to improve the photoconversion efficiency and stability of MHP-based photocatalysts, with the purpose of bringing some enlightenment to the development of stable and efficient catalytic systems (Table 3).
Table 3 Lead-free MHP-based catalysts for CO2PR with CO-dominated products
| Photocatalyst |
Light source |
System |
R
totalelectron (μmol g−1 h−1) |
Products |
Selectivity (%) |
Ref. |
| Cs3Bi2I9/Bi2WO6 |
— |
H2O (vapor) |
14.67 |
CO |
∼100 |
42
|
| Cs3Bi2Br9 |
— |
H2O (vapor) |
54.6 |
CO, H2 |
98.7 |
43
|
| Cs2AgInCl6@Ag-2 |
300 W Xe lamp |
EA |
71 |
CO, CH4 |
∼100 |
44
|
| Cs2AgBiBr6/Ce-UiO-66-H |
— |
H2O (vapor) |
623.7 |
CO |
∼100 |
45
|
| Cs2AgBiBr6/Sr2FeNbO6 |
— |
EA/H2O |
164.96 |
CO, CH4 |
∼100 |
46
|
| Cs2CuBr4 |
— |
H2O (vapor) |
179.29 |
CO, CH4 |
∼100 |
47
|
| Cs2CuBr4@KIT-6 |
300 W Xe lamp |
EA/H2O |
240.5 |
CO, CH4 |
∼100 |
66
|
| Cs2AgBiBr6 |
— |
EA |
17.5 |
CO, CH4 |
∼100 |
84
|
| Cs3Bi2I9/CeO2-3:1 |
— |
H2O (vapor) |
877.04 |
CO, CH4 |
∼100 |
86
|
| Cs3Bi2I9 |
|
|
7.3 |
CO |
∼100 |
92
|
| Cs3Bi2(Cl0.5Br0.5)9 |
— |
H2O (vapor) |
32 |
CO |
∼100 |
92
|
| 3D Cs2NaBiCl6 |
300 W Xe lamp |
H2O (vapor) |
69.4 |
CO |
∼100 |
93
|
| Cs2CuBr4/CeO2 |
300 W Xe lamp |
EA/H2O |
241.82 |
CO, CH4 |
∼100 |
100
|
| MCM-41@Cs3Bi2Br9 |
— |
H2O (vapor) |
34.48 |
CO |
∼100 |
109
|
| Cs2AgBiBr6@MCM-48 |
300 W Xe lamp, λ > 420 nm |
EA |
99.13 |
CO, CH4 |
∼100 |
110
|
| ZnSe–CsSnCl3 |
— |
TL/IPA |
128.32 |
CO, CH4 |
∼100 |
114
|
| Cs2AgBiBr6/CTF-1 |
300 W Xe lamp, λ ≥ 420 nm |
EA |
130.2 |
CO, CH4 |
∼100 |
127
|
| Cs4CuSb2Cl12 |
300 W Xe lamp |
H2O (vapor) |
351 |
CO, CH4 |
∼100 |
197
|
| Cs3Sb2Br9 |
— |
ODE/H2O |
255 |
CO |
∼100 |
198
|
| Rb3Bi2I9 |
32 W UV-lamp, λ = 305 nm |
H2O (vapor) |
17.24 |
CO, CH4 |
∼100 |
199
|
| MA3Bi2I9 |
— |
— |
9.28 |
CO, CH4 |
∼100 |
199
|
| CABB/Ni-MOF |
300 W Xe lamp |
EA |
241.14 |
CO, CH4 |
∼100 |
200
|
| Cs2AgBiBr6/Bi2WO6 |
300 W Xe lamp, λ > 420 nm |
EA/IPA |
87.66 |
CO, CH4 |
∼100 |
201
|
| Cs3Sb2I9 |
— |
H2O (vapor) |
95.7 |
CO |
∼100 |
202
|
| Cs2AgBiBr6/Ti3C2Tx |
150 W Xe-lamp, λ ≥ 400 nm |
H2O (vapor) |
50.6 |
CO, CH4, H2 |
— |
203
|
| Cs3Bi2Br9/MOF 525 Co |
300 W Xe lamp |
H2O (vapor) |
124.8 |
CO, CH4 |
∼100 |
204
|
| Cs3Sb2(Br0.7I0.3)9 |
— |
H2O (vapor) |
18.5 |
CO |
∼100 |
205
|
| Cs2TeCl6 |
300 W Xe lamp |
H2O (vapor) |
320 |
CO, CH4 |
∼100 |
206
|
| Cs2TeBr6 |
300 W Xe lamp |
H2O (vapor) |
366.8 |
CO, CH4 |
∼100 |
207
|
| Cs2AgBiBr6@SiO2 |
λ > 420 nm |
ACN/H2O |
271.76 |
CO, H2 |
93.8 |
208
|
| Cs2AgBiBr6–Cu–RGO |
— |
H2O (vapor) |
91.4 |
CO, CH4, H2 |
97.8 |
209
|
3.3 Strategies to improve the photo-conversion performance of MHPs
3.3.1 Morphological regulation.
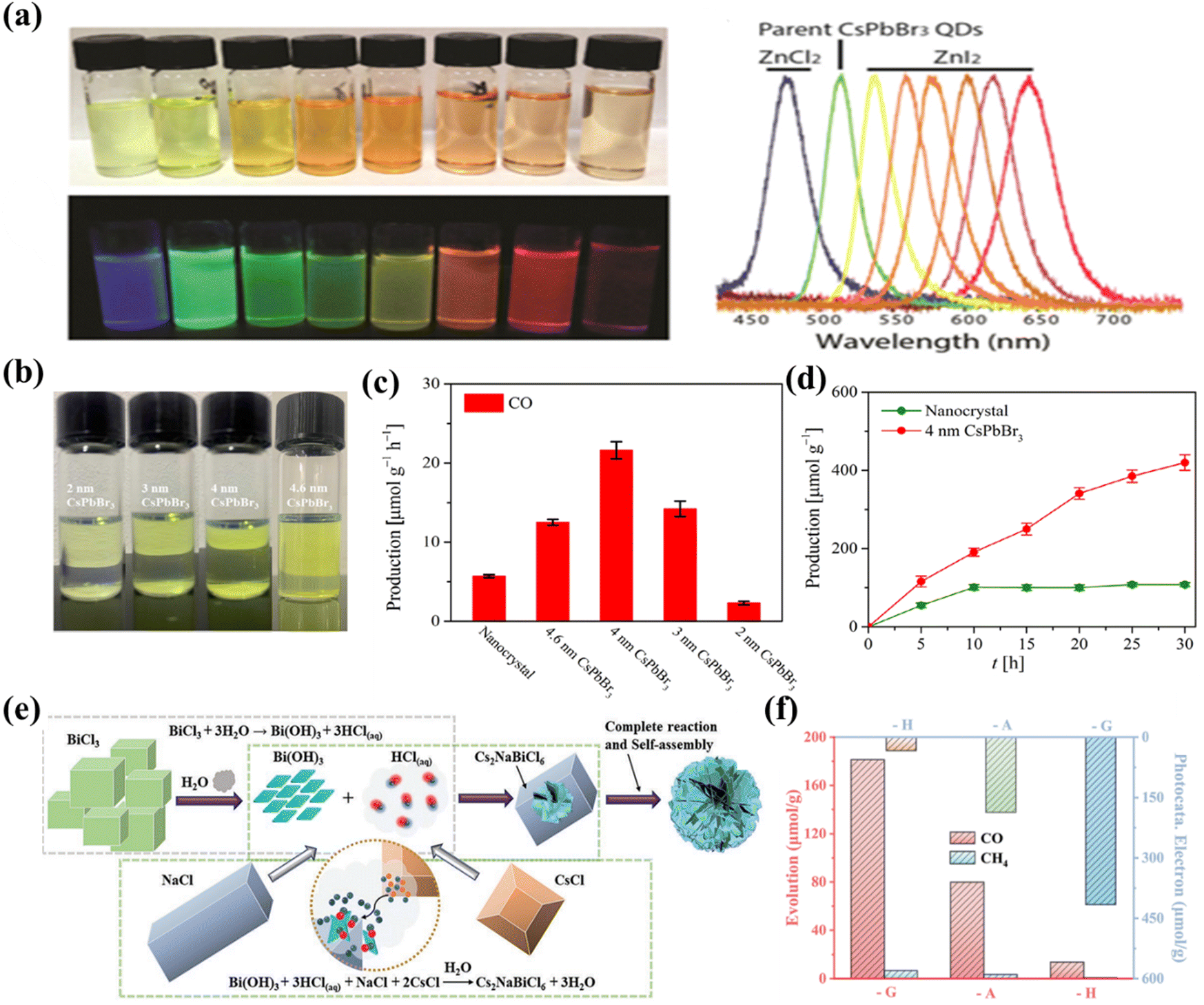
Perovskites have many different morphologies, such as 0D QDs, 1D rods, 2D nanosheets, 3D cube/star-shaped nanocrystals, etc., which can significantly affect the CO2 reduction performance. Sun et al. reported a variety of shapes of CsPbX3 materials prepared by a facile ligand-assisted method and found that CsPbX3 materials exhibit shape-dependent photoelectronic properties.87 For example, the extremely small particle size of CsPbBr3 QDs makes it significantly different from bulk materials in terms of optical and electronic properties. These 0D CsPbBr3 QDs can be easily functionalized via surface modification, with tunable luminescence wavelength when they are in solution and their forbidden bandwidth becomes narrower when they grow in aggregation (Fig. 7a). Similarly, a relevant report has demonstrated that the quantum dots are better than conventional fluorescent organic dyes in terms of tunable broad excitation and narrow emission spectra, signal brightness, high quantum yields and photostability.88 One of the properties of 2D materials is the large exciton binding energy due to dielectric shielding and reduced quantum confinement effects.89 For example, the exciton binding energy of monolayer WS2 is as high as 700 MeV. 2D halide perovskites have significant vibrational intensity, large exciton binding energy, effective radiative recombination and high photoluminescence quantum yields, which result from the large energy difference and high dielectric constant between the inorganic and organic layers.90 Moreover, the exciton binding energy of two-dimensional perovskites can be fine-tuned by inserting small molecular layers.91 In a study conducted by Wu et al., 2D CsPbBr3 nanosheets were synthesized at room temperature by varying the amount of hydrobromic acid employed in the synthesis process (as depicted in Fig. 7b), resulting in nanosheets with thicknesses of 2, 3, 4, and 4.6 nm.92 The effect of CsPbBr3 nanosheet thickness was investigated for photocatalytic CO2 reduction in water vapor and CO2 gas (gas–solid system without a sacrificial agent). It was found that the thickness of CsPbBr3 nanosheets has a significant effect on the performance of CO2 photoreduction, and the CO generation rates of 2.3, 14.2, 21.6, 12.5 and 5.7 μmol g−1 h−1 are observed for 2, 3, 4 and 4.6 nm thick CsPbBr3 nanosheets and nanocrystal CsPbBr3, respectively (Fig. 7c and d). Among these, CsPbBr3 nanosheets with a thickness of 4 nm exhibit the highest photocatalytic activity, which can be attributed to the suitable visible light absorption range and the higher number of photogenerated carriers for CO2 reduction. From another perspective, the 2D CsPbBr3 nanosheets have shorter carrier transfer distances and uncoordinated active metal sites compared to the bulk CsPbBr3 nanocrystals, favoring the transport of photogenerated carriers to the catalyst surface. In a study by Pi and his collaborators, they prepared chlorine-deficient 3D-graded Cs2NaBiCl6 porous microspheres assembled from highly crystalline nanosheets by a grinding method (Fig. 7e).93 The CO yield of gas–solid photocatalytic reduction of CO2 in the absence of sacrificial agents reached 30.22 μmol g−1 h−1. Experimental results and density functional theory (DFT) calculations indicate that the chlorine vacancies possess the triple effect of inhibiting photogenerated electron–hole complexation, enhancing CO2 adsorption, and significantly reducing the free energy barrier for the generation of the key intermediate COOH*. Moreover, the surface and defect engineering coupling of the 3D hierarchical sample brings a 12.34-fold enhancement of CO2 photoreduction activity compared to the pristine Cs2NaBiCl6 (Fig. 7f).
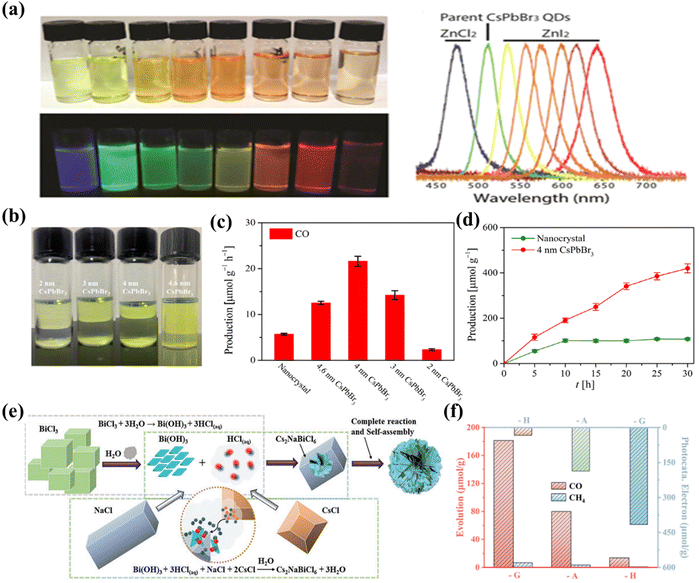 |
| | Fig. 7 (a) Anion exchange of the as-synthesized spherical CsPbBr3 quantum dots with stable dispersions in toluene under room light or excitation using an ultraviolet lamp at room temperature and PL spectra of the solutions.87 (b) Photographic images of CsPbBr3 nanosheets exposed to sunlight (top) and an ultraviolet lamp (bottom) in hexane.92 (c) The comparison of CO production with CsPbBr3 NCs and CsPbBr3 nanosheets as photocatalysts after 5 h of irradiation under a 300 W Xe lamp, with a light intensity of 100 mW cm2.92 (d) Photocatalytic evolution of CO catalyzed by CsPbBr3 NCs and 4 nm CsPbBr3 nanosheets in the CO2 and water vapor system.92 (e) A feasible growth diagram of three-dimensional hierarchical structure Cs2NaBiCl6-G.93 (f) CH4 and CO generation from CO2 photoreduction using different star nanocrystals.93 | |
3.3.2 Co-catalyst modification.
When used on their own, co-catalysts have little or no activity. When used as an additive to the primary catalyst, the addition of co-catalysts can change the catalyst surface structure, the distribution of pore structure, the valence of ions, acidity and alkalinity, etc., which can improve the overall activity, stability, selectivity and lifetime of the catalyst. In photocatalytic reactions, the rate of charge separation is one of the critical factors determining the overall efficiency. Utilizing co-catalysts to improve the photoconversion efficiency has been reported as an effective strategy in previous studies.94,95 Specifically, the roles played by co-catalysts in the photoreduction process include the following: (1) promoting the separation of photogenerated electron–hole pairs; (2) reducing the overpotential required for CO2 reduction; (3) improving the overall catalyst stability; and (4) enhancing the selectivity of CO2 and hindering other side reactions. In order to better describe the roles of co-catalysts, several typical co-catalytic reaction systems are described later.
3.3.2.1 Noble metal deposition.
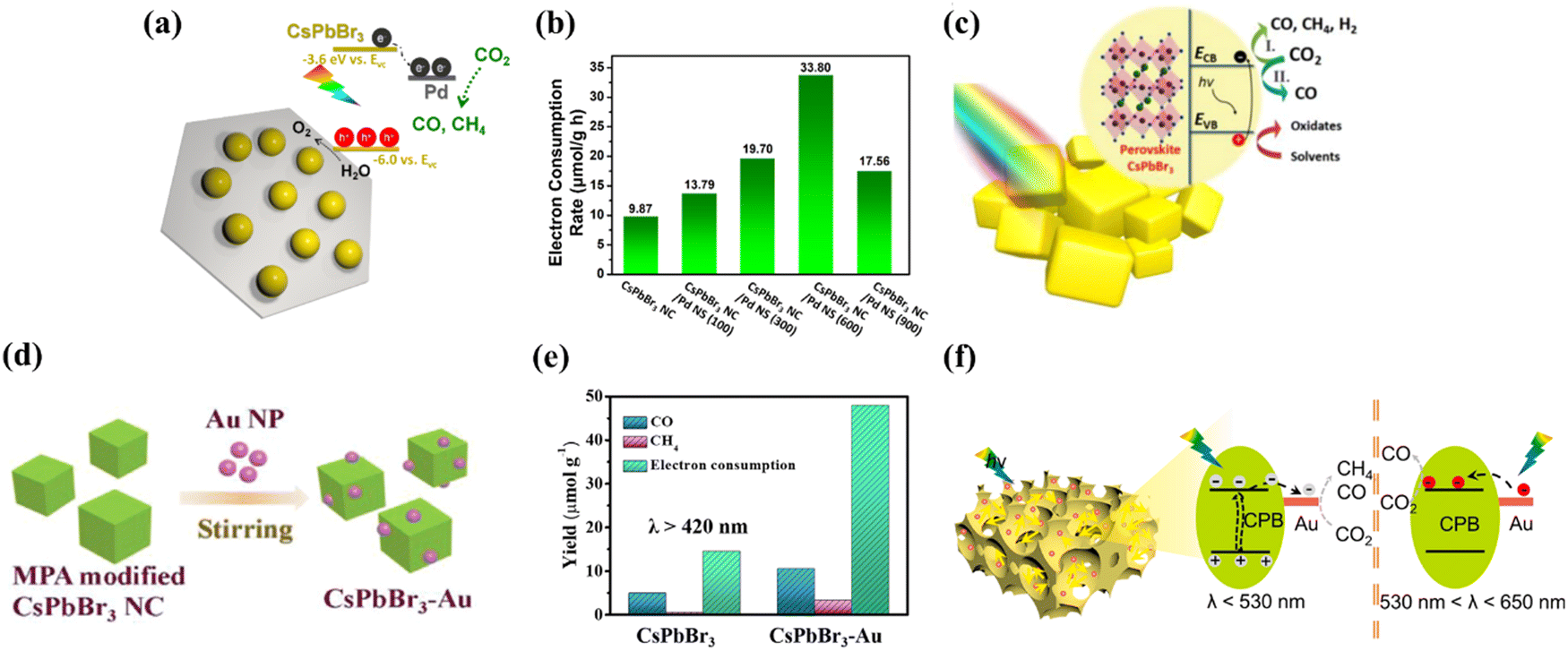
Most semiconductors require additional catalysts to achieve high CO2 reduction activity, even in the presence of a sacrificial electron donor. This is caused by both the tendency of electron–hole pairs to recombine before reaching the surface for reaction and the slow surface reaction that does not allow efficient charge consumption. With the purpose of extracting electrons to the surface, metals such as platinum are traditionally used as co-catalysts. Noble metals, in particular, act as electron sinks and provide effective proton reduction sites that greatly enhance the proton reduction reaction. Of these, Pd and Pt are widely used as typical noble metal co-catalysts for photocatalytic reactions. As an example, Kuang’ s group prepared a novel 0D CsPbBr3 nanocrystal/2D Pd nanosheet composite photocatalyst that can catalyze CO2 conversion efficiently and stably under visible light irradiation (Fig. 8a).96 The enhanced photocatalytic activity can be explained by the Pd nanosheets acting as an electron reservoir, rapidly separating electron–hole pairs in the CsPbBr3 nanocrystals via Schottky contacts and providing ideal sites for the CO2 reduction reaction. The highest electron consumption rate of the CsPbBr3 nanocrystal/Pd nanosheet composite is 33.79 μmol g−1 h−1, which is 2.43 times higher than that of the pristine CsPbBr3 nanocrystal (9.86 μmol g−1 h−1), providing a practical and versatile solution to enhance the photocatalytic performance of MHPs through semiconductor/metal design (Fig. 8b). Meanwhile, their team also reported CsPbBr3 as a photocatalyst for CO2 reduction with high stability and selectivity in non-aqueous solvents.97 In view of the crucial role of solvents in enhancing the photocatalytic reaction rate, EA was chosen as the most effective solvent. Besides, the addition of a Pt co-catalyst by photochemical deposition further improves the electron yield, highlighting the importance of optimizing the reaction medium and surface catalytic activity for superior performance (Fig. 8c). In addition to Pd and Pt, the loading of Au as a co-catalyst on the inner/outer surface also facilitates the efficient conversion of CO2 to CO. Recently, Liao and co-workers explored a novel photocatalyst (CsPbBr3–Au), which has the ability to excite wavelength-dependent photocatalytic reduction of CO2 (Fig. 8d).98 Under visible light (λ > 420 nm) illumination, photogenerated electrons in CsPbBr3 are available for injection into Au with an electron injection rate and efficiency of 2.84 × 109 s−1 and 78%, respectively. The enhanced charge separation further translates into 3.2-fold enhancement in photocatalytic CO2 reduction activity compared to pristine CsPbBr3 (Fig. 8e). Experimental results indicate that the nanocomposite has a strong local surface plasmon resonance effect in the visible region and can effectively promote the CO2 reduction reaction. Not too long ago, Huang et al. demonstrated a three-dimensionally ordered macroporous Au–CsPbBr3 (3DOM Au–CPB) composite loaded with gold nanoparticles, achieving better carrier separation performance through band bending effects at the Au/CPB interface (Fig. 8f).99 As for Bi-based perovskites, Au loading also enhances the CO2 photoreduction efficiency. For example, Fu et al. prepared a Cs3Bi2Br9/V2O5 photocatalyst that reduced CO2 to CO with a yield of 37.8 μmol g−1 h−1, which is 5.3 times higher than that of pure Cs3Bi2Br9.100 In addition, the loading of Au nanoparticles on Cs3Bi2Br9/V2O5 as a co-catalyst further improves the separation efficiency of the photogenerated charge carriers. The optimum Au–Cs3Bi2Br9/V2O5 composite exhibits a high CO yield of 98.95 μmol g−1 h−1 with a selectivity of 98.74%.
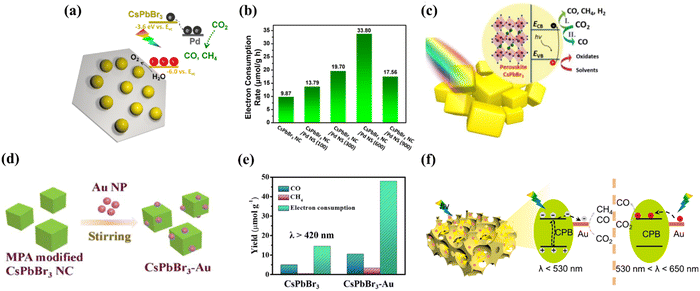 |
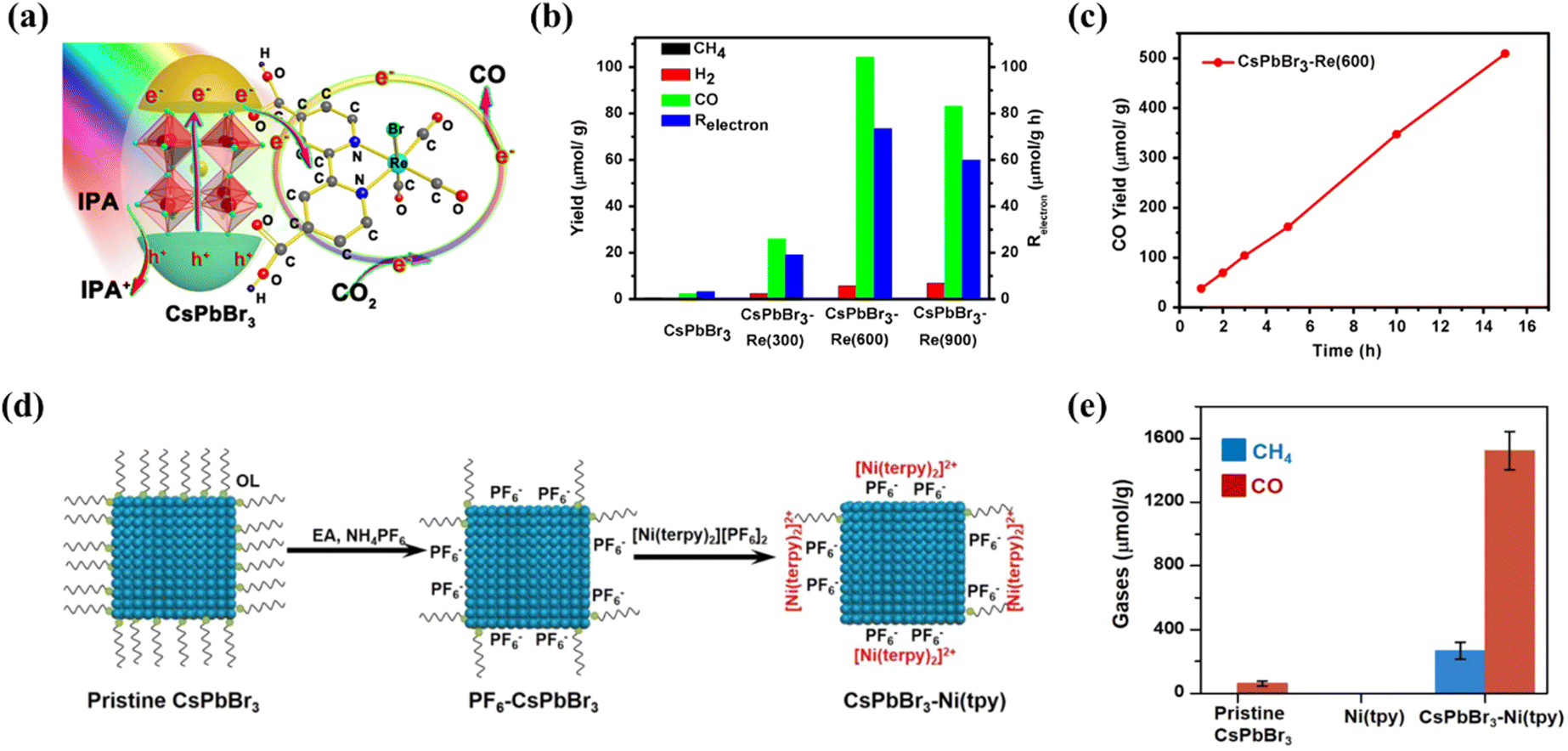
| | Fig. 8 (a) The sketch of the composite material and its corresponding band alignments.96 (b) The electron consumption rates under visible light illumination (>420 nm).96 (c) Schematic diagram of the photoreduction of CO2 on the surface of CsPbBr3 crystals with (I) and without (II) the participation of protons.97 (d) Sketch of the fabrication of CsPbBr3–Au nanocomposites; morphology characterization of the synthesized CsPbBr3–Au nanocomposite.98 (e) Photocatalytic performance: yield of the CO2 reduction products and electron consumption over CsPbBr3 and CsPbBr3–Au under illumination with λ > 420 nm.98 (f) The possible charge transfer route during the photocatalytic reaction.99 | |
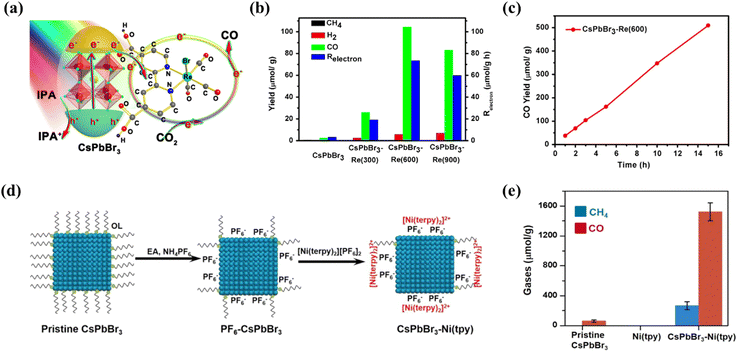 |
| | Fig. 9 (a) Schematic diagram of the working principle of the CsPbBr3–Re(CO)3Br(dcbpy) composite in CO2 reduction.101 (b) Photocatalytic CO2 reduction performances: product yield after 3 h of reaction.101 (c) Time course of CO yield of CsPbBr3–Re(600) during 15 h of continuous illumination.101 (d) Schematic of the formation of the CsPbBr3–Ni(tpy) photocatalyst.102 (e) Production of gases using various photocatalysts based on pristine CsPbBr3, pristine Ni(tpy) and CsPbBr3–Ni(tpy); error bars correspond to standard deviations based on three experiments.102 | |
3.3.3 Ion doping.
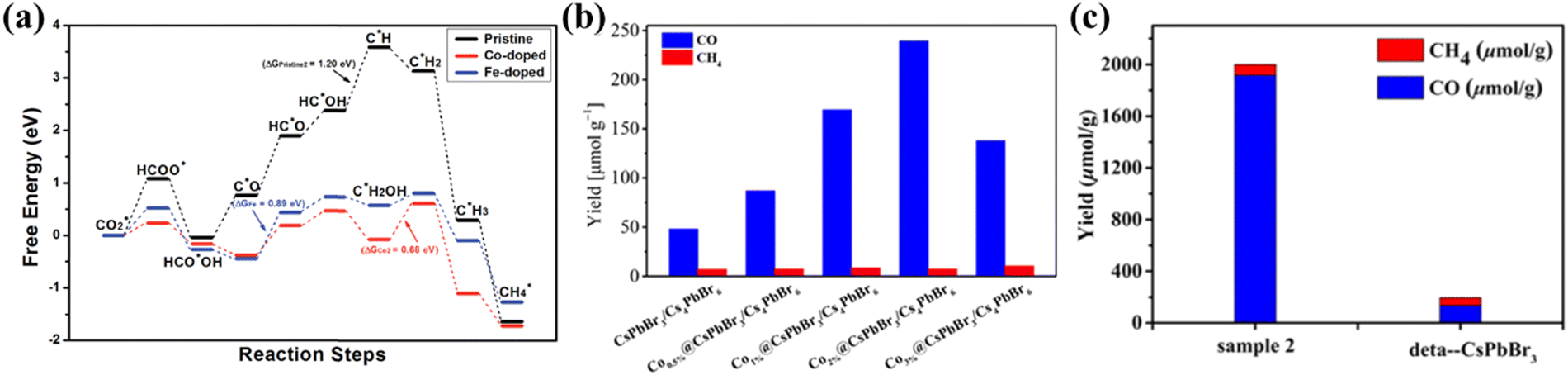
Doping is considered as an effective and facile method to improve the catalytic performance of photocatalysts.103 By doping and substituting heterogeneous cations in LHPs, their overall photoelectric performance can be significantly improved.104 Recently, Tang et al. used DFT calculations to predict that when Co and Fe atoms are doped into the perovskite, its catalytic capacity for CO2 reduction will increase.105 The basis is in the case of Co and Fe doping, the perovskite adsorbs CO2*, C*OOH and HCOO* intermediates much more strongly than the pristine perovskite (Fig. 10a). Inspired by it, some scholars have proven the theory. Dong et al. promoted the catalytic activity of perovskite NCs in visible light-driven CO2 reduction by increasing the number of catalytic sites and improving charge separation through direct doping of Co cations on the CsPbBr3/Cs4PbBr6 surface.55 Co1%@NCs doped with Co achieve higher CO yields (1385 mmol g−1) compared to the undoped NCs (678 mmol g−1) in a 15 h reaction (Fig. 10b). In addition, Mn doping is also an effective strategy. Liu's group demonstrated a Mn-doped CsPb(Br/Cl)3 mixed halide perovskite as a catalyst to improve CO2 photoreduction efficiency.106 A series of CsPb(Br/Cl)3:Mn perovskites were obtained by adjusting the Mn content. For the optimum catalyst sample, the yield of CO reached 1917 μmol g−1 which is 14.2-fold higher than that of CsPbBr3 (Fig. 10c).
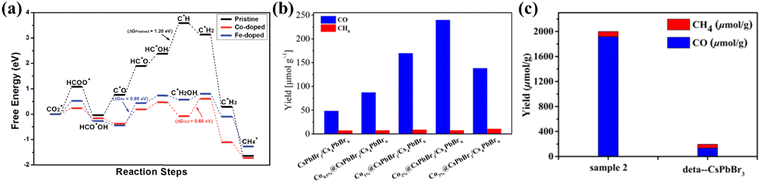 |
| | Fig. 10 (a) The free energy diagrams of the most favored paths of CO2 reduction in pristine, Co-doped and Fe-doped cases. The DG of the rate-determining step for each case is listed.105 (b) The yields of CO and CH4 generated from photocatalytic CO2 reduction in pure water based on undoped and Co-doped CsPbBr3/Cs4PbBr6 with various doping concentrations.55 (c) Comparison chart of the yields between Mn-doped optimum catalyst sample 2 and CsPbBr3.106 | |
3.3.4 Crystal plane regulation.
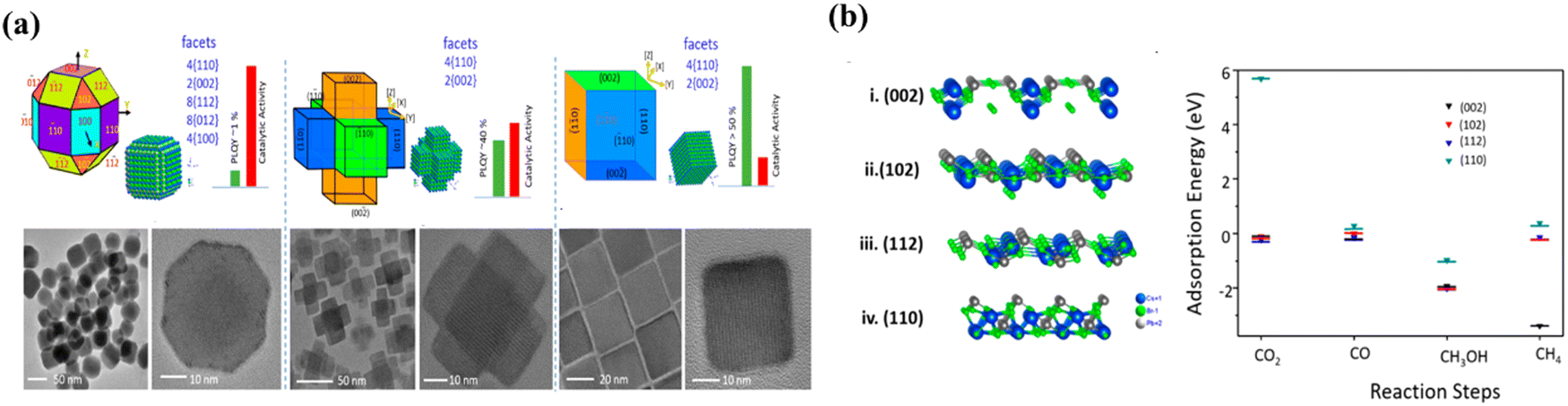
Crystal faces of LHPs also play a pivotal role in determining their catalytic activity and selectivity for CO2 reduction. Shyamal et al. prepared CsPbBr3 materials with different exposure surfaces for photocatalytic CO2 reduction.107 Polyhedral non-cubic, hexapod and cubic CsPbBr3 nanocrystals were synthesized, which are schematically shown in Fig. 11a. Despite having similar surface areas, the halogen deficient polyhedral CsPbBr3 exhibits superior CO2 reduction activity compared to the halogen-rich cubic CsPbBr3. DFT calculations indicate that the reactivity is facet-dependent with the activity order of (112) > (102) > (002) > (110). Thus, polyhedral CsPbBr3 with the (112) facet would be a more efficient photocatalyst for CO2 reduction compared to hexapod and cubic CsPbBr3 (Fig. 11b).
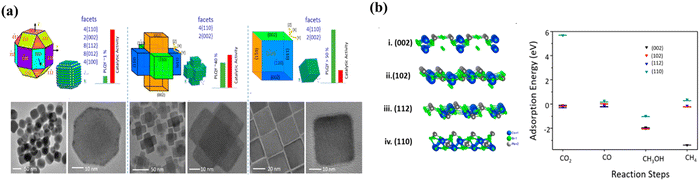 |
| | Fig. 11 (a) Models showing polyhedral noncube-, hexapod-, and cube-shaped CsPbBr3 nanostructures, respectively. In each case, facets were labeled according to the assigned viewing axis and are also listed separately. Approximate PLQYs and observed catalytic activity in CO2 reduction are provided in bar diagrams. TEM and HRTEM images of noncube-, hexapod-, and cube-shaped nanostructures, respectively.107 (b) Atomic model showing the (002), (102), (112) and (110) facets of orthorhombic CsPbBr3 (Pbnm) and adsorption energies of CO2, CO, CH3OH, and CH4 on top of (002) (black), (102) (red), (112) (blue), and (110) (cyan) facets.107 | |
3.3.5 Encapsulation.
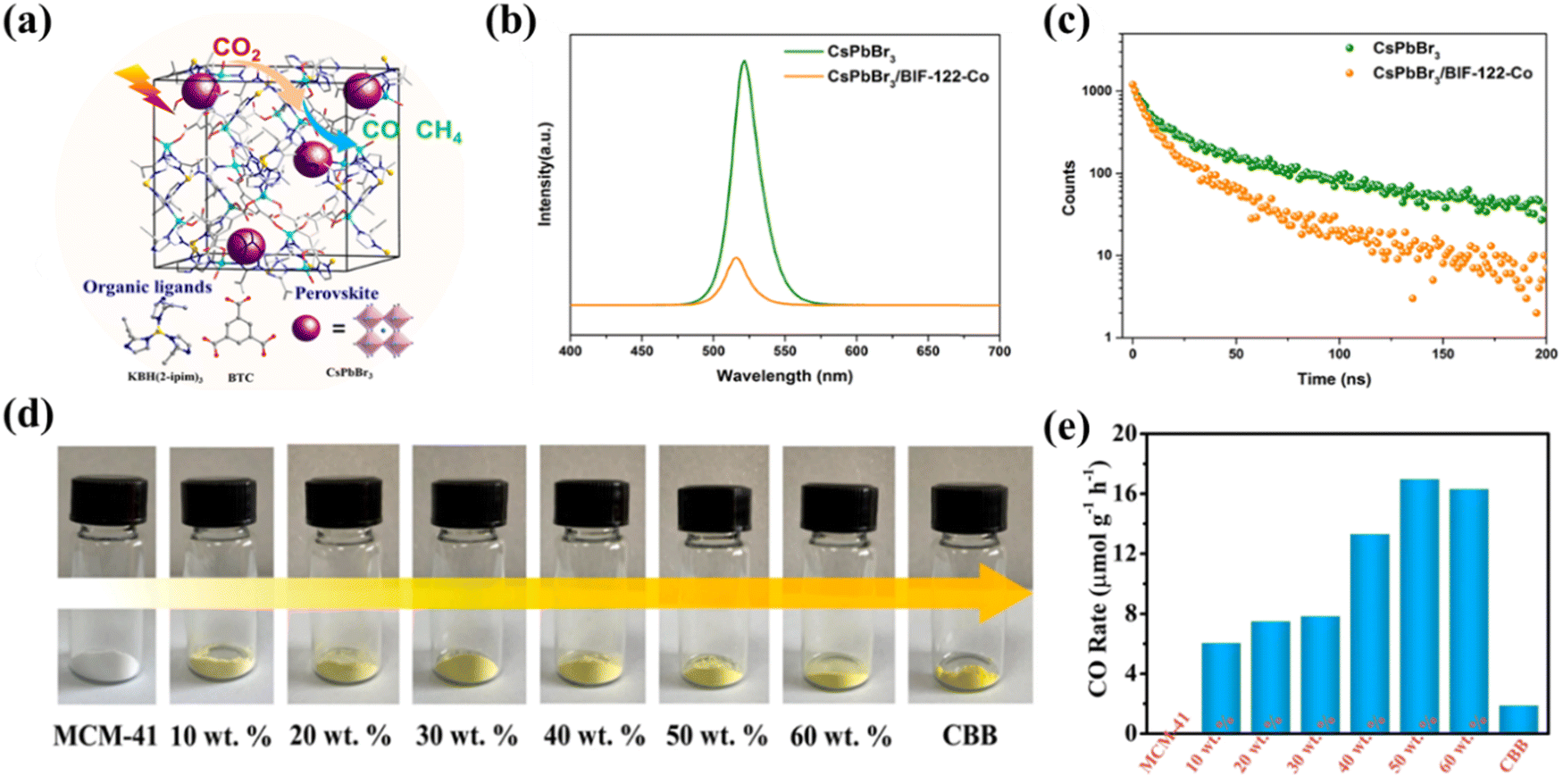
Encapsulation has been demonstrated to be an effective method to prevent the decomposition of halide perovskites and improve their stability. Chen et al. synthesized microporous crystals of BIF-122-Co and encapsulated CsPbBr3 perovskites as the main substrate to obtain a composite CsPbBr3/BIF-122-Co (Fig. 12a) with high photocatalytic CO2 reduction activity.108 PL spectroscopy and electrochemical experiments indicate (Fig. 12b and c) that photogenerated electrons in the encapsulated CsPbBr3 could be rapidly transferred to BIF-122-Co, thereby inhibiting electron–hole complexation in individual CsPbBr3. Moreover, the porous BIF-122-Co backbone in the composite also significantly improves the stability of CsPbBr3 perovskites in the photocatalytic reaction system. Mesoporous molecular sieves have received great attention as adsorbents, catalysts, and catalyst supports. Cui et al. synthesized space-restricted lead-free halide perovskite Cs3Bi2Br9 in MCM-41 molecular sieve using a simple and effective impregnation method (Fig. 12d).109 The MCM-41@CBB (Cs3Bi2Br9) composite material shows excellent performance in photocatalytic reduction of CO2 to CO under visible light irradiation and gas–solid conditions, in which CO generation rate of MCM-41@50 wt% CBB was measured to be 17.24 μmol g−1 h−1, far exceeding that of pure CBB (1.89 μmol g−1 h−1) (Fig. 12e). Similarly, very recently, Zhang's team have grown Cs2AgBiBr6 (CABB) nanocrystals in situ in MCM-48 mesoporous molecular sieve for photocatalytic CO2 reduction.110 The CABB@MCM-48 composite material shows excellent performance, far surpassing that of the original CABB. Such excellent photocatalytic performance of CABB@MCM-48 is attributed to enhanced CO2 adsorption, effective charge separation and an abundance of active sites.
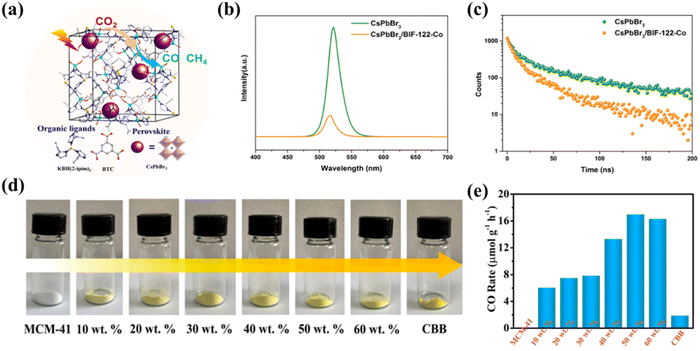 |
| | Fig. 12 (a) Schematic illustration of the surface modification of CsPbBr3 NCs.108 (b) Schematic representation of CO2 photoreduction over the CsPbBr3–Ni(tpy) photocatalyst system.108 (c) The CsPbBr3 QDs were anchored on the polymeric semiconductor g-C3N4 porous nanosheets via strong N–Br bonds.108 (d) The MCM-41, CBB and MCM-41 supported CBB with different weight ratios.109 (e) The photocatalytic performance of CBB, MCM-41 and CBB loaded in MCM-41 with different weight ratios.109 | |
3.3.6 Surface finishing.
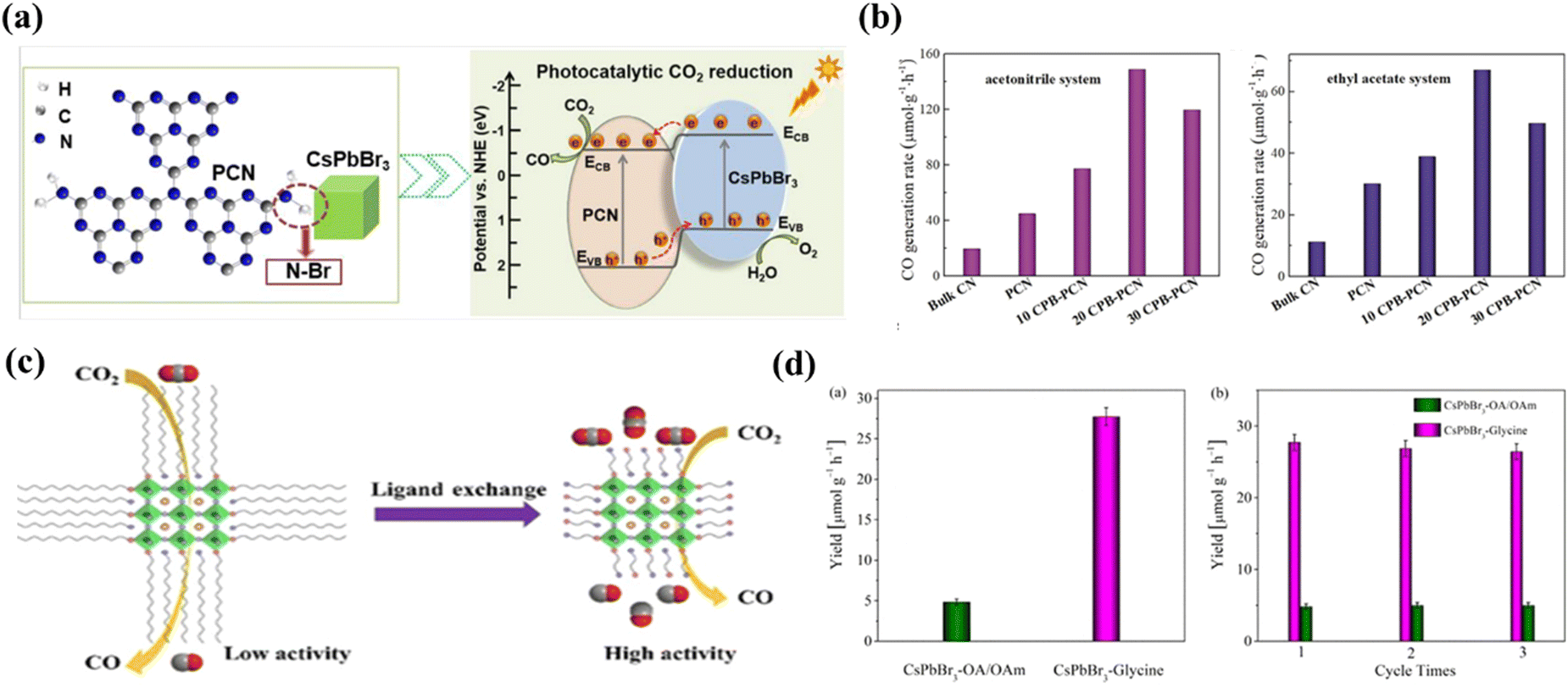
Surface properties of semiconductors play a critical role in facilitating reactant adsorption, product desorption, and interfacial charge transfer. Therefore, the development of an effective surface modification strategy for halide perovskites is fundamental to achieving optimal photocatalytic performance. The strategy is especially crucial given that the performance of these materials is highly dependent on their surface properties. Fig. 13a shows a composite photocatalyst system constructed by Xu et al. using N–Br chemical bonding to immobilize CsPbBr3 quantum dots on the surface of NHx-rich porous g-C3N4 nanosheets.111 This composite photocatalyst exhibits good stability and high yield of 149 μmol h−1g−1 in the acetonitrile/water system, which is 15-fold higher than that of the pristine CsPbBr3 quantum dots. The special N–Br bonding state led to enhanced charge separation between the two materials and improved charge carrier lifetime, contributing to excellent photocatalytic performance (Fig. 13b). Besides, ligand capping is essential for the preparation of stable MHP nanocrystals (NCs). For example, Xu et al. constructed CsPbBr3 nanocrystals with short-chain glycine as a ligand through a ligand exchange strategy.112 Photogenerated carrier separation and CO2 absorption are significantly improved due to the reduced site barrier of glycine and the presence of amine groups in the glycine without affecting the stability of the MHP NCs (Fig. 13c). The yield of CsPbBr3 nanocrystals with glycine ligands for photocatalytic CO2 conversion to CO without any organic sacrificial reagent increased significantly to ∼27.7 μmol g−1 h−1 (Fig. 13d), which is 5-fold higher than that of CsPbBr3 NCs with conventional long alkyl capped ligands (OA-OAm).
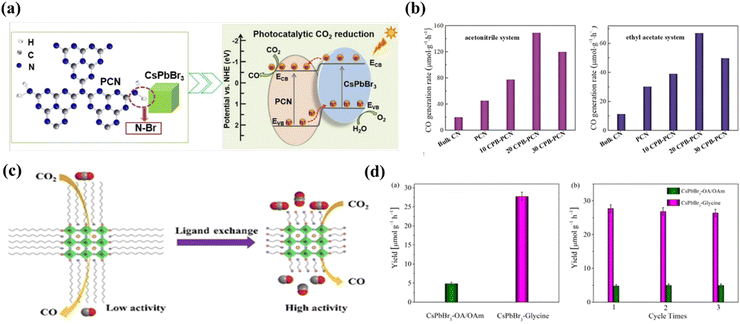 |
| | Fig. 13 (a) CsPbBr3 QDs were anchored on the polymeric semiconductor g-C3N4 porous nanosheets via strong N–Br bonds.111 (b) Generation of CO in the acetonitrile/water (left) and ethyl acetate/water system (right) over various photocatalysts.111 (c) Glycine-functionalized CsPbBr3 nanocrystals are constructed through a facile ligand-exchange strategy.112 (d) Yields of CO generated from CO2 reduction with CsPbBr3-OA/OAm and CsPbBr3–glycine NCs as photocatalysts, under Xe-lamp irradiation for 4 h with a 400 nm filter and a light intensity of 100 mWcm−2 (left) and yields of CO generated from CO2 reduction for three consecutive cycles of 4 h each with CsPbBr3-OA/OAm and CsPbBr3–glycine NCs as photocatalysts (right).112 | |
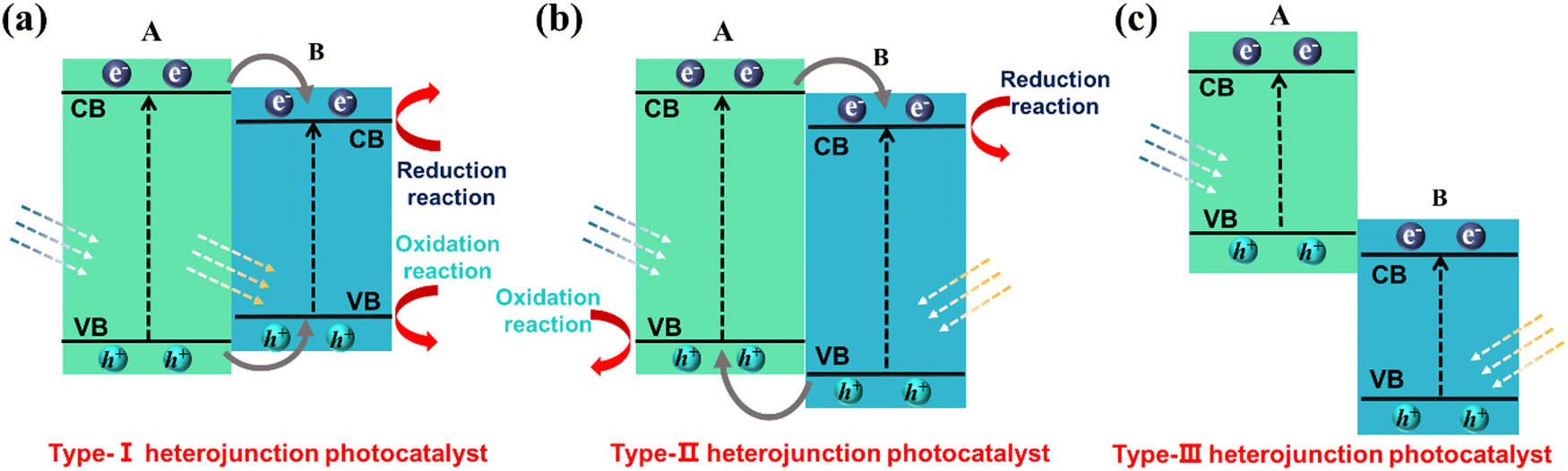
3.3.7 Heterogeneous junction engineering.
The construction of heterogeneous structures is generally considered to be an effective method that can overcome the limitations of single catalysts, namely slow electron transfer and severe radiation compounding. Heterojunctions improve the photocatalytic efficiency by optimizing the electron ‘path’ and transferring electrons and holes to minimize the loss rate of valuable electrons–holes. Specifically, by choosing two (or more) semiconductor materials, heterojunctions are formed by suitable synthesis methods. A matched energy band arrangement can significantly accelerate carrier transfer and suppress recombination. It is generally accepted that there are three types of heterogeneous junction structures: type I (spanning gap) (Fig. 14a), type II (staggered gap) (Fig. 14b) and type III (fractured gap) (Fig. 14c). Of these, type II heterojunctions are considered as the most suitable for facilitating the separation of photogenerated carriers due to the spatial separation of photogenerated electrons and holes that are transferred to different semiconductors.
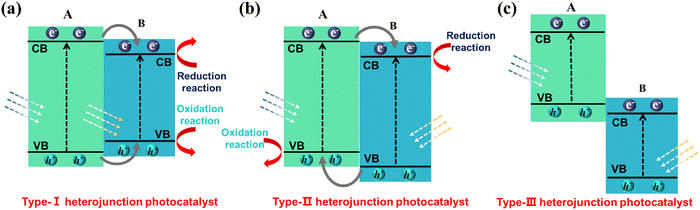 |
| | Fig. 14 Schematic illustration of electron–hole separation of three conventional heterojunctions: (a) type-I, (b) type-II and (c) type-III. | |
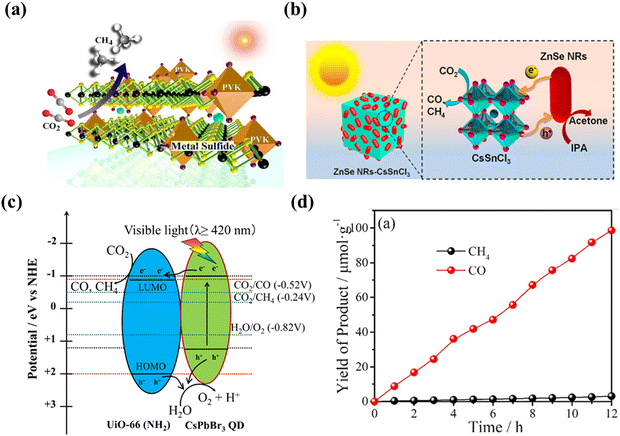 |
| | Fig. 15 (a) Schematic illustration of the photocatalytic CO2 reduction.113 (b) Schematic representation of the CO2 photoreduction process on ZnSe–CsSnCl3.114 (c) Schematic illustration of the possible mechanism of photocatalytic CO2 reduction on CsPbBr3/UiO-66(NH2).115 (d) Photocatalytic CO2 reduction into chemical fuels under a 300 W Xe lamp for 15%-CsPbBr3 QDs/UiO-66(NH2).115 | |
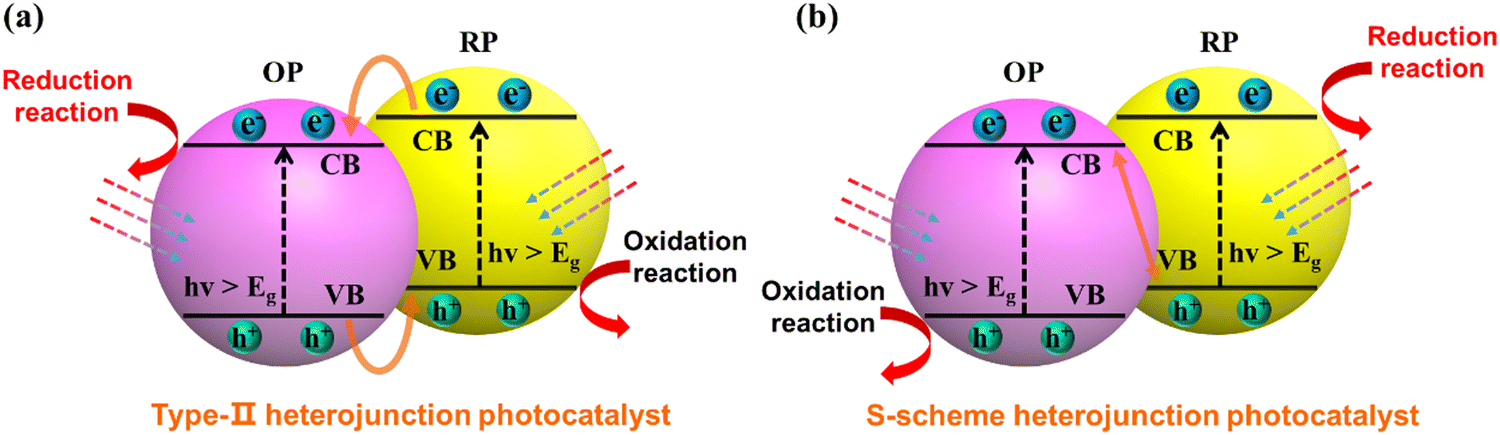
When considering the thermodynamics of a type-II heterojunction, the junction can exhibit exceptional electron–hole separation efficiency. This phenomenon can be attributed to the fact that the electrons are transferred to the conduction band with a weak reduction potential for reaction, while the holes are transferred to the valence band with a weak oxidation potential (as illustrated in Fig. 16a). From the kinetics perspective, it is important to note that the existence of original photogenerated electrons in semiconductor B can actually inhibit the continuous transfer of electrons from semiconductor A. Similarly, the presence of photogenerated holes in semiconductor A can hinder the transfer of holes from semiconductor B. This limitation ultimately restricts the improvement of carrier separation efficiency in a type-II heterojunction. Although the thermodynamic properties of type-II heterojunctions can facilitate enhanced charge separation, the kinetic limitations should be carefully considered to maximize their full potential.
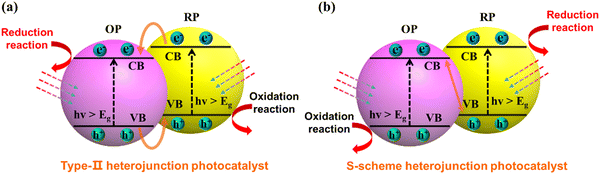 |
| | Fig. 16 Schematic illustration of electron–hole separation of a (a) conventional type-II heterojunction photocatalyst and (b) S-scheme heterojunction photocatalyst. | |
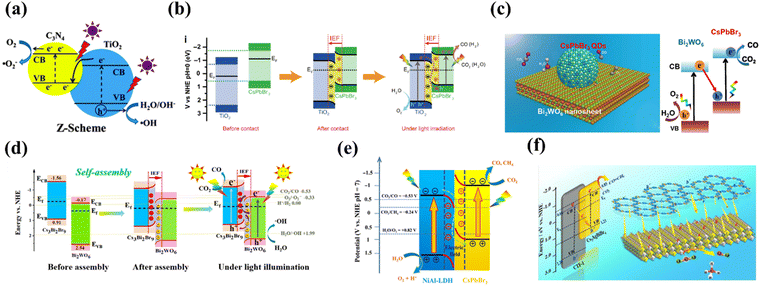 |
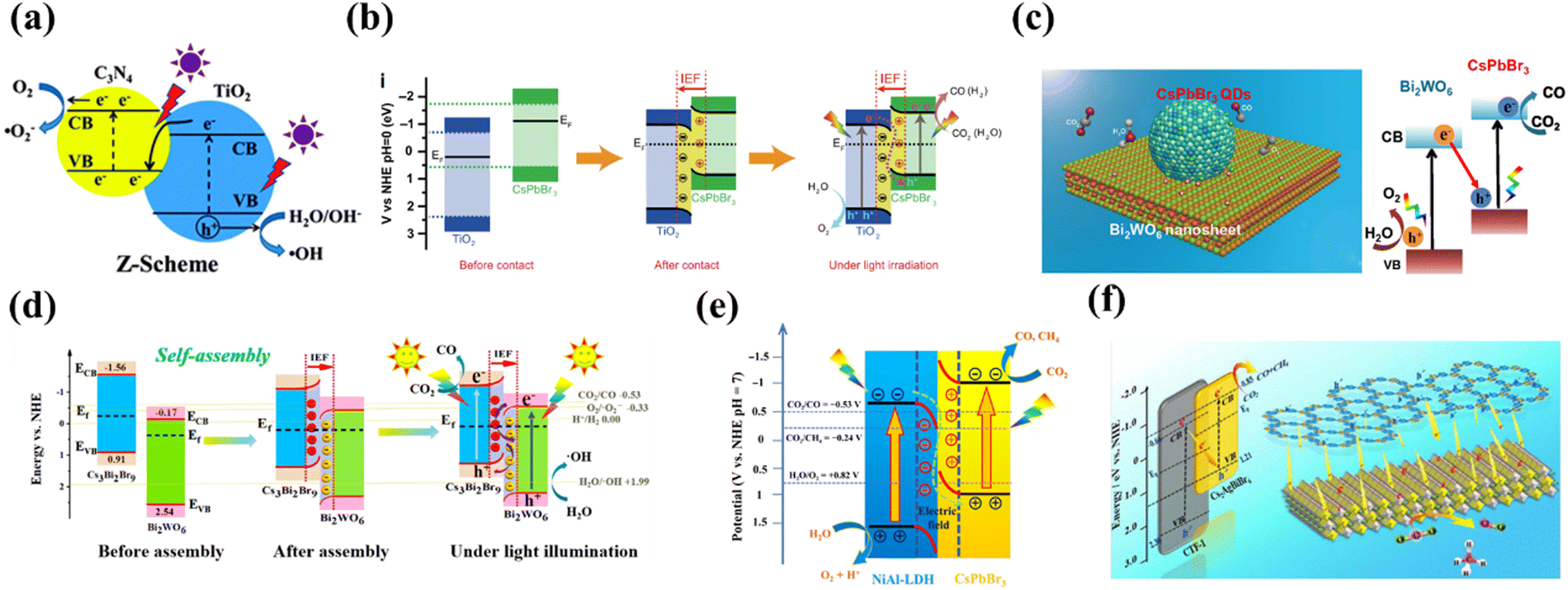
| | Fig. 17 (a) Diagram of a Z-shaped heterogeneous junction.117 (b) Schematic illustration of the TiO2/CsPbBr3 heterojunction: internal electric field (IEF) induced charge transfer, separation, and the formation of an S-scheme heterojunction under UV visible-light irradiation for CO2 photoreduction.118 (c) 0D/2D CPB/BWO of the Z-scheme photocatalytic system for CO2 reduction.121 (d) Energy band bending and IEF formation for CBB and BWO assembly before and after, which strongly promotes the S-scheme photoelectron transfer during the CRR process.122 (e) Proposed pathway for photocatalytic CO2 reduction under visible light irradiation of CsPbBr3/NiAl-LDH.125 (f) Diagram of Cs2AgBiBr6/CTF-1 photoreduction.127 | |
Not long ago, the concept of direct Z-type heterojunctions was also successfully applied to MHP-based CO2PR. For example, Yu et al. synthesized TiO2/CsPbBr3 heterojunction structures with S-type (direct Z-type) electron transfer pathways by a simple electrostatic self-assembly method, and the unique electron transfer pattern allows for a high spatial separation of electron–hole pairs to facilitate efficient CO2 photoreduction (Fig. 17b).118 More efficient charge separation between TiO2 and CsPbBr3 and enhanced photoreduction activity towards CO2 due to IEF induction were demonstrated. DFT calculations show that the work function of TiO2 is greater than that of CsPbBr3, implying that electrons are transferred from CsPbBr3 to TiO2 during hybridization, resulting in an IEF at the interface. In situ XPS analysis demonstrates that the IEF drives the photoinduced electrons in the CB of TiO2 to move into the VB of CsPbBr3, confirming the S pathway of charge transfer. Bi2WO6 is widely used as a typical semiconductor photocatalyst for wastewater treatment, air purification and CO2 reduction.119,120 Recently, Bi2WO6 has also been explored for the construction of two-component heterojunction materials for efficient CO2 photoreduction. For example, Wang et al. constructed a direct Z-type heterojunction by decorating 0D CsPbBr3 onto 2D Bi2WO6 nanosheets (Fig. 17c).121 The close contact between 0D CsPbBr3 and 2D Bi2WO6 nanosheets facilitates charge separation and transfer between the two semiconductors. Similarly, Feng's group fabricated Cs3Bi2Br9/Bi2WO6 (CBB/BWO) S-type heterojunctions with 0D/2D structures by adopting an electrostatic self-assembly method.122 The composite achieves a CO generation rate of 220.1 μmol g−1 h−1, which is 115.8 and 18.5 times higher than that of CBB PQDS and BWO NS, respectively. High performance is mainly attributed to the close heterogeneous surface contact between CBB PQDS and BWO NS, which facilitates effective space charge separation and shortens the charge transfer distance (photoelectron transfer process before and after the S-type heterojunction assembly is shown in Fig. 17d). Layered double hydroxides (LDHs), as a class of two-dimensional layered materials, have been widely used in photocatalysis due to their unique layered structure, environmental friendliness, tunable electronic band structure, flexible chemical composition and strong CO2 adsorption capacity.123,124 For instance, Zhao et al. reported that CsPbBr3 nanocrystals are activated by the layered double hydroxide nanosheets (CLDH) to boost the photocatalytic CO2 reduction (Fig. 17e).125 These CLDH without any co-catalysts and sacrificial agents show significantly enhanced CO2 photoreduction performance and the average electron consumption rate of CLDH (49.16 μ−1 h−1) is approximately 3.7 times higher than that of pure CsPbBr3. Covalent organic frameworks (COFs) are a family of highly crystalline organic semiconductors that have potential applications in a variety of fields due to their large surface area and abundant pores. As a branch of COFs, two-dimensional covalent triazine frameworks (CTFs) are very attractive because their rich triazine units have given them some special advantages, namely excellent chemical and thermal stability, high carrier mobility, and powerful CO2 adsorption and activation capabilities.126 Cs2AgBiBr6/CTF-1 (CABB/CTF-1) composites were designed by Zhang et al. through a self-assembly method (Fig. 7f), where the large surface-to-surface contact area of the 2D structure ensures intimate interfacial interactions and effective charge transfer/separation.127 Meanwhile, the periodic pore structure of CTF-1 enables the CABB/CTF-1 composite photocatalyst to provide stronger CO2 adsorption/activation capability.
In general, type-II and direct Z-scheme heterojunctions have emerged as two promising approaches for promoting the photocatalytic activity by enhancing the spatial separation of charge carriers. While direct Z-scheme heterojunctions have been shown to exhibit even higher separation efficiency of electrons and holes compared to type-II heterojunctions, there is still much place to be explored in terms of constructing MHP-based Z-scheme heterojunctions and characterizing the electron transfer pathways involved. Further research is necessary to fully understand the mechanisms in the heterojunctions and optimize their performance for practical applications.
4. CO2 photoreduction products beyond CO
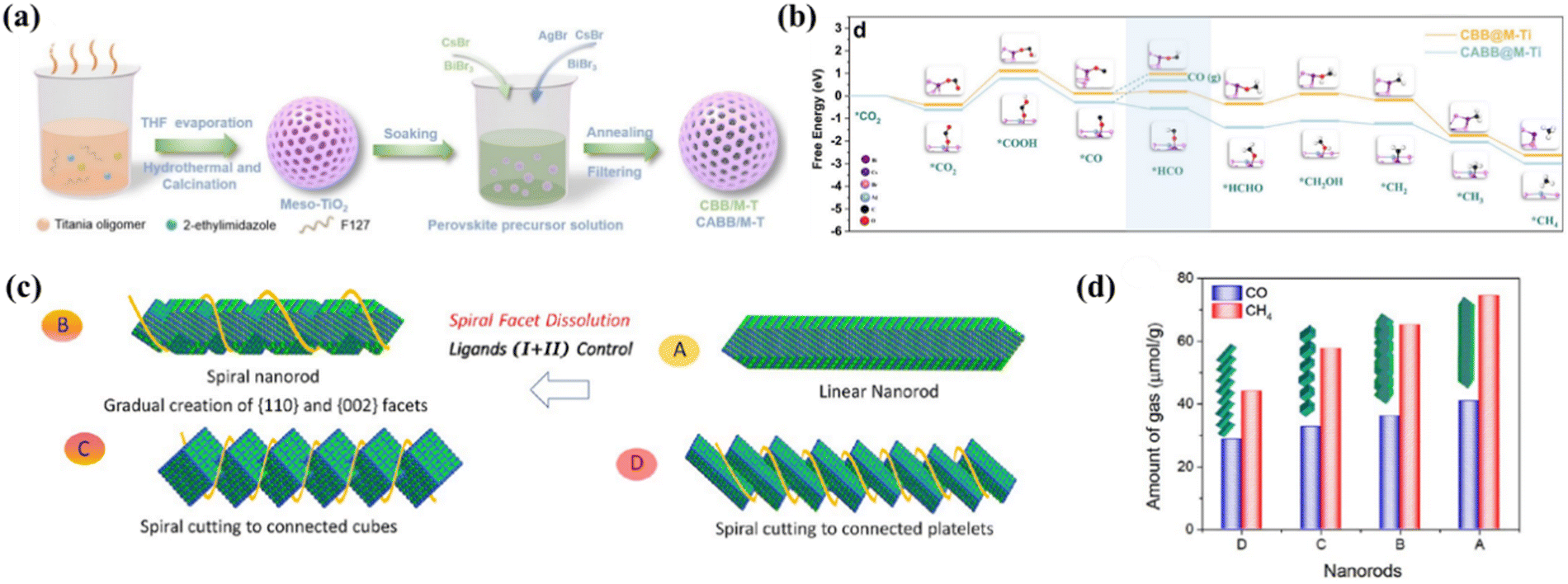
Equations for the redox reactions of possible products in the photocatalytic CO2 reduction process are listed in Table 1, which can provide useful insights into the equilibrium potential and reaction enthalpy. To date, most perovskite-based photocatalysts facilitate a two-electron reduction process to CO. Selective photocatalytic reduction of CO2 to CH4 is much more difficult to accomplish as it is an eight-electron process, but the production of CH4 is also very important in terms of the energy source.128 It is a major component of natural gas and is widely used for electricity generation, heating and fuel transportation.129 Furthermore, CH4 is a raw material for many important chemical compounds such as CH3OH, C2H4 and C3H6. These compounds are used extensively in the manufacture of plastics, fertilizers, solvents and other chemical products. Therefore, the exploration of efficient photocatalysts for the conversion of CO2 to CH4 is of great significance in both energy and chemical industries. Table 4 summarizes the recent studies on MHP-based CO2PR with CH4-dominated products. Mesoporous materials are a class of star materials with an open pore structure and a large specific surface area, making them ideal for catalytic reactions. Meanwhile, rational design of the structure of the mesoporous shell could facilitate the accelerated separation of electron–hole pairs. Recently, Sun et al. reported the synthesis of CBB@M-Ti and CABB@M-Ti by growing lead-free perovskite nanodots Cs3Bi2Br9 (CBB) and Cs2AgBiBr6 (CABB) in mesoporous titanium dioxide (M-Ti) frameworks for photocatalytic CO2 reduction (Fig. 18a).130 M-Ti and the formation of an IEF between the CBB and CABB interfaces induce the electron–hole separation. CBB@M-Ti and CABB@M-Ti show high CH4 yields of 151.1 and 144 μmol g−1 with a selectivity of 88.7% and 84.2%, which are significantly higher than those of M-Ti, CBB and CABB. DFT calculations suggest that the high selectivity of CH4 can be attributed to double adsorption-mediated hydrogenation of CO to *HCO rather than CO desorption (Fig. 18b). A special phase structure of perovskite can also increase the selectivity of the CH4 product. For instance, Bera's group synthesized helical CsPbBr3 nanorods using a prelattice of orthorhombic phase Cs2CdBr4 with Pb(II) diffusion, where helicity is achieved by controlling the composition of the alkylammonium ions in the reaction system and selectively dissolving some of the helical facets of the nanorods (Fig. 18c).131 The optimum catalyst (<3% Cd) achieves the yields of 40.81 μmol g−1 h−1 (CO) and 74.45 μmol g−1 h−1 (CH4) and CH4 selectivity as high as 87.9% (Fig. 18d). Despite these efforts on the photoconversion of CO2 to CH4 based on MHPs, higher selectivity and yield of CH4 are still required, which needs further in-depth understanding of the reaction mechanism, optimization of catalyst design and tuning of reaction conditions and surface properties.
Table 4 MHP-based catalysts for CO2PR with CH4-dominated products
| Photocatalyst |
Light source |
System |
R
totalelectron (μmol g−1 h−1) |
Products |
Selectivity (%) |
Ref. |
| Fe:CsPbBr3 |
450 W Xe-lamp |
EA/H2O |
55 |
CH4, CO |
∼100 |
55
|
| CsPbBr3/BIF-122-Co |
— |
EA/H2O |
90.67 |
CH4, CO, H2 |
57.5 |
108
|
| Cs2SnI6/SnS2 |
100 W Xe lamp, λ > 400 nm |
CH3OH/H2O |
16 |
CH4 |
∼100 |
113
|
| CsPbBr3@g-C3N4 |
— |
EA |
59.98 |
CH4, CO |
∼100 |
117
|
| Pt–CsPbBr3/Bi2WO6 |
150 W Xe lamp, AM 1.5G |
EA/IPA |
324.0 |
CH4, CO, H2 |
95.4 |
119
|
| Cs3Bi2Br9@M-Ti |
720 nm ≥ λ ≥ 420 nm |
IPA |
202.68 |
CH4, CO |
∼100 |
130
|
| Cs2AgBiBr6@M-Ti |
720 nm ≥ λ ≥ 420 nm |
IPA |
271.58 |
CH4, CO |
∼100 |
130
|
| Cd:CsPbBr3 |
450 W Xe-lamp |
EA/H2O |
338.4 |
CH4, CO |
∼100 |
131
|
| Ti3C2/CsPbBr3 QDs |
300 W Xe-lamp |
EA |
190.44 |
CH4, CO |
∼100 |
135
|
| Cu scaffold with CsPbBr3 film |
— |
EA/H2O |
251.88 |
CH4, C2H4, C2H6, C3H8, H2 |
93.27 |
141
|
| CsCuCl3 MCs |
AM 1.5G 100 mW cm−2 |
EA/IPA |
32.66 |
CH4, CO |
∼100 |
197
|
| CsPbBr3 (Cd ∼3%) star |
450 W Xe lamp, λ > 420 nm |
EA/H2O |
237.2 |
CH4, CO |
∼100 |
210
|
| CsPbBr3 NC/a-TiO2 |
150 W Xe-lamp, AM 1.5G |
EA/IPA |
64.45 |
CH4, CO, H2 |
95.5 |
211
|
| CsPbBr3@ZIF-8 |
100 W Xe lamp, AM 1.5G |
H2O (vapor) |
15.498 |
CH4, CO |
∼100 |
212
|
| CsPbBr3@ZIF-67 |
100 W Xe lamp, AM 1.5G |
H2O (vapor) |
29.630 |
CH4, CO |
∼100 |
212
|
| CsPbBr3/GO |
500 W Xe-lamp, λ > 400 nm |
H2O (vapor) |
162.6 |
CH4, H2 |
91.5 |
213
|
| CsPbBr3 NC/BZNW/MRGO |
λ > 420 nm, AM 1.5G |
H2O (vapor) |
52.02 |
CH4, CO |
∼100 |
214
|
| α-Fe2O3/amine-RGO/CsPbBr3 |
150 W Xe lamp, λ > 420 nm |
H2O (vapor) |
80.95 |
CH4, CO, H2 |
99.3 |
215
|
| CsPbBr3 NS–Cu–RGO |
— |
H2O (vapor) |
∼103 |
CH4, CO, H2 |
∼99.4 |
216
|
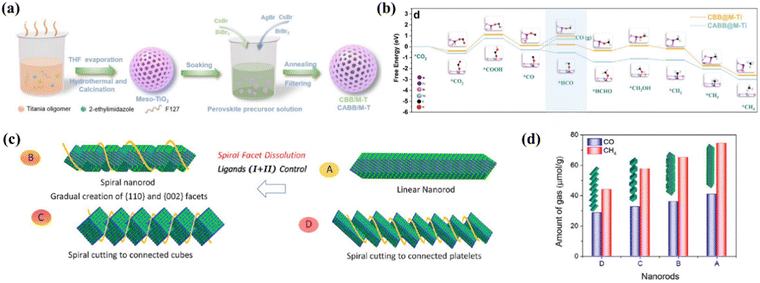 |
| | Fig. 18 (a) Schematic illustration of the synthesis of X% CBB@M-Ti and CABB@M-Ti.130 (b) Gibbs free energy pathway for the formation of CH4 and CO from CO2 over CBB@M-Ti and CABB@M-Ti.130 (c) Models showing CsPbBr3 nanorods spirally cutting to different spiral nanorods with different reaction conditions of A to D. For the case of C, models are done with normal cubes though truncated shaped were observed. All these models are schematic and were designed using Diamond Crystal Impact software. Cs atoms in all cases were kept on surfaces.130 (d) Comparison of photocatalytic CO2 reduction activity of four CsPbBr3 nanorods.131 | |
In addition to the two main products (i.e., CO and CH4), the photoreduction of CO2 to multi-carbon products appears to be more attractive and challenging. The reason is that multi-carbon products usually have a higher economic value and energy density.132 Meanwhile, compared to CO and CH4, multi-carbon compounds possess wider potential for application in the chemical, energy and material sectors.133 Despite the diversity of catalytic systems observed for C2+ products, there are hardly any experimental reports on the production of C2+ products with MHP-based photocatalytic systems.134–136 The difficulty of MHPs for the production of C2+ may be understood in that the general process of e−/h+ recombination limits the transfer of multiple electrons in a short period of time, thus leading to an energy waste that not only decreases CO2 conversion but also results in product selectivity towards C1.137 It has been found that the multiple electrons coupled to proton transfer (MECPT) reactions, along with C–C coupling steps, play a crucial role in the formation of C2+ products.138 In recent years, Cu-based catalysts have been extensively reported for the preparation of multi-carbon products by (photo)electrochemical CO2 reduction.139,140 Therefore, the utilization of Cu-based catalysts to tune the reaction selectivity toward C2+ products may be an effective choice. Recently, as a pioneering study, Choi's group reported for the first time the use of MHP-based catalysts for the generation of multi-carbon products.141 They prepared a composite photocatalyst by embedding CsPbBr3 nanocrystals in a porous Cu scaffold and applied this integrated catalyst for photocatalytic CO2 reduction reaction (Fig. 19a). As a result, ethylene (C2H4, 46.5 μmol g−1) is detected as the main product, together with the formation of methane (CH4, 18.9 μmol g−1), ethane (C2H6, 17.1 μmol g−1) and propane (C3H8, 11.3 μmol g−1) (Fig. 19b). The reason behind the generation of multi-carbon products can be associated with the copper support and the graded porous structure. For an in-depth understanding of the photocatalytic CO2 reduction mechanism, in situ ATR-IR measurements were carried out (Fig. 19c and d). The results show that the C1 reaction pathway leading to CH4 dominates on the bare planar perovskite, while the C2 reaction pathway leading to C2H4, C2H6 and C3H8 is evident on the perovskite embedded in a porous Cu support. *CHO triggers the C1 pathway of CO2 reduction on the bare planar perovskite, leading to the formation of CH4. In contrast, *OCCO generated via CO dimerization on the perovskite embedded in porous Cu stents triggers the C2 pathway. It is believed that this pioneering work will provide inspiration for the subsequent development of highly efficient MHP-based CO2PR for multi-carbon products.
 |
| | Fig. 19 (a) Comparison of the charge transport processes in a planar perovskite film and the perovskite layer embedded in a nanoporous copper scaffold.141 (b) Yield of the CO2 reduction products after 5 h of photochemical reaction. The inset image represents the relative energetic diagram of the CsPbBr3 perovskites within a porous copper scaffold, which acts as an assistant for electron transfer in photocatalytic CO2 reduction.141 (c) In situ ATR-IR spectra of photocatalytic CO2 reduction on the CsPbBr3 perovskite photocatalyst embedded in a porous copper scaffold.141 (d) In situ ATR-IR spectra of photocatalytic CO2 reduction on the CsPbBr3 perovskite photocatalyst embedded in a porous copper scaffold.141 | |
5. Conclusion and outlook
In this review, we present a timely and comprehensive review of recent advances in MHP-based CO2PR. Firstly, the review describes the structure and photoelectric properties of MHPs. Then, various strategies are summarized for improving the efficiency of CO2PR to CO, using lead-based and lead-free MHP materials. Moreover, detailed discussions are given about the MHP-based CO2PR to CH4 and C2+ products, with a focus on the design strategies for enhancing specific selectivity.
Although remarkable advances in this field have been made, several challenges still exist. In our opinion, future research efforts for MHP-based CO2 photoreduction should focus on the following aspects, as schematically shown in Fig. 20.
 |
| | Fig. 20 Future research perspectives of MHPs for photocatalytic CO2 reduction. | |
5.1 Exploiting new MHPs
5.1.1 Stable MHPs.
The physical and chemical instability of MHPs under high temperature, humidity or an oxidizing atmosphere could significantly reduce their operational lifetime, which prompts researchers to seek effective solutions to improve the stability of MHPs. Some studies have shown that partial or total replacement of Pb in MHPs with other metallic elements (e.g., Cu, Bi) has proven to be an effective strategy for improving the stability of MHPs.142,143 In addition, optimizing the crystal growth conditions, such as temperature and humidity, can help to minimize the presence of defects and impurities that may lead to degradation of the crystal structure. Moreover, special encapsulation materials (e.g., polymer films) or inorganic coatings (e.g., SiO2, TiO2, C60, r-GO and g-C3N4) can be utilized to shield MHPs from the external environment and enhance their stability.144,145 Specifically, the surface encapsulation prevents MHPs from coming into direct contact with the external environment and acts as a solid barrier to water and other polar solvents. Physical confinement also inhibits anion exchange and ion migration at the MHP surface. Replacing the long chain OA/OAm with other suitable ligands could also passivate the surface of the perovskites and prevent degradation of the MHPs, preserving their structural stability for extended time periods.112 These solutions will enable MHPs to maintain their photoelectric performance and stability, thereby extending their lifespan and improving their overall performance.
5.1.2 Environmentally friendly MHPs.
Lead toxicity is a major challenge for the long-term viability of MHPs. To address this problem, there has been ongoing search for atoms with similar radii as potential replacement of lead in MHPs. Some of the promising alternatives are Ag, Sn, Rb, Bi, In and Ge. Of these alternatives, Bi-based perovskites have shown great potential for application in CO2 photoreduction.45,86 Continuous research efforts into these alternatives are essential for the development of sustainable MHPs. In addition, the manufacturing process of some MHPs may present extra environmental impacts, including the release of polluting gases. To alleviate these concerns, it is also important to optimize the production processes and reduce harmful emissions. These approaches will help pave the way for the future development of green MHP-based photoreduction, where MHPs are produced and utilized with minimal impact on the environment.
5.1.3 Large-scale MHPs.
To date, most research studies on the photocatalytic reduction of CO2 to solar fuels are limited to the laboratory scale. On the one hand, the recovery of catalysts is both expensive and time-consuming. On the other hand, the reaction kinetics of photocatalysis are sluggish and the product conversion rates are low. In order to achieve industrial-scale production of MHP-based photocatalysts, optimization of the synthetic protocols and material composition could be considered, leading to cost-efficiency and high photoconversion efficiency.
5.2 Generating multi-carbon products
So far, the MHP-based CO2PR products are mainly C1 products (i.e., CO and CH4), while C2+ products have rarely been reported. This can be explained by the severe radiative recombination of MHPs which limits the migration of multiple electrons in a short period of time, and the slow kinetics of the C–C coupling reaction which leads to high reaction energy barriers as well as low multi-carbon product selectivity.146,147 In order to design efficient MHP-based photocatalysts and exert control over the reaction pathways for desired products, some suggestions are given: (1) exploring the potential of Cu-based photocatalysts for CO2PR. Extensive research has demonstrated that the Cu-based materials are effective in promoting C–C coupling in CO2 electroreduction for C2+ products, which could inspire the exploration of Cu-based photocatalysts for the generation of C2+ products. It is emphasized that the driving force behind C–C coupling does not stem from specific elements (e.g. Cu). Actually, the asymmetric charge distribution leads to the formation of high electron density centers which are needed for multi-electron requirements, ultimately driving the C–C coupling reactions.147 Recent studies have also demonstrated that the excellent conversion of C2+ products by Cu-based catalysts depends largely on their good adsorption capacity for most of the C2+ intermediates.148–150 This feature is conducive to causing charge polarization, which induces the C–C coupling reaction. In view of this, it is worthwhile to explore the development of composite catalysts by combining MHPs with Cu. Constructing photocatalytic systems with heterogeneous Cu species and further optimizing through charge polarization strategies may be conducive to achieving efficient CO2PR for the production of C2+ chemicals. (2) Regulatory intermediates. It is widely accepted that CO is the key intermediate in the formation of multi-carbon products through the process of dimerization (in the form of CO–CO) or coupling (in the form of CO–COH) pathways.151,152 Obviously, these steps necessitate the presence of the precursor, referred to as the steady-state adsorbed CO (*CO), in close proximity on the surface with a high coverage. Thus, enriching the *CO concentration and decorating active sites on MHPs can stabilize *CO. Subsequently, the adsorbed CO intermediates (i.e., *CO) on adjacent sites create an asymmetric charge distribution, thereby enhancing the activation of CO and promoting the formation of key intermediates. (3) Combining electro/enzymatic catalysis. In recent years, photoelectrocatalysis and photoenzymecatalysis have shown great potential to convert CO2 into multi-carbon products.153–155 In a photocatalytic system, the internal charge separation and transfer processes occur within the absorbing nanoparticles, which are driven by the built-in electric field (IEF). However, these processes often encounter significant challenges, including severe charge recombination and insufficient driving force. To address these challenges, MHPs can be decorated as photocathodes to establish a photoelectrocatalysis system. By applying an external bias, charge separation can be effectively promoted. Additionally, the implementation of an ion membrane in the photoelectrocatalysis system spatially isolates the oxidation and reduction active sites, leading to the effective suppression of the reverse reaction. In addition to photoelectrocatalysis, another promising strategy is combining the biological-enzymes for CO2PR. The biological-enzyme catalysts offer advantages such as high selectivity, environmental friendliness, mild operating conditions and facile carbon–carbon coupling, which facilitate the conversion of CO2 into valuable products.155 Recent studies have elucidated the participation of certain coenzymes in a multitude of vital metabolic reactions, where they play a facilitating role in the light-induced C–C coupling of short-chain carbon building blocks with long-chain carbons.156,157 Hence, it is anticipated that the future integration of MHPs with specific enzymes to form photoenzymecatalysis systems will instigate research interest among scholars.
5.3 Establishing a rigorous assessment method
Some reports on MHPs have reported only a low conversion rate within the range of μmol g−1 in the experimental study of CO2PR. In this case, very few detected products are most likely not from the photocatalytic CO2 reduction. Therefore, several important factors need to be considered to demonstrate that the products are generated by the photocatalytic reduction of CO2 and are not from accidental contamination or photo-oxidation of the organic system itself (e.g., EA or ACN). (1) Avoiding carbon pollution. As most MHPs are water sensitive, the reaction system is mostly chosen to be carried out in an organic system (e.g. EA or ACN), which stabilizes the structure of the MHPs and promotes the solubility of CO2. However, the organic system can bring some negative effect, such as unavoidable degradation of EA/ACN to CO/CH4 in the presence of light and the oxidation of the sacrificial agent which may also give rise to the associated C1 products.158,159 To exclude the influence of solvents, it is advisable to utilize the “solid–vapor reaction mode” for water-sensitive systems.160 Simultaneously, it is worth noting that certain preceding studies have demonstrated the enhanced photocatalytic performance through the integration of the MHP with a carbonaceous substrate. However, this approach neglects the adverse consequence whereby the photodegradation of carbonaceous catalysts may lead to the generation of a C1 product.85,191,193,198 To exclude possible contamination from the photodegradation of carbon substrates, it is critical to conduct control experiments utilizing pristine carbon substrates. Hence, to quantify the yield of photocatalysis in the presence of organic solvents or contaminants, it is necessary to carry out the reaction under inert conditions (N2 or Ar) while keeping other conditions constant. Furthermore, control experiments under dark conditions using CO2 to exclude the potential influence of chemical catalysis is equally important. To prove the effectiveness of photocatalytic CO2 reduction convincingly, the product generation rate should significantly surpass that of control experiments. (2) Tracking carbon sources. Further definitive evidence that the detected products originate from the photoreduction of CO2 requires isotopic labeling techniques using 13CO2. Briefly, the accumulation of 13Cx products should correspond to the amount of 12Cx consumed in 12CO2 under the same experimental conditions. (3) Concerning water oxidation. As mentioned above, a significant portion of the literature focuses on CO2 reduction reactions employing sacrificial agents, which cannot be regarded as authentic artificial photosynthesis. Since many of the sacrificial agents are thermodynamically spontaneous, the introduction of a light source merely accelerates the progress of the reaction. Therefore, the crucial criterion for verifying the utilization of solar energy in achieving the conversion of CO2 to chemical fuel is the occurrence of a water oxidation reaction (WOR). In other words, it is important to investigate whether simultaneous water oxidation takes place during the CO2 reduction process. In short, establishing a rigorous measurement and evaluation method will make the results more persuasive.
5.4 Advancing experimental and computational tools
A precise understanding of the catalyst structure (e.g., defects, atomic configurations and elemental arrangements) is beneficial to investigate the structure–activity correlation. To achieve this goal, a wide range of characterization techniques have been developed. Traditional ex situ characterization can only reflect the pre- and post-reaction state of a catalyst. For catalytic reactions, it is of great importance to probe the system under the real reaction conditions, such as real-time detection of the changes in the atomic structure of the catalyst, the intermediates of adsorbed molecules, the chemical state of the catalyst, and the displacement of ligand atoms. The state of the reaction intermediates is crucial to study the mechanism of the catalytic reaction. An accurate understanding of the catalyst structure can also help construct theoretical models that are more realistic, reasonable and valid. To unravel the above issues, employment of in situ/operando characterization techniques is necessary. Up to now, the real-time structures of MHPs during CO2PR are seldom explored and in-depth investigation could shed more light on this aspect. Meanwhile, theoretical simulations (e.g., machine learning and DFT calculations) have been regarded as a powerful tool to explain experimental results and predict catalytic performance, providing guidelines to speed up the development of new catalysts. Therefore, rigorous theoretical simulations in corroboration with advanced characterization techniques may be the best way to uncover the photocatalytic mechanism and explore high-efficiency photocatalytic systems.
5.5 Excavating practical applications
Conventional photocatalytic reactors are small devices and are mainly used for lab-scale proof-of-concept demonstrations. In order to achieve industrial-scale production, the development of large-scale flat-plate-reactors and reaction tanks can be considered. Besides, most of the photocatalytic conversions of CO2 focus on high-purity CO2 feed stream, requiring additional energy to capture and enrich CO2. To address this issue, future technologies need to be designed to capture CO2 directly in air and reduce it in situ to enable photoconversion at low CO2 concentrations. Furthermore, photocatalysis also needs to consider the impact of the environment on the reaction. For example, light intensity, temperature, humidity and the presence of organic substances may potentially alter the reaction performances. Future research is required to provide more detailed understanding of the impact of these environmental factors on photocatalytic reactions and the adaptation of systems to different environmental conditions.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the Natural Science Foundation for Young Scholars of Jiangsu Province (no. BK20220879), National Natural Science Foundation for Young Scholars of China (no. 22209072), and Jiangsu Specially-Appointed Professors.
References
- L. Zhao, F. Ye, D. Wang, X. Cai, C. Meng, H. Xie, J. Zhang and S. Bai, ChemSusChem, 2018, 11, 3524–3533 CrossRef CAS PubMed.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- F. Xu, K. Meng, B. Cheng, J. Yu and W. Ho, ChemCatChem, 2018, 11, 465–472 CrossRef.
- X. Zhang, X. Zhao, K. Chen, Y. Fan, S. Wei, W. Zhang, D. Han and L. Niu, Nanoscale, 2021, 13, 2883–2890 RSC.
- J. Wang, S. Lin, N. Tian, T. Ma, Y. Zhang and H. Huang, Adv. Funct. Mater., 2020, 31, 2008008 CrossRef.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS PubMed.
- C. Xie, W. Chen, S. Du, D. Yan, Y. Zhang, J. Chen, B. Liu and S. Wang, Nano Energy, 2020, 71, 104653 CrossRef CAS.
- M. Baca, W. Kukulka, K. Cendrowski, E. Mijowska, R. J. Kalenczuk and B. Zielinska, ChemSusChem, 2019, 12, 612–620 CrossRef CAS PubMed.
- S. Purohit, K. L. Yadav and S. Satapathi, Langmuir, 2021, 37, 3467–3476 CrossRef CAS PubMed.
- S. Chen, K. Roh, J. Lee, W. K. Chong, Y. Lu, N. Mathews, T. C. Sum and A. Nurmikko, ACS Nano, 2016, 10, 3959–3967 CrossRef CAS PubMed.
- M. M. Stylianakis, T. Maksudov, A. Panagiotopoulos, G. Kakavelakis and K. Petridis, Materials, 2019, 12, 859 CrossRef CAS PubMed.
- X. Hu, X. Zhang, L. Liang, J. Bao, S. Li, W. Yang and Y. Xie, Adv. Funct. Mater., 2014, 24, 7373–7380 CrossRef CAS.
- H. Wang and D. H. Kim, Chem. Soc. Rev., 2017, 46, 5204–5236 RSC.
- S. Yakunin, M. Sytnyk, D. Kriegner, S. Shrestha, M. Richter, G. J. Matt, H. Azimi, C. J. Brabec, J. Stangl, M. V. Kovalenko and W. Heiss, Nat. Photonics, 2015, 9, 444–449 CrossRef CAS PubMed.
- H. Wei, Y. Fang, P. Mulligan, W. Chuirazzi, H.-H. Fang, C. Wang, B. R. Ecker, Y. Gao, M. A. Loi, L. Cao and J. Huang, Nat. Photonics, 2016, 10, 333–339 CrossRef CAS.
- J.-P. Correa-Baena, M. Saliba, T. Buonassisi, M. Grätzel, A. Abate and W. Tress, Science, 2017, 358, 739–744 CrossRef CAS PubMed.
- N.-G. Park, Mater. Today, 2015, 18, 65–72 CrossRef CAS.
- S. Adjokatse, H.-H. Fang and M. A. Loi, Mater. Today, 2017, 20, 413–424 CrossRef CAS.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- S. A. Veldhuis, P. P. Boix, N. Yantara, M. Li, T. C. Sum, N. Mathews and S. G. Mhaisalkar, Adv. Mater., 2016, 28, 6804–6834 CrossRef CAS PubMed.
- J. Park, J. Kim, H. S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. I. Seok, Nature, 2023, 616, 724–730 CrossRef CAS PubMed.
- J. Peng, Y. Chen, K. Zheng, T. Pullerits and Z. Liang, Chem. Soc. Rev., 2017, 46, 5714–5729 RSC.
- T. C. Sum and N. Mathews, Energy Environ. Sci., 2014, 7, 2518–2534 RSC.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- B.-M. Bresolin, Y. Park and D. Bahnemann, Catalysts, 2020, 10, 709 CrossRef CAS.
- Z.-Y. Chen, N.-Y. Huang and Q. Xu, Coord. Chem. Rev., 2023, 481, 215031 CrossRef CAS.
- J. Yuan, H. Liu, S. Wang and X. Li, Nanoscale, 2021, 13, 10281–10304 RSC.
- H. Liu and S. Bansal, Mater. Today Energy, 2023, 32, 101230 CrossRef CAS.
- Z. Zhang, S. Wang, X. Liu, Y. Chen, C. Su, Z. Tang, Y. Li and G. Xing, Small Methods, 2021, 5, 2000937 CrossRef CAS PubMed.
- J. Wang, Y. Shi, Y. Wang and Z. Li, ACS Energy Lett., 2022, 7, 2043–2059 CrossRef CAS.
- E. M. Akinoglu, D. A. Hoogeveen, C. Cao, A. N. Simonov and J. J. Jasieniak, ACS Nano, 2021, 15, 7860–7878 CrossRef CAS PubMed.
- H. Huang, D. Verhaeghe, B. Weng, B. Ghosh, H. Zhang, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, Angew. Chem., Int. Ed., 2022, 61, 202203261 CrossRef PubMed.
- X. Wang, J. He, X. Chen, B. Ma and M. Zhu, Coord. Chem. Rev., 2023, 482, 215076 CrossRef CAS.
- J. Wang, J. Liu, Z. Du and Z. Li, J. Energy Chem., 2021, 54, 770–785 CrossRef CAS.
- C. Han, X. Zhu, J. S. Martin, Y. Lin, S. Spears and Y. Yan, ChemSusChem, 2020, 13, 4005–4025 CrossRef CAS PubMed.
- Q. A. Akkerman, G. Raino, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- S. Lee, G. Y. Jang, J. K. Kim and J. H. Park, J. Energy Chem., 2021, 62, 11–26 CrossRef CAS.
- M.-G. Ju, M. Chen, Y. Zhou, J. Dai, L. Ma, N. P. Padture and X. C. Zeng, Joule, 2018, 2, 1231–1241 CrossRef CAS.
- Z.-L. Liu, R.-R. Liu, Y.-F. Mu, Y.-X. Feng, G.-X. Dong, M. Zhang and T.-B. Lu, Sol. RRL, 2021, 5, 2000691 CrossRef CAS.
- J. Sheng, Y. He, J. Li, C. Yuan, H. Huang, S. Wang, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 CrossRef CAS PubMed.
- T. Chen, M. Zhou, W. Chen, Y. Zhang, S. Ou and Y. Liu, Sustainable Energy Fuels, 2021, 5, 3598–3605 RSC.
- L. Ding, F. Bai, B. Borjigin, Y. Li, H. Li and X. Wang, Chem. Eng. J., 2022, 446, 137102 CrossRef CAS.
- A. Mahmoud Idris, S. Zheng, L. Wu, S. Zhou, H. Lin, Z. Chen, L. Xu, J. Wang and Z. Li, Chem. Eng. J., 2022, 446, 137197 CrossRef CAS.
- J. Sheng, Y. He, M. Huang, C. Yuan, S. Wang and F. Dong, ACS Catal., 2022, 12, 2915–2926 CrossRef CAS.
- Y. Kim, E. Yassitepe, O. Voznyy, R. Comin, G. Walters, X. Gong, P. Kanjanaboos, A. F. Nogueira and E. H. Sargent, ACS Appl. Mater. Interfaces, 2015, 7, 25007–25013 CrossRef CAS PubMed.
- M. Kovalenko, L. Protesescu and M. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- P. Chen, W. J. Ong, Z. Shi, X. Zhao and N. Li, Adv. Funct. Mater., 2020, 30, 1909667 CrossRef CAS.
- J. Liang, D. Chen, X. Yao, K. Zhang, F. Qu, L. Qin, Y. Huang and J. Li, Small, 2020, 16, 1903398 CrossRef CAS PubMed.
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. B. Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- W. Yang, J. Noh, N. Jeon, Y. Kim, S. Ryu, J. Seo and S. Seok, Science, 2015, 348, 9272 Search PubMed.
- X. Wang, J. He, J. Li, G. Lu, F. Dong, T. Majima and M. Zhu, Appl. Catal., B, 2020, 277, 119230 CrossRef CAS.
- Y. F. Mu, W. Zhang, X. X. Guo, G. X. Dong, M. Zhang and T. B. Lu, ChemSusChem, 2019, 12, 4769–4774 CrossRef CAS PubMed.
- Y. Yang, M. Yang, D. T. Moore, Y. Yan, E. M. Miller, K. Zhu and M. C. Beard, Nat. Energy, 2017, 2, 16207 CrossRef CAS.
- Y. H. Kim, H. Cho and T. W. Lee, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11694–11702 CrossRef CAS PubMed.
- Y. Yang, Y. Yan, M. Yang, S. Choi, K. Zhu, J. M. Luther and M. C. Beard, Nat. Commun., 2015, 6, 7961 CrossRef CAS PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 341, 6225 Search PubMed.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- K. Chen, S. Schunemann, S. Song and H. Tuysuz, Chem. Soc. Rev., 2018, 47, 7045–7077 RSC.
- W.-J. Yin, T. Shi and Y. Yan, J. Phys. Chem. C, 2015, 119, 5253–5264 CrossRef CAS.
- Q. V. Le, K. Hong, H. W. Jang and S. Y. Kim, Adv. Electron. Mater., 2018, 4, 1800335 CrossRef.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- S.-H. Guo, J. Zhou, X. Zhao, C.-Y. Sun, S.-Q. You, X.-L. Wang and Z.-M. Su, J. Catal., 2019, 369, 201–208 CrossRef CAS.
- Z. Zhang, D. Li, H. Hu, Y. Chu and J. Xu, Inorg. Chem., 2023, 62, 9240–9248 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Gratzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- J. Tauc, Rev. Mod. Phys., 1957, 29, 308–324 CrossRef CAS.
- Y. H. Kim, J. S. Kim and T. W. Lee, Adv. Mater., 2019, 31, 1804595 CrossRef CAS PubMed.
- Z. H. Yan, M. H. Du, J. Liu, S. Jin, C. Wang, G. L. Zhuang, X. J. Kong, L. S. Long and L. S. Zheng, Nat. Commun., 2018, 9, 3353 CrossRef PubMed.
- H. L. Wu, X. B. Li, C. H. Tung and L. Z. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef PubMed.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS PubMed.
- S. G. Shetty, I. M. Ciobica, E. J. Hensen and R. A. van Santen, Chem. Commun., 2011, 47, 9822–9824 RSC.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2016, 2, 16185 CrossRef.
- H. Luo, S. Guo, Y. Zhang, K. Bu, H. Lin, Y. Wang, Y. Yin, D. Zhang, S. Jin, W. Zhang, W. Yang, B. Ma and X. Lu, Adv. Sci., 2021, 8, 2100786 CrossRef CAS PubMed.
- Y. Lin, Y. Shao, J. Dai, T. Li, Y. Liu, X. Dai, X. Xiao, Y. Deng, A. Gruverman, X. C. Zeng and J. Huang, Nat. Commun., 2021, 12, 7 CrossRef CAS PubMed.
- F. Hao, C. C. Stoumpos, D. H. Cao, R. P. H. Chang and M. G. Kanatzidis, Nat. Photonics, 2014, 8, 489–494 CrossRef CAS.
- M. Chen, M. G. Ju, H. F. Garces, A. D. Carl, L. K. Ono, Z. Hawash, Y. Zhang, T. Shen, Y. Qi, R. L. Grimm, D. Pacifici, X. C. Zeng, Y. Zhou and N. P. Padture, Nat. Commun., 2019, 10, 16 CrossRef CAS PubMed.
- D. Wu, Y. Tao, Y. Huang, B. Huo, X. Zhao, J. Yang, X. Jiang, Q. Huang, F. Dong and X. Tang, J. Catal., 2021, 397, 27–35 CrossRef CAS.
- Z. Chen, X. Li, Y. Wu, J. Zheng, P. Peng, X. Zhang, A. Duan, D. Wang and Q. Yang, Sep. Purif. Technol., 2022, 295, 121250 CrossRef CAS.
- T. Wang, D. Yue, X. Li and Y. Zhao, Appl. Catal., B, 2020, 268, 118399 CrossRef CAS.
- Y. Zhang, Z. Sun, Z. Wang, Y. Zang and X. Tao, Int. J. Hydrogen Energy, 2022, 47, 8829–8840 CrossRef CAS.
- H. Lv, H. Yin, N. Jiao, C. Yuan, S. Weng, K. Zhou, Y. Dang, X. Wang, Z. Lu and Y. Zhang, Small Methods, 2023, 7, 2201365 CrossRef CAS PubMed.
- L. Zhou, Y. F. Xu, B. X. Chen, D. B. Kuang and C. Y. Su, Small, 2018, 14, 1703762 CrossRef PubMed.
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS PubMed.
- Y.-X. Feng, G.-X. Dong, K. Su, Z.-L. Liu, W. Zhang, M. Zhang and T.-B. Lu, J. Energy Chem., 2022, 69, 348–355 CrossRef CAS.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- A. P. Alivisatos, W. Gu and C. Larabell, Annu. Rev. Biomed. Eng., 2005, 7, 55–76 CrossRef CAS PubMed.
- C. Huo, B. Cai, Z. Yuan, B. Ma and H. Zeng, Small Methods, 2017, 1, 1600018 CrossRef.
- J. Xing, Y. Zhao, M. Askerka, L. N. Quan, X. Gong, W. Zhao, J. Zhao, H. Tan, G. Long, L. Gao, Z. Yang, O. Voznyy, J. Tang, Z. H. Lu, Q. Xiong and E. H. Sargent, Nat. Commun., 2018, 9, 3541 CrossRef PubMed.
- M. Ahmadi, T. Wu and B. Hu, Adv. Mater., 2017, 29, 1605242 CrossRef PubMed.
- L.-Y. Wu, M.-R. Zhang, Y.-X. Feng, W. Zhang, M. Zhang and T.-B. Lu, Sol. RRL, 2021, 5, 2100263 CrossRef CAS.
- J. Pi, X. Jia, Z. Long, S. Yang, H. Wu, D. Zhou, Q. Wang, H. Zheng, Y. Yang, J. Zhang and J. Qiu, Adv. Energy Mater., 2022, 12, 2202074 CrossRef CAS.
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- Y. Sun, X. Meng, Y. Dall'Agnese, C. Dall'Agnese, S. Duan, Y. Gao, G. Chen and X. F. Wang, Nano-Micro Lett., 2019, 11, 79 CrossRef CAS PubMed.
- Y. Xu, M.-Z. Yang, H.-Y. Chen, J.-F. Liao, X.-D. Wang and D.-B. Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS.
- Y.-X. Chen, Y.-F. Xu, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, Sustainable Energy Fuels, 2020, 4, 2249–2255 RSC.
- J.-F. Liao, Y.-T. Cai, J.-Y. Li, Y. Jiang, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, J. Energy Chem., 2021, 53, 309–315 CrossRef CAS.
- R. Tang, H. Sun, Z. Zhang, L. Liu, F. Meng, X. Zhang, W. Yang, Z. Li, Z. Zhao, R. Zheng and J. Huang, Chem. Eng. J., 2022, 429, 132137 CrossRef CAS.
- Z. Zhang, D. Li, Y. Chu, L. Chang and J. Xu, J. Phys. Chem. Lett., 2023, 14, 5249–5259 CrossRef CAS.
- Z.-C. Kong, H.-H. Zhang, J.-F. Liao, Y.-J. Dong, Y. Jiang, H.-Y. Chen and D.-B. Kuang, Sol. RRL, 2019, 4, 1900365 CrossRef.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- J. Wang, T. Xia, L. Wang, X. Zheng, Z. Qi, C. Gao, J. Zhu, Z. Li, H. Xu and Y. Xiong, Angew. Chem., Int. Ed., 2018, 57, 16447–16451 CrossRef CAS PubMed.
- C. H. Lu, G. V. Biesold-McGee, Y. Liu, Z. Kang and Z. Lin, Chem. Soc. Rev., 2020, 49, 4953–5007 RSC.
- C. Tang, C. Chen, W. Xu and L. Xu, J. Mater. Chem. A, 2019, 7, 6911–6919 RSC.
- Y. W. Liu, S. H. Guo, S. Q. You, C. Y. Sun, X. L. Wang, L. Zhao and Z. M. Su, Nanotechnology, 2020, 31, 215605 CrossRef CAS PubMed.
- S. Shyamal, S. K. Dutta, T. Das, S. Sen, S. Chakraborty and N. Pradhan, J. Phys. Chem. Lett., 2020, 11, 3608–3614 CrossRef CAS PubMed.
- Z.-Y. Chen, Q.-L. Hong, H.-X. Zhang and J. Zhang, ACS Appl. Mater. Interfaces, 2021, 5, 1175–1182 Search PubMed.
- Z. Cui, P. Wang, Y. Wu, X. Liu, G. Chen, P. Gao, Q. Zhang, Z. Wang, Z. Zheng, H. Cheng, Y. Liu, Y. Dai and B. Huang, Appl. Catal., B, 2022, 310, 121375 CrossRef CAS.
- Z. Zhang, D. Li, Z. Dong, Y. Jiang, X. Li, Y. Chu and J. Xu, Sol. RRL, 2023, 7, 2300038 CrossRef CAS.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS PubMed.
- Y. Xu, W. Zhang, K. Su, Y. X. Feng, Y. F. Mu, M. Zhang and T. B. Lu, Chem. – Eur. J., 2021, 27, 2305–2309 CrossRef CAS PubMed.
- X. D. Wang, Y. H. Huang, J. F. Liao, Y. Jiang, L. Zhou, X. Y. Zhang, H. Y. Chen and D. B. Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS PubMed.
- N. Li, X. Chen, J. Wang, X. Liang, L. Ma, X. Jing, D. L. Chen and Z. Li, ACS Nano, 2022, 16, 3332–3340 CrossRef CAS PubMed.
- S. Wan, M. Ou, Q. Zhong and X. Wang, Chem. Eng. J., 2019, 358, 1287–1295 CrossRef CAS.
- A. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed.
- T. Chen, L. Liu, C. Hu and H. Huang, Chin. J. Catal., 2021, 42, 1413–1438 CrossRef CAS.
- Z. Jiang, X. Liang, H. Zheng, Y. Liu, Z. Wang, P. Wang, X. Zhang, X. Qin, Y. Dai, M.-H. Whangbo and B. Huang, Appl. Catal., B, 2017, 219, 209–215 CrossRef CAS.
- J. Wang, J. Wang, N. Li, X. Du, J. Ma, C. He and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 31477–31485 CrossRef CAS PubMed.
- Y. Feng, D. Chen, Y. Zhong, Z. He, S. Ma, H. Ding, W. Ao, X. Wu and M. Niu, ACS Appl. Mater. Interfaces, 2023, 15, 9221–9230 CrossRef CAS PubMed.
- L. Tan, S. M. Xu, Z. Wang, Y. Xu, X. Wang, X. Hao, S. Bai, C. Ning, Y. Wang, W. Zhang, Y. K. Jo, S. J. Hwang, X. Cao, X. Zheng, H. Yan, Y. Zhao, H. Duan and Y. F. Song, Angew. Chem., Int. Ed., 2019, 58, 11860–11867 CrossRef CAS PubMed.
- K. Wang, L. Zhang, Y. Su, D. Shao, S. Zeng and W. Wang, J. Mater. Chem. A, 2018, 6, 8366–8373 RSC.
- S. Zhao, Q. Liang, Z. Li, H. Shi, Z. Wu, H. Huang and Z. Kang, Nano Res., 2022, 15, 5953–5961 CrossRef CAS.
- M. Liu, K. Jiang, X. Ding, S. Wang, C. Zhang, J. Liu, Z. Zhan, G. Cheng, B. Li, H. Chen, S. Jin and B. Tan, Adv. Mater., 2019, 31, 1807865 CrossRef PubMed.
- Z. Zhang, Y. Jiang, Z. Dong, Y. Chu and J. Xu, Inorg. Chem., 2022, 61, 16028–16037 CrossRef CAS PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- K. Thampi, J. Kiwi and M. Grtzel, Nature, 1987, 327, 11 CrossRef.
- Q. M. Sun, J. J. Xu, F. F. Tao, W. Ye, C. Zhou, J. H. He and J. M. Lu, Angew. Chem., Int. Ed., 2022, 61, 202200872 CrossRef PubMed.
- S. Bera, S. Shyamal and N. Pradhan, J. Am. Chem. Soc., 2021, 143, 14895–14906 CrossRef CAS PubMed.
- A. E. Farrell, R. J. Plevin, B. T. Turner, A. D. Jones, M. O'Hare and D. M. Kammen, Science, 2006, 311, 506–508 CrossRef CAS PubMed.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219 CrossRef CAS PubMed.
- J. Li, A. Xu, F. Li, Z. Wang, C. Zou, C. M. Gabardo, Y. Wang, A. Ozden, Y. Xu, D. H. Nam, Y. Lum, J. Wicks, B. Chen, Z. Wang, J. Chen, Y. Wen, T. Zhuang, M. Luo, X. Du, T. K. Sham, B. Zhang, E. H. Sargent and D. Sinton, Nat. Commun., 2020, 11, 3685 CrossRef CAS PubMed.
- L. Kong, Z. Chen, Q. Cai, L. Yin and J. Zhao, Chin. Chem. Lett., 2022, 33, 2183–2187 CrossRef CAS.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- T. T. Zhuang, Z. Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P. N. Chen, X. L. Zheng, H. Liang, W. N. Ge, B.-J. Ye, D. Sinton, S. H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- B. N. Choi, J. Y. Seo, Z. An, P. J. Yoo and C.-H. Chung, Chem. Eng. J., 2022, 430, 132807 CrossRef CAS.
- S. Peng, Z. Yang, M. Sun, L. Yu and Y. Li, Adv. Mater., 2023, 35, e2304711 CrossRef PubMed.
- Y. Han, S. Yue and B. B. Cui, Adv. Sci., 2021, 8, e2004805 CrossRef PubMed.
- H. Zhao, K. Kordas and S. Ojala, J. Mater. Chem. A, 2023, 11, 22656–22687 RSC.
- Z. J. Li, E. Hofman, J. Li, A. H. Davis, C. H. Tung, L. Z. Wu and W. Zheng, Adv. Funct. Mater., 2017, 28, 1704288 CrossRef.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S. M. Chang and E. W. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- X. Zhang, F. Han, B. Shi, S. Farsinezhad, G. P. Dechaine and K. Shankar, Angew. Chem., Int. Ed., 2012, 51, 12732–12735 CrossRef CAS PubMed.
- H. Park, H. H. Ou, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. A, 2015, 119, 4658–4666 CrossRef CAS PubMed.
- I. Shown, H. C. Hsu, Y. C. Chang, C. H. Lin, P. K. Roy, A. Ganguly, C. H. Wang, J. K. Chang, C. I. Wu, L. C. Chen and K. H. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- X. Chang, T. Wang, P. Yang, G. Zhang and J. Gong, Adv. Mater., 2019, 31, e1804710 CrossRef PubMed.
- W. Liu, W. Hu, L. Yang and J. Liu, Nano Energy, 2020, 73, 104750 CrossRef CAS.
- N. S. Weliwatte and S. D. Minteer, Joule, 2021, 5, 2564–2592 CrossRef CAS.
- H. Salehizadeh, N. Yan and R. Farnood, Chem. Eng. J., 2020, 390, 124584 CrossRef CAS.
- S. Bajracharya, S. Srikanth, G. Mohanakrishna, R. Zacharia, D. P. Strik and D. Pant, J. Power Sources, 2017, 356, 256–273 CrossRef CAS.
- R. Das, S. Chakraborty and S. C. Peter, ACS Energy Lett., 2021, 6, 3270–3274 CrossRef CAS.
- J. S. Martin, N. Dang, E. Raulerson, M. C. Beard, J. Hartenberger and Y. Yan, Angew. Chem., Int. Ed., 2022, 61, e20220557 CrossRef PubMed.
- Y. Zhang, L. Shi, H. Yuan, X. Sun, X. Li, L. Duan, Q. Li, Z. Huang, X. Ban and D. Zhang, Chem. Eng. J., 2022, 430, 132820 CrossRef CAS.
- S. Xu, L. Yang, Y. Wei, Y. Jia, M. Hu, L. Bai, J. Zhang, X. Li, S. Wei and J. Lu, Dalton Trans., 2022, 51, 8036–8045 RSC.
- Y. H. Chen, J. K. Ye, Y. J. Chang, T. W. Liu, Y. H. Chuang, W. R. Liu, S. H. Liu and Y. C. Pu, Appl. Catal., B, 2021, 284, 119751 CrossRef CAS.
- J. C. Wang, N. Li, A. M. Idris, J. Wang, X. Du, Z. Pan and Z. Li, Sol. RRL, 2021, 5, 2100154 CrossRef CAS.
- Y. X. Chen, Y. F. Xu, X. D. Wang, H. Y. Chen and D. B. Kuang, Sustainable Energy Fuels, 2020, 4, 2249–2255 RSC.
- X. Li, J. Liu, G. Jiang, X. Lin, J. Wang and Z. Li, J. Colloid Interface Sci., 2023, 643, 174–182 CrossRef CAS PubMed.
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Chem. – Eur. J., 2017, 23, 9481–9485 CrossRef CAS PubMed.
- Q. Wang, J. Wang, J. C. Wang, X. Hu, Y. Bai, X. Zhong and Z. Li, ChemSusChem, 2021, 14, 1131–1139 CrossRef CAS PubMed.
- Y. F. Xu, M. Z. Yang, H. Y. Chen, J. F. Liao, X. D. Wang and D. B. Kuang, ACS Appl. Mater. Interfaces, 2018, 1, 5083–5089 CAS.
- Y. J. Dong, Y. Jiang, J. F. Liao, H.-Y. Chen, D. B. Kuang and C.-Y. Su, Sci. China Mater., 2022, 65, 1550–1559 CrossRef CAS.
- X. Jiang, Y. Ding, S. Zheng, Y. Ye, Z. Li, L. Xu, J. Wang, Z. Li, X. J. Loh, E. Ye and L. Sun, ChemSusChem, 2022, 15, 202102295 CrossRef PubMed.
- X. Wang, J. He, L. Mao, X. Cai, C. Sun and M. Zhu, Chem. Eng. J., 2021, 416, 128077 CrossRef CAS.
- L. Li, Z. Zhang, C. Ding and J. Xu, Chem. Eng. J., 2021, 419, 129543 CrossRef CAS.
- K. Su, G. X. Dong, W. Zhang, Z. L. Liu, M. Zhang and T. B. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 50464–50471 CrossRef CAS PubMed.
- Z. Zhang, M. Shu, Y. Jiang and J. Xu, Chem. Eng. J., 2021, 414, 128889 CrossRef CAS.
- Z. Dong, Y. Shi, Y. Jiang, C. Yao and Z. Zhang, J. CO2 Util., 2023, 72, 102480 CrossRef CAS.
- Z. Zhang, Z. Dong, Y. Jiang, Y. Chu and J. Xu, Chem. Eng. J., 2022, 435, 135014 CrossRef CAS.
- Z. Zhang, L. Li, Y. Jiang and J. Xu, Inorg. Chem., 2022, 61, 3351–3360 CrossRef CAS PubMed.
- R. Cheng, E. Debroye, J. Hofkens and M. B. J. Roeffaers, Catalysts, 2020, 10, 1352 CrossRef CAS.
- L. Ding, B. Borjigin, Y. Li, X. Yang, X. Wang and H. Li, ACS Appl. Mater. Interfaces, 2021, 13, 51161–51173 CrossRef CAS PubMed.
- Y. Xi, X. Zhang, Y. Shen, W. Dong, Z. Fan, K. Wang, S. Zhong and S. Bai, Appl. Catal., B, 2021, 297, 120411 CrossRef CAS.
- J. Hu, M. Yang, X. Ke, S. Yang, K. Wang, H. Huang, W. Wang, D. Luo, Z. Zheng, L. Huang, P. Xiao, C. Tu, Y. Min, N. Huo and M. Zhang, J. Power Sources, 2021, 481, 228838 CrossRef CAS.
- J. Wang, L. Xiong, Y. Bai, Z. Chen, Q. Zheng, Y. Shi, C. Zhang, G. Jiang and Z. Li, Sol. RRL, 2022, 6, 2200294 CrossRef CAS.
- Z. Dong, Z. Zhang, Y. Jiang, Y. Chu and J. Xu, Chem. Eng. J., 2022, 433, 133762 CrossRef CAS.
- Y. Jiang, Y. Wang, Z. Zhang, Z. Dong and J. Xu, Inorg. Chem., 2022, 61, 10557–10566 CrossRef CAS PubMed.
- D. Li, J. Zhou, Z. Zhang, Y. Jiang, Z. Dong, J. Xu and C. Yao, ACS Appl. Mater. Interfaces, 2022, 5, 6238–6247 CAS.
- Y. Jiang, C. Mei, Z. Zhang and Z. Dong, Dalton Trans., 2021, 50, 16711–16719 RSC.
- M. Xiao, M. Hao, M. Lyu, E. G. Moore, C. Zhang, B. Luo, J. Hou, J. Lipton-Duffin and L. Wang, Adv. Funct. Mater., 2019, 29, 1905683 CrossRef CAS.
- J. Zhu, Y. Zhu, J. Huang, L. Hou, J. Shen and C. Li, Nanoscale, 2020, 12, 11842–11846 RSC.
- G. Zhang, X. Ke, X. Liu, H. Liao, W. Wang, H. Yu, K. Wang, S. Yang, C. Tu, H. Gu, D. Luo, L. Huang and M. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 44909–44921 CrossRef CAS PubMed.
- Y. Gong, J. Shen, Y. Zhu, W. Yan, J. Zhu, L. Hou, D. Xie and C. Li, Appl. Surf. Sci., 2021, 545, 149012 CrossRef CAS.
- M. Shu, Z. Zhang, Z. Dong and J. Xu, Carbon, 2021, 182, 454–462 CrossRef CAS.
- N. Zhang, J. J. Li, Y. Li, H. Wang, J. Y. Zhang, Y. Liu, Y. Z. Fang, Z. Liu and M. Zhou, J. Colloid Interface Sci., 2022, 608, 3192–3203 CrossRef CAS PubMed.
- J. Bian, C. Huang and R. Q. Zhang, ChemSusChem, 2016, 9, 2723–2735 CrossRef CAS PubMed.
- S.-X. Yuan, K. Su, Y.-X. Feng, M. Zhang and T.-B. Lu, Chin. Chem. Lett., 2023, 34, 107682 CrossRef CAS.
- Y. F. Mu, W. Zhang, G. X. Dong, K. Su, M. Zhang and T. B. Lu, Small, 2020, 16, 2002140 CrossRef CAS PubMed.
- A. Pan, X. Ma, S. Huang, Y. Wu, M. Jia, Y. Shi, Y. Liu, P. Wangyang, L. He and Y. Liu, J. Phys. Chem. Lett., 2019, 10, 6590–6597 CrossRef CAS PubMed.
- D. Wu, C. Tian, J. Zhou, Y. Huang, J. Lai, B. Gao, P. He, Q. Huang and X. Tang, Carbon Neutralization, 2022, 1, 298–305 CrossRef.
- C. Lu, D. S. Itanze, A. G. Aragon, X. Ma, H. Li, K. B. Ucer, C. Hewitt, D. L. Carroll, R. T. Williams, Y. Qiu and S. M. Geyer, Nanoscale, 2020, 12, 2987–2991 RSC.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S. M. Chang and E. W. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed.
- Y. Jiang, R. Zhou, Z. Z. Zhang, Z. L. Dong and J. Y. Xu, J. Mater. Chem. C, 2023, 11, 2540 RSC.
- L. Wu, S. Zheng, H. Lin, S. Zhou, A. Mahmoud Idris, J. Wang, S. Li and Z. Li, J. Colloid Interface Sci., 2023, 629, 233–242 CrossRef CAS PubMed.
- Y. Wang, Q. Zhou, Y. Zhu and D. Xu, Appl. Catal., B, 2021, 294, 120236 CrossRef CAS.
- S. Cheng, X. Chen, M. Wang, G. Li, X. Qi, Y. Tian, M. Jia, Y. Han, D. Wu, X. Li and Z. Shi, Appl. Surf. Sci., 2023, 621, 156877 CrossRef CAS.
- N. Li, X.-P. Zhai, B. Ma, H.-J. Zhang, M.-J. Xiao, Q. Wang and H.-L. Zhang, J. Mater. Chem. A, 2023, 11, 4020–4029 RSC.
- D. Wu, B. Huo, Y. Huang, X. Zhao, J. Yang, K. Hu, X. Mao, P. He, Q. Huang and X. Tang, Small, 2022, 18, 2106001 CrossRef CAS PubMed.
- C. Tian, Q. Huang, D. Wu, J. Lai, F. Qi, N. Zhang, W. Zhang and X. Tang, Mater. Today Energy, 2022, 28, 101067 CrossRef CAS.
- Q. Huang, C. Tian, J. Lai, F. Qi, N. Zhang, J. Liu, Z. Liang, D. Wu and X. Tang, Inorg. Chem., 2022, 61, 14447–14454 CrossRef CAS PubMed.
- J. Liu, Z. Wu, F. Zhang, M. Zhao, C. Li, J. Li, B. Wen and F. Wang, Nanoscale, 2023, 15, 7023–7031 RSC.
- S. Kumar, I. Hassan, M. Regue, S. Gonzalez-Carrero, E. Rattner, M. A. Isaacs and S. Eslava, J. Mater. Chem. A, 2021, 9, 12179 RSC.
- S. Bera, A. Patra, S. Shyamal, D. Nasipuri and N. Pradhan, ACS Energy Lett., 2022, 7, 3015–3023 CrossRef CAS.
- Y. F. Xu, X. D. Wang, J. F. Liao, B. X. Chen, H. Y. Chen and D. B. Kuang, Adv. Mater. Interfaces, 2018, 5, 1801015 CrossRef.
- Z. C. Kong, J. F. Liao, Y. J. Dong, Y. F. Xu, H. Y. Chen, D. B. Kuang and C. Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. B. Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- Y. Jiang, J. F. Liao, Y. F. Xu, H. Y. Chen, X. D. Wang and D. B. Kuang, J. Mater. Chem. A, 2019, 7, 13762 RSC.
- Y. Jiang, J. F. Liao, H. Y. Chen, H. H. Zhang, J. Y. Li, X. D. Wang and D. B. Kuang, Chem, 2020, 6, 766–780 CAS.
- S. Kumar, M. Regue, M. A. Isaacs, E. Freeman and S. Eslava, ACS Appl. Energy Mater., 2020, 3, 4509–4522 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Wanlin
Guo
* and
Wanlin
Guo