
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction microenvironment control in membrane electrode assemblies for CO2 electrolysis
Chuanchuan
Yan
ab,
Dunfeng
Gao
 *a,
Juan-Jesús
Velasco-Vélez
c and
Guoxiong
Wang
*a
*a,
Juan-Jesús
Velasco-Vélez
c and
Guoxiong
Wang
*a
aState Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: dfgao@dicp.ac.cn; wanggx@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cALBA Synchrotron Light Source, Cerdanyola del Vallés, Barcelona 08290, Spain
First published on 13th September 2023
Abstract
CO2 electrolysis is an emerging and promising carbon neutrality technology, but currently suffers from challenging selectivity issues at industrially relevant reaction rates. Selectivity control in CO2 electrolysis relies on the molecular understanding and manipulation of multiple parallel reaction pathways that are equally governed by catalytically active sites and the reaction microenvironments in their vicinity. In this perspective, we summarize and discuss the latest achievements in reaction microenvironment control for active, selective, energy- and carbon-efficient CO2 electrolysis, with particular attention being paid to that in membrane electrode assembly electrolyzers operating at industrial current densities (≥200 mA cm−2). The effects and underlying catalytic mechanisms of reaction microenvironments tailored by functional organic molecules/polymers and reactant feed compositions on the activity and selectivity of CO2 electrolysis are discussed using selected examples. The efforts made to tailor acidic reaction microenvironments by controlling the transport of reactive species for carbon-efficient CO2 electrolysis are also exemplified. Finally, we illustrate current challenges and future opportunities in the mechanistic understanding and rational design of reaction microenvironments for improving CO2 electrolysis performance.
Broader contextIn the context of carbon neutrality, CO2 electrolysis has recently become a hot topic in both academia and industry, as it can electrochemically convert CO2 to valuable chemicals and fuels under ambient conditions, with water and green electricity derived from renewable energy sources. However, CO2 electrolysis suffers from complex reaction pathways and selectivity issues, especially at industrially relevant reaction rates. Rather than focusing on rational design of efficient catalytic materials, manipulating reaction microenvironments in the vicinity of catalytically active sites has been increasingly recognized as equally important as catalysts in tuning activity and selectivity of CO2 electrolysis. The reaction microenvironments are closely associated with local concentrations of reactants (e.g., CO2 and H2O) and reaction intermediates (e.g., *CO, *H, and *OCCOH) as well as other reactive species (e.g., H+, OH−, carbonate, bicarbonate, cations, and anions) at the electrode–electrolyte interface. This perspective summarizes and discusses the latest achievements in reaction microenvironment control for selective, energy- and carbon-efficient CO2 electrolysis, with a special emphasis on that in membrane electrode assembly electrolyzers operating at industrial current densities. Strategies for effective reaction microenvironment control include molecular modification with organic molecules and polymers (ionomers), adjusting reactant feed composition, as well as tailoring acidic microenvironments. |
1. Background
CO2 electrolysis powered by renewable energy is one of the emerging technologies towards carbon neutrality. It provides a green and sustainable route that converts CO2, water, and electricity to valuable chemicals and fuels such as CO, formate, methanol, and methane, as well as more valuable multicarbon (C2+) products like ethylene, acetate, ethanol, and propanol.1–3 As a complex electrocatalytic reaction involving carbon, hydrogen, and oxygen elements, CO2 electrolysis suffers from many challenges in selectivity control owing to a very broad product distribution of CO2 electroreduction, especially at industrially relevant reaction rates. The strong competition of hydrogen evolution from water electrolysis further adds complexity in controlling selectivity towards specific products. Directing CO2 electrolysis pathways on demand is of remarkable importance for highly efficient electrocatalytic CO2 conversion.Since Hori's seminal works in the 1980s,4,5 most studies on improving CO2 electrolysis performance have been devoted to the rational design and precise preparation of highly efficient catalytic materials.6,7 On the other hand, similar to the surrounding environments of metal centers in enzyme catalysis, reaction microenvironments in the vicinity of catalytically active sites have been increasingly recognized as equally important as active sites for tuning activity and selectivity of heterogeneous catalytic reactions including CO2 electrolysis.8–11 The reaction microenvironments can modulate specific transition states and control the transport of reactive species to and from catalytically active sites, via multiple physicochemical effects.9 Rationally manipulating reaction microenvironments offers an alternative strategy to break the linear scaling relationships that are widely present in heterogeneous electrocatalysis over bare metal surfaces.12 For CO2 electrolysis, the reaction microenvironments are closely associated with local concentrations of reactive species including reactants (e.g., CO2, H2O), reaction intermediates (e.g., *CO, *H, and *OCCOH) as well as other key species (e.g., H+, OH−, carbonate, bicarbonate, cations, and anions) at the electrode–electrolyte interface.13–18 Such properties are also relevant to the configurations of electrodes and electrolyzers used for CO2 electrolysis.19,20 A zero-gap membrane electrode assembly (MEA) electrolyzer with gas diffusion electrodes (GDEs) and solid polymer electrolytes has been recently recognized as the most advanced device towards practical application in modern electrochemistry, using which high current density and high energy efficiency can be simultaneously achieved.21–23 In an MEA electrolyzer, liquid electrolytes or pure water and the CO2 gas are fed to the anode and the cathode, respectively. CO2 electrolysis proceeds at the cathode in the presence of reactive species (e.g., water and cations) transported from the anode through an ion exchange membrane. Compared to the liquid–solid interfaces in traditional electrochemistry, the gas–liquid–solid three-phase electrochemical interfaces in MEA electrolyzers become much more complicated, resulting in notably distinct reaction microenvironments. In this perspective, we discuss recent advances in the reaction microenvironment control for CO2 electrolysis, with a special emphasis on that in MEA electrolyzers operating at industrial current densities that should be ≥200 mA cm−2 in order to minimize the capital-expenditure of an electrolyzer to economically compelling levels.24 Tailored reaction microenvironments created by organic molecules and polymers as well as reactant feed compositions for active and selective CO2 electrolysis, as well as acidic microenvironments for carbon-efficient CO2 electrolysis will be exemplified using selected examples (Fig. 1).
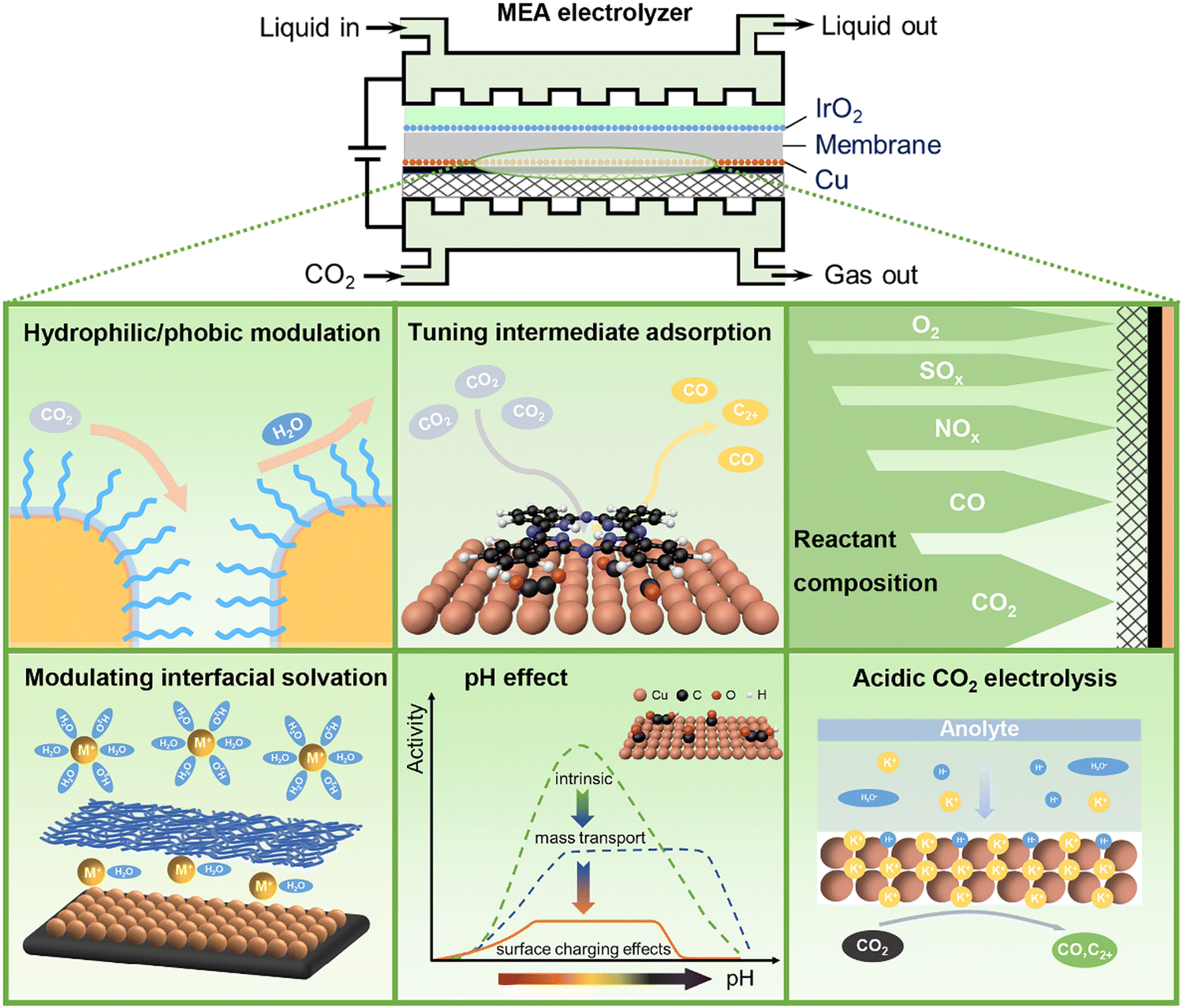 | ||
| Fig. 1 Reaction microenvironment control in MEA electrolyzers for CO2 electrolysis. | ||
2. Tailoring reaction microenvironments by molecular modifications
In the colloidal synthesis of size- and shape-selected nanoparticle catalysts, capping agents such as small molecule ligands and polymers are commonly used to maintain specific nanostructures by binding to metal atoms and minimizing surface energies.25 While ligands blocking surface sites are usually considered to be detrimental to catalysis, an increasing number of reports have indicated that some ligands with appropriate design can also play a positive catalytic role in CO2 electrolysis.26–30 The incorporation of such ligands onto catalyst surfaces does not directly introduce alternative active sites, but would create specific reaction microenvironments that can influence the activity and selectivity of CO2 electrolysis via steric and electronic effects.26–30 These molecular modifications are expected to control reaction pathways via manipulating electrode hydrophilicity/hydrophobicity, modulating interfacial cation solvation, tuning the adsorption of reaction intermediates, inducing additional chemical activation of CO2 as well as increasing ion conductivity in MEA electrolyzers.2.1 Organic molecules and polymers
As water is a reactant for both CO2 electrolysis and the hydrogen evolution reaction (HER), the relative local concentration of water versus CO2 is a key factor affecting catalytic selectivity. Water is usually in excess, while CO2 is insufficient under high current densities owing to limited gas diffusion from bulk liquid electrolytes or gas–liquid interfaces.24 Organic molecules and polymers with remarkable hydrophobic character can repel water molecules surrounding catalyst surfaces and allow CO2 gas to easily access catalyst surfaces. Mougel and co-workers developed a superhydrophobic Cu dendrite catalyst modified with 1-octadecanethiol, inspired by the gas-trapping cuticles of subaquatic spiders.31 The hydrophobic dendritic Cu surface pushes the liquid electrolyte away to form a gas–liquid–solid triple-phase boundary at the electrode, resulting in enhanced CO2 mass transport and thus an increased local CO2 concentration (Fig. 2a). The bio-inspired hydrophobic electrode achieves an ethylene Faradaic efficiency (FE) of 56% and an ethanol FE of 17%, compared to 9% and 4% on an unmodified, wettable counterpart. Hydrophobic molecules are also argued to prevent interfacial water from reorientation under the action of a cathodic electric field, resulting in unfavorable water dissociation to form protons for the HER.32 Polymers (including ionomers) are often used as binders in the preparation of catalyst layers in GDEs, and Nafion (with both hydrophilic and hydrophobic functional groups) is the most widely used binder as revealed by the knowledge from well-established fuel cell technology. Luo and co-workers further investigated two other polymer binders for Cu catalysts: polyacrylic acid (PAA, with a hydrophilic –COOH group) and fluorinated ethylene propylene (FEP, with a hydrophobic –CFx group).33 In a flow cell, the Cu–FEP electrode exhibited a peak FE of 52% for C2+ products with a partial current density of over 600 mA cm−2, followed by Cu–Nafion and Cu–PAA. The water contact angle follows the order of Cu–PAA < Cu–Nafion < Cu–FEP, while the captive bubble contact angle for the CO2 bubble increases from Cu–FEP (47°) to Cu–Nafion (73°) and Cu–PAA (117°). These results suggest that electrode hydrophobicity and CO2-philicity can be easily tuned by a thin layer of polymer binders with distinct functional groups, with the hydrophobic FEP showing the highest local CO2/H2O concentration ratio.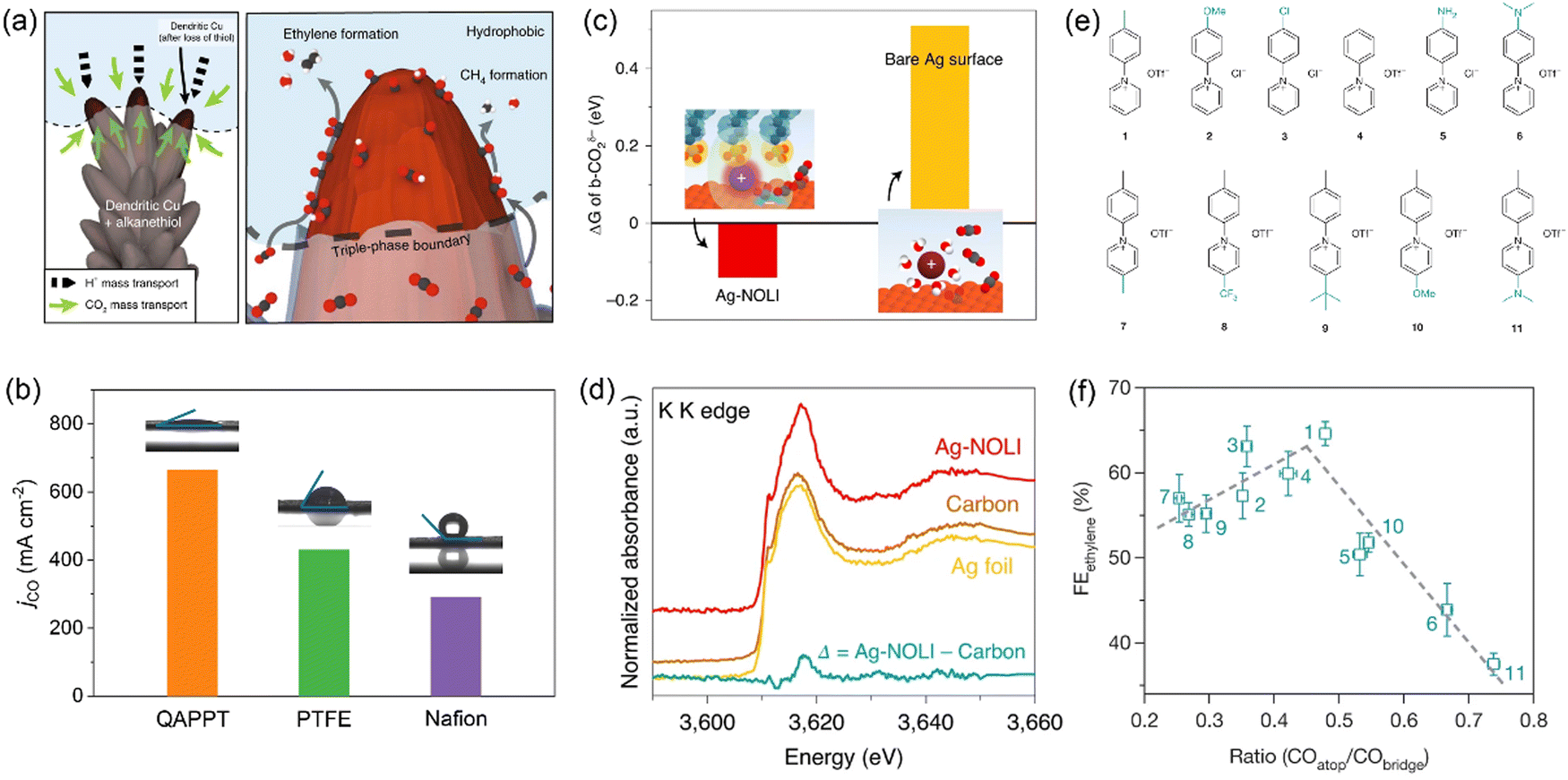 | ||
| Fig. 2 (a) Illustration of the role of hydrophobicity in promoting CO2 electroreduction over HER. Reproduced with the permission.31 Copyright 2019, Springer Nature. (b) CO partial current densities over QAPPT-, PTFE- and Nafion-modified Ni–N–C electrodes measured in an MEA electrolyzer. Reproduced with the permission.36 Copyright 2023, Wiley. (c) Free energy differences between physisorbed and chemisorbed CO2 for Ag-NOLI and bare Ag surface. (d) Potassium K-edge XANES of Ag-NOLI, Ag foil, and carbon paper. Reproduced with the permission.43 Copyright 2020, Springer Nature. (e) Molecular structures of N-arylpyridinium additives. (f) Correlation between ethylene FE and the ratio of *COatop/*CObridge over Cu electrodes modified by dimers of N-arylpyridiniums in (e). Reproduced with the permission.45 Copyright 2020, Springer Nature. | ||
On the other hand, water is not always in excess and is likely insufficient in the case of MEA electrolyzers operating at industrial current densities when a liquid electrolyte or pure water is fed to the anode and dry CO2 gas is fed to the cathode. In MEA electrolyzers, water needed for CO2 electrolysis comes from the anode through diffusion and electro-osmotic transport.34,35 While humidified CO2 is used in some cases, the major source of water is still the anolyte.33,34 The excessive consumption of water at high reaction rates would significantly decrease the water concentration at the cathode, so that the low water availability limits the reaction, especially for hydrophobic catalytic materials. Using hydrophobic Ni–N–C as a model catalyst, our group investigated the role of interfacial water in CO2 electrolysis in a custom-made alkaline MEA electrolyzer.36 The amount of interfacial water (i.e., electrode hydrophilicity/hydrophobicity) is facilely adjusted using three polymer binders: Nafion, polytetrafluoroethylene (PTFE, containing nonionic surfactants), and quaternary ammonia poly(N-methyl-piperidine-co-p-terphenyl) (QAPPT), as evidenced by in situ environmental scanning electron microscopy (ESEM) and contact angle measurements. In contrast to the Cu electrodes measured in H-cells and flow cells,31,33,37 the hydrophilic Ni–N–C–QAPPT electrode shows the highest CO partial current density, up to 665 mA cm−2, outperforming all previously reported Ni–N–C catalysts for CO2 electrolysis (Fig. 2b). In situ attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy results indicate that increasing the amount of interfacial water modulates the mechanism of CO formation from the *COO− pathway to the *COOH pathway.36 It is unclear whether the structure of interfacial water is also modified by different polymers, but it seems that the strategy that the HER can be suppressed by interfacial water structure modification with organic molecules or electrolyte additives in H-cells32,38,39 does not necessarily apply to the case of MEA electrolyzers. This exceptional finding further highlights the importance of reaction microenvironment control in MEAs.
Apart from hydrophilic/phobic modulation, organic molecules and polymers can tune activity and selectivity of CO2 electrolysis by varying local pH. Gewirth and co-workers developed a Cu–polyamine hybrid catalyst through co-electroplating and achieved an ethylene FE of 87% at −0.47 V vs. the reversible hydrogen electrode (RHE) in 10 M KOH electrolyte.40 The remarkable ethylene selectivity is attributed to the higher *CO coverage and higher local pH in the presence of polyamine retained on the Cu electrode surface as revealed by the in situ Raman spectroscopy results. Xie and co-workers employed an anti-swelling anion exchange ionomer (AEI) to manipulate the reaction microenvironment of an oxide-derived Cu nanosheet catalyst.41 The OH−-accumulated –N(CH3)3+ groups and anti-swelling backbone of AEI synergistically regulated local pH and the water amount, resulting in a C2+ FE of 85.1% at a current density of 800 mA cm−2. Bell and co-workers tailored the reaction microenvironment of a sputtered Cu catalyst using bilayer cation- and anion-conducting ionomer coatings.10 In combination with pulsed CO2 electrolysis, the tailored reaction microenvironment results in a 2.5-fold increase in the C2+ production rate with a C2+ FE as high as 90% compared with static electrolysis over bare Cu, owing to the higher local pH (via Donnan exclusion) and increased local CO2/H2O concentration ratio (via ionomer properties). Although the current densities reported in this work were low (10 to 20 mA cm−2), the fundamental understanding may also be favorable for the design of membranes and ionomers for high performance MEA electrolyzers. For instance, Mallouk and co-workers introduced a weak-acid cation exchange layer via layer-by-layer (LBL) assembly into bipolar-membrane-based MEA electrolyzers.42 Local pH measurements in MEA electrolyzers indicate a sharp pH increase from ∼2 to ∼5 within the first few bilayers and a steady pH in the bulk of the cation exchange layer. Thus, the presence of the weak-acid layer increased local pH near the catalyst layer and suppressed the HER current density without affecting CO2 electroreduction.
The ligands could improve CO2 electrolysis performance by modulating the composition, concentration, and solvation structure of interfacial cations at the electrode/electrolyte interfaces. Yang and co-workers developed a nanoparticle/ordered-ligand interlayer (NOLI) structure composed of an Ag nanoparticle surface and a detached layer of tetradecylphosphonic acid ligands in its vicinity.43 The reversible adsorption/desorption of the ligand layer creates a catalytic pocket at the metal/ligand interface, where hydrated cations are intercalated and then lose their hydration shell due to the strong electrostatic interaction between K+ and anionic phosphonate groups of the ligands (Fig. 2c and d). Thus, dehydrated K+ cations are anchored close to the metal surface, stabilizing the *COO− intermediate, consistent with the cation effects correlated with hydration free energy over bare metal surfaces.44 The modularity of NOLI is demonstrated with other anionic ligands with a long hydrocarbon chain (e.g., oleic acid) and other noble metals (e.g., Au, Pd). With GDE and a neutral electrolyte, the NOLI achieved a CO FE of 98.1% at 400 mA cm−2.
Molecular modification provides an alternative strategy of tuning the stabilities of key intermediates to facilitate desired reaction pathways. Sargent and co-workers functionalized the surface of Cu catalysts with a library of dimers of N-arylpyridiniums (Fig. 2e) through electro-dimerization.45 These adhered molecules improved the stabilization of the *COatop intermediate (Fig. 2f), thus favoring the C–C coupling pathway via the dimerization of *COatop and *CObridge. The optimal Cu catalyst modified with N-aryl-substituted tetrahydro-bipyridine oligomeric films achieved an impressive CO2-to-ethylene conversion with an ethylene FE of 72% in a flow cell and a full-cell energy efficiency of 20% in an MEA electrolyzer. As *CO is a common intermediate for the formation of C2+ products and its coverage is relatively high compared to other intermediates (e.g., *COOH, *OCCOH), many studies have been devoted to tuning the configuration and coverage of *CO adsorption by modifying Cu surfaces using ligands with versatile functional groups.46–51 The *CO intermediate can further be enriched at the metal–organic interfaces using a tandem catalyst through functionalizing Cu surfaces with a group of porphyrin-based metallic complexes capable of reducing CO2 to CO.52 The interactions between ligands and complex C2 intermediates at the molecular level would also tune relative selectivity among C2+ products after C–C coupling; however, relevant mechanistic understanding is still missing. Some adhered ligands could induce surface reconstruction, thus generating specific surface sites for tuning adsorption of reaction intermediates during CO2 electrolysis.53–55 Whether such ligands have other direct electronic and steric effects is not yet well clarified.
The ligands may chemically activate the inert CO2 molecule via specific interactions between CO2 and functional groups such as amino, pyridyl, and imidazolium.30 For instance, the cysteamine ligand can promote CO2 chemisorption through hydrogen bonding with the amino group, giving rise to 53-fold enhancement in turnover frequency (TOF) for CO production over Ag.56 By generating a highly reactive N-heterocyclic carbene (NHC) structure under reducing conditions, the imidazolium ligand can capture CO2 to form carboxylate intermediates.57 Wang and co-workers put forward a polyquinone modification strategy that activated CO2via quasi-reversible bonding between electrochemically reduced quinone groups and the CO2 molecule.58 Further incorporating the polyquinone into a Cu GDE resulted in an ethylene partial current density of 325 mA cm−2 at a cell voltage of 3.5 V in an MEA electrolyzer fed with pure water. In addition to CO2 activation, the ligands may also play a physical role as a CO2-enriching layer when dilute CO2 is fed to GDE.59,60
2.2 Bifunctional ionomers
As discussed above, ionomers such as Nafion and QAPPT have been used as binders in the preparation of catalyst layers used in MEAs. However, when liquid electrolytes (e.g., KHCO3, KOH) are fed to MEA electrolyzers,36,48 the ion conductivity of ionomers is not a governing factor for CO2 electrolysis performance. In contrast, when pure water is used, the introduction of ionically conductive ionomers into catalyst layers can remarkably enlarge the electrochemical interface, as revealed by other modern polymer electrolyte-based technologies like fuel cells. CO2 electrolysis with pure water completely avoids electrolyte consumption and corrosion issues.21 It is highly desirable to integrate ion conductivity and chemical activation of CO2 into one ionomer/polymer. Zhuang and co-workers developed such a bifunctional ionomer, namely, quaternary ammonia poly(ether ether ketone) (QAPEEK) with carbonyl groups in the polymer chain.61In situ ATR-SEIRAS measurements indicated that the carbonyl groups close to the electrode surface played a similar role to alkali metal cations, which helped in activating CO2 and stabilizing the *COO− intermediate (Fig. 3a and b). The remarkable advantage of the bifunctional QAPEEK ionomer versus other anion exchange ionomers (e.g., QAPPT, Sustainion, Fumasep, and PiperION) was verified by its drastically improved ethylene production (Fig. 3c and d). With a porous Cu catalyst, the QAPEEK ionomer helped achieving an industrial-scale ethylene partial current density of 420 mA cm−2 at a low cell voltage of 3.54 V without any electrolyte consumption.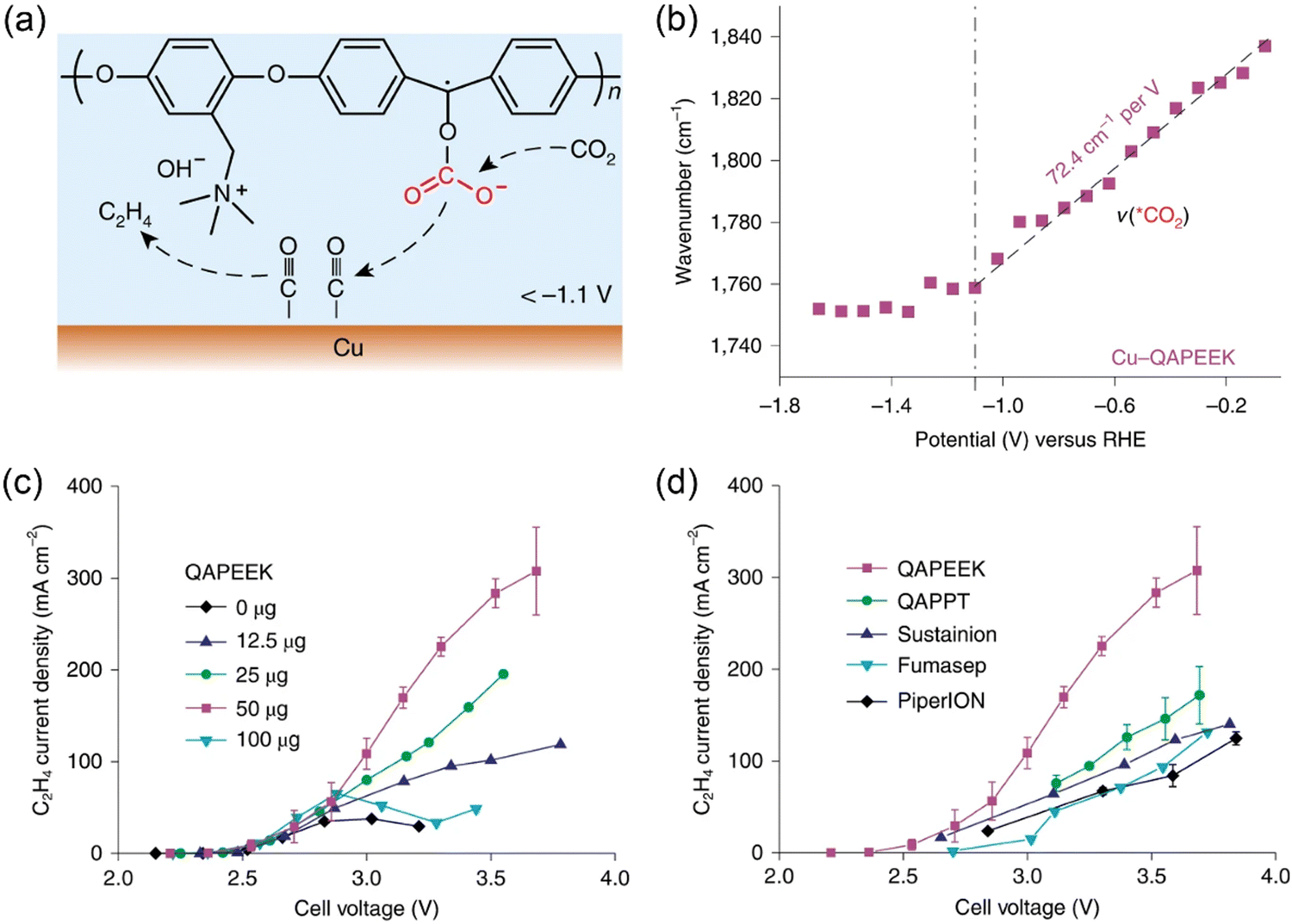 | ||
| Fig. 3 (a) Schematic illustration of QAPEEK-promoted CO2 electroreduction on Cu. (b) Potential-dependent shift of ATR-SEIRAS band of activated CO2 (*CO2) on Cu-QAPEEK. (c, d) Ethylene partial current densities of (c) Cu-QAPEEK electrodes with different amounts of QAPEEK and (d) Cu electrodes with different ionomers. Reproduced with the permission.61 Copyright 2022, Springer Nature. | ||
3. Tailoring reaction microenvironments by reactant feed compositions
While pure CO2 is usually used in fundamental studies of CO2 electrolysis, the realistic composition of flue gas streams (e.g., from coal-fired power plants) is very complicated. The CO2 concentration is only 10–25% in flue gas which contains considerable amounts of N2, unconverted O2, trace amounts of SOx and NOx as well as CO (due to incomplete combustion). Direct electrolysis of CO2 from such industrial sources is necessary but very challenging. On the other hand, the presence of these impurities in the feed would also change the reaction microenvironments for CO2 electrolysis.The reduction of O2 is thermodynamically favorable over that of CO2; so it is urgently needed to develop O2-tolerant electrodes. A polymer of intrinsic microporosity62 and a hydrated ionomer catalyst coating63 have been developed to locally slow O2 transport as a CO2-selective layer on the electrodes. The presence of O2 in the feed is also argued to facilitate CO2 electrolysis by stabilizing surface Cu oxide or oxyhydroxide species.64,65 The presence of a trace amount of NOx shows a negligible impact on CO2 electrolysis despite loss in FE due to the facile electroreduction of NOx,65,66 while SOx could poison Cu catalysts and suppress the C2+ production.65,67 Another important feature of flue gas is its low CO2 concentration which limits CO2 electrolysis rate owing to the first-order reaction kinetics. This issue stimulates a new research direction, namely, integrated CO2 capture and electroreduction technologies.59,68,69 Coating a layer of organic ligands onto catalyst surfaces to enrich CO2 and increase the local CO2 concentration in reaction microenvironments is an effective method to improve electrolysis performance of dilute CO2 feed.59
The flue gas from steel plants contains a large fraction of CO, and simultaneous conversion of CO2 and CO is desirable. The co-electrolysis of CO2/CO is rarely studied to date, and the underlying co-electrolysis mechanism is also under debate. The promoting effect of cross-coupling of CO2/CO and the major contribution from CO2 or CO alone have been proposed.70–74 These studies are conducted in H-cells with neutral electrolytes. Our group conducted CO2/CO co-electrolysis in an alkaline MEA electrolyzer where reaction microenvironments were quite different from those in H-cells.65 An obvious difference was the available CO2/CO concentration which was close to that in the CO2/CO feeds in MEAs without being affected by CO2/CO solubility. With increasing CO pressure in the feed, the major product gradually shifted from ethylene to acetate and meanwhile the current density increased drastically (Fig. 4a). Under optimized reaction conditions, C2+ FE and partial current density reached 90.0% and 3.1 A cm−2, respectively. Structural characterization, control experiments with Ar/CO co-feeds, isotopic labeling experiments, and operando Raman spectroscopy measurements (Fig. 4b–e) indicated that the feed-composition-dependent selectivity changes were not ascribed to any structural changes, but were induced by reaction microenvironments, herein, *CO coverage (by the pressure of CO) and local pH (by the ratio of CO2). It should be noted that CO2–CO cross-coupling did occur in CO2/CO co-electrolysis under MEA conditions,65 but did not improve the formation of ethylene.70,74 The insights presented in our work highlight the great importance of both reactant feed composition and electrolyzer configuration in the reaction microenvironment control for selective production of single products.
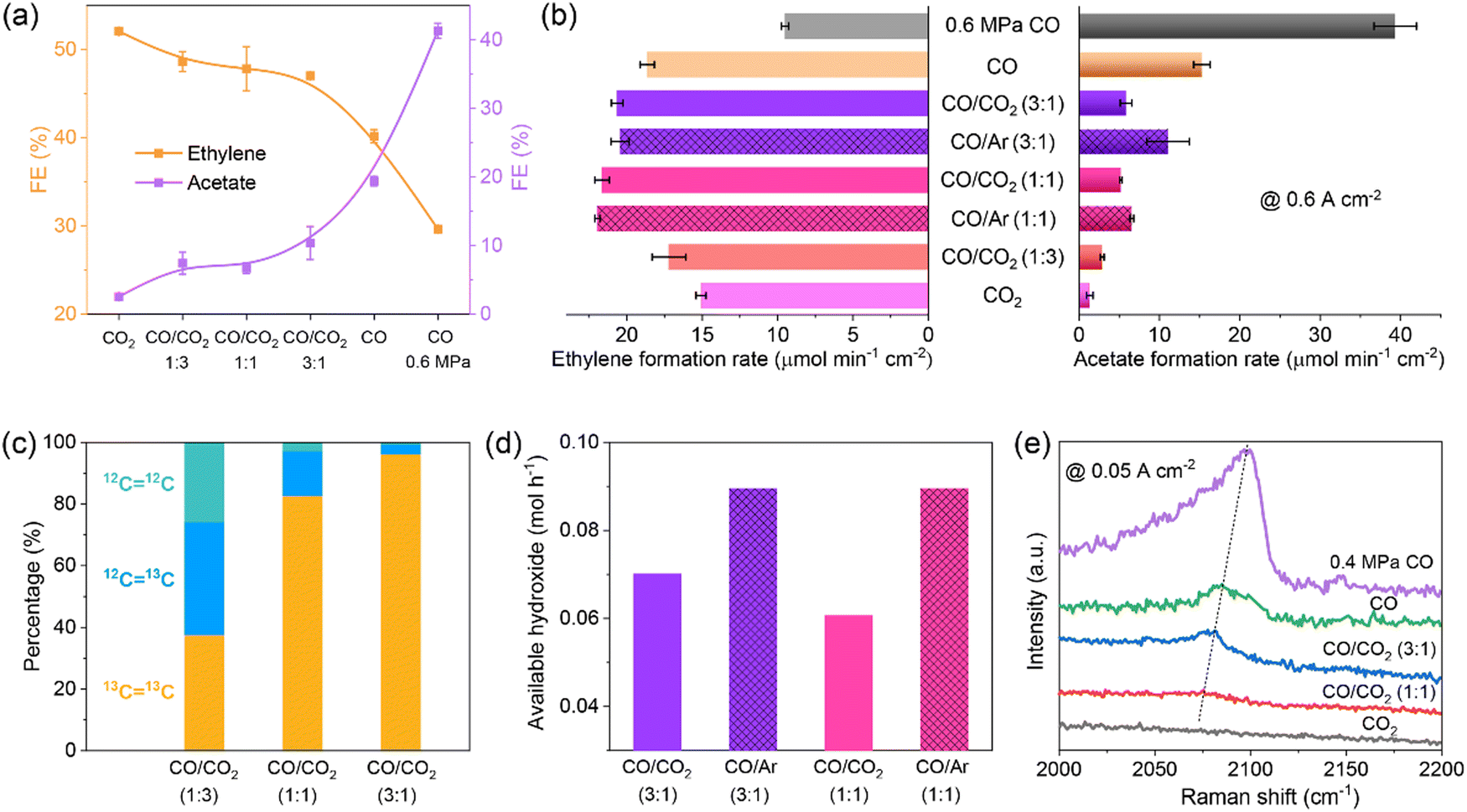 | ||
Fig. 4 (a) FEs and (b) formation rates of ethylene and acetate, (c) molar ratios of 12CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 12CH2, 12CH213CH2, and 13CH213CH2 in produced ethylene, (d) available OH− formation rates, and (e) operando *COatop Raman peaks over CuO nanosheet catalyst measured with different feeds. Reproduced with the permission.65 Copyright 2023, Springer Nature. 12CH2, 12CH213CH2, and 13CH213CH2 in produced ethylene, (d) available OH− formation rates, and (e) operando *COatop Raman peaks over CuO nanosheet catalyst measured with different feeds. Reproduced with the permission.65 Copyright 2023, Springer Nature. | ||
4. Tailoring reaction microenvironments in acidic CO2 electrolysis
A challenging issue of CO2 electrolysis in alkaline and neutral media is carbonate formation and crossover which leads to substantial CO2 loss, high cost for downstream CO2 regeneration and separation, as well as reduced long-term electrolyzer stability.75 Strategies such as rinsing GDEs with water, applying periodic regeneration voltage and using bipolar membranes (BPMs) have been proposed to alleviate the effects of carbonate formation,76–78 but failed to address this issue by eliminating its formation intrinsically. The origin of the carbonate formation is the homogeneous reaction of CO2 with OH− that is present in electrolytes or is formed as a side product of proton consumption via the reduction of CO2 and water. The latter would be the major OH− source for the carbonate formation in MEA electrolyzers operating at industrial current densities. CO2 crossover occurs via electromigration of carbonate or bicarbonate ions from the cathode to anode through an anion exchange membrane. The Bjerrum plot of the carbonate system suggests that the formation of carbonate and bicarbonate can be completely suppressed at a pH lower than 4. Therefore, CO2 electrolysis in acidic media holds great promise in achieving carbon-efficient CO2 conversion. Given the remarkable interfacial pH increase upon applying high current densities as revealed by the reaction–diffusion modelling results,79 reaction media should be strongly acidic, ideally with a bulk pH lower than 2, in order to fully eliminate the carbonate formation. However, such harsh reaction conditions give rise to extra difficulty in selectivity control owing to facile HER and unfavorable C–C coupling.Many attempts have been made to improve acidic CO2 electrolysis by tailoring reaction microenvironments, for instance, creating a locally alkaline environment and increasing the cation concentration near catalyst surfaces.80–83 Sinton and co-workers developed a cation-carrying and proton-blocking catalyst adlayer to suppress HER and promote CO2 electrolysis to C2+ products.84 The adlayer was composed of insulating polymer nanoparticles (IPN, imine- and carbonyl-functionalized covalent organic frameworks) and perfluorinated sulfonic-acid (PFSA) ionomers, and it can restrict proton transport flux to cation-conducting hydrophilic nanochannels and enrich K+ near the Cu catalyst surface (Fig. 5a). The resulting high local alkalinity and cation-enriched reaction microenvironment enabled a C2+ FE of 75% and a single-pass CO2 utilization of 45% towards C2+ at 200 mA cm−2 in a slim flow cell. The asymmetric ion migration–adsorption strategy has also been verified via modifying catalyst surfaces with covalent organic frameworks and organic additives.85,86 In addition to manipulating local cation concentrations, such molecular modifications were also expected to exhibit other direct microenvironment effects by interacting with intermediates, similar to the case of alkaline electrolysis discussed above (Section 2). Our group developed an acidic MEA electrolyzer using K2SO4 + H2SO4 anolytes with pH 0–2 and used HER-inactive Ni–N–C as a model catalyst.87 By optimizing pH, K+ concentration and CO2 pressure, acidic CO2 electrolysis achieved a CO FE of 95% at a total current density of 500 mA cm−2 at pH 0.5 and the CO2 loss can be reduced by 86% at 300 mA cm−2, compared with alkaline CO2 electrolysis (Fig. 5b–d). By further adjusting the flow rate of input CO2, the single-pass CO2 utilization reached as high as 85%, breaking the upper limit of 50% for CO production in alkaline CO2 electrolysis. The impressive CO selectivity at high current densities was attributed to the presence of high-concentration K+ cations that migrated from the anolyte. Li and co-workers also achieved a CO FE of ∼80% and a single-pass CO2 utilization of ∼90% in an acidic MEA electrolyzer using Cs+ cations and Ag catalyst.88 It seemed that high-concentration cations were indispensable for efficient acidic CO2 electrolysis. A couple of mechanisms on cation effects have been proposed, such as interfacial electric field, buffering interfacial pH, stabilizing intermediates and non-electric field effect.18 All the proposed reaction mechanisms were positively correlated with cation concentration. Further investigations on producing C2+ products in acidic MEA electrolyzers are still needed, due to relatively facile HER performance of Cu catalysts and the limited local cation concentration in MEA configurations. It should be noted that for acidic CO2 electrolysis in anolyte-fed MEA electrolyzers, salt deposition still occurred at an extended electrolysis period, especially when using high-concentration cations due to the transfer of cations from the anolyte to cathode through membranes.87 Replacing alkali metal cations with organic cations or cationic functional groups/ionomers likely achieved sustainable CO2 electrolysis under acidic conditions or even with pure water.61,89–91 This strategy has recently shown great promise, but whether the catalytic capability of these organic cations is comparable to that of alkali metal cations is still not yet clear. Given that CO does not react with OH− to form carbonate and C2+ selectivity of CO electrolysis is higher, tandem acidic/alkaline electrolysis, namely, connecting an acidic MEA electrolyzer (CO2 electrolysis to CO) and an alkaline MEA electrolyzer (CO electrolysis to C2+), has been proposed and demonstrated as an energy- and carbon-efficient route for CO2 electrolysis to C2+ products.87
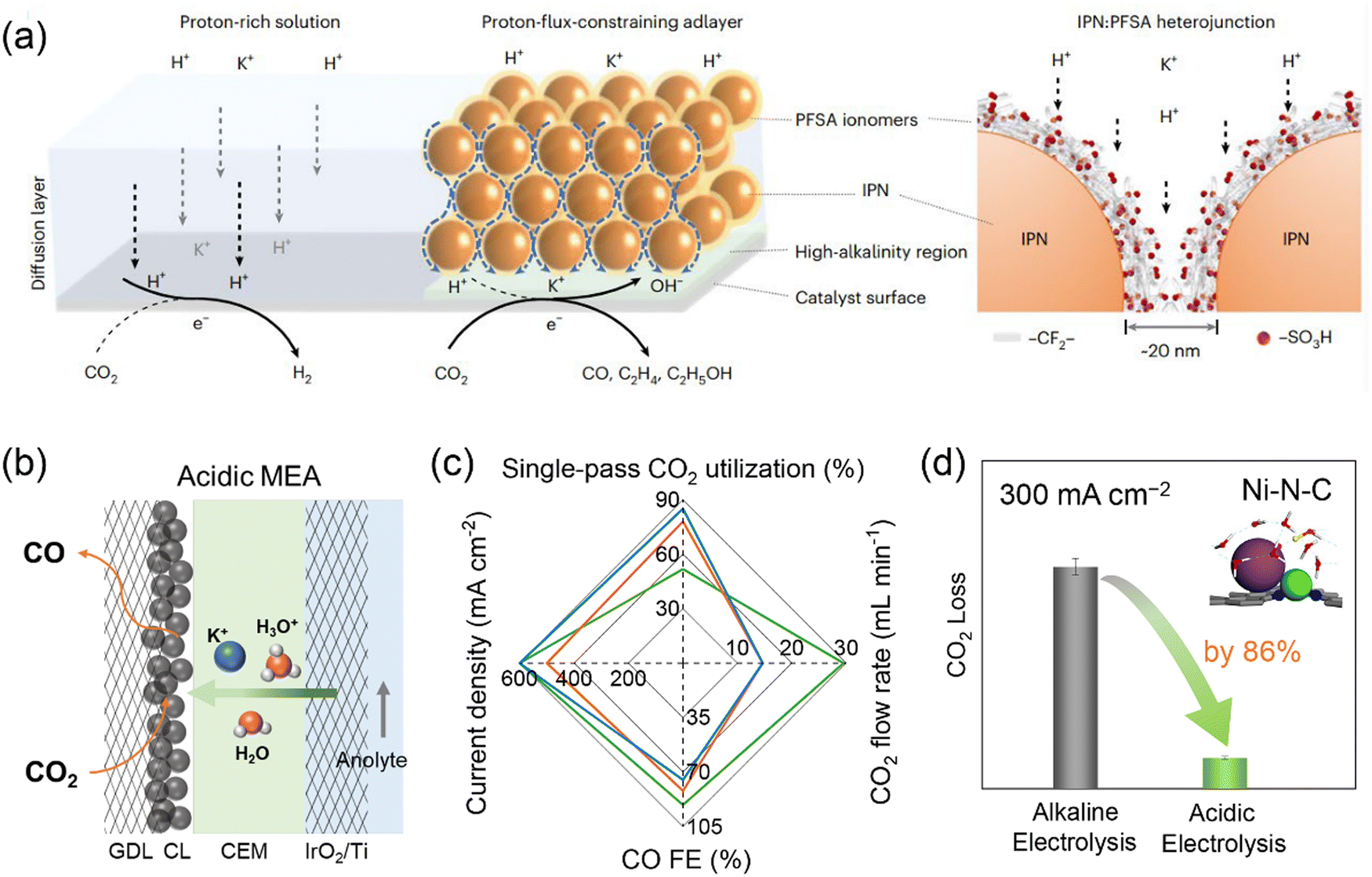 | ||
| Fig. 5 (a) Reaction microenvironment control in strong acid via proton-flux-constraining ionomer adlayer design. Reproduced with the permission.84 Copyright 2023, Springer Nature. (b) Schematic representation of acidic MEA electrolyzer. (c) Acidic CO2 electrolysis performance over Ni–N–C catalyst. (d) Comparison of CO2 loss in acidic electrolysis (0.5 M K2SO4 + H2SO4 anolyte with pH 0.5, 0.5 MPa CO2) and alkaline electrolysis (1 M KOH anolyte, 0.5 MPa CO2) over Ni–N–C catalyst. Reproduced with the permission.87 Copyright 2023, Royal Society of Chemistry. | ||
5. Conclusions and outlook
In this perspective, we have summarized and discussed the fundamentals and strategies of tuning reaction microenvironments for active, selective, energy- and carbon-efficient CO2 electrolysis, with particular attention being paid to those examples using MEA electrolyzers towards practical applications. The effects and underlying mechanisms of reaction microenvironments created by molecular modifications with organic molecules/polymers and tuned by reactant feed compositions on the activity and selectivity of CO2 electrolysis are illustrated in detail. The efforts to rationally control the transport of H+/OH− and cations to modify acidic reaction microenvironments for carbon-efficient CO2 electrolysis are also discussed. The presented examples and associated mechanistic insights highlight future opportunities and design strategies towards reaction microenvironment control for improved activity and selectivity of CO2 electrolysis.Despite the recent achievements, the intrinsic underlying effects of reaction microenvironments around catalytically active sites are not understood in depth. For instance, molecular modification of catalyst surfaces is the most widely studied case of reaction microenvironment control, but multiple likely effects (e.g., due to hydrophobicity, local pH, intermediate stabilization, and ion transport) often co-exist in one catalyst system and are difficult to be decoupled. Although control experiments can be performed to rule out contributions of some factors, it is hardly realistic to precisely control ligand structures, catalyst surface structures as well as their complicated interactions, especially under industrially relevant harsh conditions. Currently, the enhancement in C2+ production has been demonstrated with the aid of tailored reaction microenvironments of Cu catalysts and has been mainly ascribed to the enrichment and activation of CO2 and the improved adsorption of key intermediates such as *CO.10,36,43,45,61 However, how the organic ligands interact with more complex intermediates beyond *CO2 and *CO at the molecular level is still not yet clarified. The precise molecular control over specific interactions between ligands and intermediates would be favorable for tuning selectivity among C2+ products and generating specific C2+ products.92 Advanced operando spectroscopy and microscopy characterization with sufficient temporal and spatial resolution would help in resolving the exact structures of catalysts and ligands in these systems.42,93–97
In addition to initial states and conditions, reaction microenvironments are also dynamic and closely associated with reaction rates. As the mass transport of reactive species (e.g., CO2, H2O, H+, OH−, carbonate, bicarbonate, cations, and intermediates) at the mesoscale in the reaction microenvironments significantly influences reaction kinetics, their concentration profiles at the electrode/electrolyte interface should be accurately determined via experimental measurements under reaction conditions and/or multiphysics simulations using reasonable reaction–diffusion models.35,79,98–102 The reaction microenvironments in MEAs (e.g., membrane, ionomer, and catalyst layer structure) are more complex, but currently not well-investigated yet, adding extra degrees of freedom for activity and selectivity control, especially in acidic CO2 electrolysis. We believe that revealing the nature of electrocatalytic interfaces at the mesoscale would help in designing customizable reaction microenvironments for active, selective, energy- and carbon-efficient CO2 electrolysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1504603), the National Natural Science Foundation of China (22002155, 22372171, 22125205, and 92045302), the Fundamental Research Funds for the Central Universities (20720220008), the Natural Science Foundation of Liaoning Province (2021-MS-022), the Liao Ning Revitalization Talents Program, the Dalian Institute of Chemical Physics (DICP I202203) and the Photon Science Center for Carbon Neutrality.References
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS.
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave, X. X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrodi, M. Escudero-Escribano, J. V. F. de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Y. Huo, Y. J. Hwang, C. Janaky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. P. Kuhl, W. H. Lee, Z. Q. Liang, X. Liu, S. C. Ma, M. Ma, H. S. Oh, M. Robert, B. Roldan Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastian-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. J. Xu, H. Yadegari and Y. X. Zhou, J. Phys. Energy, 2022, 4, 042003 CrossRef.
- D. F. Gao, W. J. Li, H. Y. Wang, G. X. Wang and R. Cai, Trans. Tianjin Univ., 2022, 28, 245–264 CrossRef CAS.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 1695–1698 CrossRef CAS.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 897–898 CrossRef CAS.
- Y. Y. Birdja, E. Perez-Gallent, M. C. Figueiredo, A. J. Gottle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- D. F. Gao, R. M. Aran-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- H. B. Li, J. P. Xiao, Q. Fu and X. H. Bao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5930–5934 CrossRef CAS PubMed.
- A. R. Riscoe, C. J. Wrasman, A. A. Herzing, A. S. Hoffman, A. Menon, A. Boubnov, M. Vargas, S. R. Bare and M. Cargnello, Nat. Catal., 2019, 2, 852–863 CrossRef CAS.
- C. Kim, J. C. Bui, X. Y. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- J. X. Guo, Y. Zheng, Z. P. Hu, C. Y. Zheng, J. Mao, K. Du, M. Jaroniec, S. Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- Y. Lai, N. B. Watkins, A. Rosas-Hernandez, A. Thevenon, G. P. Heim, L. Zhou, Y. S. Wu, J. C. Peters, J. M. Gregoire and T. Agapie, ACS Cent. Sci., 2021, 7, 1756–1762 CrossRef CAS PubMed.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- J. C. Bui, C. Kim, A. J. King, O. Romiluyi, A. Kusoglu, A. Z. Weber and A. T. Bell, Acc. Chem. Res., 2022, 55, 484–494 CrossRef CAS PubMed.
- A. N. Xu, N. Govindarajan, G. Kastlunger, S. Vijay and K. R. Chan, Acc. Chem. Res., 2022, 55, 495–503 CrossRef CAS PubMed.
- B. W. Deng, M. Huang, X. L. Zhao, S. Y. Mou and F. Dong, ACS Catal., 2022, 12, 331–362 CrossRef CAS.
- J. J. Lv, R. N. Yin, L. M. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. L. Jin, X. Wang and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207252 CrossRef CAS.
- Y. W. Rong, J. Q. Sang, L. Che, D. F. Gao and G. X. Wang, Acta Phys.-Chim. Sin., 2023, 39, 2212027 Search PubMed.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- D. F. Gao, P. F. Wei, H. F. Li, L. Lin, G. X. Wang and X. H. Bao, Acta Phys.-Chim. Sin., 2021, 37, 2009021 Search PubMed.
- Z. L. Yin, H. Q. Peng, X. Wei, H. Zhou, J. Gong, M. M. Huai, L. Xiao, G. W. Wang, J. T. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455–2462 RSC.
- L. Ge, H. Rabiee, M. R. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 CAS.
- Z. Zhang, X. Huang, Z. Chen, J. J. Zhu, B. Endrodi, C. Janaky and D. H. Deng, Angew. Chem., Int. Ed., 2023, 62, e202302789 CrossRef CAS PubMed.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- J. G. Wang, F. Q. Zhang, X. W. Kang and S. W. Chen, Curr. Opin. Electrochem., 2019, 13, 40–46 CrossRef CAS.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- D. H. Nam, P. De Luna, A. Rosas-Hernandez, A. Thevenon, F. W. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- M. Jun, D. Kim, M. Kim, M. Kim, T. Kwon and K. Lee, ACS Omega, 2022, 7, 42655–42663 CrossRef CAS PubMed.
- Q. S. Zhu, C. J. Murphy and L. R. Baker, J. Am. Chem. Soc., 2022, 144, 2829–2840 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- S. J. Mu, L. Li, R. J. Zhao, H. L. Lu, H. L. Dong and C. H. Cui, ACS Appl. Mater. Interfaces, 2021, 13, 47619–47628 CrossRef CAS PubMed.
- T. H. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli, W. Luo and A. Zuttel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS.
- W. Choi, S. Park, W. Jung, D. H. Won, J. Na and Y. J. Hwang, ACS Energy Lett., 2022, 7, 939–945 CrossRef CAS.
- W. Choi, Y. Choi, E. Choi, H. Yun, W. Jung, W. H. Lee, H. S. Oh, D. H. Won, J. Na and Y. J. Hwang, J. Mater. Chem. A, 2022, 10, 10363–10372 RSC.
- P. F. Wei, H. F. Li, R. T. Li, Y. Wang, T. F. Liu, R. Cai, D. F. Gao, G. X. Wang and X. H. Bao, Small, 2023, 19, 202300856 Search PubMed.
- A. Kormányos, B. Endrődi, Z. Zhang, A. Samu, L. Mérai, G. F. Samu, L. Janováka and C. Janáky, EES Catal., 2023, 1, 263–273 RSC.
- Z. Q. Zhang, S. Banerjee, V. S. Thoi and A. S. Hall, J. Phys. Chem. Lett., 2020, 11, 5457–5463 CrossRef CAS PubMed.
- N. Mohandas, T. N. Narayanan and A. Cuesta, ACS Catal., 2023, 13, 8384–8393 CrossRef CAS.
- X. Y. Chen, J. F. Chen, N. M. Alghoraibi, D. A. Henckel, R. X. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- Y. Zhao, X. L. Zu, R. H. Chen, X. D. Li, Y. W. Jiang, Z. Q. Wang, S. M. Wang, Y. Wu, Y. F. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446–10454 CrossRef CAS PubMed.
- Z. F. Yan, J. L. Hitt, Z. C. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed.
- D. Kim, S. Yu, F. Zheng, I. Roh, Y. F. Li, S. Louisia, Z. Y. Qi, G. A. Somorjai, H. Frei, L. W. Wang and P. D. Yang, Nat. Energy, 2020, 5, 1032–1042 CrossRef CAS.
- V. J. Ovalle, Y. S. Hsu, N. Agrawal, M. J. Janik and M. M. Waegele, Nat. Catal., 2022, 5, 624–632 CrossRef CAS.
- F. W. Li, A. Thevenon, A. Rosas-Hernández, Z. Y. Wang, Y. L. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. H. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Z. Tao, Z. Q. Liang, M. C. Luo, X. Wang, H. H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. G. C. Li, Z. J. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- Y. Shi, K. Sun, J. Shan, H. Li, J. Gao, Z. Chen, C. Sun, Y. Shuai and Z. Wang, ACS Catal., 2022, 12, 8252–8258 CrossRef CAS.
- P. Ding, H. Y. An, P. Zellner, T. F. Guan, J. Y. Gao, P. Muller-Buschbaum, B. M. Weckhuysen, W. van der Stam and I. D. Sharp, ACS Catal., 2023, 13, 5336–5347 CrossRef CAS PubMed.
- A. Ozden, F. W. Li, F. P. G. de Arquer, A. Rosas-Hernandez, A. Thevenon, Y. H. Wang, S. F. Hung, X. Wang, B. Chen, J. Li, J. Wicks, M. C. Luo, Z. Y. Wang, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, ACS Energy Lett., 2020, 5, 2811–2818 CrossRef CAS.
- M. S. Xie, B. Y. Xia, Y. W. Li, Y. Yan, Y. H. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- L. Fan, C. Y. Liu, P. Zhu, C. Xia, X. Zhang, Z. Y. Wu, Y. Y. Lu, T. P. Senftle and H. T. Wang, Joule, 2022, 6, 205–220 CrossRef CAS.
- Q. Chang, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, N. S. Marinkovic, A. H. A. Park, S. Kattel and J. G. Chen, JACS Au, 2022, 2, 214–222 CrossRef CAS PubMed.
- F. W. Li, Y. G. C. Li, Z. Y. Wang, J. Li, D. H. Nam, Y. Lum, M. C. Luo, X. Wang, A. Ozden, S. F. Hung, B. Chen, Y. H. Wang, J. Wicks, Y. Xu, Y. L. Li, C. M. Gabardo, C. T. Dinh, Y. Wang, T. T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- H. L. Wu, J. Li, K. Qi, Y. Zhang, E. Petit, W. S. Wang, V. Flaud, N. Onofrio, B. Rebiere, L. Q. Huang, C. Salameh, L. Lajaunie, P. Miele and D. Voiry, Nat. Commun., 2021, 12, 7210 CrossRef CAS PubMed.
- X. R. He, J. W. Chen, Y. F. Xu, Y. Shen, Y. F. Zeng, J. Y. Zhu, B. J. Xu and C. Wang, Nano Res., 2023, 16, 4554–4561 CrossRef CAS.
- C. F. Cheng, T. F. Liu, Y. Wang, P. F. Wei, J. Q. Sang, J. Q. Shao, Y. P. Song, Y. P. Zang, D. F. Gao and G. X. Wang, J. Energy Chem., 2023, 81, 125–131 CrossRef CAS.
- Z. J. Wang, L. N. Wu, K. Sun, T. Chen, Z. H. Jiang, T. Cheng and W. A. Goddard, J. Phys. Chem. Lett., 2018, 9, 3057–3061 CrossRef CAS PubMed.
- J. Tamura, A. Ono, Y. Sugano, C. C. Huang, H. Nishizawa and S. Mikoshiba, Phys. Chem. Chem. Phys., 2015, 17, 26072–26078 RSC.
- J. M. Li, F. F. Li, C. Liu, F. Y. Wei, J. Gong, W. Z. Li, L. W. Xue, J. L. Yin, L. Xiao, G. W. Wang, J. T. Lu and L. Zhuang, ACS Energy Lett., 2022, 7, 4045–4051 CrossRef CAS.
- Y. Y. Cheng, J. Hou and P. Kang, ACS Energy Lett., 2021, 6, 3352–3358 CrossRef CAS.
- J. C. Wang, T. Cheng, A. Q. Fenwick, T. N. Baroud, A. Rosas-Hernandez, J. H. Ko, Q. Gan, W. A. Goddard and R. H. Grubbs, J. Am. Chem. Soc., 2021, 143, 2857–2865 CrossRef CAS PubMed.
- W. Z. Li, Z. L. Yin, Z. Y. Gao, G. W. Wang, Z. Li, F. Y. Wei, X. Wei, H. Q. Peng, X. T. Hu, L. Xiao, J. T. Lu and L. Zhuang, Nat. Energy, 2022, 7, 835–843 CrossRef CAS.
- X. Lu, Z. Jiang, X. L. Yuan, Y. S. Wu, R. Malpass-Evans, Y. R. Zhong, Y. Y. Liang, N. B. McKeown and H. L. Wang, Sci. Bull., 2019, 64, 1890–1895 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, J. J. Zhong, C. P. O'Brien, C. M. Gabardo, C. McCallum, J. Li, C. T. Dinh, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2020, 13, 554–561 RSC.
- M. He, C. S. Li, H. C. Zhang, X. X. Chang, J. G. G. Chen, W. A. Goddard, M. J. Cheng, B. J. Xu and Q. Lu, Nat. Commun., 2020, 11, 3844 CrossRef CAS PubMed.
- P. F. Wei, D. F. Gao, T. F. Liu, H. F. Li, J. Q. Sang, C. Wang, R. Cai, G. X. Wang and X. H. Bao, Nat. Nanotechnol., 2023, 18, 299–306 CrossRef CAS PubMed.
- B. H. Ko, B. Hasa, H. Shin, E. Jeng, S. Overa, W. Chen and F. Jiao, Nat. Commun., 2020, 11, 5856 CrossRef CAS PubMed.
- W. Luc, B. H. Ko, S. Kattel, S. Li, D. Su, J. G. G. Chen and F. Jiao, J. Am. Chem. Soc., 2019, 141, 9902–9909 CrossRef CAS PubMed.
- A. Prajapati, R. Sartape, M. T. Galante, J. H. Xie, S. L. Leung, I. Bessa, M. H. S. Andrade, R. T. Somich, M. V. Reboucas, G. T. Hutras, N. Diniz and M. R. Singh, Energy Environ. Sci., 2022, 15, 5105–5117 RSC.
- G. Lee, A. S. Rasouli, B. Lee, J. Zhang, D. H. Won, Y. C. Xiao, J. P. Edwards, M. G. Lee, E. D. Jung, F. Arabyarmohammadi, H. Liu, I. Grigioni, J. Abed, T. Alkayyali, S. Liu, K. Xie, R. K. Miao, S. Park, R. Dorakhan, Y. Zhao, C. P. O’Brien, Z. Chen, D. Sinton and E. H. Sargent, Joule, 2023, 7, 1277–1288 CrossRef CAS.
- X. L. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- N. S. R. Cuellar, C. Scherer, B. Kackar, W. Eisenreich, C. Huber, K. Wiesner-Fleischer, M. Fleischer and O. Hinrichsen, J. CO2 Util., 2020, 36, 263–275 CrossRef.
- M. Moradzaman and G. Mul, J. Phys. Chem. C, 2021, 125, 6546–6554 CrossRef CAS PubMed.
- J. Gao, A. Bahmanpour, O. Krocher, S. M. Zakeeruddin, D. Ren and M. Gratzel, Nat. Chem., 2023, 15, 705–713 CrossRef CAS PubMed.
- W. Gao, Y. Xu, L. Fu, X. Chang and B. Xu, Nat. Catal., 2023 DOI:10.1038/s41929-023-01002-6.
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed.
- B. De Mot, M. Ramdin, J. Hereijgers, T. J. H. Vlugt and T. Breugelmans, ChemElectroChem, 2020, 7, 3839–3843 CrossRef CAS.
- Y. Xu, J. P. Edwards, S. J. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O'Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- K. Xie, R. K. Miao, A. Ozden, S. J. Liu, Z. Chen, C. T. Dinh, J. E. Huang, Q. C. Xu, C. M. Gabardo, G. Lee, J. P. Edwards, C. P. O'Brien, S. W. Boettcher, D. Sinton and E. H. Sargent, Nat. Commun., 2022, 13, 3609 CrossRef CAS PubMed.
- Y. Xie, P. F. Ou, X. Wang, Z. Y. Xu, Y. C. Li, Z. Y. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. H. Wang, T. X. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- C. P. O'Brien, R. K. Miao, S. J. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C. T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- Z. S. Ma, Z. L. Yang, W. C. Lai, Q. Y. Wang, Y. Qiao, H. L. Tao, C. Lian, M. Liu, C. Ma, A. L. Pan and H. W. Huang, Nat. Commun., 2022, 13, 7596 CrossRef CAS PubMed.
- X. D. Sheng, W. X. Ge, H. L. Jiang and C. Z. Li, Adv. Mater., 2022, 34, e202201295 Search PubMed.
- Z. Jiang, Z. S. Zhang, H. Li, Y. R. Tang, Y. B. Yuan, J. Zao, H. Z. Zheng and Y. Y. Liang, Adv. Energy Mater., 2023, 13, e202203603 Search PubMed.
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao, P. Ou, T. Alkayyali, S. Zhang, J. Ning, Y. Liang, Y. Xu, M. Fan, Y. Chen, J. E. Huang, K. Xie, J. Zhang, C. P. O’Brien, F. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CrossRef.
- A. Ozden, J. Li, S. Kandambeth, X. Y. Li, S. J. Liu, O. Shekhah, P. F. Ou, Y. Z. Finfrock, Y. K. Wang, T. Alkayyali, F. P. G. de Arquer, V. S. Kale, P. M. Bhatt, A. H. Ip, M. Eddaoudi, E. H. Sargent and D. Sinton, Nat. Energy, 2023, 8, 179–190 CrossRef CAS.
- W. X. Nie, G. P. Heim, N. B. Watkins, T. Agapie and J. C. Peters, Angew. Chem. Int. Ed., 2023, 62, e202216102 CrossRef CAS PubMed.
- H. F. Li, H. B. Li, P. F. Wei, Y. Wang, Y. P. Zang, D. F. Gao, G. X. Wang and X. H. Bao, Energy Environ. Sci., 2023, 16, 1502–1510 RSC.
- B. B. Pan, J. Fan, J. Zhang, Y. Q. Luo, C. Shen, C. Q. Wang, Y. H. Wang and Y. G. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS.
- J. Li, X. Li, C. M. Gunathunge and M. M. Waegele, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9220–9229 CrossRef CAS PubMed.
- S. Weng, W. L. Toh and Y. Surendranath, J. Am. Chem. Soc., 2023, 145, 16787–16795 CrossRef CAS PubMed.
- M. Fan, J. E. Huang, R. K. Miao, Y. Mao, P. Ou, F. Li, X. Li, Y. Cao, Z. Zhang, J. Zhang, Y. Yan, A. Ozden, W. Ni, Y. Wang, Y. Zhao, Z. Chen, B. Khatir, C. P. O’Brien, Y. Xu, Y. C. Xiao, G. I. N. Waterhouse, K. Golovin, Z. Wang, E. H. Sargent and D. Sinton, Nat. Catal., 2023, 6, 763–772 CrossRef CAS.
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. Tamadoni Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Nat. Energy, 2023, 8, 891–900 CrossRef CAS.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Aran-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kuhl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- K. Ye, G. Zhang, B. Ni, L. Guo, C. Deng, X. Zhuang, C. Zhao, W.-B. Cai and K. Jiang, eScience, 2023, 3, 100143 CrossRef.
- R. V. Mom, L.-E. Sandoval-Diaz, D. F. Gao, C.-H. Chuang, E. A. Carbonio, T. E. Jones, R. Arison, D. Ivanov, M. Hävecker, B. Roldan Cuenya, R. Schlögl, T. Lunkenbein, A. Knop-Gericke and J.-J. Velasco-Vélez, ACS Appl. Mater. Interfaces, 2023, 15, 30052–30059 CrossRef CAS PubMed.
- A. Bohme, J. C. Bui, A. Q. Fenwick, R. Bhide, C. N. Feltenberger, A. J. Welch, A. J. King, A. T. Bell, A. Z. Weber, S. Ardo and H. A. Atwater, Energy Environ. Sci., 2023, 16, 1783–1795 RSC.
- X. Y. Wang, C. Y. Guo, B. W. Zhu, D. Z. Xiao, D. F. Gao, Z. Liu and F. Yang, J. Chem. Phys., 2023, 158, 204701 CrossRef CAS PubMed.
- L. C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2020, 13, 3592–3606 RSC.
- E. F. Johnson, E. Boutin, S. Liu and S. Haussener, EES Catal., 2023, 1, 704–719 RSC.
- H. Mistry, F. Behafarid, R. Reske, A. S. Varela, P. Strasser and B. Roldan Cuenya, ACS Catal., 2016, 6, 1075–1080 CrossRef CAS.
- H. Simonson, W. E. Klein, D. Henckel, S. Verma, K. C. Neyerlin and W. A. Smith, ACS Energy Lett., 2023, 8, 3811–3819 CrossRef CAS.
- X. Wang, Q. Chen, Y. Zhou, Y. Tan, Y. Wang, H. Li, Y. Chen, M. Sayed, R. A. Geioushy, N. K. Allam, J. Fu, Y. Sun and M. Liu, Nano Res., 2023 DOI:10.1007/s12274-023-5910-9.
| This journal is © The Royal Society of Chemistry 2024 |