
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystalline carbon nitrides for photocatalysis
Han
Li
ab,
Bei
Cheng
ac,
Jingsan
Xu
 *d,
Jiaguo
Yu
e and
Shaowen
Cao
*ac
*d,
Jiaguo
Yu
e and
Shaowen
Cao
*ac
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: swcao@whut.edu.cn
bSchool of Materials Science and Engineering, Hubei University of Automotive Technology, Shiyan 442020, P. R. China
cHubei Technology Innovation Center for Advanced Composites, Wuhan University of Technology, Wuhan 430070, P. R. China
dSchool of Chemistry and Physics, Queensland University of Technology, Brisbane, QLD 4000, Australia. E-mail: jingsan.xu@qut.edu.au
eLaboratory of Solar Fuel, Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, P. R. China
First published on 6th January 2024
Abstract
Photocatalysis is considered as an effective approach to address energy and environmental issues. Carbon nitride (CN) is a promising metal-free semiconductor photocatalyst because of its unique properties such as tunable electronic band structure, facile/cheap synthesis and high chemical stability. However, the pristine CN prepared by the traditional thermal polymerization method is usually an amorphous or semi-crystalline conjugated bulk with a high density of structural defects, resulting in its moderate photocatalytic activity. Increasing the crystallinity of CN is an effective strategy to enhance its photocatalytic activity, and a few methods have been proposed, including high-temperature and high-pressure treatment, ionothermal method, solvothermal synthesis and microwave-assisted thermal polymerization. This review summarizes recent advances in the preparation of crystalline carbon nitrides (CCNs) and the design of CCNs-based photocatalysts in terms of nanostructure design, molecular structure engineering and construction of CCNs-based heterojunctions. In addition, their applications in a range of photocatalysis fields such as water splitting, carbon dioxide reduction, degradation of pollutants, organic synthesis and H2O2 production are reviewed. Finally, the concluding remarks are presented as well as challenges and prospects for future development of CCNs-based photocatalysts.
 Han Li | Han Li received his BE degree from North China University of Water Resources and Electric Power. Then he obtained his MS and PhD degrees in Materials Science and Engineering from Wuhan University of Technology. Now he works at the School of Materials Science and Engineering, Hubei University of Automotive Technology. His current research focuses on photocatalytic H2O2 production, CO2 reduction, water splitting, and environmental purification. |
 Bei Cheng | Bei Cheng received her BS degree and MS degree from Tongji University, and her PhD degree from Wuhan University of Technology (WUT). In 2010, she became a Professor at WUT. Her research interests include semiconductor photocatalysis (such as photocatalytic hydrogen production, CO2 reduction, H2O2 evolution, and so on) and the fabrication and application of environmental cleaning materials. |
 Jingsan Xu | Jingsan Xu received his BS degree in Materials Science and Engineering in 2008 from Jilin University, and his PhD degree in 2013 from Shanghai Institute of Ceramics, Chinese Academy of Sciences. Then he finished the postdoctoral work from 2013 to 2016 in the Max Planck Institute of Colloids and Interfaces. He commenced as an ARC DECRA fellow at QUT in 2016 was promoted to full professor in 2021. His research field is focused on solar energy harvesting, catalysis and interfaces. One topic he is particularly interested in is light-driven chemical reactions, such as organic synthesis, supramolecular assembling, crystal transformations. |
 Jiaguo Yu | Jiaguo Yu received his BS degree and MS degree in Chemistry from Central China Normal University and Xi'an Jiaotong University, respectively, and his PhD degree in Materials Science from Wuhan University of Technology (WUT). In 2000, he became a Professor at WUT. In 2021, he moved to China University of Geoscience (Wuhan). His research interests include semiconductor photocatalysis, photocatalytic hydrogen production, CO2 reduction, perovskite solar cells, electrocatalysis, and so on. He is a foreign member of Academia Europaea (2020), foreign fellow of the European Academy of Sciences (2020), and fellow of the Royal Society of Chemistry (2015). |
 Shaowen Cao | Shaowen Cao received his BS degree in Geochemistry in 2005 from University of Science and Technology of China, and his PhD degree in Materials Chemistry & Physics in 2010 from Shanghai Institute of Ceramics, Chinese Academy of Sciences. Then he was a Research Fellow at Nanyang Technological University until Feb 2014. From Mar 2018 to Feb 2020, he was a Visiting Scientist at Max Planck Institute of Colloids and Interfaces. He is now a professor at Wuhan University of Technology. His-current research interests include the design and fabrication of photocatalytic materials for energy and environmental applications. |
Broader contextThe excessive consumption of fossil fuel has not only led to energy shortages, but also resulted in environmental pollution. Semiconductor-based photocatalysis is recognized as an effective strategy to alleviate the above issues since it can directly convert clean solar energy into valuable chemical fuels and degrade pollutants. Carbon nitride (CN) has emerged as a promising photocatalyst due to its unique properties including tunable electronic structure, facile/cheap synthesis and high chemical stability. However, pristine CN suffers from limited light absorption and severe charge recombination, thus resulting in moderate photocatalytic activity. Improving the crystallinity of CN has been demonstrated as an effective approach to enhance the photocatalytic performance because of the improved charge transfer, reduced bulk defects and enhanced light absorption. This review summarizes recent advances in the preparation of crystalline carbon nitrides (CCNs) and the design of CCNs-based photocatalysts in terms of nanostructure design, molecular structure engineering and construction of CCNs-based heterojunctions. In addition, their applications in a range of photocatalysis fields are reviewed. Finally, the challenges and prospects for future development of CCNs-based photocatalysts were also discussed. We hope that this review could provide valuable insights for designing highly efficient CCNs-based photocatalysts for energy and environmental applications. |
1. Introduction
Semiconductor-based photocatalysis is recognized as an effective solution to the energy shortage and environmental pollution since it can directly convert clean solar energy into valuable chemical fuels such as hydrogen and hydrocarbon, selectively synthesize organic compounds, and degrade organic pollutants.1–6 In the past decades, various semiconductors (such as TiO2,7–9 Fe2O3,10 ZnO,11 Bi2WO6,12 BiVO4,13 CdS,14etc.) have been investigated as photocatalysts for solar-to-chemical energy conversion. However, the large band gap of metallic oxide semiconductors restricts the applications of photocatalysis due to the insufficient utilization of sunlight. While metal sulfides have also been considered due to their narrow band gap and appropriate conduction band positions, their further applications were limited by the disadvantages including chemical instability due to photo-corrosion and self-oxidation.15 Therefore, the development of photocatalysts with visible-light activity and chemical stability has become a popular research topic and major task for a long time.Polymeric carbon nitride has emerged as a promising candidate photocatalyst since 2009 due to its extraordinary properties, such as non-toxicity, low cost, easy preparation, high physicochemical stability, and well-matched band structure,16 enabling its widespread applications in photocatalytic water splitting, carbon dioxide reduction, pollutant degradation, organic synthesis, etc.17–22 Unfortunately, the pristine CN prepared by traditional thermal polymerization is usually an amorphous or semi-crystalline conjugated polymer with high-density of structural defects, leading to its moderate photocatalytic activity. The low crystallinity caused by incomplete polymerization leads to the formation of hydrogen bonds in the covalent CN framework. The intralayer hydrogen bonds in pristine CN hinder the in-plane electron conduction, resulting in poor intralayer transport. Theoretically, increasing the crystallinity of CNs not only enhances the transport of charge carriers and reduces the recombination rate of electron–hole pairs, but also extends the π-conjugated system and improves the light absorption capability. Therefore, reducing structural defects by increasing the crystallinity of CNs is a critical strategy to optimize their photocatalytic performance.23–26 Notably, the kinetic hindrance is a dominant issue during bulk condensation, which leads to incomplete polymerization. Hence, it is highly desirable to promote the mass and heat transfer to improve the crystallinity. To this end, considerable effort has been made to prepare CCNs, including high-temperature and high-pressure strategy, ionothermal route, solvothermal approach and microwave-assisted thermal synthesis.
Up to now, there has been numerous publications on the design of CCNs-based photocatalysts for various applications, including several specific and inspiring reviews.21,23,27,28 However, an overarching understanding on the preparation, design, mechanism and applications of CCNs-based photocatalysts is still lacking. Hence, we present a comprehensive review of the latest significant progress in the preparation, design, and applications of CCNs-based photocatalysts. Specifically, the development history of CNs will be introduced first, followed by the synthesis method and design of CCNs-based photocatalysts. Then the applications of CCNs-based photocatalysts in water splitting, carbon dioxide reduction, pollutant removal and organic synthesis, and H2O2 production are presented. Finally, the concluding criticism and perspectives on the challenges and future development of CCNs-based photocatalysts are briefly discussed.
2. Discovery and development history of CNs
To gain a comprehensive understanding of CNs, it is necessary to discuss their development history (Fig. 1a), which dates back to 1834, when Berzelius first synthesized a linear polymer, named “melon” by Liebig.29 A more in-depth understanding of the composition and structure of these compounds was described by Franklin in 1922.30 Pauling and Sturdivant proposed in 1937 that the coplanar tri-s-triazine unit was the basic structural unit of these polymeric derivatives, which was confirmed by X-ray studies.31 Later, Redemann and Lucas32 deduced that the 21 molecules of 2,5,8-triamino-tris-s-triazine, that is, C126H21N175 may be the oligomeric condensation product of carbon nitride, which was earlier described by Franklin. Based on these findings, it can be concluded that it is not possible to assign a single structure to melon, as it is a mixture of molecules of different sizes and nanostructures.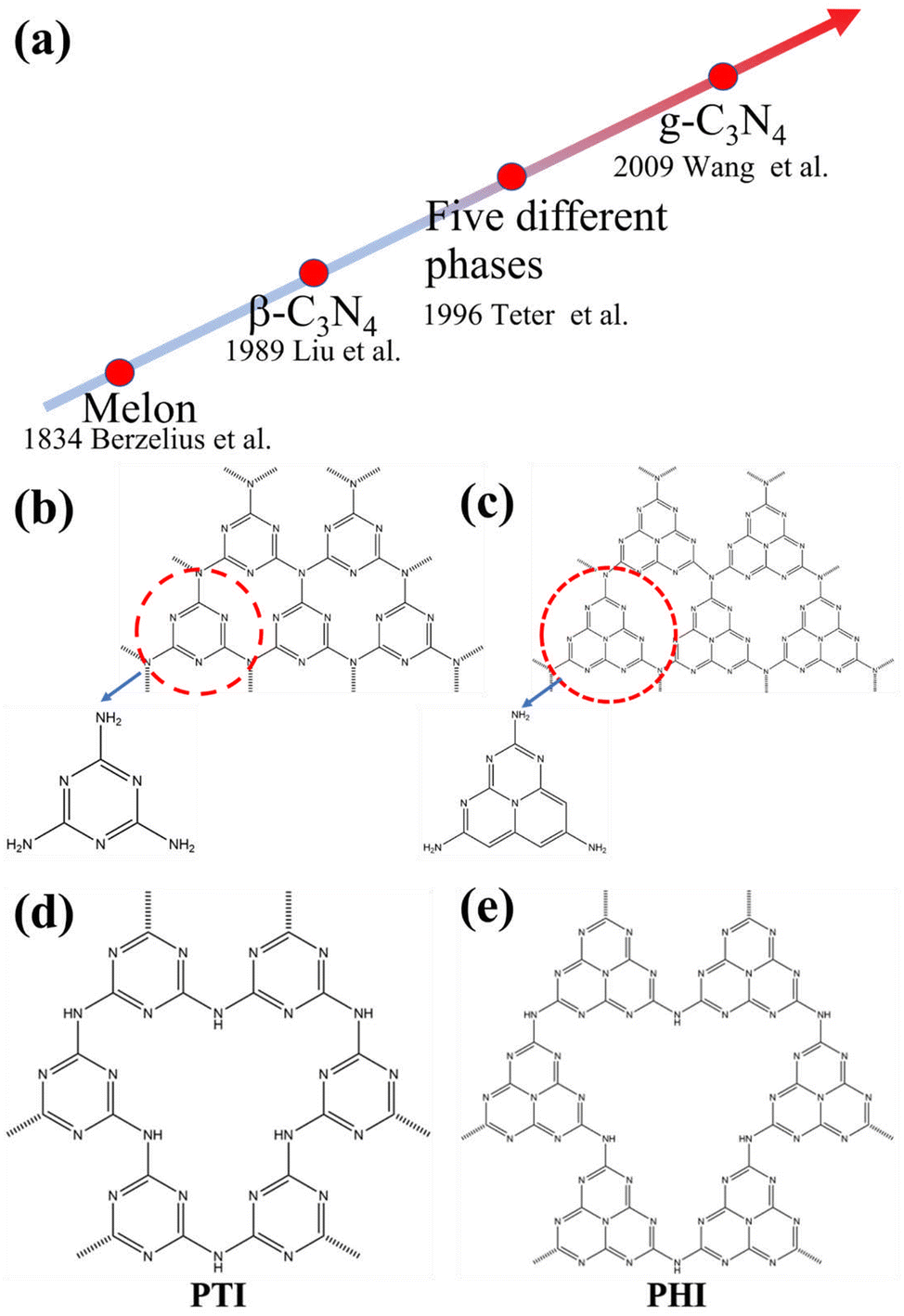 | ||
| Fig. 1 (a) The roadmap of the development of carbon nitrides. (b) Triazine and (c) tri-s-triazine (heptazine) structures of g-C3N4. (d) Structures of poly(triazine imide) (PTI) and (e) poly(heptazine imide) (PHI). | ||
This melon-based CNs were forgotten for a long time as an unproven species, owning to chemical inertness and poor solubility. Surprisingly, interest in CNs was revived in 1989, as it was theoretically predicted that by replacing Si with C in β-Si3N4, the sp3-bonded C3N4 phase (β-C3N4) exhibited extremely high bulk modulus and hardness values, which could compare with, even exceed that of diamond.33 Later, theoretical investigations conducted by Teter and Hemley further confirmed this prediction. Meanwhile, five different phases of CNs were predicted, including α-C3N4, β-C3N4, cubic C3N4, pseudocubic C3N4 and graphitic C3N4 (g-C3N4).34 These interesting results not only motivated experimental preparation of the hypothetical phase, but also stimulated further theoretical studies on CNs. However, the preparation of single-phase β-C3N4 was proved to be extremely challenging due to its low thermodynamic stability.35 Further theoretical research confirmed that g-C3N4, with a graphite-like layered structure, is the most stable allotrope under ambient conditions.36–38 However, the characterization of g-C3N4 is a challenging task due to its low crystallinity and high degree of disorder. Owing to the lack of experimental data, discussions about the actual existence of ideal g-C3N4 and its possible molecular structure are prevalent. It has been pointed out that the two basic structural units for the construction of g-C3N4 isomers are the triazine (C3N3) and tri-s-triazine/heptazine (C6N7) rings, respectively (Fig. 1b and c).36,38–41 Moreover, the heptazine rings was energetically favorable over the triazine-based units.42,43 The heptazine rings were interconnected through bridging imide bonds (Fig. 1c), and recent studies have demonstrated that the pyrolysis of cyanamide, dicyandiamide, or melamine yields a melon polymer composed of melem subunits,42–52 indicating that this unit is the most robust and stable building block. Therefore, the heptazine-based g-C3N4 has been extensively investigated in many fields, particularly in catalysis,53,54 where the discovery of melon-based carbon nitride as a metal-free polymeric photocatalyst for water splitting was first proposed by Wang et al. in 2009.16 Since then, the research on g-C3N4 as a metal-free polymeric conjugated photocatalyst has been nothing short of explosive. However, the pristine CN prepared by the traditional thermal polymerization method is a melon-based structure, which is amorphous or semi-crystalline and exhibits moderate photocatalytic activity.
In principle, increasing the crystallinity of the photocatalysts not only enhances the mobility of charge carriers and reduce recombination centers, but also extends the π-conjugated system and improves the light absorption capacity, thus enhancing the photocatalytic performance. Therefore, various approaches have been developed to synthesize CCNs. The actual crystal structure of CCNs were further confirmed because of their high crystallinity. Then, their crystal structure (Fig. 1d and e) was proposed and confirmed by density functional theory (DFT) calculations and experimental results. Kawaguchi et al. proposed a PTI-based CCNs structure (Fig. 1d), consisting of a two-dimensional (2D) plane framework of triazine units connected through NH groups.39 While the PTI-based CCNs synthesized by molten salt method contains metal ions embedded in the interlayer or void. Schnick and co-workers presented the crystal structure of the PHI-based CCNs (Fig. 1e), which was composed of six heptazine subunits arranged in a hexagonal configuration and bridged by imide-bonds.55 This connection scheme introduced a large pore space in the planner network compared with g-C3N4. Similarly, the alkali metal ions in the framework of PHI-based CCNs are generally located in the voids formed by the six heptazine units during the molten salt process. Moreover, the advantages of CCNs are summarized as follows: (1) the high crystallinity of CCNs could decrease the number of defects, thereby reducing the recombination centers of charge carriers. (2) The high crystallinity of CCNs could narrow the band gap due to the extended π-conjugated system. (3) The high crystallinity of CCNs could decresae the interlayer distance, which enhances interlayer exciton dissociation and improves the charge transport (conductivity).
3. Properties of CNs and CCNs
3.1. Thermal stability
Thermal gravimetric analysis (TGA) of the CN proved that the CN can remain stable up to 600 °C and above.45 A significant endothermal peak is observed at 630 °C, as well as a continuous weight loss due to the breakdown of CN. The complete decomposition of CNs at 750 °C confirmed its high thermal stability.16,56 The decomposition temperature of the CN could vary slightly depending on the different preparation procedures in literature,39,45,57,58 probably due to the different degrees of polycondensation.3.2. Chemical stability
The strong van der Waals force between the CN layers makes them prone to agglomerate and therefore insoluble in most solvents. No detectable dissolubility or reactivity of CNs was observed in conventional solvents, including H2O, C2H5OH, DMF, THF, diethyl ether, and toluene.58 To investigate the stability and durability of CNs in different solvents, CNs powder was dispersed in H2O, acetone, ethanol, pyridine, acetonitrile, dichloromethane, DMF, acetic acid, and 0.1 M sodium hydroxide aqueous solution for 30 days. After that, the dispersed CNs were dried at 80 °C for 10 hours. Subsequently, the FTIR spectra of the impregnated CNs were measured and showed almost no change compared with the fresh ones, indicating their good chemical stability and excellent durability. In addition, CNs treated with a certain concentration of acid could form a colloidal dispersion or a true solution,59,60 which however is reversible. The photochemical stability of CCNs was considered to be high in most previous studies. Nevertheless, Zou and co-workers reported that the photocatalytic performance of CCN nanotube was decreased because of the loss of K, Na, and Cl ions.61 Rcently, Hou and co-workers proposed that the PHI-based CCN was unstable during the photocatalytic process due to the Na+ loss.62 Interestingly, the addition of NaCl could improve the stability. Therefore, it is reasonable to assume that the CCNs are suitable for photocatalytic reactions in seawater. In short, the photochemical stability of CCNs needs to be further investigated and the focus could be the relationship between the leaching of metal ions and the stability of photocatalytic activity.3.3. Optical and electronic properties
The optical properties of CNs were tested by UV-vis diffuse reflectance spectroscopy and photoluminescence. As revealed by the theoretical calculation, the CNs are an n-type semiconductor featuring a bandgap up to 5 eV, depending mainly on the structural or geometrical diversity.63 In fact, conventional CNs exhibit strong bandgap adsorption around 420 nm, which is in accordance with the light yellow color. Preparation approaches, including various precursors and polymerization temperature, may marginally affect the absorption edge of CNs, mainly due to different crystallinity, crystal structures, packing, particle size and defects.16,56,64,65 Generally, the pristine CNs exhibits blue luminescence at room temperature. The photoluminescence (PL) was observed between 430 nm and 550 nm, with a peak around 470 nm. Time-resolved photoluminescence (TRPL) was used to investigate the charge transfer dynamics, exhibiting an average lifetime of ∼5 ns.66As revealed by the UV-vis diffuse reflectance spectroscopy, the CN possesses a bandgap of ca. 2.7 eV. While the conductive band (CB) and valence band (VB) are positioned at ca. −1.1 eV (vs. normal hydrogen electrodes) and ca. +1.6 eV (vs. normal hydrogen electrodes), respectively. This appropriate electronic band structure makes CNs a promising candidate for applications in solar energy conversion.
4. Synthesis and characterization of CCNs
Despite the in-depth research on the structure of CNs, the preparation of CCNs is still challenging. As we all know, the kinetic hindrance is the predominant issue during the condensation process, which results in incomplete polymerization. To solve this problem, many strategies have been developed to promote the mass and heat transfer. Early attempts to prepare CCNs were based on thin film growth techniques using various physical and chemical vapor deposition approaches.67–71 Considering that most photocatalysts are powder-based, in this review, the synthesis methods of CCNs are focused on bulk synthesis, as shown in Fig. 2.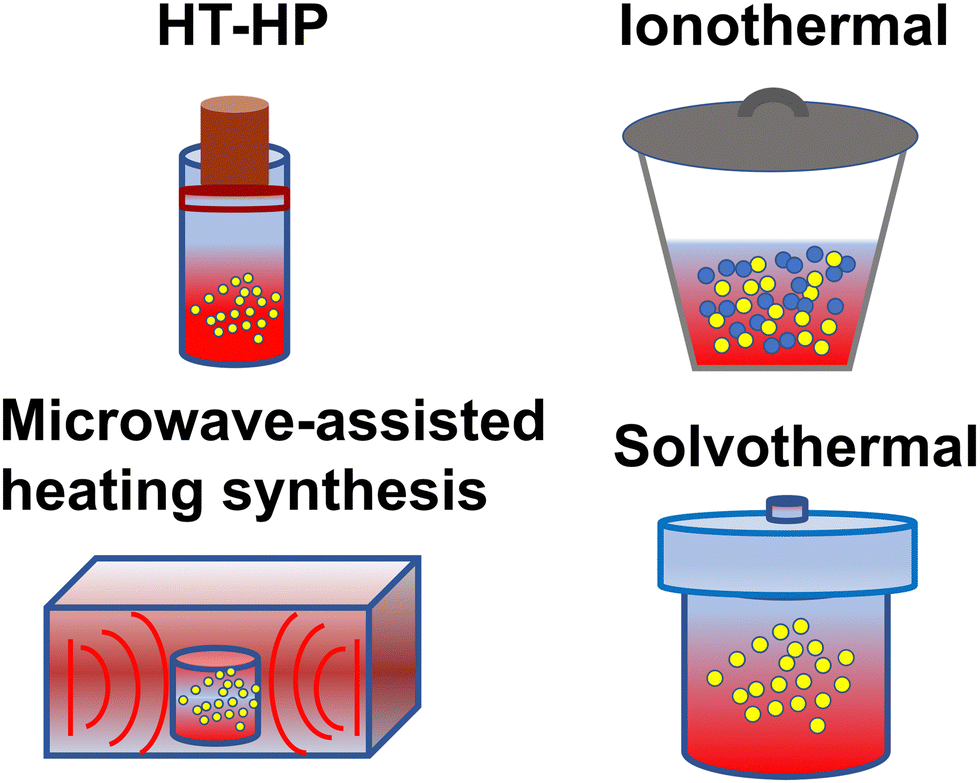 | ||
| Fig. 2 Schematic diagram of synthesis method: high temperature-high pressure method, ionothermal route, microwave-assisted heating synthesis, and solvothermal approach. | ||
4.1. High temperature-high pressure method
Inspired by the high-pressure synthesis of diamond from graphite, early attempts to prepare CCNs adopted the same approach. Weill and co-workers reported a new model of graphitic C3N4 with different carbon vacancies and layered structures.72 The CCN was synthesized in a belt-type apparatus at 3 GPa and 800 °C using melamine as the precursor and hydrazine as the nitriding solvent. The XRD pattern of the sample indicates that it was an orthorhombic structure that could not be indexed according to the model proposed by Teter and Hemley.34 Moreover, all the expected peaks of orthorhombic symmetry were absent and the relative intensities did not agree with the calculated results, mainly due to the effect of the internal pressure, which may lead to reorientation of the specimens. Wolf and co-workers reported another significant research for the preparation of CCN by the high temperature and high pressure (HT–HP) approach.73 This novel CCN, containing stoichiometric amounts of HCl, was prepared through a solid-state reaction at 1.0–1.5 GPa and 500–550 °C using melamine and cyanuric chloride as precursors. This product had a two-dimensional C6N9H3 framework belonging to the hexagonal space group (P63/m), which was similar to the structure proposed by Kouvetakis et al.,74 but lacking one-third of the triazine rings (Fig. 3a). The vacancy was occupied by Cl− ions and the N atoms in the framework were protonated to maintain charge balance. The electron energy-loss spectroscopy (EELS) revealed that both the C and N atoms were sp2-hybridized (Fig. 3b). The electron diffraction pattern confirmed the hexagonal symmetry. Riedel and co-workers obtained a novel CCN phase with a well-defined structure and chemical composition by the HT–HP method.75 In this study, C2N4H4 was used as the single-source precursor, and the synthesis was performed at 1700–2300 °C and above 27 GPa. The electron energy-loss spectroscopy (EELS) indicates that the C atoms and N atoms were sp3-hybridized (Fig. 3c). The as prepared CCN featured a 3D carbon nitride network and exhibited a defective wurtzite-type structure, which was confirmed by the electron-diffraction results (Fig. 3d) and DFT studies. In summary, even with some shortcomings like high temperature nitrogen loss, the HT–HP strategy is still an efficient approach to synthesize CCNs. More importantly, some precursors were proved to contribute to the synthesis of CCNs, but the application of CCNs synthesized by the HT–HP approach requires further research.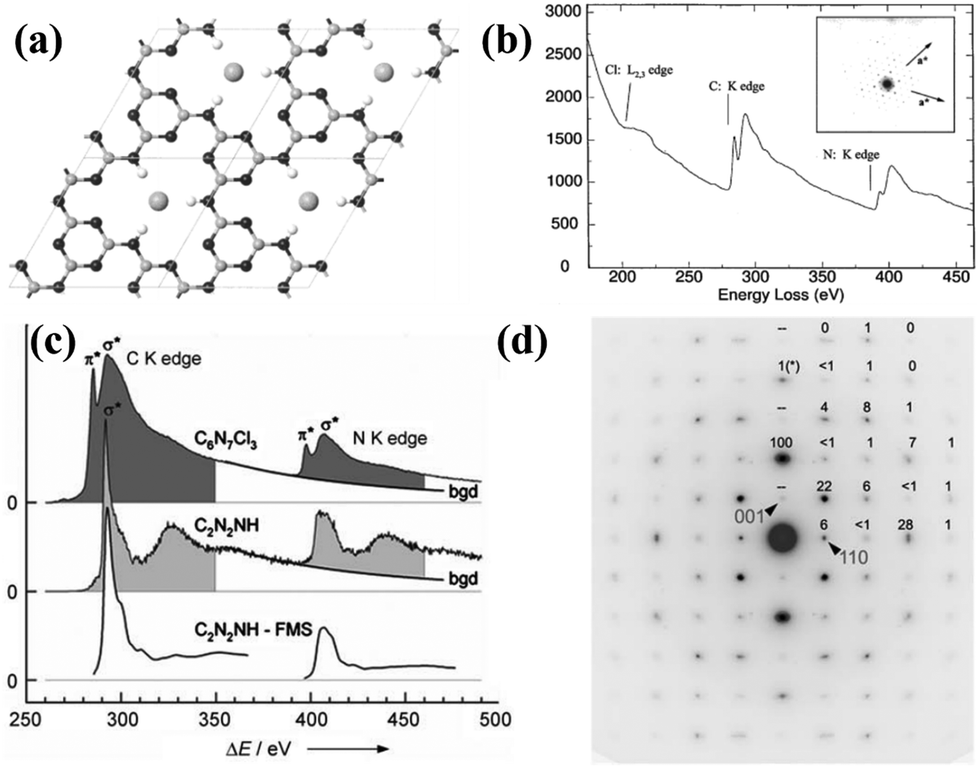 | ||
Fig. 3 (a) Proposed molecular structure of C6N9H3·HCl. (b) EELS spectrum of C6N9H3·HCl and the corresponding electron diffraction pattern (inset). Reproduced with permission.73 Copyright 2001, American Chemical Society. (c) EELS spectrum of C and N K edges of the C6N7Cl3 and C2N2(NH). (d) SAED pattern along the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10] zone of the orthorhombic cell of dwur-C2N2(NH). Reproduced with permission.75 Copyright 2007, Wiley-VCH. 10] zone of the orthorhombic cell of dwur-C2N2(NH). Reproduced with permission.75 Copyright 2007, Wiley-VCH. | ||
4.2. Ionothermal (molten salt) method
Kinetic barriers are a major problem in solid-state reactions, while a liquid environment would be more conducive to heat and mass transfer. Motivated by the application of the eutectic salt mixtures in organic syntheses and electrochemical processes, Thomas and co-workers first used ionothermal approach to synthesize CCN.46 LiCl and KCl (45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55 wt%) were utilized as a good solvent because of its high-temperature stability, non-corrosiveness, good solvation of precursors and intermediates, and more importantly its melting point below the polycondensation point of s-heptazine. The XRD pattern (Fig. 4a) of CCN shows two sharp peaks at 12.1 and 26.5°, which was different from the melon-based CN synthesized by the traditional bulk condensation method. The obtained CCN had a hexagonal prism morphology due to the preferential growth in the z-direction, confirming the single crystal property (Fig. 4a). The high crystallinity was further determined by high resolution transmission electron microscopy (HRTEM) image, the lattice fringes were clearly observed (Fig. 4b) and the crystal plane spacing measured by selected area electron diffraction (SAED) was consistent with the XRD results. However, the location of the residual chloride ions was unclear, which needs further investigation. Schnick and co-workers conducted a detailed structural characterization of CCN synthesized by ionothermal method using LiCl/KCl as solvent.76 The solid-state nuclear magnetic resonance (NMR) spectroscopy, FTIR spectrum, and 15N cross-polarization with polarization inversion (CPPI) experiments confirmed that the CCN was a 2D network constructed from imide-bridged triazine units rather than heptazine building blocks. Thus, the CCN was identified as poly(triazine imide) (PTI). The reason for this CCN consisting of triazine is probably because the molten salt destabilizes or even decomposes the heptazine units at higher temperatures, since the use of heptazine-based precursors, such as melem, also results in the formation of PTI at 600 °C. Several strong X-ray reflections of PTI suggest that the salt melting method leads to its high crystallinity. Interestingly, the interlayer spacing of 0.336 nm determined by the XRD result was significantly larger than that of other graphite-like CN condensation products, such as melon,77 PHI55 and C6N9H3·HCl.73 This broadening of the interlayer spacing could be attributed to the incorporation of Cl ions. The HRTEM and SAED results (Fig. 4c) revealed the hexagonal symmetry and graphite-like stacking. The Cl− ions were located at the in-plane voids and intercalated between the layers, while Li+ ions also occupied the in-plane vacancy and were distributed around chloride ions for charge compensation. Hence, the CCN was also known as PTI/Li+Cl− due to the molecular structure and chemical composition (Fig. 4d).
:55 wt%) were utilized as a good solvent because of its high-temperature stability, non-corrosiveness, good solvation of precursors and intermediates, and more importantly its melting point below the polycondensation point of s-heptazine. The XRD pattern (Fig. 4a) of CCN shows two sharp peaks at 12.1 and 26.5°, which was different from the melon-based CN synthesized by the traditional bulk condensation method. The obtained CCN had a hexagonal prism morphology due to the preferential growth in the z-direction, confirming the single crystal property (Fig. 4a). The high crystallinity was further determined by high resolution transmission electron microscopy (HRTEM) image, the lattice fringes were clearly observed (Fig. 4b) and the crystal plane spacing measured by selected area electron diffraction (SAED) was consistent with the XRD results. However, the location of the residual chloride ions was unclear, which needs further investigation. Schnick and co-workers conducted a detailed structural characterization of CCN synthesized by ionothermal method using LiCl/KCl as solvent.76 The solid-state nuclear magnetic resonance (NMR) spectroscopy, FTIR spectrum, and 15N cross-polarization with polarization inversion (CPPI) experiments confirmed that the CCN was a 2D network constructed from imide-bridged triazine units rather than heptazine building blocks. Thus, the CCN was identified as poly(triazine imide) (PTI). The reason for this CCN consisting of triazine is probably because the molten salt destabilizes or even decomposes the heptazine units at higher temperatures, since the use of heptazine-based precursors, such as melem, also results in the formation of PTI at 600 °C. Several strong X-ray reflections of PTI suggest that the salt melting method leads to its high crystallinity. Interestingly, the interlayer spacing of 0.336 nm determined by the XRD result was significantly larger than that of other graphite-like CN condensation products, such as melon,77 PHI55 and C6N9H3·HCl.73 This broadening of the interlayer spacing could be attributed to the incorporation of Cl ions. The HRTEM and SAED results (Fig. 4c) revealed the hexagonal symmetry and graphite-like stacking. The Cl− ions were located at the in-plane voids and intercalated between the layers, while Li+ ions also occupied the in-plane vacancy and were distributed around chloride ions for charge compensation. Hence, the CCN was also known as PTI/Li+Cl− due to the molecular structure and chemical composition (Fig. 4d).
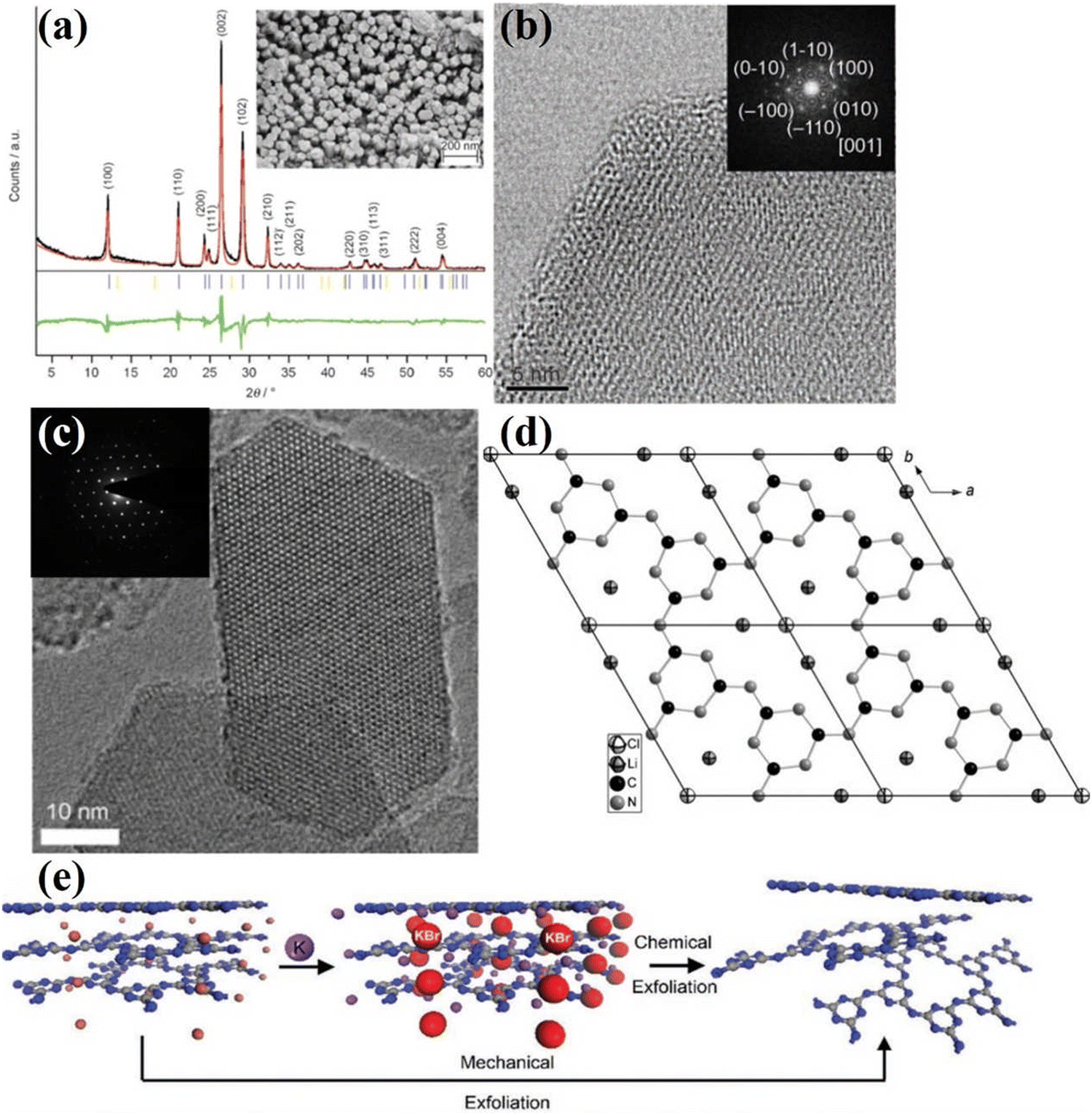 | ||
| Fig. 4 (a) XRD pattern of CCN and SEM image of CCN (inset). (b) HRTEM image along the [001] axis and the corresponding SAED (inset). Reproduced with permission.46 Copyright 2008, Wiley-VCH. (c) HRTEM image of PTI/Li+Cl− and the corresponding SAED pattern of the hk0 plane (inset). (d) Projection of the structure of single-layer PTI/Li+Cl− along the c axis. Reproduced with permission.76 Copyright 2011, Wiley-VCH. (e) Schematic diagram of the preparation approach from PTI/Br to PTI nanosheet. Reproduced with permission.79 Copyright 2013, Wiley-VCH. | ||
Considering that interlayer distance and electronic properties can be modified by ionic intercalation, Bojdys and co-workers prepared CCN with adjustable interlayer distances by changing the eutectic melt.78 Compared with the PTI/Li+Cl− (0.338 nm) prepared in KCl/LiCl eutectic mixture, the as-synthesized poly(triazine imide) intercalated by bromide ions exhibited an expanded interlayer distance (0.352 nm). While the PTI/F showed a reduced gallery height due to smaller anions. Similar to the processing of bulk graphite into graphene, the bulk PTI could be mechanically and chemically exfoliated into thin sheets. According to this hypothesis, Bojdys and co-workers acquired a thin nanosheet from the PTI/Br by Scotch-tape exfoliation (Fig. 4e).79 The thickness of the obtained nanosheet was determined by the AFM analysis, corresponding to the stacking of 10–100 layers. The PTI/Br could also be exfoliated by gas-phase metal potassium treatment. Once the K(PTI/Br) encountered water, K would react with water and generate H2, which was released between the layers, thus promoting the exfoliation process. XRD and SAED patterns show that the obtained nanosheet are predominantly crystalline.
Overall, the ionothermal route is a facile, eco-friendly and powerful approach for the synthesis of CCNs. Moreover, the CCNs synthesized by ionothermal approach generally contained alkali metal ions and exhibited distinct properties and high activity for photocatalysis. However, the underlying formation mechanism of CCNs in molten salt is still not well understood.
4.3. Solvothermal method
Considering the easy loss of nitrogen during high-temperature synthesis, the solvothermal route, which was generally performed at intermediate temperatures, had gained much attention.80–88 Bai and co-workers synthesized graphite-like CCN by reacting CCl4 with NH4Cl at 400 °C through the solvothermal route.81 From the sharp peak of the XRD pattern (Fig. 5a), which was consistent with the theoretical results, it could be confirmed that the sample was graphite-like C3N4 with high crystallinity, which was further proved by the clear diffraction spots (Fig. 5b) in the SAED pattern. Zhu and co-workers reported a solvothermal route for the synthesis of CCN using the C3N3Cl3 and Li3N as precursors in benzene at 355 °C for 12 h.82 The high intensity peaks of α-C3N4 and β-C3N4 appeared in the XRD pattern (Fig. 5c), indicating that the CCN consisted mainly of crystalline a-C3N4 and β-C3N4. The EELS (Fig. 5d) revealed that the carbon atom was mainly sp3 bonded in the sample, further confirming the formation of α-C3N4 and β-C3N4 crystalline phases. Moreover, benzene could effectively dissolve and disperse C3N3Cl3 and activate Li3N, which facilitated the crystallization of CN under supercritical conditions. In the same year, Xie and co-workers prepared well-crystallized graphitic CN via a benzene-thermal method by reacting C3N3Cl3 with NaNH2 at 180–220 °C for 8–12 h.83 The sharp (002) peak in the XRD pattern (Fig. 5e) proved the good crystallinity of CN. Moreover, the XPS result further confirmed that the atomic N/C ratio was in good agreement with the C3N4 stoichiometry. The SAED results (Fig. 5f) indicate that the diffraction ring can be indexed to (002) reflection, which corresponds to the graphitic structure of polycrystalline feature, proving the successful preparation of CCN. Cao and co-workers developed an acetonitrile-promoted solvothermal route for the synthesis of CCN with cyano and carboxyl groups.88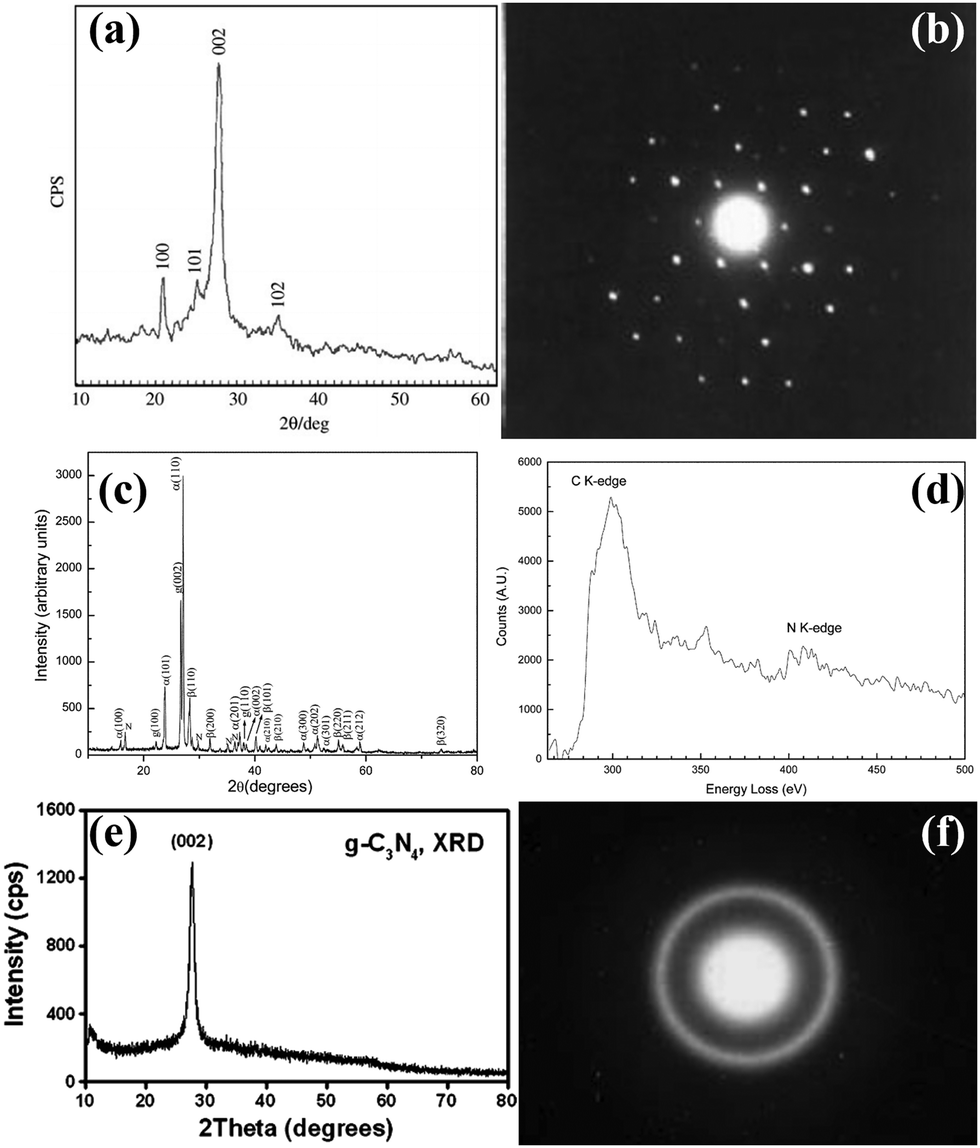 | ||
| Fig. 5 (a) XRD pattern of g-C3N4 nanocrystals. (b) The SAED of g-C3N4 single crystal. Reproduced with permission.81 Copyright 2003, Elsevier. (c) XRD pattern of the CCN. (d) EELS spectrum of C and N K-edges of CCN. Reproduced with permission.82 Copyright 2003, The Royal Society of Chemistry. (e) XRD pattern of g-C3N4 nanocrystallites. (f) The SAED pattern of g-C3N4 nanocrystallites. Reproduced with permission.83 Copyright 2003, Elsevier. | ||
In summary, the solvothermal method is a convenient way for the large-scale synthesis of novel nanostructured CCNs. Note that the solvothermal methods sometimes require high temperture and harsh chemicals, which could be further optimized. In addition, the synthesis mechanism of this novel nanostructured CCN prepared by solvothermal method deserves further investigation.
4.4. Microwave (MW)-assisted heating synthesis
Although there are numerous publications on the synthesis of CCNs, the synthetic methods usually required high temperature and long reaction time (several hours). Therefore, an effective synthetic strategy for the rapid synthesis of CCN is highly desirable. Xue and co-workers developed a rapid synthesis strategy known as “microwave-assisted heating synthesis”, enabling the preparation of CCN within minutes.89 In this approach, CuO was utilized as the microwave (MW) absorber, which intensively absorbed MW and induced rapid heating within few minutes. Evidently, the peak intensities of the obtained product (Fig. 6a) were higher than those of the pristine CN, confirming the enhanced crystallinity, which was further confirmed by the HRTEM image. Very distinct lattice fringes with a distance of 0.33 nm, corresponding to (002) facets, were observed (Fig. 6b). It is well known that a supramolecular preorganization is a self-templating approach where a preorganized network formed by hydrogen bonding, ensuring the perfect alignment of CN during the polymerization process. Motivated by this idea, Yuan and co-workers designed a rapid and facile strategy for the preparation of highly crystalline CN with decreased structural defects by combining the microwave-assisted thermolysis strategy with a supramolecular preorganization approach.90 In contrast to the broad and weak XRD pattern of the CN540 sample, an intense and narrow diffraction peak was observed in CN10, suggesting the improved crystallinity (Fig. 6c). Moreover, the peak at 27.5° was moved to a higher angle as the reaction time was extended (Fig. 6c, right), indicating a decrease of the interlayer distance, confirming that the polycondensation was enhanced by the microwave-assisted thermolysis route. More importantly, the lifetime of the charge carriers in CN10 was reduced due to the rapid charge transfer, further confirming the improved crystallinity (Fig. 6d). Overall, the MW-assisted heating route is a facile, rapid, and robust approach for the synthesis of CCNs. It has several advantages. First of all, it is a time and energy saving method. The preparation process can be shortened to 15 minutes. Secondly, this is a general approach which can be applied to other nitrogen-rich organic precursors.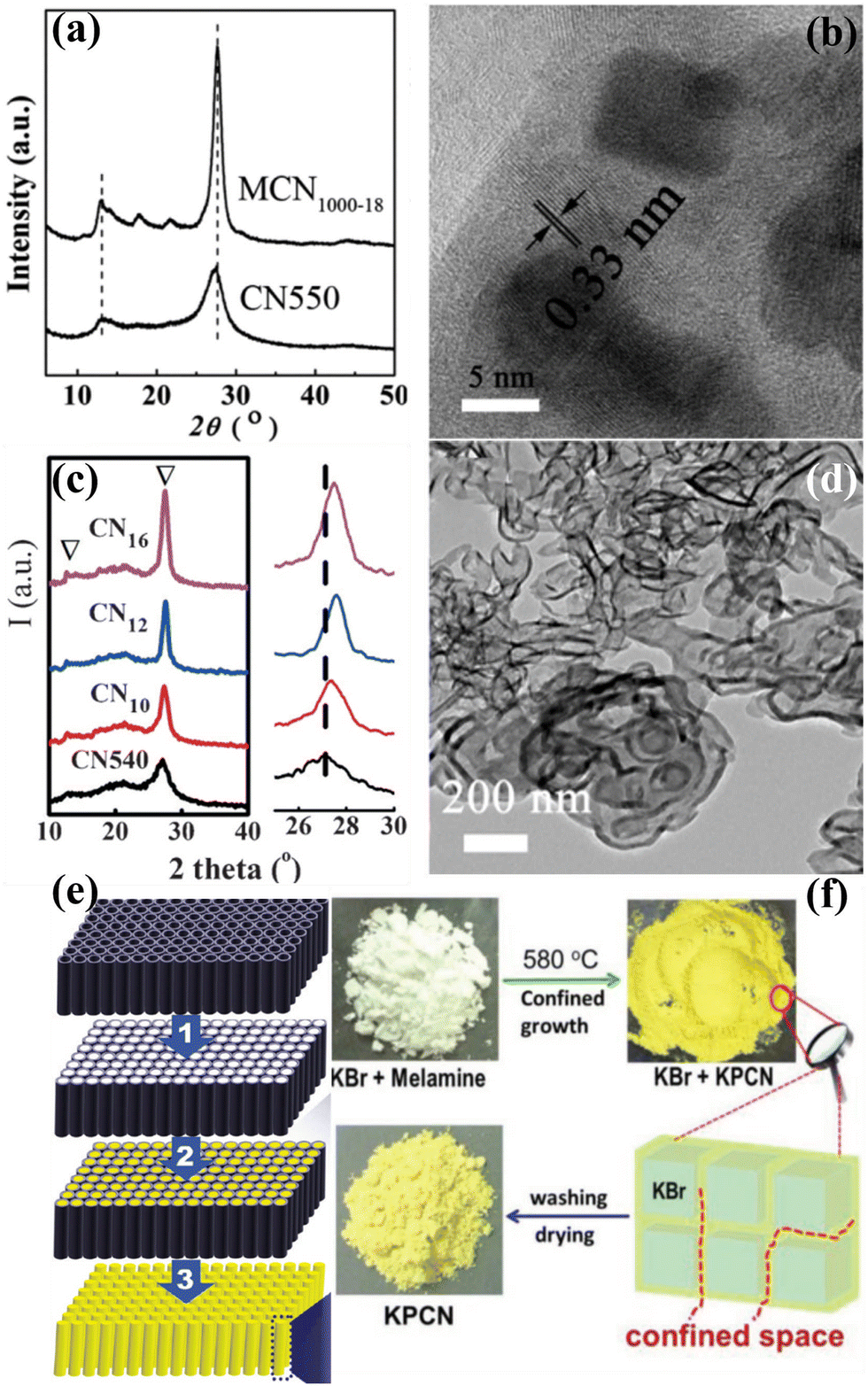 | ||
| Fig. 6 (a) XRD patterns of the MCN1000-18 and CN550. (b) HRTEM image of the MCN1000-18. Reproduced with permission.89 Copyright 2014, The Royal Society of Chemistry. (c) XRD patterns of CNt (t = 10, 12, and 16) and CN540. (d) TEM image of CN16.90 Copyright 2016, Wiley-VCH. (e) Schematic representation of the synthesis of CCN nanorods through nanoconfinement method. Reproduced with permission.92 Copyright 2011, American Chemical Society. (f) Schematic diagram of the preparation of KPCN from KBr confined growth. Reproduced with permission.95 Copyright 2019, Wiley-VCH. | ||
For comparison, the advantages and disadvantages of different methods for synthesizing CCNs are summarized in Table 1.
| Method | Advantages | Disadvantages |
|---|---|---|
| HT–HP | Adjustable carbon/nitrogen ratio | Expensive equipment; high temperature; high pressure |
| Ionothermal (molten salt) | Cheap equipment; easy operation; large-scale preparation | Complicated procedure for washing samples |
| Solvothermal | Cheap equipment; easy operation | Harsh feedstock; high pressure |
| Microwave (MW)-assisted heating | Large-scale preparation; easy operation; time and energy saving | Uncontrollable temperature |
4.5. Other methods
Yin et al. developed a distinct method to synthesize the single-crystal β-C3N4 nanorods through mechano-chemical reaction and subsequent thermal annealing under NH3 gas flow.91 Li and co-workers synthesized high crystalline CN nanorods through a nanofabrication strategy which could overcome the kinetic restrictions of bulk polycondensation (Fig. 6e).92 The nanoconfinement strategy was achieved by thermal polymerization of cyanamide within the nanochannels of anodic alumni oxide (AAO) template, resulting in improved orientation and crystallinity. Zhang and co-workers developed a simple approach to prepare CCN through protonating specific intermediates during the thermal condensation process, which could promote the solid-state polymerization kinetics.93 Hu and co-workers prepared CCN nanosheets through an alternating cooling and heating strategy.94 Su and co-workers fabricated K cations intercalated CCN via a salt-template-confined growth route (Fig. 6f).95 The solid KBr could act as a solid template and provide a confined space, thus leading to the confined growth of CCN within the KBr crystal to achieve the crystalline structure. Similarly, KCl and NaCl were used as a template to provide a confined space between the salt crystals to effectively guide the growth of CCN.96,97 Huang and co-workers proposed a new approach of engineering hydrogen bonding interactions within the CN framework of melons (doping-induced) to promote the crystallinity of CN.98 This conception was realized by the removal of some amino groups and the use of co-doped B and F atoms to connect the melons via sodium fluoroborate-assisted heat polymerization. Moreover, the CCN quantum dots could be prepared by a solid-phase reaction strategy99 or “dry hydrothermal” method.100 The ground mixture of urea and sodium citrate was thermally treated in an autoclave at 180 °C for 1 h or 140 °C for 3 h to synthesize CCN quantum dots.4.6. Characterization of CCNs
Generally, the conventional characterization methods for the crystal structure of CCNs are XRD and HRTEM analyses. Due to the different building blocks of CNNs, the XRD patterns could distinguish different crystal structure of CCNs. The (100) peak of PTI-based CCN is located at ∼12°, corresponding to the in-plane pores. While the (100) peak of PHI-based CCN is located at ∼8.1°, which corresponds to a larger in-plane spacing. Moreover, as revealed by the HRTEM images, the lattice fringes of PTI-based CCN is around 0.73 nm, while the lattice spacing of PHI-based CCN is approximately 1.1 nm. Overall, the XRD and HRTEM analyses could only provide the average structural information. In recent years, solid-state NMR spectroscopy101 and neutron diffraction102 have provided the structure information of local and light element of CCNs. However, real-space details and atomic resolution imaging of the local configuration of CCNs is still challenging. Very recently, integrated differential phase contrast scanning transmission electron microscopy (iDPC-STEM) is an emerging phase imaging technique based on center of mass shift of the convergent beam electron diffraction patterns by utilizing the four-quadrant detector.103 Through analyzing the scattered electrons, the x and y components of offset vectors of DPC image were obtained.104 Through two-dimensional integration, the iDPC image was obtained, enabling us to realize the low dose imageing of the light element beam-sensitive materials with atomic resolution and high signal-to-noise ratio.105 Therefore, the atomic-resolution imaging of the true spatial structure of CCNs was directly visualized by the iDPC-STEM.106–108 For example, He and co-workers revealed the atomic structure (including the light elements) and atomic electric field of the PTI crystal with the low dose imaging by the DPC-STEM technique.107 As displayed in the iDPC images (Fig. 7a and b), the six triazine units with highly ordered atomic structures were interconnected to build the 2D honeycomb network and chloride ions occupied the central triangular pore. Moreover, the location of Li and H atoms with three configurations could be determined by the iDPC-STEM and corresponding simulated images (Fig. 7c–f). The H atoms were surrounded by bridging N atoms and the lithium ions were used to balance the charge of bridged imide groups. Furthermore, the atomic coupling electric field between the Li and neighboring atoms was directly visualized by the atomic electric field imaging (Fig. 7g and h). Meanwhile, the DFT calculations confirmed that the Li–N ionic bonds were formed, which not only extended the π-conjugated electronic structure, but also created an additional charge transfer channel to facilitate in-plane charge migration (Fig. 7i). Therefore, the charge migration route was schematically presented in Fig. 7i. Besides, the Li ions could be intercalated into the layers, which also confirmed by the NMR and XPS results.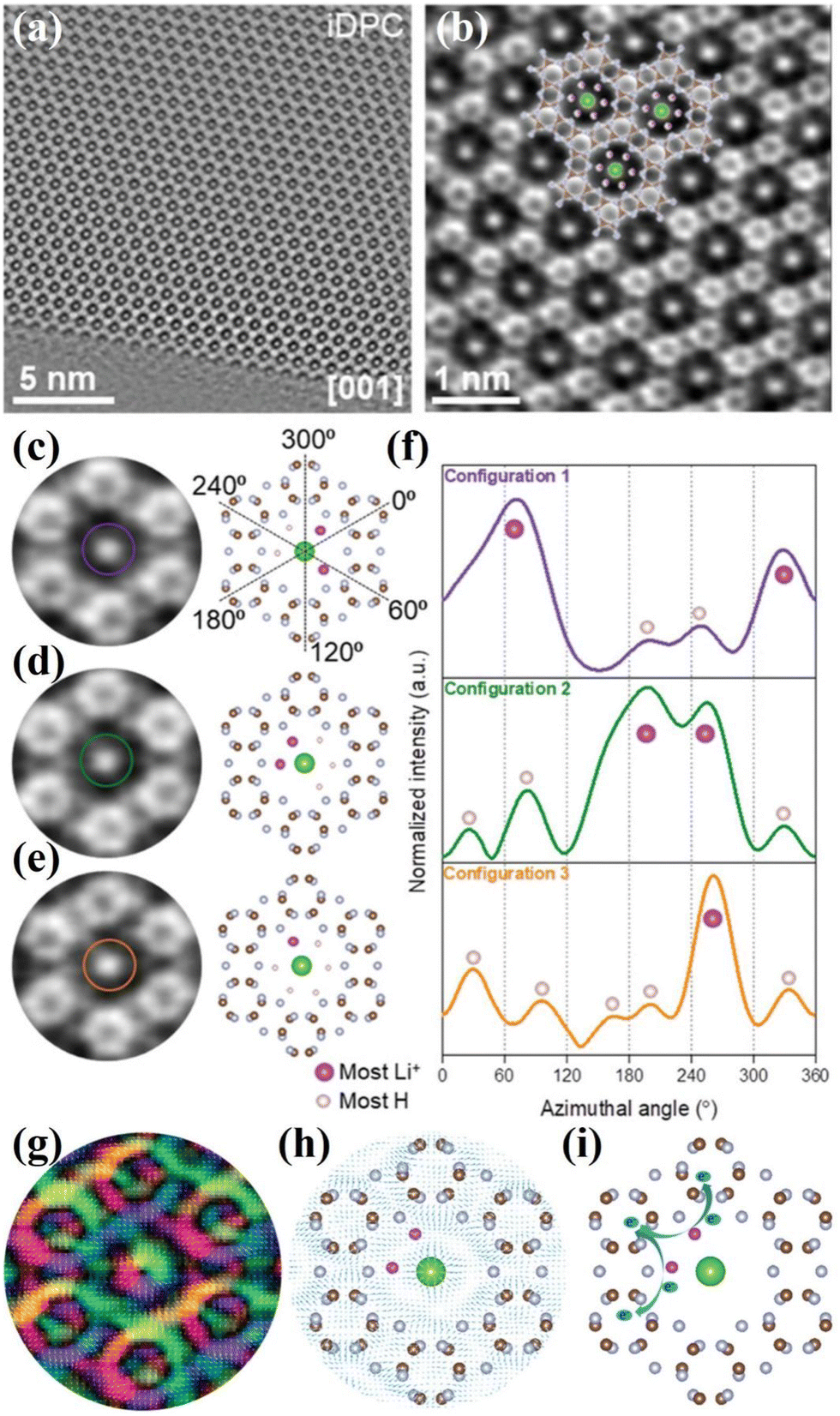 | ||
| Fig. 7 (a) iDPC image of PTI along the [001] zone axis. (b) Enlarged iDPC image. (c)–(e) Three Li/H configurations in the intralayer cavities detected in iDPC and the corresponding atomic structure models. (f) Intensity profiles following the defined circles in (c)–(e). Dashed lines in (c) guide the azimuthal angles of intensity profile in (f). (g) Magnified color map with the arrow vector. (h) The arrow vector map overlapped with the atomic structure of PTI. (i) Schematic diagram of the migration of photoexcited electrons between lithium ions and the triazine units. Reproduced with permission.107 Copyright 2021, Wiley-VCH. | ||
5. Design of CCNs-based photocatalysts
5.1. Nanostructure design
Considering that the photocatalytic process is an interfacial reaction, the photocatalytic activity largely depends on the shape, size and nanostructure of CCNs. The nanostructure of CCNs can be designed to obtain high specific surface area, enhanced electron transport property, reduced migration distance and abundant surface active sites, all of which are beneficial to improve the photocatalytic activity. Until now, various nanostructures of CCNs such as nanosheets, nanotubes, nanorods and nanofibers have been prepared by many ingenious approaches. The following is a detailed description of various strategies for designing nanostructures of CCNs.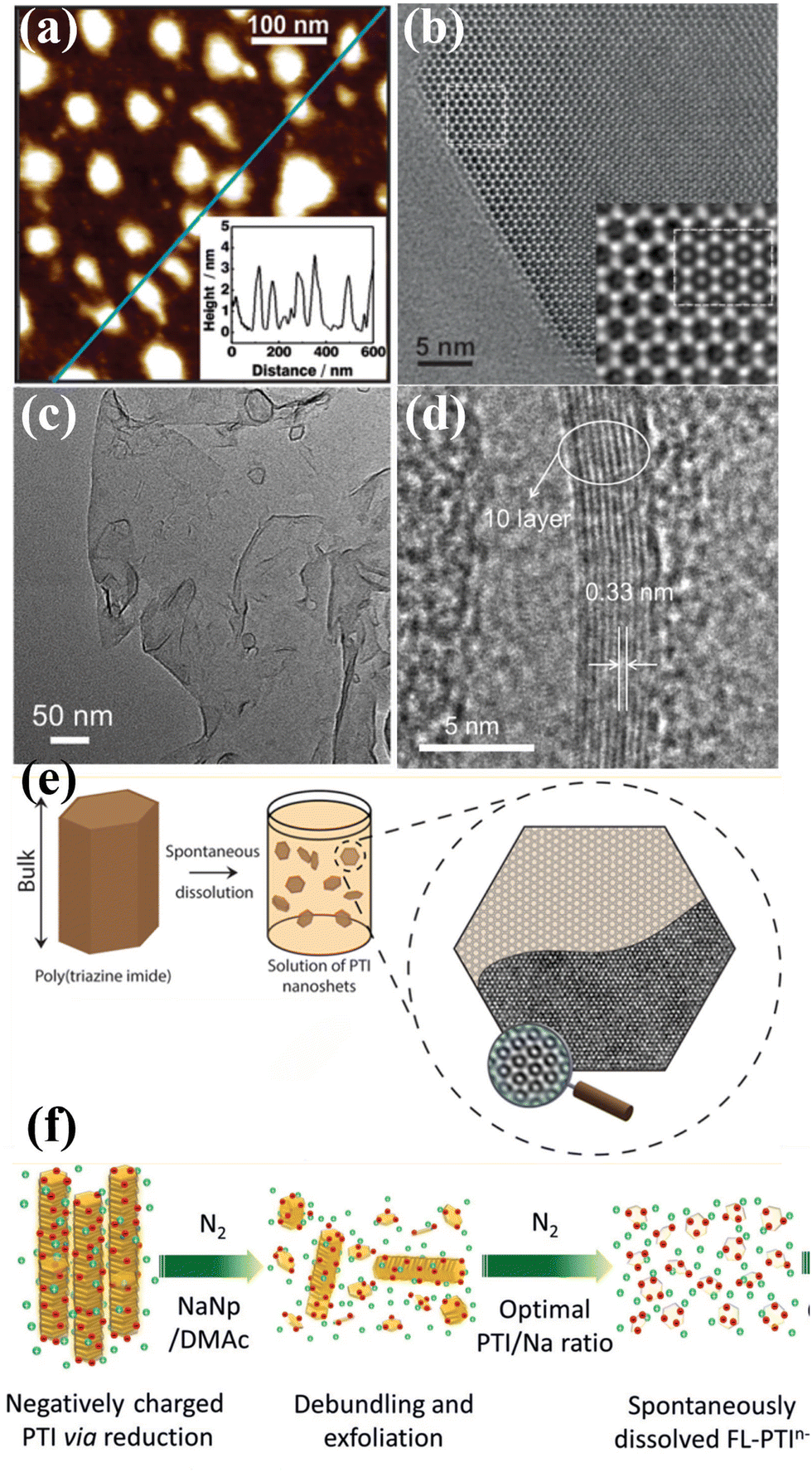 | ||
| Fig. 8 (a) AFM image of the PTI nanosheets and corresponding height image (inset). (b) HRTEM image of PTI nanosheet and simulation (inset). Reproduced with permission.65 Copyright 2014, American Chemical Society. (c) TEM image of the CCN nanosheets. (d) HRTEM image of CCN nanosheets. Reproduced with permission.112 Copyright 2017, Wiley-VCH. (e) Schematic illustration of the PTI nanosheet prepared by spontaneous dissolution. Reproduced with permission.109 Copyright 2017, American Chemical Society. (f) Schematic illustration of charging and exfoliation of PTI. Reproduced with permission.110 Copyright 2018, Wiley-VCH. | ||
McMillan and co-workers demonstrated that the 3D bulk PTI-based CCN was delaminated into a defect-free, hexagonal two-dimensional nanosheets by a thermodynamically driven dissolution process without any chemical or physical intervention (Fig. 8e).109 The bulk CCN spontaneously dissolved in select polar aprotic solvents due to the free energy gain from the solvent coordination effect. Interestingly, the obtained nanosheet are stacked in several layers rather than monolayer, suggesting that the interlayer interactions are energetically comparable to those between the solvent and nanosheet surfaces. Mattevi and co-workers reported a framework charging route for the fast and high yield preparation of PTI nanosheets through sodium naphthalide (NaNp)/DMAc reduction (Fig. 8f).110 NaNp was used as an electron charge transfer agent, while DMAc was expected to be an excellent room-temperature solvent for anionic monolayer/few-layer CCNs. The negative charge from NaNp could be rapidly transferred to PTI, leading to short-range Coulombic repulsion, which resulted in the exfoliation of PTI into solvated few-layer sheets. Agrawal and co-workers prepared monolayer PTI-based CCN nanosheets in anhydrous dimethylacetamide (DMAc) by a simple solvent exfoliation route.111 The DMAc played an essential role in exfoliation due to the strong interaction of carbonyl oxygen of DMAc with Li+, which weakened the interlayer interaction.
Despite there are numerous research publications related to the liquid exfoliation of bulk CCN, this process requires a long sonication processing time (10–16 h). To address these bottlenecks concerning the exfoliation of bulk CCN, thermal exfoliation has emerged as a fast and effective strategy.113,116 For example, Zhang and co-workers proposed a simple but effective strategy for the high-yield synthesis of CCN nanosheets with a specific surface area of up to 34 m2 g−1 by two-step calcination.113 The structure of the nanosheet was confirmed by the TEM image, and a clear crystal lattice with a distance of 3.30 Å is observed in the HRTEM image, indicating its high crystallinity. The electrochemical impedance spectra (EIS) analysis indicates that the electron-transfer resistance of CCN nanosheets was lower than that of bulk CN. Also, an obvious enhancement of photocurrent was observed for CCN nanosheets compared with bulk CN, which indicated the effective separation of charge carriers and enhanced photoinduced charge mobility in the CCN nanosheets. Moreover, the TRPL spectra shows a prolonged lifetime of charge carriers, indicating a higher electron mobility. Thus, the prepared CCN nanosheets showed outstanding photocatalytic performance, with a hydrogen generation rate of 9577.6 μmol h−1 g−1, 15.5 times that of bulk CN.
In summary, the preparation of CCN nanosheets has been achieved by various exfoliation strategies. The superiority of the CCN nanosheets could be attributed to the increased specific surface area, enhanced electron transfer capability, prolonged lifetime of charge carriers, and improved charge separation efficiency.
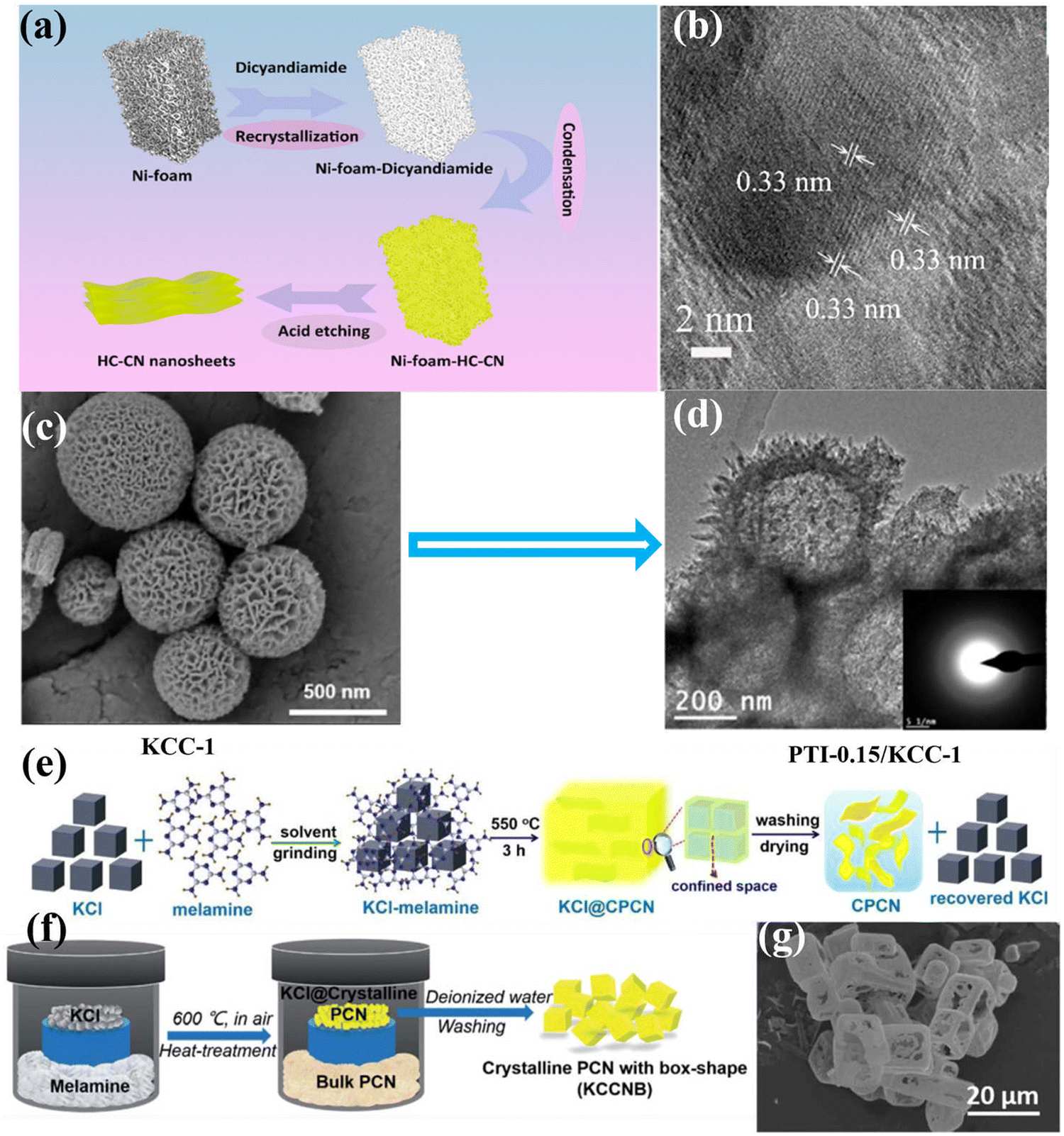 | ||
| Fig. 9 (a) Schematic diagram of the fabrication of HC–CN photocatalyst. (b) HRTEM image of HC–CN. Reproduced with permission.117 Copyright 2018, American Chemical Society. (c) SEM image of KCC-1. (d) TEM image of PTI-0.15/KCC-1. Reproduced with permission.118 Copyright 2015, American Chemical Society. (e) Schematic diagram of the synthesis of CCN nanosheet by the solid salt confinement method. Reproduced with permission.96 Copyright 2019, Elsevier. (f) Schematic diagram of the vapor-deposition strategy. (g) SEM image of KCCNB-30.122 Copyright 2020, Royal Society of Chemistry. | ||
In addition, mesoporous photocatalysts can be designed to improve the photocatalytic performance by increasing the specific surface and abundant active sites, as well as promoting the separation of photogenerated carriers. Recently, Yu and co-workers fabricated highly ordered and porous CCN through a one-step template-mediated strategy, that is, a bifunctional NaHCO3 template-mediated approach.119 Specifically, NaHCO3 served as a gaseous template to promote the creation of the porous structures, while the resulting Na2CO3 could act as high-temperature solvent to facilitate the crystallization of porous CCN. Besides the porous structure and high crystallinity, the cyano groups were also introduced to the surface of porous CCN, which could serve as effective active sites for H2 generation. As a result, the specific surface area of the porous CCN could be up to 15.34 m2 g−1, which was 3.5 times larger than that of bulk CN. Moreover, the clear lattice fringe with a distance of 0.32 nm was observed in the HRTEM image, confirming its high crystallinity. Owning to the great crystallinity, cyano-group defects and porous structures, the porous CCN exhibited a remarkably increased H2 evolution rate, which was more than two times higher than that of bulk CN.
In the process of preparing CCN by salt-assisted synthesis strategy, the salt could be used as a template. Zou and co-workers fabricated an intercalated CCN nanotube through the molten salt route.61 In general, these salts could be used as templates as their crystals have cubic morphology. Before the temperature reached the melting point of the mixed salt, thermal polymerization had occurred, and the polymerization intermediate had grown on the interface of the cubic chloride crystals, thus forming the nanotubes. By intercalating the alkali metal ions into the layers of CN, the migration and separation of photoinduced carriers was dramatically improved. Accordingly, the intercalated CCN nanotubes showed high photocatalytic hydrogen production activity, reaching 0.346 mmol h−1 within the first hour, 34.6 times higher than that of the bulk CN. Wang and co-workers fabricated the CCN nanosheets by a solid-salt-assisted growth strategy (Fig. 9e).96 The solid salt acted as a structure-directing template to steer the orderly growth of CCN within the restricted space of the KCl crystals. Therefore, the exfoliation of bulk CCN was achieved due to the confinement effect of salt and the interaction of K+ ions. As expected, the CCN nanosheets exhibited significantly improved photocatalytic activity compared with bulk CN. This improved activity could be ascribed to the high crystallinity and shortened interlayer distance, which could improve the light harvesting capability and boost the transfer of photoexcited carriers. Quan and co-workers synthesized (3D) branched CCN nanoneedles by combining a post-synthesis ionothermal approach with a quenching method.121 The melted eutectic salts were used as an induction reagent to enhance the crystallinity and created 3D structures. Interestingly, the prepared nanoneedle with sharp curvature could evoke a nanoscale lightning rod-like effect to accelerate directional charge transfer, thus suppressing the recombination of photoexcited charge carriers. As expected, the 3D branched CCN nanoneedles showed outstanding photocatalytic activity for both H2 generation (0.27 mmol h−1) and H2O2 production (0.33 mmol h−1), which was 85 and 23 times than that of the pristine CN, respectively. Shu and co-workers fabricated K+ doped heptazine-based CCN with box-shape through vapor-deposition approach using KCl as a template (Fig. 9f).122 The obtained K+ doped CCN box had a diameter of ca. 10 μm, with a wall thickness of several hundred nanometers, which was confirmed by the SEM image (Fig. 9g). Due to the special box shape, the incident visible light could be reflected and absorbed multiple times, thus improving the light utilization. As a result, the K+ doped CCN showed an improved photocatalytic H2 generation performance, which was 33 times than that of bulk CN. This enhanced performance could be ascribed to the improved light absorption in the box shape, and the effective charge separation and transfer due to the doping of K and high crystallinity.
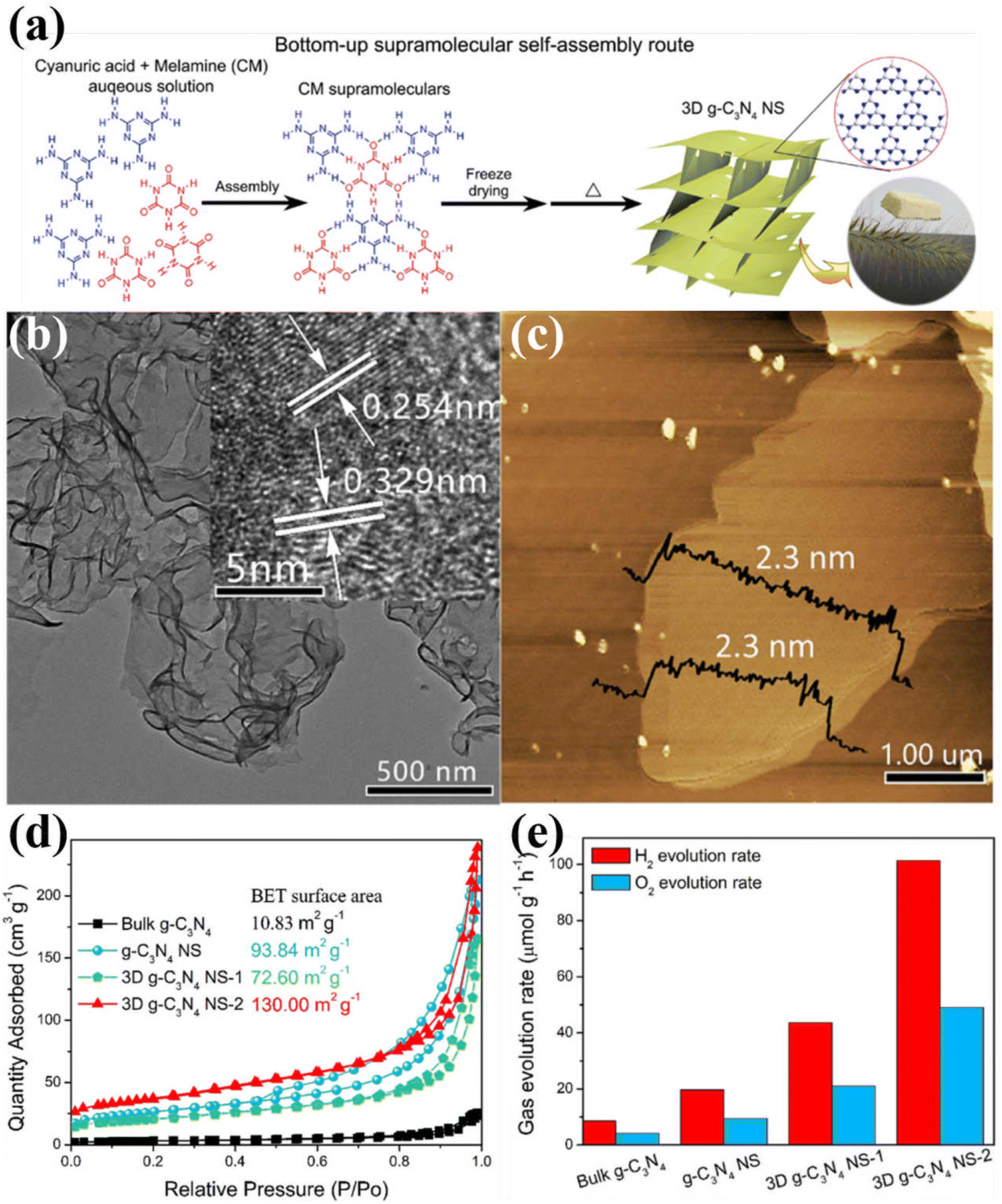 | ||
| Fig. 10 (a) Schematic diagram of the fabrication of 3D g-C3N4 NS through the bottom-up supramolecular self-assembly strategy. (b) TEM image (HRTEM image inset) and (c) AFM image of 3D g-C3N4 NS-2. (d) N2 adsorption–desorption isotherms of the samples. (e) Photocatalytic OWS activity over bulk g-C3N4, g-C3N4 NS, and 3D g-C3N4 NS. Reproduced with permission.124 Copyright 2019, Elsevier. | ||
To sum up, nanostructure design is one of the promising strategies to modulate the physical and chemical features of CCNs. Various morphologies of CCN from 1D to 2D, 3D nanoarchitectures were prepared by different synthesis methods. These nanostructured CCNs offer great advantages in terms of charge transfer, light absorption, and abundant active sites. For CCN with hollow structure, improved light absorption was achieved, which could improve the light utilization. In addition, 3D nanostructures of CCN have a large specific surface area which provided an additional pathway for electron transport. More importantly, combining supramolecular self-assembly strategies with solvothermal methods is highly desirable in the design and development of nanostructured CCNs.
5.2. Molecular structure engineering
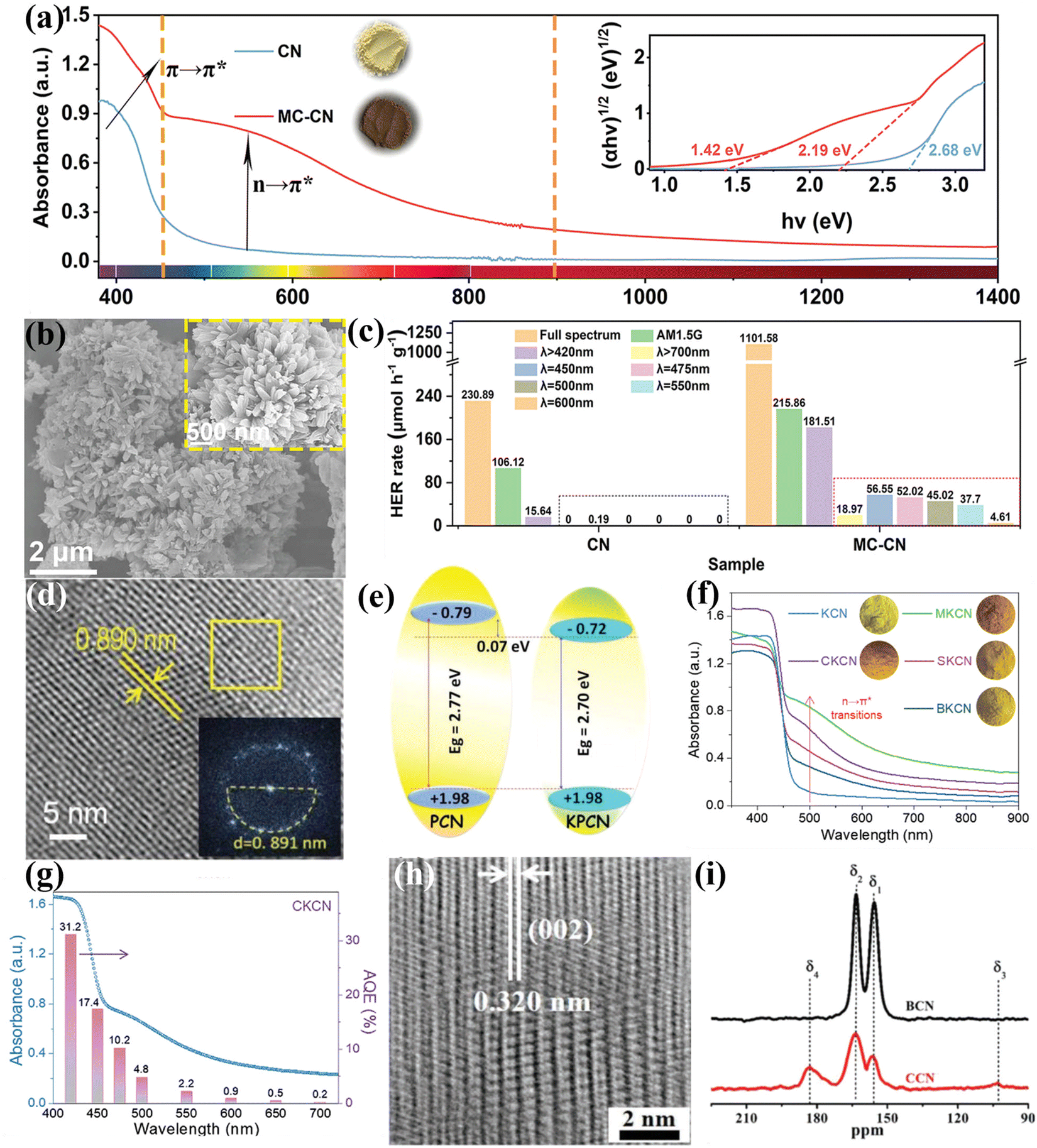 | ||
| Fig. 11 (a) UV-vis diffuse reflectance spectra (DRS) of CN and MC-CN and the corresponding Kudelka–Munk plots (inset). (b) SEM and high magnification SEM images (inset) of MC-CN. (c) Wavelength dependence of the photocatalytic H2 generation rate of CN and MC-CN. Reproduced with permission.129 Copyright 2022, Wiley-VCH. (d) HRTEM image and FFT pattern (inset) of KPCN. (e) Schematic representation of the band structures of PCN and KPCN. Reproduced with permission.95 Copyright 2019, Wiley-VCH. (f) UV-vis-NIR DRS spectra of KCN, MKCN, CKCN, SKCN, and BKCN samples. (g) Wavelength dependence of AQE of CKCN. Reproduced with permission.131 Copyright 2022, Wiley-VCH. (h) HRTEM image of CCN. (i) 13C NMR of BCN and CCN. Reproduced with permission.88 Copyright 2019, Wiley-VCH. | ||
Besides heteroatom doping, self-doping has also been developed by recent works.134–136 For example, our group developed an acetonitrile-promoted solvothermal route for the synthesis of CCN with cyano and carboxyl groups.88 The high crystallinity was confirmed by the HRTEM image (Fig. 11h). The NMR studies revealed that the defects were most likely located at the structural terminations of the CCN framework (Fig. 11i). Moreover, the introduction of surface defects not only improved visible light absorption, but also increased CO2 binding affinity and improved charge transfer and electron storage. Therefore, the as-prepared CCN with surface defects showed superior photocatalytic CO2 reduction performance and selectivity in the gas phase. Wang and co-workers found that the CCN with defects achieved a better charge separation because of the built-in electric field.134
In conclusion, elemental doping plays a key role in modulating the band structure of CCNs. In most cases, elemental doping not only narrowed the bandgap, but also improved the separation of charge carriers because of the built-in electric field, thereby improving the photocatalytic activity. Although many studies on heteroatom-doped CCNs have been successfully developed, there is still an urgent need to control the distribution and concentration of doping, such as gradient doping.
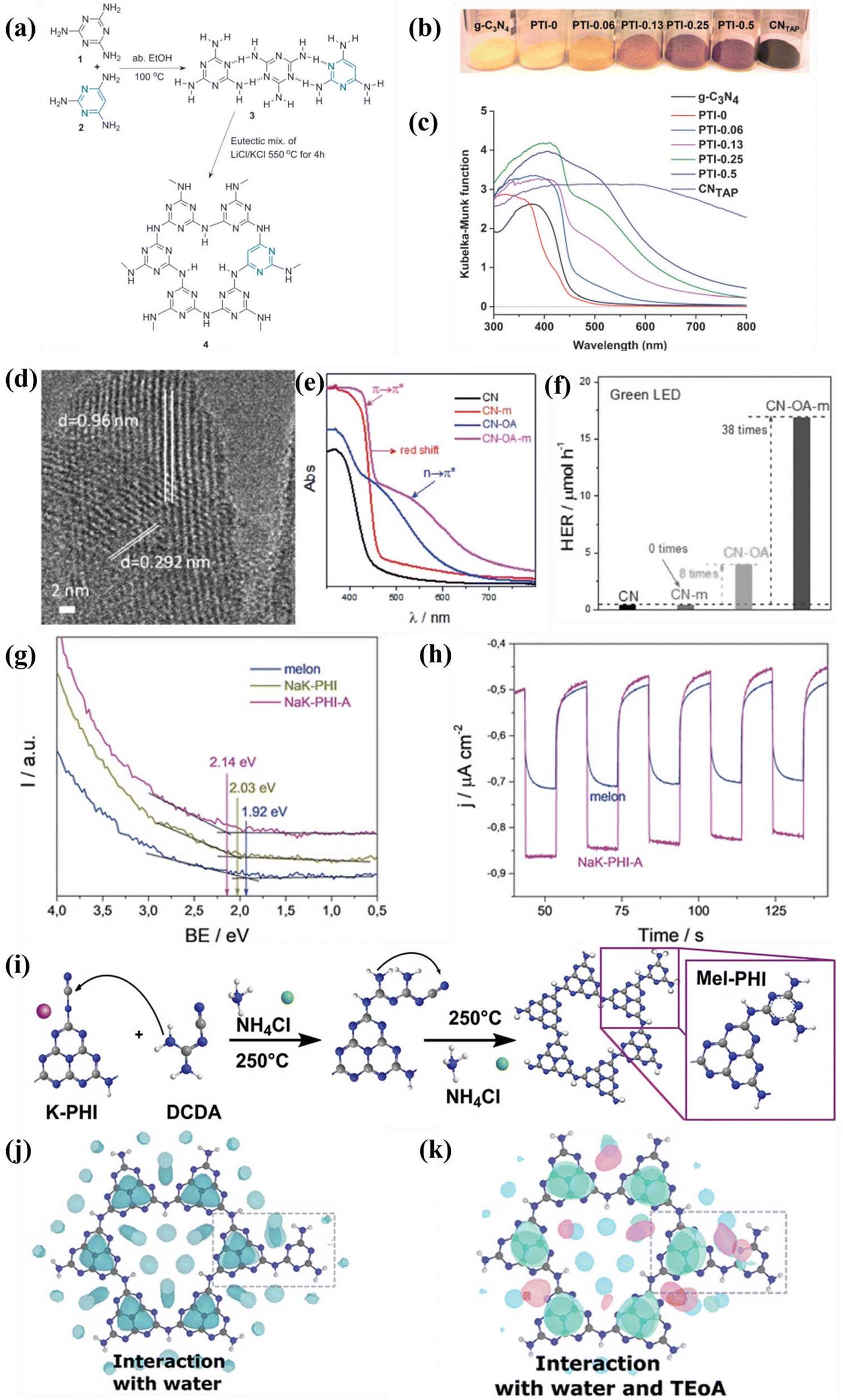 | ||
| Fig. 12 (a) Schematic illustration of the synthesis of TAP-incorporated PTI. (b) Color and (c) UV-vis DRS of the as-obtained samples. Reproduced with permission.140 Copyright 2014, Wiley-VCH. (d) HRTEM image of CN-OA-m. (e) UV-vis DRS of the as-prepared samples. (f) Photocatalytic H2 generation rates of the samples irradiated with green LED. Reproduced with permission.141 Copyright 2017, Wiley-VCH. (g) UPS spectra of melon, NaK-PHI, and NaK-PHI-A. (h) Photocurrent curves of melon and NaK-PHI-A. Reproduced with permission.142 Copyright 2019, Wiley-VCH. (i) Schematic illustration for the preparation of Mel-PHI. (j) Interaction of the Mel-PHI with H2O molecules. (k) Interaction of Mel-PHI with TEOA and H2O. Reproduced with permission.143 Copyright 2021, Wiley-VCH. | ||
Overall, molecular doping not only modulated the bandgap of CCNs, but also delocalized the π electrons. Doping with a very small amounts of organic additives could significantly modulate the bandgap and electronic structures. However, the doping sites of organic monomer were still obscured, which need further investigation.
0) facets, as schematically illustrated in Fig. 13b. The high-resolution aberration-corrected integrated differential phase contrast (AC-iDPC) image (Fig. 13c and d) further confirms the high degree of atomic ordering of the PTI/Li+Cl−. Furthermore, C, N, and Cl atoms could be distinctly distinguished without tedious image simulation. Therefore, the atomic structure of PTI/Li+Cl− crystals was presented and confirmed. The reactive facets were identified by photodeposition of co-catalysts, which could be used as a probe. Interestingly, both the photoexcited electrons and holes accumulated on the {1010} facets, confirming by the selective photodeposition of the Pt or Co. Therefore, increasing the prismatic surface areas (reactive facets) could dramatically improve the activity of photocatalytic OWS (Fig. 13e). Notably, the apparent quantum yield (AQY) of PTI-550 reached 8% at 365 nm. Although crystal facet engineering had been achieved, the intrinsic mechanisms of the molecular structure, nucleation and crystallization of PTI-based CCN during the ionothermal process remain unclear. Therefore, the same group used microscopic characterization techniques to reveal the molecular conjugation and crystal growth processes from mesoscale to nanoscale by various sample processing methods.147 Combining various characterization techniques, the crystallization mechanism of PTI at the molecular scale in molten salts was proposed in Fig. 13f. The heptazine precursors were depolymerized into triazine units, and subsequently PTI-based frameworks was constructed by introducing Li+ and Cl− ions. During the crystal precipitation process, the KCl cubes served as a nucleation center and template to guide the orientation and orderly stacking of PTI monomers, thereby producing agglomerated PTI crystals. More importantly, it was found that the aggregation of PTI crystals decreased the exposed area of the active center, which should result in a significant decrease in photocatalytic activity. Therefore, to optimize the photocatalytic performance, it is highly desirable to prevent aggregation and enhance the dispersity by controlling the crystallization fronts, that is, the weight proportion of precursor and salt (Fig. 13g–i). Consequently, when the weight ratio of salt to precursor was 1:10, the best photocatalytic activity was realized due to the large exposed reactive facet and good dispersibility (Fig. 13i).
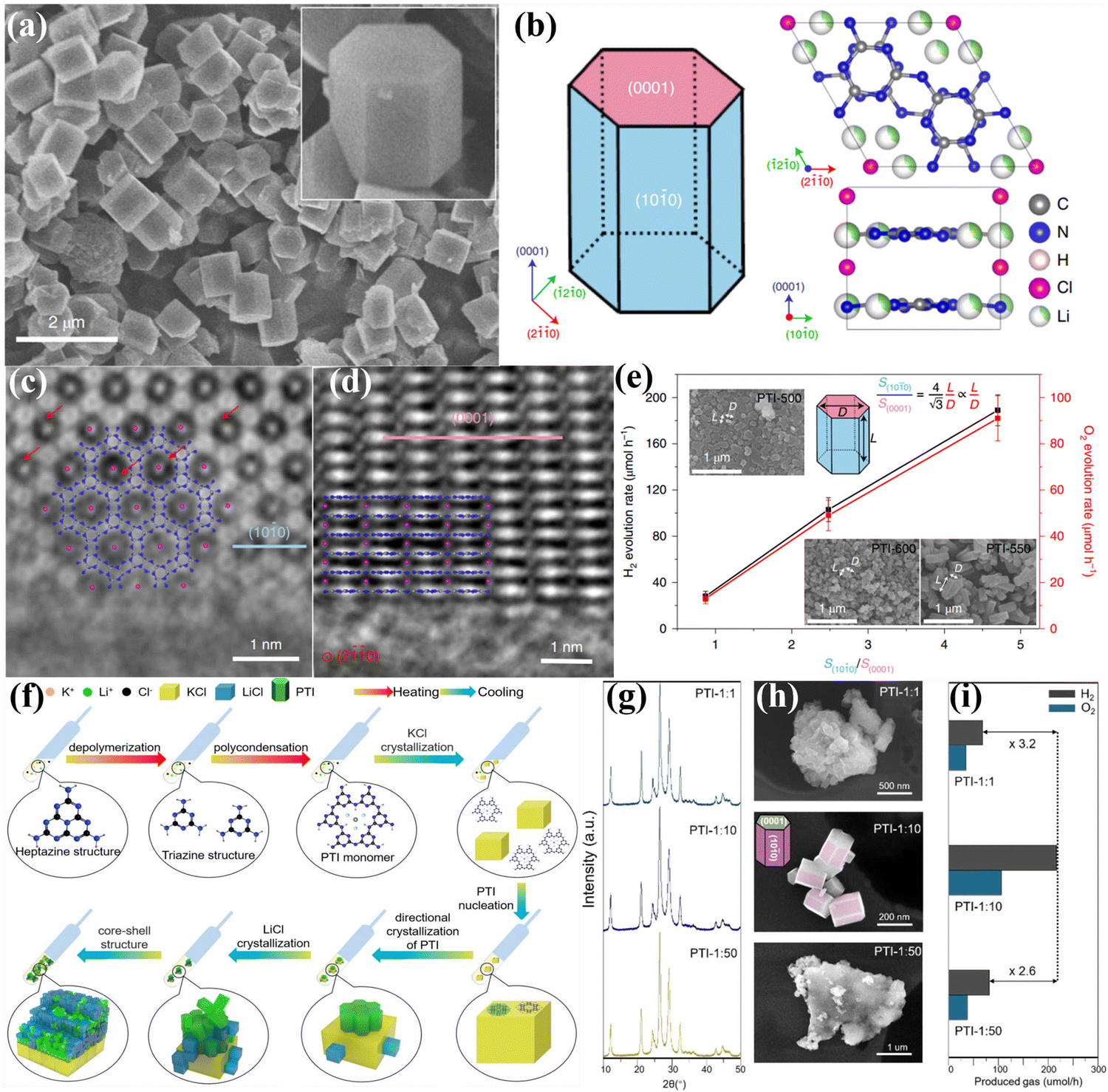 | ||
| Fig. 13 (a) SEM image of PTI/Li+Cl− and the magnified view of the crystal (inset). (b) Schematic representation of the PTI/Li+Cl− crystals and the atomic structure of PTI/Li+Cl−. (c) AC-iDPC image of PTI/Li+Cl− crystal aligned along the [0001] direction. (d) AC-iDPC image of PTI/Li+Cl− along the [20] zone axis. (e) The photocatalytic activities on different ratio of {100} and {0001} of the samples prepared at different temperatures. Reproduced with permission.106 Copyright 2020, Springer Nature. (f) Schematic representation of the molecular conjugation and directional crystallization mechanism of PTI crystals during molten salt process. (g) XRD patterns, (h) SEM images and (i) photocatalytic OWS activities of PTI samples synthesized by tailoring the weight ratio of precursor and salt. Reproduced with permission.147 Copyright 2023, Wiley-VCH. | ||
5.3. Development of CCNs-based heterojunctions photocatalysts
Construction of CCNs-based heterojunction has become an attractive strategy to improve the photocatalytic performance of CCNs by boosting the separation of photoexcited electrons and holes. The CCNs-based heterojunction can be classified into isotype heterojunction, semiconductor/CCN heterojunction, carbon/CCN heterojunction and metal/CCN heterojunction. The isotype heterojunction constructed between two different crystalline phases of the same substance (known as crystal-phase heterojunctions) was an useful approach to enhance the photocatalytic performance,148–150 which in this section was categorized as amorphous/crystalline CN heterojunction151–155 and heptazine/triazine-based CCN heterojunction.156–161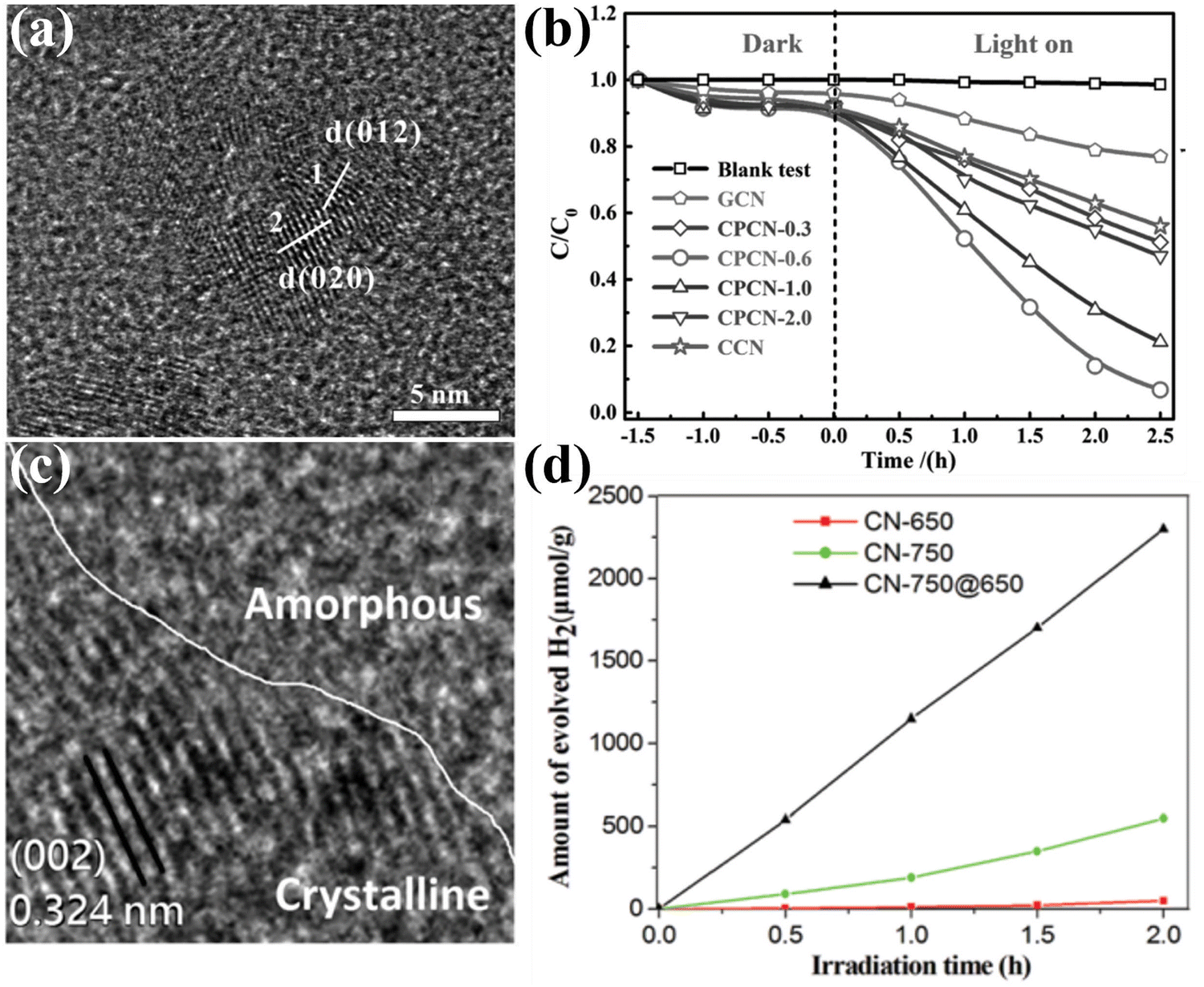 | ||
| Fig. 14 (a) HRTEM image of CPCN. (b) Photocatalytic degradation performance of the samples. Reproduced with permission.151 Copyright 2017, Wiley-VCH. (c) TEM image of CN-750@650. (d) Photocatalytic H2 generation rates of the samples under visible light (λ > 420 nm). Reproduced with permission.155 Copyright 2019, Royal Society of Chemistry. | ||
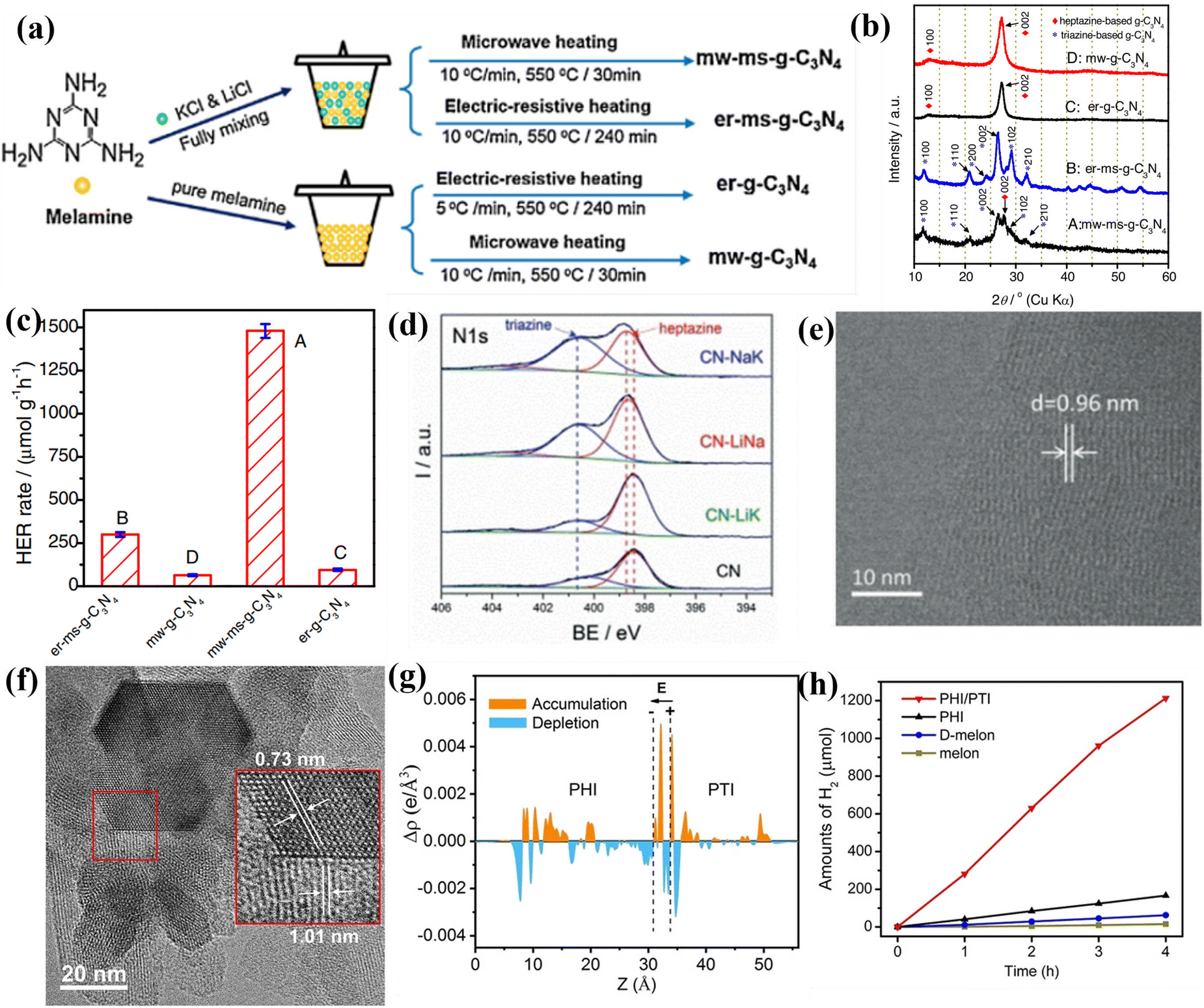 | ||
| Fig. 15 (a) Schematic illustration of the synthesis of g-C3N4 photocatalysts. (b) XRD patterns of the samples. (c) Photocatalytic H2 production rates of the samples. Reproduced with permission.156 Copyright 2017, Elsevier. (d) High-resolution N1s XPS spectra of CN, CN-LiNa, CN-LiK, and CN-NaK. (e) HRTEM image of CN-NaK. Reproduced with permission.159 Copyright 2018, Wiley-VCH. (f) HRTEM image of PHI/PTI. (g) The average charge density difference over the PHI/PTI heterojunction. (h) Photocatalytic H2 production performances of the samples. Reproduced with permission.162 Copyright 2022, Wiley-VCH. | ||
Although the above studies had confirmed the triazine-/heptazine-based isotype heterojunction, they did not provide a clear interface for phase heterojunctions. Quan and co-workers developed a secondary growth method for the preparation of CCN phase heterojunction (triazine/heptazine), in which the triazine-based CCN was vertically aligned on heptazine-based CCN.158 The XRD pattern confirmed the coexistence of triazine-based CCN and heptazine-based CCN. The HRTEM analysis further demonstrated the phase heterojunction, which consisted of well-matched lattice fringes between (002) plane of heptazine-based and triazine-based CCN, respectively. Owning to the increased crystallinity and accelerated charge separation, the triazine-/heptazine-based CCN exhibited higher photocatalytic performance than that of PTI and melon-based CN. Wang and co-workers fabricated a triazine/heptazine-based CCN with D–A heterostructure in eutectic salts (NaCl/KCl).159 This NaCl/KCl mixture had a much higher melting point of 652 °C compared with the formerly used KCl/LiCl mixture, which resulted in the CN solids being only partial “dissolution” and forming triazine-based PTI. The high-resolution XPS displayed a significant increase in the peak of a more electron-poor N atom during the molten salt treatment process, suggesting the co-existence of triazine and heptazine building blocks (Fig. 15d). The clear lattice fringes demonstrated the high crystallinity, confirming that the post-condensation in appropriate salt mixtures could modulate the polycondensation process (Fig. 15e). Despite the reduced crystallinity, the CCN still exhibited excellent hydrogen production performance because the well-designed D–A structure accelerated the transfer of charge carriers at the interface. In another study, Xiang and co-workers prepared a crystalline isotype heptazine-/triazine-based heterojunction through molten salt approach using preheated melamine as the precursor.161 The heterojunction structure and interfacial interactions were first confirmed by first principle calculations. Due to the high crystallinity and heterojunction structure, the optimized sample showed an excellent photocatalytic hydrogen generation rate of 890 μmol h−1 g−1, and an AQY of 26.7% at 420 nm.
Recently, Wang and co-workers synthesized PHI/PTI heterojunction with a semi-coherent interface by a simple two-step molten salt method.162 The HRTEM image demonstrates that the semi-coherent interface was constructed by two lattice fringes with spacing of 1.01 and 0.73 nm, which corresponded to the (10–10) plane of PHI and PTI, respectively (Fig. 15f). Moreover, the DFT calculations demonstrates that the electrons redistribution was observed across the semi-coherent interface, thereby establishing an intrinsic electric field and regulating the charge flow transport from PHI to PTI (Fig. 15g). As a result, the PHI/PTI presented a remarkably enhanced photocatalytic H2 generation performance of 348.5 μmol h−1 because of the promoted charge separation and transfer (Fig. 15h).
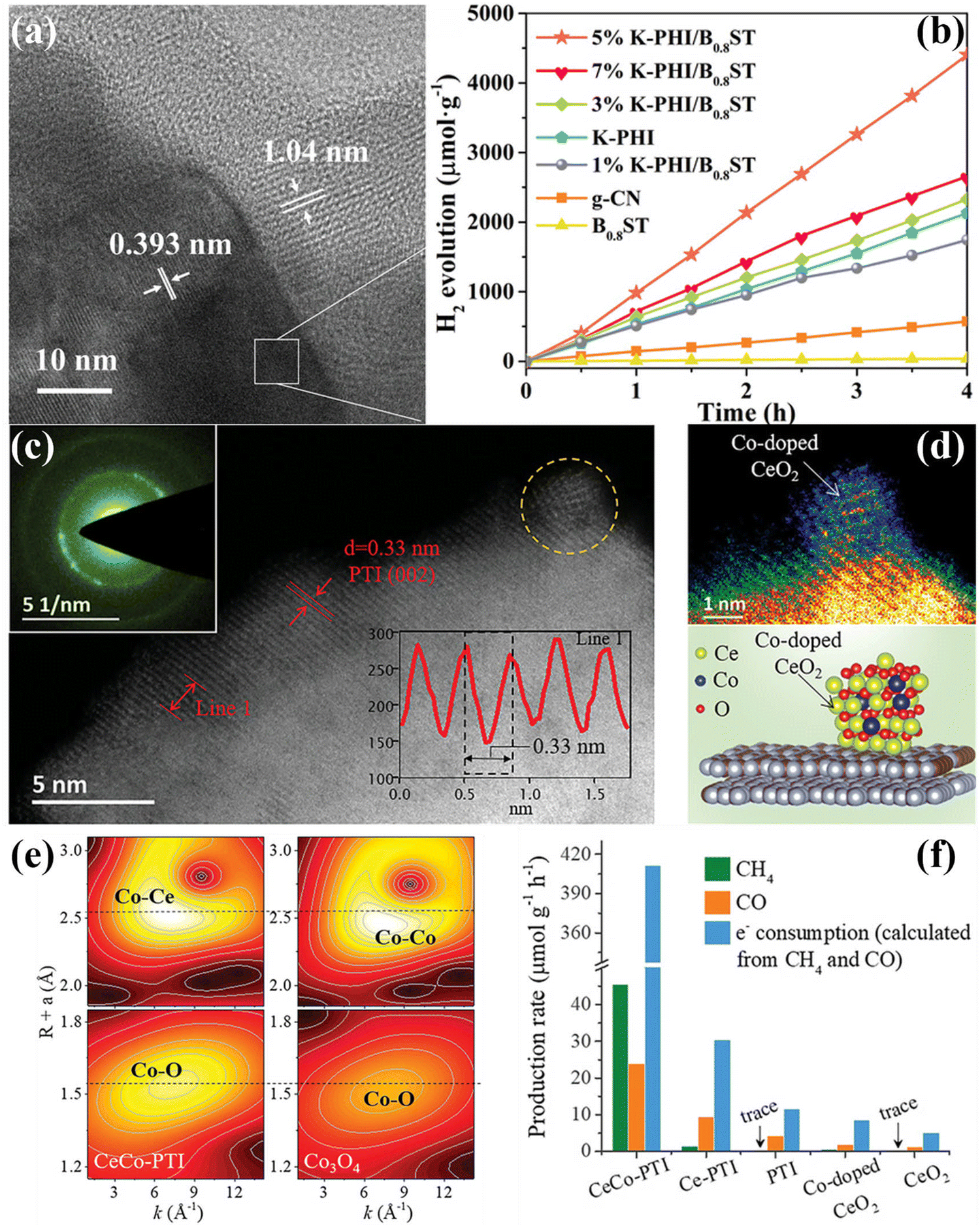 | ||
| Fig. 16 (a) HRTEM image of 5% K-PHI/B0.8ST. (b) Photocatalyitc H2 production rates of the samples irradiated with visible-light (λ > 420 nm). Reproduced with permission.173 Copyright 2022, Wiley-VCH. (c) and (d) AC HAADF-STEM image of CeCo-PTI. The structure model of CeCo-PTI is displayed at the bottom right. (e) Wavelet transform (WT) contour plots of CeCo-PTI (left) and Co3O4 (right). (f) CH4 and CO production rate of the samples, as well as the equivalent electron consumption. Reproduced with permission.174 Copyright 2022, Wiley-VCH. | ||
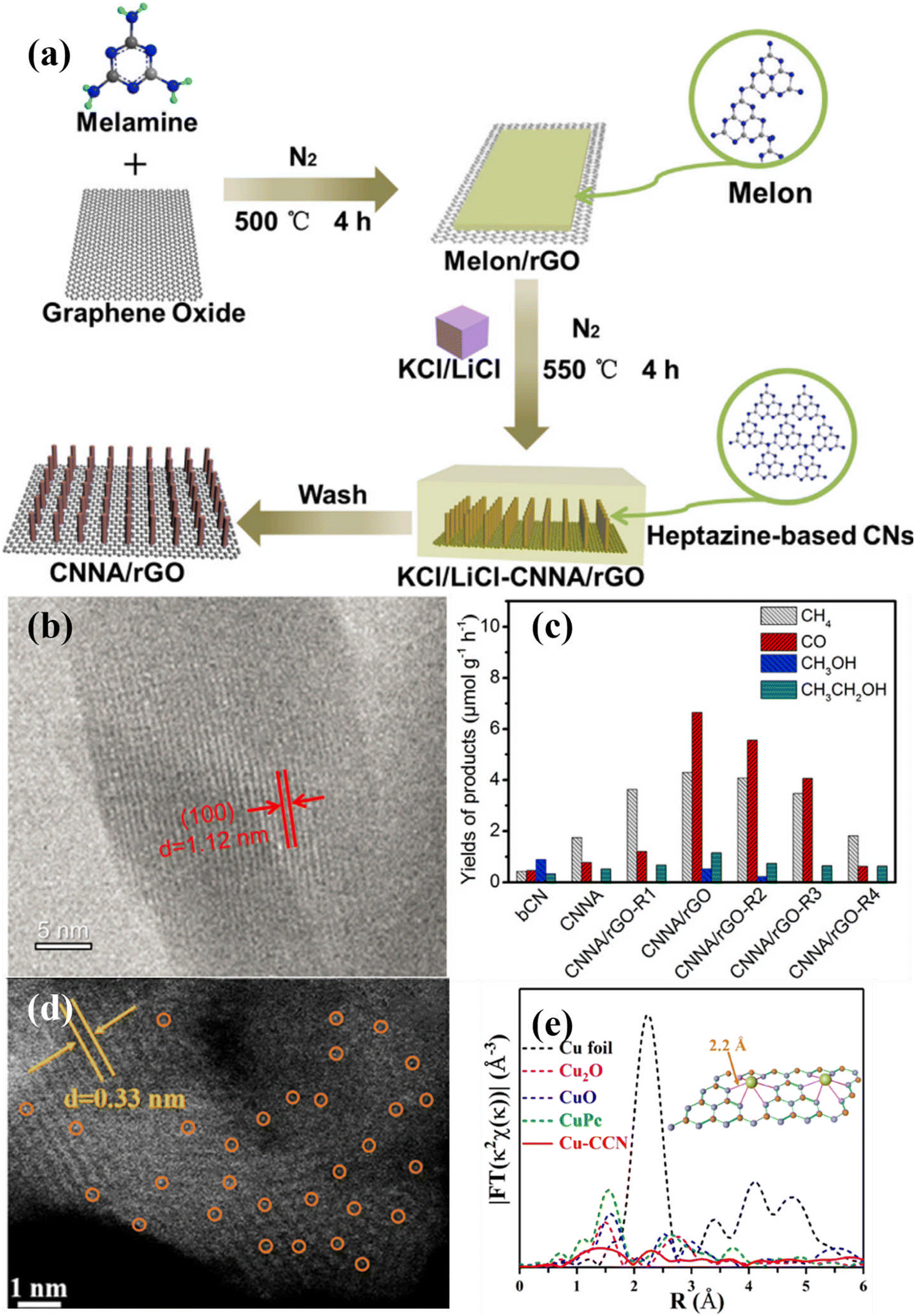 | ||
| Fig. 17 (a) Schematical illustration of the preparation of CNNA/rGO. (b) HRTEM image of CN nanorod in CNNA/rGO. (c) Photocatalytic CO2 reduction activities of bCN, CNNA, CNNA/rGO-R4, and CNNA/rGO. Reproduced with permission.175 Copyright 2019, Elsevier. (d) HAADF-STEM image of Cu–CCN. (e) FT-EXAFS of the samples.184 Copyright 2020, American Chemical Society. | ||
6. Application of CCNs-based photocatalysts
In this section, we will focus on recent progress of CCNs-based photocatalysts for photocatalytic applications such as photocatalytic water splitting, photocatalytic CO2 reduction, photocatalytic degradation of pollutants, selective organic syntheses and H2O2 production. In the following subsections, various photocatalytic applications of CCNs-based photocatalysts are briefly outlined. Some selected examples are described based on the latest findings.6.1. Photocatalytic water splitting
Direct water splitting for H2 generation using photocatalysts and sunlight is a sustainable technology that conversion of solar energy into valuable hydrogen energy. Theoretically, for CCNs photocatalyst, overall water splitting could be achieved on account of suitable CB and VB positions.106,124,185–188 For example, Wang and co-workers achieved overall water splitting by using crystalline PTI as photocatalyst without sacrificial reagents.185 The in situ photodeposition of Pt/Co on the PTI not only improved the photocatalytic performance, but also adjusted the ratio of H2 and O2 to 2:1 by tuning the amount of Co cocatalyst. The AQY at 380 nm was calculated to be 2.1%. The low efficiency of overall water splitting is presumably because of the kinetic restriction of the oxygen evolution reaction. Motivated by this work, the same group further investigated the photoactivity of overall water splitting by using Co and Pt co-catalysts to identify the reactive facets of single crystal PTI/Li+Cl−.106 It was found that both the reduction and oxidation reactions preferred to occur on the same {1010} crystal plane, which was distinct from the spatial separation of reduction and oxidation reactions for BiVO4 and TiO2 photocatalyst. Based on this finding, increasing the prismatic surface area (reactive facets) could dramatically improve the activity of photocatalytic overall water splitting. Moreover, it was demonstrated that the lower defect densities could lead to superior photocatalytic performance. Motivated by this research, the same group developed a fully condensed poly (triazine imide) photocatalysts with expanded π-conjugated system and low density structural defects through a mild ionothermal strategy with LiCl/NaCl.189 The melting point of LiCl/NaCl is about 552 °C, closing to the polymerization temperature, thereby providing moderate reaction condition and preventing the depolymerization of triazine monomer. The binding energies of C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in PTI-LiNa showed a positive shift of 0.35 eV, implying that the completely condensed nitrogen species were formed (Fig. 18a). The PTI-LiNa displayed the best photocatalytic overall water splitting performance with the H2 and O2 generation rate of 273 and 135 μmol h−1 because of the effective exciton dissociation and the promotion of the transfer and migration of photogenerated charge carrier (Fig. 18b).
C in PTI-LiNa showed a positive shift of 0.35 eV, implying that the completely condensed nitrogen species were formed (Fig. 18a). The PTI-LiNa displayed the best photocatalytic overall water splitting performance with the H2 and O2 generation rate of 273 and 135 μmol h−1 because of the effective exciton dissociation and the promotion of the transfer and migration of photogenerated charge carrier (Fig. 18b).
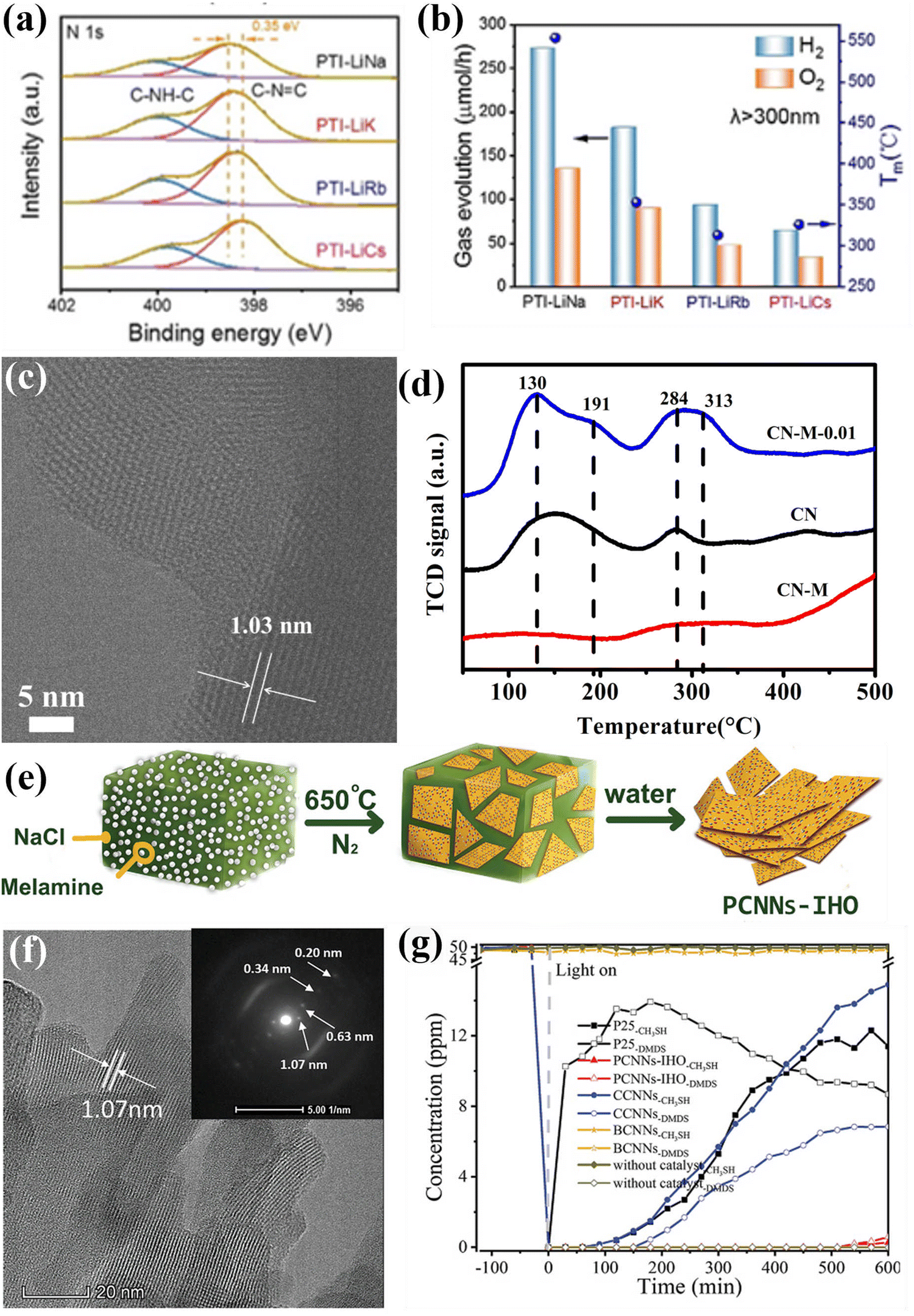 | ||
| Fig. 18 (a) High-resolution N1s XPS spectra of PTI-LiNa, PTI-LiK, PTI-LiRb and PTI-LiCs. (b) Photocatalytic overall water splitting performances of PTI/Li+Cl− prepared in different eutectic salts along with their melting points. Reproduced with permission. Copyright 2022, Wiley-VCH. (c) HRTEM image of CN-M-0.01. (d) CO2 TPD measurements. Reproduced with permission.224 Copyright 2020, Royal Society of Chemistry. (e) Schematical diagram of the preparation of PCNNs-IHO through on-surface polymerization. (f) HRTEM image of PCNNs-IHO and SAED pattern of the (100) plane (inset). (g) Photocatalytic CH3SH mineralization performance of PCNNs-IHO, CCNNs, BCNNs, and P25. Reproduced with permission.97 Copyright 2021, Wiley-VCH. | ||
Recently, the use of CCNs-based photocatalysts for hydrogen production half reaction has been intensively investigated, as this is a less difficult reaction than photocatalytic overall water splitting. However, low efficiency still hinders its practical application. Therefore, several modification strategies of pristine CCNs, including nanostructure design, molecular structure engineering and heterojunction construction have been systematically studied to optimize the photocatalytic activity. The latest progress using CCNs-based photocatalysts for photocatalytic H2 generation are summarized in Table 2 without detailed discussion. It is demonstrated that CCNs prepared from various precursors64,140,141,190 and molten salts61,64,115,191,192 exhibited different photocatalytic activities due to different crystal structures of the products. In general, the specific morphology and microstructure of CCNs could promote charge separation and transfer, as well as provide more active sites, thereby enhancing the photocatalytic hydrogen evolution activity. For example, numerous CCNs nanosheet65,94,112–114,123,124 have been proven to be effective in enhancing the photocatalytic H2 production performance. Moreover, Lotsch and co-workers investigated how particle size and surface functional groups affect the photocatalytic performance.193 The particle size and surface defects were systematically modulated by ultrasonication approach. It was demonstrated that optimal amount of defect not only accelerated the charge separation, but also facilitated the loading of Pt and reduced its size, thus boosting the photocatalytic H2 production activity. Apart from the nanostructure design, the defect engineering and molecular doping could narrow the bandgap and boost charge separation, thereby improving the photocatalytic hydrogen production performance, as detailed in Section 4.2. For instance, Zhang and co-workers proposed the doping of oxamide molecular into the PHI-based CCN to enhance the photocatalytic H2 production performance, which was 38 times higher than that of CN under green LED light irradiation.141 Lotsch and co-workers investigated the influence of ionic conductivity on photocatalytic activity by adjusting various counterions in the PHI framework.194 It was indicated that the ions influenced the stacking of 2D layers, thereby affecting the ionic conductivity, which was the bottleneck of electron stabilization and extraction. Benefiting from the highest ionic conductivity, the Na-PHI exhibited the best photocatalytic H2 production performance. Apart from the molecular structure engineering, construction of CCNs-based heterojunctions is another effective approach for improving the photocatalytic performance by accelerating the separation and transfer of charge carriers. For instance, Wang and co-workers fabricated a triazine/heptazine-based CCN with D–A heterostructure to improve the photocatalytic H2 production performance by promoting interfacial charge carrier transfer.159
| Photocatalysts | Cocatalyst | Reactant solution | Light source | Activity (μmol g−1 h−1), stability and quantum efficiency | Ref. |
|---|---|---|---|---|---|
| CCN intercalation compound | 3 wt% Pt | 100 mL of TEOA (10 vol%) and saturated salt solution | 300 W, Xe lamp, >420 nm | 5020.0, 50% decrease in activity under 20 h irradiation (5 h per run), 21.2% 420 nm | 61 |
| PTI/Li+Cl− | 5 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >300 nm | 800.0, N/A, N/A | 64 |
| PTI nanosheets | 2.2 wt% Pt | 10 ml of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 1750.0, retained after 6 cycles, 1.3% 400 nm | 65 |
| PTI-0.13 | 3 wt% Pt | 25 ml of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 8160.0, N/A, 7.0% 420 nm | 140 |
| LiK-CCN | 3 wt% Pt | 38 mL of TEOA (10 vol%) | 50 W white LED array, >410 nm | 812.0, slight drop under 72 h irradiation (12 h per run), N/A | 190 |
| PTI-0.15/KCC-1 | 3 wt% Pt | 25 mL of TEOA (10 vol%) | 300 W, Xe lamp, 385–740 nm | 1320.0, stable after 4 cycles (10 h per cycle), 16.9% 420 nm | 118 |
| MoS2/PHI | N/A | 38 mL of TEOA (10 vol%) | 50 W white LED array, >410 nm | 53.6, sustained over 65 h, 0.2% 420 nm | 198 |
| ZnO QDs/PTI | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, ≥420 nm | 440.0, stable after 12 h irradiation, 1.27% 420 nm | 164 |
| PHI-based CCN | 3 wt% Pt | 100 mL of TEOA (10 vol%) and K2HPO4 (0.01mol) | 300 W, Xe lamp, ≥420 nm | 15400.0, high stability, 50.7% 420 nm |
115 |
| PTI/PHI-based CCN | 3 wt% Pt | 50 mL of TEOA (10 vol%) | 300 W, Xe lamp, ≥420 nm | 1480.0, no obvious decrease in activity after 3 cycles (4 h per cycle), 10.7% 420 nm | 156 |
| PHIK-0.5 | 3 wt% Pt | 38 mL of TEOA (10 vol%) | 50 W white LED array, >410 nm | 656.0, N/A, N/A | 199 |
| PTI-based CCN | 1 wt% Pt and 9 wt% Co | 100 mL of methanol (10 vol%) | 300 W, Xe lamp | 649.0, N/A, 2.1% 380 nm | 185 |
| PHI-Mg | 1.57 wt% Pt | TEOA | 50 W white LED array | 1255.0, N/A, N/A | 200 |
| CN-OA-m | 3 wt% Pt | 38 mL of TEOA (10 vol%) | 50 W green LED light, 525 ± 1.0 nm | 340.0, no evident decrease in activity after four runs, 10.0% 525 nm | 141 |
| Na-PHI | 0.51wt% Pt | 38 mL of TEOA (10 vol%) | 50 W white LED array | 1344.0, N/A, N/A | 192 |
| CCN nanorods | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 1680.0, N/A, N/A | 92 |
| CCN nanosheets | 3 wt% Pt | 100 mL of methanol (10 vol%) | >420 nm | 1060.0, no evident change in activity after 3 cycles (4 h per run), 8.57% 420 nm | 112 |
| PTI/PHI-based CCN | 1 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 4813.2, N/A, N/A | 157 |
| CCN nanosheets | 3 wt% Pt | 10 mL of TEOA (10 vol%) | 300 W, Xe lamp, >400 nm | 2570.0, no detectable decresase in activity after 4 cycles, N/A | 114 |
| Amorphous/crystalline CN | 2 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 1179.0, no obvious activity decay after 4 cycles (2 h per cycle), N/A | 152 |
| Amorphous/crystalline CN | N/A | 80 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 310.0, stable after 3 cycles, 1.86% 420 nm | 154 |
| CCN | 0.5 wt% Pt | 10 mL of TEOA (15 vol%) | 300 W, Xe lamp, >420 nm | 2000.0, no obvious activity decay after 4 cycles (3 h per cycle), 5.6% 420 nm | 89 |
| CCN-ATZ-NaK | 3 wt% Pt | 38 mL of TEOA (10 vol%) and 0.01mol K2HPO4 | 50 W white LED light, >420 nm | 13000.0, no evident decrease in activity, 65.0% 420 nm |
201 |
| PTI/PHI-based CCN | 3 wt% Pt | 38 mL of TEOA (10 vol%) and NaCl 3 wt% | 50 W white LED light, >420 nm | 11720.0, no evident decrease in activity after 4 runs (4 h per run), 60% 420 nm |
159 |
| CCN nanosheets | 3 wt% Pt | 300 mL of TEOA (10 vol%) | 300 W, Xe lamp, >400 nm | 808.5, no obvious activity decay after 4 runs (4 h per run), 6.17% 420 nm | 117 |
| CCN | 3 wt% Pt | 100 mL of methanol (10 vol%) | 300 W, Xe lamp, >420 nm | 660.0, no obvious activity decay after 6 cycles (4 h per cycle), 6.8% 420 nm | 202 |
| PTI/PHI-based CCN | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 2880.0, no obvious activity decay after 4 cycles (4 h per cycle), 12.9% 420 nm | 158 |
| S-doped CCN | 1 wt% Pt | 80 mL of TEOA (20 vol%) | 300 W, Xe lamp, >420 nm | 1511.2, no obvious activity decay after 5 cycles (3 h per cycle), 3.9% 420 nm | 128 |
| Holey CCN nanosheets | 0.2 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 1631.6, no obvious activity decay after 4 cycles (4 h per cycle), 2.1% 420 ± 15 nm | 123 |
| CCN nanosheets | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp AM1.5 G | 9577.6, retained in 5 cycles (2.5 h per cycle), 9.01% 420 nm | 113 |
| Single crystalline melem | 1 wt% Pt | 80 mL of TEOA (10 vol%) | 300 W, Xe lamp, >400 nm | 74.6, N/A, N/A | 203 |
| NaK-PHI-A | 3 wt% Pt | 38 mL of TEOA (10 vol%) | 100 W white LED light | 6840.0, almost no change in activity after 4 runs (4 h per run), N/A | 142 |
| CCN | 3 wt% Pt | 20 mL of TEOA (5 vol%) | Blue LED lamp (450 nm) | 2100.0, N/A, 13.0% 450 nm | 204 |
| Defect-CCN | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, ≥455 nm | 1280.0, no significant activity decay after 4 cycles (4 h per cycle), 3.5% 420 nm | 134 |
| 3D porous CCN | 1 wt% Pt and 3 wt% IrO2 | 100 ml distilled water | 300 W, Xe lamp, ≥420 nm | 101.4, no notable activity decay after 100 h reaction, 1.4% 420 nm | 124 |
| CCN | 1 wt% Pt | 50 mL of TEOA (10 vol%) | 300 W, Xe lamp, 420–780 nm | 1356.0, no significant variation after 5 cycles (3 h per cycle), 11.4% 420 nm | 96 |
| Zn-doped PHI-based CCN | 3 wt% Pt | 100 mL of methanol (10 vol%) | 300 W, Xe lamp, >420 nm | 1500.0, retained in 4 cycles (4 h per cycle), 12.6% 420 nm | 130 |
| N-GQDs /PTI/ZnO-QDs | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, ≥420 nm | 1360.0, maintained for 4 cycles (3 h per cycle), 3.37% 420 nm | 165 |
| Cyanamide defect-CCN | 3 wt% Pt | 80 mL of TEOA (10 vol%) | 300 W, Xe lamp, ≥420 nm | 2690.0, maintained for 4 cycles (5 h per cycle), 42.0% 420 nm | 135 |
| Defective CCN/ZnIn2S4 | 1 wt% Pt | 100 mL of TEOA (20 vol%) | 300 W, Xe lamp, ≥420 nm | 1650.0, retained in 3 runs (6 h per run), 18.2% 420 nm | 166 |
| Nitrogen vacancy modified CCN | 2 wt% Pd | 100 mL of methanol (10 vol%) | 300 W, Xe lamp, >420 nm | 552.7, no significant decrease in activity after 5 cycles (3 h per cycle), N/A | 144 |
| WOx/CCN | N/A | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 2180.0, remained basically stable after 4 cycles (3 h per cycle), N/A | 168 |
| CCN nanosheets | 0.5 wt% Pt | 20 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 830.0, slight decrease in activity after 4 cycles (4 h per cycle), N/A | 205 |
| CCN | 1 wt% Pd/SiO2 | 5 mol L−1 of ethanol, 3 mol L−1 of lactic acid | 1000 W, Xe lamp, >400 nm | 520.0, N/A, 22% 405 nm | 206 |
| CCN | 3 wt% Pt | 80 mL of TEOA (10 vol%) | 350 W, Xe lamp | 683.54, no obvious decrease in activity after 4 cycles (3 h per cycle), 6.6% 420 nm | 207 |
| CCN | 0.5 wt% Pt | 20 mL of TEOA (15 vol%) | 300 W, Xe lamp, ≥420 nm | 2025.0, N/A, N/A | 90 |
| Heptazine-/triazine-based CCN | 3 wt% Pt | 80 mL of TEOA (10 vol%) | 350 W, Xe lamp | 890.0, no obvious activity decay after 4 cycles (3 h per cycle), 26.7% 420 nm | 161 |
| KPCN | 1 wt% Pt | 150 mL of TEOA (10 vol%) | 300 W, Xe lamp, 420–780 nm | 1188.0, slightly decrease in activity after 4 cycles (3 h per cycle), 8.6% 420 ± 5 nm | 208 |
| CCN | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 339.4, no significant decrease in activity after 5 cycles (4 h per cycle), 3.8% 420 nm | 209 |
| CCN nanosheet | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 4060.0, almost no change in activity after 4 runs (4 h per run), N/A | 116 |
| CCN with box-shape | 1mL H2PtCl6·6H2O solution | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp | 1948.0, no noticeable decrease in activity after 3 cycles (4 h per cycle), 2.09% 420 nm | 122 |
| Porous CCN | 1 wt% Pt | 80 mL of lactic acid (10 vol%) | four LED lights 3 W 420 nm | 1010.0, highly stable during the 4 cycles (2 h per cycle), 1.56% 420 nm | 119 |
| K+ and cyano-group co-doped CCN | 3 wt% Pt | 50 ml of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 4125.0, no significant change in activity after 3 cycles (4 h per cycle), N/A | 210 |
| PTI/Li+Cl− | 1 wt% Pt 0.5 wt% Co | 100 ml deionized water | 300 W, Xe lamp, >300 nm | 1890.0, stable after 96 h reaction, 8.0% 365 nm | 106 |
| Cyano-group modified CCN | 1 wt% Pt | 80 mL of lactic acid (10 vol%) | Four LED lights 3 W 420 nm | 758.8, stable during cycle tests, 1.17% 420 nm | 120 |
| CCN hollow spheres | 3 wt% Pt | 80 mL of bisphenol A (10 mg L−1) | 350 W, Xe lamp | 151.2, no significant decrease in activity after 4 cycles (3 h per cycle), 1.53% 420 nm | 125 |
| O and K co-doped CCN | 3 wt% Pt | 50 mL of TEOA (20 vol%) | 300 W, Xe lamp, >420 nm | 7844.0, no obvious decrease in activity after 6 cycles (5 h per cycle), 20.5% 420 nm | 211 |
| K-doped CCN | 3 wt% Pt | 50 mL of TEOA (20 vol%) | 300 W, Xe lamp, >420 nm | 5238.0, 8.4% decrease in activity after 5 cycles (2 h per cycle), 25.7% 420 nm | 212 |
| Nanostructured K-PHI | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp | 2161.0, stable in initial 2 cycles but significantly decrese in the third cycle, 2.32% 420 nm | 213 |
| Ultrathin CCN with D–A structure | 3 wt% Pt | 100 mL of TEOA (20 vol%) | 300 W, Xe lamp, >420 nm | 5442.74, no obvious change in activity after 8 runs within 40 h, 23.3% 420 nm | 214 |
| Na-PHI | 8 wt% Pt | 10 mL of methanol (10 vol%) | 300 W, Xe lamp AM1.5 G | 2400 ± 100, N/A, N/A | 194 |
| Hierarchical CCN arrays | 3 wt% Pt | 50 mL of TEOA (20 vol%) | 300 W, Xe lamp, >420 nm | 758.8, stable during 5 cycles (5 h per cycle), 60.0% 420 nm | 215 |
| K-PHI/B0.8ST | 1 wt% Pt | 100 mL of TEOA (20 vol%) | 300 W, Xe lamp, >420 nm | 1087.4, no obvious decay in activity after 4 cycles (4 h per cycle), 8.05% 420 nm | 173 |
| CCN homojunction | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 3550.2, no significant decrease in activity after 4 cycles (3 h per cycle), 25.0% 420 nm | 216 |
| K+ implanted CCN | 3 wt% Pt | 50 ml of TEOA (10 vol%) | 300 W, Xe lamp, 420–780 nm | 4008.0, 11% decrease in activity after 5 cycles (2 h per cycle), 26.1% 420 nm | 217 |
| Single atom Co loaded on CCN | 1 wt% Pt | 20 ml of lactic acid (20 vol%) | 300 W, Xe lamp, ≥420 nm | 32100.0, slight decrease in activity after 12 h reaction, 49.5% 420 nm |
218 |
| Carbon fiber/CCN | 1 wt% Pt | 100 mL of TEOA (10 vol%) | 3 W LED light, 420 nm | 6398.0, slight decrease in activity after 4 cycles (2 h per cycle), 3.88% 420 nm | 219 |
| single-atom Co coordinated CCN/PTI | 1 wt% Pt | 30 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 3538.0, no obvious change in activity after 8 cycles (3 h per cycle), 20.88% 430 nm | 220 |
| Red CCN nanosheet | 1.1 wt% Pt | 33 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 14665.0, N/A, N/A |
221 |
| ReS2/CCN | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 3460.0, slightly decrease in activity after 3 cycles (5 h per cycle), 5.42% 400 nm | 222 |
| MC-CN | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 181.51, no apparent activity decay after 5 cycles (3 h per cycle), 3.26% 400 nm | 129 |
| Ca and K co-doped CCN | 3 wt% Pt | 50 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 5190.0, 9.7% decrease in activity after 4 cycles (5 h per cycle), 31.2% 420 nm | 131 |
| PHI/PTI | 3 wt% Pt | 100 mL of TEOA (10 vol%) | 300 W, Xe lamp, >420 nm | 6970.0, slight decrease in activity after 12 runs (4 h per run), 42.0% 420 nm | 162 |
However, the conventional H2 production half-reaction is achieved by using various sacrificial reagents, which increases cost and wastes the energy of holes. To address this issue, the coupling of organic synthesis with H2 evolution reaction has emerged as an appealing strategy, which not only makes full use of photoexcited electron and hole to realize sustainability targets, but also enhances the performance of photocatalytic H2 production. Recently, some organic substrates coupled with H2 evolution have been reported on CCNs-based photocatalysts, including bisphenol A195 and cellulose.196
In addition to the hydrogen production reaction, the half reaction of O2 produced from water oxidation is a complex multi-electron transfer process requiring a large overpotential. The CCN has also been shown as a promising catalyst for photocatalytic water oxidation. In a recent work, Wang and co-workers developed a van der Waals homojunction of Zn-PHI/PHI for photocatalytic O2 evolution through a ternary molten salt approach.197 The Zn doped PHI generated during the calcination process contributed to the formation of homojunction, which created an hole migration channels and promoted charge separation. Consequently, the AQY of the optimized photocatalyst reached up to 3.6% at 420 nm.
6.2. Photocatalytic CO2 reduction
With the rapid industrialization and economic development, the concentration of greenhouse gases, i.e., carbon dioxide in the atmosphere, is increasing. Therefore, it is desirable to reduce its concentration through capture or conversion. Inspired by the photosynthesis of natural green plants, artificial photosynthesis, photocatalytic conversion of CO2 into hydrocarbon fuels has been recognized as an appealing technology to mitigate greenhouse effect and address global energy shortage. The photocatalytic reduction of CO2 is a complex multistep process, as shown in eqn (1)–(8),223 the possible products and corresponding redox potentials (versus normal hydrogen electrode (NHE) at pH 7) are listed below| CO2 + e− → CO2˙ E0redox = −1.9 V | (1) |
| CO2 + 2H+ + 2e−→ CO + H2O E0redox = −0.53 V | (2) |
| CO2 + 2H+ + 2e−→ HCOOH E0redox = −0.61 V | (3) |
| CO2 + 4H+ + 4e− → HCHO + H2O E0redox = −0.48 V | (4) |
| CO2 + 6H+ + 6e− → CH3OH + H2O E0redox = −0.38 V | (5) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O E0redox = −0.24 V | (6) |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O E0redox = −0.33 V | (7) |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O E0redox = −0.32 V | (8) |
Based on the above reaction process, the photocatalytic CO2 reduction is a proton-assisted multi-electron reaction process, which affects the selectivity of products. Moreover, the CO2 molecules are extremely difficult to be activated to participate in the reactions due to their stable linear structure and chemical inertness. Meanwhile, the proton donor for CO2 reduction should be provided by H2O. Thus, the hydrogen production via reducing H2O is a competitive reaction for photocatalytic CO2 reduction, resulting in a low selectivity and activity for CO2 reduction. These factors make it difficult to realize high efficiency and good selectivity for photocatalytic CO2 reduction. In order to deal with these challenges, some key factors need to be considered, such as light absorption, CO2 adsorption and activation, separation and transfer of charges, and products desorption.
It is well known that improving light absorption is essential for enhancing photocatalytic performance. Wang and co-workers synthesized PHI-based CCN through the molten salt approach using a new stable monomer with high nitrogen content (5-aminotetrazole).201 The molten salt method could modulate the crystallinity, grain boundary structure and chemical property to improve the light absorption. Due to the well-constructed PHI structure, improved light absorption and optimized electronic properties, the CCN exhibited excellent photocatalytic performance.
From the viewpoint of thermodynamics, transferring the first electron to the CO2 molecule to activate CO2 is indeed unfavorable due to a very negative reduction potential of 1.9 V (see eqn (1)). Therefore, improving the CO2 activation ability of CCNs is an efficient approach to enhance the photocatalytic activity.88,184,224 Based on the above discussion, our group designed a defect-modified CCN to improve the performance of photocatalytic CO2 reduction.224 As shown in Fig. 18c, HRTEM image reveals clear lattice fringes with a d spacing of 1.03 nm, confirming the high crystallinity, suggesting that the crystallinity was well maintained by KOH treatment. The CO2 temperature-programmed desorption measurement (Fig. 18d) showed that the desorption peak shifted to a higher temperature, suggesting that the defect was beneficial to the adsorption and activation of CO2, thus lowering the activation energy barrier. The defect-modified CCN exhibited better photocatalytic performance than CCN due to the high crystallinity and optimal activation sites.
The selectivity of the CO2 photoreduction reaction products is influenced by CO2 adsorption. For example, our group found that the cyano and carboxyl groups not only greatly enhanced the adsorption and activation of CO2, but also improved the charge transfer.88 Due to the strong chemisorption of CO2, the defective CCN exhibited good photocatalytic CO2 reduction performance with a total generation rate of 12.07 μmol h−1 g−1 and selectivity (91.5%), which was better than most previously reported CN photocatalysts. Xiang and co-workers prepared a single Cu atom/CCN photocatalyst (Cu–CCN) by molten salt and reflux approach.184 The introduction of single Cu atoms served as reaction centers for CO2 adsorption and activation. DFT calculation revealed that the reduction of CO2 to CO is an energy favorable reaction compared with the CO2 reduction to CH4. Consequently, the Cu–CCN presented improved photocatalytic CO2 reduction activity and high selectivity (nearly 100%) for the conversion of CO2 to CO. Taking advantage of CO2 adsorption and charge transfer, Liu and co-workers developed tungsten doped CCN by solvothermal method for CO2 conversion.225 Owning to the synergetic modulation of photogenerated electron capture and enrichment, as well as the adsorption and activation of CO2 and CO intermediates, the W-doped CCN showed excellent photocatalytic CO2 reduction performance and high selectivity for hydrocarbons.
The effective separation of photoexcited charge carriers is beneficial to improve the photocatalytic CO2 reduction activity.226 Furthermore, considering that the CO2 reduction is a multi-electron involved process, coupling the cocatalyst, such as graphene, and single atom to promote the transfer and collection of electrons is an efficient strategy to improve the CO2 reduction performance. For instance, our group constructed a 1D/2D heterojunction (CNNA/rGO) of CCN nanoarrays (CNNA) aligned on 2D graphene sheet through an ionothermal method.175 Due to the heterojunction effect and ordered 1D structure, the CNNA/rGO exhibited greatly improved CO2 adsorption capacity. Moreover, the charge transfer and accumulation were greatly improved after the introduction of rGO. The CNNA/rGO exhibited efficient photocatalytic reduction performance due to the selective CO2 binding, improved charge transport, enhanced light absorption as well as promoted exciton splitting. Xiang and co-workers designed dual-single-atoms of Pt–Cu on N-vacancy-rich CCN for efficient and selective CO2 reduction.227 The dual single atoms not only integrated the advantages of platinum and copper atoms, but also promoted the charge transfer and migration.
The conventional reaction system discussed above usually couples CO2 reduction half-reaction with pure water oxidation half-reaction. However, the water oxidation half-reaction suffers from sluggish kinetics and large overpotential, resulting in low catalytic efficiency. Therefore, replacing the H2O oxidation half-reaction with a thermodynamically favorable organic synthesis to simultaneously produce two valuable chemicals is an attractive approach compared with the conventional CO2 reduction system. For instance, Sun and co-workers proposed the co-doping of K and S into CCN through a salt-template-assisted incorporation approach for simultaneous photoreforming of biomass and CO2 reduction.133 The K and S co-doping could improve the visible-light absorption and accelerate the charge transport and separation. Consequently, the K and S co-doped CCN presented superior photocatalytic performance with a CO generation rate of 16.27 μmol g−1 h−1 and lactic acid yield (78.07%).
6.3. Photocatalytic degradation of pollutants
With the rapid development of industrialization and population growth, a large amount of toxic and hazardous pollutants has been released into the environment, posing a great threat to human health and the ecological environment. In order to achieve the sustainable development of human society, photocatalytic degradation of pollutants has been demonstrated to be a promising green approach to address the environmental problems. The mechanism of photocatalytic degradation is as follows. Under light irradiation, photogenerated electrons could reduce the O2 adsorbed on the surface of photocatalyst to generate superoxide radicals (˙O2−), which would contribute to the formation of ˙OH radicals, 1O2, and H2O2. Meanwhile, in some cases, the photoexcited holes may react with OH− on the surface to generate ˙OH radicals. These main active species (e−, h+, ˙O2−, 1O2, H2O2, ˙OH) in the above process could oxidize organic pollutants into CO2 and H2O.Recently, the CCNs-based photocatalysts have been proved to be effective in degrading various pollutants, including rhodamine B (RhB),151,152,168,171,228–232 methylene blue (MB),160,229,233 methyl orange (MO),98 phenol,98,229 tetracycline,172,234 pharmaceuticals and personal care products,235 17α-ethinylestradiol,236 soluble U(VI).237 For instance, Liang and co-workers reported that the tetragonal CCN hollow tubes exhibited efficient photocatalytic activity in degrading methylene blue and phenol, which could be attributed to the high crystallinity, unique one-dimensional hollow structure and large specific surface area.229 Li and co-workers investigated the photocatalytic degradation of pharmaceuticals and personal care products (PPCPs) by CCN.235 They proposed the route of direct two-electron reduction on the CCN surface to produce H2O2 and subsequent conversion to ˙OH radicals, rather than the conventional single-electron reduction reaction on bulk CN. Due to the improved charge separation and electron transfer ability, the CCN exhibited superior photocatalytic activity towards PPCPs-type pollutants. To obtain an effective charge separation ability, Guo and co-workers fabricated a CoP/CCN composite photocatalyst via a facile solvothermal approach for the photodegradation of tetracycline.172 HRTEM image confirms the high crystallinity, suggesting reduced structural defects. Furthermore, CoP acted as the cocatalyst to accelerate the separation of the charge carriers. As a result, the designed composites showed superior photocatalytic performance. The photocatalytic reduction of soluble U(VI) around the clock was achieved by using K+ and cyano group co-modified PHI.237 The K+ could stabilize the electrons and generate long-lived radicals, enabling the photocatalytic reduction of U(VI) in the dark. Besides the photocatalytic degradation of pollutants in the liquid phase, CCN have also been intensively investigated for the photocatalytic removal of gas phase pollutants, including NO238,239 and CH3SH.97 For instance, Wang and co-workers prepared in-plane highly ordered CCN nanosheets (PCNNs-IHO) through on-surface polymerization approach for the mineralization of CH3SH (Fig. 18e).97 As revealed by the HRTEM image, a clear lattice spacing of 1.07 nm corresponding to the in-plane stacking was observed, suggesting the enhanced in-plane ordering (Fig. 18f). Moreover, the NaCl not only acted as unique support to guide the assembly of two-dimension nanosheets, but also served as a reactant to afford Na+ incorporation to obtain structural base sites. Owning to the structural basicity, the PCNNs-IHO showed excellent photocatalytic performance for the mineralization of CH3SH to SO42− (Fig. 18g).
The aforementioned work demonstrated that the CCNs-based photocatalysts presented outstanding photocatalytic degradation performance since their high crystallinity could effectively separate and transfer the charge carriers.
6.4. Photocatalytic organic synthesis
Solar-driven organic transformation is a promising technology for the synthesis of many important compounds. Recently, CCNs-based photocatalysts for selective organic synthesis under mild conditions have been intensively investigated. For example, Huang and co-workers demonstrated that a conversion rate of 5.81% from cyclohexane to cyclohexanone and a selectivity over 99.9% could be achieved by using the PTI as a photocatalyst without any other oxidants, initiators or solvents.240 Wang and co-workers reported that the conversion of benzyl alcohol to benzaldehyde with 90% selectivity was achieved using CCN as a photocatalyst to activate O2.241 This enhanced photocatalytic activity could be ascribed to the synergistic effect of effective electron–hole separation and increased surface reaction kinetics. Su and co-workers reported the selective deuteration of alkenes and alkynes by the in situ generated D-species in heavy water splitting using Pd nanoparticles modified KPCN.95 Pieber and co-workers coupled Ni catalyst with CN-OA-m photocatalyst for selective C–O cross-couplings of carboxylic acids with aryl halides.242 As a continuation of the above research, the same group further extended this method to dual nickel/photocatalytic C–O coupling.243 Hu and co-workers designed CCN with surface nitrogen vacancies and cyano groups for isomerization of glucose to fructose with a selectivity of 60%.244 Wang and co-workers reported the preparation of Fe-PHI with atomically dispersed Fe–N4C active center by ion exchange method for selective C–H bond oxidation to valuable carbonyl compounds.245 Savateev and co-workers reported a series of research proving the efficient photocatalytic organic conversion on K-PHI photocatalysts, including the selective oxidation of benzyl alcohol to benzaldehyde and 1,4-dihydropyridines to pyridines,246 oxidative cyclization N-acylhydrazones to 1,3,4-oxadiazoles,247 photocatalytic Kindler reaction,248 oxidative C–H thiolation of toluenes to dibenzyldisulfanes,249 cyclodimerization of the chalcones to cyclopentanoles,250 halogenation of aromatic hydrocarbons,251 tetrahydroisoquinoline and chalcones to polyaromatic heterocycles,252 [3+2]-cycloadditions reaction,253 chromoselective synthesis of sulfonyl chlorides and sulfonamides,254 and so on. It was found that the conversion and selectivity of benzyl alcohol to benzaldehyde were excellent under relatively mild reaction conditions when elemental sulfur (S8) and K-PHI were used as sacrificial electron acceptor and photocatalyst, respectively.246 Recently, Savateev and co-workers described a cascade of energy-transfer reactions for the preparation of various oxadiazoles-1,2,4 and isoxazoles via dipolar [3+2]-cycloaddition.253 The crystal structure of the potassium poly(heptazine imide) (K-PHI) was refined by the XRD data and TEM image (Fig. 19a and b). The high crystallinity was confirmed by the HRTEM image (Fig. 19c). Interestingly, once excited with visible light, the K-PHI could undergo crossover between singlet–triplet intersystem, thereby providing a reasonably long-lived triplet excited state. Then, the triplet excited states could sensitize singlet oxygen (1O2), and, in turn, employ 1O2 as a starting point for the preparation of up to 25 different N-rich heterocycles (Fig. 19d). Moreover, the same group reported the selective synthesis of three different products (arylchlorides, sulfonyl chlorides and diaryldisulfides) from S-arylthioacetates by changing the excitation light, using K-PHI as the catalyst (Fig. 19e).254 The mechanism of the chromoselective oxidation reaction is presented in Fig. 19f. Specifically, the diaryldisulfides was produced by the green or red light irradiation, which was not sufficient to separate excitons and only participated in the energy transfer to O2. The formation of sulfenyl chloride was achieved by the blue light irradiation, enabling electron transfer to O2 and thioacetate. When irradiated with 365 or 410 nm light, the two-electron oxidation of Cl− occurred and resulted in the over-chlorination of sulfenyl chloride, which was unstable and decomposed to chlorobenzene.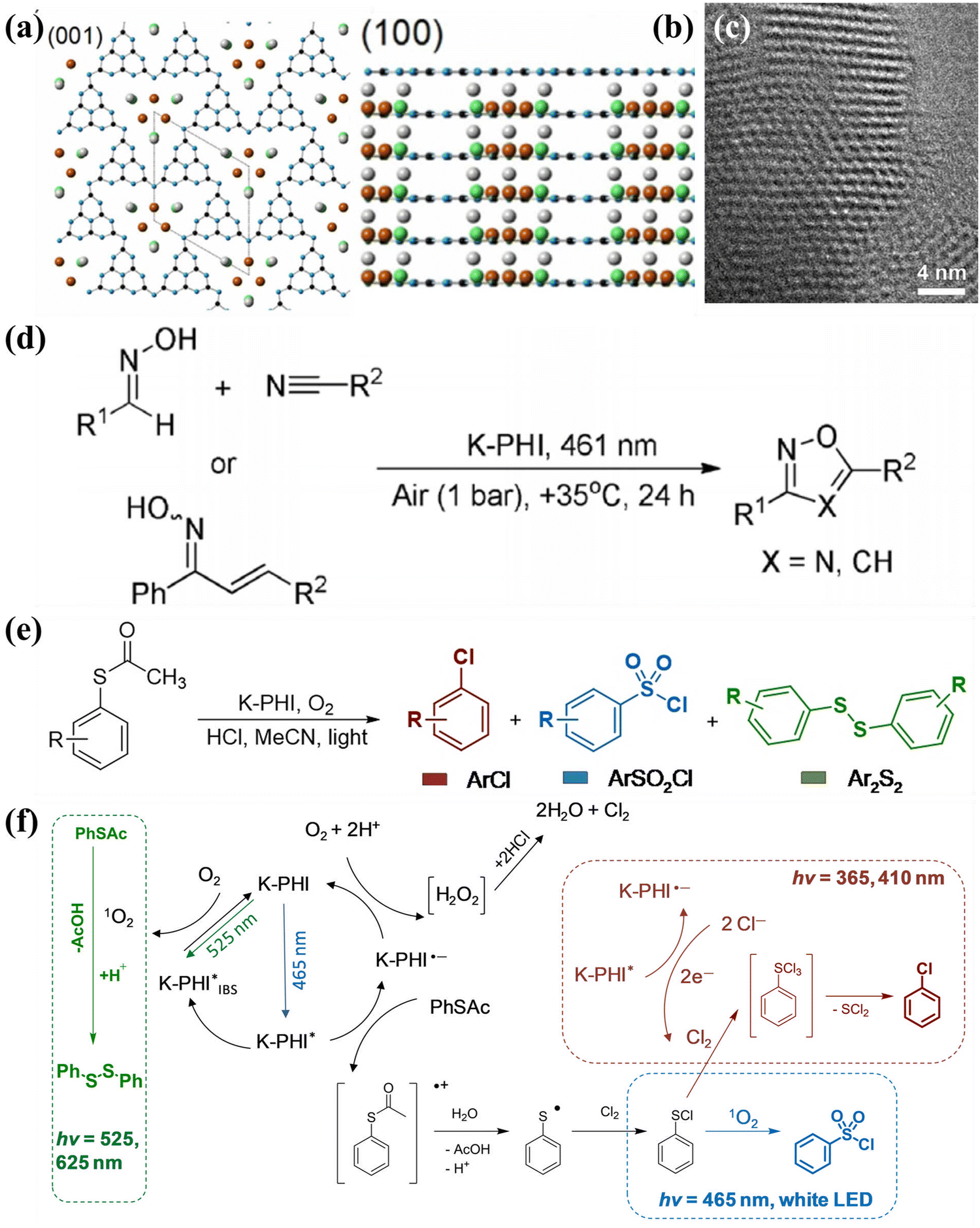 | ||
| Fig. 19 (a) and (b) Crystal structure of KPHI revealed by the XRD data. (c) HRTEM image of K-PHI. (d) Photocatalytic synthesis of oxadiazoles-1,2,4 and isoxazoles. Reproduced with permission.253 Copyright 2020, Wiley-VCH. (e) Three possible products of S-arylthioacetates oxidation by K-PHI. (f) The chromoselective oxidation mechanism for the production of sulfonyl chlorides, arylchlorides, and disulfides from thio-derivatives. Reproduced with permission.254 Copyright 2021, Wiley-VCH. | ||
6.5. Photocatalytic hydrogen peroxide (H2O2) production
H2O2 is a valuable and mild oxidizing chemical extensively used in paper bleaching, environmental remediation, sterilization, energy conversion/storage, and fine chemical synthesis.255,256 The conventional and prevailing method for industrial production of hydrogen peroxide is the anthraquinone process, which requires intense energy consumption and produces hazardous waste. Therefore, it is highly desirable to develop an eco-friendly and sustainable route for H2O2 synthesis. Photocatalytic production of H2O2 from H2O and O2 is a green and attractive approach because it requires only renewable sunlight as the energy input. The process of artificial photosynthesis of H2O2 could be divided into the five main routes257–261 (Fig. 20): (I) The direct two-electron O2 reduction process, (II) sequential single-electron O2 reduction route, (III) sequential electrons and holes transfer pathway, (IV) the direct two-electron H2O oxidation reaction, (V) sequential single-electron H2O oxidation pathway. Therefore, the possible intermediates and corresponding redox potentials (versus normal hydrogen electrode (NHE) at pH = 0) for the production of H2O2via different pathways are presented in eqn (9)–(14).262| O2 + e− → ˙O2− E0redox = −0.33 V | (9) |
| ˙O2− + 2H+ + e− → H2O2 E0redox = +1.44 V | (10) |
| ˙O2− + h+ → 1O2 E0redox = +0.34 V | (11) |
| O2+ 2H+ + 2e−→ H2O2 E0redox = +0.68 V | (12) |
| H2O+ 2h+ → H+ + ˙OH E0redox = +2.73 V | (13) |
| 2H2O+ 2h+ → H2O2+ 2H+ E0redox = −1.76 V | (14) |
 | ||
| Fig. 20 Reaction pathways of the photosynthesis of H2O2 through the O2 reduction or H2O oxidation reaction. | ||
Recently, CCNs-based photocatalysts have emerged as a candidate for efficient artificial photosynthesis of H2O2.263–265 For instance, Yu and co-workers fabricated ion-intercalated CCN (CN-KCl/KI) through a facile ionothermal approach for photosynthesis of H2O2.266 The ionothermal method not only enhanced the crystallinity of CN, but also resulted in the intercalation of K+ and I−. The doping of I− reduces the band gap and improves the light absorption. While the intercalation of K+ not only accelerates the interlayer carrier transport and separation, but also enhances the selectivity for the two-electron O2 reduction pathway. Benefiting from the synergistic effect of crystallinity tuning and dual doping engineering, the CN-KCl/KI shows an excellent H2O2 generation rate of 13.1 mmol g−1 h−1 at 400 nm with the AQY of 23.6%.
7. Conclusion and outlook
In conclusion, CCNs have been a popular research topic in photocatalysis due to their extraordinary features such as high photogenerated carrier transfer capacity, improved light absorption ability and low electron–hole pair recombination rate. In this review, we summarized the recent developments of CCNs and CCNs-based photocatalysts, focusing on the preparation methods and design of such photocatalysts with remarkably improved photocatalytic performance and high photogenerated carrier separation capability. This review discusses the preparation of CCNs based on high-temperature and high-pressure routes, simple ionothermal (molten salt) approach, solvothermal method and microwave-assisted heating synthesis. The applications of CCNs-based photocatalysts in the field of solar energy conversion, such as photocatalytic water splitting, photocatalytic CO2 reduction, photocatalytic pollutant degradation, photocatalytic organic synthesis and photocatalytic H2O2 production, are introduced. Despite the above significant progress in CCNs-based photocatalysts, there are still several important challenges to be addressed:(1) During the synthesis of CCNs by ionothermal (molten salt) method, ions could be used as a “template” to induce different morphologies, but the deep mechanism is still unclear and more experiments and discussions should be conducted.
(2) The applications of CCN prepared by the solvothermal and microwave-assisted thermal synthesis route are relatively isolated.
(3) Although the crystallinity and visible light utilization have been significantly improved, the specific surface area of CCN is low and the exposed active sites are limited.
(4) More attention should be paid to the template, supramolecular chemistry strategy and solvothermal approach to tune the morphology and microstructure. There is an urgent need to develop more supramolecular precursors to synthesize CCN with various nanostructures.
(5) Even though the performance of CCNs-based photocatalysts has been greatly improved, the enhancement mechanism is not deep and specific enough. Some characterization techniques and DFT calculations should be applied in this field, such as surface photo-voltage microscopy (SPVM), Kelvin probe force microscopy (KPFM) and calculation of built-in electric field.
(6) The surface functionalization of CCNs needs to be further explored to optimize photocatalytic performance.
(7) To further advance the performance of photocatalytic reduction of CO2, a more comprehensive understanding of the reaction process is necessary. From this perspective, in situ/operando techniques would be extremely useful tools to investigate the reaction process and intermediates. For instance, in situ Fourier transform infrared spectroscopy (FTIR) and operando Raman can uncover the intermediates of the reaction.
(8) Although the ionothermal (molten salt) method is an effective approach for large-scale production of CCNs, other efficient synthesis methods need to be further explored. Moreover, compared with metal-based photocatalysts, the CCNs would be suitable for photocatalytic reactions in seawater, including the production of H2 and H2O2 in seawater.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2022YFE0114800), and the National Natural Science Foundation of China (22278324 and 52073223).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Y. Fang, Y. Zheng, T. Fang, Y. Chen, Y. Zhu, Q. Liang, H. Sheng, Z. Li, C. Chen and X. Wang, Sci. China: Chem., 2020, 63, 149–181 CrossRef CAS.
- P. Kuang, M. Sayed, J. Fan, B. Cheng and J. Yu, Adv. Energy Mater., 2020, 10, 1903802 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS PubMed.
- C. Yang, B. Cheng, J. Xu, J. Yu and S. Cao, EnergyChem, 2023 DOI:10.1016/j.enchem.2023.100116.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 CrossRef PubMed.
- H. Li, Q. Song, S. Wan, C.-W. Tung, C. Liu, Y. Pan, G. Luo, H. M. Chen, S. Cao, J. Yu and L. Zhang, Small, 2023, 19, 2301711 CrossRef CAS PubMed.
- X. Zhou, Q. Xu, W. Lei, T. Zhang, X. Qi, G. Liu, K. Deng and J. Yu, Small, 2014, 10, 674–679 CrossRef CAS PubMed.
- E. S. Jang, J.-H. Won, S.-J. Hwang and J.-H. Choy, Adv. Mater., 2006, 18, 3309–3312 CrossRef CAS.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- C. Bie, B. Zhu, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2019, 31, 1902868 CrossRef CAS PubMed.
- T. Di, Q. Xu, W. Ho, H. Tang, Q. Xiang and J. Yu, ChemCatChem, 2019, 11, 1394–1411 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- Y. Fang and X. Wang, Chem. Commun., 2018, 54, 5674–5687 RSC.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- A. Savateev, I. Ghosh, B. Koenig and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS PubMed.
- H. Li, S. Tao, S. Wan, G. Qiu, Q. Long, J. Yu and S. Cao, Chin. J. Catal., 2023, 46, 167–176 CrossRef CAS.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 CrossRef CAS PubMed.
- Z. Zhou, Y. Zhang, Y. Shen, S. Liu and Y. Zhang, Chem. Soc. Rev., 2018, 47, 2298–2321 RSC.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- Y. Wang, S. Z. F. Phua, G. Dong, X. Liu, B. He, Q. Zhai, Y. Li, C. Zheng, H. Quan, Z. Li and Y. Zhao, Chem, 2019, 5, 2775–2813 CAS.
- A. Savateev and M. Antonietti, ChemCatChem, 2019, 11, 6166–6176 CrossRef CAS.
- B. Yan, Z. Chen and Y. Xu, Chem. – Asian J., 2020, 15, 2329–2340 CrossRef CAS PubMed.
- J. Liebig, Ann. Pharm., 1834, 10, 1–47 CrossRef.
- E. C. Franklin, J. Am. Chem. Soc., 1922, 44, 486–509 CrossRef CAS.
- L. Pauling and J. H. Sturdivant, Proc. Natl. Acad. Sci. U. S. A., 1937, 23, 615–620 CrossRef CAS PubMed.
- C. E. Redemann and H. J. Lucas, J. Am. Chem. Soc., 1940, 62, 842–846 CrossRef CAS.
- A. Y. Liu and M. L. Cohen, Science, 1989, 245, 841–842 CrossRef CAS PubMed.
- D. M. Teter and R. J. Hemley, Science, 1996, 271, 53–55 CrossRef CAS.
- D. C. Nesting and J. V. Badding, Chem. Mater., 1996, 8, 1535–1539 CrossRef CAS.
- A. Y. Liu and R. M. Wentzcovitch, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 10362–10365 CrossRef CAS PubMed.
- J. Ortega and O. F. Sankey, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 2624–2627 CrossRef CAS PubMed.
- J. E. Lowther, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 11683–11686 CrossRef CAS.
- M. Kawaguchi and K. Nozaki, Chem. Mater., 1995, 7, 257–264 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- E. Kroke, M. Schwarz, E. Horath-Bordon, P. Kroll, B. Noll and A. D. Norman, New J. Chem., 2002, 26, 508–512 RSC.
- J. Sehnert, K. Baerwinkel and J. Senker, J. Phys. Chem. B, 2007, 111, 10671–10680 CrossRef CAS PubMed.
- E. Horvath-Bordon, E. Kroke, I. Svoboda, H. Fuess and R. Riedel, New J. Chem., 2005, 29, 693–699 RSC.
- T. Komatsu and T. Nakamura, J. Mater. Chem., 2001, 11, 474–478 RSC.
- M. J. Bojdys, J.-O. Mueller, M. Antonietti and A. Thomas, Chem. – Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed.
- B. Jurgens, E. Irran, J. Senker, P. Kroll, H. Muller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef PubMed.
- A. Sattler, S. Pagano, M. Zeuner, A. Zurawski, D. Gunzelmann, J. Senker, K. Mueller-Buschbaum and W. Schnick, Chem. – Eur. J., 2009, 15, 13161–13170 CrossRef CAS PubMed.
- B. V. Lotsch and W. Schnick, Chem. Mater., 2006, 18, 1891–1900 CrossRef CAS.
- B. V. Lotsch and W. Schnick, Chem. – Eur. J., 2007, 13, 4956–4968 CrossRef CAS PubMed.
- T. Komatsu, J. Mater. Chem., 2001, 11, 799–801 RSC.
- T. Komatsu, J. Mater. Chem., 2001, 11, 802–805 RSC.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chem. Commun., 2006, 4530–4532 RSC.
- M. Doblinger, B. V. Lotsch, J. Wack, J. Thun, J. Senker and W. Schnick, Chem. Commun., 2009, 1541–1543 RSC.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- D. R. Miller, J. J. Wang and E. G. Gillan, J. Mater. Chem., 2002, 12, 2463–2469 RSC.
- E. G. Gillan, Chem. Mater., 2000, 12, 3906–3912 CrossRef CAS.
- Y. Zhang, A. Thomas, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 50–51 CrossRef CAS PubMed.
- Z. Zhou, J. Wang, J. Yu, Y. Shen, Y. Li, A. Liu, S. Liu and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 2179–2182 CrossRef CAS PubMed.
- H. Gao, S. Yan, J. Wang, Y. A. Huang, P. Wang, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2013, 15, 18077–18084 RSC.
- Q. Deng, H. Li, W. Hu and W. Hou, Angew. Chem., Int. Ed., 2023, 62, e202314213 CrossRef CAS PubMed.
- M. Deifallah, P. F. McMillan and F. Cora, J. Phys. Chem. C, 2008, 112, 5447–5453 CrossRef CAS.
- Y. Ham, K. Maeda, D. Cha, K. Takanabe and K. Domen, Chem. – Asian J., 2013, 8, 218–224 CrossRef CAS PubMed.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- C. M. Niu, Y. Z. Lu and C. M. Lieber, Science, 1993, 261, 334–337 CrossRef CAS PubMed.
- K. M. Yu, M. L. Cohen, E. E. Haller, W. L. Hansen, A. Y. Liu and I. C. Wu, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 5034–5037 CrossRef CAS PubMed.
- Z. M. Ren, Y. C. Du, Y. X. Qiu, J. D. Nu, Z. F. Ying, X. X. Xiong and F. M. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 5274–5277 CrossRef CAS PubMed.
- Z. J. Zhang, S. S. Fan and C. M. Lieber, Appl. Phys. Lett., 1995, 66, 3582–3584 CrossRef CAS.
- Y. F. Zhang, Z. H. Zhou and H. L. Li, Appl. Phys. Lett., 1996, 68, 634–636 CrossRef CAS.
- I. Alves, G. Demazeau, B. Tanguy and F. Weill, Solid State Commun., 1999, 109, 697–701 CrossRef CAS.
- Z. H. Zhang, K. Leinenweber, M. Bauer, L. A. J. Garvie, P. F. McMillan and G. H. Wolf, J. Am. Chem. Soc., 2001, 123, 7788–7796 CrossRef CAS PubMed.
- J. Kouvetakis, A. Bandari, M. Todd, B. Wilkens and N. Cave, Chem. Mater., 1994, 6, 811–814 CrossRef CAS.
- E. Horvath-Bordon, R. Riedel, P. F. McMillan, P. Kroll, G. Miehe, P. A. van Aken, A. Zerr, P. Hoppe, O. Shebanova, I. McLaren, S. Lauterbach, E. Kroke and R. Boehler, Angew. Chem., Int. Ed., 2007, 46, 1476–1480 CrossRef CAS PubMed.
- E. Wirnhier, M. Doeblinger, D. Gunzelmann, J. Senker, B. V. Lotsch and W. Schnick, Chem. – Eur. J., 2011, 17, 3213–3221 CrossRef CAS PubMed.
- B. V. Lotsch, M. Doeblinger, J. Sehnert, L. Seyfarth, J. Senker, O. Oeckler and W. Schnick, Chem. – Eur. J., 2007, 13, 4969–4980 CrossRef CAS PubMed.
- S. Y. Chong, J. T. A. Jones, Y. Z. Khimyak, A. I. Cooper, A. Thomas, M. Antonietti and M. J. Bojdys, J. Mater. Chem. A, 2013, 1, 1102–1107 RSC.
- M. J. Bojdys, N. Severin, J. P. Rabe, A. I. Cooper, A. Thomas and M. Antonietti, Macromol. Rapid. Commun., 2013, 34, 850–854 CrossRef CAS PubMed.
- H. Montigaud, B. Tanguy, G. Demazeau, I. Alves and S. Courjault, J. Mater. Sci., 2000, 35, 2547–2552 CrossRef CAS.
- Y. J. Bai, B. Lu, Z. G. Liu, L. Li, D. L. Cui, X. G. Xu and Q. L. Wang, J. Cryst. Growth, 2003, 247, 505–508 CrossRef CAS.
- Q. Lv, C. B. Cao, C. Li, J. T. Zhang, H. X. Zhu, X. Kong and X. F. Duan, J. Mater. Chem., 2003, 13, 1241–1243 RSC.
- Q. X. Guo, Y. Xie, X. J. Wang, S. C. Lv, T. Hou and X. M. Liu, Chem. Phys. Lett., 2003, 380, 84–87 CrossRef CAS.
- Q. Luv, C. B. Cao, J. T. Zhang, C. Li and H. S. Zhu, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 633–636 CrossRef CAS.
- Y. L. Gu, L. Y. Chen, L. Shi, J. H. Ma, Z. H. Yang and Y. T. Qian, Carbon, 2003, 41, 2674–2676 CrossRef CAS.
- C. Li, X. Yang, B. Yang, Y. Yan and Y. Qian, Mater. Chem. Phys., 2007, 103, 427–432 CrossRef CAS.
- J. Zhang, W. Liu, X. Li, B. Zhan, Q. Cui and G. Zou, Mater. Res. Bull., 2009, 44, 294–297 CrossRef CAS.
- P. Xia, M. Antonietti, B. Zhu, T. Heil, J. Yu and S. Cao, Adv. Funct. Mater., 2019, 29, 1900093 CrossRef.
- Y.-P. Yuan, L.-S. Yin, S.-W. Cao, L.-N. Gu, G.-S. Xu, P. Du, H. Chai, Y.-S. Liao and C. Xue, Green Chem., 2014, 16, 4663–4668 RSC.
- Y. Guo, J. Li, Y. Yuan, L. Li, M. Zhang, C. Zhou and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 14693–14697 CrossRef CAS PubMed.
- L. W. Yin, Y. Bando, M. S. Li, Y. X. Liu and Y. X. Qi, Adv. Mater., 2003, 15, 1840–1844 CrossRef CAS.
- X.-H. Li, J. Zhang, X. Chen, A. Fischer, A. Thomas, M. Antonietti and X. Wang, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS.
- J. Wang, Y. Shen, Y. Li, S. Liu and Y. Zhang, Chem. – Eur. J., 2016, 22, 12449–12454 CrossRef CAS PubMed.
- S. Kang, L. Zhang, M. He, Y. Zheng, L. Cui, D. Sun and B. Hu, Carbon, 2018, 137, 19–30 CrossRef CAS.
- C. Qiu, Y. Xu, X. Fan, D. Xu, R. Tandiana, X. Ling, Y. Jiang, C. Liu, L. Yu, W. Chen and C. Su, Adv. Sci., 2019, 6, 1801403 CrossRef PubMed.
- Y. Xu, X. He, H. Zhong, D. J. Singh, L. Zhang and R. Wang, Appl. Catal., B, 2019, 246, 349–355 CrossRef CAS.
- F. S. Guo, B. Hu, C. Yang, J. S. Zhang, Y. D. Hou and X. C. Wang, Adv. Mater., 2021, 33, 2101466 CrossRef CAS PubMed.
- Y.-Y. Li, B.-X. Zhou, H.-W. Zhang, S.-F. Ma, W.-Q. Huang, W. Peng, W. Hu and G.-F. Huang, Nanoscale, 2019, 11, 6876–6885 RSC.
- J. Zhou, Y. Yang and C. Y. Zhang, Chem. Commun., 2013, 49, 8605–8607 RSC.
- Z. Xing, K. T. Dong, N. Pavlopoulos, Y. X. Chen and L. Amirav, Angew. Chem., Int. Ed., 2021, 60, 19413–19418 CrossRef CAS PubMed.
- H. Schlomberg, J. Kroger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CrossRef CAS PubMed.
- M. Pauly, J. Kroger, V. Duppel, C. Murphey, J. Cahoon, B. V. Lotsch and P. A. Maggard, Chem. Sci., 2022, 13, 3187–3193 RSC.
- Y. Lin, M. Zhou, X. Tai, H. Li, X. Han and J. Yu, Matter, 2021, 4, 2309–2339 CrossRef CAS.
- M. Ma, X. Zhang, L. Xu, X. Chen, L. Wang, T. Cheng, F. Wei, J. Yuan and B. Shen, Adv. Mater., 2023, 35, 2300653 CrossRef CAS PubMed.
- H. Xiong, H. Wang, X. Chen and F. Wei, ACS Catal., 2023, 13, 12213–12226 CrossRef CAS.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- W. Wang, J. Cui, Z. Z. Sun, L. Xie, X. K. Mu, L. M. Huang and J. Q. He, Adv. Mater., 2021, 33, 2106359 CrossRef CAS PubMed.
- H. Y. Chi, C. L. Chen, K. N. Zhao, L. F. Villalobos, P. A. Schouwink, L. Piveteau, K. P. Marshall, Q. Liu, Y. Han and K. V. Agrawal, Angew. Chem., Int. Ed., 2022, 61, e202207457 CrossRef CAS PubMed.
- T. S. Miller, T. M. Suter, A. M. Telford, L. Picco, O. D. Payton, F. Russell-Pavier, P. L. Cullen, A. Sella, M. S. P. Shaffer, J. Nelson, V. Tileli, P. F. McMillan and C. A. Howard, Nano Lett., 2017, 17, 5891–5896 CrossRef CAS PubMed.
- J. Jia, E. R. White, A. J. Clancy, N. Rubio, T. Suter, T. S. Miller, K. McColl, P. F. McMillan, V. Brazdova, F. Cora, C. A. Howard, R. V. Law, C. Mattevi and M. S. P. Shaffer, Angew. Chem., Int. Ed., 2018, 57, 12656–12660 CrossRef CAS PubMed.
- L. F. Villalobos, M. T. Vandat, M. Khchoune, Z. Nadizadeh, M. Mensi, E. Oveisi, D. Campi, N. Marzari and K. V. Agrawal, Sci. Adv., 2020, 6, eaay9851 CrossRef CAS PubMed.
- H. Ou, L. Lin, Y. Zheng, P. Yang, Y. Fang and X. Wang, Adv. Mater., 2017, 29, 1700008 CrossRef PubMed.
- W. Iqbal, B. Qiu, Q. Zhu, M. Xing and J. Zhang, Appl. Catal., B, 2018, 232, 306–313 CrossRef CAS.
- Y. Wang, X. Hou, J. Zhang, T. Xu, S. Liu and B. Liu, ChemPhotoChem, 2018, 2, 490–497 CrossRef CAS.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- X. Zhou, Y. Shi, W. Xu, Y. Wang, Y. Zhang, Y. Wang, Y. Wu, N. Wu, Y. Sun, Y. Du and W. Zhong, J. Alloys Compd., 2020, 827, 154307 CrossRef CAS.
- W. Xing, W. Tu, Z. Han, Y. Hu, Q. Meng and G. Chen, ACS Energy Lett., 2018, 3, 514–519 CrossRef CAS.
- M. K. Bhunia, S. Melissen, M. R. Parida, P. Sarawade, J.-M. Basset, D. H. Anjum, O. F. Mohammed, P. Sautet, T. Le Bahers and K. Takanabe, Chem. Mater., 2015, 27, 8237–8247 CrossRef CAS.
- X. Wu, H. Ma, W. Zhong, J. Fan and H. Yu, Appl. Catal., B, 2020, 271, 118899 CrossRef CAS.
- H. Yu, H. Ma, X. Wu, X. Wang, J. Fan and J. Yu, Sol. RRL, 2020, 5, 2000372 CrossRef.
- Z. Zeng, X. Quan, H. Yu, S. Chen and S. Zhang, J. Catal., 2019, 375, 361–370 CrossRef CAS.
- W. Wang, Z. Shu, J. Zhou, D. Meng, Z. Zhao and T. Li, J. Mater. Chem. A, 2020, 8, 6785–6794 RSC.
- Q. Liu, X. Wang, Q. Yang, Z. Zhang and X. Fang, Appl. Catal., B, 2018, 225, 22–29 CrossRef CAS.
- X. Chen, R. Shi, Q. Chen, Z. Zhang, W. Jiang, Y. Zhu and T. Zhang, Nano Energy, 2019, 59, 644–650 CrossRef CAS.
- Y. Li, D. Zhang, J. Fan and Q. Xiang, Chin. J. Catal., 2021, 42, 627–636 CrossRef CAS.
- Q. Liang, Z. Li, Z.-H. Huang, F. Kang and Q.-H. Yang, Adv. Funct. Mater., 2015, 25, 6885–6892 CrossRef CAS.
- X. Xiao, Y. Gao, L. Zhang, J. Zhang, Q. Zhang, Q. Li, H. Bao, J. Zhou, S. Miao, N. Chen, J. Wang, B. Jiang, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2003082 CrossRef CAS PubMed.
- H. Wang, Y. Bian, J. Hu and L. Dai, Appl. Catal., B, 2018, 238, 592–598 CrossRef CAS.
- B. Y. Zhai, H. G. Li, G. Y. Gao, Y. Wang, P. Niu, S. L. Wang and L. Li, Adv. Funct. Mater., 2022, 32, 2207375 CrossRef CAS.
- Y. Wang, X. Zhou, W. Xu, Y. Sun, T. Wang, Y. Zhang, J. Dong, W. Hou, N. Wu, L. Wu, B. Zhou, Y. Wu, Y. Du and W. Zhong, Appl. Catal., A, 2019, 582, 117118 CrossRef CAS.
- G. Q. Zhang, Y. S. Xu, M. Rauf, J. Y. Zhu, Y. L. Li, C. X. He, X. Z. Ren, P. X. Zhang and H. W. Mi, Adv. Sci., 2022, 9, 2201677 CrossRef CAS PubMed.
- Y. Xu, M. Fan, W. Yang, Y. Xiao, L. Zeng, X. Wu, Q. Xu, C. Su and Q. He, Adv. Mater., 2021, 33, 2101455 CrossRef CAS PubMed.
- Z. Liu, J. Ma, M. Hong and R. Sun, ACS Catal., 2023, 13, 2106–2117 CrossRef CAS.
- W. Ren, J. Cheng, H. Ou, C. Huang, M.-M. Titirici and X. Wang, ChemSusChem, 2019, 12, 3257–3262 CrossRef CAS PubMed.
- J. Yuan, Y. Tang, X. Yi, C. Liu, C. Li, Y. Zeng and S. Luo, Appl. Catal., B, 2019, 251, 206–212 CrossRef CAS.
- Y. Ding, S. Maitra, C. Wang, S. Halder, R. Zheng, T. Barakat, S. Roy, L.-H. Chen and B.-L. Su, Interdiscip. Mater., 2022, 1, 213–255 CrossRef.
- S. Wan, J. Xu, S. Cao and J. Yu, Interdiscip. Mater., 2022, 1, 294–308 CrossRef.
- F. X. Yin, P. Q. Qin, J. S. Xu and S. W. Cao, Acta Phys. -Chim. Sin., 2023, 39, 2212062 Search PubMed.
- M. Chang, Z. Pan, D. Zheng, S. Wang, G. Zhang, M. Anpo and X. Wang, ChemSusChem, 2023, 16, e202202255 CrossRef CAS PubMed.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- G. Zhang, G. Li, Z.-A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- G. Zhang, M. Liu, T. Heil, S. Zafeiratos, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 14950–14954 CrossRef CAS PubMed.
- J. Kröger, A. Jiménez-Solano, G. Savasci, P. Rovó, I. Moudrakovski, K. Küster, H. Schlomberg, H. A. Vignolo-González, V. Duppel, L. Grunenberg, C. B. Dayan, M. Sitti, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2021, 11, 2003016 CrossRef.
- X. Liu, B. Jing, G. Lun, Y. Wang, X. Wang, C. Fang, Z. Ao and C. Li, Chem. Commun., 2020, 56, 3179–3182 RSC.
- S. Bai, L. Wang, Z. Li and Y. Xiong, Adv. Sci., 2017, 4, 1600216 CrossRef PubMed.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- X. Liang, S. Xue, C. Yang, X. Ye, Y. Wang, Q. Chen, W. Lin, Y. Hou, G. Zhang, M. Shalom, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216434 CrossRef CAS PubMed.
- T. Kawahara, Y. Konishi, H. Tada, N. Tohge, J. Nishii and S. Ito, Angew. Chem., Int. Ed., 2002, 41, 2811–2813 CrossRef CAS PubMed.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, R.-Q. Sun and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145–10149 CrossRef CAS PubMed.
- Q. Liang, Z. Li, Y. Bai, Z.-H. Huang, F. Kang and Q.-H. Yang, Small, 2017, 13, 1603182 CrossRef PubMed.
- Z. Liu, G. Wang, H.-S. Chen and P. Yang, Chem. Commun., 2018, 54, 4720–4723 RSC.
- Z. Jiang, X. Zhang, J. Wang, L. Chen, H.-S. Chen and P. Yang, Chem. Commun., 2018, 54, 13519–13522 RSC.
- M. Z. Rahman, J. Moffatt and N. Spooner, Mater. Horiz., 2018, 5, 553–559 RSC.
- X. Zhang, J.-P. Veder, S. He and S. P. Jiang, Chem. Commun., 2019, 55, 1233–1236 RSC.
- H. Liu, D. Chen, Z. Wang, H. Jing and R. Zhang, Appl. Catal., B, 2017, 203, 300–313 CrossRef CAS.
- A. Jin, Y. Jia, C. Chen, X. Liu, J. Jiang, X. Chen and F. Zhang, J. Phys. Chem. C, 2017, 121, 21497–21509 CrossRef CAS.
- Z. Zeng, H. Yu, X. Quan, S. Chen and S. Zhang, Appl. Catal., B, 2018, 227, 153–160 CrossRef CAS.
- G. Zhang, L. Lin, G. Li, Y. Zhang, A. Savateev, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 9372–9376 CrossRef CAS PubMed.
- A. Jin, X. Liu, M. Li, Y. Jia, C. Chen and X. Chen, ACS Sustainable Chem. Eng., 2019, 7, 5122–5133 CrossRef CAS.
- Y. Li, F. Gong, Q. Zhou, X. Feng, J. Fan and Q. Xiang, Appl. Catal., B, 2020, 268, 118381 CrossRef CAS.
- J. Zhang, X. C. Liang, C. Zhang, L. H. Lin, W. D. Xing, Z. Y. Yu, G. G. Zhang and X. C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202210849 CrossRef CAS PubMed.
- R. Paul, Q. Zhai, A. K. Roy and L. Dai, Interdiscip. Mater., 2022, 1, 28–50 CrossRef.
- H. Zhang, F. Liu, Z. Mou, X. Liu, J. Sun and W. Lei, Chem. Commun., 2016, 52, 13020–13023 RSC.
- H. Zhang, Y. Cao, L. Zhong, X. Cao, J. He, J. Sun and W. Lei, Appl. Surf. Sci., 2019, 485, 361–367 CrossRef CAS.
- M. Wang, S. Huang, X. Pang, M. Song, C. Du and Y. Su, Sustainable Energy Fuels, 2019, 3, 3422–3429 RSC.
- X. Yan, G. Ning and P. Zhao, Catalysts, 2019, 9, 55 CrossRef.
- X. Zhang, S. He and S. P. Jiang, Carbon, 2020, 156, 488–498 CrossRef CAS.
- G. Chen, Z. Zhou, B. Li, X. Lin, C. Yang, Y. Fang, W. Lin, Y. Hou, G. Zhang and S. Wang, J. Environ. Sci., 2023 DOI:10.1016/j.jes.2023.05.028.
- G. Chen, F. Wei, Z. Zhou, B. Su, C. Yang, X. F. Lu, S. Wang and X. Wang, Sustainable Energy Fuels, 2023, 7, 381–388 RSC.
- N. A. Rodriguez, A. Savateev, M. A. Grela and D. Dontsova, ACS Appl. Mater. Interfaces, 2017, 9, 22941–22949 CrossRef CAS PubMed.
- F. Guo, X. Huang, Z. Chen, H. Sun and L. Chen, Chem. Eng. J., 2020, 395, 125118 CrossRef CAS.
- M. Wang, Z. Zhang, Z. Chi, L. L. Lou, H. Li, H. Yu, T. Ma, K. Yu and H. Wang, Adv. Funct. Mater., 2023, 33, 2211565 CrossRef CAS.
- L. Cheng, X. Y. Yue, J. J. Fan and Q. J. Xiang, Adv. Mater., 2021, 34, 2200929 CrossRef PubMed.
- Y. Xia, Z. Tian, T. Heil, A. Meng, B. Cheng, S. Cao, J. Yu and M. Antonietti, Joule, 2019, 3, 2792–2805 CrossRef CAS.
- F. Yang, D. Deng, X. Pan, Q. Fu and X. Bao, Natl. Sci. Rev., 2015, 2, 183–201 CrossRef CAS.
- S. Cao, F. Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747–4765 RSC.
- G. Vile, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. Lopez and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef CAS PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- S. Cao, H. Li, T. Tong, H.-C. Chen, A. Yu, J. Yu and H. M. Chen, Adv. Funct. Mater., 2018, 28, 1802169 CrossRef.
- L. Zhang, R. Long, Y. Zhang, D. Duan, Y. Xiong, Y. Zhang and Y. Bi, Angew. Chem., Int. Ed., 2020, 59, 6224–6229 CrossRef CAS PubMed.
- J. Wang, T. Heil, B. Zhu, C.-W. Tung, J. Yu, H. M. Chen, M. Antonietti and S. Cao, ACS Nano, 2020, 14, 8584–8593 CrossRef CAS PubMed.
- H. Li, B. Zhu, B. Cheng, G. Luo, J. Xu and S. Cao, J. Mater. Sci. Technol., 2023, 161, 192–200 CrossRef.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- L. Lin, C. Wang, W. Ren, H. Ou, Y. Zhang and X. Wang, Chem. Sci., 2017, 8, 5506–5511 RSC.
- Y. F. Shao, X. Q. Hao, S. D. Lu and Z. L. Jin, Chem. Eng. J., 2023, 454, 140123 CrossRef CAS.
- M. Liu, G. Zhang, X. Liang, Z. Pan, D. Zheng, S. Wang, Z. Yu, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304694 CrossRef CAS PubMed.
- Q. Wang, G. Zhang, W. Xing, Z. Pan, D. Zheng, S. Wang, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307930 CrossRef CAS PubMed.
- M. Liu, C. Wei, H. Zhuzhang, J. Zhou, Z. Pan, W. Lin, Z. Yu, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113389 CrossRef CAS PubMed.
- D. Dontsova, S. Pronkin, M. Wehle, Z. Chen, C. Fettkenhauer, G. Clavel and M. Antonietti, Chem. Mater., 2015, 27, 5170–5179 CrossRef CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Cora, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CrossRef CAS.
- Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 29, 1700555 CrossRef PubMed.
- J. Kroger, A. Jimenez-Solano, G. Savasci, V. W. H. Lau, V. Duppel, I. Moudrakovski, K. Kuster, T. Scholz, A. Gouder, M. L. Schreiber, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Adv. Funct. Mater., 2021, 31, 2102468 CrossRef.
- J. Kroger, F. Podjaski, G. Savasci, I. Moudrakovski, A. Jimenez-Solano, M. W. Terban, S. Bette, V. Duppel, M. Joos, A. Senocrate, R. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Adv. Mater., 2022, 34, 2107061 CrossRef CAS PubMed.
- Y. Li, D. N. Zhang, J. J. Fan and Q. J. Xiang, Chin. J. Catal., 2021, 42, 627–636 CrossRef CAS.
- D. B. Nimbalkar, V. Nguyen, C. Y. Shih and H. S. Teng, Appl. Catal., B, 2022, 316, 121601 CrossRef CAS.
- Z. M. Pan, M. Zhao, H. Y. Zhuzhang, G. G. Zhang, M. Anpo and X. C. Wang, ACS Catal., 2021, 11, 13463–13471 CrossRef CAS.
- D. Dontsova, C. Fettkenhauer, V. Papaefthimiou, J. Schmid and M. Antonietti, Chem. Mater., 2016, 28, 772–778 CrossRef CAS.
- A. Savateev, S. Pronkin, J. D. Epping, M. G. Willinger, C. Wolff, D. Neher, M. Antonietti and D. Dontsova, ChemCatChem, 2017, 9, 167–174 CrossRef CAS.
- A. Savateev, S. Pronkin, M. G. Willinger, M. Antonietti and D. Dontsova, Chem. – Asian J., 2017, 12, 1517–1522 CrossRef CAS PubMed.
- G. Zhang, G. Li, T. Heil, S. Zafeiratos, F. Lai, A. Savateev, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS PubMed.
- L. Lin, W. Ren, C. Wang, A. M. Asiri, J. Zhang and X. Wang, Appl. Catal., B, 2018, 231, 234–241 CrossRef CAS.
- R. Lei, B. Du, X. Lai, J. Wu, Z. Zhang, S. Liu, R. Wu, X. Li, B. Song and J. Jian, J. Mater. Chem. A, 2019, 7, 13234–13241 RSC.
- Y. Dai, Q. Bu, R. Sooriyagoda, P. Tavadze, O. Pavlic, T. Lim, Y. Shen, A. Mamakhel, X. Wang, Y. Li, H. Niemantsverdriet, B. B. Iversen, F. Besenbacher, T. Xie, J. P. Lewis, A. D. Bristow, N. Lock and R. Su, J. Phys. Chem. Lett., 2019, 10, 5381–5386 CrossRef CAS PubMed.
- L. Cui, X. Hou, H. Du and Y. Yuan, Appl. Surf. Sci., 2020, 501, 144259 CrossRef CAS.
- V. V. Shvalagin, G. V. Korzhak, S. Y. Kuchmiy, M. A. Skoryk, O. V. Selyshchev and D. R. T. Zahn, J. Photochem. Photobiol., A, 2020, 390, 112295 CrossRef CAS.
- Y. Li, D. Zhang, X. Feng and Q. Xiang, Chin. J. Catal., 2020, 41, 21–30 CrossRef CAS.
- Y. Xu, C. Qiu, X. Fan, Y. Xiao, G. Zhang, K. Yu, H. Ju, X. Ling, Y. Zhu and C. Su, Appl. Catal., B, 2020, 268, 118457 CrossRef CAS.
- L. Wang, Y. Hong, E. Liu, Z. Wang, J. Chen, S. Yang, J. Wang, X. Lin and J. Shi, Int. J. Hydrogen Energy, 2020, 45, 6425–6436 CrossRef CAS.
- P. Deng, L. Shi, H. Wang and W. Qi, Colloids Surf., A, 2020, 601, 125023 CrossRef CAS.
- G. Q. Zhang, Y. S. Xu, C. X. He, P. X. Zhang and H. W. Mi, Appl. Catal., B, 2021, 283, 119636 CrossRef CAS.
- G. Q. Zhang, Y. S. Xu, D. F. Yan, C. A. X. He, Y. L. Li, X. Z. Ren, P. X. Zhang and H. W. Mi, ACS Catal., 2021, 11, 6995–7005 CrossRef CAS.
- W. B. Wang, Z. Shu, Z. H. Liao, J. Zhou, D. W. Meng, T. T. Li, Z. L. Zhao and L. Xu, Chem. Eng. J., 2021, 424, 130332 CrossRef CAS.
- H. N. Che, C. M. Li, C. X. Li, C. B. Liu, H. J. Dong and X. H. Song, Chem. Eng. J., 2021, 410, 127791 CrossRef CAS.
- Z. Z. Sun, H. Z. Dong, Q. Yuan, Y. Y. Tan, W. Wang, Y. B. Jiang, J. Y. Wan, J. W. Wen, J. J. Yang, J. Q. He, T. Cheng and L. M. Huang, Chem. Eng. J., 2022, 435, 134865 CrossRef CAS.
- Z. H. Yu, X. Y. Yue, J. J. Fan and Q. J. Xiang, ACS Catal., 2022, 12, 6345–6358 CrossRef CAS.
- G. Q. Zhang, J. Y. Zhu, Y. S. Xu, C. Yang, C. X. He, P. X. Zhang, Y. L. Li, X. Z. Ren and H. W. Mi, ACS Catal., 2022, 12, 4648–4658 CrossRef CAS.
- Y. J. Wang, D. J. Xie, G. Wang, Y. S. Wu, R. Shi, C. Zhou, X. F. Meng and T. R. Zhang, Nano Energy, 2022, 104, 107938 CrossRef CAS.
- F. Lin, S. Zhou, G. H. Wang, J. Wang, T. Y. Gao, Y. R. Su and C. P. Wong, Nano Energy, 2022, 99, 107432 CrossRef CAS.
- S. H. Shen, J. Chen, Y. Q. Wang, C. L. Dong, F. Q. Meng, Q. H. Zhang, Y. L. Huangfu, Z. Lin, Y. C. Huang, Y. R. Li, M. T. Li and L. Gu, Sci. Bull., 2022, 67, 520–528 CrossRef CAS PubMed.
- S. F. An, Y. K. Guo, X. Y. He, P. Gao, G. J. Hou, J. G. Hou, C. S. Song and X. W. Guo, Appl. Catal., B, 2022, 310, 121323 CrossRef CAS.
- T. Y. Yang, Y. Y. Shao, J. D. Hu, J. F. Qu, X. G. Yang, F. Y. Yang and C. M. Li, Chem. Eng. J., 2022, 448, 137613 CrossRef CAS.
- X. Li, J. Wen, J. Low, Y. Fang and J. Yu, Sci. China Mater., 2014, 57, 70–100 CrossRef.
- H. Li, B. Zhu, S. Cao and J. Yu, Chem. Commun., 2020, 54, 5641–5644 RSC.
- Y. Liang, X. Wu, X. Liu, C. Li and S. Liu, Appl. Catal., B, 2022, 304, 120978 CrossRef CAS.
- H. Li, F. Li, J. Yu and S. Cao, Acta Phys. -Chim. Sin., 2021, 37, 2010073 Search PubMed.
- L. Cheng, P. Zhang, Q. Y. Wen, J. J. Fan and Q. J. Xiang, Chin. J. Catal., 2022, 43, 451–460 CrossRef CAS.
- J. Wu, X. Ji, X. Yuan, Z. Zhao, Y. Li, B. Wen, H. Zhang, D. Yu, Y. Zhao and Y. Tian, Chem. Mater., 2019, 31, 9188–9199 CrossRef CAS.
- L. Tian, J. Li, F. Liang, J. Wang, S. Li, H. Zhang and S. Zhang, Appl. Catal., B, 2018, 225, 307–313 CrossRef CAS.
- Y. Yu and J. Wang, Ceram. Int., 2016, 42, 4063–4071 CrossRef CAS.
- Q. Liang, Z.-H. Huang, F. Kang and Q.-H. Yang, ChemCatChem, 2015, 7, 2897–2902 CrossRef CAS.
- C. Fettkenhauer, X. Wang, K. Kailasam, M. Antonietti and D. Dontsova, J. Mater. Chem. A, 2015, 3, 21227–21232 RSC.
- L. H. Amorin, V. Y. Suzuki, N. H. de Paula, J. L. Duarte, M. A. Toledo da Silva, C. A. Taft and F. D. A. La Porta, New J. Chem., 2019, 43, 13647–13653 RSC.
- Y. Duan, J. Li, X. Shang, D. Jia, C. Li and S. Liu, J. Mater. Sci., 2020, 55, 13675–13686 CrossRef CAS.
- Y. Wang, B. Jing, F. Wang, S. Wang, X. Liu, Z. Ao and C. Li, Water Res., 2020, 180, 115925 CrossRef CAS PubMed.
- V. Vinoth Kumar, D. Avisar, V. Lakshmi Prasanna, Y. Betzalel and H. Mamane, J. Hazard. Mater., 2020, 398, 122880 CrossRef PubMed.
- J. J. Wang, P. Li, Y. Wang, Z. Y. Liu, D. Q. Wang, J. J. Liang and Q. H. Fan, Adv. Sci., 2023, 10, 2205542 CrossRef CAS PubMed.
- Z. Y. Gu, Z. T. Cui, Z. J. Wang, T. R. Chen, P. Sun and D. W. Wen, J. Mater. Sci. Technol., 2021, 83, 113–122 CrossRef CAS.
- M. Zhou, L. B. Zeng, R. Li, C. Yang, X. Qin, W. K. Ho and X. C. Wang, Appl. Catal., B, 2022, 317, 121719 CrossRef CAS.
- Y. Zhang, L. Hu, C. Zhu, J. Liu, H. Huang, Y. Liu and Z. Kang, Catal. Sci. Technol., 2016, 6, 7252–7258 RSC.
- M. Zhou, P. Yang, R. Yuan, A. M. Asiri, M. Wakeel and X. Wang, ChemSusChem, 2017, 10, 4451–4456 CrossRef CAS PubMed.
- B. Pieber, J. A. Malik, C. Cavedon, S. Gisbertz, A. Savateev, D. Cruz, T. Heil, G. Zhang and P. H. Seeberger, Angew. Chem., Int. Ed., 2019, 58, 9575–9580 CrossRef CAS PubMed.
- C. Cavedon, A. Madani, P. H. Seeberger and B. Pieber, Org. Lett., 2019, 21, 5331–5334 CrossRef CAS PubMed.
- J. Wang, H. Zhao, B. C. Zhu, S. Larter, S. W. Cao, J. G. Yu, M. G. Kibria and J. G. Hu, ACS Catal., 2021, 11, 12170–12178 CrossRef CAS.
- M. A. R. da Silva, I. F. Silva, Q. Xue, B. T. W. Lo, N. V. Tarakina, B. N. Nunes, P. Adler, S. K. Sahoo, D. W. Bahnemann, N. Lopez-Salas, A. Savateev, C. Ribeiro, T. D. Kuhne, M. Antonietti and I. F. Teixeira, Appl. Catal., B, 2022, 304, 120965 CrossRef CAS.
- A. Savateev, D. Dontsova, B. Kurpil and M. Antonietti, J. Catal., 2017, 350, 203–211 CrossRef CAS.
- B. Kurpil, K. Otte, M. Antonietti and A. Savateev, Appl. Catal., B, 2018, 228, 97–102 CrossRef CAS.
- B. Kurpil, B. Kumru, T. Heil, M. Antonietti and A. Savateev, Green Chem., 2018, 20, 838–842 RSC.
- A. Savateev, B. Kurpil, A. Mishchenko, G. Zhang and M. Antonietti, Chem. Sci., 2018, 9, 3584–3591 RSC.
- B. Kurpil, Y. Markushyna and A. Savateev, ACS Catal., 2019, 9, 1531–1538 CrossRef CAS.
- Y. Markushyna, C. Teutloff, B. Kurpil, D. Cruz, I. Lauermann, Y. Zhao, M. Antonietti and A. Savateev, Appl. Catal., B, 2019, 248, 211–217 CrossRef CAS.
- B. Kurpil, K. Otte, A. Mishchenko, P. Lamagni, W. Lipinski, N. Lock, M. Antonietti and A. Savateev, Nat. Commun., 2019, 10, 945 CrossRef PubMed.
- A. Savateev, N. V. Tarakina, V. Strauss, T. Hussain, K. ten Brummelhuis, J. M. Sanchez Vadillo, Y. Markushyna, S. Mazzanti, A. P. Tyutyunnik, R. Walczak, M. Oschatz, D. M. Guldi, A. Karton and M. Antonietti, Angew. Chem., Int. Ed., 2020, 59, 15061–15068 CrossRef CAS PubMed.
- Y. Markushyna, C. M. Schusslbauer, T. Ullrich, D. M. Guldi, M. Antonietti and A. Savateev, Angew. Chem., Int. Ed., 2021, 60, 20543–20550 CrossRef CAS PubMed.
- C. Yang, S. Wan, B. Zhu, J. Yu and S. Cao, Angew. Chem., Int. Ed., 2022, 61, e202208438 CrossRef CAS PubMed.
- J. Cheng, S. Wan and S. Cao, Angew. Chem., Int. Ed., 2023, 62, e202310476 CrossRef CAS PubMed.
- G. G. Kramarenko, S. G. Hummel, S. M. Martin and G. R. Buettner, Photochem. Photobiol., 2006, 82, 1634–1637 CrossRef CAS PubMed.
- H. Mattila, S. Khorobrykh, V. Havurinne and E. Tyystjärvi, J. Photochem. Photobiol., B, 2015, 152, 176–214 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- S. Zhao and X. Zhao, Appl. Catal., B, 2019, 250, 408–418 CrossRef CAS.
- H. Cheng, J. Cheng, L. Wang and H. X. Xu, Chem. Mater., 2022, 34, 4259–4273 CrossRef CAS.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Small, 2022, 18, 2104561 CrossRef CAS PubMed.
- J. D. Hu, C. Chen, H. B. Yang, F. Y. Yang, J. F. Qu, X. G. Yang, W. Sun, L. M. Dai and C. M. Li, Appl. Catal., B, 2022, 317, 121723 CrossRef CAS.
- H. Guo, C. G. Niu, C. Liang, H. Y. Niu, Y. Y. Yang, H. Y. Liu, N. Tang and H. X. Fang, Chem. Eng. J., 2021, 409, 128030 CrossRef CAS.
- P. Sharma, T. J. A. Slater, M. Sharma, M. Bowker and C. R. A. Catlow, Chem. Mater., 2022, 34, 5511–5521 CrossRef CAS PubMed.
- L.-L. Liu, F. Chen, J.-H. Wu, J.-J. Chen and H.-Q. Yu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2215305120 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |