
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Harnessing single-atom catalysts for CO2 electroreduction: a review of recent advances
Chang
Chen†
a,
Jiazhan
Li†
c,
Xin
Tan†
a,
Yu
Zhang
a,
Yifan
Li
d,
Chang
He
a,
Zhiyuan
Xu
a,
Chao
Zhang
*b and
Chen
Chen
 *a
*a
aEngineering Research Center of Advanced Rare Earth Materials, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: cchen@mail.tsinghua.edu.cn
bMOE International Joint Laboratory of Materials Microstructure, Institute for New Energy Materials and Low-Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: czhang@email.tjut.edu.cn
cState Key Laboratory of Chemical Resource Engineering, College of Chemistry, Beijing University of Chemical Technology, Beijing 100029, China
dDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, AB T6G1H9, Canada
First published on 13th October 2023
Abstract
Electrochemical CO2 reduction is an effective pathway to convert CO2 into valuable fuels and chemicals, which provides a potential alternative to fossil fuel resources and plays a notable role in mitigating environmental issues and energy crises. The feasibility of the CO2 reduction reaction (CO2RR) hinges on the development of catalysts that feature high activity, selectivity, and stability. As a new research frontier, single-atom catalysts (SACs) have shown immense potential in the field of CO2 reduction by virtue of their unique geometric/electronic structures, and have also provided new opportunities for atomic-level understanding of structure–function relationships. Therefore, this review aims to outline recent advances of SACs for CO2RR. We start by introducing the current research status and general synthesis strategies of SACs, and then shift our focus to analyzing the various regulation strategies and deciphering the structure–function relationships of SACs in the CO2RR. Finally, we propose future directions and opportunities for CO2RR-oriented SACs, while also highlighting potential challenges that may be encountered along the way.
Broader contextExcessive emission of CO2, which causes environmental degradation and energy crises, has caused panic in the scientific community and the general population. Therefore, the development of effective CO2 conversion and utilization technologies is urgent and of great significance. One promising approach is the use of clean energy to drive electrochemical CO2 reduction, which can help mitigate the negative effects of CO2 and generate high-value-added products. However, the extreme inertness of linear CO2 molecules and the competing hydrogen evolution reaction severely affect their catalytic activity and selectivity. Fortunately, single-atom catalysts (SACs), with their unique geometric/electronic structures, have emerged as a frontier research area for the CO2 reduction reaction (CO2RR) and have achieved significant progress. To accelerate the research progress of SACs in the CO2RR, it is necessary to summarize and review the current state of this field and provide guidance for future research. This article first introduces the basic parameters of the CO2RR and summarizes the mainstream reaction mechanisms for generating C1 and C2 products. Then, the current development status of SACs is outlined, along with common synthesis strategies. The application of SACs in the CO2RR is then highlighted, including analyzing the various regulation strategies and deciphering the structure–function relationships of SACs in the CO2RR. Finally, future potential directions and challenges confronted by SACs in the realm of the CO2RR are also contemplated. |
1. Introduction
The continuous consumption of fossil fuels has led to excessive emission of CO2 into the Earth's atmosphere, which has consequently caused climate change and environmental crises, and provoked great concern in the scientific community and the general population.1–5 Besides, as CO2 accounts for over 96% of the Martian atmosphere, utilizing CO2 resources for Mars exploration is also a new research direction for future human space programs.6 Thus, effective CO2 conversion and utilization technologies are pressing and important grand research challenges in maintaining carbon neutrality balance and alleviating energy shortages. To address the CO2 issue, different routes have been proposed: (1) capturing and storing CO2;3,7–9 (2) improving the efficiency of fossil fuel combustion and reducing emissions;10 and (3) chemically converting CO2 to form high-value-added products.11–16 Among these options, the electrochemical CO2 reduction reaction (CO2RR) has emerged as one of the most promising routes to addressing the issues of recycling CO2 and producing valuable chemical products.17–20 Furthermore, the CO2RR can be driven by renewable (but intermittent) energy sources (such as wind, solar, and tidal) without extra CO2 emission, and the reduction process can be easily performed by adjusting the potential range and current densities under ambient pressure and temperature.17,21,22However, the CO2RR suffers from two stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds (bond energy, 806 kJ mol−1) in the extremely inert linear CO2 molecule, generally leading to a low energy efficiency with high overpotentials.23,24Table 1 summarizes the standard potentials E0 (with respect to the standard hydrogen electrode, SHE) of CO2RR processes and the competitive hydrogen evolution reaction (HER) in aqueous media.17,18,25 Specifically, a significant external driving force is needed to initiate the single-electron reduction of CO2 to the key intermediate *CO2− during the CO2RR process (CO2 + e− → *CO2−), or to undergo the proton-coupled electron transfer (PCET) process and produce different hydrocarbons and oxygenates, including (but not limited to) CO, HCOOH, HCHO, CH3OH, CH4, C2H4, and C2H5OH.26,27 In addition, thermodynamically, the reduction potentials for the main reduction products mentioned above in the CO2RR typically fall within the range of ±0.2 V from that of the HER.17 However, the presence of a high barrier for CO2 activation and the involvement of multiple electron transfer steps in the reaction pathways of the CO2RR result in sluggish kinetics and side reactions involving the HER. As a result, the CO2RR usually shows high overpotentials and poor selectivity. In this regard, developing efficient and highly selective electrocatalysts to promote CO2RR technology towards industrial applications is the key to addressing the aforementioned challenges.20,24
O double bonds (bond energy, 806 kJ mol−1) in the extremely inert linear CO2 molecule, generally leading to a low energy efficiency with high overpotentials.23,24Table 1 summarizes the standard potentials E0 (with respect to the standard hydrogen electrode, SHE) of CO2RR processes and the competitive hydrogen evolution reaction (HER) in aqueous media.17,18,25 Specifically, a significant external driving force is needed to initiate the single-electron reduction of CO2 to the key intermediate *CO2− during the CO2RR process (CO2 + e− → *CO2−), or to undergo the proton-coupled electron transfer (PCET) process and produce different hydrocarbons and oxygenates, including (but not limited to) CO, HCOOH, HCHO, CH3OH, CH4, C2H4, and C2H5OH.26,27 In addition, thermodynamically, the reduction potentials for the main reduction products mentioned above in the CO2RR typically fall within the range of ±0.2 V from that of the HER.17 However, the presence of a high barrier for CO2 activation and the involvement of multiple electron transfer steps in the reaction pathways of the CO2RR result in sluggish kinetics and side reactions involving the HER. As a result, the CO2RR usually shows high overpotentials and poor selectivity. In this regard, developing efficient and highly selective electrocatalysts to promote CO2RR technology towards industrial applications is the key to addressing the aforementioned challenges.20,24
| Half-cell reaction | Standard potentials |
|---|---|
| 2H+ + 2e−→ H2 | −0.42 |
| CO2 + e− → *CO2− | −1.9 |
| CO2 + 2H+ + 2e− → CO + H2O | −0.52 |
| CO2 + 2H+ + 2e− → HCOOH | −0.61 |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.51 |
| CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | −0.34 |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | −0.33 |
During the past decades, a broad class of electrocatalysts has been developed for the CO2RR, including metals,28–31 metal oxides,32–37 metal alloys,38–41 molecular compounds,42 metal phosphides/chalcogenides,43,44 and metal-free carbon-based materials.45–47 Most notably, single-atom catalysts (SACs) have emerged as a new family of catalytic materials by virtue of their distinctive structural properties and exceptional catalytic performances.48,49 As a new class of frontier materials, the SACs feature maximized atomic utilization with their active sites isolated with each other but interacting with the support.50 In 2011, Qiao et al. reported a single-atom platinum catalyst supported on iron oxide nanocrystallites (Pt1/FeOx), and the concept of a “single-atom” catalyst was proposed for the first time.51 Since then, the concept was quickly accepted by the scientific community and the scope of relevant materials has broadened significantly. Various materials have been explored as the support (such as carbon materials,52,53 MXenes,54 oxides,55 and carbides56), and the elements for the metal active centers have nearly covered all the non-radioactive metals in the periodic table.57,58 The CO2RR over SACs can be traced back to the 1970s, when molecular catalysts (cobalt/nickel phthalocyanines) were used as the active sites.59 With the development of synthesis technology, characterization technology (high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) and X-ray absorption spectroscopy (XAS), in particular), and theoretical computational chemistry, the establishment of the structure–function relationship between the structure of SACs and the performance of the CO2RR has made great progress. Specifically, SACs have adjustable coordination environments and structures, making it easier to obtain uniform catalytic active centers. Furthermore, SACs can also serve as an ideal bridge between homogeneous and heterogeneous catalysis.48–50 For the CO2RR, the reaction process typically involves multiple PCET steps accompanied by complex intermediates, and SACs with well-defined structures have become an ideal research model. This could help to identify the reaction mechanism and to understand the structure–function relationship of SACs in the CO2RR, guiding us to develop new advanced electrocatalysts. For example, SACs featuring Ag, Mn, Fe, and Ni single-atom active sites often exhibit excellent CO selectivities.27 In the process of the rational design of CO2RR catalysts, researchers have found that the generation of non-CO products (such as C2+ products) is very difficult and complex.60 Because of the isolation and simplicity of their active sites, SACs are not so competent in some cases, prompting researchers to develop dual-atom catalysts or multi-atom catalysts (DACs or MACs) with more than one type of active sites.61,62 There could be a synergistic effect between the dual-atom sites or multi-atom sites, providing more possibilities and pathways for the generation of high value-added products.63–67
Compared with other types of heterogeneous catalysts, SACs have unique advantages in geometric/electronic structures, which gives them great potential to become marvellous catalysts for the CO2RR. In this review, as shown in Fig. 1, we use the metaphor of an alchemical furnace as the main concept, with three supporting pillars representing the general synthesis strategies for SACs. The four different “elixirs” within the furnace chamber represent the diverse regulation strategies for SACs in CO2RR applications. Eventually, the reactant CO2 undergoes transformations within this alchemical furnace to produce various products. At the end of this review, we present an outlook on the future directions, opportunities, and challenges in the field of SACs for the CO2RR.
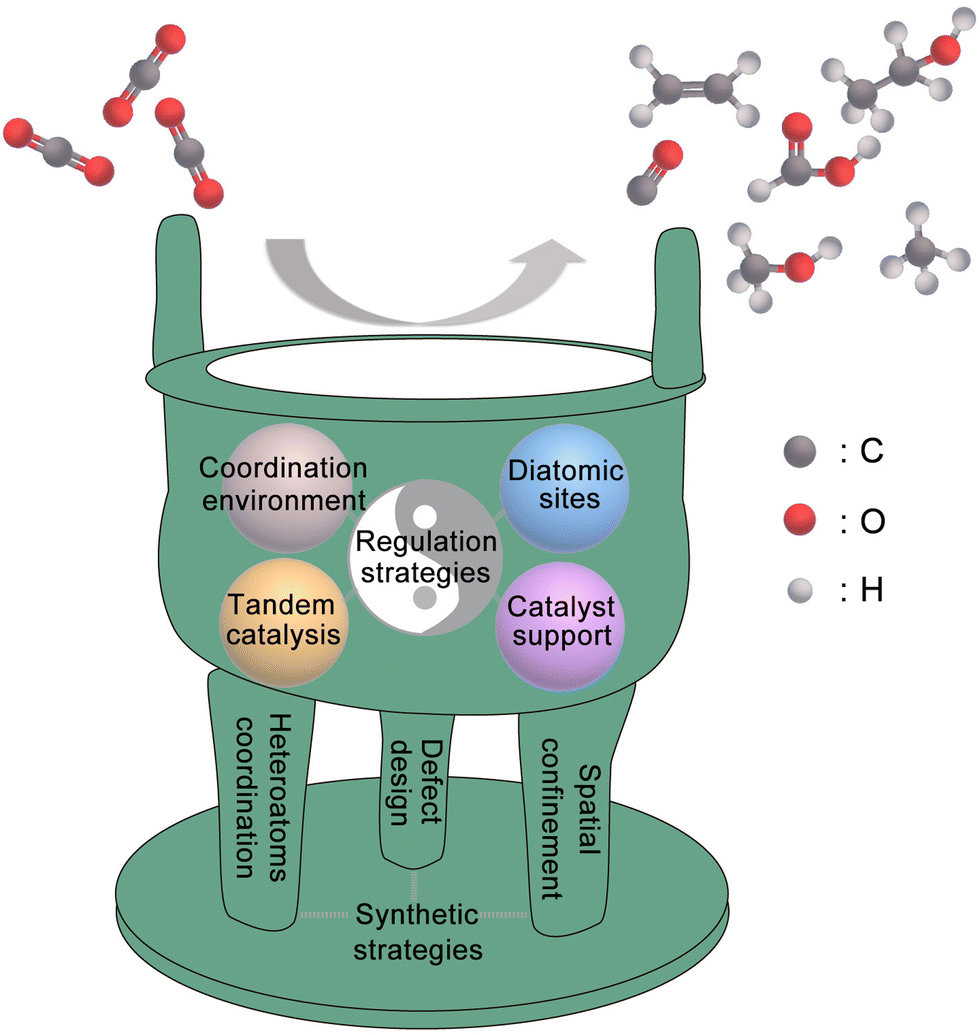 | ||
| Fig. 1 Schematic diagram of the SACs for the CO2RR in this review (including the general synthesis strategies for SACs and the regulation strategies of SACs for the CO2RR). | ||
2. Fundamental parameters and reaction mechanisms of the CO2RR
2.1 Fundamental parameters of the CO2RR
It is necessary to introduce the fundamental parameters before proceeding further with this review. At present, the standard metrics used to evaluate and compare the performance of CO2RR electrocatalytic materials mainly include the following: faradaic efficiency (FE), onset potential, current density, partial current density, stability, turnover frequency (TOF).25,68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1); Q represents the total amount of charge during the CO2RR process. By analysing the FE, the difference in selectivity of different catalysts for the CO2RR can be directly compared.
485 C mol−1); Q represents the total amount of charge during the CO2RR process. By analysing the FE, the difference in selectivity of different catalysts for the CO2RR can be directly compared.
2.2 Reaction mechanisms of the CO2RR
The CO2RR is a rather complex process involving multiple PCET steps, with different reaction pathways associated with 2–18 electron transfers, resulting in a wide distribution of products, including C1 products (such as CO, HCOOH, CH3OH, HCHO, and CH4), and C2+ products (such as C2H4, C2H5OH, CH3COOH, and CH3COCH3). Great efforts have been made both experimentally and theoretically to elucidate the CO2RR mechanism, but there is still a lack of consensus on some reaction steps, and their reaction mechanisms are still not fully understood. More in situ characterization techniques, even more advanced coupled multi in situ characterization techniques, as well as theoretical calculations are needed to further decipher the CO2RR reaction mechanism. Here, we summarize the possible reaction pathways of C1 products and the main C2 products based on the current prevailing understanding of CO2RR mechanisms, so as to design high-activity, high-selectivity, and high-stability SAC catalysts rationally.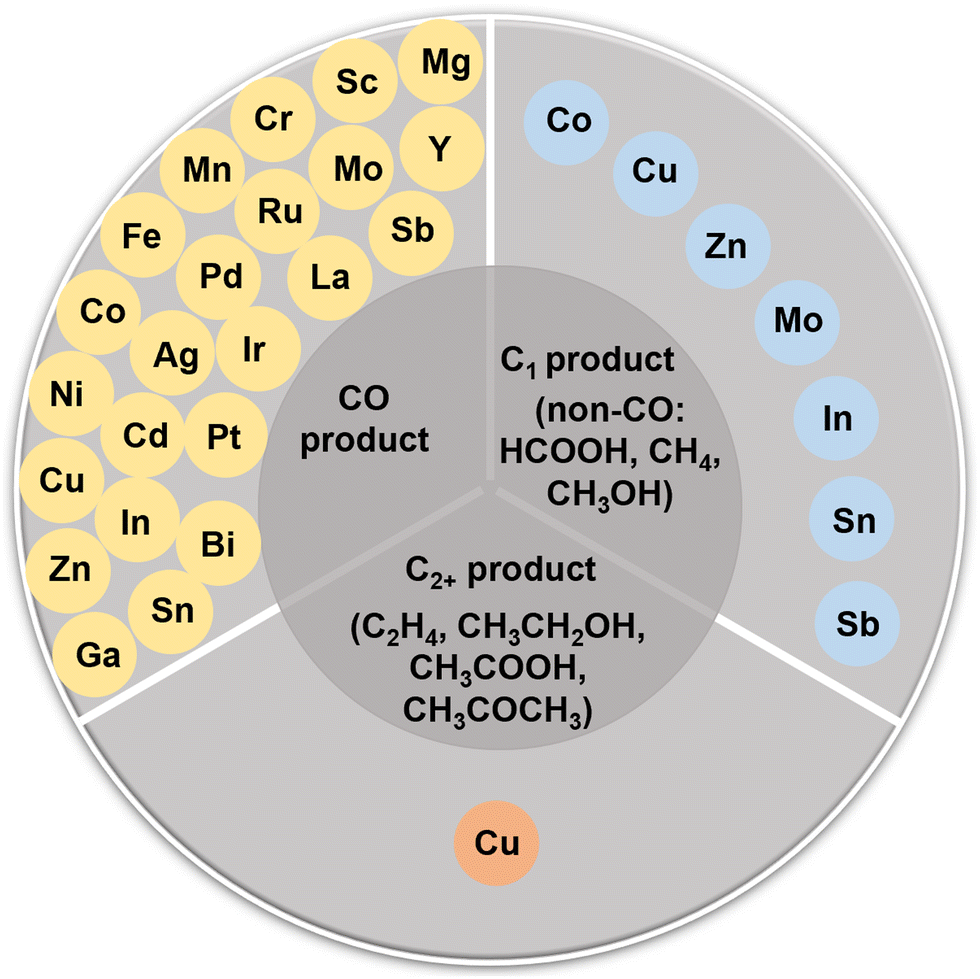 | ||
| Fig. 2 Schematic diagram of the reported elements in SACs for CO2RR. | ||
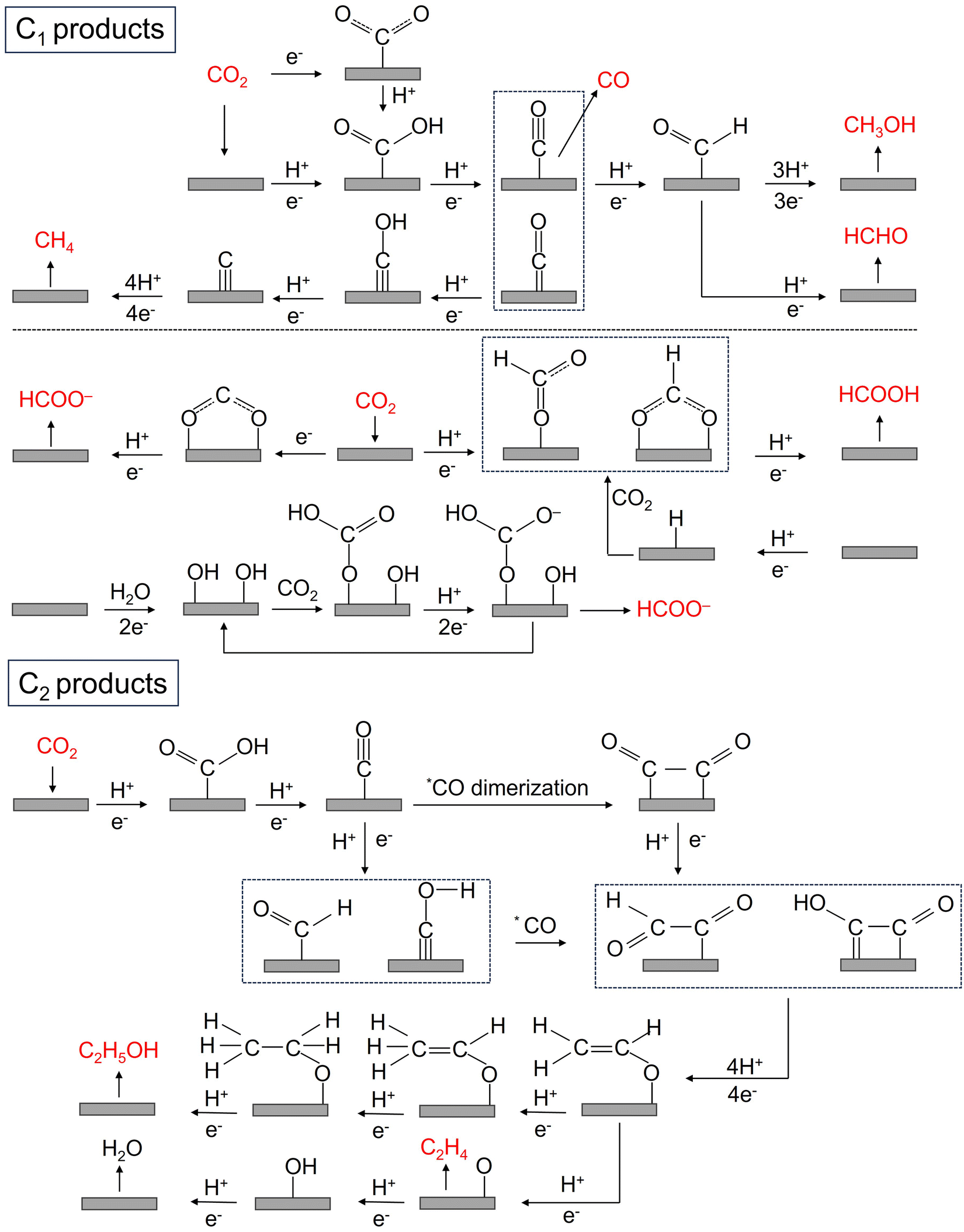 | ||
| Fig. 3 The possible reaction pathways of C1 and C2 products in the CO2RR. | ||
Unlike other C1 products, the generation of HCOOH or HCOO− has a reaction pathway of its own. The generation of HCOOH or HCOO− may be achieved through an intermediate, which is believed to be mainly through the binding of oxygen atoms to the catalyst surface, and the generation of this intermediate may have two different pathways: (1) CO2− radicals bind to the catalyst surface and react with adjacent water molecules or protons to generate HCOO− or HCOOH; (2) CO2 is directly inserted into metal–hydrogen or metal–hydroxyl bonds to form intermediates, which further yields HCOOH or HCOO−.70,77
3. Synthesis strategies and characterization techniques of SACs
3.1 Synthesis strategies of SACs
The term “SACs” was coined on the basis of the characteristics of their active centers; however, in reality, individual atoms cannot exist in isolation but instead rely on a supporting material.48,49 Owing to their high surface energy, isolated metal atoms have a tendency to aggregate into thermodynamically more stable metal clusters. Hence, the interaction between the support and single atoms is crucial in preventing the aggregation of individual atoms. Since the concept of “single-atom-catalysts” emerged in 2011,51 various strategies have been proposed to synthesize SACs with high metal loadings and high CO2RR performances. In this section, we mainly discuss the challenges and current strategies for increasing the metal loading and tuning the electronic structure of SACs, which are categorized into “heteroatom coordination strategy”, “spatial confinement strategy” and “defect design strategy”. In addition, some other advanced synthesis strategies of SACs are also introduced.Many studies have already shown that the physicochemical state of SACs can be optimized by adjusting the coordination number, coordination structure and coordination environment of the metal single-atom centers, thus further improving their CO2RR performance. The coordination number of the central metal atoms can modulate the electronic structure of the catalyst and thus alter the catalytic performance. Wang et al. reported two Co-based catalysts with different coordination numbers, denoted as Co–N4 and Co–N2. The catalysts were prepared by adjusting the pyrolysis temperature to control the extent of volatilization (Fig. 4a).86 Zhang et al. devised a post-synthetic metal substitution (PSMS) strategy in combination with the characteristic that Zn-based nitrogen-doped carbon is easily converted into a Zn–N3 structure at high temperature.87 The uniformly dispersed Ni–N3–C with a metal loading of 0.85 wt. % was obtained, which had a lower *COOH free energy than Ni–N4–C. The coordination structure of SACs can also affect the CO2RR performance. By replacing part of the Zn2+ sites on nitrogen-doped carbon nanotubes with Ni2+, Hou et al. developed Ni SACs for the efficient conversion of CO2 to CO.88 By direct pyrolysis of metal–organic frameworks (MOFs) assembled with Fe and Ni-doped ZnO nanoparticles, Jiao et al. precisely constructed a novel Fe1–Ni1–N–C catalyst, with neighboring Fe and Ni single-atom pairs decorated on a nitrogen-doped carbon support (Fig. 4b).89 The unique architecture of the carriers facilitates the design of the local coordination environment for SAC active sites. Using covalent organic frameworks (COFs) as supports, the metal active sites in SACs can be uniformly dispersed on the multidimensional structure with a high specific surface area, thus the catalysts typically display excellent conductivity and conduction efficiency.90–92 MOFs have great potential for application in the CO2RR because of their well-defined metal nodes, tunable organic ligands and ordered pore structure.93,94 The preparation of SACs using MOFs can follow two strategies. First, catalytic organometallic complexes or inorganic metal species are immobilized into MOF pores. Second, the single metal sites are intrinsically created into metal nodes and metallolinkers within frameworks during and before the synthesis process. With MOF carbonization and by firmly embedding single atoms into the carbon skeleton through robust metal–heteroatom (N, O, or S) coordination bonds, SACs with exceptional features and applications can be obtained.95
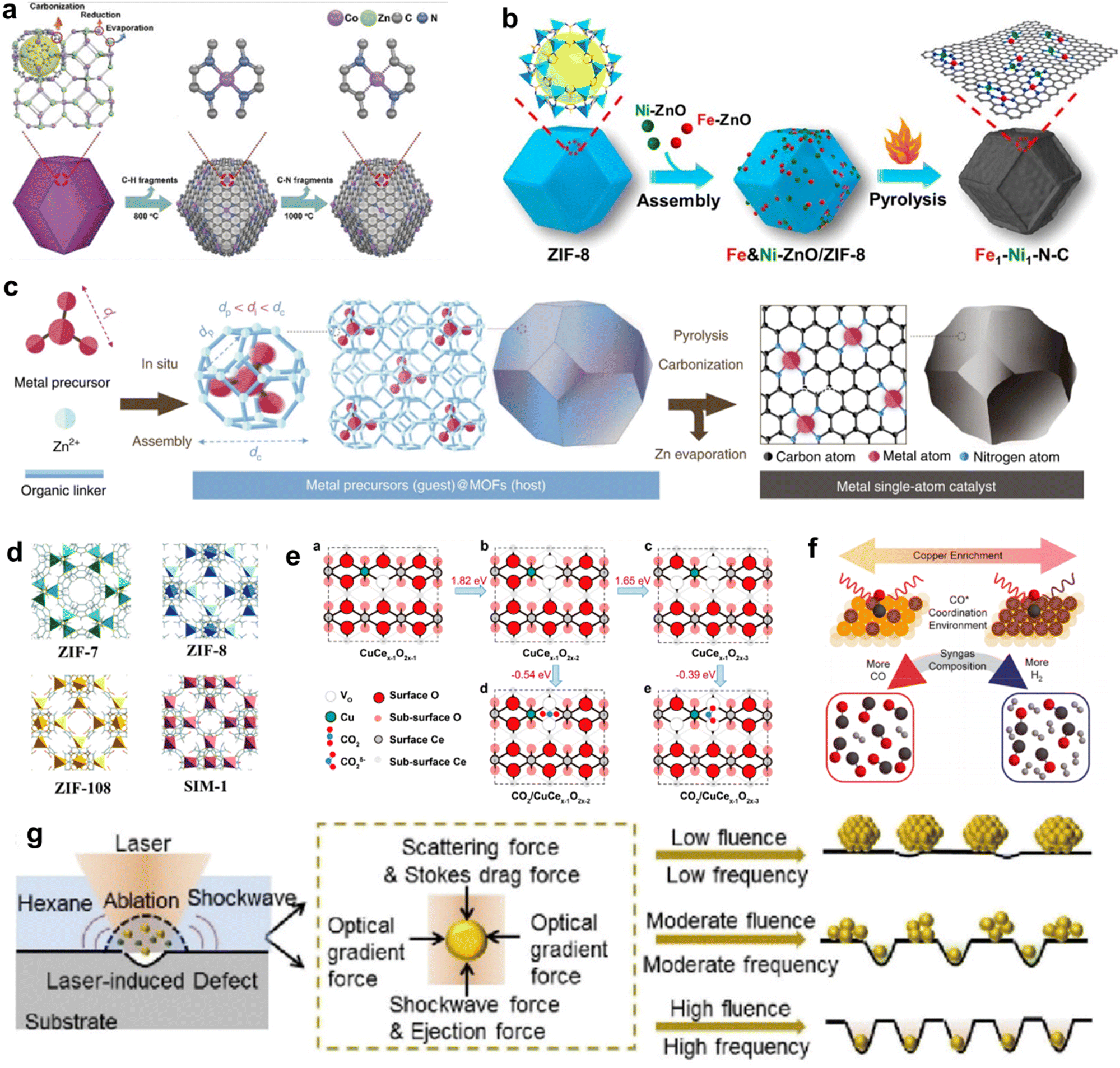 | ||
| Fig. 4 (a) The formation process of Co–N4 and Co–N2.86 (b) Schematic illustration for the construction of Fe1–Ni1–N–C with neighbouring Fe and Ni single-atoms based on ZIF-8.89 (c) Scheme of the host–guest strategy for the fabrication of isolated metal single-atom-site catalysts.100 (d) The model structures of ZIF catalysts.102 (e) Theoretical calculations of the most stable structures of Cu-doped CeO2(110) and their effects on CO2 activation.106 (f) Scheme depicting the relationship between the Cu-enriched Au surface, in situ characterization of CO* coordination, and syngas composition.113 (g) Schematic diagram depicting the synthesis of Pt nanoparticles, Pt clusters, and Pt single atoms under laser planting with low, moderate, and high fluences and frequencies, respectively.118 | ||
In recent years, a variety of metal SACs have been successfully synthesized using the coordination design strategy. However, the relationship between the coordination structure of SACs and the performance of CO2RR is not sufficiently clear. To design CO2RR electrocatalysts with high activity and selectivity, the structure–performance relationships need to be further investigated.
In summary, the spatial confinement strategy could significantly suppress the migration and aggregation of metal atoms within the porous carrier, while the synergy between the metal single-atom sites and the shape-selective carrier improves the CO2RR performance. The key issue in using this strategy is the selection of suitable metal precursors and support materials with uniform cage/pore sizes to ensure metal species confinement.
In general, the construction of atomically dispersed metal catalysts through defect engineering has proven to be an effective strategy. The type and quantity of defects significantly affect the stability and electronic structure of single atom active centers.20 Therefore, facile and effective methods of defect construction still need to be developed. In addition, it is of great significance to investigate the formation mechanism of isolated catalytic centers at defect sites for the design of SAC catalyst configuration and the synthesis of high-loading SACs.
Chemical vapor deposition is a novel method for synthesizing single atoms, Qu et al. developed a gas-migration/NH3-mediated strategy to convert bulk materials (Cu, Co or Ni) directly into single atoms.116 At high temperatures, ammonia molecules were used as a medium to drag metal atoms out of bulk metal to form gas-phase complexes, then the complex molecules were captured by defects on the nitrogen-rich carbon support, forming isolated metal sites. Considering that the corrosiveness of NH3 would pose stringent requirements on the equipment, a new gas-migration strategy that employed volatile metal oxides as a single atom source was demonstrated.117 At high temperatures, metal oxides were evaporated to generate gaseous species, which were transported onto nitrogen-doped carbon and anchored by defects to form isolated metal atom/NC catalysts. Recently, a novel one-step laser-planting strategy was employed to create a set of high-density SACs on various supports, including carbon black, graphene quantum dots, metals, and oxides (Fig. 4g).118 The laser pulses created defects on the support and simultaneously decomposed the precursors into metal atoms, which were subsequently immobilized on the as-produced defects via electronic interaction. Using this strategy, a high loading of single atoms of 41.8 wt.% has been achieved.
In addition to the above-mentioned synthesis methods, a few other special strategies have been proposed including nanoparticles becoming single atoms via solid diffusion,119–121 solid-state atom diffusion,122,123in situ thermal atomization methods,119 electrospinning polymer nanofibers,124 and crosslinking and self-assembly of graphene quantum dots.125
3.2 Characterization techniques of SACs
Upon providing an overview of the synthesis strategies for SACs, determining the geometric structure and electronic environment of SACs is crucial for investigating their structure–function relationship in CO2RR applications. Primarily, ascertaining the geometric structure enables the provision of compelling evidence to discern between SACs and nanoparticle catalysts, thus illuminating the intricate coordination configuration inherent in SACs. Concomitantly, delving into the electronic environment facilitates the acquisition of critical data concerning the electron orbitals and oxidation states of catalytic active centers. While a plethora of techniques exist for material characterization, such as X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), Raman, and others, their applicability may fall short in fulfilling the exigencies of atomic-resolution structural characterization of SACs. In contrast, characterization techniques tailored expeditiously for resolving the geometric/electronic structures of SACs predominantly involve AC-HAADF-STEM, XAS, and Mössbauer spectroscopy. Herein, we will not delve into the details of each characterization technique, but rather focus on discussing the distinct characterization techniques tailored for SACs.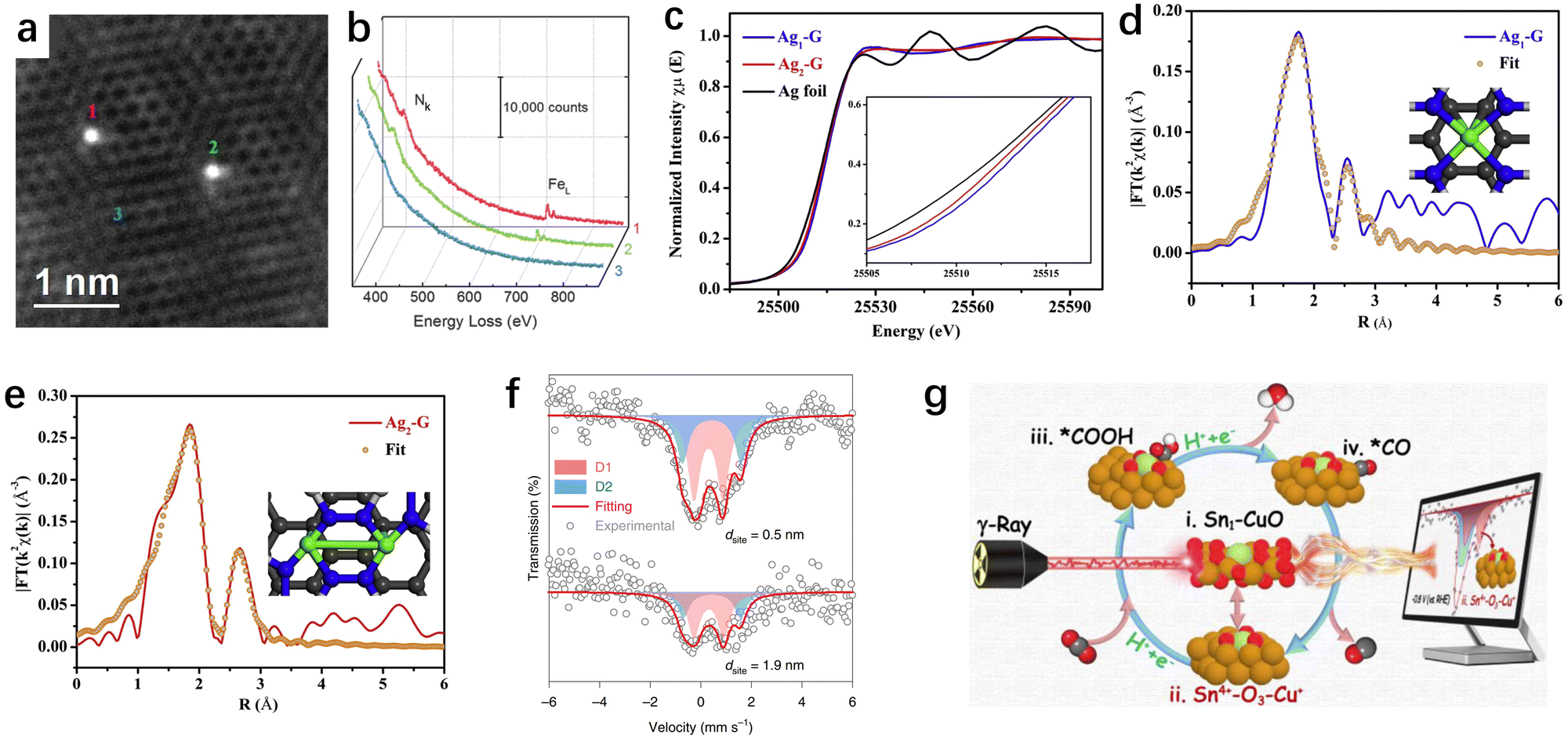 | ||
| Fig. 5 (a) AC-HAADF-STEM image of individual Fe atoms (labelled 1, 2, and 3) in a few-layer graphene sheet.126 (b) EEL spectra of the N k-edge and Fe L-edge acquired from single atoms (1 and 2) and few-layer graphene (3).126 (c) Ag K-edge XANES spectra (inset is the expanded view of the rising edge).127 EXAFS R-space fitting curves for Ag1–G (d) and Ag2–G (e), respectively. The inset of (d) and (e) is the schematic model of Ag1–G and Ag2–G, respectively (Ag in green, N in blue, and C in gray).127 (f) Iron-57 Mössbauer spectroscopy of two catalysts with dsite = 0.5 and 1.9nm. The curves were offset for clarity.129 (g) 119Sn Mössbauer spectroscopy as a probe technique to track the catalyst's metastable species.130 | ||
4. Structure–function relationship of SACs in electrochemical CO2RRs
4.1 Coordination environment regulation
The coordination environment of SACs can directly influence the electronic and geometric structure of the metal active site, thereby altering the adsorption/desorption processes of key intermediates in the CO2RR. Additionally, different coordination environments can result in distinct microenvironments for the CO2RR, such as interfacial water and local pH, playing a significant regulatory role in the reaction process. In this section, we primarily focus on the discussion of two types of coordination regulations: in-plane and out-of-plane coordination.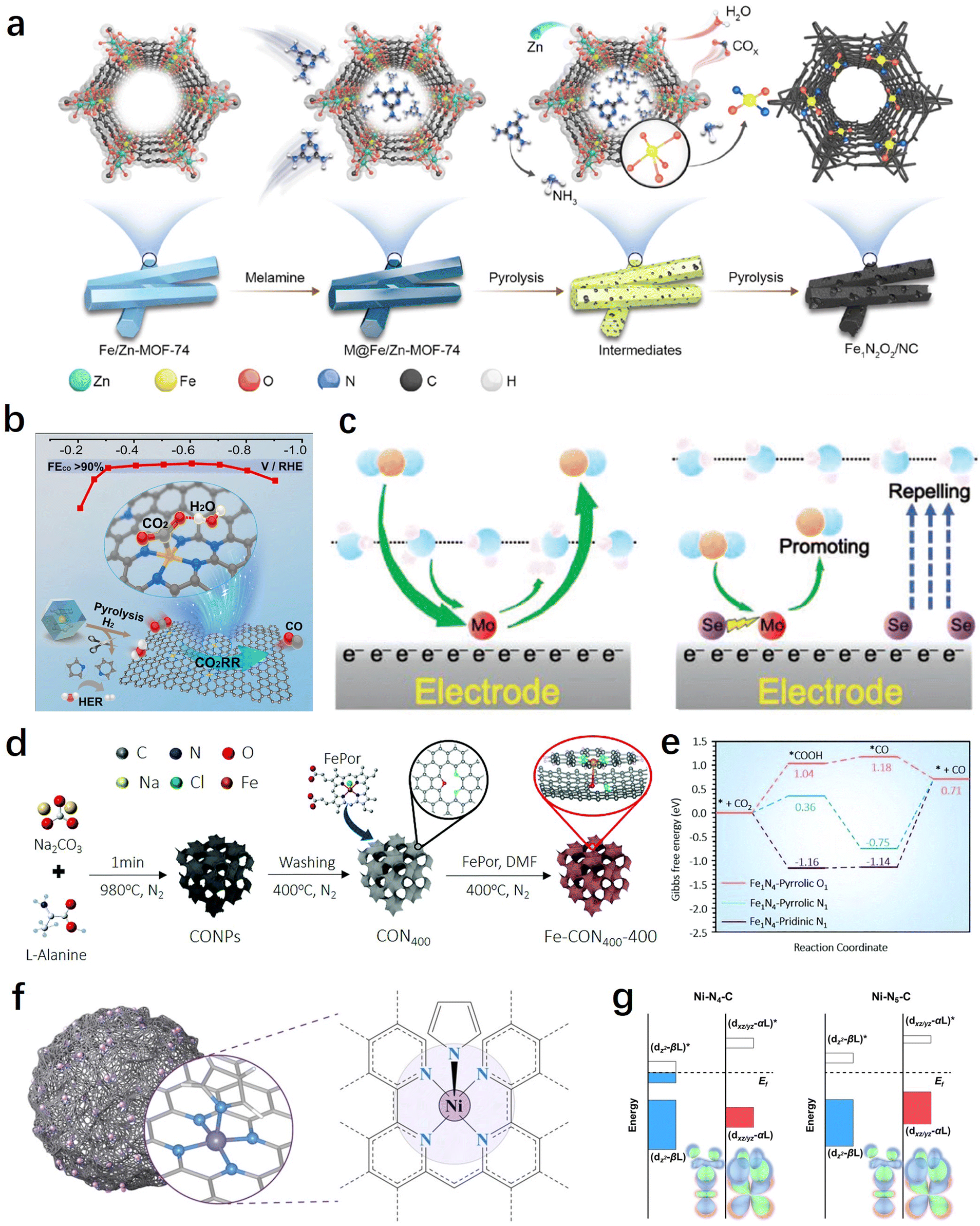 | ||
| Fig. 6 (a) Schematic illustration of the preparation of Fe1N2O2/NC.132 (b) Synthesis method and CO2RR performance of H2–FeN4/C.135 (c) Schematic diagram of the reaction mechanism of MoSA–SeSA.136 (d) Schematic of the synthetic process for well-designed iron single-atomic catalysts.138 (e) CO2RR pathways on Fe sites of different catalyst configurations.138 (f) Schematic of the single-atom nanozyme Ni–N5–C.140 (g) Orbital interactions between Ni centers (dz2, dxz/yz) and adsorbed COOH on Ni–N4–C and Ni–N5–C.140 | ||
H2O is a molecule that directly participates in the reaction, generating H through dissociation for the PCET process. Therefore, it is crucial to regulate the interfacial water for efficient CO2RR.134 In this regard, besides directly adjusting the electronic structure of the metal center by optimizing the first coordination sphere, controlling the interfacial microenvironment through non-coordinating heteroatom doping also has significant importance in the development of high-performance SACs. For example, in order to promote the synergistic adsorption and activation of CO2 and H2O, Liu et al. constructed a SASC with FeN4/graphitic N atomic interface sites through a H2-assisted pyrolysis strategy, which achieved a CO faradaic efficiency of over 90% within the range of −0.3 to −0.8 V.135Operando attenuated total reflection surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) and the first-principle calculations indicate that the presence of graphitic N sites facilitates the co-adsorption of H2O, thereby promoting the activation of CO2 and its conversion into the key intermediate *COOH (Fig. 6b). In another work, Sun et al. achieved efficient electrochemical CO2 to CO conversion on Mo SACs through the incorporation of Se into the carbon support.136 The study demonstrates that the introduction of neighbouring Se (6.0 Å) can effectively improve the reaction thermodynamics of the MoN4 sites through long-range electronic interactions, promoting *CO desorption. On the other hand, the remaining Se far away from Mo SACs (>8.5 Å) can improve the electrode/electrolyte interface by repelling water molecules to suppress the hydrogen evolution reaction (HER), thereby enhancing the selectivity of the CO2RR (Fig. 6c).
From the above results, it can be observed that high selectivity in electrochemical CO2-to-CO conversion has been achieved through the coordination structure and electronic modulation of SACs. However, there are still limitations in terms of current density and catalyst durability. Recently, Huang et al. made a breakthrough in achieving high selectivity, high current density, and sustained catalyst durability through axial coordination engineering.140 Inspired by natural metalloenzymes, they designed and constructed a single-atom nanoenzyme with a Ni–N5–C structure featuring axial N coordination (Fig. 6f). Compared to the traditional planar Ni–N4 coordination structure, the square-pyramidal NiN5 site's dz2 and dxz/yz orbital energy levels were increased and decreased, respectively (Fig. 6g). This facilitates the adsorption and activation of CO2 molecules and the desorption of CO, greatly enhancing the reaction kinetics. Therefore, the synthesized catalyst achieved a 99.6% CO faradaic efficiency (turnover frequency of 69.7 s−1) at an ultra-high current density of 1.23 A cm−2, and exhibited exceptional stability for 100 hours, surpassing significantly many other reported SACs. In addition to the coordination of N and O, elements such as S, Cl, Br, and I have also demonstrated promising potential in axial coordination engineering.141,142 Overall, the proposal and development of axial coordination regulation will contribute to further investigations of the mechanism of single-atom catalysts, as well as expanding the strategies and methods for the regulation of SACs.143,144
4.2 Diatomic sites regulation
In recent years, DACs have gained extensive attention as extensions of SACs.145 Compared with SACs, DACs possess more complex and flexible active sites, enabling them to achieve better catalytic performance. This is particularly important for the CO2RR involving various reaction intermediates. When all intermediates are adsorbed on a single active site, it becomes challenging to achieve optimal adsorption states for each intermediate. By constructing bimetallic active sites, the adsorption energy of different intermediates can be optimized, thereby breaking this linear relationship and achieving greater catalytic performance. Furthermore, the construction of diatomic sites has potential for achieving synergistic adsorption and activation of both CO2 and H2O, thereby further enhancing reaction kinetics. In this section, we will categorize DACs into homonuclear diatomic sites and heteronuclear diatomic sites for discussion.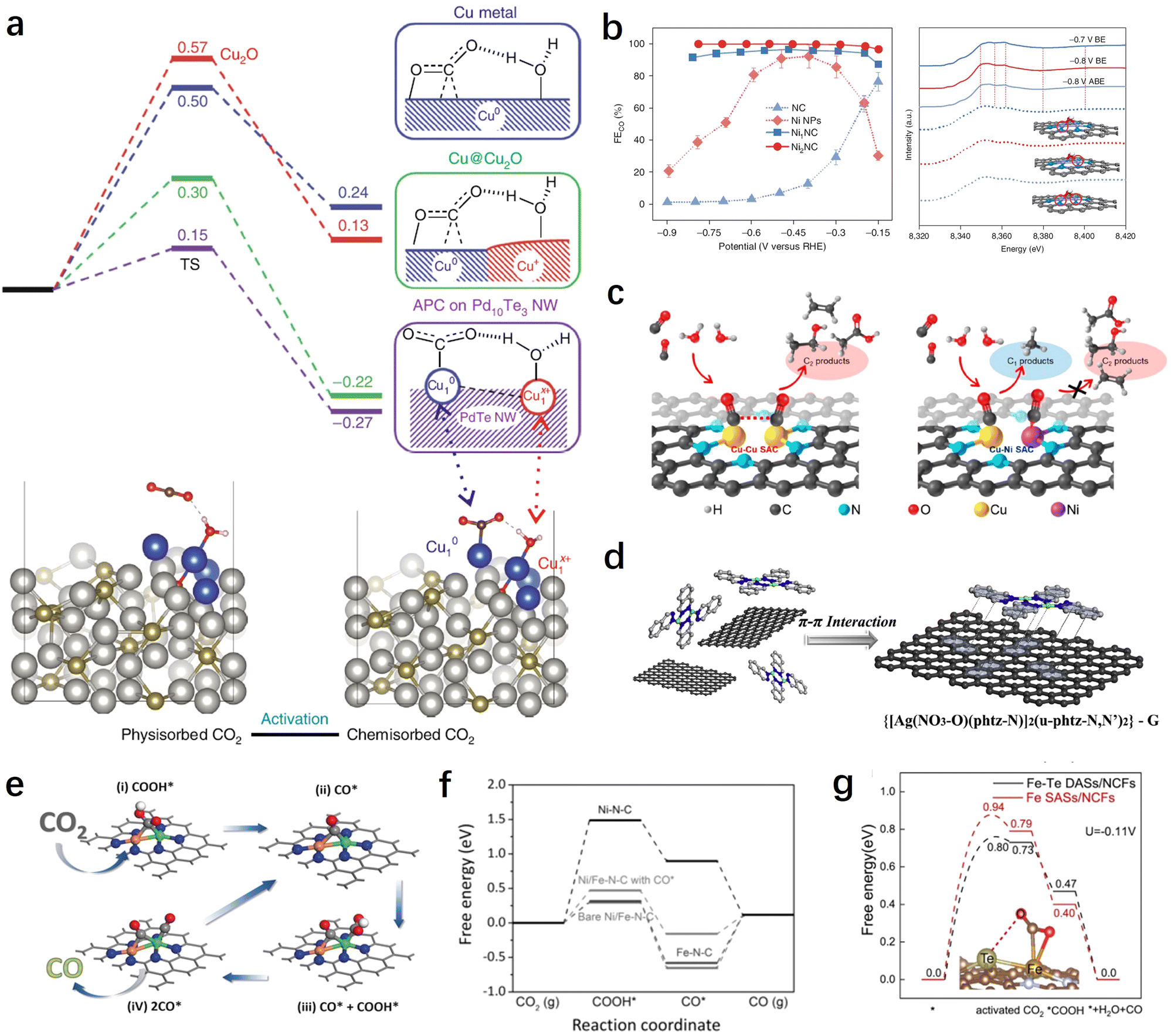 | ||
| Fig. 7 (a) Free energy profiles for the CO2 activation mode of Cu-APC catalysts.111 (b) CO2RR performance and XANES spectra of Ni2NC catalysts.96 (c) Schemes showing the structures and products for the CO2RR on dual Cu–Cu and Cu–Ni SACs.146 (d) Schematic illustration of the synthesis process of Ag2–G catalysts.127 (e) The catalytic mechanism on diatomic NiFe catalysts based on the optimized structures of adsorbed intermediates COOH* and CO*.147 (f) Calculated free energy diagrams for the CO2RR yielding CO on different catalysts.147 (g) The free energy diagram of the CO2RR on Fe–Te DASs/NCFs.149 | ||
In addition, DACs have demonstrated superior performance and advantages over SACs in the synthesis of multi-carbon products. For instance, by constructing dual-atomic Cu sites, Li et al. successfully achieved efficient production of C2 products (mainly ethylene and acetic acid) on DACs with a high faradaic efficiency of 91% and a partial current density of 90 mA cm−2.146 Theoretical calculations suggest that the neighboring Cu sites can facilitate the carbon–carbon coupling process of CO. Interestingly, when one of the Cu atoms is replaced by a Ni atom, this highly efficient carbon–carbon coupling pathway is terminated as a result of the excessive CO adsorption by the Ni atom (Fig. 7c). This finding emphasizes the importance of compatibility between bimetallic centers for the CO2RR process.
Despite some breakthroughs in the activity and selectivity of DACs for the CO2RR, there are still difficulties and challenges in the precise synthesis and controllable preparation of dual-atomic sites. To this end, Li et al. have developed a dual-core metal molecular pre-synthesis and post-adsorption strategy.127 They first synthesized a well-defined binuclear Ag complex and then obtained DACs with AgN3–AgN3 sites through the π–π interaction between the complex and graphene, as well as low-temperature treatment (Fig. 7d). These AgN3–AgN3 sites demonstrated greater advantages in synergistic adsorption and activation of CO2 compared to single Ag sites.
Modulating the activation mode of CO2 molecules through synergistic effects between neighboring atoms is an attractive yet challenging strategy. Pan et al. utilized neighboring Te atoms to induce and regulate the Fe site and constructed Fe–Te diatomic sites for synergistic CO2RRs.149 The results demonstrate that the low-valent Feδ+ forms bonds with one carbon and one oxygen atom of the CO2 molecule, while the adjacent Teδ+ acts as an electron donor, regulating the electronic structure of the Feδ+ site and stabilizing the other oxygen atom of the CO2 molecule. This enhances the 2πu orbital interaction of CO2 and makes the CO2 molecule more prone to bending, thereby reducing the activation barrier and improving the CO2RR performance (Fig. 7g). In addition to transition metals and main group metals, rare earth metals have shown significant potential in modulating the catalytic activity of metal sites and activating CO2 due to their unique electronic structure and catalytic properties. Therefore, designing and constructing heteronuclear diatomic sites containing rare earth elements show promise for further tuning the reaction pathways and performance of CO2RRs. For instance, Liang et al. synthesized atomically dispersed InCe/NC catalysts using a one-step method.150 Experimental results demonstrated that the introduction of Ce promotes the electrochemical conversion of CO2 to formate, while the conversion of CO and the hydrogen are suppressed. Theoretical calculations revealed that the incorporation of single Ce atoms not only significantly facilitates electron transfer but also optimizes the In-5p orbitals, enhancing the adsorption energy of the key intermediate *OCHO on the In sites along the formate pathway.
4.3 Tandem catalytic regulation
Currently, electrochemical CO2 reduction for the production of simple 2e− products such as CO or formate has achieved high selectivity and activity, particularly when utilizing single-atom catalysts. However, significant challenges persist in the production of hydrocarbons or alcohols from the CO2RR. It is generally understood that the conversion of CO2 to multi-electron deep reduction products typically involves a two-step sequential process:151 (1) CO2 reduction to CO; (2) further reduction of CO to obtain deep reduction products. The electrochemical CO2 reduction process comprises multiple coupled or consecutive proton-electron transfer steps, and the design and synthesis of tandem catalytic systems using single-atom catalysts as CO-forming or water activation sites show significant potential for facilitating the production of high-value products in these two steps.The potential matching between different materials needs to be carefully considered prior to designing such a tandem catalytic system, where the high selectivity generation of CO and subsequent C–C coupling processes need to be simultaneously achieved at a specific potential. Generally, for Cu-based catalysts, C–C coupling often occurs at relatively negative potentials (>−1 V vs. RHE). Therefore, it is essential for SACs to have a broad potential range while maintaining a high CO faradaic efficiency. According to reports, Ni SACs have been shown to maintain a CO selectivity of over 97% even at −2.4 V vs. RHE, making them an ideal candidate material for these tandem catalytic systems.140 For instance, Yin et al. prepared hybrid tandem catalysts by combining Ni single atoms loaded on high surface area carbon supports with Cu nanowires (Fig. 8a).154In situ SEIRAS studies revealed that the incorporation of Ni single atoms significantly enhanced the enrichment of CO on the Cu surface, thereby facilitating the C–C coupling process. By adjusting the ratio of hybrid components, the optimized catalyst achieved a 66% ethylene faradaic efficiency in a flow-cell device, which was improved by five times compared to pure copper. Similarly, Meng et al. prepared a non-precious metal-based tandem electrocatalyst, PTF(Ni)/Cu, by anchoring Ni single atoms and loading Cu nanoparticles onto a porphyrin-based triazine framework.155 During the CO2RR process, the single-atom Ni efficiently reduces CO2 to the intermediate CO, which is immediately converted to ethylene through highly efficient C–C coupling reactions catalyzed by nearby Cu particles. Therefore, compared to the non-tandem catalyst PTF/Cu (triazine framework with no metal Ni atoms in the porphyrin center) that mainly produces methane, the faradaic efficiency of ethylene has increased by fivefold (−1.1 V vs. RHE) (Fig. 8b).
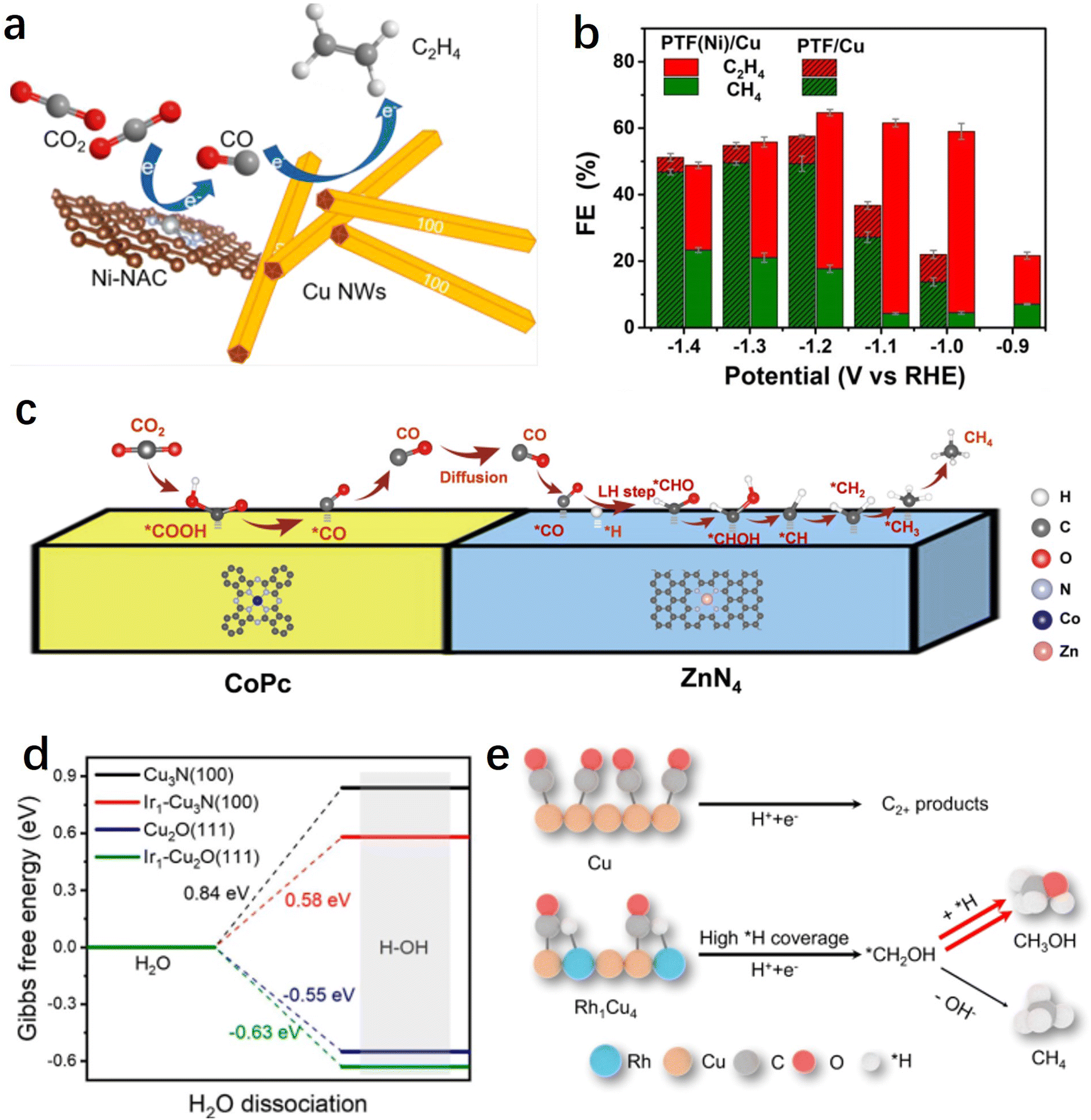 | ||
| Fig. 8 (a) Schematic diagrams of the tandem systems of Cu/Ni-NAC hybrid catalysts.154 (b) Faradaic efficiency of C2H4 and CH4 at different potentials on PTF(Ni)/Cu and PTF/Cu.155 (c) A proposed reaction mechanism for the production of CH4 over CoPc@Zn–N–C.158 (d) Calculated ΔG for the H2O dissociation process.162 (e) Mechanism of CH3OH formation on the Rh1Cu4 catalyst.161 | ||
Potential matching determines the compatibility of different components, while spatial distribution determines the efficiency of the tandem reaction.156 The generated CO on SACs often needs to spill over to adjacent Cu sites, and if the distance between them is too far, the tandem process will be hindered. Wang et al. systematically studied the influence of spatial distribution between two components on the tandem reaction process.157 They chose CoPC molecules, known as excellent CO-producing small molecule catalysts, and Cu nanocubes (Cucub) as a model, and prepared two distinct mixed catalysts. One catalyst was CoPC–Cucub/C, where CoPC and Cucub shared an interface; the other was CoPC–C/Cucub, where CoPC was initially loaded onto carbon black and subsequently mixed with Cucub. Electrochemical tests revealed that CoPC–Cucub/C exhibited nearly double the selectivity for C2 products compared to Cucub/C, whereas the faradaic efficiency of CoPC–C/Cucub decreased by half. This outcome elucidates the surface transport mechanism of CO intermediates between the two components of the tandem catalyst, highlighting the importance of their relative spatial distribution for achieving efficient tandem reactions. In addition to the tandem systems between single atoms and metal Cu, tandem catalytic pathways can also be formed between different single-atom sites. For instance, Lin et al. reported a tandem catalytic system using CoPC and Zn–N–C for the deep hydrogenation reduction of CO2 to produce methane.158 The research results demonstrated that CO2 was initially reduced to CO on CoPC, then diffused to Zn–N–C, where it undergoes further reduction to CH4 at the ZnN4 site (Fig. 8c). Compared to using CoPC or Zn–N–C alone, the tandem catalyst showed more than 100-fold increase in the CH4/CO production rate ratio.
On the reaction pathway of CO2 reduction to CH4, the further hydrogenation of the intermediate *CO is often considered the rate-determining step, while the slow water dissociation kinetics limits the rate of proton supply, thereby impeding the electrochemical methanation of CO2. To address this issue, Chen et al. successfully introduced single-atom Ir into copper-based catalysts, resulting in the synthesis of Ir single-atom-doped Cu3N/CuO hybrid materials.162 This hybrid catalyst exhibits exceptionally high methane selectivity and activity (with a 75% methane Faraday efficiency at a current density of 320 mA cm−2), surpassing catalysts lacking Ir single-atom doping. Experimental and theoretical results indicate that Ir single atoms facilitate the dissociation of water into protons and supply them to Cu3N/CuO sites for the protonation of *CO to *CHO, significantly accelerating the kinetics of CO2 methanation (Fig. 8d). It has been indicated that methanol and methane products share a common intermediate, *CHO. After overcoming the energy barrier of the rate-determining step along the C1 pathway (i.e., *CO-to-*CHO), the critical branching step for generating methanol or methane involves either continued hydrogenation (resulting in *CHOH, *CH2OH) or dehydroxylation (leading to *CH, *CH2, etc.).163,164 Therefore, further optimizing the *H adsorption on the catalyst surface shows promise for enhancing methanol selectivity. Building on this insight, Zhang et al. synthesized a Rh-doped Rh1Cu4 single-atom alloy catalyst.161 DFT results demonstrate that isolated Rh sites enhance *H coverage on the Rh1Cu4 catalyst and facilitate the hydrogenation of *CH2OH, leading to methanol production. The Rh1Cu4 catalyst exhibits the highest methanol partial current density of 111.7 ± 12.8 mA cm−2 with a Faraday efficiency of 46.2 ± 5.3%. Furthermore, comparative experiments reveal that the introduction of Rh clusters results in excessive *H coverage on the catalyst surface, leading to a decrease in methanol selectivity and an increase in hydrogen production (Fig. 8e). This further shows the advantage of single-atom sites as water activation or *H adsorption sites.
In summary, SACs can promote the supply of CO and protons during the CO2RR process by facilitating the generation of CO and the dissociation of water. Subsequently, these intermediate products can be captured by another active site in the tandem catalyst, forming *CO and *Had species, leading to the acceleration of the rate-determining steps involving C–C coupling and PCET.
4.4 Catalyst support regulation
Choosing the appropriate support material is essential to providing strong binding through ion or covalent interactions, thus securely immobilizing single atoms. Additionally, the specific characteristics of the support can strongly influence the electronic states of SACs, further impacting the catalytic performance of the CO2RR. The majority of SACs used for the CO2RR are carbon-based due to their excellent conductivity. Metals, metal oxides, and other supports with unique structural properties and coordination environments can offer more possibilities for tuning the performance of SACs.We are well aware that copper is the only metal that exhibits appreciable activity towards the multi-electron transfer hydrocarbon products. In particular, Cu SACs exhibited distinct performance compared to non-Cu-based SACs in the generation of products such as methane, ethylene, acetone, etc.82 However, Cu SACs usually undergo reconstruction involving atomic re-arrangement and compositional change during the CO2RR process, and this uncontrollable electrochemical reconstruction not only leads to the inherently poor stability of Cu-based SACs, but also makes the investigation of real active sites and the establishment of structure-performance relationships shrouded in fog. Traditional carbon-based supports have shown some limitations in stabilizing atomically dispersed metal Cu.165 Therefore, there is an urgent need to develop novel supports that can stabilize copper-based SACs to advance the field of CO2RRs. Recently, inspired by traditional Cu/SiO2 heterogeneous catalysts, Tan et al. have developed a method to stabilize atomic Cu by constructing a Cu–O–Si interface (Fig. 9a).166 The as-prepared CuSiOx catalyst achieved an efficient electrochemical CO2 methanation with a high faradaic efficiency of CH4 (72.5% at −1.27 V vs. RHE) and excellent stability. To address the issue of poor conductivity of the oxide-based support, the authors also assembled a flow-cell device and achieved a remarkable CO2-to-CH4 conversion rate of 0.22 μmol cm−2 s−1. More importantly, in situ X-ray absorption spectrum experiments indicated that the atomic Cu–O–Si interfacial sites exhibited ultra-high electrochemical stability and reconstruction-resistance over a broad potential window or long-time potentiostatic electrolysis, which attribute to the strong interactions between Cu and silica (Fig. 9b). Using CeO2 as a support to enhance the stability of Cu is also a feasible approach. Zhou et al. reported a CeOx-stabilized Cu2+ strategy (Cu–CeOx) in which CeO2 acts as a self-sacrificing component to protect the atomic Cu2+ active species.167 They found that after CO2RRs, the concentration of Ce3+ in Cu–CeOx increased significantly, as evidenced by in situ Raman spectroscopy experiments, indicating that electrons preferentially reduce Ce4+ to Ce3+ in Cu–CeOx. Meanwhile, the newly formed Ce3+/Ce4+ couple acts as a conductive network to enhance the conductivity of the overall solid solution. It can accelerate electron transfer and suppress the accumulation of electrons at Cu2+ sites, preventing them from self-reduction.
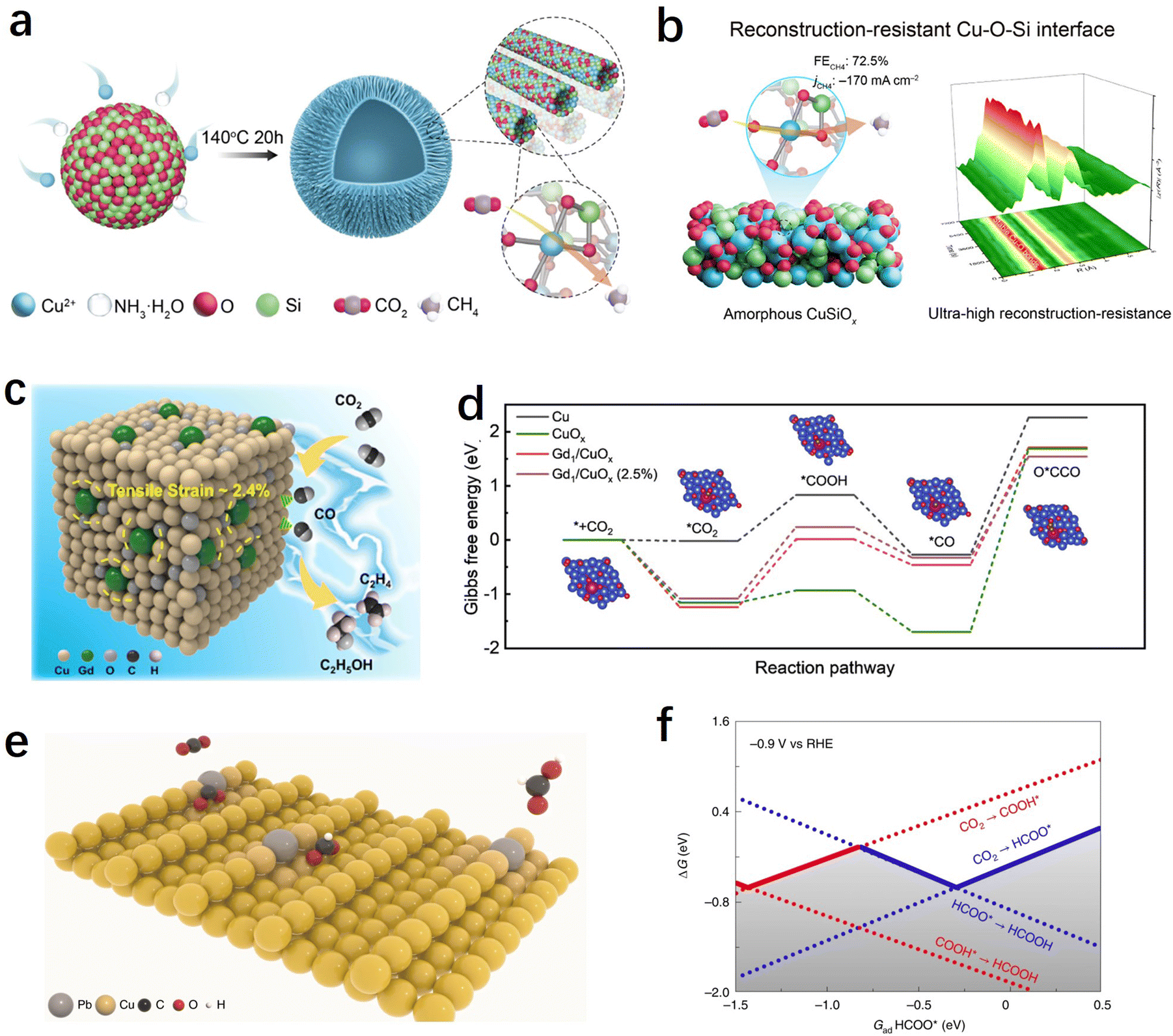 | ||
| Fig. 9 (a) Schematic illustration of the CuSiOx synthetic process.166 (b) The table of contents for the CuSiOx catalyst during the CO2RR (including the schematic illustration and characterization of the reconstruction-resistance, and the CO2RR performance).166 (c) Illustration of C2+ product generation promoted by stress engineering in Gd/CuOx.171 (d) Gibbs free energy diagrams of CO2 reduction to O*CCO on surfaces of Cu, CuOx, Gd1/CuOx, and Gd1/CuOx.171 (e) Schematic illustration of CO2 conversion into HCOOH over Pb1Cu single-atom alloys.38 (f) One-dimensional RPD for the CO2RR. The solid lines show the GRPD-limiting steps for the more favoured COOH* path and the HCOO* path.38 | ||
Oxygen vacancies or Lewis acid sites on metal oxides can also be utilized to modulate the activity of SACs. For instance, Wang et al. found that a single Cu atom substituted on the CeO2(110) surface could stably enrich three oxygen vacancies around each Cu site, leading to an efficient CO2 adsorption and activation center that promotes methane production.106 Chen et al. have demonstrated through theoretical predictions that the Lewis acid sites in metal oxides such as Al2O3 and Cr2O3 not only facilitate the activation of CO2 but also lower the energy barrier for the *CH4O intermediate, providing a favourable pathway for methane production.168 Based on these findings, the authors synthesized Cu SACs supported on ultrathin Al2O3 nanosheets with strong Lewis acid sites. The catalyst exhibited an excellent electrochemical CO2–CH4 conversion performance, with a methane faradaic efficiency of 62% at −1.2 V vs. RHE. Yin et al. demonstrated that strong binding sites for CO2 adsorption can be formed at the Cu–CeO2 interface, promoting CO2 activation and conversion into *CO intermediates on the nearby Cu surface.169 As the Cu loading increases, the CO2 reduction product can be tailored from C1 to C2+.
Using Cu-based materials as supports (such as metallic copper or copper oxides) and introducing atomically dispersed heteroatoms into Cu is a highly effective approach for modulating the activity of Cu-based catalysts. Previous researches have indicated that Cu+ sites played a crucial role in improving the selectivity of C2+ products.170 It can not only enhance the CO adsorption and facilitate C–C coupling reactions but also stabilize C2 intermediates. However, Cu+ was found to be unstable under negative potentials during the CO2RR process. Feng et al. synthesized Gd-doped CuOx catalyst (Gd1/CuOx) using a simple one-step solvent thermal method.171 The research findings demonstrated that atomically dispersed Gd can stabilize Cu+ and induce tensile strain in the catalyst. The prepared Gd1/CuOx exhibited excellent performance in CO2-to-C2+ product conversion, achieving a faradaic efficiency of 81.4% for C2+ products at −0.8 V. Experimental and computational results suggested that Gd doping improved the stability of the crucial intermediate O*CCO, reduced the energy barrier for C–C coupling, and resulted in high CO2RR-to-C2+ product performance (Fig. 9c and d). Zheng et al. synthesized a Pb-alloyed Cu catalyst, denoted as Pb1Cu, by incorporating Pb single atoms onto the surface of metallic copper (Fig. 9e).38 The introduction of Pb had a significant impact on the protonation process during the CO2RR. Rather than occurring on the oxygen site, the initial protonation was observed to preferentially take place on the carbon site of CO2 when Pb was present. This led to the formation of the *HCOO pathway instead of the *COOH pathway, resulting in Pb1Cu demonstrating exceptional formate activity with an ultra-high faradaic efficiency of approximately 96% at current densities exceeding 1 A cm−2 (Fig. 9f). Moreover, the researchers utilized the Pb1Cu catalyst to construct a solid electrolyte reactor, enabling the continuous production of pure formic acid solution for a remarkable duration of 180 hours, while maintaining a high current density above 100 mA cm−2. These findings highlight the potential of Pb1Cu as a promising catalyst for the CO2RR and its application in the efficient and sustained production of formic acid.
In conclusion, adjusting the type, composition, and structure of the supports can optimize the electronic and geometric properties of SACs, enhancing the catalytic activity and stability. This holds significant importance for the efficient, stable, and sustainable development of SACs for the CO2RR.
5. Conclusions and perspectives
The unique geometric/electronic structures of SACs qualify them as a new family of catalytic materials and a new frontier in the research field of CO2RRs. In this review, we introduced the research status of SACs and summarized the synthesis strategies for SACs. Furthermore, we briefly introduced the fundamental parameters in the CO2RR and attempted to summarize the possible reaction mechanisms for generating C1 and C2 products in the CO2RR. Most importantly, we focused on discussing the structure–function relationship between the structure of SACs and the performance of CO2RR, mainly manifested in the following four aspects of regulation: coordination environment regulation, diatomic sites regulation, tandem catalytic regulation, and catalyst support regulation. Although significant progress has been made in the synthesis of SACs as well as in the application of SACs in the CO2RR and the establishment of their structure–function relationships, there are still certain limitations or challenges to the practical application of SACs in CO2RRs. Summarized below are some potential research directions and challenges for SACs in CO2RR applications in the future (Fig. 10). | ||
| Fig. 10 Schematic illustration of future directions and challenges of SACs for the CO2RR. | ||
1. Precise synthesis and characterization of SACs
The precise synthesis of SACs has always been a challenge for their application in various fields, including the CO2RR. Metal single atoms are often fixed by the coordination of heteroatoms on the support owing to their high surface free energy. At present, the preparation of SACs often resorts to high-temperature calcination steps, which may lead to a certain degree of randomness in the generation of SACs. Many studies have shown that minor alterations in calcination temperature can lead to major changes in the coordination environment of SACs, and needless to say, their catalytic activity also changes accordingly. Therefore, when a high-temperature calcination step is used to prepare SACs, strict control of calcination conditions is necessary to ensure the repeatability of the preparation of different batches of catalysts. Obviously, a more precise strategy for preparing SACs has always been pursued, especially for the preparation of high-loading SACs, which currently faces greater difficulties. In addition, as the research on SACs in different applications deepens, there is increasing interest on multi-site catalysts (such as DACs). However, research on DACs is currently still in its early stages, and many basic concepts and standards have yet to be established. For instance, the characterization of SACs mainly depends on AC-HAADF-STEM and XAS technologies. AC-HAADF-STEM records the projected images of metal single atoms under the incident electron beam, and cannot distinguish the spatial arrangement of metal single atoms in close proximity; on the other hand, XAS records the average signal of the test sample, and cannot separate the information of a single active site. The limitations of these characterization techniques are particularly prominent in the analysis of DACs. Especially in the AC-HAADF-STEM images of DACs, the spatial arrangement of adjacent metal atoms cannot be determined by simply reading the high-contrast bright spots in the projected image while ignoring their spatial relationship along the incident electron beam. In this regard, one of our recent works attempts to identify “true/false” DACs by using in situ rotation operation in AC-HAADF-STEM characterization.172 Therefore, more precise synthesis strategies and more advanced characterization methods need to be further explored, as the precise synthesis and characterization of SACs are prerequisites for understanding their structure–function relationship.2. Rational design of SACs for producing non-CO products
As summarized in this review, considerably satisfactory CO selectivity has been achieved from the CO2RR over a number of SACs, such as Fe, Ni, Mn, and Ag, but it still poses a great challenge for high-value-added products beyond CO. There are two main reasons for this: (1) on the one hand, a single metal active site has moderate binding strength to the intermediate of CO2RR, especially *COOH and *CO. Thus, *CO has a moderate desorption energy barrier, which is beneficial for the production of CO. (2) On the other hand, the atomically dispersed metal active sites are isolated from each other, leading to the physical separation of the intermediates, thus blocking further C–C coupling. Owing to its complex reaction mechanism and diverse reaction pathways, it is rather difficult to precisely control the distribution of the reaction products in the CO2RR. In order to expand the product range of the CO2RR and to obtain high-value-added products, two promising strategies could be explored. One is to rationally design DACs with multiple reaction active sites, which can provide mutually related synergistic active sites. The interrelated multiple active sites could provide a heteroatomic interface for asymmetric C–C coupling, which is expected to yield specific multi-carbon (C2+) products. Another effective strategy is to construct tandem catalytic systems. The general principle is to combine SACs with high CO selectivity with Cu-based catalysts to achieve continuous conversion of CO2 to CO and further to multi-carbon products, thus achieving high C2+ Faraday efficiencies. This method can promote the transfer of reaction intermediates and electrons, thereby improving reaction efficiency and selectivity. In addition, the tandem catalytic system lessens the difficulties in the catalyst design, and makes the catalytic reaction more controllable and predictable.3. Deciphering the catalytic mechanisms of CO2RRs
At present, significant progress has been made in both the synthesis of SACs and their application in CO2RRs. However, owing to the diverse reaction pathways and complex reaction intermediates, the dynamic process and reaction mechanism of the CO2RR are still difficult to understand. The catalytic activity of the CO2RR is highly dependent on its active sites; therefore, the structure of the active site and its dynamic evolution during the catalytic reaction process are crucial. However, conventional characterization methods often detect the state of catalysts or catalytic reactions at a certain point in time but cannot monitor their dynamic evolution process. Therefore, it would be very helpful to use in situ technologies to identify the dynamic evolution of active sites and capture catalytic reaction intermediates, but this also requires more stringent test conditions. Currently, in situ tests are often conducted separately, such as in situ XAS, in situ TEM, and in situ Raman. It is expected that more advanced coupled in situ test techniques could be developed in the future to facilitate in-depth exploration of reaction mechanisms. In addition, machine learning has made significant progress in the preparation of nanomaterials and its application in the CO2RR, and it is expected to be introduced into the construction and performance prediction of SACs in the future. Machine learning can achieve high-throughput material performance prediction and assist in analyzing catalytic mechanisms, thus greatly reducing the trial-and-error costs.4. Industrial-level applications
The industrial application of SACs in the CO2RR is mainly focused on two aspects: large-scale synthesis of SACs and industrial-level performance of SACs in the CO2RR. At present, a number of methods have been developed in the laboratory for the preparation of SACs, but a significant proportion of these methods are rather complex and cumbersome. The complex or harsh preparation conditions would result in poor repeatability of SACs preparation, thereby limiting its large-scale production. Moreover, apart from considerations regarding its repeatability, large-scale production of SACs also needs to balance economic benefits, which drives the preparation of SACs towards simpler, more efficient, and more reliable directions. On the other hand, in addition to achieving industrial-level preparation, SACs also need to meet industrial-level test conditions and performance indicators. As we already know, the low solubility of CO2 in aqueous electrolytes greatly constrains its mass transport at high current densities, which is also a bottleneck for the industrialization of the CO2RR. To address these issues, it has been attempted to combine SACs with gas diffusion layers and the assembly of membrane electrode components in flow cells, which helps to meet the requirements of industrial-level reactions and has made some notable progress thus far. However, reactors often fail during operation owing to various problems such as catalyst aggregation and membrane flooding, and these issues still need to be addressed one by one in future device design. In addition, the stability of SACs is also one of the evaluation metrics for their potentials of industrialization. Currently, the research on SACs in the CO2RR is mainly focused on catalytic activity and selectivity, while stability tests often fail to meet the industrial requirements. Therefore, it is necessary to strengthen the research on catalyst stability in the future, to explore the deactivation mechanism of catalysts, and to design efficient SACs that feature balanced activity, selectivity, and stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFF0500503), the National Natural Science Foundation of China (21925202, U22B2071), and the International Joint Mission On Climate Change and Carbon Neutrality.Notes and references
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2016, 16, 16–22 CrossRef PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS.
- D. R. Feldman, W. D. Collins, P. J. Gero, M. S. Torn, E. J. Mlawer and T. R. Shippert, Nature, 2015, 519, 339–343 CrossRef CAS PubMed.
- H.-Y. Kuang, Y.-X. Lin, X.-H. Li and J.-S. Chen, J. Mater. Chem. A, 2021, 9, 20857–20873 RSC.
- A. Seiff and D. B. Kirk, J. Geophys. Res., 1977, 82, 4364–4378 CrossRef CAS.
- V. G. Dovì, F. Friedler, D. Huisingh and J. J. Klemeš, J. Cleaner Prod., 2009, 17, 889–895 CrossRef.
- Z. Yuan, M. R. Eden and R. Gani, Ind. Eng. Chem. Res., 2015, 55, 3383–3419 CrossRef.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- H. Herring, Energy, 2006, 31, 10–20 CrossRef.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- M. Bevilacqua, J. Filippi, H. A. Miller and F. Vizza, Energy Technol., 2015, 3, 197–210 CrossRef CAS.
- Y. Li, S. H. Chan and Q. Sun, Nanoscale, 2015, 7, 8663–8683 RSC.
- W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, e2001848 CrossRef.
- H. Gu, J. Wu and L. Zhang, Nano Res., 2022, 15, 9747–9763 CrossRef CAS.
- T. N. Nguyen, M. Salehi, Q. V. Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- X. Tan, Z. Zhuang, Y. Zhang, K. Sun and C. Chen, Chem. Commun., 2023, 59, 2682–2696 RSC.
- S. Wang, L. Wang, D. Wang and Y. Li, Energy Environ. Sci., 2023, 16, 2759–2803 RSC.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- Y. Wang, Y. Liu, W. Liu, J. Wu, Q. Li, Q. Feng, Z. Chen, X. Xiong, D. Wang and Y. Lei, Energy Environ. Sci., 2020, 13, 4609–4624 RSC.
- S. G. Han, D. D. Ma and Q. L. Zhu, Small Methods, 2021, 5, e2100102 CrossRef.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- Y. Cheng, S. Yang, S. P. Jiang and S. Wang, Small Methods, 2019, 3, 1800440 CrossRef.
- C. Cao, D. D. Ma, J. Jia, Q. Xu, X. T. Wu and Q. L. Zhu, Adv. Mater., 2021, 33, e2008631 CrossRef.
- C. J. Peng, G. Zeng, D. D. Ma, C. Cao, S. Zhou, X. T. Wu and Q. L. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 20589–20597 CrossRef CAS PubMed.
- Y. Zhao, X. Tan, W. Yang, C. Jia, X. Chen, W. Ren, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2020, 59, 21493–21498 CrossRef CAS PubMed.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- D. Zhong, Z. J. Zhao, Q. Zhao, D. Cheng, B. Liu, G. Zhang, W. Deng, H. Dong, L. Zhang, J. Li, J. Li and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 4879–4885 CrossRef CAS.
- W. Zhang, Q. Qin, L. Dai, R. Qin, X. Zhao, X. Chen, D. Ou, J. Chen, T. T. Chuong, B. Wu and N. Zheng, Angew. Chem., Int. Ed., 2018, 57, 9475–9479 CrossRef CAS.
- A. Eilert, F. S. Roberts, D. Friebel and A. Nilsson, J. Phys. Chem. Lett., 2016, 7, 1466–1470 CrossRef CAS PubMed.
- L.-P. Yuan, W.-J. Jiang, X.-L. Liu, Y.-H. He, C. He, T. Tang, J. Zhang and J.-S. Hu, ACS Catal., 2020, 10, 13227–13235 CrossRef CAS.
- Z. Chen, M.-R. Gao, N. Duan, J. Zhang, Y.-Q. Zhang, T. Fan, J. Zhang, Y. Dong, J. Li, Q. Liu, X. Yi and J.-L. Luo, Appl. Catal., B, 2020, 277, 119252 CrossRef CAS.
- T. Yuan, Z. Hu, Y. Zhao, J. Fang, J. Lv, Q. Zhang, Z. Zhuang, L. Gu and S. Hu, Nano Lett., 2020, 20, 2916–2922 CrossRef CAS PubMed.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C. W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS.
- H. Xie, Y. Wan, X. Wang, J. Liang, G. Lu, T. Wang, G. Chai, N. M. Adli, C. Priest, Y. Huang, G. Wu and Q. Li, Appl. Catal., B, 2021, 289, 119783 CrossRef CAS.
- T. Shen, S. Wang, T. Zhao, Y. Hu and D. Wang, Adv. Energy Mater., 2022, 12, 2201823 CrossRef CAS.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS.
- M. E. Ahmed, S. Adam, D. Saha, J. Fize, V. Artero, A. Dey and C. Duboc, ACS Energy Lett., 2020, 5, 3837–3842 CrossRef CAS.
- J. Xu, S. Lai, M. Hu, S. Ge, R. Xie, F. Li, D. Hua, H. Xu, H. Zhou, R. Wu, J. Fu, Y. Qiu, J. He, C. Li, H. Liu, Y. Liu, J. Sun, X. Liu and J. Luo, Small Methods, 2020, 4, 2000567 CrossRef CAS.
- Z. Chen, X. Zhang, M. Jiao, K. Mou, X. Zhang and L. Liu, Adv. Energy Mater., 2020, 10, 1903664 CrossRef CAS.
- X. Ma, J. Du, H. Sun, F. Ye, X. Wang, P. Xu, C. Hu, L. Zhang and D. Liu, Appl. Catal., B, 2021, 298, 120543 CrossRef CAS.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew. Chem., Int. Ed., 2020, 59, 11123–11129 CrossRef CAS PubMed.
- W. Zhu and C. Chen, Chem, 2019, 5, 2737–2739 CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- S. Zhang, X. Ao, J. Huang, B. Wei, Y. Zhai, D. Zhai, W. Deng, C. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 9691–9698 CrossRef CAS PubMed.
- N. Fu, X. Liang, Z. Li, W. Chen, Y. Wang, L. Zheng, Q. Zhang, C. Chen, D. Wang, Q. Peng, L. Gu and Y. Li, Nano Res., 2020, 13, 947–951 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- Q. Li, Z. Li, Q. Zhang, L. Zheng, W. Yan, X. Liang, L. Gu, C. Chen, D. Wang, Q. Peng and Y. Li, Nano Res., 2020, 14, 1435–1442 CrossRef.
- J. Yang, W. H. Li, S. Tan, K. Xu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19085–19091 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158–159 RSC.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- H. Liu, H. Rong and J. Zhang, ChemSusChem, 2022, 15, e202200498 CrossRef CAS.
- Q. An, J. Jiang, W. Cheng, H. Su, Y. Jiang and Q. Liu, Small Methods, 2022, 6, e2200408 CrossRef PubMed.
- Y. Ying, X. Luo, J. Qiao and H. Huang, Adv. Funct. Mater., 2020, 31, 2007423 CrossRef.
- Y. Hu, Z. Li, B. Li and C. Yu, Small, 2022, 18, e2203589 CrossRef.
- Y. N. Gong, C. Y. Cao, W. J. Shi, J. H. Zhang, J. H. Deng, T. B. Lu and D. C. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202215187 CrossRef CAS PubMed.
- W. Zhang, Y. Chao, W. Zhang, J. Zhou, F. Lv, K. Wang, F. Lin, H. Luo, J. Li, M. Tong, E. Wang and S. Guo, Adv. Mater., 2021, 33, e2102576 CrossRef PubMed.
- F. Pan and Y. Yang, Energy Environ. Sci., 2020, 13, 2275–2309 RSC.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- R. Kortlever, J. Shen, K. J. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2019, 10, 1754–1768 CrossRef.
- S. Vijay, W. Ju, S. Brückner, S.-C. Tsang, P. Strasser and K. Chan, Nat. Catal., 2021, 4, 1024–1031 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS.
- Z. Pan, K. Wang, K. Ye, Y. Wang, H.-Y. Su, B. Hu, J. Xiao, T. Yu, Y. Wang and S. Song, ACS Catal., 2020, 10, 3871–3880 CrossRef CAS.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef.
- T. T. H. Hoang, S. Ma, J. I. Gold, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2017, 7, 3313–3321 CrossRef CAS.
- B. Zhang, B. Zhang, Y. Jiang, T. Ma, H. Pan and W. Sun, Small, 2021, 17, e2101443 CrossRef PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 131, 15242–15247 CrossRef.
- P. Shao, W. Zhou, Q. L. Hong, L. Yi, L. Zheng, W. Wang, H. X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687–16692 CrossRef CAS PubMed.
- Q. Liu, J. Ma and C. Chen, Chin. J. Catal., 2022, 43, 898–912 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H. L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- Y. Hou, Y.-L. Liang, P.-C. Shi, Y.-B. Huang and R. Cao, Appl. Catal., B, 2020, 271, 118929 CrossRef CAS.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS.
- Q. Cao, L.-L. Zhang, C. Zhou, J.-H. He, A. Marcomini and J.-M. Lu, Appl. Catal., B, 2021, 294, 120238 CrossRef CAS.
- Y. Zhou, L. Chen, L. Sheng, Q. Luo, W. Zhang and J. Yang, Nano Res., 2022, 15, 7994–8000 CrossRef CAS.
- Y. Zhu, X. Yang, C. Peng, C. Priest, Y. Mei and G. Wu, Small, 2021, 17, e2005148 CrossRef.
- S. Chen, W. H. Li, W. Jiang, J. Yang, J. Zhu, L. Wang, H. Ou, Z. Zhuang, M. Chen, X. Sun, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202114450 CrossRef CAS.
- Y. S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed.
- S. Mallakpour, F. Sirous and C. M. Hussain, Top. Curr. Chem., 2021, 380, 7 CrossRef.
- Q. Hao, H.-x Zhong, J.-z Wang, K.-h Liu, J.-m Yan, Z.-h Ren, N. Zhou, X. Zhao, H. Zhang, D.-x Liu, X. Liu, L.-w Chen, J. Luo and X.-b Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef.
- J. Fonseca, T. Gong, L. Jiao and H.-L. Jiang, J. Mater. Chem. A, 2021, 9, 10562–10611 RSC.
- Y. Yun, H. Zeng, L. Li, H. Li, S. Cheng, N. Sun, M. Li, H. Sheng, S. Hu, T. Yao and M. Zhu, Adv. Mater., 2023, 35, e2209561 CrossRef PubMed.
- B. Xu, H. Wang, W. Wang, L. Gao, S. Li, X. Pan, H. Wang, H. Yang, X. Meng, Q. Wu, L. Zheng, S. Chen, X. Shi, K. Fan, X. Yan and H. Liu, Angew. Chem., Int. Ed., 2019, 131, 4965–4970 CrossRef.
- Z. Li, Y. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C. T. He, C. Peng, W. C. Cheong, Q. Li, M. Zhang, Z. Chen, N. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X. M. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- X. Jiang, H. Li, J. Xiao, D. Gao, R. Si, F. Yang, Y. Li, G. Wang and X. Bao, Nano Energy, 2018, 52, 345–350 CrossRef CAS.
- Z. Li, D. He, X. Yan, S. Dai, S. Younan, Z. Ke, X. Pan, X. Xiao, H. Wu and J. Gu, Angew. Chem., Int. Ed., 2020, 132, 18731–18736 CrossRef.
- N. Mohd Adli, W. Shan, S. Hwang, W. Samarakoon, S. Karakalos, Y. Li, D. A. Cullen, D. Su, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2020, 133, 1035–1045 CrossRef.
- G. Hai, X. Xue, S. Feng, Y. Ma and X. Huang, ACS Catal., 2022, 12, 15271–15281 CrossRef CAS.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- C. Jia, S. Li, Y. Zhao, R. K. Hocking, W. Ren, X. Chen, Z. Su, W. Yang, Y. Wang, S. Zheng, F. Pan and C. Zhao, Adv. Funct. Mater., 2021, 31, 2107072 CrossRef CAS.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, e1808135 CrossRef PubMed.
- H. Zou, G. Zhao, H. Dai, H. Dong, W. Luo, L. Wang, Z. Lu, Y. Luo, G. Zhang and L. Duan, Angew. Chem., Int. Ed., 2023, 62, e202217220 CrossRef CAS PubMed.
- G. Shi, Y. Xie, L. Du, X. Fu, X. Chen, W. Xie, T. B. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203569 CrossRef CAS PubMed.
- J. Jiao, R. Lin, S. Liu, W. C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S. F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS.
- Z. Ma, T. Wan, D. Zhang, J. A. Yuwono, C. Tsounis, J. Jiang, Y. H. Chou, X. Lu, P. V. Kumar, Y. H. Ng, D. Chu, C. Y. Toe, Z. Han and R. Amal, ACS Nano, 2023, 17, 2387–2398 CrossRef CAS PubMed.
- M. B. Ross, C. T. Dinh, Y. Li, D. Kim, P. De Luna, E. H. Sargent and P. Yang, J. Am. Chem. Soc., 2017, 139, 9359–9363 CrossRef CAS.
- N. Xuan, J. Chen, J. Shi, Y. Yue, P. Zhuang, K. Ba, Y. Sun, J. Shen, Y. Liu, B. Ge and Z. Sun, Chem. Mater., 2018, 31, 429–435 CrossRef.
- Z. Wang, J. Yang, J. Cao, W. Chen, G. Wang, F. Liao, X. Zhou, F. Zhou, R. Li, Z. Q. Yu, G. Zhang, X. Duan and Y. Wu, ACS Nano, 2020, 14, 6164–6172 CrossRef CAS PubMed.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- Z. Yang, B. Chen, W. Chen, Y. Qu, F. Zhou, C. Zhao, Q. Xu, Q. Zhang, X. Duan and Y. Wu, Nat. Commun., 2019, 10, 3734 CrossRef PubMed.
- B. Wang, X. Zhu, X. Pei, W. Liu, Y. Leng, X. Yu, C. Wang, L. Hu, Q. Su, C. Wu, Y. Yao, Z. Lin and Z. Zou, J. Am. Chem. Soc., 2023, 145, 13788–13795 CrossRef CAS PubMed.
- J. Yang, Z. Qiu, C. Zhao, W. Wei, W. Chen, Z. Li, Y. Qu, J. Dong, J. Luo, Z. Li and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 14095–14100 CrossRef CAS PubMed.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS.
- S. Wei, A. Li, J. C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W. C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- C. Zhao, Y. Wang, Z. Li, W. Chen, Q. Xu, D. He, D. Xi, Q. Zhang, T. Yuan, Y. Qu, J. Yang, F. Zhou, Z. Yang, X. Wang, J. Wang, J. Luo, Y. Li, H. Duan, Y. Wu and Y. Li, Joule, 2019, 3, 584–594 CrossRef CAS.
- Y. Qu, L. Wang, Z. Li, P. Li, Q. Zhang, Y. Lin, F. Zhou, H. Wang, Z. Yang, Y. Hu, M. Zhu, X. Zhao, X. Han, C. Wang, Q. Xu, L. Gu, J. Luo, L. Zheng and Y. Wu, Adv. Mater., 2019, 31, e1904496 CrossRef.
- H. Yang, Q. Lin, Y. Wu, G. Li, Q. Hu, X. Chai, X. Ren, Q. Zhang, J. Liu and C. He, Nano Energy, 2020, 70, 104454 CrossRef CAS.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. T. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed.
- H. Chung, D. Cullen, D. Higgins, B. Sneed, E. Holby, K. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- Y. Li, C. Chen, R. Cao, Z. Pan, H. He and K. Zhou, Appl. Catal., B, 2020, 268, 118747 CrossRef CAS.
- U. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, 1805623 CrossRef.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- R. Chen, J. Zhao, Y. Li, Y. Cui, Y. Lu, S. Hung, S. Wang, W. Wang, G. Huo, Y. Zhao, W. Liu, J. Wang, H. Xiao, X. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2023, 145, 20683–20691 CrossRef CAS PubMed.
- K. Li, W. Wang, H. Zheng, X. Wang, Z. Xie, L. Ding, S. Yu, Y. Yao and F. Zhang, Mater. Today Phys., 2021, 19, 100427 CrossRef CAS.
- D. Zhao, K. Yu, P. Song, W. Feng, B. Hu, W.-C. Cheong, Z. Zhuang, S. Liu, K. Sun, J. Zhang and C. Chen, Energy Environ. Sci., 2022, 15, 3795–3804 RSC.
- Z. Zhang, J. Zhu, S. Chen, W. Sun and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215136 CrossRef CAS.
- H. Xiao, W. A. Goddard, 3rd, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed.
- C. Liu, Y. Wu, K. Sun, J. Fang, A. Huang, Y. Pan, W.-C. Cheong, Z. Zhuang, Z. Zhuang, Q. Yuan, H. L. Xin, C. Zhang, J. Zhang, H. Xiao, C. Chen and Y. Li, Chem, 2021, 7, 1297–1307 CAS.
- K. Sun, K. Yu, J. Fang, Z. Zhuang, X. Tan, Y. Wu, L. Zeng, Z. Zhuang, Y. Pan and C. Chen, Adv. Mater., 2022, 34, e2206478 CrossRef PubMed.
- J. Masana, J. Xiao, H. Zhang, X. Lu, M. Qiu and Y. Yu, Appl. Catal., B, 2023, 323, 122199 CrossRef CAS.
- Z. Chen, A. Huang, K. Yu, T. Cui, Z. Zhuang, S. Liu, J. Li, R. Tu, K. Sun, X. Tan, J. Zhang, D. Liu, Y. Zhang, P. Jiang, Y. Pan, C. Chen, Q. Peng and Y. Li, Energy Environ. Sci., 2021, 14, 3430–3437 RSC.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- J. R. Huang, X. F. Qiu, Z. H. Zhao, H. L. Zhu, Y. C. Liu, W. Shi, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202210985 CrossRef CAS.
- J.-X. Peng, W. Yang, Z. Jia, L. Jiao and H.-L. Jiang, Nano Res., 2022, 15, 10063–10069 CrossRef CAS.
- C. Chen, Z. Chen, J. Zhong, X. Song, D. Chen, S. Liu, W. Cheong, J. Li, X. Tan, C. He, J. Zhang, D. Liu, Q. Yuan, C. Chen, Q. Peng and Y. Li, Nano Res., 2023, 16, 4211–4218 CrossRef CAS.
- X. Liu, Y. Liu, W. Yang, X. Feng and B. Wang, Chemistry, 2022, 28, e202201471 CrossRef CAS PubMed.
- B. Huang, S. Huang, C. Lu, L. Li, J. Chen, T. Hu, D. Lützenkirchen-Hecht, K. Yuan, X. Zhuang and Y. Chen, CCS Chem., 2022, 1–12, DOI:10.31635/ccschem.022.202202241.
- N. Qiu, J. Li, H. Wang and Z. Zhang, Sci. China Mater., 2022, 65, 3302–3323 CrossRef CAS.
- S. Li, A. Guan, C. Yang, C. Peng, X. Lv, Y. Ji, Y. Quan, Q. Wang, L. Zhang and G. Zheng, ACS Mater. Lett., 2021, 3, 1729–1737 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- J. D. Yi, X. Gao, H. Zhou, W. Chen and Y. Wu, Angew. Chem., Int. Ed., 2022, 61, e202212329 CrossRef CAS PubMed.
- Y. Pan, C. Liu, N. Zhang, M. Li, M. Wang, X. Yang, H.-C. Chen, Y. Zhang, W. Hu, W. Yan, H. M. Chen, S. Liu, H. Xiao, J. Li and C. Chen, Chem Catal., 2023, 3, 100610 CrossRef CAS.
- Z. Liang, L. Song, M. Sun, B. Huang and Y. Du, Nano Res., 2023, 16, 8757–8764 CrossRef CAS.
- W. Zheng, X. Yang, Z. Li, B. Yang, Q. Zhang, L. Lei and Y. Hou, Angew. Chem., Int. Ed., 2023, e202307283 CAS.
- F. Li, Y. Li, Z. Wang, J. Li, D. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. Gabardo, C. Dinh, Y. Wang, T. Zhuang, D. Sinton and E. Sargent, Nat. Catal, 2020, 3, 75–82 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero, T. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- Z. Yin, J. Yu, Z. Xie, S. W. Yu, L. Zhang, T. Akauola, J. G. Chen, W. Huang, L. Qi and S. Zhang, J. Am. Chem. Soc., 2022, 144, 20931–20938 CrossRef CAS.
- D. L. Meng, M. D. Zhang, D. H. Si, M. J. Mao, Y. Hou, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 25485–25492 CrossRef CAS PubMed.
- B. Cao, F.-Z. Li and J. Gu, ACS Catal., 2022, 12, 9735–9752 CrossRef CAS.
- M. Wang, A. Loiudice, V. Okatenko, I. D. Sharp and R. Buonsanti, Chem. Sci., 2023, 14, 1097–1104 RSC.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- H. Deng, C. Guo, P. Shi and G. Zhao, ChemCatChem, 2021, 13, 4325–4333 CrossRef CAS.
- T. Zhang, B. Yuan, W. Wang, J. He and X. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202302096 CrossRef CAS.
- J. Zhang, P. Yu, C. Peng, X. Lv, Z. Liu, T. Cheng and G. Zheng, ACS Catal., 2023, 13, 7170–7177 CrossRef CAS.
- S. Chen, Z. Zhang, W. Jiang, S. Zhang, J. Zhu, L. Wang, H. Ou, S. Zaman, L. Tan, P. Zhu, E. Zhang, P. Jiang, Y. Su, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 12807–12815 CrossRef CAS PubMed.
- H. Xiao, T. Cheng and W. Goddard, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. Goddard, J. Phys. Chem. Lett., 2015, 6, 4767–4773 CrossRef CAS PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvao Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS.
- X. Tan, K. Sun, Z. Zhuang, B. Hu, Y. Zhang, Q. Liu, C. He, Z. Xu, C. Chen, H. Xiao and C. Chen, J. Am. Chem. Soc., 2023, 145, 8656–8664 CAS.
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed.
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou, Y. Q. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 7325–7331 CrossRef CAS PubMed.
- J. Yin, Z. Gao, F. Wei, C. Liu, J. Gong, J. Li, W. Li, L. Xiao, G. Wang, J. Lu and L. Zhuang, ACS Catal., 2022, 12, 1004–1011 CrossRef CAS.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed.
- J. Feng, L. Wu, S. Liu, L. Xu, X. Song, L. Zhang, Q. Zhu, X. Kang, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 9857–9866 CrossRef CAS PubMed.
- C. Chen, Y. Li, A. Huang, X. Liu, J. Li, Y. Zhang, Z. Chen, Z. Zhuang, Y. Wu, W. Cheong, X. Tan, K. SUn, Z. Xu, D. Liu, Z. Wang, K. Zhou and C. Chen, J. Am. Chem. Soc., 2023, 145, 21273–21283 CrossRef CAS PubMed.
Footnote |
| † Chang Chen, Jiazhan Li, and Xin Tan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |